
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Factors influencing the chiral imprinting in perovskite nanoparticles
Nazifa
Tabassum
 a,
Brian P.
Bloom
a,
Gouranga H.
Debnath
*b and
David H.
Waldeck
*a
a,
Brian P.
Bloom
a,
Gouranga H.
Debnath
*b and
David H.
Waldeck
*a
aDepartment of Chemistry, University of Pittsburgh, Pittsburgh, Pennsylvania 15260, USA. E-mail: gouranga.debnath@jainuniversity.ac.in
bCentre for Nano and Material Sciences, Jain University, Bangalore, Karnataka 562112, India. E-mail: dave@pitt.edu
First published on 7th November 2024
Abstract
Chiral perovskites have emerged as a new class of nanomaterials for manipulation and control of spin polarized current and circularly polarized light for applications in spintronics, chiro-optoelectronics, and chiral photonics. While significant effort has been made in discovering and optimizing strategies to synthesize different forms of chiral perovskites, the mechanism through which chirality is imbued onto the perovskites by chiral surface ligands remains unclear. In this minireview, we provide a detailed discussion of one of the proposed mechanisms, electronic imprinting from a chiral ligand.
Introduction
Since the seminal work by Moon and coworkers, which reported chiroptical signatures in two-dimensional hybrid organic–inorganic perovskite (HOIP) films,1 interest in the development and understanding of chirality in perovskite materials has grown dramatically.2,3 This interest stems from the additional functionality that chirality imbues into the optoelectronic properties of perovskites, i.e., their tunable emission over the entire visible spectral region,4 large defect tolerance,5 near unity photon emission quantum yields,6 and high charge carrier mobility.7 Therefore, in addition to achiral perovskites being at the forefront of sources for light emitting diodes (LEDs),8 lasers,9 and photovoltaics,10 chiral perovskites could become the next-generation nanomaterials for opto-spintronic devices,11 circularly polarized light sources12,13 and detectors;14 and they provide a platform for examining the chiral induced spin selectivity effect15 through demonstrations of spin polarized charge transport,16–18 as well as spin dependent photogalvanic and photovoltaic responses in optoelectronic devices.19The general strategies employed to generate colloidal chiral perovskite nanoparticles (NPs) can be broadly classified into two main categories: (a) a direct synthetic approach in which the chiral ligands, along with the cationic and anionic precursors in the presence of appropriate surface functionalizing ligands and solvents, are reacted to form the chiral perovskite NPs, and (b) a post-synthetic modification of achiral perovskite NPs with chiral ligands.20–22 Note that direct synthetic strategies can involve ligand assisted precipitation, sonochemical synthesis, non-solvent crystallization, slow evaporation crystallization, cooling crystallization, or anti-solvent vapor assisted crystallization. The reader is directed to recent review articles which discuss these different synthetic approaches for inducing chirality in perovskite NPs in more detail.11,23–26 Understanding the origin and mechanism of chirality in perovskites is paramount for maximizing their performance in applications. Thus, the synthetic approaches and the resulting morphologies ought to play a critical role in this regard.
The major mechanisms that have been proposed for the induction of chirality in perovskites include the following:
• Surface distortion of the perovskite from interaction with the chiral ligand: surface distortions have been hypothesized to explain the circular dichroism (CD) and circularly polarized luminescence (CPL) activity of R- and S-α-octylamine functionalized cesium lead bromide (CsPbBr3) NPs.27 This idea was later corroborated by computational studies which showed that chiral ligands attached to the surface of perovskite NPs can cause noncentrosymmetric distortion of the surface lattice, penetrating up to five unit cells, thereby inducing chiro-optical properties.28
• Chiral crystal structure mediated by the chiral organic ligands: the report by Moon and coworkers (vide supra) represents this category where the chiral spacers i.e. R- or S-methylbenzylamine (MBA) positioned between layers of lead iodide frameworks in a two-dimensional perovskite imparts chirality to the whole crystal structure.1 Similar findings were shown by Chiu and coworkers on 2D HOIPs synthesized with a series of para substituted halogen organic spacers [(p-XMBA)2PbI4, where X = F, Cl, Br, and I], where a direct correlation of the spacing between the inorganic layers and the halogen–halogen interaction between organic cations and inorganic sheets was shown to modulate the CD and CPL signals.29
• Chiral assemblies of perovskites: in 2018, Shi et al. reported strong tunable CPL emission over the visible range from a co-assembly of achiral CsPbX3 (X = Cl, Br, and I) perovskite NCs with the chiral organic lipid N,N′-bis(octadecyl)-L-glutamic diamide (LGAm) and its enantiomer DGAm.30 While CD features were not observed due to scattering from the gel, the two enantiomers of the composite gel produced mirror image CPL spectra that could be modulated through temperature. Similar chirality transfer has been reported in other composite assemblies of achiral perovskite NPs with chiral matrices, such as chiral amino-acid containing gels,31 chiral metal–organic frameworks,32 and liquid crystal films33,34 among others.
• Electronic interaction between a chiral ligand and an achiral perovskite: in 2018, we showed that using a combination of chiral R- or S-phenylethylammonium and achiral octylamine ligands for the direct synthesis of methylammonium lead bromide perovskite nanoplatelets (NPLs) resulted in the generation of chiral NPLs.35 The temperature and concentration dependent CD spectra of the NPLs showed that the chiro-optical activity results from chiral imprinting on the electronic states of the quantum confined NPLs and not explicitly from aggregation or surface structural distortion phenomena. Subsequent reports on the generation of 3D quantum confined chiral CsPbBr3 nanocubes corroborated the effect of electronic coupling on chiral imprinting in inorganic metal halide perovskite quantum dots.20–22Fig. 1 outlines the different chiral induction mechanisms proposed by researchers to date. Although these examples provide a loose guide about the origin and mechanism of chirality in perovskite NPs, a more thorough understanding is necessary to guide materials development.
