
Click catalysis and DNA conjugation using a nanoscale DNA/silver cluster pair†
Caleb J.
Setzler
 and
Jeffrey T.
Petty
*
and
Jeffrey T.
Petty
*
Department of Chemistry, Furman University, Greenville, SC 29613, USA. E-mail: jeff.petty@furman.edu
First published on 11th September 2024
Abstract
DNA-bound silver clusters are most readily recognized by their strong fluorescence that spans the visible and near-infrared regions. From this suite of chromophores, we chose a green-emitting Ag106+ bound to C4AC4TC3GT4 and describe how this DNA/cluster pair is also a catalyst. A DNA-tethered alkyne conjugates with an azide via cycloaddition, an inherently slow reaction that is facilitated through the joint efforts of the cluster and DNA. The Ag106+ structure is the catalytic core in this complex, and it has three distinguishing characteristics. It facilitates cycloaddition while preserving its stoichiometry, charge, and spectra. It also acidifies its nearby alkyne to promote H/D exchange, suggesting a silver–alkyne complex. Finally, it is markedly more efficient when compared with related multinuclear DNA–silver complexes. The Ag106+ is trapped within its C4AC4TC3GT4 host, which governs the catalytic activity in two ways. The DNA has orthogonal functional groups for both the alkyne and cluster, and these can be systematically separated to quench the click reaction. It is also a polydentate ligand that imprints an elongated shape on its cluster adduct. This extended structure suggests that DNA may pry apart the cluster to open coordination sites for the alkyne and azide reactants. These studies indicate that this DNA/silver cluster pair work together with catalysis directly driven by the silver cluster and indirectly guided by the DNA host.
Introduction
Size dictates the chemical and optical properties of noble metal nanomaterials, with distinctive behaviors emerging at nanometer scales.1 For example, while gold in its bulk form is inert, its nanoparticles catalyze CO oxidation with a sharp 100-fold jump in efficiency for sizes ≲6 nm.2,3 These small nanoparticles are active because large surface areas expose coordination sites for exogenous reagents.4 This surface chemistry and catalysis are now more precisely controlled using noble metal nanoclusters.5,6 These are more precisely described as nanoscale molecules because they have a well-defined number of metals and ligands, organized as a metal core in a ligand shell.7 Now, size becomes a less relevant metric, and the stoichiometry, structure, charge, and coordination environment dictate catalytic efficiency.8 Their catalysis is being studied and optimized through a fruitful collaboration of experimental and theoretical studies. A suite of synthetic methods can manipulate both the metal core and ligand shell at the atomic level.9 X-ray diffraction along with a diverse set of analytical tools can atomically map these complexes.10 High-level theoretical calculations are used to develop models to understand and optimize catalysis.11 Here, we describe a nanoscale molecule comprised of a silver cluster within DNA, and this complex collectively catalyzes a cycloaddition reaction.Transition metal cations are the linchpins of alkyne–azide cycloadditions because they catalyze these click reactions with rates that are 107–108 faster than the original Huisgen reaction.12–15 These catalyzed reactions are efficient at room temperature and in dilute solutions and are consequently used to link modular units and synthesize novel nanomaterials.16–19 A metal center facilitates stepwise annulation by coordinating an alkyne and azide in its open sites.20 This metal also activates these pendant ligands because it is an electrophile. For example, Cu+ coordinates across alkyne π-bonds to withdraw electron density and acidify the terminal proton.15 A nucleophilic acetylide then attacks its neighboring azide, and the resulting C–N bond sets the foundation for an eventual triazole ring closure.13 While a single Cu+ facilitates alkyne–azide cycloadditions, multinuclear complexes are more efficient. Two copper coordination sites are supported by kinetic studies that establish a second-order rate law with respect to copper.21 A dimeric Cu(I) catalyst is supported because a copper acetylide assembles with a Cu(I) complex and exchanges 63Cu and 65Cu isotopes.22 The pendant ligands freely migrate between the two coordination centers. The diverse end-on and side-on coordination modes of copper acetylides may underlie this enhanced activity because these interlinked complexes readily evolve into copper-triazole intermediates.23–25 To precisely control metal stoichiometry, molecular and nanoscale complexes have been synthesized.
Click reactions are widely used because they can be adapted and optimized for diverse reaction conditions, so these ‘black box’ approaches have motivated the search for more precisely defined catalysts.19,26–28 For example, di-nuclear Cu+ complexes are bridged by a bidentate carbene and labile acetate, and these complexes are effective homogeneous catalysts in a range of solvents.29 Larger nanoscale molecules offer new opportunities to explore the metal stoichiometry, structure, and coordination environment. One such nanocluster complex incorporates eight Cu+ that are internally linked by acetylides and peripherally capped by carbenes.30 The acetylides link copper atoms via both σ and π bonds, and the complex fluctuates with rapidly exchanging ligands. A Cu20 complex is concentrically organized around a partially reduced Cu42+ core with an outer shell having 12 acetylide ligands bridging 16 Cu+.31 The acetylides chemically exchange with exogenous alkynes to yield a mixture of triazole products. A Cu58 cluster is a click catalyst whose activity is enhanced by excising a single copper atom.32 This Cu57 may be more active because the copper atoms and ligands around the vacated site reorganize. Mixed Au/Cu and Ag/Cu clusters reveal that electronic synergism facilitates alkyne/azide annulations.33,34
Besides Cu(I), other transition metals catalyze click reactions, and we consider a silver molecule.13,35,36 Again highlighting size-dependent properties, these molecular forms of silver are fluorophores that are ∼1010 and ∼102 brighter than bulk and nanoparticle forms of silver, respectively.37,38 These fluorophores have diverse spectra that have been revealed using DNA.39 Single-stranded oligonucleotides coordinate silvers via their electron-rich, heterocyclic nucleobases, and multiple nucleobases frame binding pockets for a specific multinuclear cluster. DNA is programmable because its sequence and structure encode specific chromophores with spectra that span the visible and near-infrared regions and brightnesses that vary by ∼103.40,41 Here, we consider this spectroscopic DNA code from a chemical perspective. Silver clusters are protected within their DNA shell, but this matrix is permeable, as illustrated by cluster reactions with oxidizing and reducing agents.42–45 Here, we study a DNA/silver cluster pair that catalyzes alkyne–azide cycloadditions, a favored and selective reaction that is not perturbed by the diverse functional groups in DNA.46 We first describe the DNA scaffold that shares both a silver cluster and an alkyne. The DNA-tethered alkyne reacts with azides without changing the nearby cluster, and a covalent triazole linkage was confirmed by etching the cluster from its DNA matrix. The cluster/DNA complex in D2O reveals that the cluster acidifies its neighboring alkyne, possibly via a silver–alkyne complex. The click reaction efficiency depends on the cluster–alkyne proximity, which was controlled by inserting thymine spacers in the DNA strand. The solution pH also regulates the reaction efficiency. Collectively, these experiments show how a DNA/silver cluster pair work together to catalyze alkyne–azide cycloadditions.
Experimental
Synthesis
The desalted oligonucleotides C4AC4TC3GT4 and Hx-C4AC4TC3GT4, where Hx is 1-hexyne attached at the 5′ phosphate position, were purchased from Integrated DNA Technologies and dissolved in deionized water before use. Molar extinction coefficients were calculated based on the nearest-neighbor approximation,47 and the concentrations of these DNA stock solutions were determined using the Lambert–Beer law. In a typical synthesis, 150 μL of sodium cacodylate buffer solution (1 mM, pH = 7) that contained 30 μM C4AC4TC3GT4 (1) and 300 μM AgNO3 (10 molar equivalents![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was used. The mixture was first heated (∼80 °C) for 5 min and then cooled to room temperature. Then an aqueous solution of NaBH4 was added (5 molar equivalents:1) and vortexed. The buffers were acetic acid/acetate (pH = 5), cacodylic acid/cacodylate (pH = 6–7), and boric acid/borate (pH = 7.5 and 8). The azides were N3–C3H6–NH2, N3–C6H12O2–C10H16N3O2S (Lumiprobe) and N3–C3H6–OH (Synthonix). D2O (Cambridge Isotopes) was used for H/D exchange.
