
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanoscale assembly of enantiomeric supramolecular gels driven by the nature of solvents†
Tómas A.
Gudmundsson
 ab,
Geethanjali
Kuppadakkath
a,
Dipankar
Ghosh
a,
Manuel
Ruether
b,
Annela
Seddon
c,
Rebecca E.
Ginesi
d,
James
Doutch
e,
Dave J.
Adams
d,
Thorfinnur
Gunnlaugsson
b and
Krishna K.
Damodaran
*a
ab,
Geethanjali
Kuppadakkath
a,
Dipankar
Ghosh
a,
Manuel
Ruether
b,
Annela
Seddon
c,
Rebecca E.
Ginesi
d,
James
Doutch
e,
Dave J.
Adams
d,
Thorfinnur
Gunnlaugsson
b and
Krishna K.
Damodaran
*a
aDepartment of Chemistry, Science Institute, University of Iceland, Dunhagi 3, 107 Reykjavík, Iceland. E-mail: krishna@hi.is
bSchool of Chemistry and Trinity Biomedical Sciences Institute (TBSI) and Advanced Materials and BioEngineering Research (AMBER) Centre, Trinity College Dublin, The University of Dublin, Dublin 2, D02 PN40, Ireland
cSchool of Physics, HH Wills Physics Laboratory, Tyndall Avenue, University of Bristol, Bristol, BS8 1TL, UK
dSchool of Chemistry, University of Glasgow, Glasgow, G12 8QQ, UK
eISIS Pulsed Neutron and Muon Source, Harwell Science and Innovation Campus, Didcot, OX11 0QX, UK
First published on 3rd April 2024
Abstract
Understanding the key parameters that control the self-assembly process is critical to predict self-assembly modes in multi-component systems, which will lead to the development of nanofibrous materials with tuneable properties. Enantiomeric amino acid-based low-molecular-weight gelators (LMWGs) were mixed in polar (polar protic) and aromatic apolar (aromatic) solvents and compared to their individual counterparts to probe the effect of solvent polarity on the self-assembly process. Scanning electron microscopy (SEM) reveals that xerogels of individual components display hollow needles in polar protic solvents, while chiral coils are observed in aromatic solvents. In contrast, the multi-component gel displays hollow needle morphologies in both solvents, indicating similar morphologies in polar protic solvents but an entirely different nanostructure for the individual gel networks in aromatic solvents. PXRD experiments performed on the dried gels showed that the nature of the solvents plays a vital role in the co-assembly process of multi-component gels. The self-assembly modes and the gel state structure of the gels are analysed by wide-angle X-ray diffraction (WAXS) and small-angle neutron diffraction (SANS), which reveals that the mixed gel undergoes different co-assembly modes depending on the nature of the solvent systems. This study shows that different co-assembly modes can be achieved for structurally similar components by varying the solvent polarity, demonstrating the importance of solvent choice in the self-assembly process of multi-component gels.
Introduction
Natural processes rely on complex multi-component systems built upon simple building blocks such as fatty acids, sugars, or amino acids.1 These small components self-assemble to form functional large-scale architectures, e.g., the DNA double-helix, or protein-based ion channels in cell membranes. This has prompted researchers to mimic nature's design to develop functional materials by employing the self-assembly of synthetic multi-component systems.2 Supramolecular gels based on low molecular weight gelators (LMWGs)3–10 are a fascinating class of soft materials because the bulk properties of these materials depend on the nanoscale self-assembly of the gelators, which can be tuned by manipulating the self-assembly process.11–16 Multi-component systems based on LMWGs possess properties beyond their single-component counterparts. Additionally, the properties of these systems can be tuned depending on the proportional composition of the individual components.17–21 There is no surprise that the development of such multi-component gels has received increased interest in recent times, as they can display a wide range of potential applications such as oil spill recovery,22 anti-microbial gels,23 drug delivery,24,25 and catalysis.26Multi-component gels are obtained by the interaction of two or more components, such as gelators or gelators, with non-gelating organic compounds/metal ions.27 In mixed-gelator systems, the spatial orientation of the individual gelators and the dynamic nature of the non-covalent interactions play a vital role in the self-assembly process. Mixing individual gelators could lead to destructive interference resulting in collapse, or constructive interactions can lead to self-sorting or co-assembled fibres.28,29 In co-assembled networks, gelators may exhibit a statistical distribution in the gel network termed “random co-assembly”, as opposed to an organised distribution “specific co-assembly”. Mixing structurally similar gelators can lead to favourable interactions between components, resulting in improved gel properties.30,31 For example, enantiomeric mixtures can form more thermodynamically and kinetically stable gels due to a more compact gelator packing, as previously shown by our group in multi-component enantiomeric LMWGs based on bis(urea) compounds tagged with phenylalanine methyl esters.30
Enantiomeric LMWGs based on amino acid derivatives take their inspiration from natural building blocks and are ideal candidates for designing mixed gels owing to their availability in enantiomeric and racemic forms, easily modifiable functional groups, and are open to applications in vivo due to their biocompatibility.30–34 Multi-component gels based on amino acids obtained by mixing enantiomeric gels display tuneable properties.35–37 We have reported the crystallographic evidence of the self-assembly process in multi-component gels based on amino acid derivatives and showed that the structural similarity of the enantiomers could lead to specific co-assembly in multi-component gels.31 The choice of solvent plays an important role in the nature and mode of aggregation of multi-component gels,38,39 where self-assembly modes can be tuned by solvent polarity. For example, polar solvents compete with polar moieties in the gelator, causing the binding strength of the gelator to decrease.40
The C3-symmetric supramolecular platform benzene-1,3,5-tricarboxamide (BTA)41–45 has been thoroughly studied for its tendency to form nanofibers leading to gelation in a wide range of solvents,46–49 which is ideal for studying the effect of solvent polarity on the self-assembly process. We have therefore chosen to explore the gelation properties of amino acid derived BTA in various solvents. The solid-state structural analysis of amino acid derivatives of BTA indicated that the self-assembly was dictated by hydrogen bonding along with π–π stacking, and the enantiomers often display helices with opposite handedness.42,50–52 In this work, we have tagged BTA with enantiomeric methyl valinate (MVBTA) to study the effect of solvents on the self-assembly of multi-component LMWGs. Various functionalities, such as polar amino acid ester moieties, an aromatic centre, and large non-polar groups, may facilitate different self-assembly pathways in different solvents. In contrast, single enantiomers of MVBTA have been studied before,53,54 including gelation in ionic liquids,55 but studies of multi-component MVBTA have not yet been reported.
