
Amino-modified 2-oxazoline copolymers for complexation with DNA†
Natalia
Oleszko-Torbus
 *a,
Barbara
Mendrek
a,
Wojciech
Wałach
a,
Agnieszka
Fus-Kujawa
b,
Violeta
Mitova
c,
Neli
Koseva
d and
Agnieszka
Kowalczuk
*a
*a,
Barbara
Mendrek
a,
Wojciech
Wałach
a,
Agnieszka
Fus-Kujawa
b,
Violeta
Mitova
c,
Neli
Koseva
d and
Agnieszka
Kowalczuk
*a
aCentre of Polymer and Carbon Materials, Polish Academy of Sciences, M. Curie-Sklodowskiej 34, 41-819 Zabrze, Poland. E-mail: noleszko@cmpw-pan.pl; akowalczuk@cmpw-pan.pl
bDepartment of Molecular Biology and Genetics, School of Medical Sciences in Katowice, Medical University of Silesia in Katowice, Medyków 18, 40-752 Katowice, Poland
cInstitute of Polymers, Bulgarian Academy of Sciences, Georgi Bonchev Str. Bl. 103A, 1113 Sofia, Bulgaria
dBulgarian Academy of Sciences, 1 “15 Noemvri” Str., Sofia, 1040, Bulgaria
First published on 19th January 2024
Abstract
Copolymers of 2-isopropyl-2-oxazoline (iPrOx) with 2-methyl-2-oxazoline (MetOx) and 2-(3-butenyl)-2-oxazoline (ButEnOx) were synthesized via cationic ring opening polymerization (CROP). In the next step, the copolymers were modified to obtain primary and secondary amino groups in the substituents or in the main chain. Studies aimed to prove how the position of the amino groups in 2-oxazoline-derived copolymers influences their ability to condense DNA into so-called polyplexes. The post-polymerization modifications of the copolymers were done using hydrolysis and a thio-click reaction. The physicochemical characterization of the obtained copolymers and polyplexes was performed and then biological experiments followed to assess the cytotoxicity and transfection of the copolymers with secondary amino groups in the side chains. The results from differential scanning calorimetry (DSC) and wide-angle X-ray scattering (WAXS) have shown that the obtained copolymers did not contain a crystalline fraction, which makes it possible to apply them as nucleic acid carriers. The measurements of the sizes of the obtained polyplexes evidenced that the presence of primary amino groups in the substituent (POxN1pendant) impedes DNA binding, despite the positive zeta potential. On the other hand, the polymers with secondary amines, regardless the position (in the main chain for POxN2main or as a substituent in the case of POxN2pendant), form complexes with DNA to give nanostructures with smaller sizes than polymers with H2NR displaying a negative zeta potential in the studied range of N/P values (from 3 to 10). The biological experiments have shown that the copolymer with a HNR2 group in the pendant chain is nontoxic for HT-1080 cells in the range of concentrations from 5 to 100 μg mL−1 and is able to transfect those cells.
Introduction
Three main requirements are posed for gene delivery vectors nowadays: to efficiently complex nucleic acids, to condense the bulky structure of DNA or RNA to appropriate scales for cellular internalization, and to protect the genetic material from enzymatic degradation and the external environment.1 Non-viral gene delivery vectors based on polymers, known as polyplexes, are of great interest as they offer structural and chemical versatility in terms of modifying physicochemical properties, lower immunogenicity, better stability during storage and lower costs of manufacturing in comparison to viral carriers. Polyplexes are also able to efficiently interact with the cell surface, trigger intracellular uptake and therefore deliver the introduced nucleic acid to its site of action.2,3 Polymers that may interact electrostatically with the negatively charged phosphate backbone of nucleic acids are mainly those that carry repetitive positive charges in their structure.4,5 Typically, the amino groups present in these macromolecules are protonated at physiological pH and interact with DNA or RNA. Among cationic polymers, polyethylenimine (PEI), poly(N,N′-dimethylaminoethyl methacrylate) (PDMAEMA) and poly(L-lysine) have been mainly used for preparation of complexes with DNA.2,6–12 There are also other polymeric systems known for the preparation of polyplexes.13–17 In the design of polyplexes, great emphasis is put on the ability of macromolecules to electrostatically bind and condense nucleic acids into nanometer sized particles. Many studies have shown that among others the polymer molar