
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanochemical synthesis and application of mixed-metal copper–ruthenium HKUST-1 metal–organic frameworks in the electrocatalytic oxygen evolution reaction†
Linda
Sondermann
a,
Quentin
Smith
ab,
Till
Strothmann
 a,
Annette
Vollrath
a,
Thi Hai
Yen Beglau
a and
Christoph
Janiak
*a
a,
Annette
Vollrath
a,
Thi Hai
Yen Beglau
a and
Christoph
Janiak
*a
aInstitut für Anorganische Chemie und Strukturchemie, Heinrich-Heine-Universität Düsseldorf, 40204 Düsseldorf, Germany. E-mail: janiak@uni-duesseldorf.de
bDepartment of Chemistry, Virginia Polytechnic Institute and State University, 24061 Blacksburg, USA
First published on 14th May 2024
Abstract
Novel electrode materials for electrocatalytic hydrogen generation are investigated for increasing the activity of expensive noble-metal components. Here various mixed-metal copper–ruthenium combinations of the metal–organic framework (MOF) HKUST-1 (HKUST = Hong Kong University of Science and Technology, with the formula [Cu3(BTC)2(H2O)3]n (BTC = benzene-1,3,5-tricarboxylate)) as CuxRu-BTC were synthesized through a mechanochemical method. This mechanochemical method allowed for gram-scale synthesis of the mixed-metal MOFs in a one-hour synthesis time. Characterization through powder X-ray diffraction (PXRD), N2-adsorption, scanning electron microscopy (SEM), thermogravimetric analysis (TGA), and Fourier transform infrared (FT-IR) spectroscopy confirmed the formation of a MOF with the HKUST-1 topology, albeit with lower porosity compared to neat HKUST-1. The synthesized MOFs were tested as precursor materials for catalysts for the oxygen evolution reaction (OER) and performed comparably to the industry standard ruthenium oxide (RuO2). An overpotential (η) of 314 mV (RuO2η = 312 mV), a Tafel slope (b) of 55 mV dec−1 (RuO2b = 47 mV dec−1) was achieved which in combination with a charge-transfer resistance (RCT) of 13.6 Ω (RuO2RCT = 52.8 Ω) and a faradaic efficiency (FE) of 70% (RuO2 FE = 66%) supports the derived catalyst from Cu10Ru-BTC with an intimate mixture of copper and ruthenium at the nanoscale to be effective for the OER having lower ruthenium content than RuO2. All derived catalysts from the CuxRu-BTC samples and RuO2 showed good stability in a chronopotentiometric measurement over 12 h.
Introduction
Global warming has heavily influenced the research and development of renewable energy sources like wind and solar, making them more efficient and affordable, with solar energy alone seeing an increase in efficiency from 10% in 2010 to over 23% in 2023.1,2 However, the intermittent nature of renewable energy sources causes fluctuations in energy production and makes it difficult to meet the energy demands at times.3 It is clear then that excess clean energy must be stored for later use when production does not reach demand. The definition of green hydrogen has arisen in response to these recent developments. Hydrogen is capable of being both a zero-emission fuel and a general energy storage medium. Green hydrogen is produced through the electrolysis of water using renewable energy sources such as solar or wind.4 Water electrolysis is thermodynamically a very energy-intensive process and consists of two half-reactions: the hydrogen evolution reaction (HER) and the oxygen evolution reaction (OER), whose chemical reactions in alkaline media can be given as follows, HER: 4H2O + 4e− → 2H2 + 4OH−, OER: 4OH− → O2 + 4e− + 2H2O with ΔE° = 1.23 V. In practice, the voltage required to achieve a practical current density (e.g. ∼10 mA cm−2) is higher than 1.23 V, the difference is defined as the overpotential. The overpotential needed to overcome the kinetic activation energy increases the theoretically needed energy input. To maximize hydrogen production from renewable energy, limiting the overpotential of both half-reactions is important. The OER is particularly inefficient and slow due to its four-electron transfer, resulting in a large energy loss in the reaction.5–7 Ruthenium oxide (RuO2) or iridium oxide (IrO2) catalyze the OER with low overpotential.8–10 Although effective, the large-scale application of these materials is infeasible due to the concomitant large amount of precious metals necessary.Efforts to make more cost-effective water-splitting electrocatalysts have included, inter alia, metal–organic frameworks (MOFs).11–14 MOFs are highly variable coordination networks consisting of organic linkers and metal nodes.14 MOFs are also of interest for many applications like gas storage and separation, vapor sorption, catalysis, biomedical applications, chemical sensors and devices involving electronic and ionic conduction.15 However, many of these MOFs are very difficult to apply on a large scale due to difficulties in scaling up the synthesis. Mechanochemical synthesis of MOFs offers a promising way to prepare MOFs with little to no solvent and significantly less time.16–18 Currently, the three predominant forms of mechanochemical synthesis are neat grinding, liquid-assisted grinding, and ion-liquid-assisted grinding. Neat grinding (NG) simply involves the milling of reactants with no added solvent. Liquid-assisted grinding (LAG) incorporates a small amount of solvent into the mixture to drive the reaction. Ion-liquid assisted grinding (iLAG) uses a small amount of liquid and salt to enable the reaction.16,19–22 In the mechanochemical synthesis of MOFs, LAG and iLAG are most prevalent.23 The liquid medium in LAG and iLAG assists in the deprotonation of the linkers stabilizes intermediates and the pores of the MOF.24,25 It has been reported that mechanochemical synthesis of MOFs can greatly reduce reaction times from several hours or days to 15 min.26 In addition, a mechanochemical experiment by Gao et al. found that up to 20 g of the MOF Zn-atz-ipa could be produced in a single batch in just 20 min.27 This yield is much greater than the usual amount obtained through a solvothermal batch synthesis with standard reaction vessels. This increased space-time-yield elevates the potential for MOFs in industrial applications.28 The mechanochemical synthesis of the prototypical MOF HKUST-1 (HKUST = Hong Kong University of Science and Technology) with the formula [Cu3(BTC)2(H2O)3]n (BTC = benzene-1,3,5-tricarboxylate) has been well-established.
HKUST-1 (also named Cu-BTC or MOF-199) is a MOF composed of {Cu2(OOC–)4} paddle-wheel clusters as secondary building units (SBUs) with the Cu2 units coordinated by four BTC linkers.29–34 Labile solvent molecules coordinate the two axial positions at the Cu2 handle. HKUST-1 comprises two kinds of pores and windows in a cubic 3D network which is traversed by channels along the a, b, and c axis (Fig. S1†). However, HKUST-1 displays low activity in the OER.35 Mixed-metal analogues of MOFs have shown promising results in catalyzing water oxidation, with small amounts of Ru incorporated into the structure greatly lowering the overpotential and increasing the efficiency of the reaction when compared to their parent MOFs.36–38 Electrode materials derived from mixed-metal MOFs offer a way to reduce the amount of precious metal necessary and are of interest due to the synergy of uniformly distributed two or more metal ion types at the nanoscale.39 Both Ru(II) and Ru(III) can form carboxylate-bridged paddle-wheel units and can, thus, be incorporated in the HKUST-1 framework.40–42 For charge-neutrality, Ru(III) would have a terminal hydroxido or chlorido ligand. Here, we describe the mechanochemical synthesis of Ru containing HKUST-1 and tested these compounds as precatalysts in various electrochemical experiments to determine their activity in the OER.
Results and discussion
The HKUST-1 and CuxRu-BTC MOFs were mechanochemically synthesized through liquid-assisted grinding (LAG). The reactants were loaded into two 25 mL ball mill vessels with two 10 mm zinc oxide (ZnO) balls and 400 μL of methanol each (see experimental section for details). The vessels were placed into the ball mill and allowed to run for 60 min at a frequency of 30 Hz (Fig. 1). | ||
| Fig. 1 Schematic representation of the synthesis of the mixed-metal CuxRu-BTC MOFs. | ||
The synthesis of mixed-metal MOFs typically results in the incorporation of metals at a different ratio than what was set with the stoichiometry of the reactants. This makes it important to determine the metal ratio post-synthetically, e.g., through atomic absorption spectroscopy (AAS) (Table 1). Often scanning electron microscopy energy-dispersive X-ray spectroscopy (SEM-EDX) (Table 1 and Fig. 2) and X-ray photoelectron spectroscopy (XPS) are also used for element quantification. The EDX spectra (Fig. S2†) and the XPS survey scan spectra (Fig. S16†) revealed the presence of the elements C, O, Cu and Ru and with XPS also Cl (in EDX the RuLα peak overlaps with ClKα). Both methods are, however, surface weighted. In EDX the emitted X-rays give a 1–2 μm depth analysis. In XPS the detected photoelectrons can only escape from a 1–10 nm (70–110 Å) thin surface layer of the sample. Further, EDX as an X-ray spectroscopy needs standards for peak identification and quantification due to stronger matrix effects. For XPS the ratio of the elements is best determined at the same orbital level. Otherwise, if determined from different orbital levels their relative amounts have to be determined with empirical relative sensitive factors (RSF) which increases the error.43
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ru metal ratios
:Ru metal ratios
| Sample | Theor. from synthesis | AASa | SEM-EDXb | XPSb,c |
|---|---|---|---|---|
|
a Atomic absorption spectroscopy (AAS): Weighted samples were dissolved in aqua regia. The solution was filtered carefully and diluted with ultrapure water to a volume of 25 mL for the AAS. For the determination of the Ru content 1 mL of a LaCl3 × 7H2O solution (c = 10 g L−1 in 10% HCl) was added to 9 mL of the analysis solution. To determine the Cu content the analyses solutions (without addition of LaCl3 × 7H2O solution) were further diluted with ultrapure water (1:50) for the AAS measurements. The experimental concentration values are given in Table S3.