Results and discussion
Synthesis of MVBTA
The enantiomeric R,R,R-MVBTA (1R) and S,S,S-MVBTA (1S) isomers of C3-symmetric benzene-1,3,5-tricarboxamide tagged with methyl valinate were synthesised by reacting the corresponding methyl valinate (R- or S-) hydrochloride enantiomer with benzene-1,3,5-tricarbonyl trichloride in dry dichloromethane (Scheme 1). The ester derivatives of amino acids were selected due to their better solubility in a wide range of solvents, such as polar, non-polar, aromatic hydrocarbons, hydrogen bonding, mixed aqueous solvents, etc. Circular dichroism (CD) experiments were performed in 0.0125 wt/v% solutions of 1R and 1S in ethanol (Fig. S1, see ESI†) to confirm the preservation of the chirality of the enantiomers. | ||
| Scheme 1 Synthesis of 1R and 1S enantiomers of MVBTA. | ||
Solvent screening
The gels were screened for gelation in various solvents by dissolving an appropriate amount of the gelator by heating and then sonicating until opacity was observed. The solutions were left to settle undisturbed overnight, and gelation was determined as successful by an inversion test. Multi-component gels (1R + 1S) were generated following the same procedure with an equimolar mixture of 1R and 1S.The solvents that were successfully gelled by MVBTA were categorised based on their non-covalent interactions. Polar solvents with hydrogen-bond donating capabilities were designated as “polar protic” solvents, including various alcohols and aqueous (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) solvent mixtures. Apolar aromatic solvents able to interact with the slightly electron-deficient π-cloud of MVBTA were designated as “aromatic” solvents. Meanwhile, a third category of polar solvents with only hydrogen-bond accepting capabilities was designated as “polar aprotic” solvents (Table S1, see ESI†). Gelation was unsuccessful in apolar aliphatic solvents and was excluded from the study. The inherent differences in the non-bonding interactions between the solvents and the gelator will enable us to investigate the effect of solvents in the self-assembly of LMWGs. The comparison of the gels indicated that the physical appearance of the gels depended on the type of solvent; gels from polar protic solvents were opaque and had the consistency of soaked wool, and gels from aromatic solvents were translucent and more traditionally “gel-like” (Fig. 1).
:1, v/v) solvent mixtures. Apolar aromatic solvents able to interact with the slightly electron-deficient π-cloud of MVBTA were designated as “aromatic” solvents. Meanwhile, a third category of polar solvents with only hydrogen-bond accepting capabilities was designated as “polar aprotic” solvents (Table S1, see ESI†). Gelation was unsuccessful in apolar aliphatic solvents and was excluded from the study. The inherent differences in the non-bonding interactions between the solvents and the gelator will enable us to investigate the effect of solvents in the self-assembly of LMWGs. The comparison of the gels indicated that the physical appearance of the gels depended on the type of solvent; gels from polar protic solvents were opaque and had the consistency of soaked wool, and gels from aromatic solvents were translucent and more traditionally “gel-like” (Fig. 1).
 | ||
| Fig. 1 Gels of 1S, 1R and 1R + 1S obtained from methanol (left) and mesitylene (right) at 2.0 wt/v%, respectively. | ||
Critical gel concentration (CGC)
The CGC of a gel is the minimum percentage of gelator required for gelation in a given solvent and is a metric for the gelation efficiency of the network. The CGC was determined for each gel, revealing an interesting difference between the individual enantiomeric gels and multi-component gels across solvent types (Table 1). The CGC of 1R and 1S gels was found to be lower than the 1R + 1S gels in polar protic solvents, but a reverse trend was observed in aromatic solvents.| Group | Solvent | 1R | 1S | 1R + 1S |
|---|---|---|---|---|
| a The CGC (wt/v%) table with more solvents is available in ESI (Table S1†). | ||||
| Polar protic | Methanol | 1.7 | 1.7 | 1.9 |
| Ethanol | 0.7 | 0.9 | 1.4 | |
| n-Propanol | 1.2 | 1.0 | 2.3 | |
| n-Butanol | 1.2 | 1.0 | 2.0 | |
| DMF/H2O (1:1, v/v) |
0.8 | 0.8 | 1.2 | |
| DMSO/H2O (1:1, v/v) |
0.8 | 0.8 | 1.3 | |
| Aromatic | Benzene | 1.4 | 1.4 | 1.3 |
| Toluene | 1.5 | 1.5 | 1.2 | |
| o-Xylene | 1.2 | 1.1 | 0.6 | |
| m-Xylene | 1.2 | 1.1 | 0.5 | |
| p-Xylene | 1.2 | 1.3 | 0.5 | |
| Mesitylene | 1.2 | 1.2 | 0.8 | |
A deeper look revealed interesting relationships between the solvent structure and the CGC. For polar protic solvents, methanol led to inefficient gelation in single-component gels. However, the CGC lowered significantly as the carbon-chain length increased to two for ethanol and remained relatively stable beyond that, showing the importance of apolar interactions for gelation in this system. Comparison of the CGC values in aromatic solvents showed that di- and tri-alkyl substitution in the benzene-derivatives (o-, m-, & p-xylene and mesitylene) resulted in lower CGC value of the mixed gels more than in benzene and toluene. This may have been due to the positive inductive effect of the methyl groups, where π-clouds of more substituted benzene rings are more electron rich, leading to stronger π–π interactions with the electron deficient π-cloud of BTA. A reverse trend observed was for the polar aromatic solvents with electron-withdrawing groups (chlorobenzene and nitrobenzene), which indicated that the electron density of the solvent π-cloud affected the self-assembly modes in gel network formation. This prompted us to investigate the stability of the gels towards thermal and mechanical perturbation to follow up on these observations.