:1) was used. The mixture was first heated (∼80 °C) for 5 min and then cooled to room temperature. Then an aqueous solution of NaBH4 was added (5 molar equivalents:1) and vortexed. The buffers were acetic acid/acetate (pH = 5), cacodylic acid/cacodylate (pH = 6–7), and boric acid/borate (pH = 7.5 and 8). The azides were N3–C3H6–NH2, N3–C6H12O2–C10H16N3O2S (Lumiprobe) and N3–C3H6–OH (Synthonix). D2O (Cambridge Isotopes) was used for H/D exchange.
Optical characterization
Absorption spectra of DNA–Ag cluster conjugates were collected on a Cary 50 UV-Vis spectrophotometer (Varian), and steady-state emission spectra were collected on a Fluoromax-3 spectrofluorometer (Jobin–Yvon Horiba). Fluorescence quantum yields (QY) were measured by following well-established protocols using fluorescein as the standard (QY = 95%).48 Time-correlated single-photon counting measured the fluorescence lifetimes and anisotropies of these nanoclusters. Samples were excited by a pulsed 470 nm laser (PicoQuant) at a rate of 20 MHz using a FluoTime 300 (PicoQuant) photoluminescence spectrometer. The excitation beam was vertically polarized, and its power was fine-tuned to achieve a detection rate of fewer than 5 photons per 100 pulses (<5 × 105 Hz). The emission was collected at right-angled geometry with the emission polarizer set to the magic angle (∼55°), and spectrally filtered using the monochromator. The instrument response function (IRF) was collected using colloidal silica (Aldrich), and the FWHM of the IRF was ∼150 ps. The kinetics of fluorescence decay was extracted through IRF reconvolution fitting of the measured decay (EasyTau). Fluorescence anisotropy measurements were conducted using vertically (V) polarized excitation along with vertically (V) or horizontally (H) polarized emission.49 The G-factor accounts for the detection efficiency of vertically and horizontally polarized emission and was measured using fluorescein. The fluorescence decays under two different configurations, IVV(t) and IVH(t), were reconvolved with the IRF to calculate the anisotropy decay, r(t), and rotational correlation time, τc (EasyTau).Mass spectrometry
The stoichiometry and charge of DNA–Ag cluster conjugates were characterized by electrospray ionization mass spectrometry (Q-TOF G2-S, Waters). DNA–silver clusters were dialyzed to remove low molecular weight impurities such as Na+. The samples were diluted with 100× volumes of 1 mM ammonium acetate and then reconcentrated by centrifugal dialysis using2 kDa cutoff filters (VivaSpin 20). The samples were diluted to ∼0.3 μM with 1 mM ammonium acetate and then infused via a syringe pump operated at 20 μL min−1. The spectra were collected in the negative ion mode with a capillary voltage of −2.7 kV, a sampling cone voltage of −15 V, an extraction cone voltage of 10 V, a cone gas flow of 45 L h−1, and a desolvation gas flow of 450 L h−1. The source temperature was 80 °C, and the desolvation temperature was 150 °C. Mass calibration was performed using aggregates of sodium iodide in the 400 < m/z < 2000 range. The spectra were analyzed using MassLynx V4.1.Results
Neighbors in the shared DNA scaffold
C4AC4TC3GT4 (1) assembles a silver cluster and alkyne via two distinct binding sites (Fig. 1A). This sequence was chosen because it selectively forms a single silver chromophore with λex/λem = 490/555 nm, and its multidentate binding site has been characterized by varying the DNA sequence (Fig. 1B).50 The four thymine spacers at the 3′ terminus can be removed; however, shorter variants of C4AC4TC3G no longer favor the green emitting cluster, indicating that this 14-nucleobase tract is the minimal, core-binding site.43,50–52 This sequence folds around the cluster with the central thymine functioning as a hinge.49 The DNA was further modified with a 1-hexyne molecule at its 5′ phosphate position, and this covalently modified strand is henceforth referred to as Hx-1. In summary, C4AC4TC3GT4 (1) is a scaffold that shares an alkyne and a green-emitting silver cluster. The cluster is a silver molecule that was characterized using its mass and optical spectra.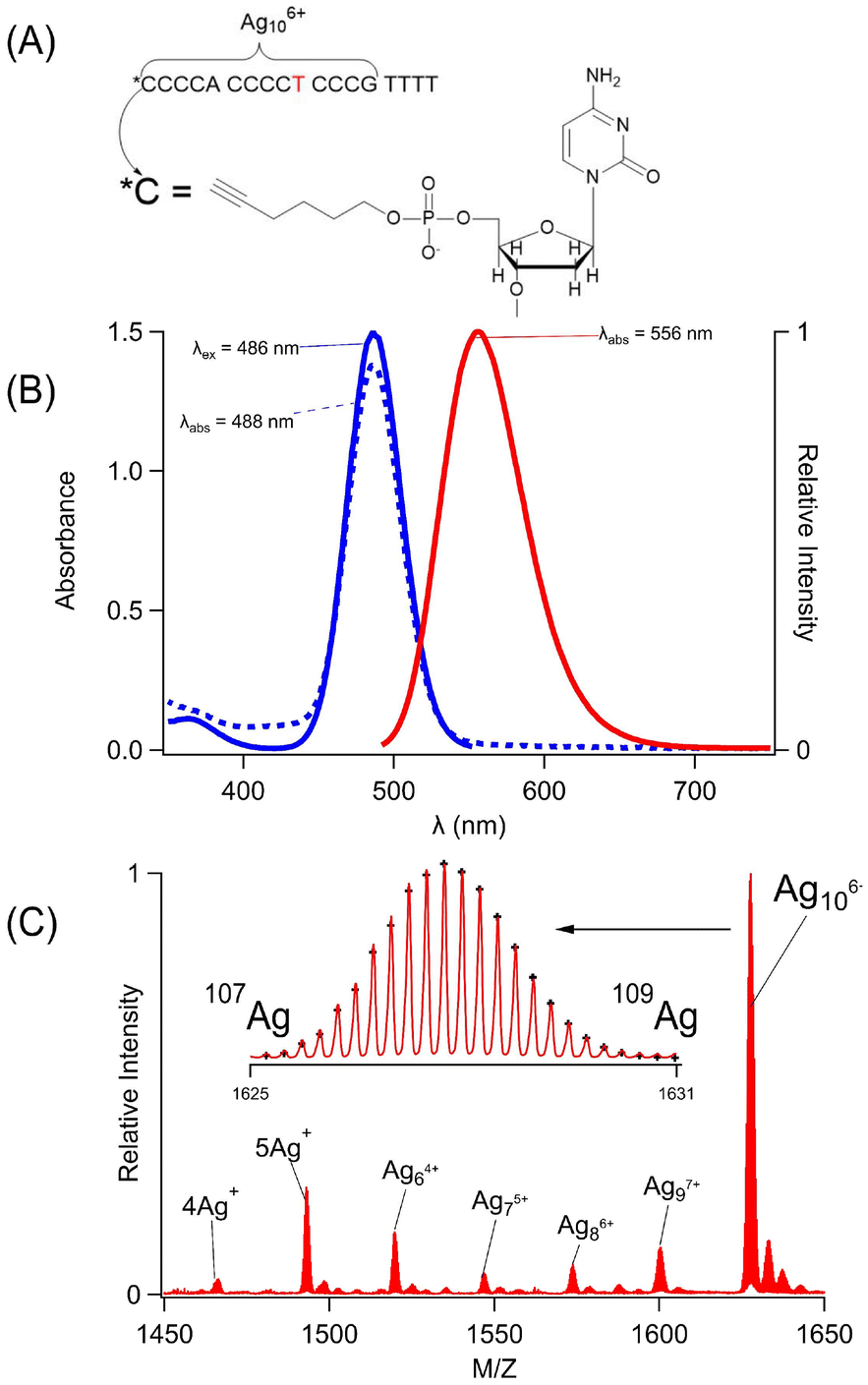 | ||
| Fig. 1 (A) The model of Hx-C4AC4TC3GT4 with 1-hexyne attached at the 5′ phosphate (Hx-1) position and Ag106+ bound to the C4AC4TC3G sub-sequence. The central T highlighted in red emphasizes a folding site. (B) Absorption (dotted blue), excitation (solid blue), and emission (solid red) spectra of Ag106+ bound to C4AC4TC3GT4. Coincidence of the absorption and excitation spectra suggests that a single cluster forms. (C) Mass spectra of the −4 state of Hx-1 with its Ag adducts. The dominant species is Ag106+. The inset shows the 107Ag:109Ag isotopic distribution, by which the molecular formula was determined. | ||