mass, topology, type of cationic functionality and charge density as well as polyplex surface charge and its conformation in the solution influenced the cytotoxicity and efficiency of transfection of such carriers.4,7,18–21 It was also shown that during polyplex preparation, it is extremely important to apply the appropriate amounts of polymer and nucleic acid (so-called N/P ratio), as this strongly affects the zeta potential (ζ) values and the sizes of formed polyplexes. By adjustment of these parameters, the efficient condensation of the bulky structure of nucleic acids into stable complexes with polymers could be achieved. Unfortunately, cationic polymers at high concentrations are known to be toxic and disruptive to the biological membranes of cells. The solution to this problem may be the copolymerization of cationic monomers with ethylene glycol, which frequently results in the decrease of cytotoxicity and the increase of transfection efficiency.4,20 As people are increasingly sensitized to PEG, alternative polymers for preparation of polyplexes must be identified. In the last decade, there has been great progress in the reported potential use of poly(2-substituted-2-oxazoline)s (POxs) in biomedical areas, as they are often considered as PEG alternatives for biomedical purposes.22–25 Due to the fact that these polymers possess in their structure various nitrogen containing groups, they have been used for the preparation of polyplexes. POxs applied to build polyplexes usually contain amino groups generated within the main chain, or introduced into the substituents.POxs with amino groups in the main chain can be obtained via partial cleavage (hydrolysis) of the amide bonds between the backbone and substituents, leading to ethyleneimine (EI) units containing secondary (HNR2) amines.26–33 POxs with pendant amino groups could be synthesized via cationic ring opening polymerization (CROP) of 2-oxazoline with a protected amino group present in the substituent at position 2 of the oxazoline ring, followed by the removal of the protecting group after the synthesis.34,35 Polymerization conditions for 2-oxazolines with substituents containing a protected group must be chosen more carefully than for 2-alkyl-2-oxazolines. CROP of 2-oxazolines with protected amino groups in the substituent initiated by macroinitiators in the form of salts of N-methyl-2-methyl-2-oxazolinium triflate, N-methyl-2-ethyl-2-oxazolinium tosylate or N-propargyl-2-methyl-2-oxazolinium tosylate led to (co)polymers containing, after deprotection, pendant primary (H2NR)34,35 or secondary amines.36 On the other hand, by using methyl triflate as the initiator, a mixture of undefined low molar mass products was obtained.34 POxs containing pendant amino groups could also be synthesized by post-polymerization modifications.37 By using this way, it was possible to obtain POxs with pendant primary and tertiary (NR3) amines. To date, POxs with pendant secondary amino groups have not been obtained on the way of post-polymerization modification.
It is known that binding of nucleic acids by functionalized POxs is dependent on the type of amino group present in the polymer structure.38 Schubert et al.37 compared the activity of polyplexes based on the copolymers of 2-methyl-2-oxazoline (MetOx) and 2-(9-decenyl)-2-oxazoline (DecEnOx) or 2-(3-butenyl)-2-oxazoline (ButEnOx) modified with pendant primary and tertiary amines. It was found that generally, POxs with pendant H2NR were more suitable for an efficient binding and protection of DNA than copolymers containing NR3 as substituents; however, a high amine content induced the cytotoxicity of the studied polyplexes (above 40 mol% in the case of copolymers based on ButEnOx). A similar conclusion was drawn by Kabanov et al.,39 who showed that polyplexes made with diethylenetriamine-containing POxs transfect macrophages significantly better than those made with a tris(2-aminoethyl)amine-based copolymer. In general, POxs with secondary amines, in the main chain or in the substituents, seem to have the best ability to complex with DNA, among amino-functionalized POxs.36,40 Nontoxic polyplex nanoparticles with a hydrodynamic diameter (Dh) less than 140 nm and of good stability upon storage and dilution were obtained from block copolymers of MetOx and 2-(N-methyl) aminomethyl-2-oxazoline, having pendant