Thermal stability (Tgel)
The thermal stabilities of the gels were evaluated across the solvent types by evaluating the gel–sol transition temperature (Tgel).8 All gels were prepared at the same concentration in order to compare the stabilities under similar conditions. Surprisingly, all gels were destabilised upon mixing regardless of the solvent type despite the relative differences in the CGC for the mixed gels (Table 2). Additionally, this was in contrast to previous observations where mixed gels based on C2-symmetric benzene-1,4-dicarboxamide analogue in similar solvent systems displayed enhanced thermal stability.31 Therefore, these results indicate that the symmetry of the linker plays an important role in the thermal stability of the multi-component gels. When looking within solvent categories, the Tgel of gels from polar protic solvents showed that the stability of all gels increased with longer carbon-chain solvents. The efficient gelation in the mixed solvent mixtures (DMF/water or DMSO/water) may be attributed to the insolubility of the compound in the antisolvent (water). The thermal stabilities of gels in polar protic solvents can be significantly tuned by mixing. Similar observations were made in aromatic solvents, where increased substitution led to greater stability. Interestingly, m-substituted xylene showed a marked increase in stability over o- and p-substitution, revealing the importance of steric interactions in the self-assembly. The thermal stability of gels in mesitylene could not be determined due to the degradation of the silicon oil used for the experiments observed soon after 160.0 °C.| Group | Solvent | wt/v% | 1R | 1S | 1R + 1S |
|---|---|---|---|---|---|
| Polar protic | Methanol | 2.5 | 68 | 69 | 66 |
| Ethanol | 2.5 | 76 | 74 | 68 | |
| n-Propanol | 2.5 | 88 | 86 | 70 | |
| n-Butanol | 2.5 | 98 | 96 | 81 | |
| n-Pentanol | 2.5 | 104 | 101 | 90 | |
| DMF/H2O (1:1, v/v) |
2.5 | 116 | 118 | 99 | |
| Aromatic | Toluene | 1.5 | 142 | 144 | 133 |
| o-Xylene | 1.5 | 150 | 151 | 143 | |
| m-Xylene | 1.5 | 155 | 156 | 148 | |
| p-Xylene | 1.5 | 152 | 150 | 144 | |
The reversible nature of the sol-to-gel phase transition of supramolecular gels can be analysed by dynamic scanning calorimetry (DSC), which provides insight into the self-assembly of multi-component gels.56 Gel network formation is observed in the DSC cooling cycle as an exothermic peak representing the sol-to-gel transition. We analysed the phase transition of the individual and mixed enantiomeric gels in n-butanol at 2.1 wt/v%, a polar protic solvent with Tgel below the boiling point of the solvent for accurate measurements (Fig. S2–S4, see ESI†). 1R and 1S displayed sharp exothermic signals at 75.0 °C and 80.0 °C, respectively. However, the multi-component 1R + 1S gels displayed a broad signal ranging from 32.0 to 64.0 °C with an exothermic maximum at 44.0 °C, indicating the formation of a polydisperse system presumably arising from co-assembled networks. The enthalpy of the formation was calculated and compared across gels. The difference in the enthalpy (kJ mol−1) was not statistically significant; t(7) = 0.2343, p = 0.82 for 1R + 1S (M = −0.799, SD = 0.021) and the individual gels (M = −0.799, SD = 0.142), which indicated the self-assembled structures remained structurally similar upon mixing. Unfortunately, the analysis in aromatic solvents was not performed as the Tgel was higher than the boiling point of the gelling solvents in all cases. We have selected the most common solvents (methanol and ethanol) and solvent mixtures (DMF/water and DMSO/water) from the polar protic category to evaluate the physical properties. We selected solvents such as toluene, one of the xylenes (o-xylene) and mesitylene with different alkyl substitutions for the aromatic solvent category.
Rheology
Rheology has been used to study the stiffness, deformation, and flow characteristics of gels, providing important insight into the structural characteristics of the gel network.57 Rheological studies were performed to confirm the formation of viscoelastic materials such as gels and to complement the relative thermal stabilities of the 1R, 1S, and 1R + 1S with their respective mechanical strengths. Initially, the viscoelastic region of the gel was determined by oscillatory amplitude sweep experiments (Fig. S5–S7, see ESI†), which reflects the minimum force required to initiate flow characteristics to the gels. The effect of mixing gels on the mechanical stability was evaluated by performing the oscillatory frequency sweep of 1R, 1S and 1R + 1S gels in o-xylene, toluene, mesitylene, MeOH, EtOH and in aqueous solutions of DMF and DMSO at 3.0 wt/v% (Fig. S8–S13, see ESI†).For each gel, the storage modulus (G′) and loss modulus (G′′) values were constant under a frequency range of 0.1–10.0 Hz in all solvents, confirming the gel characteristics. The difference in the elastic modulus was compared for the enantiomeric and mixed gels across the two solvent systems to analyse the effect of each solvent on the mechanical properties of mixed gels, which indicated that mechanical strength was similar for individual and mixed gels in both the solvent categories (Fig. 2).
 | ||
| Fig. 2 Frequency sweep performed for 1R, 1S and 1R + 1S in DMF/H2O at 3.0 wt/v%, similar results were observed across all gels (see ESI†). | ||
Nanofibre morphology
The morphologies of 1R, 1S and 1R + 1S gels in polar protic and aromatic solvents were analysed by imaging their respective xerogels (Fig. S14–S19, see ESI†). SEM images of 1R and 1S xerogels from methanol displayed a hollow needle morphology with widths ranging from 1.0 to 10.0 μm (Fig. S14, see ESI†). While the mixed gels (1R + 1S) displayed similar hollow needle-like morphologies, the needles were thinner, with diameters ranging from 0.2 to 2.0 μm (Fig. 3a) and similar needle like morphologies were observed for ethanol xerogels (Fig. S15, see ESI†). Banerjee et al. showed that the morphology of drop-cast aqueous solutions of enantiomeric MVBTA displayed opposite helicity using transmission electron microscopy (TEM).53 This prompted us to investigate the morphology of the gel fibres in the presence of water. The morphologies of the xerogels from the enantiomeric and mixed gels prepared from DMF/H2O (1:1, v/v) displayed hollow needle-like morphologies (Fig. 3b & Fig. S16, see ESI†) and similar results were observed for DMSO/H2O (1:1, v/v) xerogels (Fig. S17, see ESI†).