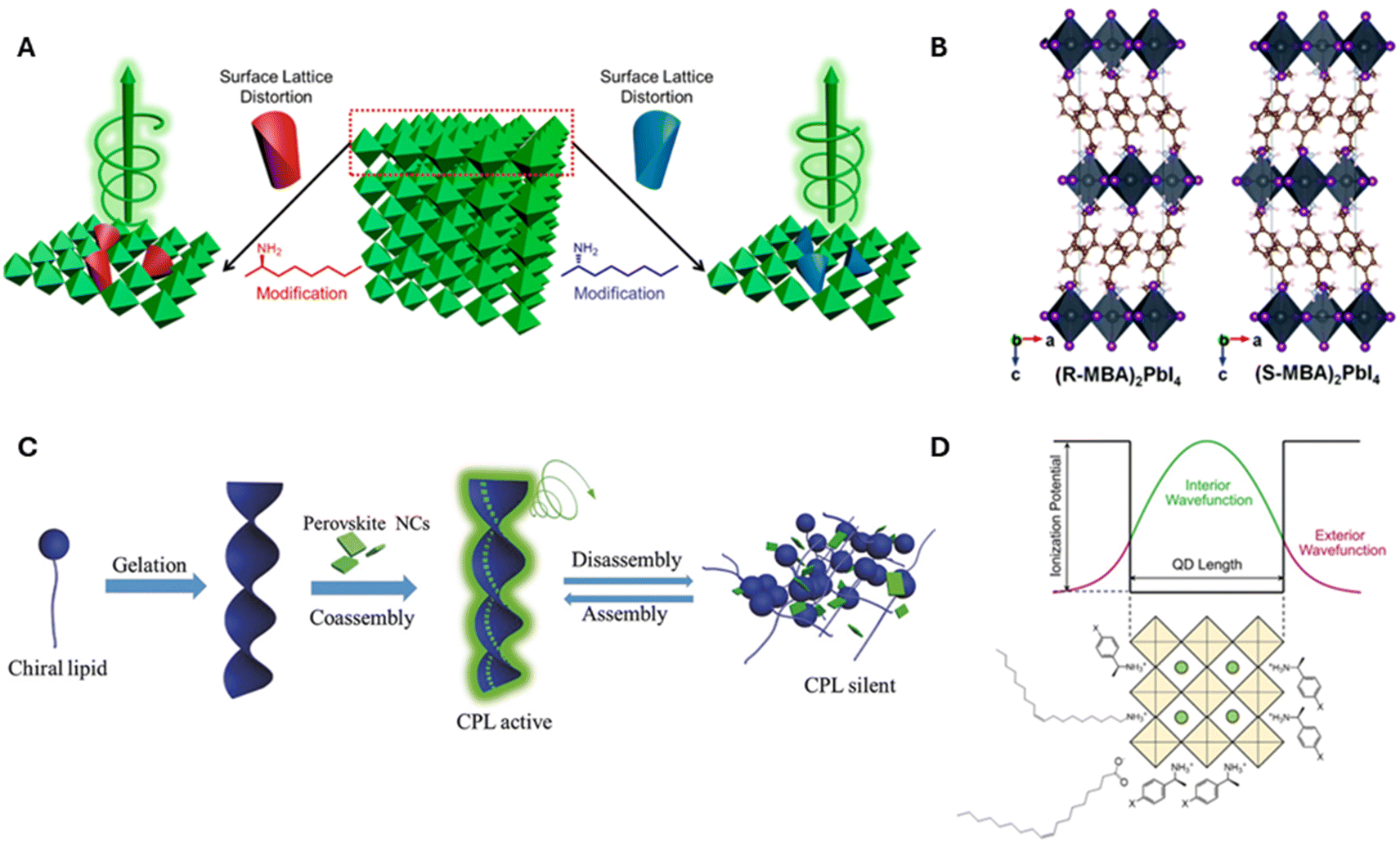 | ||
| Fig. 1 Panel A shows a schematic diagram of chirality induction through surface distortion in R- and S-α-octylamine capped CsPbBr3 NPs. Panel B shows an example of a chiral crystalline structure that can manifest upon incorporation of R and S-MBA organic cations into lead iodide perovskite octahedral sheets. Panel C shows an illustration for the induction of chirality in perovskites through the co-assembly of achiral CsPbX3 NCs onto helical lipid structures. Panel D shows a schematic diagram for electronic chiral imprinting between chiral surface ligands and 2 nm sized CsPbBr3 NPs through wavefunction hybridization. Panel A is adapted from ref. 27 with permission; Copyright 2019, American Chemical Society. Panel B is adapted from ref. 1 with permission; Copyright 2017, Royal Society of Chemistry. Panel C is reproduced from ref. 30 with permission; Copyright 2018, John Wiley and Sons. Panel D is reproduced from ref. 22; Copyright 2024, Springer Nature/the authors. | ||
In this minireview, we emphasize experimental studies into how electronic interactions influence chiral imprinting. We begin with a discussion on the effect of halide composition and perovskite stoichiometry (section 2.1) and then delve into the role of ligand identity (section 2.2) before discussing how ligand coverage and orientation affects chiroptical properties (section 2.3) and the importance of quantum confinement (section 2.4) on chiral imprinting. These experimental discussions are linked to model-based understanding of electronic imprinting which dominated the theoretical literature until the past few years, during which more realistic quantum chemical approaches have begun to appear.
2.1 Sensitivity of chiral imprinting on perovskite composition and stoichiometry
The impressive color tunability over the visible region demonstrated for achiral perovskites, achieved through choice of halide, has been shown to manifest for chiral perovskites and their CD spectra. Fig. 2A shows absorbance (left) and corresponding CD spectra (right) for the direct synthesis of methylammonium (MA) lead halide perovskite NPLs synthesized with chiral phenylethylamine (PEA) ligands.36 The first excitonic transition of the 100% Cl perovskite, ∼350 nm, can be shifted to ∼450 nm with a 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :80 I:Br halide composition and maintain its strong chiroptical features. Note that deviations in the classical bisignate structure of the CD signal occur for some of the mixed halide compositions and are attributed to deviations in packing of the Pb octahedra leading to orientational effects of the chiral ligands (see section 2.3 for more detail). Similar tunability of the perovskite's chiroptical properties can be achieved through halide exchange. Jiang et al. synthesized cesium lead bromide NPLs and exchanged the native ligands with R/S-α-PEA-X and R/S-β-PEA-X (X = Cl, I) salts. The halide exchange with iodine and chlorine resulted in a red and blue shift of the first excitonic peak, respectively, with the magnitude of the shift determined by the amount of salt added; see Fig. 2B and C.37
:80 I:Br halide composition and maintain its strong chiroptical features. Note that deviations in the classical bisignate structure of the CD signal occur for some of the mixed halide compositions and are attributed to deviations in packing of the Pb octahedra leading to orientational effects of the chiral ligands (see section 2.3 for more detail). Similar tunability of the perovskite's chiroptical properties can be achieved through halide exchange. Jiang et al. synthesized cesium lead bromide NPLs and exchanged the native ligands with R/S-α-PEA-X and R/S-β-PEA-X (X = Cl, I) salts. The halide exchange with iodine and chlorine resulted in a red and blue shift of the first excitonic peak, respectively, with the magnitude of the shift determined by the amount of salt added; see Fig. 2B and C.37