Hx-1 mimicked 1 because it formed the same silver molecule, whose stoichiometry and charge were measured by electrospray ionization mass spectrometry (Fig. 1C, 1S and Table 1S†). As with the parent strand, Hx-1 was combined with Ag+ ions, which were chemically reduced to yield a range of adducts with 4–10 silvers. Ag10 dominated, and it is a charged adduct that partially neutralizes the DNA.38,49,50,53Hx-1/Ag10 complexes with net −4 and −5 charges were identified, each with characteristic isotopic distributions due to the 51.8% 107Ag:48.2% 109Ag mixture in naturally occurring silver (Fig. 1C-inset and 1S†). The m/z values and the intensities of 49 isotopologue peaks were analyzed to yield the molecular formulas [(C175H221N53O113P18)−10(Ag10)6+]−4 and [(C175H220N53O113P18)−11(Ag10)6+]−5 (Table 1S†).53 Without the silver adduct, the −4 and −5 charged strands alone would have H227 and H226, respectively, so each complex is missing 6 H+. The reason for this deficit lies with the phosphate backbone. It acts like a buffer because its H+ ions are labile and displaced by the cationic silver cluster. The 6 fewer H+ ions suggests that the cluster is Ag106+. Both ions had Ag106+ adducts, consistent with a cluster that dictates protonation of the phosphate backbone. Oxidation states of other DNA-bound silver clusters have been measured by both mass spectrometry in the gas phase and XANES measurements in solution, yielding the same charge.54 Based on its +6 oxidation state, the Ag106+ has 4 Ag0 atoms, which are responsible for its green fluorescence.55 The optical spectra are described next.
The coordination environment in Hx-1 was further interrogated using the absorption and fluorescence spectra of the Ag106+ adduct. This chromophore is a sensitive reporter because it has diverse spectra, ranging from a weakly emissive, λmax = 400 nm chromophore to a 60-fold brighter, λmax = 490 nm fluorophore.56,57 Given these large changes, the Ag106+ spectra can discern differences between the 1 and Hx-1 hosts (Fig. 2S†). The absorption spectra have similar λmax values and absorbances, thus indicating that the clusters reside in comparable binding sites and form with similar efficiencies. The absorption and excitation maxima overlap, suggesting that a single cluster develops within their respective binding sites. The emission spectra have the same λmax = 556 nm, and the fluorescence quantum yields are similar being 24 ± 4% for 1/Ag106+ and 30 ± 4% for Hx-1/Ag106+. The respective intensity-weighted average fluorescence lifetimes are 2.05 ± 0.03 ns and 2.23 ± 0.04 ns, again supporting similar electronic environments. The fluorescence decays were further utilized to compare the shapes of the DNA–cluster complexes. During fluorescence relaxation, the emission depolarizes as the complexes rotate to yield rotation correlation times of 2.50 ± 0.05 ns and 2.64 ± 0.09 ns using the strands without and with the hexyne, respectively. These times are similar to the heterodimeric (C4AC4T + C3GT4)/Ag106+ complex and suggests that both Hx-1 and 1 are folded at the central thymine.49,58 Thus, the mass and optical spectra demonstrate that the same Ag106+ fluorophore forms with both Hx-1 and 1.
Catalytic cluster
The Ag106+ and hexyne are nearby in their shared DNA scaffold and yet appear to be decoupled, as the same Ag106+ adduct has nearly identical spectra using C4AC4TC3GT4 without and with 1-hexyne appended. However, from the vantage of the alkyne, it now reacts with azides, a reaction that is prohibitively slow without the cluster.12 Two smaller aliphatic and one larger biotin-based azides were used: N3–C3H6–NH2, N3–C3H6–OH, and N3–C6H12O2–C10H16N3O2S (biotin) (Fig. 2A, B, and 3S†). Azide complexes with Hx-1/Ag106+ were identified in the mass spectra, and their formulas were derived from the isotopologue peaks in the −4 and −5 states of these complexes. Based on the m/z values and the isotopologue intensity distributions, the following formulas were derived for the −4 and −5 charged ions of each complex: [(C178H229O113N57P18)−10(Ag10)6+]−4 and [(C178H228O113N57P18)−11(Ag10)6+]−5 for N3–C3H6–NH2 (Table 2S†), [(C178H228O114N56P18)−10(Ag106+)]−4 and [(C178H227O114N56P18)−11(Ag106+)]−5 for N3–C3H6–OH (Table 3S†), and [(C191H249O117N59SP18)−10(Ag106+)]−4 and [(C191H248O117N59SP18)−11(Ag106+)]−5 for N3–C6H12O3–biotin (Table 4S†). These formulas show Ag106+ is preserved when the DNA-tethered hexyne reacts with the azides. Furthermore, the coordination sites are conserved, as indicated