 | ||
| Fig. 3 SEM images of mixed MVBTA xerogels (2.0 wt/v%) in (a) methanol, (b) DMF/water (1:1, v/v), (c) mesitylene and (d) o-xylene. | ||
Dried fibres obtained from gels in polar protic solvents did not express chirality, unlike those formed through the evaporation of dilute solutions, probably due to the differences in sample preparation. These results showed that mixing enantiomers in polar protic solvents did not significantly affect the morphology of the gel fibres. Analysis of the SEM images of 1R and 1S xerogels from mesitylene proved to be radically different from the xerogels of polar protic solvents. SEM images of the enantiomeric gels prepared from mesitylene displayed chiral helices and impressive coils, and the helicity was reversed for the opposite enantiomer (Fig. 4).
 | ||
| Fig. 4 SEM images of the enantiomeric xerogels of MVBTA (at 2.0 wt/v%) in mesitylene (top) and o-xylene (bottom). | ||
The coils consisted of approximately 0.1 μm thick tapes that twisted into larger structures with thickness ranging from 0.2 to 1.0 μm. The fibres were twisted into some interesting shapes, where the samples often showed a mixture of thick coils, telephone cord-like species and twisted helices. In some cases, the morphology of the fibres resembled wrapped cords on twisted fibres (Fig. S18, see ESI†). The helicity of the fibres was reversed when the opposite enantiomeric gelator was analysed. However, any sign of chirality disappeared upon mixing the enantiomers, resulting in fibres with tape-like and needle morphologies (Fig. 3c). Similar morphologies were observed for the enantiomeric gels (Fig. 4) and the mixed gels (Fig. 3d) in o-xylene. The nanostructures of the gel network could be significantly tuned by solvent choice, from the expression of chirality to its erasure in polar solvents or mixed systems. The comparisons of the morphologies of enantiomeric and mixed xerogels obtained from polar hydrogen-bonding and non-polar aromatic solvents show a clear difference in their hierarchical structures. These results indicate that the gelators display similar characteristics and self-assembly patterns in solvents from similar solvent categories.
Solid state 13C-NMR
Solid-state (ss) NMR spectroscopy was used to compare the packing patterns of the gelator molecule in the xerogel states. The nanostructures of the gels were probed by drying the gels to measure the chemical environment of the resulting xerogel. We have performed the ss-13C-NMR experiments of the 1R, 1S, and 1R + 1S xerogels obtained from polar protic (MeOH, EtOH and DMF/water) and aromatic solvent gels (mesitylene and o-xylene) at 2.0 wt/v% to analyse the effect of the solvent in the self-assembly of multi-component gels (Fig. S20–S24, see ESI†). The signals observed in the 1R and 1S spectra retained near-identical chemical shifts; however, additional signals appeared after mixing, indicating the presence of different environments that may result from co-assembled networks.Powder X-ray diffraction (PXRD)
Powder X-ray diffraction (PXRD) of the xerogels obtained from polar protic and aromatic solvents were analysed to gain insight into the packing modes of LMWGs (Fig. S25–S31, see ESI†). PXRD patterns help to identify the role of various non-bonding interactions in gels by comparing the PXRD patterns of the individual xerogels with the mixed enantiomeric gels obtained from polar protic and aromatic solvents.58 The PXRD patterns of dried enantiomeric gels (1R and 1S) were superimposable in both solvent types and akin to the ss-NMR results. The PXRD patterns of the mixed enantiomeric gels displayed different patterns compared to the individual enantiomeric xerogels in the corresponding solvents. These results show that a new packing domain was observed in the 1R + 1S mixed xerogels, indicative of co-assembly. The PXRD patterns of 1R + 1S from a particular solvent category were nearly identical, but the PXRD patterns of 1R + 1S from different solvent categories (polar protic and aromatic) were not matching, indicating the formation of a different co-assembled network depending on the solvent categories (Fig. S32, see ESI†). These results indicate that the nature of the solvents plays a vital role in the self-assembly process of multi-component gels.Gel-state scattering
X-ray and neutron scattering are powerful techniques to examine the nano- and micro-structures of supramolecular assemblies by measuring their scattering intensities at small or wide angles.59 Wide-angle X-ray scattering (WAXS) data were collected for 1R, 1S and 1R + 1S in each solvent category to verify the observations made in the xerogel state, as drying can cause artefacts (Fig. 5 and Fig. S33, see ESI†).60 The packing modes of 1R and 1S in methanol and DMF/H2O showed distinct signals which were near-identical, as would be expected for enantiomeric species. The WAXS data for the mixed 1R + 1S gels displayed a slightly different pattern compared to the individual enantiomers, signifying a different packing mode, which fit well with the observations obtained from PXRD.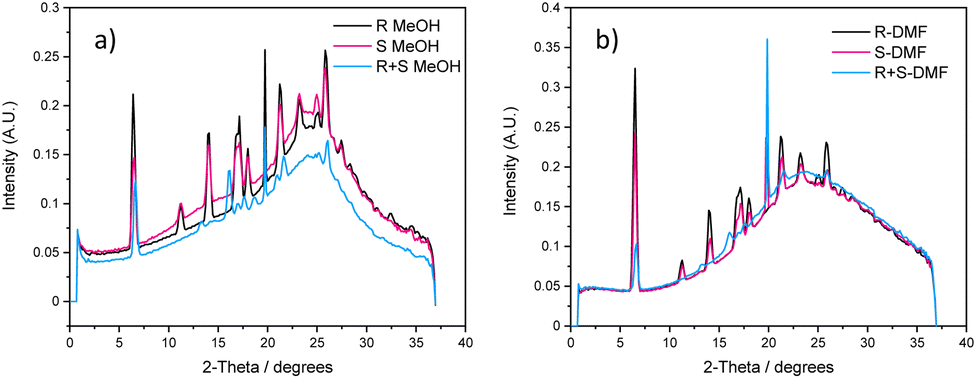 | ||
| Fig. 5 Overlaid WAXS for 2.0 wt/v% MVBTA gels made in (a) methanol and (b) in DMF/water (1:1, v/v). | ||
The experiments repeated using gels from mesitylene gave similar results. 1R and 1S were of poor resolution due to the polydispersity of the sample (as observed in SEM), yielding nearly no discrete signals. Upon mixing, the order was improved in 1R + 1S, and discrete signals appeared (Fig. S33, see ESI†), indicating a different type of packing mode, presumably due to the better packing arising from co-assembled enantiomers.