b Surface-weighted spectroscopies, see text.
c Based on Cu 2p and Ru 3p orbitals together with relative sensitive factors (RSF) because of the different orbitals.43
|
||||
| Cu10Ru-BTC | 10:1 |
10:1 |
11:1 |
6:1 |
| Cu7Ru-BTC | 8:1 |
7:1 |
8:1 |
4:1 |
| Cu6Ru-BTC | 6:1 |
6:1 |
7:1 |
3:1 |
 | ||
| Fig. 2 SEM images (first row) and EDX elemental mapping for Cu (second row) and Ru (third row) for (a) Cu10Ru-BTC, (b) Cu7Ru-BTC and (c) Cu6Ru-BTC. Further SEM images and the SEM-EDX spectra are displayed in Fig. S2 to S6†. | ||
According to the AAS measurements, the intended 10:1, 8:1 and 6:1 Cu:Ru ratio were indeed incorporated into the samples within the margin of error. The close to theoretical Cu:Ru ratios found in the samples is explained by a similar reactivity, that is reaction rate of the used metal salts with H3BTC to form the metal-carboxylate linkages and paddle-wheel units. The lower incorporation ratio of the 8:1 Cu:Ru sample can be rationalized by the smaller size of the Ru3+ ions causing strain in the framework and making it less favorable for Ru atoms to be integrated. This was supported by the AAS results (Table 1) as well as previous literature pertaining to mixed-metal MOFs. SEM-EDX shows a homogeneous distribution of Cu and Ru inside the samples (Fig. 2).
The sample names are based on the experimentally found incorporated molar ratios from the AAS measurements. An attempted synthesis with a 3:1 Cu:Ru ratio resulted in an amorphous product (Fig. S10†).
The structure of the synthesized compounds was confirmed through powder X-ray diffraction (PXRD). The diffractograms of the CuxRu-BTC samples in Fig. 3 match with the diffractogram of mechanochemically synthesized HKUST-1 and its simulation. The slightly higher noise in the diffractograms of the CuxRu-BTC samples indicates smaller crystallite sizes and/or coordination defects (see below). The PXRDs give no indication of residual (crystalline) metal salts or ligand remaining from the starting materials in the products (Fig. S11†). The lower intensity of the {002} reflection at 6.7°, can be explained (i) from the flat sample holder and the Bragg–Brentano geometry for the measurement. At low angle this geometry broadens the X-ray beam spot on the sample such that only a fraction of the diffracted radiation reaches the detector giving lower than expected intensities for reflection below ∼7°. (ii) The (002) = (020) = (200) lattice planes pass through and stems from the Cu atoms (Fig. S1d†). It is conceivable that the incorporation of Ru will slightly distort the lattice such that the metal atoms will lie above and below this lattice plane. (iii) The encapsulation of nano guests in the pores of HKUST-1 may decrease the intensity of the low angle peak as was seen for the (110) peak of ZIF-8 at 2θ = 7.3° with gradually higher loading of C60@ZIF-8.44 Schlichte et al. also observed a lowered intensity of the first reflex depending on the variations in the degree of hydration of HKUST-1.30 Additionally, Kathuria et al. found that the reduced intensity or even missing first reflex in the PXRD pattern is in relation to the loss of the fcc crystal structure of HKUST-1.45 Through our CH elemental analysis (Table S2†) we know that solvent molecules are in our MOF samples and it would be possible that mechanochemically synthesized MOFs with the incorporation of Ru into the structure could slightly distort the lattice as indicated above in (ii).
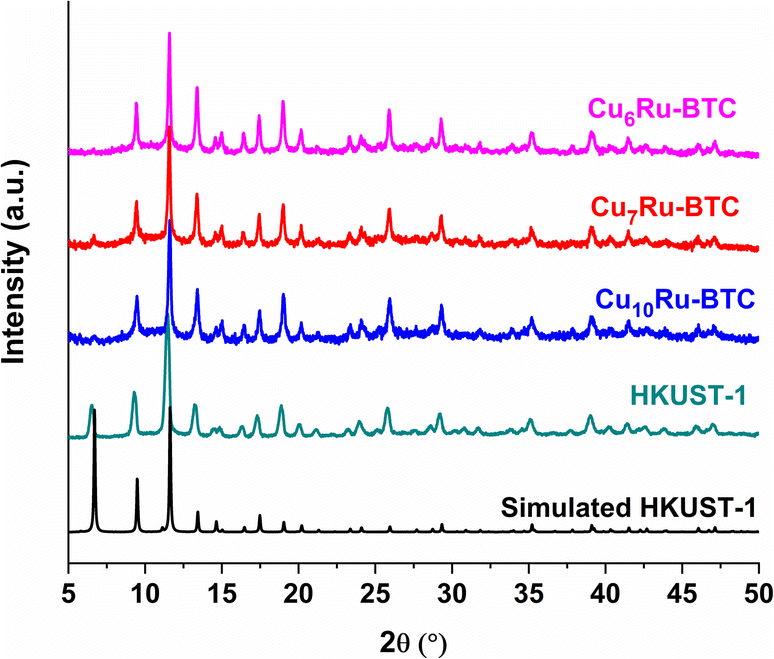 | ||
| Fig. 3 PXRD patterns of Cu6Ru-BTC, Cu7Ru-BTC, Cu10Ru-BTC, mechanochemically synthesized HKUST-1 and simulated HKUST-1 (CCDC no. 112954). | ||
Thermogravimetric analysis (TGA) of the as-synthesized samples shows an initial mass loss before 100 °C that can be attributed to the loss of (coordinated) solvent from the pores of the structure. For the TGA the samples had not been activated before. A second mass loss step occurs around 300 °C, which agrees with literature values (see Fig. S12† and accompanying text for details).34
The Fourier transform infrared (FT-IR) spectra (Fig. S13†) of the as-synthesized mixed-metal MOFs align well with the mechanochemically synthesized HKUST-1 and with the literature spectra of HKUST-1 from solvothermal synthesis.32 The broad band from 3600 cm−1 to 3000 cm−1 can be attributed to O–H vibrations from coordinated water or methanol solvent in the structure, coming from the crystal water of the reactants or the methanol for LAG. There was no sample activation for FT-IR.
Using N2-adsorption and desorption isotherms (Fig. 4a) at 77 K the Brunauer–Emmett–Teller (BET) surface areas (SBET) and the porosity of the synthesized MOFs were determined. The isotherms are a composite of IUPAC type I and probably type II with an H3 or H4 hysteresis loop (there is no characteristic plateau as for type IV).46 The pronounced uptake at low relative pressure stems from the filling of the micropores. The type II branch is given by non- or macroporous adsorbents and derives here from the texture effect of the physisorption in the meso- and macroporous voids of the aggregated crystallites which are obtained in the mechanochemical procedure (cf.Fig. 2). The larger HKUST-1 crystals from solvothermal synthesis give the expected type I isotherm.29 H3 loops are given by aggregates and also if the macropores are not completely filled with the pore condensate.46 The mechanochemically synthesized HKUST-1 exhibited a BET surface area of 1027 m2 g−1, which falls within the range of previously solvothermal and mechanochemically synthesized HKUST-1 (SBET = 630–1720 m2 g−1).47–50 The mixed-metal analogues all exhibited surface areas lower than that of HKUST-1. The values were 603 m2 g−1, 611 m2 g−1, and 859 m2 g−1 for Cu10Ru-BTC, Cu7Ru-BTC and Cu6Ru-BTC respectively. The trend of a decreasing SBET with integration of Ru into the HKUST-1 structure was rationalized by Gotthardt et al.32 to arise from coordination defects also because of the size difference between Ru(III) and Cu(II). The total pore volumes based on the adsorption were found to be 0.46 cm3 g−1, 0.47 cm3 g−1 and 0.55 cm3 g−1 (at p/p0 = 0.90) for Cu10Ru-BTC, Cu7Ru-BTC, and Cu6Ru-BTC respectively. The total pore volume of mechanochemically synthesized HKUST-1 was determined to 0.54 cm3 g−1. The main pore size distribution maxima below 1 nm (10 Å) (Fig. 4b) are in agreement with the pore window size of 9 Å (Fig. S1†). The small maxima above 2 nm indicate some mesopore contribution. The coordination defects in the CuxRu-BTC samples will also lead to smaller crystallites as was indicated through the noise of the diffractograms (Fig. 3).