in similar absorption spectra (Fig. 4S†). Our subsequent results focus on N3–C3H6–NH2. While Hx-1 hosts other silver adducts, only the Hx-1/Ag106+ complex associates with this azide, and the efficiency of labelling was calculated by integrating peak areas for all DNA/silver complexes (Fig. 2C, red vs. black peaks). The integrity of the N3–C3H6–NH2/DNA/cluster complex was challenged by diluting a sample with 100× volumes of solution to dissociate weakly bound complexes and then reconcentrated using centrifugal dialysis. The absorption spectra remain consistent with the same λmax and absorbance, indicating that the cluster is stable in its azide-tagged DNA host (Fig. 5S†). Also, the reaction efficiencies are similar, so we conclude that only the DNA-bound Ag106+, and not leached Ag0/Ag+, facilitate the reaction (Fig. 5S†).31,59 Thus, the DNA-bound cluster facilitates but remains unchanged by alkyne–azide coupling.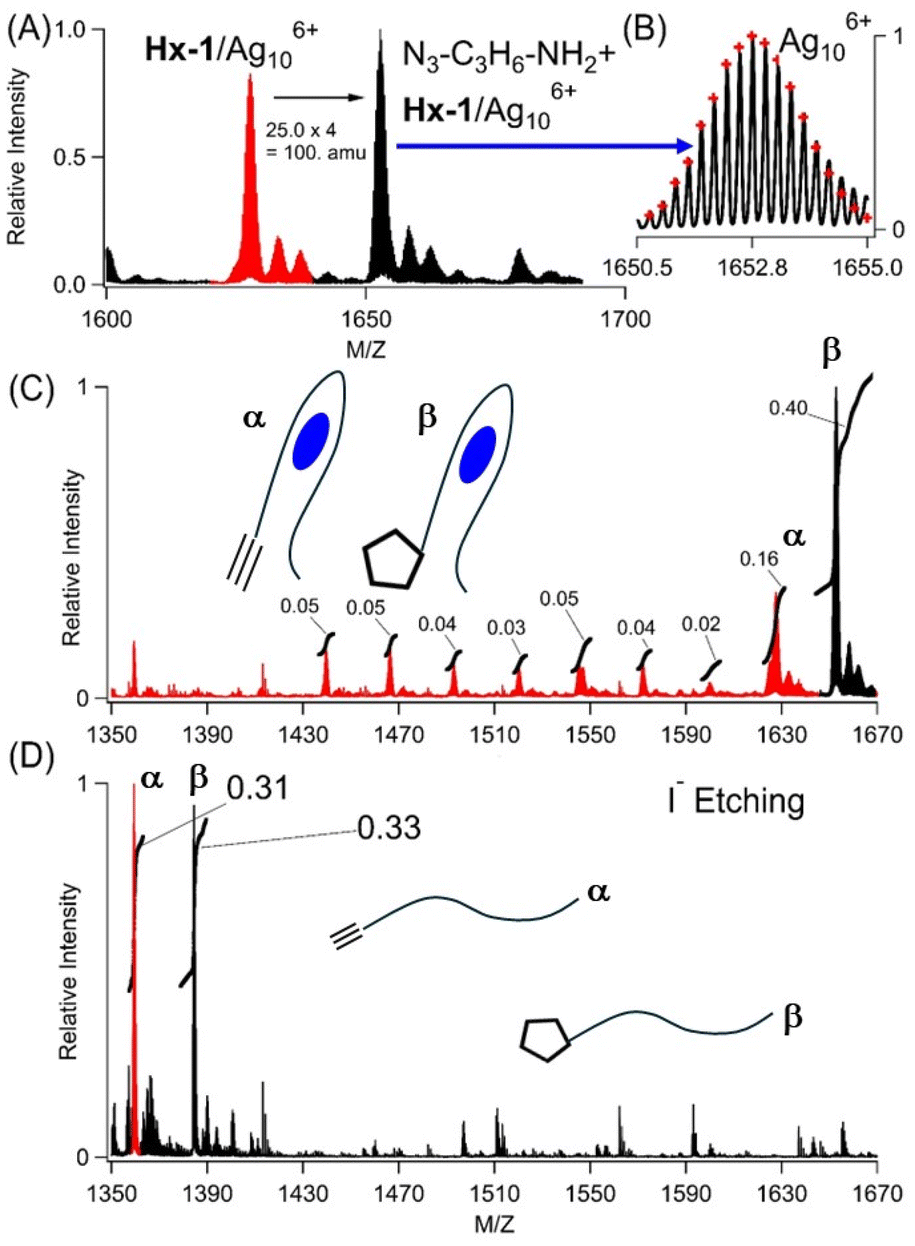 | ||
| Fig. 2 (A) Mass spectra of the −4 charged ion of Hx-1/Ag106+ (red) after reacting with N3–C3H6–NH2 to form a new complex with the expected mass difference of 100 amu (black). (B) Expanded view of the Hx-1/Ag106+/N3–C3H6–NH2 isotopic distribution by which the molecular formula was determined (see Table 2S†). (C) Mass spectra of Hx-1/Ag106+ (red peak, α label) and Hx-1/Ag106+/N3–C3H6–NH2 (black peak, β label) before etching. Relative integrated areas are included, and these were used to calculate the labelling efficiency of Hx-1/Ag106+/N3–C3H6–NH2vs. all the other DNA/silver complexes. (D) Mass spectra of Hx-1 (red peak, α label) and Hx-1/N3–C3H6–NH2 (black peak, β label) after etching. The relative integrated peak areas are included. The m/z ranges match in (C) and (D) to emphasize that silver atoms are removed from DNA using I−. | ||
Ag106+ was used as a starting point to create other possible silver-laden DNA catalysts.44 This chromophore has 4 Ag0 atoms that provide a photochemical handle by which Ag106+ can be selectively excited and degraded by irradiating at 490 nm.44 The photodestruction quantum yield was ∼10−4, so the sample was irradiated for an extended period. As the Ag40 absorption at 490 nm diminished, a new higher energy transition due to Ag20 developed at 340 nm (Fig. 6SC†).60–64 However, this photochemical change is more interesting than a simple Ag40 → Ag20 conversion because the mass spectra identified two distributions of photoproducts (Fig. 6SA and 6SB†). As expected from the λ = 490 → 340 nm shift, the DNA coordinated to Ag20, but multiple clusters with different numbers of Ag+ were identified – Ag64+, Ag75+ (dominant species), Ag86+, and Ag97+. Irradiation also produced purely oxidized 4 and 5 Ag+ adducts, which are devoid of Ag0 and thus spectrally silent. We used a sample irradiated for a short time to collectively evaluate how the DNA-bound Ag+, Ag20, and Ag40 species react with N3–C3H6–NH2. Despite their high abundance and redundancy, the Ag+ and Ag20 complexes did not react and only the Ag106+ adduct promoted alkyne–azide coupling (see red peaks in Fig. 6SA†). Thus, the alkyne reaction with azides is specific for Ag106+, and the reaction products were characterized by extracting the cluster from the DNA.
Three experiments indicate that Ag106+ facilitates cycloaddition to yield a covalently linked triazole, as shown using N3–C3H6–NH2 with Hx-1/Ag106+. First, this azide remained integrated with the DNA after Ag106+ was removed. Silver clusters were etched by I−, presumably because AgI(s) has such a small Ksp = 10−18. With 1 equivalent of iodide:silver, the Ag106+ absorption at 490 nm was quenched, signalling that silvers were no longer associated with the DNA (Fig. 7S†). In support of this, two strands devoid of silvers emerged in the mass spectra: Hx-1 alone and with N3–C3H6–NH2 (Fig. 2C). The latter supports alkyne–azide cycloaddition between the DNA-tethered alkyne and N3–C3H6–NH2, a union that is selective and strongly thermodynamically favored (ΔG ∼ −45 kcal).12,16 The areas of these two peaks were integrated to give a labelling efficiency of 52%, comparable to the value of 48% before etching (Fig. 2D vs. 2C). Inactive DNA-bound Ag+ and Ag20 adducts were further supported by the low labelling efficiency of a photolyzed sample (Fig. 8SB†). Second, N3–C3H6–NH2 was added to 1/Ag106+ but did not form a stable complex (Fig. 9S†). Third, after reaction with N3–C3H6–NH2 and then I−, the denuded DNA strands were dialyzed against 100× and 10000× volumes of solution (Fig. 10S†). The relative amounts of unlabelled and labelled DNA did not change, further supporting a stable, covalently linked triazole.