As further confirmation, small-angle neutron scattering (SANS) was performed to compare the packing between enantiomers and mixed gels (Fig. S34 and S35, see ESI†). Gels were prepared in DMSO-d6/D2O as representatives of polar protic solvent gels. Similar scattering was observed for 1S and 1R + 1S and the latter scattered slightly stronger, but the scattering pattern was weaker for 1R. The data for 1R can be best fit to a combined cylinder and power law model (∼4.0), whilst the data for 1S and 1R + 1S were best fit by a power law (∼4.0) alone (Table S2, see ESI†). Although these data imply that similar scattering was observed for 1S and 1R + 1S, it is hard to interpret this alone, because, looking at the scattering intensities of 1R + 1S, this would possibly be consistent with a system that was scattering as a mixture of 1S and 1R. Since 1R scatters were so weak, the scattering of 1S dominated the system, and therefore, the scattering intensities would possibly be consistent with the co-assembled network in 1R + 1S. In toluene-d8, the scattering of 1R + 1S was entirely different from 1R and 1S (Table S3, see ESI†), which is substantiated by the SEM images. These data strongly imply a co-assembled network in 1R + 1S because the scattering of 1R + 1S was different from what would be expected from a mixture of the two components.
Conclusions
Enantiomeric gels based on amino acid derivatives (1R and 1S) were synthesised by tagging valine methyl esters to the tripodal BTA, and multi-component gels 1R + 1S were prepared by mixing the enantiomeric gels. We have shown that the gelation properties of the individual and multi-component gels in various solvent systems depended on the type and nature of the solvents. The morphologies of the xerogels from polar protic solvents did not show a drastic difference, but distinct morphologies were observed for individual and multi-component gels in aromatic solvents, indicating a different co-assembly mode compared to polar protic solvents. PXRD experiments revealed that self-assembly modes in individual gels were not affected by the nature of the solvent, but different co-assembly modes were observed in enantiomeric multi-component systems depending on the solvent category. The shape and packing modes of the gel fibres in the gel state were analysed by WAXS and SANS in both solvent systems. These results confirmed that the mixed enantiomeric gels formed a different co-assembled network in polar protic and aromatic solvents. Analysis of the results suggest that the co-assembly modes of multi-component systems in the gel state can be tuned depending on the nature of the solvents, which shows the impressive tunability of the bulk properties of multi-component gels by simple solvent selection. This study shows that structural similar components (enantiomers) can display different co-assembly modes and will add to the ongoing efforts to unravel the factors dictating the self-assembly modes in the multi-component system, enabling us to design a multi-component system with predictable properties.Experimental
Materials and methods
All starting materials were purchased from commercial sources and were used as supplied. Anhydrous dichloromethane (DCM) was obtained by distilling over CaH2. Anhydrous methanol and ethanol were obtained by distilling over Mg turnings in the presence of iodine.Synthesis
S,S,S-methyl valinate BTA (1S): yield: 2.15 g, 90.0%. 1H NMR (400 MHz, CDCl3) δ 8.39 (s, 3H), 7.04 (d, J = 7.4 Hz, 3H), 4.78 (dd, J = 8.6, 5.3 Hz, 3H), 3.80 (s, 9H), 2.35–2.23 (m, 3H), 1.02 (dd, J = 6.8, 3.5 Hz, 18H). 13C NMR (100 MHz, CDCl3) δ 172.54, 165.78, 135.03, 128.86, 58.17, 52.57, 31.68, 19.28, 18.35. MS (ESI) calcd for C27H39N3O9 549.26; found 572.2579 [M + Na]+. CHN analysis found: C, 59.0; N, 7.2; H, 7.4. Calc. for C27H39N3O9: C, 59.0; N, 7.65; H, 7.15%.
R,R,R-methyl valinate BTA (1R): yield: 1.93 g, 80.6%. 1H NMR (400 MHz, CDCl3) δ 8.41 (s, 3H), 6.97 (d, J = 8.5 Hz, 3H), 4.78 (dd, J = 8.6, 5.2 Hz, 3H), 3.79 (s, 9H), 2.35–2.23 (m, 3H), 1.02 (dd, J = 6.8, 4.2 Hz, 18H). 13C NMR (100 MHz, CDCl3) δ 172.48, 165.76, 135.08, 128.86, 58.16, 52.57, 31.70, 19.27, 18.33. MS (ESI) calcd for C27H39N3O9 549.26; found 572.2579 [M + Na]+. CHN analysis found: C, 59.0; N, 7.2; H, 7.5. Calc. for C27H39N3O9: C, 59.0; N, 7.65; H, 7.15%.