HNR2 groups in an amount of about 20 mol%.36 In turn, for the majority of systems based on partially hydrolyzed POxs containing HNR2 groups in the main chain (EI units), the aggregation of the polymers in the solution was required, prior to the complexation with DNA. The self-organization proceeded either at room temperature, due to the amphiphilic character of POx chains, leading to micelle-like structures,41 or at elevated temperatures resulting in the formation of the so-called mesoglobules.12,40,42,43 In some cases, polyplexes based on aggregated POxs with EI units were unstable when the temperature was lowered, which was attempted to prevent, among other things, by creating a shell of mesoglobules composed of poly(N-isopropylacrylamide) (PNIPAM).40 On the other hand, there are also known EI-based POx systems, where the aggregation of the macromolecules did not occur before complexing with nucleic acids.44,45
A key aspect in the activity of polyplexes is their stability; thus, it is important that the complexes remain dissolved in solution and do not precipitate, for example, due to crystallization of the polymer. In our previous studies,46 we have shown that hydrolyzed 2-isopropyl-2-oxazoline-based copolymers did not crystallize, exhibited a change in the ζ potential with variation in temperature and were non-toxic to HT-1080 cells, thus appearing to be a good material for the preparation of new polymeric vectors applicable in gene therapy. Therefore, in this study we aimed to investigate how the position of the amino groups in 2-isopropyl-2-oxazoline (iPrOx) copolymers (in the substituents or in the main chain) affects the efficiency of DNA complexation and its condensation into polyplexes of scales appropriate for effective cellular internalization. Additionally, we wanted to compare the ability of POxs with pendant H2NR or HNR2 amines to form complexes with DNA. To achieve these goals, three iPrOx-based copolymers of a similar content of amines were obtained: with primary amino groups in the substituents, with secondary amino groups in the substituents, and with secondary amino groups in the main chain. Having in mind the potential cytotoxicity, we wanted the amount of amines in the copolymers not to exceed 20 mol%, which at the same time, based on literature data, seems to be sufficient for effective DNA binding. The precursor of POxs with primary and secondary amino groups in the substituents was the copolymer of iPrOx with MetOx and ButEnOx. MetOx was chosen as the comonomer to improve the water solubility, which is limited for P(iPrOx-co-ButEnOx). Due to the presence of vinyl double bonds of 2-(3-butenyl)-2-oxazoline, it was possible to introduce appropriate amines into the copolymer, through the so-called thio-click reaction, with the use of aminothiols. For the first time, a polyoxazoline containing secondary amino groups in substituents was obtained by post-polymerization modification. Importantly, the presence of modified ButEnOx units was expected to increase the transfection efficiencies of resulting polyplexes, as long alkyl substituents are known to cause endocytosis of the polymer carriers through interactions with the cell membrane.4 The precursor of POx with secondary amino groups in the main chain (EI units) was the copolymer of iPrOx and MetOx. A selective partial hydrolysis of P(iPrOx-co-MetOx) was carried out, and a copolymer with HNR2 amines in the main chain was obtained, of similar content to that in the case of POxs with pendant secondary amino groups. All the obtained POxs containing amino groups were characterized in terms of their ability to crystallize. In the case of the copolymer of iPrOx with MetOx and ButEnOx, it was verified how long, rigid, aliphatic substituents derived from ButEnOx affected the thermal and crystalline properties of the copolymers. The ability of macromolecules to self-organize in water was also studied, as their aggregation may affect the availability of amino groups required to interact with negatively charged nucleic acids. Research on the ability of the obtained copolymers to interact with DNA was carried out. The size of the complexes, and their zeta potential value, morphology, stability as a function of time, and cytotoxicity were investigated. The most promising polyplex was subjected to model transfection studies.