Enhanced acidity
Hx-1 becomes more acidic due to its Ag106+ adduct, and the acidic site was identified using H/D exchange. The DNA/cluster complex has nearly identical absorption spectra in H2O and D2O, suggesting that the clusters were formed with similar efficiencies in the same coordination environment (Fig. 11S†). To isolate the effect of the cluster, it was extracted from its DNA host using CN−, which readily complexes with Ag+ to form Ag(CN)2−. With K = 1018, this CN− and its Ag(CN)2− complex ion are expected to overwhelm any affinity of DNA for silvers, as supported by quenched Ag106+ absorption (Fig. 11S†). This sample was finally washed with 100 volumes of H2O (Fig. 3A – case I). Swapping D2O with this large amount of H2O will reprotonate the plethora of naturally acidic and basic functional groups in a DNA strand.47 However, the alkyne appended onto 1 is inherently more basic with pKa ∼ 26, thus resisting reprotonation.12 Mass spectra show denuded strands with net −5, −4, and −3 charges, and the m/z values and the isotope distributions of the isotopologue peaks show that the molecular formula has exchanged a hydrogen with deuterium, i.e. C175H230DN53O113P18 (DNA with zero net charge) (Fig. 3B, 12SA and Table 5S†). Three other experiments firmly established the alkyne deuteration site. First, Hx-1/Ag106+ was prepared in H2O, and the mass spectra show the expected fully protonated, bare DNA (Fig. 3A – case II, 12SB and Table 6S†). Second, C4AC4TC3GT4/Ag106+ without the hexyne was prepared in D2O, yet the silver-free DNA was fully protonated (Fig. 3A – case III, 12SC and Table 7S†). As a further control, Hx-1 without Ag106+ in D2O shows no deuteration (Fig. 3A – case IV, 12SD and Table 8S†). These experiments suggest that Ag106+ coordinates with and acidifies its neighboring alkyne.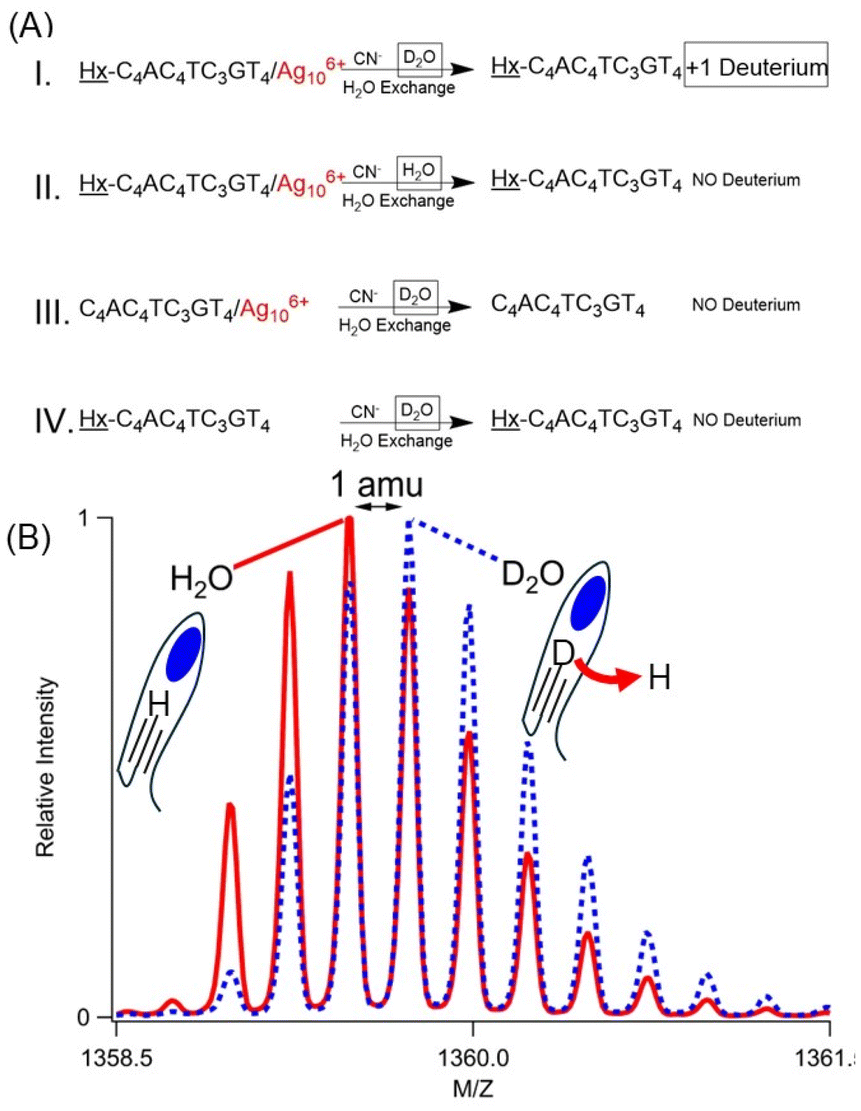 | ||
| Fig. 3 (A) Reaction conditions used to study deuteration of the alkyne. Complexes were synthesized in either D2O or H2O (indicated by boxes). Strands with or without the hexyne (indicated by underlining) and strands with and without the cluster (indicated by red text) were compared. CN− etched the silver cluster, then samples were washed with H2O to reprotonate exchangeable sites in the DNA. Denuded strands were then analyzed by mass spectrometry. (B) Mass spectra the −4 charged ion of Hx-1/Ag106+ prepared in D2O (blue dotted lines) and H2O (red solid lines), etched with CN−, and washed with H2O (cases I and II, respectively, above). A shift of 1 amu suggests that the former replaces 1 hydrogen for a deuterium atom. | ||
Reaction efficiency
Hx-1 anchors both the cluster and alkyne, and their proximity controls the reaction efficiency. This DNA is modular because it can be lengthened to separate its two adducts. Thymine spacers were inserted because they bind poorly with silvers and do not perturb a cluster coordination site.51,52Hx-Tx-1 strands with x = 0, 1, 2, 3, and 4 thus hold Ag106+ in its favored C4AC4TC3G binding site while still keeping the hexyne on the 5′ terminus. Considering DNA as a linear polymer, these longer strands progressively distanced the alkyne from its neighboring cluster. All five oligonucleotides formed the same Ag106+ adducts, whose similar λmax ∼ 490 nm and absorbances suggest that the clusters were developed with comparable efficiencies (Fig. 13S†). Thus, the added thymine spacers were innocuous, and the cluster remained confined and thus progressively more distant from the 5′-terminal hexyne. These DNA/Ag106+ complexes reacted differently with N3–C3H6–NH2, as the reaction efficiencies were highest without an added thymine (x = 0 strand) but diminished as more thymine spacers were added (Fig. 4 and 14S†). Thus, these results suggest the DNA is modular and can be lengthened to temper the click reaction.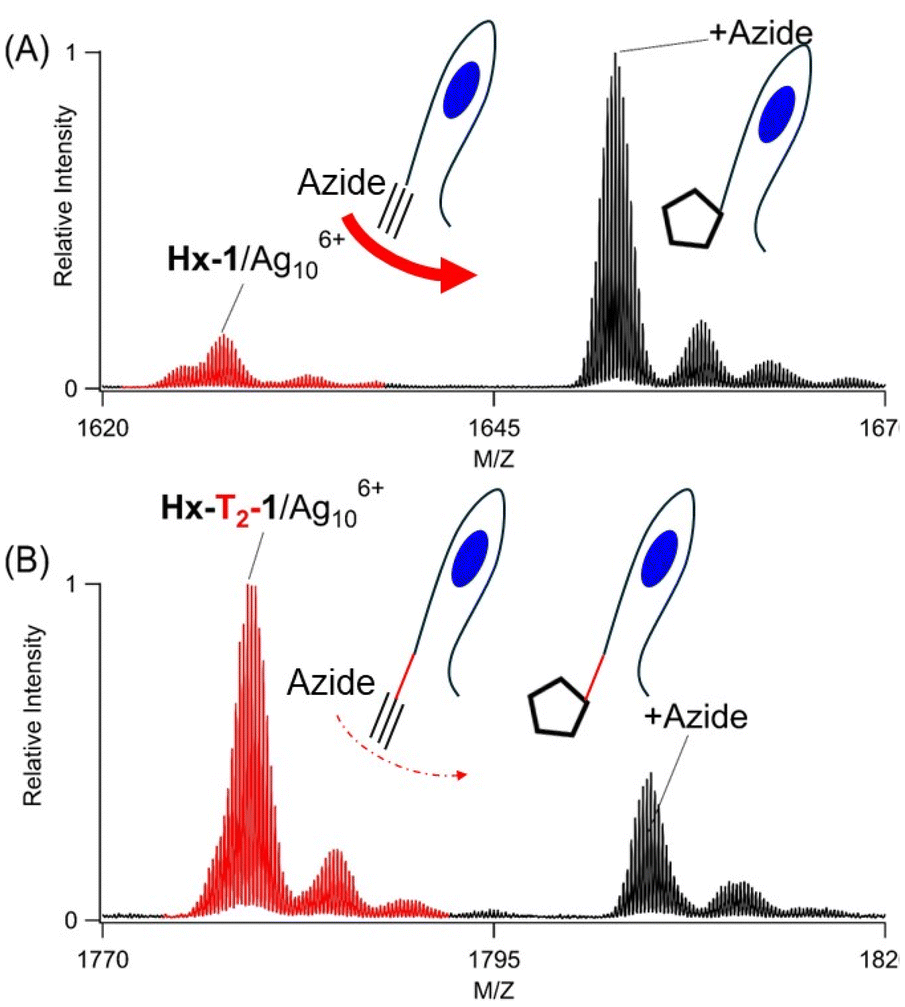 | ||
| Fig. 4 Mass spectra of the −4 charged ion of Hx-Tx-1Ag106+ with x = 0 and 2 (A and B, respectively.). After reaction with N3–C3H6–NH2, new azide complexes form (black peaks) from their Hx-Tx-1/Ag106+ precursors (red peaks). Reaction efficiencies decrease with the number of thymines in the Tx spacers. | ||
Conditions relevant to click reactions were also considered. Oxidation of redox active metals can render incompetent catalysts.26,27 With Hx-1/Ag106+, solutions purged with O2vs. N2 showed no differences in conversion, suggesting that O2 does not affect the Ag106+ catalyst (Fig. 15S†).42 The solution pH can also influence a click reaction through deprotonation of terminal alkynes and reprotonation of Cu-triazolyl intermediates. Over the range from pH = 5–8.5, the same Ag106+ adducts formed with Hx-1 with little variance in their optical spectra (Fig. 16S and 17S†). However, strong differences emerged with the addition of N3–C3H6–NH2. The conversion efficiency increases ∼25-fold over this pH range, with a sharp jump at pH = 6–7. An enhanced reaction was also observed at higher pH for Cu(I) coordinated with benzimidazole ligands.65 As with these polydentate amine-based ligands, DNA is a rich reservoir of acidic and basic heteroatoms that may regulate deprotonation and reprotonation steps in a click reaction.66
Discussion