Gelation details
Critical gel concentration (CGC)
Gelation was attempted in various solvents, starting at 1.0 wt/v% and adding 1.0 wt/v% until a gel formed. A gel was prepared at the lowest necessary concentration for a specific solvent, then the additional solvent was added in portions, and the gelation was repeated until the excess solvent was left on top of the gel. The solvent was decanted, the CGC was calculated by weight, and a new gel was prepared to confirm the value. The experiment was repeated as necessary.T gel experiment
Enantiopure and mixed gels were prepared at 2.5 wt/v% in polar protic solvents and 1.5 wt/v% in aromatic solvents and left to stand for 24 hours. A spherical glass ball (108 mg, 3.0 mm diameter) was carefully placed on top of the gel, and the vial was lowered into an oil bath. The oil bath was gradually heated and the temperature at which the ball touched the bottom of the vial was recorded as Tgel.Differential scanning calorimetry (DSC)
The DSC experiments were performed using a PerkinElmer DSC 8000 instrument calibrated against zinc and indium. Gels made at 2.1 wt/v% in 2-butanol were weighed and placed into 60.0 μL stainless steel pans with a pressure threshold of 24 bar. The cooling cycle spectra were collected by rapidly heating the samples to 110 °C and holding them for 20 minutes before cooling to 20.0 °C at 5.0 °C min−1. The process was repeated for a total of 3 cycles.Rheology
The rheology experiments were performed using an Anton Paar Modular Compact Rheometer 302 with a 25.0 mm stainless-steel parallel plate. Gels of 1R, 1S and 1R + 1S were prepared at 3.0 wt/v% and left to stand for over 24 hours. The linear viscoelastic region was determined by an amplitude sweep from 0.01% to 100%, and the viscoelastic properties of the gels were evaluated by oscillatory measurements, using a frequency sweep starting at 10.0 Hz going down to 0.1 Hz with a 0.0125% deformation.Scanning electron microscopy (SEM)
Gels were prepared at 2.0 wt/v% and left undisturbed for 24 hours. The gels were filtered by suction, and the residue was dried in the fume hood overnight. The xerogels were spread over a conductive carbon adhesive and gold-coated in an Edwards Sputter Coater S150B before being examined in a Zeiss Supra 25 FE-SEM with an SE2 detector.NMR & MS experiments
The 1H- and 13C-NMR spectra were recorded on a Bruker Avance III 400 spectrometer (1H-NMR: 400.23 MHz, 13C-NMR: 100.63 MHz). Solid-state NMR spectra were recorded on a Bruker 400 Advance HD (1H-NMR: 400.13 MHz, 13C-NMR: 100.61 MHz) with a 3.2 mm HX CP/MAS probe and the samples were prepared by creating gels at 2.0 wt/v%, filtering and drying them to obtain the corresponding xerogels. All mass spectra were recorded on a Bruker ESI Compact spectrometer.Powder X-ray diffraction (PXRD)
The xerogels of 1R, 1S and 1R + 1S were obtained by filtering and drying 2.0 wt/v% gels of the respective compounds in MeOH, EtOH, DMF/water (1:1, v/v), DMSO/water (1:1, v/v), toluene, o-xylene and mesitylene. The xerogels were placed on a glass slide and PXRD was measured in a PANalytical instrument, with Cu-anode, within 2θ range of 4.0–60.0°, at a step size of 0.02°.
Wide-angle X-ray scattering (WAXS)
Samples were prepared in stainless steel flat cells between two 7.0 μm thick mica windows (Xenocs). Data were collected on a Ganesha 300XL SAXS/WAXS instrument (Xenocs) in a Q range of 0.07 Å−1 ≤Q ≤ 2.8 Å−1, and for an exposure time of 600 s. Data were corrected for absolute intensity, transmission, and thickness.Small-angle neutron scattering (SANS)
Gels of 1R, 1S and 1R + 1S were obtained at 1.3 wt/v% in DMSO-d6/D2O (1:1, v/v), or at 1.5 wt/v% in toluene-d8 using the gelation procedure outlined above. SANS measurements were performed using the SANS2D instrument (ISIS, Rutherford Appleton Laboratory, Didcot, UK) under experiment RB2310028, using a wavelength band of 0.9 to 13 Å to access a q range of 0.004 to 0.7 Å−1. Gels were measured in 2 mm path length UV spectrophotometer grade quartz cuvettes (Hellma). These were placed in a temperature-controlled sample rack during the measurements.
The data was reduced to 1D scattering curves of intensity vs. Q using the facility provided software. Electronic backgrounds were subtracted, the full detector images for all data were normalised and scattering from the empty cell was subtracted. The scattering from DMSO-d6/D2O (1:1, v/v) and toluene-d8 were also measured and subtracted from the data using the Mantid software package installed inside the ISIS virtual machines, IDAaaS.61 This data was then fitted to the appropriate models using the SasView software package (https://www.sasview.org/). The best fit was determined as the one which fit well to the data and had the lowest χ2 value.
Author contributions
T.A.G and K.K.D developed the methodology, and T.A.G performed the synthesis and characterisation of the compounds, gelation properties and the characterisation of gels. T.A.G and K.K.D did the formal analysis. K.K.D validated the results and provided the conceptualisation for the project. M.R and T.A.G performed the DSC measurements, and M.R performed the ss-13C-NMR. A.S performed WAXS measurements and analysis, R.G, J.D and D.J.A performed SANS measurements and analysis. D.G and G.K performed PXRD measurements. T.A.G wrote the original draft and K.K.D, A.S, D.J.A and T.G. reviewed and edited the draft. K.K.D carried out the supervision, project administration and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the University of Iceland Research Fund and Icelandic Research Fund (IRF-228902-051) Rannís Iceland for funding. T. G and T. A. G thank Science Foundation Ireland (SFI) funded Advanced Materials and BioEngineering Research (AMBER) Centre for funding (Research Centres Phase 2 12/RC/2278_P2), and Advanced Microscopy Laboratory (AML), Trinity College Dublin for access to SEM. D. G thanks the University of Iceland for the post-doctoral grant. R. G. thanks the EPSRC (EP/R51322/1) for funding. The Ganesha X-ray scattering apparatus used for this research was purchased under EPSRC Grant “Atoms to Applications” (EP/K035746/1). This work benefitted from the SasView software, originally developed by the DANSE project under NSF award DMR-0520547. We acknowledge STFC beamtime allocation RB2310028 on SANS2D, https://doi.org/10.5286/ISIS.E.RB RB2310028 at ISIS Neutron and Muon Source, Didcot, UK. We thankfully acknowledge Dr Sigríður Jónsdóttir, the University of Iceland and Dr John E. O'Brien, Trinity College Dublin for NMR spectroscopy.References
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS PubMed
.