Experimental
Chemicals
Isobutyronitrile (99.6%, Aldrich), 2-aminoethanol (99%, Aldrich), cadmium acetate (>98%, Fluka), methyl 4-nitrobenzenesulfonate (99%, Aldrich), 4-pentenoic acid (98%, J&K Scientific GmbH), N-hydroxysuccinimide (98%, Aldrich), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDAC) (>99%, Carl Roth GmbH), CaH2 (95%, Sigma-Aldrich), diethyl ether (pure for analysis, POCH), magnesium sulfate (min. 99.5%, anhydrous, Alfa Aesar), NaOH (pure for analysis, Chempur), KOH (85%, POCH), cysteamine (>98%), 2-(butylamino)ethanethiol (97%, Aldrich), thioacetic acid (97%, Alfa-Aesar), 2,2-dimethoxy-2-phenylacetophenone (DMPA) (99%, Aldrich) and benzophenone (>99%, Fluka) were used as received. Acetonitrile (for HPLC, POCH, Gliwice, Poland) and dichloromethane (pure for analysis, POCH) were dried over CaH2 and distilled under a dry argon atmosphere. Methanol (min. 99.85%, gradient grade for HPLC, ChemSolute) was heated over magnesium flakes and distilled under argon. THF (pure for analysis, POCH) was distilled from a Na/K alloy. MetOx (98%, Aldrich, Steinheim, Germany) was dried over KOH, distilled, dried over CaH2 and distilled again. iPrOx was synthesized according to Witte and Seeliger.47 ButEnOx was synthesized according to Schlaad.48Biological materials
A deoxyribonucleic acid sodium salt from salmon testes (DNA, 1.3 × 106 Da, Sigma-Aldrich) was used as received for the analysis of the sizes, zeta potential and morphology of polyplexes. The plasmid DNA pMetLuc2-control vector (pDNA) (4781 base pairs, Clontech, Warszawa, Poland) was used for the cytotoxicity tests of polyplexes and for the transfection studies. pDNA isolation was performed using the Plasmid DNA Maxi Kit (Omega Bio-Tek, Norcross, GA, USA). The Ready-To-Glow™ Luciferase Reporter System was obtained from TakaraBio (Saint-Germain-en-Laye, France) and used according to the manufacturer's instructions. Human fibrosarcoma cells HT-1080 (ATCC#CCL-121, the American Type Culture Collection, Manassas, VI, USA) were used for the analysis of cytotoxicity of copolymers and polyplexes, as well as for the transfection studies. The cells were cultured in a cell culture medium consisting of Dulbecco's Modified Eagle's Medium (DMEM) (Sigma Aldrich, Germany) supplemented with 10% FBS, 1% L-glutamine, 1000 U mL−1 penicillin, 100 μg mL−1 streptomycin and 250 μg mL−1 amphotericin B at 37 °C and 5% CO2.Copolymerizations and modifications of copolymers
Copolymers were obtained via CROP initiated by 4-nitrobenzenesulfonate.49–51 The total ratio of the initial monomers (MetOx, iPrOx and ButEnOx) to the initiator concentration was chosen to be 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for all syntheses. Polymerizations were carried out at 70 °C for 5 days for full conversion of the monomers (checked by gas chromatography or high-performance liquid chromatography). Then, water was added, the mixture was kept for 10 minutes at room temperature under stirring, the excess acetonitrile was evaporated, and the obtained polymer was dried by lyophilization. Two polymeric precursors of a gradient chain microstructure were obtained; P(iPrOx70-co-MetOx15-co-ButEnOx15) and P(iPrOx83-co-MetOx17). P(iPrOx70-co-MetOx15-co-ButEnOx15) was then modified with the appropriate aminothiols (cysteamine or 2-(butylamino)ethanethiol) according to the procedure described in a previous study,48 obtaining the POxN1pendant and POxN2pendant, respectively. The optimization of the reaction is described in the ESI.† Reactions were carried out in a mixture of dry THF and methanol (50:50 v/v). As an UV light source, 2 × 15 W lamp (Uvitec) of λ = 254 nm was used. The copolymer solution was dialyzed against methanol (MWCO: 1 kDa), the last dialysis was performed against water, and then the aqueous solution of the polymer was lyophilized. P(iPrOx83-co-MetOx17) was hydrolyzed according to our previous studies46 and POxN2main was obtained.
:1 for all syntheses. Polymerizations were carried out at 70 °C for 5 days for full conversion of the monomers (checked by gas chromatography or high-performance liquid chromatography). Then, water was added, the mixture was kept for 10 minutes at room temperature under stirring, the excess acetonitrile was evaporated, and the obtained polymer was dried by lyophilization. Two polymeric precursors of a gradient chain microstructure were obtained; P(iPrOx70-co-MetOx15-co-ButEnOx15) and P(iPrOx83-co-MetOx17). P(iPrOx70-co-MetOx15-co-ButEnOx15) was then modified with the appropriate aminothiols (cysteamine or 2-(butylamino)ethanethiol) according to the procedure described in a previous study,48 obtaining the POxN1pendant and POxN2pendant, respectively. The optimization of the reaction is described in the ESI.† Reactions were carried out in a mixture of dry THF and methanol (50:50 v/v). As an UV light source, 2 × 15 W lamp (Uvitec) of λ = 254 nm was used. The copolymer solution was dialyzed against methanol (MWCO: 1 kDa), the last dialysis was performed against water, and then the aqueous solution of the polymer was lyophilized. P(iPrOx83-co-MetOx17) was hydrolyzed according to our previous studies46 and POxN2main was obtained.