 | ||
| Fig. 4 (a) N2-sorption isotherms at 77 K (adsorption filled circles; desorption empty circles) and (b) NLDFT-derived pore size distributions of Cu6Ru-BTC, Cu7Ru-BTC, Cu10Ru-BTC and mechanochemically synthesized HKUST-1 (model cylindrical pores at metal oxide). | ||
Based on work from Gotthardt et al. we assume that Cu can be exchanged with Ru and there is a partial substitution of Cu2+ with Ru3+ ions resulting in mixed-metal paddle-wheel structures.32 If Ru fragments would be only incorporated into the pores, the expectation would be that the BET surface area and pore volume of Cu6Ru-BTC with the highest Ru content should have been the lowest, which was not the case. Further, matrix-assisted laser-desorption/ionization time-of-flight-mass spectroscopy (MALDI-TOF-MS) revealed fragments containing CuxRu isotope patterns (x = 1–3) (Fig. S14 and S15†).
To determine the oxidation states of the metals XPS was carried out. High-resolution XPS spectra (HR-XPS spectra) of C, O, Cl, Cu and Ru were recorded (Fig. 5, S17 and S18†). HR-XPS spectra of the Cu 3p region (Fig. S17†) and closer analysis of the Cu 3p3/2 orbital (Fig. S18a†) confirm the sole presence of Cu in oxidation state +2 (Cu2+ species) in all samples.51 The spectral region for C 1s and Ru 3d3/2 and Ru 3d5/2 overlap and deconvoluted spectra of C 1s and Ru 3d (Fig. 5a) show peaks at 288.1, 285.6 and 284.3 eV for HKUST-1, which were assigned to the linker BTC.52 For the CuxRu-BTC mixed-metal samples an additional peak at ∼286.8 eV for Ru 3d3/2 is added to the fit and a (shoulder) peak at around 282.5 eV can be attributed to Ru3+ 3d5/2 with the C 1s peaks all shifted to slightly higher binding energies.52,53 The HR-XPS spectra of Ru 3p orbital states in the binding energy region from 460 to 490 eV further support the incorporation of Ru solely in the oxidation state +3 (Ru3+) into the structure (Fig. 5b). The peaks at 486.3 and 464.1 eV correspond to Ru3+ 3p1/2 and Ru3+ 3p3/2 species.52,53 Peaks for Ru(II) would be expected at 281.7–280.2 eV for 3d5/2, 285.5 eV for 3d3/2 and 462.5–461.2 eV for 3p3/2.52,54
 | ||
| Fig. 5 HR-XPS spectra of (a) C 1s and Ru 3d and (b) Ru 3p for Cu10Ru-BTC, Cu7Ru-BTC, Cu6Ru-BTC and mechanochemically synthesized HKUST-1. | ||
Electrochemical data
To test the effectiveness of the samples catalyzing the oxygen evolution reaction, OER, multiple experiments were performed. Nickel foam (NF) was used as a support material for the working electrode. A NF sheet was accurately cut into 1 × 1 cm pieces. The NF pieces were used without any pretreatment as a substrate for the sample-containing ink. In the following it is implied that the CuxRu-BTC MOFs are precursor materials to the catalytic species which form in the alkaline medium where many MOFs are of low stability.39,55 The decomposition of MOFs in an alkaline aqueous medium usually leads to metal oxides/hydroxides.56–58 MOFs are seen as good precursor materials to these active metal oxide/hydroxide electrocatalyst to achieve a defined mixed-metal composition with a uniform distribution of the metals on the nano-level.39,57–59The overpotential (η) was derived from the linear sweep voltammetry (LSV) plots in Fig. 6a, which showed the Cu10Ru-BTC sample performed best, i.e., had the lowest overpotential before the stability test. The stability of the electrocatalysts was also tested by both 1000 cyclic voltammetry cycles (CVs) and 12 h of chronopotentiometry (Fig. S19†). Following 1000 CVs the Cu10Ru-BTC sample decreased slightly in current density, giving a slight increase in overpotential, while the Cu7Ru-BTC and Cu6Ru-BTC samples improved considerably in performance. This can be attributed to the samples still being activated during the stability test cycles. The small peak seen around 1.4 V vs. RHE for each sample can most likely be attributed to nickel oxidation (Ni2+ → Ni3+) present from the NF. As mentioned previously a current density of 10 mA cm−2 was the benchmark for determining the overpotential of the samples. This value corresponds to the current density of a 10% efficient solar-to-fuel conversion device under sun illumination.57 A comparison of the overpotentials of each sample both before and after the stability test is as follows: Cu10Ru-BTC (η = 308 mV → 314 mV), Cu7Ru-BTC (η = 339 mV → 319 mV), Cu6Ru-BTC (η = 376 mV → 327 mV), and HKUST-1 (η = 432 mV → 325 mV) (Fig. 6b and Table S5†). Comparing these samples to RuO2 revealed promising results. In our experiment the overpotentials of RuO2 before and after the stability test (η = 344 mV → 312 mV) were in accordance with literature values60 and the “after” values were highly comparable to those of the Cu10Ru-BTC and Cu7Ru-BTC samples.