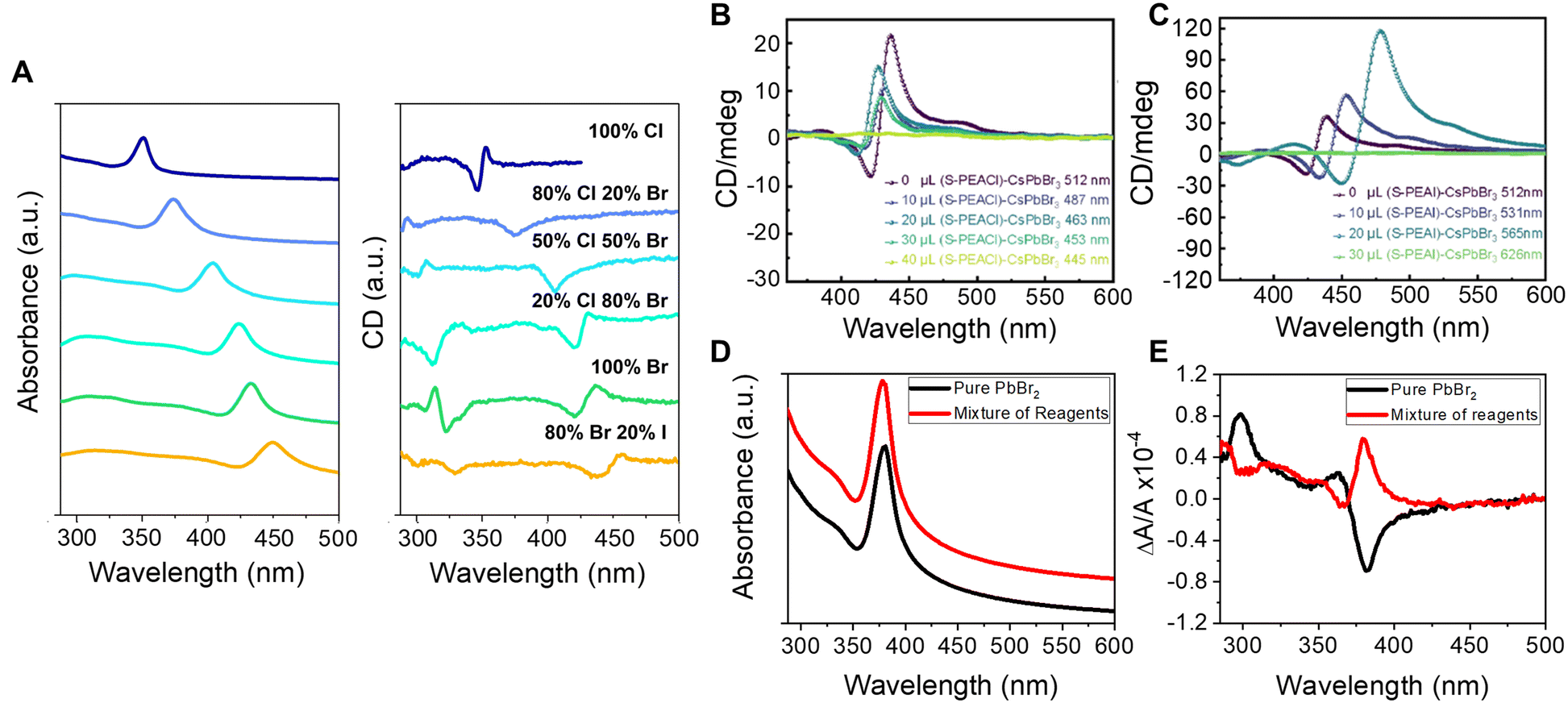 | ||
| Fig. 2 Panel A shows absorbance (left) and CD spectra (right) of methylammonium lead halide perovskite NPLs with different halide compositions. Panels B & C show CD spectra of CsPbBr3 perovskite NPLs upon ligand exchange using S-PEA-Cl (B) and S-PEA-I (C) salts. Panels D & E show absorbance and CD spectra, respectively, of methylammonium lead halide NPLs containing 71% chloride and 29% bromide ions. The black spectra are for NPLs synthesized by mixing pure lead bromide and chloride ligand precursor salts in the desired proportions, and the red spectra represent NPLs synthesized with 71:29 Cl:Br stoichiometric ratio of all reagents. Note, the absorbance data are offset for clarity. Panels A, D, & E are adapted from ref. 36 with permission; Copyright 2022, University of Pittsburgh. Panels B & C are reproduced from ref. 37 with permission; Copyright 2022, American Chemical Society. | ||
It is important to note that the chiral imprinting appears to persist independent of the perovskite NP's shape and the cation identity; studies report chiroptical activity in nanocubes,20–22 nanorods, and nanowires,38 as well as in systems comprising formamidinium39 and cesium40,41 cations in place of methyl ammonium. Elucidating trends between the CD response and stoichiometry/composition of perovskites, however, remains challenging. In part, this can be attributed to the lack of appropriate studies which consider factors known to affect the intensity of the CD, e.g., ligand coverage and orientation (section 2.3); however, data also need to be reported in CD units which account for the changes in the extinction coefficient of perovskites with different composition and stoichiometry to make valid comparisons.
Further complications to understanding chiroptical properties arise in perovskites with mixed halide compositions. Consider for example a methylammonium lead halide NPL with a 71:29 percent ratio of Cl:Br synthesized in two ways: (1) where all of the reagents, e.g. PbX2, MAX, and PEAX, comprise a 71:29 ratio of Cl to Br, and (2) PbBr2 is used as the sole Br-source and MACl and PEACl are used as the cation and ligand salt, respectively. Fig. 2D shows overlapping absorption spectra for the two synthetic approaches, those with the 71:29 stoichiometric ratio of all reagents (red) and those made with a pure PbBr2 source (black), and indicate that the halide composition for the different preparations is approximately the same.36 Interestingly, however, an opposite CD-response is observed despite the same enantiomeric form of the ligand, R-PEA, for the synthesis (Fig. 2E). We posit that the behavior is related to differences in the homogeneity of the NPL surface leading to changes in the ligand packing, akin to that shown for chiral CdSe nanoparticles with different crystalline structure showing opposite Cotton effects;42 however, more rigorous studies are necessary to quantify the ligand coverage, orientation, and structural ordering of the perovskites to validate this hypothesis. Regardless of the underlying cause, this study uniquely highlights the sensitivity of chiral imprinting to perovskite composition and preparation.