- H. Li, Z.-J. Yao, D. Liu and G.-X. Jin, Coord. Chem. Rev., 2015, 293–294, 139–157 CrossRef CAS
- S. Banerjee, R. K. Das and U. Maitra, J. Mater. Chem., 2009, 19, 6649–6687 RSC
- M. de Loos, B. L. Feringa and J. H. van Esch, Eur. J. Org. Chem., 2005, 3615–3631 CrossRef CAS
- M. George and R. G. Weiss, Acc. Chem. Res., 2006, 39, 489–497 CrossRef CAS PubMed
- D. K. Kumar and J. W. Steed, Chem. Soc. Rev., 2014, 43, 2080–2088 RSC
- J. W. Steed, Chem. Soc. Rev., 2010, 39, 3686–3699 RSC
- G. Yu, X. Yan, C. Han and F. Huang, Chem. Soc. Rev., 2013, 42, 6697–6722 RSC
- D. K. Smith, Soft Matter, 2024, 20, 10–70 RSC
- D. J. Adams, J. Am. Chem. Soc., 2022, 144, 11047–11053 CrossRef CAS PubMed
- X. Cao, A. Gao, J.-t. Hou and T. Yi, Coord. Chem. Rev., 2021, 434, 213792 CrossRef CAS
- P. Verma, A. Singh, F. A. Rahimi, P. Sarkar, S. Nath, S. K. Pati and T. K. Maji, Nat. Commun., 2021, 12, 7313 CrossRef CAS PubMed
- M. Godoy-Gallardo, M. Merino-Gómez, L. C. Matiz, M. A. Mateos-Timoneda, F. J. Gil and R. A. Perez, ACS Biomater. Sci. Eng., 2023, 9, 40–61 CrossRef CAS PubMed
- P. R. A. Chivers and D. K. Smith, Nat. Rev. Mater., 2019, 4, 463–478 CrossRef CAS
- C. L. Esposito, P. Kirilov and V. G. Roullin, J. Controlled Release, 2018, 271, 1–20 CrossRef CAS PubMed
- S. Panja and D. J. Adams, Chem. Soc. Rev., 2021, 50, 5165–5200 RSC
- C. Ferreiro-Córdova, E. Del Gado, G. Foffi and M. Bouzid, Soft Matter, 2020, 16, 4414–4421 RSC
- E. R. Cross, S. Sproules, R. Schweins, E. R. Draper and D. J. Adams, J. Am. Chem. Soc., 2018, 140, 8667–8670 CrossRef CAS PubMed
- E. R. Draper, E. G. B. Eden, T. O. McDonald and D. J. Adams, Nat. Chem., 2015, 7, 848 CrossRef CAS PubMed
- J. Raeburn and D. J. Adams, Chem. Commun., 2015, 51, 5170–5180 RSC
- J. A. Foster, R. M. Edkins, G. J. Cameron, N. Colgin, K. Fucke, S. Ridgeway, A. G. Crawford, T. B. Marder, A. Beeby, S. L. Cobb and J. W. Steed, Chem. – Eur. J., 2014, 20, 279–291 CrossRef CAS PubMed
- L. Yan, G. Li, Z. Ye, F. Tian and S. Zhang, Chem. Commun., 2014, 50, 14839–14842 RSC
- C. C. Piras, C. S. Mahon and D. K. Smith, Chem. – Eur. J., 2020, 26, 8452–8457 CrossRef CAS PubMed
- M. Putti, T. Mes, J. Huang, A. W. Bosman and P. Y. W. Dankers, Biomater. Sci., 2020, 8, 163–173 RSC
- A. K. Patterson and D. K. Smith, Chem. Commun., 2020, 56, 11046–11049 RSC
- J. Zhu, R. Wang, R. Geng, X. Zhang, F. Wang, T. Jiao, J. Yang, Z. Bai and Q. Peng, RSC Adv., 2019, 9, 22551–22558 RSC
- L. E. Buerkle and S. J. Rowan, Chem. Soc. Rev., 2012, 41, 6089–6102 RSC
- E. R. Draper and D. J. Adams, Chem. Soc. Rev., 2018, 47, 3395–3405 RSC
- C. Colquhoun, E. R. Draper, E. G. B. Eden, B. N. Cattoz, K. L. Morris, L. Chen, T. O. McDonald, A. E. Terry, P. C. Griffiths, L. C. Serpell and D. J. Adams, Nanoscale, 2014, 6, 13719–13725 RSC
- D. A. Tómasson, D. Ghosh, Z. Kržišnik, L. H. Fasolin, A. A. Vicente, A. D. Martin, P. Thordarson and K. K. Damodaran, Langmuir, 2018, 34, 12957–12967 CrossRef PubMed
- D. Ghosh, A. D. Farahani, A. D. Martin, P. Thordarson and K. K. Damodaran, Chem. Mater., 2020, 32, 3517–3527 CrossRef CAS
- K. Kodama, M. Obata, S. Sugimura, H. Yuhara and T. Hirose, Chem. – Eur. J., 2023, 29, e202202692 CrossRef CAS PubMed
- K. D. C. Magdato and M. C. Paderes, Soft Mater., 2023, 21, 251–260 CrossRef CAS
- A. Croitoriu, L. E. Nita, A. G. Rusu, A. Ghilan, M. Bercea and A. P. Chiriac, Polymers, 2022, 14, 3354 CrossRef CAS PubMed
- R. K. Das, R. Kandanelli, J. Linnanto, K. Bose and U. Maitra, Langmuir, 2010, 26, 16141–16149 CrossRef CAS PubMed
- S. Cicchi, G. Ghini, L. Lascialfari, A. Brandi, F. Betti, D. Berti, P. Baglioni, L. Di Bari, G. Pescitelli, M. Mannini and A. Caneschi, Soft Matter, 2010, 6, 1655–1661 RSC
- J. N. Loos, C. E. Boott, D. W. Hayward, G. Hum and M. J. MacLachlan, Langmuir, 2021, 37, 105–114 CrossRef CAS PubMed