Fig. 6c displays the Tafel plots for the samples. The Tafel slope shows the applied voltage increase necessary to increase the current density by a factor of ten and also gives insight into the mechanism of the reaction by indicating the rate determining step according to eqn (1)–(4) (M = active site).58 The Tafel slope b is calculated using the Tafel equation (η = a + b × log(j)).61 The values for b were as follows: Cu10Ru-BTC 55 mV dec−1, Cu7Ru-BTC 53 mV dec−1, Cu6Ru-BTC 63 mV dec−1, HKUST-1 54 mV dec−1 and RuO2 47 mV dec−1. For the Cu10Ru-BTC and the Cu7Ru-BTC sample the rate determining step with Tafel slopes of 55 mV dec−1 and 53 mV dec−1 is in between those of eqn (2) and (3) values. For the Cu6Ru-BTC sample (b = 63 mV dec−1) the rate determining step is the metal hydroxide deprotonation (respectively hydroxide adsorption/water desorption) according to eqn (2). The value gathered for RuO2 (b = 47 mV dec−1) is slightly higher than the literature value (b = 40 mV dec−1), which suggests that the rate determining step for the RuO2 catalyst the metal oxide oxidation given in eqn (3).62
| M + OH− ⇄ MOH + e− (b = 120 mV dec−1) | (1) |
| MOH + OH− ⇄ MO− + H2O (b = 60 mV dec−1) | (2) |
| MO− → MO + e− (b = 45 mV dec−1) | (3) |
| 2MO → 2M + O2 (b = 19 mV dec−1) | (4) |
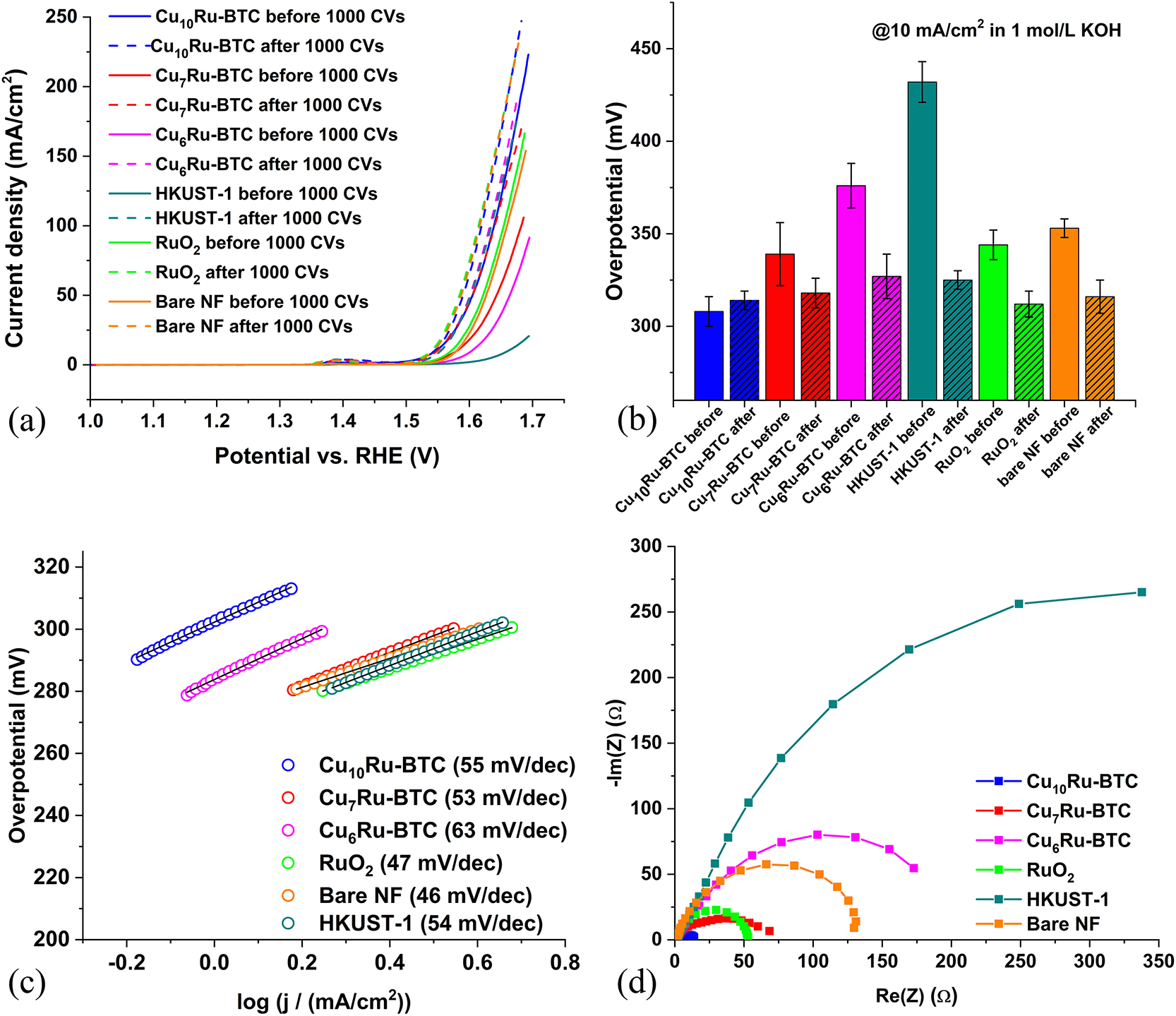 | ||
| Fig. 6 (a) LSV curves before and after 1000 CVs of Cu6Ru-BTC, Cu7Ru-BTC, Cu10Ru-BTC, HKUST-1, bare NF, and RuO2. (b) Overpotentials calculated from LSV curves in (a). (c) Tafel plots for Cu6Ru-BTC, Cu7Ru-BTC, Cu10Ru-BTC, HKUST-1, bare NF, and RuO2. (d) Nyquist plots gathered from EIS for Cu6Ru-BTC, Cu7Ru-BTC, Cu10Ru-BTC, HKUST-1, bare NF and RuO2. | ||
Using electrochemical impedance spectroscopy (EIS), a value for the charge transfer resistance of the electrocatalyst can be found. Charge transfer resistance (RCT) is a metric that shows the resistance to transfer electrons during a redox reaction. This value is inversely proportional to the charge transfer rate, making it important for a good catalyst to have a low RCT.63 The values gathered for the mixed-metal samples varied drastically by the amount of Ru incorporated into the structure. The Nyquist plot (Fig. 6d) was gathered in the frequency range of 0.01 Hz to 10 kHz at an alternating current (AC) potential amplitude of 10 mV at 1.5 V (vs. RHE). The plot was fitted to the simple Randle cell model to determine the RCT.64 The Cu6Ru-BTC sample performed the worst from the mixed-metal samples with a RCT of 204.5 Ω, Cu7Ru-BTC performed better with a RCT of 56.1 Ω, and Cu10Ru-BTC performed much better with a RCT of 13.6 Ω. The value for Cu10Ru-BTC was also found to be lower than the RuO2 sample with an RCT of 52.8 Ω, which matches closely with literature values.65
Fig. 7a shows CV plots of Cu10Ru-BTC used for the determination of the electrochemical surface area (ECSA) and Fig. 7b displays the current vs. scan rate plot gathered from the CV plots in Fig. 7a. The ECSA of a catalyst can give insight into the OER and super capacitive performance of a given catalyst. A higher ECSA for a catalyst correlates with a higher number of redox-active sites making it more effective in the OER.66 The ECSA was obtained using the double-layer capacitance method where the current of the sample in a non-faradaic range was collected at varying scan rates. The current at 1.10 V was then plotted against the scan rate and the slope of this graph resulted in the double layer capacitance (CDL).66 The ECSA is calculated from eqn (5), with the specific capacitance (CS) being 40 μF cm−2 in 1 mol L−1 KOH solution according to the literature.67 The mechanochemically synthesized samples recorded an ECSA of 0.091, 0.096, and 0.100 cm2 for Cu6Ru-BTC, Cu7Ru-BTC, and Cu10Ru-BTC, respectively (Table S6†). The Cu10Ru-BTC sample performed similarly to the RuO2 sample which recorded an ECSA of 0.094 cm2.
| ECSA = CDL/CS | (5) |
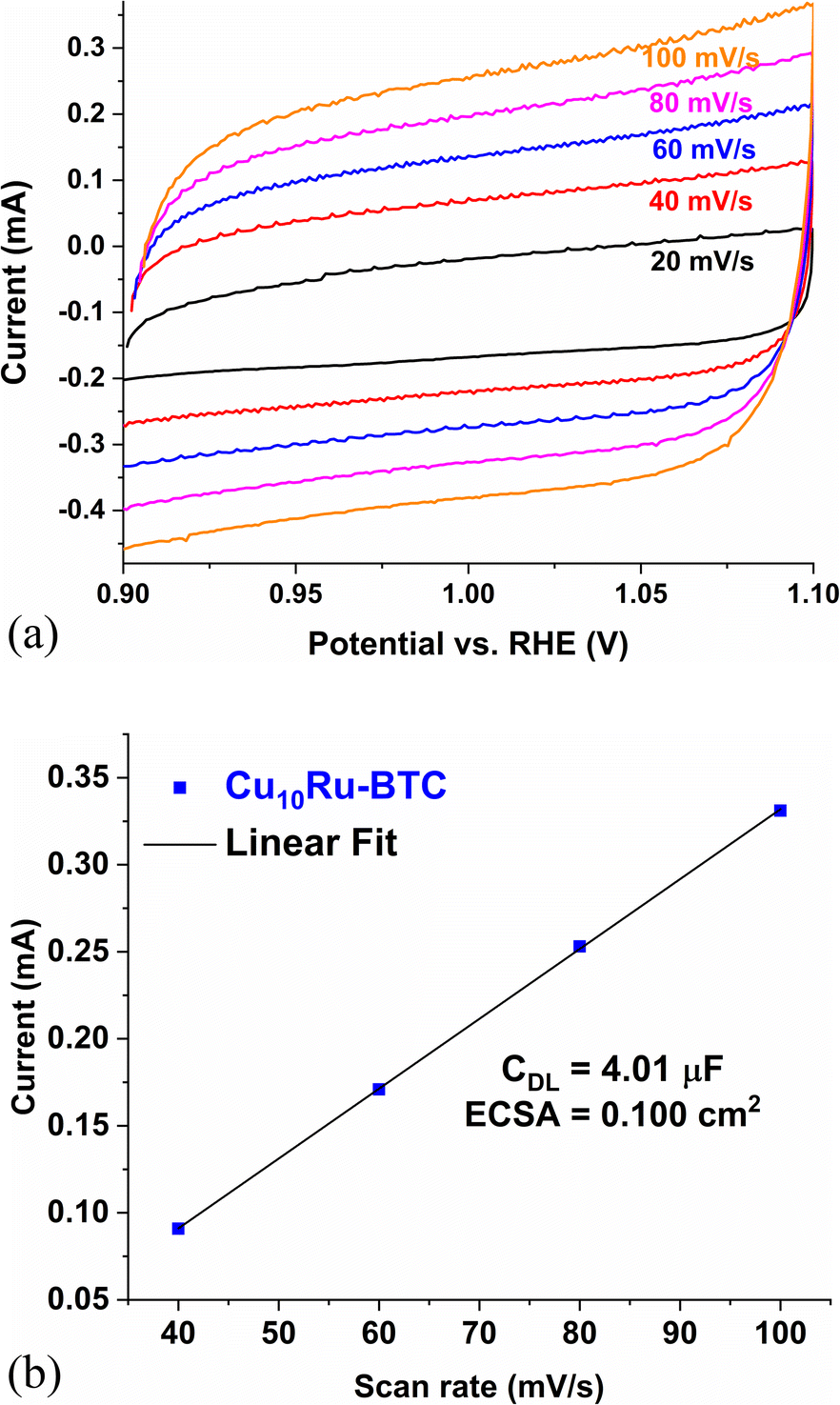 | ||
| Fig. 7 (a) CV plots of Cu10Ru-BTC with different scan rates used for the calculation of the ECSA; (b) current vs. scan rate plot obtained from the CVs in (a) (CDL = double layer capacitance, ECSA = electrochemical surface area). | ||
The faradaic efficiency (FE) was calculated by the method of Galán-Mascarós et al.68 (see Experimental section for details). Cu10Ru-BTC reached the highest FE with 70% from the tested samples, but the others are closely following with Cu7Ru-BTC FE = 68%, Cu6Ru-BTC FE = 66%, HKUST-1 FE = 64%, RuO2 FE = 66% and bare NF FE = 61% (Table S7†).