Together, Hx-1 and Ag106+ catalyze an alkyne–azide click reaction, and each component in this pair has distinct roles. Ag106+ is the catalyst, and we discuss three of its characteristics – it is preserved while facilitating the click union, it acidifies its neighboring alkyne, and it is distinctly active compared to related complexes. The activity of this catalyst is regulated by the DNA host, which serves two roles – it segregates the cluster within the larger DNA host and it imprints an elongated cluster shape. We now discuss these five characteristics of this nanoscale catalyst.Ag106+ unites a DNA-tethered alkyne with an azide without changing its stoichiometry, charge, and spectra. However, click reactions conserve atoms, so mass spectra cannot distinguish whether an azide reacts with the alkyne or simply coordinates with silvers. To consider these two possibilities, iodide was added upon completing the reaction. This halide degrades the cluster to quench the Ag106+ absorption and strip the DNA of silvers. Two bare Hx-1 strands are identified – without and with the azide. With the silver coordination sites now missing, we conclude that the azide is covalently linked as a triazole. In support of this, the Hx-1/azide conjugate survives 10000× dilution, thus supporting a stable, covalent linkage that resists dissociation. Also, without the DNA-bound alkyne, 1/Ag106+ does not form a stable complex with azides. Thus, Ag106+ catalyzes cycloaddition, and its role as a catalyst is suggested by studies in D2O.
Transition metals catalyze alkyne–azide cycloadditions in steps, beginning with the deprotonation of a terminal alkyne. Ag106+ may follow a similar catalytic path because it acidifies its neighboring alkyne, thereby effecting the H/D exchange in D2O. By analogy with other Cu(I) and Ag(I) catalysts, a deprotonated alkyne converts into a metal acetylide, a strong nucleophile that can attack an azide to set the first C–N bond for a triazole.36,67 Further experiments are underway to evaluate this proposed mechanism.12 Beyond a mononuclear complex, silver acetylides can be multinuclear with both end-on and side-on coordination to give a rich and diverse array of structures.25,68 To better understand the nuclearity of the Hx-1/Ag106+ catalyst, we studied pared down versions of this complex.
Relative to mononuclear complexes, multinuclear click catalysts are more effective, so smaller analogs of Hx-1/Ag106+ were studied. The 4 Ag0 units in Ag106+ were selectively irradiated to yield DNA complexes with less Ag0–Ag20 (Ag64+, Ag75+, Ag86+, and Ag97+) and purely Ag+ (3–5 Ag+) adducts. However, these multinuclear derivatives of Ag106+ are incompetent catalysts, behaving as if the DNA was completely bare. They may be inactive because of their structure. Their wide distribution of silvers suggests that the preexisting Hx-1 binding site is indiscriminate and has little preference for a particular adduct. Thus, these photoproducts may be loosely bound with ill-defined structures, so they may be poor catalysts. We are searching for truncated oligonucleotides better suited for these photofragments. For example, complementary strands can shorten long DNA binding sites to favor smaller clusters.57 Metal oxidation may also underlie the tepid activity of the Ag20 and Ag+ adducts with Hx-1. Dinuclear copper complexes have dynamic coordination environments whose ligands readily exchange because copper oxidation weakens π-backbonding and ligand coordination.22,69 One avenue to better understand the metal oxidation might be to change the balance of Ag+ and Ag0 in a cluster. Specifically, Ag117+ is also a green-emitting fluorophore like Ag106+, but it has an additional Ag+ that could alter the activity of this cluster.32,42,70 We are also studying other DNA/silver cluster complexes. In summary, Ag106+ is the catalytic core of the Hx-1/Ag106+ complex and has three characteristics: it joins the alkyne with azides via cycloaddition, it acidifies and possibly activates the alkyne, and it is markedly more active than related DNA–silver complexes. The click reaction evolves within the confines of the Hx-1 scaffold, and we now discuss how it guides these reactions.
The green-emitting Ag106+ exists because it is trapped and shielded inside Hx-1, and this C4AC4TC3G coordination site is a discrete unit in a larger DNA polymer. In addition, the DNA was further derivatized with 1-hexyne at its 5′ terminal phosphate position, yielding a dual-labeled Hx-1/Ag106+ conjugate. These adducts are independent and were separated by inserting thymine spacers. Furthermore, these binding sites are independent, so the alkyne and cluster adduct can be separated by inserting thymine spacers, neutral spacers that bind poorly with silver. With these longer Hx-Tx-1 strands, the cluster does not spill out of its binding site, as signified by the consistent mass and optical spectra for these variants. As the number of thymine spacers increases, the click conversion efficiency drops. Considering the DNA to be a linear polymer, increasing the distance between the alkyne and the cluster thus tempers the click reaction with azides. Besides phosphates, specific nucleobases can also be alternative sites to derivatize a DNA. For example, X-ray diffraction studies show that the 3′-terminal adenine in (CACCTAGCGA)2-Ag16 is not associated with the cluster and can be labeled with peptides and proteins.71,72 Also, fluorescence anisotropy studies show that the central thymine in C4AC4TC3GT4 (1) is a folding site where the strand can be broken.49 The resulting heterodimer reassembles to form the same green-emitting Ag106+ as the contiguous C4AC4TC3GT4. Thus, we suggest that specific sites could be derivatized in C4AC4TC3GT4/Ag106+ to better understand its click catalysis. DNA is not only a scaffold that tunes the reaction efficiency but is also a polydentate ligand that shapes its cluster adduct.