Formation of polyplexes for measurements of sizes and zeta potential
A suitable amount of DNA (from salmon testes) solution in water (c = 5 mg mL−1) was added to a fixed volume of POx solution in water (2 mL, c = 7.5 mg mL−1) to achieve the desired nitrogen/phosphate (N/P) ratios. The obtained solutions were mixed and incubated at room temperature for 30 min. Polyplexes prepared by this method were used for the DLS and zeta potential measurements without filtering.Measurements
The molar mass and molar mass dispersity (Đ) of the copolymers were determined using a SEC MALLS system with a multiangle laser light scattering detector (DAWN EOS, Wyatt Technologies, Santa Barbara, CA, USA, λ = 658 nm) and a refractive index detector (Dn-1000 RI WGE DR Bures, Dallgow, Germany, λ = 620 nm). Measurements were carried out in N,N′-dimethylformamide (DMF) (POCH, Gliwice, Poland) (with 5 mmol L−1 of LiBr; flow rate of 1 mL min−1) using 100 Å, 1000 Å and 3000 Å GRAM columns (Polymer Standards Service, Mainz, Germany). The composition of the copolymers was analyzed by 1H NMR. The spectra were recorded in CDCl3 or D2O using a Bruker Ultrashield spectrometer (Bruker, Billerica, MA, USA) operating at 600 MHz. The thermal properties of copolymers were analyzed by differential scanning calorimetry. DSC measurements were carried out using a TA–DSC Q2000 apparatus (TA Instruments, New Castle, DE, USA) under a nitrogen atmosphere with a flow rate of 50 mL min−1. The measurements were taken in the range from 0 to 200 °C. The heating rate was 10 °C min−1. The data were collected and then analyzed using Universal Analysis 2000 with Universal V4.5a software (TA Instruments, New Castle, DE, USA). The crystalline properties of copolymers were analyzed by wide-angle X-ray scattering (WAXS) analysis. A diffractometer TUR–M62 (VEB TuR, Dresden, Germany) equipped with an HZG–3 goniometer (VEB TuR, Dresden, Germany) with Cu Kα radiation was used. Calculations of the intensities and positions of peaks were carried out using WAXSFIT software (WAXSFIT, Bielsko Biała, Poland). Turbidimetric measurements were carried out with the use of Specord 200 plus ultraviolet-visible spectrophotometer (Analytik Jena, Jena, Germany) equipped with a programmable thermocontroller. The cloud point temperature (TCP) value was defined as the temperature at which the transmittance of the copolymer solutions reached 50% of its initial value. The critical aggregation concentration (cac) was determined using trans-1,6-diphenyl-1,3,5-hexatriene (DPH) as a probe, according to.52 Dynamic light scattering (DLS) measurements were performed with the use of a Brookhaven BI-200 goniometer with a digital autocorrelator (BI-9000 AT, Brookhaven Instruments, New York, USA) and vertically polarized laser light (Brookhaven Instruments, New York, USA) operating at 35 mW and λ = 637 nm. The autocorrelation functions were analyzed using the constrained regularized algorithm CONTIN. The measurements were made at a 90° angle at 25 °C in at least triplicate. Before DLS analysis, solutions of copolymers were passed through membrane filters with the nominal pore size of 0.22 μm (Graphic Controls, DIA-Nielsen, Düren, Germany); the polyplexes were not filtered. Zeta potential (ζ) measurements were performed in triplicate with the use of a Zetasizer Nano ZS 90 (Malvern Instruments, Malvern, UK) in a disposable folded capillary cell. ζ was calculated from the electrophoretic mobility, u, employing the Helmholtz–Smoluchowski equation (u = εζ/η, where ε is the dielectric constant of the solvent and η is the viscosity of the solvent). Cryogenic transmission electron microscopy (cryo-TEM) images were obtained using a Tecnai F20 TWIN microscope (FEI Company), equipped with a field emission gun operating at an acceleration voltage of 200 kV. Images were recorded on an Eagle 4k HS camera (FEI Company) and processed with TIA software (FEI Company).Cytotoxicity of the POxN2pendant polymer and its polyplex