In consideration of the obtained electrocatalytic results a positive, synergistic effect between copper and ruthenium can be seen. This synergistic effect between copper and ruthenium was also observed in materials used in other applications like Li–CO2 batteries, low-temperature ammonia oxidation and propylene epoxidation.69–71 Petrucci et al.72 has shown that incorporating copper in a ruthenium catalyst enhances the capacitive response of the materials, promotes electron transfer reversibility and exhibits an increased corrosion resistance.
From the tested samples the catalyst derived from Cu10Ru-BTC exhibits the highest activity for the OER, even though it has a lower amount of ruthenium than other samples. It was also shown from Zhang et al.73 that a too heavy loading of RuO2 would not result in an advantage in capacitance anymore, which leads us to believe that for our samples the best performing ratio of copper and ruthenium was achieved with Cu10Ru-BTC.
It is common for MOFs to act as sacrificial precursor catalysts in electrochemical reactions, in particular in an aqueous alkaline medium, with transformation of the MOF to a metal oxide/hydroxide in or on a carbon framework.39,56–58 This not only gives an intimate metal alloying in the oxide/hydroxide but also introduces varying and tunable morphologies into the structure that allows for increased access to active sites in the catalyst material. Therefore, it is important to investigate the structure of the substance after being introduced to the chemical conditions during the electrochemical study. To achieve this, the MOF powder samples were soaked in 1 mol L−1 KOH for 24 h and the product investigated by PXRD. As seen in Fig. 8 and S24†, in aqueous KOH HKUST-1 and the CuxRu-BTC samples deconstructed to form a sample which contained reflexes that align well with CuO simulated reflexes.
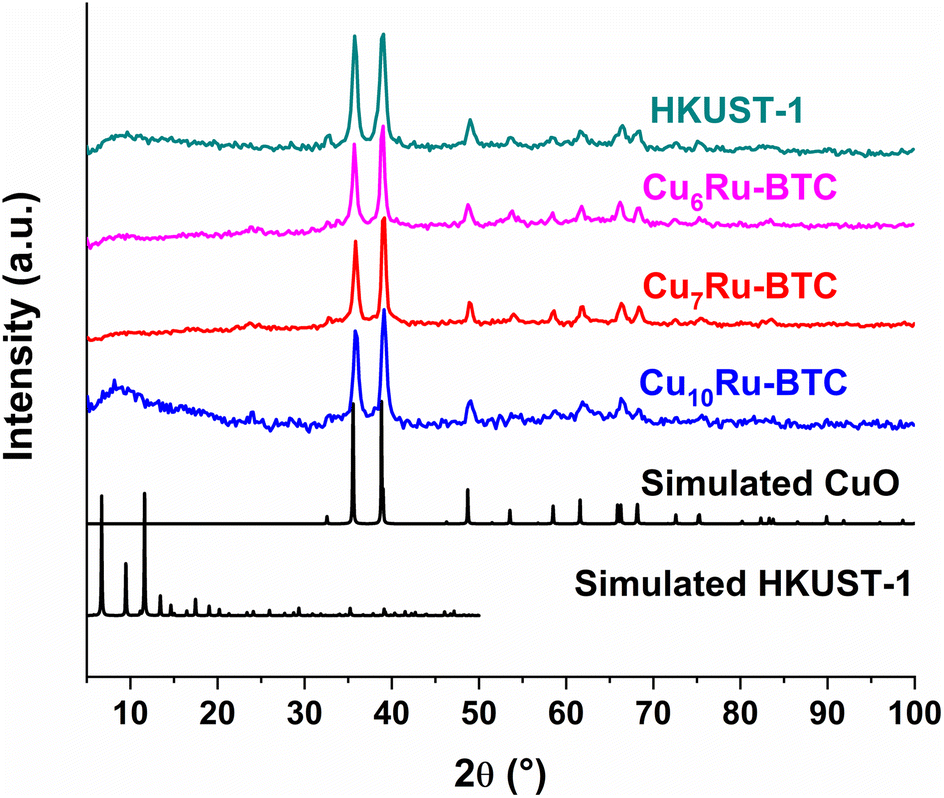 | ||
| Fig. 8 PXRD patterns of mechanochemically synthesized HKUST-1, Cu6Ru-BTC, Cu7Ru-BTC, Cu10Ru-BTC, following 24 h in 1 mol L−1 KOH solution, simulated CuO (COD: 15-26990) and simulated HKUST-1 (CCDC no. 112954). | ||
To assess the possible fate of the Ru component from the CuxRu-BTC samples, we soaked the educt RuCl3·H2O in the 1 mol L−1 KOH electrolyte for 24 h to test how it will change similar to as we did with the mechanochemically synthesized CuxRu-BTC MOFs (in which the Ru content was too low for post-mortem PXRD detection). The comparison of the resulting diffractogram with the simulations for different RuO2 polymorphs suggests the formation of tetragonal RuO2 (Fig. S25†).
In order to address the effect of the second metal at the nanoscale in the OER performance of bimetallic CuxRu-BTC, we also compared the activity for the electrocatalyst derived from a physical mixture of CuO and RuO2, which probably form from CuxRu-BTC in KOH. CuO:RuO2 mixtures at molar ratios of 10:1, 8:1 and 6:1 were prepared from commercial CuO and the “RuO2” obtained from the respective KOH treatments and tested electrochemically. From the LSV curves (Fig. S21a†) and the resulting overpotentials at 10 mA cm−2 and 50 mA cm−2 before and after 1000 CVs (Fig. S21b and Table S8†) it can be seen that the physical mixtures of the oxides performed worse than the electrocatalysts resulting from the mechanochemically synthesized MOFs. The magnitude of activation, indicated by the difference in the overpotential of the samples, was less than in the case of the MOFs. The Tafel slopes of the physical oxide mixtures were also higher (Fig. S21c and Table S8†) and the charge transfer resistance was much higher for the physical oxide mixtures (Fig. S21d and Table S8†), than the CuxRu-BTC MOF derived materials (Table S5†) which indicates a reduced activity in comparison to the MOF samples. In case of the performed chronopotentiometry (Fig. S22†) all physically-mixed metal oxide samples showed an increasing potential over the measurement time of 12 h, suggesting that the physical mixtures are not as stable as the resulting products from the mechanochemically synthesized samples. The arising ECSAs were also lower for the physical mixtures than in the MOF samples (Fig. S23, Tables S8 and S5†).
Our study and comparison demonstrate that a bimetallic MOF improves the electrocatalytic performance of the derived catalyst by giving an intimate and uniform mixture of the involved metals at the nanoscale. One thing to note here again is that the 10:1 CuO:RuO2 physical mixture seems to be the sample with the highest activity, similar to what we observed for the MOF-derived samples. Following the electrochemical results, we recorded the faradaic efficiency (FE) for the 10:1 CuO to RuO2 mixture (FEwith correction = 50%, FEwithout correction = 66%). The FE was even however even lower than for bare nickel foam, which prompted us to not further determine the FE for the other mixtures.