2.2 Ligand identity and chiral imprinting
The influence of chiral ligand identity on chiroptical properties manifests in n = 2, methylammonium lead halide NPLs with R/S-2-octylammonium (R- or S-2OA) and R/S-naphthylethylammonium (R- or S-NEA) ligand shells.43 While the absorption profiles of the R- or S-NEA and R-or S-2OA NPLs show similar first excitonic transitions (Fig. 3A), their CD spectra show a markedly different response (Fig. 3B and C). The R- (dotted line) and S-NEA capped NPLs (solid line) show a bisignate feature in the 400–500 nm region in which the peak and trough have approximately the same area (Fig. 3B); however, the R- (dotted line) and S-2OA NPLs (solid line) at the same excitonic transition exhibit very different areas (Fig. 3C). These findings are consistent with other reports in which changes in the CD structure of methylammonium lead bromide NPLs is observed with 4-dimethylbenzylamine, methylbenzylamine, ethylbenzylamine, and cyclohexylethylamine ligands.44 Note that, similar ligand identity dependent changes in the bisignate lobes of R/S-NEA and R/S-2OA was also observed in 2 nm chiral CsPbBr3 nanocubes synthesized through ligand exchange and therefore imply that the perovskite – ligand interaction is important for observing the phenomenon and not the synthetic method.20 While an exact mechanism responsible for these observations has yet to be identified, researchers have attributed the effect to ligand binding affinity, ligand binding geometry, π–π stacking, or long range ordering on the nanomaterial surface being induced by the chiral ligands.44–48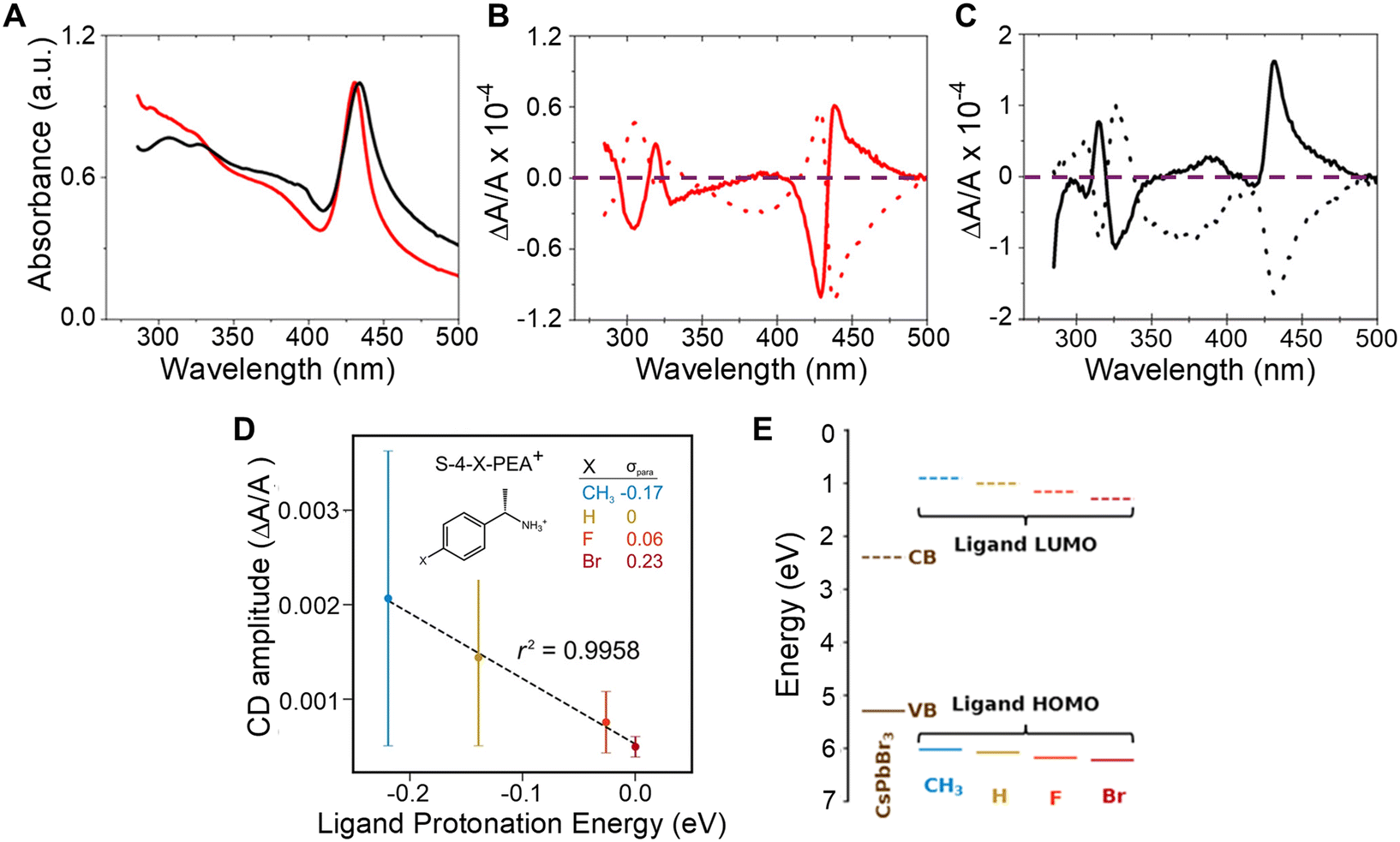 | ||
| Fig. 3 Panel A shows UV-visible absorption spectra of CH3NH3Pb2Br7 NPLs capped with chiral 2OA (black) and chiral NEA (red) ligands. Panels B and C show CD spectra of the R- (dotted) and S-NEA (solid) CH3NH3Pb2Br7 NPLs and R-(dotted) and S-2OA (solid) CH3NH3Pb2Br7 NPLs respectively. The purple dashed line demarcates the position in the plots where the CD intensity is zero. Panel D shows the correlation between CD amplitudes and ligand protonation energy in R or S-4-X-PEABr capped CsPbBr3 NPs. Panel E shows the variation in energy level alignment between CsPbBr3 NPs and R-/S-4-X-PEA+-Br− ion pair complexes. Panel A–C are reproduced from ref. 43; Copyright 2022 American Chemical Society. Panels D & E are adapted from ref. 22; Copyright 2024, Springer Nature/the authors. | ||
In addition to affecting the shape of the chiroptical features, the difference between the peak and the trough of the bisignate feature in the CD spectrum is affected by the ligand identity. Such an effect was observed for chiral 2 nm sized CsPbBr3 NPs capped with R/S-4-X-phenethylammonium bromide (R/S-4-X-PEABr, where X = CH3, H, F, Br).22 Here, the amplitude of the CD spectra of the NPs capped with R/S-4-X-PEABr showed a variation in intensity which followed the trend CH3 > H > F > Br and were found to correlate well with the Hammett parameter (σpara), i.e. logarithm of the ratio of the ionization equilibrium constant of benzoic acid with CH3, F, and Br at the para position to that with H at the para position, a metric for the withdrawal of electron density from the ammonium group across the phenyl ring and the ligand protonation energy (Fig. 3D). Note that, the large error bars in Fig. 3D are associated with synthetic batch-to-batch variation in the amplitude of the CD; however systematic trends with ligand protonation energy are observed for each synthetic batch. The relationship between CD amplitude and ligand protonation energy could arise from an increase (or decrease) in wavefunction overlap between the molecular orbitals of the chiral ligand and those on the perovskite NP and/or differences in the resonance between the electronic state energies of the perovskite and the ligands. An estimation of the energy alignment between the perovskite valence band (VB) and conduction band (CB) to that of the HOMO and LUMO of the bound R/S-4-X-PEABr are shown in Fig. 3E, and show that the HOMO approaches resonance with the valence band of the perovskite as the electron donating character of the functional group improves.22 A more thorough understanding of this trend necessitates additional experimental and theoretical studies involving a more robust class of ligands with larger variations in wavefunction overlap and electronic state energies.