Bare silver clusters have multiple interconverting isomers, and a DNA strand can select and favor specific isomers. For example, a single-stranded oligonucleotide coordinates a weakly emissive Ag106+ adduct, but this DNA hybridizes with a short complementary strand to now favor a strongly emissive Ag106+.56 This switch reverses when the complement denatures. We now consider the shape of the Ag106+ adduct bound to Hx-1. This complex is a member of a larger class of Ag40-based chromophores with green emission whose structures have been considered from a variety of perspectives.38,40,55 EXAFS studies of a DNA-bound Ag106+ show limited metal–metal vs. metal–DNA coordination when compared to a related weakly fluorescent cluster.54,73 UV-based DNA footprinting identifies large segments of the DNA host that are protected from photodegradation by the Ag106+ adduct, again contrasting with localized binding and protection by a weakly fluorescent cluster.74 Time-resolved infrared spectra show that the C4AC4TC3GT4 strand used in this study chelates with its Ag106+ adduct using multiple nucleobases.75 X-ray diffraction studies of an Ag117+ unit show a rod-like structure with distinct Ag6 and Ag5 subunits that follow the contour of the DNA polymer.70 Quantum mechanical models predict cluster spectra based on rod-like shapes.38,55 We propose that Ag106+ bound to Hx-1 also adopts an elongated shape and is dispersed within its DNA matrix, favoring metal–ligand over metal–metal coordination. An important question is the relative strength of nucleobase–silver coordination and competition with exogenous reagents, and prior studies indicate that reagents can penetrate and access open sites on the cluster.8,32,44,45
Conclusion
Single-stranded oligonucleotides encode the spectra of silver molecules via their sequence and structure, and this templated synthesis is illustrated by the C4AC4TC3GT4 strand which preferentially forms Ag106+. Besides being a strong fluorophore with green emission, this cluster also catalyzes click reactions between a DNA-tethered alkyne and azides. We conclude that DNA is a scaffold that guides catalysis in two ways. First, it segregates the cluster into a discrete, orthogonal binding pocket that leaves the larger DNA polymer open to be further functionalized. We hope to probe the nanoscale DNA/cluster catalyst by site-specifically modifying the DNA. Second, DNA imprints the cluster shape and thus prescribes the coordination environment. We hope to modify the DNA polymer to fine-tune how click reagents access open coordination sites and bind with exposed silvers. Our overall conclusion is that silver molecules are effective click catalysts, and their activity can be directed by a DNA scaffold.Author contributions
Jeffrey Petty and Caleb Setzler contributed equally to this work. Both authors conducted the experiments, analyzed the data, and wrote the manuscript. All authors have read and approved the final version of the manuscript.Data availability
The data supporting this article are included as part of the ESI.†Conflicts of interest
No conflicts to declare.Acknowledgements
The authors thank the National Science Foundation (CHE-1611451 and CHE-2002910) and the Furman Advantage program. This work was supported in part by the National Science Foundation EPSCoR Program under NSF Award No. OIA-1655740. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect those of the National Science Foundation.References
- P. P. Edwards and J. M. Thomas, Angew. Chem., Int. Ed., 2007, 46, 5480–5486 CrossRef CAS PubMed.
- M. Haruta, T. Kobayashi, H. Sano and N. Yamada, Chem. Lett., 2006, 16, 405–408 CrossRef.
- G. C. Bond and D. T. Thompson, Gold Bull., 2000, 33, 41–50 CrossRef CAS.
- N. Lopez, T. V. W. Janssens, B. S. Clausen, Y. Xu, M. Mavrikakis, T. Bligaard and J. K. Nørskov, J. Catal., 2004, 223, 232–235 CrossRef CAS.
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801–802, 10.1039/C39940000801.
- R. L. Whetten, J. T. Khoury, M. M. Alvarez, S. Murthy, I. Vezmar, Z. L. Wang, P. W. Stephens, C. L. Cleveland, W. D. Luedtke and U. Landman, Adv. Mater., 1996, 8, 428–433 CrossRef CAS.
- R. Jin, Nanoscale, 2015, 7, 1549–1565 RSC.
- M. F. Matus and H. Häkkinen, Nat. Rev. Mater., 2023, 8, 372–389 CrossRef CAS.
- Y. Du, H. Sheng, D. Astruc and M. Zhu, Chem. Rev., 2020, 120, 526–622 CrossRef CAS PubMed.
- P. D. Jadzinsky, G. Calero, C. J. Ackerson, D. A. Bushnell and R. D. Kornberg, Science, 2007, 318, 430–433 CrossRef CAS PubMed.
- O. Lopez-Acevedo, K. A. Kacprzak, J. Akola and H. Häkkinen, Nat. Chem., 2010, 2, 329–334 CrossRef CAS PubMed.
- F. Himo, T. Lovell, R. Hilgraf, V. V. Rostovtsev, L. Noodleman, K. B. Sharpless and V. V. Fokin, J. Am. Chem. Soc., 2005, 127, 210–216 CrossRef CAS PubMed.
- C. Wang, D. Ikhlef, S. Kahlal, J.-Y. Saillard and D. Astruc, Coord. Chem. Rev., 2016, 316, 1–20 CrossRef CAS.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- D. Honcharenko, K. Druceikaite, M. Honcharenko, M. Bollmark, U. Tedebark and R. Strömberg, ACS Omega, 2021, 6, 579–593 CrossRef CAS PubMed.
- N. Z. Fantoni, A. H. El-Sagheer and T. Brown, Chem. Rev., 2021, 121, 7122–7154 CrossRef CAS PubMed.
- S. I. Presolski, V. P. Hong and M. G. Finn, Curr. Protoc. Chem. Biol., 2011, 3, 153–162 CrossRef PubMed.
- J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302–1315 RSC.
- V. O. Rodionov, V. V. Fokin and M. G. Finn, Angew. Chem., Int. Ed., 2005, 44, 2210–2215 CrossRef CAS PubMed.
- B. T. Worrell, J. A. Malik and V. V. Fokin, Science, 2013, 340, 457–460 CrossRef CAS PubMed.
- B. F. Straub, Chem. Commun., 2007, 3868–3870, 10.1039/B706926J.
- M. Ahlquist and V. V. Fokin, Organometallics, 2007, 26, 4389–4391 CrossRef CAS.
- A. K. Gupta and A. Orthaber, Chem. – Eur. J., 2018, 24, 7536–7559 CrossRef CAS PubMed.
- M. G. Finn and V. V. Fokin, in Catalysis without Precious Metals, 2010, pp. 235–260. DOI:10.1002/9783527631582.ch10.
- V. O. Rodionov, S. I. Presolski, D. Díaz Díaz, V. V. Fokin and M. G. Finn, J. Am. Chem. Soc., 2007, 129, 12705–12712 CrossRef CAS PubMed.
- P. S. Donnelly, S. D. Zanatta, S. C. Zammit, J. M. White and S. J. Williams, Chem. Commun., 2008, 2459–2461, 10.1039/B719724A.
- R. Berg, J. Straub, E. Schreiner, S. Mader, F. Rominger and B. F. Straub, Adv. Synth. Catal., 2012, 354, 3445–3450 CrossRef CAS.
- A. Makarem, R. Berg, F. Rominger and B. F. Straub, Angew. Chem., Int. Ed., 2015, 54, 7431–7435 CrossRef CAS PubMed.
- A. W. Cook, Z. R. Jones, G. Wu, S. L. Scott and T. W. Hayton, J. Am. Chem. Soc., 2018, 140, 394–400 CrossRef CAS PubMed.
- C. Dong, R.-W. Huang, A. Sagadevan, P. Yuan, L. Gutiérrez-Arzaluz, A. Ghosh, S. Nematulloev, B. Alamer, O. F. Mohammed, I. Hussain, M. Rueping and O. M. Bakr, Angew. Chem., Int. Ed., 2023, 62, e202307140 CrossRef CAS PubMed.
- Y. Fang, K. Bao, P. Zhang, H. Sheng, Y. Yun, S.-X. Hu, D. Astruc and M. Zhu, J. Am. Chem. Soc., 2021, 143, 1768–1772 CrossRef CAS PubMed.
- J.-P. Gao, F.-Q. Zhang and X.-M. Zhang, Adv. Sci., 2024, 2400377 CrossRef CAS PubMed.
- J. T. Petty, J. Zheng, N. V. Hud and R. M. Dickson, J. Am. Chem. Soc., 2004, 126, 5207–5212 CrossRef CAS PubMed.