Experimental
Synthesis of HKUST-1
The synthesis of HKUST-1 used copper acetate, [Cu2(CH3CO2)4 × H2O] (1.62 g, 8.11 mmol) and benzene-1,3,5-tricarboxylic acid (H3BTC) (0.86 g, 4.09 mmol). The reactants were placed into two 25 mL ball mill vessels with two 10 mm ZnO balls each. Then, 400 μL of methanol was added to each of the vessels, and the contents were thoroughly mixed through shaking the closed vessels by hand until the fluid was worked in (Fig. S26b†). This was done to prevent clumping during the ball milling. The vessels were then mounted in the ball mill and allowed to run for 60 min at a frequency of 30 Hz. The by-product acetic acid was identified by its smell. The product was allowed to dry in air before washing with methanol (30 mL) and dried again in air. A yield of 1.66 g (67%) was achieved. For the CH analysis and estimation of the solvent content see Table S2†.Synthesis of Cu10Ru-BTC, Cu7Ru-BTC and Cu6Ru-BTC
The preparation of the mixed metal MOFs was identical to that of HKUST-1, but with a 10:1, 8:1 or 6:1 molar ratio of Cu:Ru. Therefore, 1.180 g, 1.155 g or 1.114 g [Cu2(CH3CO2)4 × H2O] (5.92 mmol, 5.79 mmol or 5.58 mmol) and 0.133 g, 0.163 g or 0.209 g RuCl3 × 3H2O (0.59 mmol, 0.72 mmol or 0.93 mmol) were mixed. The mixture reacted under identical conditions resulting in dark blue powders and the smell of acetic acid. Once again, the powders were allowed to dry in air, then washed with methanol (30 mL) and dried again in air. Yield Cu10Ru-BTC: 1.43 g (66%); yield Cu7Ru-BTC: 1.39 g (64%); yield Cu6Ru-BTC: 1.29 g (59%). For the CH analysis and estimation of the solvent content see Table S2†.
Instrumentation
The mechanochemical syntheses were performed using a Retsch MM400 mixer mill (Fig. S26†). We used 25 mL stainless-steel vessels (Fig. 1 and S17b†) because they were the only available option which we had for the larger amounts we put into the synthesis. We did not detect any chromium residue and only small amounts of iron residue by SEM-EDX (Fig. S2†) and TEM-EDX (Fig. S7–S9†).PXRDs were gathered using a Bruker D2 Phaser powder diffractometer with a power of 300 W and acceleration voltage of 30 kV at 10 mA using Cu-Kα radiation (λ = 1.5418 Å). The diffractograms were collected on a low background flat silicon sample holder and analyzed using the Match 3.11 software. The measurement range for the samples was from 5 to 50° 2θ with a scan speed of 2 s per step and 0.057° (2θ) step size.
FT-IR spectra were recorded on a Bruker TENSOR 37 IR spectrometer in KBr from 4000 to 400 cm−1.
N2-sorption measurements were performed with a micro200B automatic volumetric gas sorption analyzer from 3P at 77 K. The sorption isotherms were evaluated with the PAS_3P_10.06.06 software. Prior to the measurements the materials were activated under vacuum (<10−3 kPa) at 120 °C for 5 h. BET surface areas were determined from the N2-adsorption isotherms and the pore size distributions were derived by non-local density functional theory (NLDFT) calculations based on N2 at 77 K with the model “cylindrical pores at metal oxide”.
TGA was gathered on a Netzsch TG 209 F3 Tarsus device equipped with an aluminum crucible. A heating rate of 10 K min−1 under a N2 atmosphere was used.
CH elemental analyses were collected with a Vario Micro Cube from Elementar Analysentechnik.
AAS was conducted with a PinAAcle 900T from PerkinElmer. Exactly weighted samples (∼7 mg) were dissolved in 4 mL of aqua regia in a microwave-assisted digestion and then diluted with ultrapure water to a volume of 25 mL. For the determination of the Ru content 1 mL of a LaCl3 × 7H2O solution (c = 10 g L−1 in 10% HCl) was added to 9 mL of the analysis solution. To determine the Cu content the analyses solutions (without addition of LaCl3 × 7H2O solution) were further diluted with ultrapure water (1:50) for the AAS measurements.
SEM images were obtained with a JEOL JSM-6510 LV QSEM advanced electron microscope with a LaB6 cathode at 20 kV. The microscope was equipped with a Bruker Xflash 410 silicon drift detector and the Bruker ESPRIT software for EDX analysis. The Al and Au found in the EDX spectra can be attributed to the Al sample holder and the sputtering of the sample with gold before the investigation.
Transmission electron microscopy (TEM) images of the MOF samples before the electrochemical tests were recorded on a FEI Tecnai G2 F20 electron microscope operated at 200 kV accelerating voltage equipped with a Gatan UltraScan 1000P detector. TEM samples were prepared by drop-casting the dispersed (in ethanol) material on 200 μm carbon-coated gold grids.74
XPS was done with a ULVAC-PHI VersaProbe II microfocus spectrometer with an Al-Kα X-ray source (1486.8 eV). As reference for the binding energy scale the Cu 2p3/2 signal at 932.4 eV of a Cu standard was used, since the commonly used C 1s signal and the Ru 3d signal overlap. Only for the HKUST-1 sample the C 1s signal at 284.4 eV was used as reference for the binding energy scale. The spectra were evaluated using the Casa XPS software, version 2.3.19PR1.0.
Matrix-assisted laser-desorption/ionization time-of-flight (MALDI-TOF) mass spectra were measured on a Bruker UltrafleXtreme with Dithranol (DIT) as the matrix.
The electrochemical measurements were performed with a SP-50e Potentiostat from BioLogic Science Instruments and having a three-electrode setup. The reference electrode was a reversible hydrogen electrode (RHE) from Gaskatel (Kassel, Germany), the counter electrode was a Pt plate, and the working electrode was ink-coated nickel foam (NF). A NF sheet was accurately cut into 1 × 1 cm pieces. The NF pieces were used without any pretreatment. The activity of the samples was tested by drop casting the MOF sample onto the NF via a prepared ink. The ink consisted of 2.5 mg of MOF sample, 0.5 mL of EtOH and 20 μL of Nafion (5 wt%) and was sonicated for 30 min. 0.1 mL of the ink was drop-casted onto the NF to give a catalyst loading of 0.5 mg cm−2. The RuO2 electrode as a benchmark was prepared using the same method. The OER measurements were conducted in 1 mol L−1 KOH electrolyte that was purged with N2 for 10 min. The samples were activated by cycling the working electrode between 1.0 V and 1.7 V vs. RHE at a scan rate of 100 mV s−1. The cycling stability was measured by comparing the LSV curves after the activation protocol of 1000 cycles. The potential applied to the ohmic resistance was automatically corrected by the SP-50e Potentiostat. The voltage range for all LSV measurements was from 1.0 V to 1.7 V vs. RHE with a scan rate of 2 mV s−1. EIS data was gathered in a frequency range of 0.01 Hz to 10 kHz at a potential of 1.5 V vs. RHE. ECSA was measured by collecting the CV plots of each sample at varying scan rates of 20 mV s−1, 40 mV s−1, 60 mV s−1, 80 mV s−1, and 100 mV s−1. At least three measurements were done for each sample to determine the overpotentials, Tafel slopes, RCT and ECSA in order to reduce the experimental contingency error and the averaged results were displayed in the figures and tables.