- P. Xing, H. P. Tham, P. Li, H. Chen, H. Xiang and Y. Zhao, Adv. Sci., 2018, 5, 1700552 CrossRef PubMed
- P. Xing, C. Yang, Y. Wang, S. Z. F. Phua and Y. Zhao, Adv. Funct. Mater., 2018, 28, 1802859 CrossRef
- A. R. Hirst and D. K. Smith, Langmuir, 2004, 20, 10851–10857 CrossRef CAS PubMed
- S. Varela-Aramburu, G. Morgese, L. Su, S. M. C. Schoenmakers, M. Perrone, L. Leanza, C. Perego, G. M. Pavan, A. R. A. Palmans and E. W. Meijer, Biomacromolecules, 2020, 21, 4105–4115 CrossRef CAS PubMed
- S. Cantekin, T. F. A. de Greef and A. R. A. Palmans, Chem. Soc. Rev., 2012, 41, 6125–6137 RSC
- O. Kotova, R. Daly, C. M. G. dos Santos, M. Boese, P. E. Kruger, J. J. Boland and T. Gunnlaugsson, Angew. Chem., Int. Ed., 2012, 51, 7208–7212 CrossRef CAS PubMed
- A. D. Lynes, C. S. Hawes, K. Byrne, W. Schmitt and T. Gunnlaugsson, Dalton Trans., 2018, 47, 5259–5268 RSC
- R. Daly, O. Kotova, M. Boese, T. Gunnlaugsson and J. J. Boland, ACS Nano, 2013, 7, 4838–4845 CrossRef CAS PubMed
- L. N. J. de Windt, Z. Fernández, M. Fernández-Míguez, F. Freire and A. R. A. Palmans, Chem. – Eur. J., 2022, 28, e202103691 CrossRef CAS PubMed
- Y. Ishigaki, M. Yamanaka, T. Hagio and R. Ichino, ChemistrySelect, 2023, 8, e202300805 CrossRef CAS
- B. Wu, L. Liu, L. Zhou, J. R. Magana, M. M. R. M. Hendrix, J. Wang, C. Li, P. Ding, Y. Wang, X. Guo, I. K. Voets, M. A. Cohen Stuart and J. Wang, J. Colloid Interface Sci., 2022, 608, 1297–1307 CrossRef CAS PubMed
- S. Hafeez, M. C. Decarli, A. Aldana, M. Ebrahimi, F. A. A. Ruiter, H. Duimel, C. van Blitterswijk, L. M. Pitet, L. Moroni and M. B. Baker, Adv. Mater., 2023, 35, 2301242 CrossRef CAS PubMed
- A. Karmakar, C. L. Oliver, A. E. Platero-Prats, E. Laurila and L. Öhrström, CrystEngComm, 2014, 16, 8243–8251 RSC
- A. Desmarchelier, B. G. Alvarenga, X. Caumes, L. Dubreucq, C. Troufflard, M. Tessier, N. Vanthuyne, J. Idé, T. Maistriaux, D. Beljonne, P. Brocorens, R. Lazzaroni, M. Raynal and L. Bouteiller, Soft Matter, 2016, 12, 7824–7838 RSC
- A. J. Savyasachi, O. Kotova, S. Shanmugaraju, S. J. Bradberry, G. M. Ó'Máille and T. Gunnlaugsson, Chem, 2017, 3, 764–811 CAS
- P. P. Bose, M. G. B. Drew, A. K. Das and A. Banerjee, Chem. Commun., 2006, 3196–3198, 10.1039/B606371C
- F. Perlitius, A. Walczak, M. Čonková, G. Markiewicz, J. Harrowfield and A. R. Stefankiewicz, J. Mol. Liq., 2022, 367, 120511 CrossRef CAS
- Y. Ishioka, N. Minakuchi, M. Mizuhata and T. Maruyama, Soft Matter, 2014, 10, 965–971 RSC
- J. N. Loos, F. D'Acierno, U. Vijay Mody and M. J. MacLachlan, ChemPlusChem, 2022, 87, e202200026 CrossRef CAS PubMed
- A. Dawn and H. Kumari, Chem. – Eur. J., 2018, 24, 762–776 CrossRef CAS PubMed
- D. Ghosh, I. Lebedytė, D. S. Yufit, K. K. Damodaran and J. W. Steed, CrystEngComm, 2015, 17, 8130–8138 RSC
- D. McDowall, D. J. Adams and A. M. Seddon, Soft Matter, 2022, 18, 1577–1590 RSC
- D. J. Adams, Gels, 2018, 4, 32 CrossRef PubMed
- O. Arnold, J. C. Bilheux, J. M. Borreguero, A. Buts, S. I. Campbell, L. Chapon, M. Doucet, N. Draper, R. Ferraz Leal, M. A. Gigg, V. E. Lynch, A. Markvardsen, D. J. Mikkelson, R. L. Mikkelson, R. Miller, K. Palmen, P. Parker, G. Passos, T. G. Perring, P. F. Peterson, S. Ren, M. A. Reuter, A. T. Savici, J. W. Taylor, R. J. Taylor, R. Tolchenov, W. Zhou and J. Zikovsky, Nucl. Instrum. Methods Phys. Res., Sect. A, 2014, 764, 156–166 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Details of gelation studies and gel characterisation such as rheology, CD, SEM images, solid-state 13C-NMR, and DSC measurements. Comparison of X-Ray Powder Diffraction (XRPD) pattern and the details of WAXS/SANS. See DOI: https://doi.org/10.1039/d4nr00204k |
| This journal is © The Royal Society of Chemistry 2024 |