2.3 Impact of ligand coverage and orientation on chiral imprinting
To study the impact of ligand coverage on chiral imprinting, researchers explored the evolution of chiroptical activity in methylammonium lead halide perovskite NPLs co-passivated by chiral phenethylammonium (PEA) and achiral octylamine (OA) ligands.43 By systematically controlling the ratio of chiral PEA to achiral OA ligands, the evolution of chiral imprinting with ligand coverage was varied. Fig. 4A shows that the CD intensity, at the first excitonic transition, for Br (green) and Cl (blue) perovskite NPLs increases monotonically with increasing ratio of chiral ligands before saturating at high chiral ligand densities. To describe the phenomenon, the authors invoked a model in which a chiral electrostatic potential from surface ligands perturbs the perovskite's electronic wavefunction. Their calculations show that the saturation point for chiral imprinting depends sensitively on the size of the exciton, an intrinsic property of the NPL quantum confinement, relative to the size of the chiral ligands, i.e. larger excitons reach a CD saturation point at lower PEA:OA ratio compared to smaller excitons. A similar trend in chiral imprinting with chiral ligand coverage was observed for methylammonium lead bromide NPLs passivated by α-4-dimethylbenzylamine (DMBA).44Fig. 4B shows the CD intensity, quantified as the g-factor of CD, increases with increasing fraction of chiral ligands added to solution. Note that a subsequent decrease in the magnitude of CD intensity is shown at high ligand concentrations; however, this behavior is attributed to increased NPL polydispersity leading to destructive interference of the Cotton effects. Note that, 2 nm sized CsPbBr3 nanocubes, in which the chirality is achieved through ligand exchange, also exhibit similar behavior.20
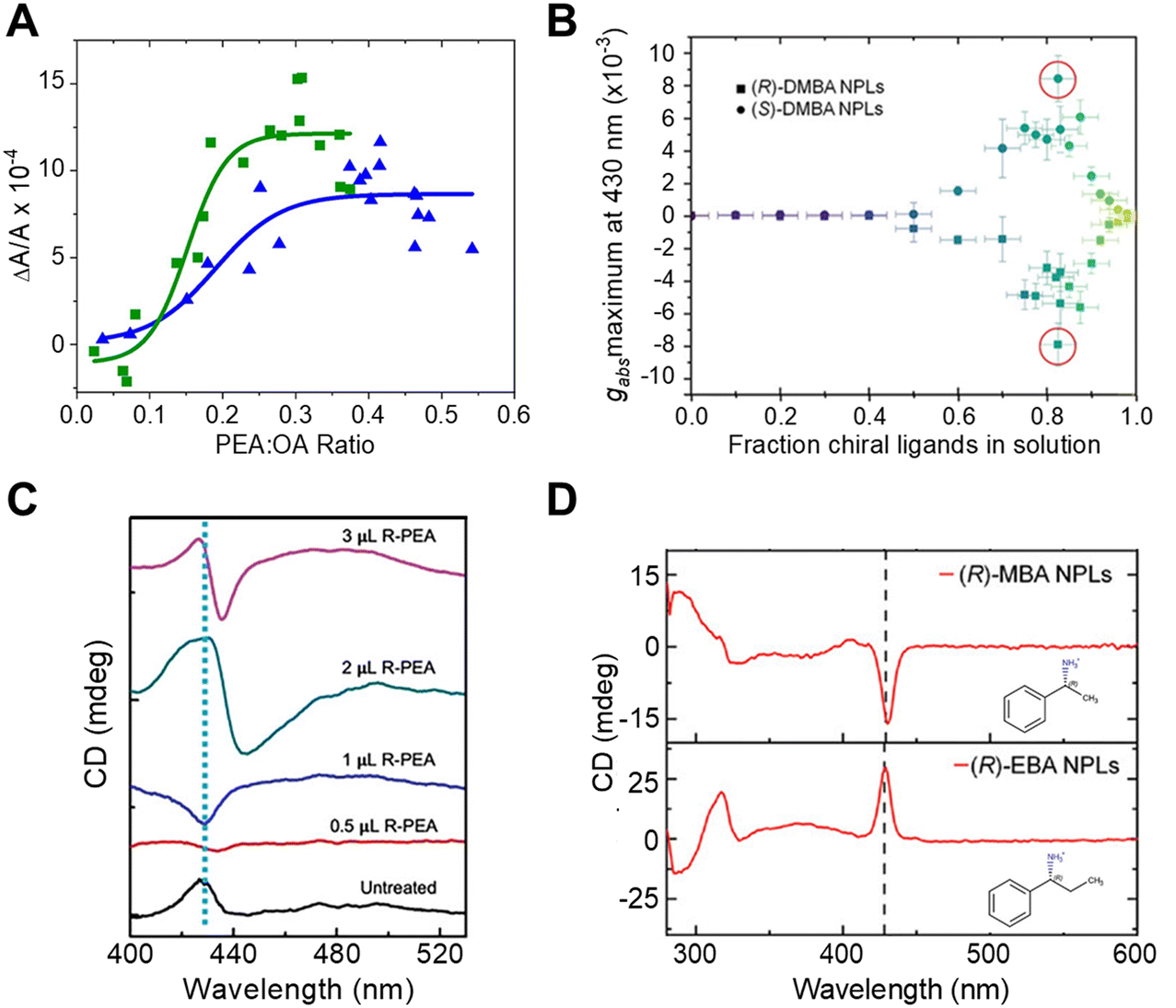 | ||
| Fig. 4 Panel A shows the change in CD intensity, measured at the first excitonic transition, of methylammonium lead bromide (green) and methylammonium lead chloride (blue) NPLs with different ratios of chiral R-PEA to achiral OA ligand coverage. Panel B shows the dependence of CD dissymmetry or g-factor (gabs) due to chiral imprinting on the fraction of chiral DMBA ligands added to methylammonium lead bromide NPLs. Panel C shows changes in the shape of the CD peak at the excitonic transition of CsPbBr3 NPLs with an increasing amount of chiral R-PEA ligand. Panel D depicts the CD spectra of methylammonium lead bromide NPLs passivated with R-MBA (top) and R-EBA (bottom) ligands. Panel A is reproduced from ref. 43; Copyright 2022 American Chemical Society. Panels B and D are adapted from ref. 44 with permission; Copyright 2022, John Wiley and Sons. Panel C is adapted from ref. 40 with permission; Copyright 2023, John Wiley and Sons. | ||
The orientation of chiral ligands bound to the perovskite can also strongly influence chiral imprinting. For instance, measurements on methylammonium lead halide NPLs passivated with OA and PEA showed reversible changes in the sign of Cotton effects as a function of solution temperature.43 At elevated temperatures, the OA ligands were shown to dissociate from the NPL surface, resulting in orientational changes of the bound chiral ligands. TD-DFT calculations corroborate this conclusion; changing the angle between the phenyl ring of the chiral PEA ligand and a perovskite cluster was shown to invert the CD response. A similar phenomenon of CD inversion with changing chiral ligand concentrations in n = 2 cesium lead bromide NPLs passivated by chiral PEA ligands has also been reported.40Fig. 4C shows that the CD spectrum at the perovskite's lowest energy excitonic transition (∼430 nm) changes from positive to negative upon addition of 0.5 μL of R-PEA, before achieving a bisignate line-shape at 2 μL addition of R-PEA.40 The effect of ligand orientation on chiral imprinting can also be inferred from studies made using different chiral ligands. Hubley et al. reported CD spectra for n = 2 methylammonium lead bromide NPLs with different chiral ligands passivating the surface (Fig. 4D).44 Here, changing the methyl group in R-α-methylbenzylamine (R-MBA) ligands to an ethyl group, α-ethylbenzylamine (R-EBA), inverts the CD signal at the NPL first excitonic transition. These changes in the CD features are attributed to different geometrical configuration and orientation of the chiral ligands on the NPL surface.