As for the cytotoxicity tests of polyplexes, plasmid DNA (pDNA) was used. One day before the cytotoxicity assay, HT-1080 cells were seeded in 0.5 mL of cell culture medium in 24 well plates at a density of 8 × 104 per well. In the following day, the cell culture medium was supplemented with tested polymer (sample POxN2pendant) to the desired final concentrations: 0 (control) 5, 10, 20, 30, 40, 50, 60, 70, 80, 90 and 100 μg mL−1, or with tested polyplex (DNA-POxN2pendant) to the desired N/P ratios: 3, 7, 10, 16, and 32. Cells were incubated with the polymer or polyplex for 24 hours. After incubation time, the culture medium with the polymer/polyplex was removed. Wells were washed with pre-warmed phosphate-buffered saline (PBS, Sigma Aldrich). Subsequently, PBS was removed and 200 μL of pre-warmed alamarBlue reagent in cell culture medium to a final concentration of 10% in each well. Then, the cells were incubated under standard conditions at 37 °C and 5% CO2 for 1 hour. Subsequently, 100 μl of the mixture from each well was transferred to a new well of a 96-well TPP plate (PerkinElmer, Waltham, MA, USA) and the fluorescence emission was monitored at 590 nm using a VICTOR™ Multilabel Plate Reader (PerkinElmer, USA) with a 560 nm excitation source. The cell viability was assessed based on the percent of live cells compared to the control cells not treated with the polymer/polyplex.Determination of the luciferase activity with plasmid DNA polyplexes
For the transfection studies, pDNA was used. The HT-1080 cells were incubated in DMEM complete growth medium in 24-well plates at a density of 2 × 104 cells per well up to 18 hours prior to the transfection. Subsequently, complexes of the POxN2pendant and pDNA (0.5 mg per well) were prepared, incubated for 30 minutes at room temperature and added to the cells in the culture medium. The cultures were incubated for 24 hours at 37 °C. Following incubation, the medium was aspirated, the fresh medium was added and the cells were incubated for an additional 24 hours. To detect the activity of secreted luciferase, 50 μL of culture medium was collected and measured using a Ready-To-Glow™ Secreted Luciferase Reporter Assay kit (TakaraBio) according to the manufacturer's instruction. The transfection efficiency was determined in 96-well plates (TPP) with a 1s exposition time on a VICTOR™ Multilabel Plate Reader (PerkinElmer).Results
Copolymers
POxs with pendant H2NR or HNR2 were synthesized via addition of aminothiols, cysteamine or 2-(butylamino)ethanethiol, respectively, to vinyl double bonds present in the substituents of P(iPrOx70-co-MetOx15-co-ButEnOx15), according to the procedure described in a previous study.48 The efficiency of the modification reactions was monitored by 1H NMR, where the decrease of the intensity of signals from the protons of –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 at ∼5.00 ppm and ∼5.90 ppm in relation to the signal intensities from the protons of the methylene groups of the copolymer main chain at 3.50 ppm was observed (Fig. S2A in the ESI†). The obtained copolymer with pendant primary amino groups was designated as the POxN1pendant, and POx with pendant secondary amino groups was designated as POxN2pendant.
CH2 at ∼5.00 ppm and ∼5.90 ppm in relation to the signal intensities from the protons of the methylene groups of the copolymer main chain at 3.50 ppm was observed (Fig. S2A in the ESI†). The obtained copolymer with pendant primary amino groups was designated as the POxN1pendant, and POx with pendant secondary amino groups was designated as POxN2pendant.
POx with secondary amino groups in the main chain was obtained by selective partial hydrolysis of the copolymer P(iPrOx83-co-MetOx17), according to a procedure described in a previous study.46 The efficiency of the hydrolysis was monitored by 1H NMR. Decreasing of the intensity of the signal in the spectral range of 1.95–2.11 ppm assigned to protons of the methyl group of MetOx in relation to the signal intensities from the protons of the methylene groups of the copolymer main chain in the range from 3.30 to 3.70 ppm was observed (Fig. S2B in the ESI†). The resulting copolymer was designated POxN2main. Copolymers of oxazolines with a similar content of primary and secondary amino groups in the substituents and with secondary amino groups in the main chain (11–15 mol%) were obtained. A simplified scheme of the structure of the copolymers is shown in Fig. 1. A synthetic scheme showing the post-polymerization modifications and the chemical structures of the initial polymers is shown in Fig. S3 in the ESI.†
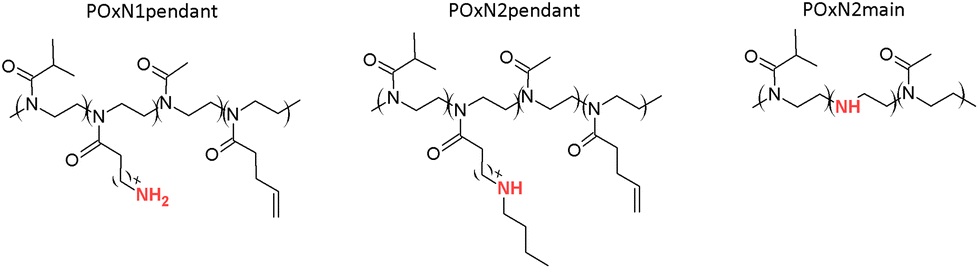 | ||
| Fig. 1 A general scheme of the structure of the copolymers. | ||
The detailed characterization of the copolymers is summarized in Table 1.