Chronopotentiometry was conducted following the above electrochemical measurements. The current density was held at 10 mA cm−2 for 12 h. The FE was calculated according to the method of Galán-Mascarós et al.,68 with the exception of using a constant current of 50 mA cm−2 instead of 10 mA cm−2. An Ocean Optics NeoFOX sensor system coupled with a FOSPOR probe was used to monitor the O2 level in the electrochemical cell. A two-point calibration was used for the FOSPOR probe with N2 atmosphere (0% O2) as one calibration point and ambient air (21% O2) as the other calibration point. Initially dissolved O2 in the electrolyte was removed by continuous purging with N2 gas for at least 1 h before beginning the chronopotentiometry measurement. Eqn (6) was used to calculate the molar amount of O2 evolved during the chronopotentiometric measurement, which presumes the applicability of the ideal gas law for the analyzed gas mixture at atmospheric pressure:
| nO2,exp = (% O2,det × Ptotal × Vgas/R × T)/100 | (6) |
The theoretically generated faradaic oxygen is determined by eqn (7):
| nO2,far = Q/ne × F | (7) |
485C mol−1). The FE (expressed in percentage) is obtained through the subsequent eqn (8):| FE = 100 × nO2,exp/nO2,far | (8) |
Conclusions
To summarize, Ru-containing analogues of HKUST-1 were synthesized using a mechanochemical approach and tested for their activity in the OER in comparison to RuO2. Through the mechanochemical method a fast gram-scale synthesis of the mixed-metal MOFs in just one-hour synthesis time could be achieved. The catalyst derived from Cu10Ru-BTC had an overpotential of η = 314 mV as well as a low charge-transfer resistance, RCT of 13.6 Ω which outperforms the RuO2 reference sample, which had an overpotential of η = 312 mV after the stability test as well as a RCT of 52.8 Ω. The Tafel slope of the Cu10Ru-BTC sample did not perform as well, achieving a value of 55 mV dec−1 (RuO2b = 47 mV dec−1) indicating a lower performance at higher current densities. Incorporation of Ru into the HKUST-1 structure through mechanochemical methods proved to lead to an effective precursor to the actual Cu/Ru-oxide/hydroxide electrocatalyst that can be produced in bulk with reduced precious metal usage. As smaller amounts of Ru proved to be better for the derived electrocatalyst properties we will continue to investigate the incorporation of even lower Ru quantities into HKUST-1. Further research into the mechanochemical synthesis of OER electrocatalysts offers a promising alternative to low scale solvothermal batch chemistry.Author contributions
Conceptualization, C. J. and L. S.; methodology, L. S.; validation, L. S. and Q. S.; formal analysis, L. S. and Q. S.; investigation, L. S., Q. S., A. V., T. S. and T. H. Y. B., resources, C. J.; data curation, L. S.; writing—original draft, L. S. and Q. S.; writing—review & editing, C. J.; visualization, L. S.; supervision, C. J.; project administration, C. J.; funding acquisition, C. J. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Note added after first publication
This article replaces the version published on 14 May 2024, which contained an error in Figure 6 .Acknowledgements
We thank Ms. Birgit Tommes for IR and Ms. Annette Ricken for the AAS measurements, Mr Lukas Eisenhuth and Mr Jonas Fabrizi for the TEM images as well as the Center for Molecular and Structural Analytics at Heinrich Heine University (CeMSA@HHU) for recording the mass spectrometric data. The research was funded by the Deutsche Forschungsgemeinschaft (DFG) within the Priority Program SPP 1928/2 COORNETs (grant Ja466/43-1).References
- S. Corona, Innovative Solutions in the Rooftop Photovoltaic Field: Techno-Economical Feasibility and Scenarios Simulation, Master thesis, Politecnico di Torino, 2023 Search PubMed.
- X. Qian, J. Phys.: Conf. Ser., 2021, 2044, 012042, DOI:10.1088/1742-6596/2044/1/012042.
- K. van der Wiel, L. P. Stoop, B. R. H. van Zuijlen, R. Blackport, M. A. van den Broek and F. M. Selten, Renewable Sustainable Energy Rev., 2019, 111, 261–275, DOI:10.1016/j.rser.2019.04.065.
- A. K. Sarker, A. K. Azad, M. G. Rasul and A. T. Doppalapudi, Energies, 2023, 16, 1556, DOI:10.3390/en16031556.
- M. Chatenet, B. G. Pollet, D. R. Dekel, F. Dionigi, J. Deseure, P. Millet, R. D. Braatz, M. Z. Bazant, M. Eikerling, I. Staffell, P. Balcombe, Y. Shao-Horn and H. Schäfer, Chem. Soc. Rev., 2022, 51, 4583–4762, 10.1039/d0cs01079k.
- X. Wang, H. Zhong, S. Xi, W. S. V. Lee and J. Xue, Adv. Mater., 2022, 34, 2107956, DOI:10.1002/adma.202107956.
- S. Abdpour, L. Rademacher, M. N. A. Fetzer, T. H. Y. Beglau and C. Janiak, Solids, 2023, 4, 181–200, DOI:10.3390/solids4030012.
- K. P. J. Gustafson, A. Shatskiy, O. Verho, M. D. Kärkäs, B. Schluschass, C.-W. Tai, B. Åkermark, J.-E. Bäckvall and E. V. Johnston, Catal. Sci. Technol., 2017, 7, 293–299, 10.1039/c6cy02121b.
- M. N. Iqbal, A. F. Abdel-Magied, H. N. Abdelhamid, P. Olsén, A. Shatskiy, X. Zou, B. Åkermark, M. D. Kärkäs and E. V. Johnston, ACS Sustainable Chem. Eng., 2017, 5, 9651–9656, DOI:10.1021/acssuschemeng.7b02845.
- A. Mills, P. A. Duckmanton and J. Reglinski, Chem. Commun., 2010, 46, 2397–2398, 10.1039/b925784e.
- R. Jaryal, R. Kumar and S. Khullar, Coord. Chem. Rev., 2022, 464, 214542, DOI:10.1016/j.ccr.2022.214542.
- K. Meyer, M. Ranocchiari and J. A. van Bokhoven, Energy Environ. Sci., 2015, 8, 1923–1937, 10.1039/c5ee00161g.
- S. Navalón, A. Dhakshinamoorthy, M. Álvaro, B. Ferrer and H. García, Chem. Rev., 2023, 123, 445–490, DOI:10.1021/acs.chemrev.2c00460.
- C. Janiak and J. K. Vieth, New J. Chem., 2010, 34, 2366–2388, 10.1039/c0nj00275e.
- R. Freund, O. Zaremba, G. Arnauts, R. Ameloot, G. Skorupskii, M. Dincă, A. Bavykina, J. Gascon, A. Ejsmont, J. Goscianska, M. Kalmutzki, U. Lächelt, E. Ploetz, C. S. Diercks and S. Wuttke, Angew. Chem., Int. Ed., 2021, 60, 23971–24001, DOI:10.1002/anie.202106259.
- D. Chen, J. Zhao, P. Zhang and S. Dai, Polyhedron, 2019, 162, 59–64, DOI:10.1016/j.poly.2019.01.024.
- T. Stolar and K. Užarević, CrystEngComm, 2020, 22, 4511–4525, 10.1039/d0ce00091d.
- S. Głowniak, B. Szczęśniak, J. Choma and M. Jaroniec, Mater. Today, 2021, 46, 109–124, DOI:10.1016/j.mattod.2021.01.008.
- T. Friščić, Chem. Soc. Rev., 2012, 41, 3493–3510, 10.1039/c2cs15332g.
- N. Shan, F. Toda and W. Jones, Chem. Commun., 2002, 2372–2373, 10.1039/b207369m.
- T. Friščić, S. L. Childs, S. A. A. Rizvi and W. Jones, CrystEngComm, 2009, 11, 418–426, 10.1039/b815174a.
- T. Friščić, C. Mottillo and H. M. Titti, Angew. Chem., Int. Ed., 2020, 59, 1018–1029, DOI:10.1002/anie.201906755.
- T. Friščić, J. Mater. Chem., 2010, 20, 7599–7605, 10.1039/c0jm00872a.
- J. R. Mason, J. N. Weyrich and H. Yang, Sustainability, 2022, 14, 9150, DOI:10.3390/su14159150.
- T. Stolar, L. Batzdorf, S. Lukin, D. Žilić, C. Motillo, T. Friščić, F. Emmerling, I. Halasz and K. Užarević, Inorg. Chem., 2017, 56, 6599–6608, DOI:10.1021/acs.inorgchem.7b00707.
- C.-A. Tao and J.-F. Wang, Crystals, 2021, 11, 15, DOI:10.3390/cryst11010015.
- T. Gao, H.-J. Tang, S.-Y. Zhang, J.-W. Cao, Y.-N. Wu, J. Chen, Y. Wang and K.-J. Chen, J. Solid State Chem., 2021, 303, 122547, DOI:10.1016/j.jssc.2021.122547.
- P. A. Julien, C. Mottillo and T. Friščić, Green Chem., 2017, 19, 2729–2747, 10.1039/c7gc01078h.
- S. S. Y. Chui, S. M. F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148–1150, DOI:10.1126/SCIENCE.283.5405.1148.
- K. Schlichte, T. Kratzke and S. Kaskel, Microporous Mesoporous Mater., 2004, 73, 81–88, DOI:10.1016/j.micromeso.2003.12.027.
- P. Krawiec, M. Kramer, M. Sabo, R. Kunschke, H. Fröde and S. Kaskel, Adv. Eng. Mater., 2006, 8, 293–296, DOI:10.1002/adem.200500223.
- M. A. Gotthardt, R. Schoch, S. Wolf, M. Bauer and W. Kleist, Dalton Trans., 2015, 44, 2052–2056, 10.1039/c4dt02491e.
- F. Gul-E-Noor, B. Jee, M. Mendt, D. Himsl, A. Pöppl, M. Hartmann, J. Haase, H. Krautscheid and M. Bertmer, J. Phys. Chem. C, 2012, 116, 20866–20873, DOI:10.1021/jp3054857.
- M. Klimakow, P. Klobes, A. F. Thünemann, K. Rademann and F. Emmerling, Chem. Mater., 2010, 22, 5216–5221, DOI:10.1021/cm1012119.
- H. Liu, L.-G. Gong, C.-X. Wang, C.-M. Wang, K. Yu and B.-B. Zhou, J. Mater. Chem. A, 2021, 9, 13161–13169, 10.1039/d1ta01503f.