2.4 Effect of quantum confinement on chiral imprinting
Understanding the role of quantum confinement on chiral imprinting requires studies that operate in a regime where ligand coverage effects and orientation (section 2.3) do not influence the CD response. While Kim et al. were the first to show a size-dependent change in the CD intensity for chiral FAPbBr3 NPs,39 the authors manipulated the size of the NPs through addition of 2OA during synthesis and conclusions regarding the role of quantum confinement are difficult to draw.In later work, we explored how the CD intensity of cesium lead bromide NPs, passivated by chiral β-methylphenethylammonium bromide (MPEABr) ligands, changes with the size of the perovskite NP.21Fig. 5A shows changes in the CD intensity at the region of the perovskite's first excitonic transition for 2 nm (red), 4 nm (blue), 5 nm (purple), and 6 nm (green) edge length perovskite nanocubes. An ∼10-fold decrease in CD intensity is observed between 2 nm and 4 nm sized NPs, and a ∼100-fold decrease in CD intensity is observed between 2 nm and 6 nm sized NPs. While differences in chiral ligand coverage gave rise to some fluctuations in the CD intensity (denoted by the y-axis error bars), these variations were small compared to the changes in the average CD intensity as a function of quantum confinement. In addition to the decrease in CD intensity with increasing size, asymmetry in the area of the peak and trough also manifests. Note that contributions from structural distortion to the overall chiroptical response were observed; however, in the strongly quantum confined regime, the predominance of CD intensity came from the electronic chiral imprinting. Attempts to reconcile the exponential size dependence of CD intensity with a coupled oscillator model failed. Rather, treating the chiral ligand shell as an extended dipole/charge distribution on the perovskite or considering a dynamic coupling of magnetic and electronic transition dipoles of the perovskite and the ligands was necessary.
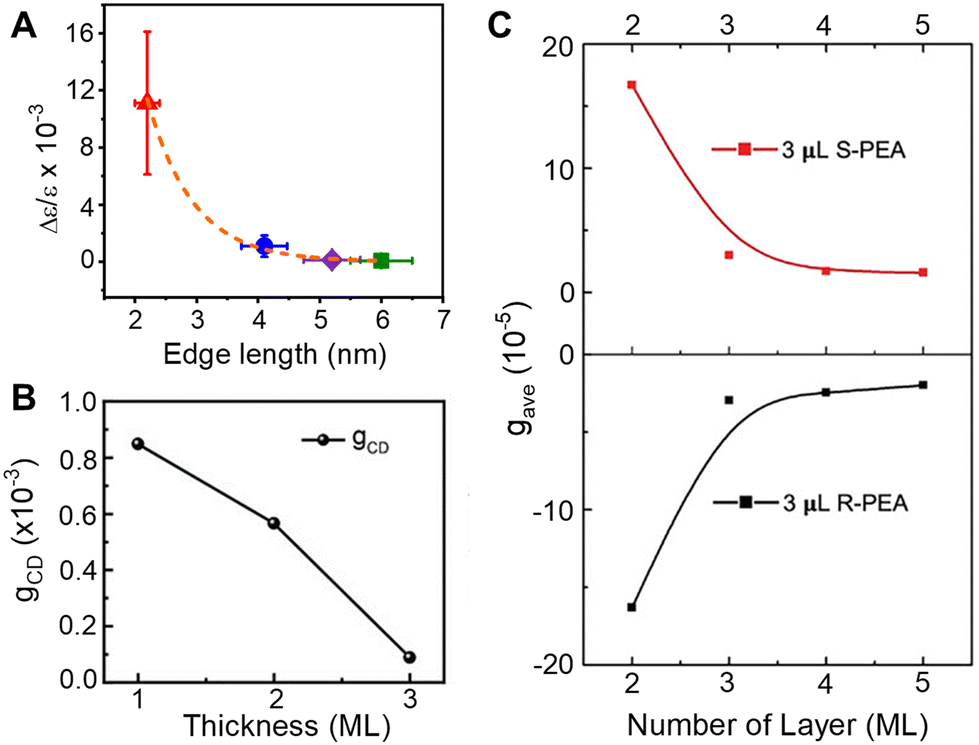 | ||
| Fig. 5 Panel A shows the change in CD intensity, represented by a g-factor term Δε/ε, with changes in the size of chiral CsPbBr3 nanocubes. Note, the size of the particles is indicated by the edge length. Panel B shows changes in the CD intensity of CsPbBr3 NPLs passivated by chiral MPEA ligands as a function of monolayer (ML) thickness. Panel C shows changes in CD intensity of CsPbBr3 NPLs passivated by S-PEA (red) and R-PEA (black) ligands as a function of ML thickness. Panel A is reproduced from ref. 21; Copyright 2023, Royal Society of Chemistry. Panel B is reproduced from ref. 49 with permission; Copyright 2023, John Wiley and Sons. Panel C is reproduced from ref. 40 with permission; Copyright 2023, John Wiley and Sons. | ||
A similar decrease in the CD intensity as a function of quantum confinement has also been reported for CsPbBr3 NPLs.49Fig. 5B shows the CD intensity for 1, 2, and 3 monolayer (ML) NPLs passivated with MPEA ligands, and Fig. 5C shows the CD intensity of 2, 3, 4, and 5 ML thick NPLs passivated with PEA ligands.40 In the latter case, an exponential size dependence on the CD intensity was observed and the authors attribute the response to a combination of electronic chiral imprinting interactions as well as contributions from surface chiral defects. While deconvoluting structural distortion-mediated CD from electronic effects on chiral imprinting remains challenging, systematic studies on 2D methylammonium lead halide chiral perovskites formed in size-controlled nanopores highlight a valuable platform for investigating the mechanistic features of chiral imprinting.50 The authors conclude that dipolar electronic interactions between the chiral ligand and the inorganic perovskite framework plays a larger role in chirality transfer than that arising from structural distortions. The contrast between the dominant electronic chiral imprinting mechanism in confined 2D perovskites versus the structural chirality seen in “bulk” 2D perovskites51 further emphasizes the role of size confinement in the chiral imprinting mechanism.
Conclusion
Chiral perovskites offer tremendous opportunities for spintronic and optoelectronic applications; however, a thorough understanding of the underlying mechanism(s) through which chirality is imprinted on perovskites is needed to predict and manipulate their chiro-optical properties. Considerable research in this regard has been performed on other semiconductor nanoparticles, namely II–VI quantum dots (QDs),52–55 and the understanding gained from those studies should prove useful for guiding future work on perovskites. For instance, Ben-Moshe et al. showed that the CD spectral shape in chiral CdS and CdSe QDs correlates well with the excitonic transitions of the QDs, and attributed the phenomenon to hybridization between the QD valence states with the HOMO orbitals of the chiral ligands.56 Subsequent time-dependent density functional theory calculations corroborate this model,42 and can explain the experimentally observed changes in chiro-optical properties with QD size,57 chiral ligand type,58–60 and shell thickness.61To date, theoretical studies of chiral imprinting on perovskite NPs argue that (1) ligand induced structural distortions are responsible for chirality in perovskites,27,28,49 analogous to that shown for 2D hybrid organic–inorganic perovskites;51,62,63 or (2) wavefunction hybridization or dipolar coupling between the chiral ligand shell and the perovskite leads to chiral induction.22,37,43,64 The preponderance of experimental studies imply that multiple mechanisms can occur simultaneously.21 Although theoretical models have been able to rationalize experimental trends for chiral imprinting,22,43ab initio quantum chemistry calculations on realistic nanoparticle sizes is lacking. DFT approaches are beginning to reach experimentally relevant sizes, however; and recent studies have identified a number of different mechanisms for electronic imprinting, which include electronic hybridization between the ligand and NP exciton,37 static ligand dipole to exciton transition dipole coupling,64 and modulation in the density of electronic states.65 Such first principles calculations, which can reproduce experimentally observed Cotton effects and trends at relevant wavelengths, promise to reveal the mechanistic features responsible for electronic chiral imprinting.