- J. McNulty and K. Keskar, Eur. J. Org. Chem., 2012, 2012, 5462–5470 CrossRef CAS.
- O. A. Yeshchenko, I. M. Dmitruk, A. A. Alexeenko, M. Y. Losytskyy, A. V. Kotko and A. O. Pinchuk, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 235438 CrossRef.
- D. Schultz, K. Gardner, S. S. R. Oemrawsingh, N. Markešević, K. Olsson, M. Debord, D. Bouwmeester and E. Gwinn, Adv. Mater., 2013, 25, 2797–2803 CrossRef CAS PubMed.
- A. Gonzàlez-Rosell, C. Cerretani, P. Mastracco, T. Vosch and S. M. Copp, Nanoscale Adv., 2021, 3, 1230–1260 RSC.
- C. I. Richards, S. Choi, J.-C. Hsiang, Y. Antoku, T. Vosch, A. Bongiorno, Y.-L. Tzeng and R. M. Dickson, J. Am. Chem. Soc., 2008, 130, 5038–5039 CrossRef CAS PubMed.
- H. C. Yeh, J. Sharma, J. J. Han, J. S. Martinez and J. H. Werner, Nano Lett., 2010, 10, 3106–3110 CrossRef CAS PubMed.
- J. T. Petty, D. Lewis, S. Carnahan, D. Kim and C. Couch, J. Phys. Chem. B, 2022, 126, 3822–3830 CrossRef CAS PubMed.
- J. T. Petty, S. Carnahan, D. Kim and D. Lewis, J. Chem. Phys., 2021, 154, 244302 CrossRef CAS PubMed.
- C. J. Setzler, C. A. Arrington, D. Lewis and J. T. Petty, J. Phys. Chem. B, 2023, 127, 10851–10860 CrossRef CAS PubMed.
- D. Lewis, C. Setzler, P. M. Goodwin, K. Thomas, M. Branham, C. A. Arrington and J. T. Petty, J. Phys. Chem. C, 2023, 127, 10574–10584 CrossRef CAS PubMed.
- C. S. McKay and M. G. Finn, Chem. Biol., 2014, 21, 1075–1101 CrossRef CAS PubMed.
- V. A. Bloomfield, D. M. Crothers and I. Tinoco Jr., Nucleic Acids: Structures, Properties, and Functions, University Science Books, Sausaltio, CA, 2000 Search PubMed.
- G. A. Crosby and J. N. Demas, J. Phys. Chem., 1971, 75, 991–1024 CrossRef CAS.
- C. He, P. M. Goodwin, A. I. Yunus, R. M. Dickson and J. T. Petty, J. Phys. Chem. C, 2019, 123, 17588–17597 CrossRef CAS.
- J. T. Petty, M. Ganguly, A. I. Yunus, C. He, P. M. Goodwin, Y.-H. Lu and R. M. Dickson, J. Phys. Chem. C, 2018, 122, 28382–28392 CrossRef CAS.
- S. M. Copp, P. Bogdanov, M. Debord, A. Singh and E. Gwinn, Adv. Mater., 2014, 26, 5839–5845 CrossRef CAS PubMed.
- B. Sengupta, C. M. Ritchie, J. G. Buckman, K. R. Johnsen, P. M. Goodwin and J. T. Petty, J. Phys. Chem. C, 2008, 112, 18776–18782 CrossRef CAS PubMed.
- K. Koszinowski and K. Ballweg, Chem. – Eur. J., 2010, 16, 3285–3290 CrossRef CAS PubMed.
- J. T. Petty, O. O. Sergev, M. Ganguly, I. J. Rankine, D. M. Chevrier and P. Zhang, J. Am. Chem. Soc., 2016, 138, 3469–3477 CrossRef CAS PubMed.
- S. M. Copp, D. Schultz, S. Swasey, J. Pavlovich, M. Debord, A. Chiu, K. Olsson and E. Gwinn, J. Phys. Chem. Lett., 2014, 5, 959–963 CrossRef CAS PubMed.
- J. T. Petty, O. O. Sergev, D. A. Nicholson, P. M. Goodwin, B. Giri and D. R. McMullan, Anal. Chem., 2013, 85, 9868–9876 CrossRef CAS PubMed.
- M. Ganguly, C. Bradsher, P. Goodwin and J. T. Petty, J. Phys. Chem. C, 2015, 119, 27829–27837 CrossRef CAS PubMed.
- D. J. E. Huard, A. Demissie, D. Kim, D. Lewis, R. M. Dickson, J. T. Petty and R. L. Lieberman, J. Am. Chem. Soc., 2019, 141, 11465–11470 CrossRef CAS PubMed.
- L. D. Pachón, J. H. van Maarseveen and G. Rothenberg, Adv. Synth. Catal., 2005, 347, 811–815 CrossRef.
- J. Belloni, M. Mostafavi, H. Remita, J.-L. Marignier and A. Marie-Odile Delcourt, New J. Chem., 1998, 22, 1239–1255 RSC.
- N. M. Dimitrijevic, D. M. Bartels, C. D. Jonah, K. Takahashi and T. Rajh, J. Phys. Chem. B, 2001, 105, 954–959 CrossRef CAS.
- B. G. Ershov, E. Janata, A. Henglein and A. Fojtik, J. Phys. Chem., 1993, 97, 4589–4594 CrossRef CAS.
- M. L. Rodríguez-Sánchez, M. J. Rodríguez, M. C. Blanco, J. Rivas and M. A. López-Quintela, J. Phys. Chem. B, 2005, 109, 1183–1191 CrossRef PubMed.
- S. A. Mitchell, G. A. Kenney-Wallace and G. A. Ozin, J. Am. Chem. Soc., 1981, 103, 6030–6035 CrossRef CAS.
- V. O. Rodionov, S. I. Presolski, S. Gardinier, Y.-H. Lim and M. G. Finn, J. Am. Chem. Soc., 2007, 129, 12696–12704 CrossRef CAS PubMed.
- F. David, C. Setzler, A. Sorescu, R. L. Lieberman, F. Meilleur and J. T. Petty, J. Phys. Chem. Lett., 2022, 13, 11317–11322 CrossRef CAS PubMed.
- E. Boz and N. Ş. Tüzün, Dalton Trans., 2016, 45, 5752–5764 RSC.
- W. Wang, X.-Y. Zhai and L. Zhao, Inorg. Chem., 2023, 62, 1414–1422 CrossRef CAS PubMed.
- R. Berg and B. F. Straub, Beilstein J. Org. Chem., 2013, 9, 2715–2750 CrossRef PubMed.
- V. Rück, V. A. Neacşu, M. B. Liisberg, C. B. Mollerup, P. H. Ju, T. Vosch, J. Kondo and C. Cerretani, Adv. Opt. Mater., 2024, 12, 2301928 CrossRef.
- V. Rück, N. K. Mishra, K. K. Sørensen, M. B. Liisberg, A. B. Sloth, C. Cerretani, C. B. Mollerup, A. Kjaer, C. Lou, K. J. Jensen and T. Vosch, J. Am. Chem. Soc., 2023, 145, 16771–16777 CrossRef PubMed.
- C. Cerretani, H. Kanazawa, T. Vosch and J. Kondo, Angew. Chem., Int. Ed., 2019, 58, 17153–17157 CrossRef CAS PubMed.
- J. T. Petty, M. Ganguly, I. J. Rankine, D. M. Chevrier and P. Zhang, J. Phys. Chem. C, 2017, 121, 14936–14945 CrossRef CAS.
- M. S. Blevins, D. Kim, C. M. Crittenden, S. Hong, H.-C. Yeh, J. T. Petty and J. S. Brodbelt, ACS Nano, 2019, 13, 14070–14079 CrossRef CAS PubMed.
- Y. Zhang, C. He, J. T. Petty and B. Kohler, J. Phys. Chem. Lett., 2020, 11, 8958–8963 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr02938k |
| This journal is © The Royal Society of Chemistry 2024 |