- D. Liu, C. Wang, Z. Zhou, C. Ye, R. Yu, C. Wang and Y. Du, Inorg. Chem. Front., 2022, 9, 6158–6166, 10.1039/d2qi01764d.
- T. Noor, L. Yaqoob and N. Iqbal, ChemElectroChem, 2021, 8, 447–483, DOI:10.1002/celc.202001441.
- X. Zhao, B. Pattengale, D. Fan, Z. Zou, Y. Zhao, J. Du, J. Huang and C. Xu, ACS Energy Lett., 2018, 3, 2520–2526, DOI:10.1021/acsenergylett.8b01540.
- S. Abdpour, M. N. A. Fetzer, R. Oestreich, T. H. Y. Beglau, I. Boldog and C. Janiak, Dalton Trans., 2024, 53, 4937–4951, 10.1039/d3dt03695b.
- J. Xia, X. Liu, H. Zhou, L. Pu, J. Zhang, X. Li, F. Li, L. Long, S. Li and Y. He, Catal. Sci. Technol., 2021, 11, 7905–7913, 10.1039/d1cy01505b.
- M. G. B. Drew, P. Higgins and G. M. McCann, J. Chem. Soc., Chem. Commun., 1987, 1385–1387, 10.1039/C39870001385.
- W. R. Heinz, D. Staude, D. Mayer, H. Bunzen and R. A. Fischer, Dalton Trans., 2021, 50, 5226–5235, 10.1039/d1dt00118c.
- A. G. Shard, J. Vac. Sci. Technol., A, 2020, 38, 041201, DOI:10.1116/1.5141395.
- V. Martinez, B. Karadeniz, N. Biliškov, I. Lončarić, S. Muratović, D. Žilić, S. M. Avdoshenko, M. Roslova, A. A. Popov and K. Užarević, Chem. Mater., 2020, 32, 10628–10640, DOI:10.1021/acs.chemmater.0c03796.
- A. Kathuria, M. G. Abiad and R. Auras, Polym. Int., 2013, 62, 1144–1151, DOI:10.1002/pi.4478.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069, DOI:10.1515/pac-2014-1117.
- Y. Chen, X. Mu, E. Lester and T. Wu, Prog. Nat. Sci.: Mater. Int., 2018, 28, 584–589, DOI:10.1016/j.pnsc.2018.08.002.
- W. W. Lestari, M. Adreane, C. Purnawan, H. Fansuri, N. Widiastuti and S. B. Rahardjo, IOP Conf. Ser.: Mater. Sci. Eng., 2016, 107, 012030, DOI:10.1088/1757-899X/107/1/012030.
- N. Stock and S. Biswas, Chem. Rev., 2012, 112, 933–969, DOI:10.1021/cr200304e.
- K.-S. Lin, A. K. Adhikari, C.-N. Ku, C.-L. Chiang and H. Kuo, Int. J. Hydrogen Energy, 2012, 37, 13865–13871, DOI:10.1016/j.ijhydene.2012.04.105.
- M. C. Biesinger, Surf. Interface Anal., 2017, 49, 1325–1334, DOI:10.1002/sia.6239.
- P. Guo, H. Cheng, X. Zeng, L. Tao, B. Peng, S. Zhang, R. A. Fischer and M. Muhler, ChemPlusChem, 2022, 87, e202200303, DOI:10.1002/cplu.202200303.
- NIST X-Ray Photoelectron Spectroscopy Database, NIST Standard Reference Database Number 20, National Institute of Standards and Technology, Gaithersburg MD, 20899, 2000, DOI:10.18434/T4T88K, (retrieved [27.12.2023]).
- D. J. Morgan, Surf. Interface Anal., 2015, 47, 1072–1079, DOI:10.1002/sia.5852.
- J. Huo, Y. Wang, L. Yan, Y. Xue, S. Li, M. Hu, Y. Jiang and Q.-G. Zhai, Nanoscale, 2020, 12, 14514–14523, 10.1039/d0nr02697b.
- S. Öztürk, G.-h. Moon, A. Spieß, E. Budiyanto, S. Roitsch, H. Tüysüz and C. Janiak, ChemPlusChem, 2021, 86, 1106–1115, DOI:10.1002/cplu.202100278.
- L. Sondermann, W. Jiang, M. Shviro, A. Spieß, D. Woschko, L. Rademacher and C. Janiak, Molecules, 2022, 27, 1241, DOI:10.3390/molecules27041241.
- T. H. Y. Beglau, L. Rademacher, R. Oestreich and C. Janiak, Molecules, 2023, 28, 4464, DOI:10.3390/molecules28114464.
- L. Yang, G. Zhu, H. Wen, X. Guan, X. Sun, H. Feng, W. Tian, D. Zheng, X. Cheng and Y. Yao, J. Mater. Chem. A, 2019, 7, 8771–8776, 10.1039/c9ta00819e.
- T. Qiu, Z. Liang, W. Guo, S. Gao, C. Qu, H. Tabassum, H. Zhang, B. Zhu, R. Zou and Y. Shao-Horn, Nano Energy, 2019, 58, 1–10, DOI:10.1016/j.nanoen.2018.12.085.
- T. Shinagawa, A. T. Garcia-Esparza and K. Takanabe, Sci. Rep., 2015, 5, 13801, DOI:10.1038/srep13801.
- R. L. Doyle, I. J. Godwin, M. P. Brandon and M. E. G. Lyons, Phys. Chem. Chem. Phys., 2013, 15, 13737–13783, 10.1039/c3cp51213d.
- Y. P. Sánchez, A. Santos and P. R. Bueno, J. Phys. Chem. C, 2022, 126, 3151–3162, DOI:10.1021/acs.jpcc.1c07801.
- S. Anantharaj and S. Noda, ChemElectroChem, 2020, 7, 2297–2308, DOI:10.1002/celc.202000515.
- C. Zhou, X. Chen, S. Liu, Y. Han, H. Meng, Q. Jiang, S. Zhao, F. Wei, J. Sun, T. Tan and R. Zhang, J. Am. Chem. Soc., 2022, 144, 2694–2704, DOI:10.1021/jacs.1c11675.
- A. Roy, A. Ray, S. Saha, M. Ghosh, T. Das, M. Nandi, G. Lal and S. Das, Int. J. Energy Res., 2021, 45, 16908–16921, DOI:10.1002/er.6885.
- P. Connor, J. Schuch, B. Kaiser and W. Jaegermann, Z. Phys. Chem., 2020, 234, 979–994, DOI:10.1515/zpch-2019-1514.
- J. Yu, S. Giancola, B. Khezri, D. Nieto-Castro, J. Redondo, F. Schiller, S. Barja, M. C. Spadaro, J. Arbiol, F. A. Garcés-Pineda and J. R. Galán-Mascarós, EES Catal., 2023, 1, 765–773, 10.1039/d3ey00101f.
- Z. Zhang, C. Yang, S. Wu, A. Wang, L. Zhao, D. Zhai, B. Ren, K. Cao and Z. Zhou, Adv. Energy Mater., 2019, 9, 1802805, DOI:10.1002/aenm.201802805.
- D. Chakraborty, C. Danvad Damsgaard, H. Silva, C. Conradsen, J. Lind Olsen, H. W. P. Carvalho, B. Mutz, T. Bligaard, M. J. Hoffmann, J.-D. Grunwaldt, F. Studt and I. Chorkendorff, Angew. Chem., 2017, 129, 8837–8841, DOI:10.1002/anie.201703468.
- W. Long, Q. Zhai, J. He, Q. Zhang, W. Deng and Y. Wang, ChemPlusChem, 2012, 77, 27–30, DOI:10.1002/cplu.201100050.
- E. Petrucci, F. Porcelli, M. Orsini, S. De Santis and G. Sotgiu, Metals, 2022, 12, 316, DOI:10.3390/met12020316.
- C. Zhang, Y. Xie, M. Zhao, A. E. Pentecost, Z. Ling, J. Wang, D. Long, L. Ling and W. Qiao, ACS Appl. Mater. Interfaces, 2014, 6, 9751–9759, DOI:10.1021/am502173x.
- M. Luysberg, M. Heggen and K. Tillmann, J. Large– Scale Res. Facil., 2016, 2, A77, DOI:10.17815/jlsrf-2-138.
Footnote |
| † Electronic supplementary information (ESI) available: EDX spectra, SEM images, PXRD, TGA, IR, porosity parameters, XPS, electrochemical data, PXRD after KOH treatment, elemental analysis. See DOI: https://doi.org/10.1039/d4mr00021h |
| This journal is © The Royal Society of Chemistry 2024 |