Much progress has been made with regards to understanding the factors that affect electronic chiral imprinting in perovskite NPs. It is now clear that the choice of chiral ligand and its subsequent orientation and coverage, when bound to the perovskite, strongly influence the chiro-optical response. Moreover, quantum confinement is shown to be critical for imprinting chirality through electronic interactions. A more thorough understanding of chirality in perovskites, however, requires more rigorous studies which can deconvolute the different mechanisms discussed here.
Data availability
This manuscript is a mini-review and uses data that has been already published as journal articles or PhD theses.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
D. H. W. acknowledges support from the U.S. Department of Energy (Grant No. ER46430) and G. H. D. acknowledges Jain University for support (Grant No. JU/MRP/CNMS/21/2022). We also would like to thank Dr W. Dunlap-Shohl for fruitful discussions.References
- J. Ahn, E. Lee, J. Tan, W. Yang, B. Kim and J. Moon, Mater. Horiz., 2017, 4, 851–856 RSC.
- T. He, J. Li, X. Li, C. Ren, Y. Luo, F. Zhao, R. Chen, X. Lin and J. Zhang, Appl. Phys. Lett., 2017, 111, 151102 CrossRef.
- S. Ma, J. Ahn and J. Moon, Adv. Mater., 2021, 33, 2005760 CrossRef CAS.
- J. Shamsi, A. S. Urban, M. Imran, L. De Trizio and L. Manna, Chem. Rev., 2019, 119, 3296–3348 CrossRef CAS PubMed.
- H. Huang, M. I. Bodnarchuk, S. V. Kershaw, M. V. Kovalenko and A. L. Rogach, ACS Energy Lett., 2017, 2, 2071–2083 CrossRef CAS.
- F. Di Stasio, S. Christodoulou, N. Huo and G. Konstantatos, Chem. Mater., 2017, 29, 7663–7667 CrossRef.
- L. M. Herz, Annu. Phys. Rev. Chem., 2016, 67, 65–89 CrossRef CAS.
- J. Cui, Y. Liu, Y. Deng, C. Lin, Z. Fang, C. Xiang, P. Bai, K. Du, X. Zuo, K. Wen, S. Gong, H. He, Z. Ye, Y. Gao, H. Tian, B. Zhao, J. Wang and Y. Jin, Sci. Adv., 2021, 7, eabg8458 CrossRef CAS PubMed.
- H. Dong, C. Zhang, X. Liu, J. Yao and Y. S. Zhao, Chem. Soc. Rev., 2020, 49, 951–982 RSC.
- A. K. Jena, A. Kulkarni and T. Miyasaka, Chem. Rev., 2019, 119, 3036–3103 CrossRef.
- J. Ma, H. Wang and D. Li, Adv. Mater., 2021, 33, 2008785 CrossRef.
- Y.-H. Kim, Y. Zhai, H. Lu, X. Pan, C. Xiao, E. A. Gaulding, S. P. Harvey, J. J. Berry, Z. V. Vardeny, J. M. Luther and M. C. Beard, Science, 2021, 371, 1129 CrossRef PubMed.
- M. P. Hautzinger, X. Pan, S. C. Hayden, J. Y. Ye, Q. Jiang, M. J. Wilson, A. J. Phillips, Y. Dong, E. K. Raulerson, I. A. Leahy, C.-S. Jiang, J. L. Blackburn, J. M. Luther, Y. Lu, K. Jungjohann, Z. V. Vardeny, J. J. Berry, K. Alberi and M. C. Beard, Nature, 2024, 631, 307–312 CrossRef PubMed.
- C. Chen, L. Gao, W. Gao, C. Ge, X. Du, Z. Li, Y. Yang, G. Niu and J. Tang, Nat. Commun., 2019, 10, 1927 CrossRef PubMed.
- B. P. Bloom, Y. Paltiel, R. Naaman and D. H. Waldeck, Chem. Rev., 2024, 124, 1950–1991 CrossRef PubMed.
- J. M. Abendroth, D. M. Stemer, B. P. Bloom, P. Roy, R. Naaman, D. H. Waldeck, P. S. Weiss and P. C. Mondal, ACS Nano, 2019, 13, 4928–4946 CrossRef PubMed.
- Z. Huang, B. P. Bloom, X. Ni, Z. N. Georgieva, M. Marciesky, E. Vetter, F. Liu, D. H. Waldeck and D. Sun, ACS Nano, 2020, 14, 10370–10375 CrossRef CAS.
- H. Lu, J. Wang, C. Xiao, X. Pan, X. Chen, R. Brunecky, J. J. Berry, K. Zhu, M. C. Beard and Z. V. Vardeny, Sci. Adv., 2019, 5, eaay0571 CrossRef CAS.
- J. Wang, H. Lu, X. Pan, J. Xu, H. Liu, X. Liu, D. R. Khanal, M. F. Toney, M. C. Beard and Z. V. Vardeny, ACS Nano, 2021, 15, 588–595 CrossRef.
- G. H. Debnath, Z. N. Georgieva, B. P. Bloom, S. Tan and D. H. Waldeck, Nanoscale, 2021, 13, 15248–15256 RSC.
- N. Tabassum, Z. N. Georgieva, G. H. Debnath and D. H. Waldeck, Nanoscale, 2023, 15, 2143–2151 RSC.
- W. A. Dunlap-Shohl, N. Tabassum, P. Zhang, E. Shiby, D. N. Beratan and D. H. Waldeck, Sci. Rep., 2024, 14, 336 CrossRef CAS PubMed.
- T. He, Y. Cui, J. Li and Y. Gao, Adv. Quantum Technol., 2023, 6, 2300178 CrossRef CAS.
- Y. Dong, Y. Zhang, X. Li, Y. Feng, H. Zhang and J. Xu, Small, 2019, 15, e1902237 CrossRef.
- Q. Wei and Z. Ning, ACS Mater. Lett., 2021, 3, 1266–1275 CrossRef CAS.
- B. Liang, L. Zhang, Y. Jiang, S. Chen and M. Yuan, J. Mater. Chem. C, 2023, 11, 4993–5008 RSC.
- W. Chen, S. Zhang, M. Zhou, T. Zhao, X. Qin, X. Liu, M. Liu and P. Duan, J. Phys. Chem. Lett., 2019, 10, 3290–3295 CrossRef.
- Y.-H. Kim, R. Song, J. Hao, Y. Zhai, L. Yan, T. Moot, A. F. Palmstrom, R. Brunecky, W. You, J. J. Berry, J. L. Blackburn, M. C. Beard, V. Blum and J. M. Luther, Adv. Funct. Mater., 2022, 32, 2200454 CrossRef.
- J.-T. Lin, D.-G. Chen, L.-S. Yang, T.-C. Lin, Y.-H. Liu, Y.-C. Chao, P.-T. Chou and C.-W. Chiu, Angew. Chem., Int. Ed., 2021, 60, 21434–21440 CrossRef.
- Y. Shi, P. Duan, S. Huo, Y. Li and M. Liu, Adv. Mater., 2018, 30, e1705011 CrossRef.
- R. Cao, X. Yang, Y. Wang and Y. Xiao, Nano Res., 2022, 16, 1459–1464 CrossRef.
- C. Zhang, Z. S. Li, X. Y. Dong, Y. Y. Niu and S. Q. Zang, Adv. Mater., 2022, 34, e2109496 CrossRef PubMed.
- C. T. Wang, K. Chen, P. Xu, F. Yeung, H. S. Kwok and G. Li, Adv. Funct. Mater., 2019, 29, 1903155 CrossRef.
- X. Yang, M. Zhou, Y. Wang and P. Duan, Adv. Mater., 2020, 32, e2000820 CrossRef PubMed.
- Z. N. Georgieva, B. P. Bloom, S. Ghosh and D. H. Waldeck, Adv. Mater., 2018, 30, 1800097 CrossRef.
- Z. N. Georgieva, Chiral Perovskites: Generating and Investigating Chiro-optical Response in Inorganic and Hybrid Perovskite Nanomaterials, Doctor of Philosophy, University of Pittsburgh, 2022 Search PubMed.
- S. Jiang, Y. Song, H. Kang, B. Li, K. Yang, G. Xing, Y. Yu, S. Li, P. Zhao and T. Zhang, ACS Appl. Mater. Interfaces, 2022, 14, 3385–3394 CrossRef CAS.
- D. Zhu, B. Tang, Y. Wu, A. S. Portniagin, H. Liu, Q. Liu, E. V. Ushakova and A. L. Rogach, J. Phys. Chem. C, 2024, 128, 8689–8697 CrossRef CAS.
- Y.-H. Kim, Y. Zhai, E. A. Gaulding, S. N. Habisreutinger, T. Moot, B. A. Rosales, H. Lu, A. Hazarika, R. Brunecky, L. M. Wheeler, J. J. Berry, M. C. Beard and J. M. Luther, ACS Nano, 2020, 14, 8816–8825 CrossRef CAS PubMed.
- B. Tang, S. Wang, H. Liu, N. Mou, A. S. Portniagin, P. Chen, Y. Wu, X. Gao, D. Lei and A. L. Rogach, Adv. Opt. Mater., 2024, 12, 2301524 CrossRef CAS.
- A. Das, S. Ghosal, K. Marjit, S. K. Pati and A. Patra, J. Phys. Chem. Lett., 2024, 15, 7822–7831 CrossRef CAS.
- U. Tohgha, K. K. Deol, A. G. Porter, S. G. Bartko, J. K. Choi, B. M. Leonard, K. Varga, J. Kubelka, G. Muller and M. Balaz, ACS Nano, 2013, 7, 11094–11102 CrossRef CAS.
- Z. N. Georgieva, Z. Zhang, P. Zhang, B. P. Bloom, D. N. Beratan and D. H. Waldeck, J. Phys. Chem. C, 2022, 126, 15986–15995 CrossRef.
- A. Hubley, A. Bensalah-Ledoux, B. Baguenard, S. Guy, B. Abécassis and B. Mahler, Adv. Opt. Mater., 2022, 10, 2200394 CrossRef CAS.
- V. A. Kuznetsova, E. Mates-Torres, N. Prochukhan, M. Marcastel, F. Purcell-Milton, J. O'Brien, A. K. Visheratina, M. Martinez-Carmona, Y. Gromova, M. Garcia-Melchor and Y. K. Gun'ko, ACS Nano, 2019, 13, 13560–13572 CrossRef CAS PubMed.
- X. Gao, X. Zhang, W. Yin, H. Wang, Y. Hu, Q. Zhang, Z. Shi, V. L. Colvin, W. W. Yu and Y. Zhang, Adv. Sci., 2019, 6, 1900941 CrossRef CAS.
- S. W. Im, H.-Y. Ahn, R. M. Kim, N. H. Cho, H. Kim, Y.-C. Lim, H.-E. Lee and K. T. Nam, Adv. Mater., 2020, 32, 1905758 CrossRef CAS.
- M. M. Green, M. P. Reidy, R. D. Johnson, G. Darling, D. J. O'Leary and G. Willson, J. Am. Chem. Soc., 1989, 111, 6452–6454 CrossRef.
- Q. Cao, R. Song, C. C. S. Chan, Z. Wang, P. Y. Wong, K. S. Wong, V. Blum and H. Lu, Adv. Opt. Mater., 2023, 11, 2203125 CrossRef.
- S. Ma, Y.-K. Jung, J. Ahn, J. Kyhm, J. Tan, H. Lee, G. Jang, C. U. Lee, A. Walsh and J. Moon, Nat. Commun., 2022, 13, 3259 CrossRef.
- M. K. Jana, R. Song, H. Liu, D. R. Khanal, S. M. Janke, R. Zhao, C. Liu, Z. V. Vardeny, V. Blum and D. B. Mitzi, Nat. Commun., 2020, 11, 4699 CrossRef.
- V. Kuznetsova, Y. Gromova, M. Martinez-Carmona, F. Purcell-Milton, E. Ushakova, S. Cherevkov, V. Maslov and Y. K. Gun'ko, Nanophotonics, 2020, 10, 797–824 CrossRef.
- S. Jiang and N. A. Kotov, Adv. Mater., 2022, 35, 2108431 CrossRef.
- L. Xiao, T. An, L. Wang, X. Xu and H. Sun, Nano Today, 2020, 30, 100824 CrossRef.
- F. P. Milton, J. Govan, M. V. Mukhina and Y. K. Gun'ko, Nanoscale Horiz., 2016, 1, 14–26 RSC.
- A. Ben-Moshe, A. Teitelboim, D. Oron and G. Markovich, Nano Lett., 2016, 16, 7467–7473 CrossRef.
- A. Ben-Moshe, D. Szwarcman and G. Markovich, ACS Nano, 2011, 5, 9034–9043 CrossRef.
- K. Varga, S. Tannir, B. E. Haynie, B. M. Leonard, S. V. Dzyuba, J. Kubelka and M. Balaz, ACS Nano, 2017, 11, 9846–9853 CrossRef PubMed.
- M. Puri and V. E. Ferry, ACS Nano, 2017, 11, 12240–12246 CrossRef.
- G. Li, X. Fei, H. Liu, J. Gao, J. Nie, Y. Wang, Z. Tian, C. He, J. L. Wang, C. Ji, D. Oron and G. Yang, ACS Nano, 2020, 14, 4196–4205 CrossRef PubMed.
- F. Purcell-Milton, A. K. Visheratina, V. A. Kuznetsova, A. Ryan, A. O. Orlova and Y. K. Gun'ko, ACS Nano, 2017, 11, 9207–9214 CrossRef.
- S. Apergi, G. Brocks and S. Tao, J. Phys. Chem. Lett., 2023, 14, 11565–11572 CrossRef PubMed.
- G. K. Inui, R. Besse, J. E. Gonzalez and J. L. F. Da Silva, Phys. Chem. Chem. Phys., 2024, 26, 16719–16731 RSC.
- A. Forde, D. Ghosh, D. Kilin, A. C. Evans, S. Tretiak and A. J. Neukirch, J. Phys. Chem. Lett., 2022, 13, 686–693 CrossRef.
- A. Forde, A. C. Evans, W. Nie, S. Tretiak and A. J. Neukirch, Nano Lett., 2024, 24, 9276–9282 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2024 |