Based on differential scanning calorimetry and wide-angle X-ray scattering analysis, we concluded that all studied copolymers containing ButEnOx (P(iPrOx70-co-MetOx15-co-ButEnOx15), POxN1pendant and POxN2pendant) did not contain a crystalline fraction. Exo- and endothermic peaks, derived from crystallization and melting, were not observed in DSC curves of copolymers (Fig. S4 in the ESI†). Also, prolonged annealing of the copolymers at 150 °C, which potentially would promote chain ordering and crystallization, did not result in the formation of crystalline fraction. Diffraction peaks were not observed in the WAXS curve for the exemplary POxN2pendant (Fig. 2), confirming the conclusion drawn from DSC that the amorphous phase is predominant.
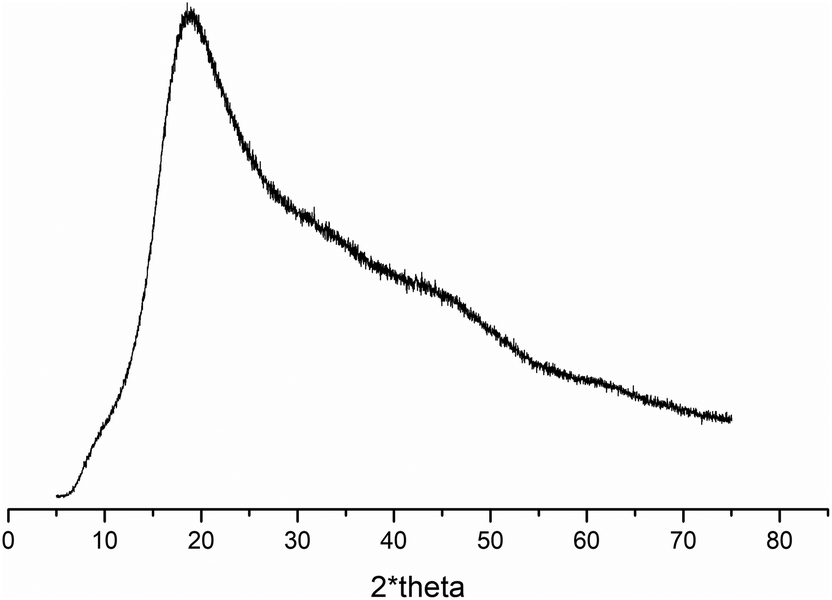 | ||
| Fig. 2 X-ray diffraction curve of POxN2pendant. | ||
Similar to the obtained copolymers containing ButEnOx, POxN2main with EI units also did not exhibit the ability to crystallize in bulk, as was described in our previous studies.46
We further studied the behavior of copolymers in an aqueous solution and if this affects their crystallization. To verify whether changes in the organization of the chains occur as a result of prolonged annealing of the aqueous copolymer solutions, the sizes of the structures were studied. Based on the spectrophotometric measurements with the use of DPH, it was observed that in the range of certain concentrations of copolymers in water, there was an increase in the absorption signal at wavelengths of 300–400 nm, which means that solubilization of the hydrophobic probe took place. The exemplary cac curve of the POxN2pendant is shown in Fig. S5B in the ESI.† It is known that at certain concentrations, polymer chains containing domains with low affinity for water organize themselves in solution to form structures that are capable of solubilizing hydrophobic substances. This means that the studied copolymers, due to their gradient microstructure, may behave similar to amphiphilic block copolymers in aqueous solution and aggregate to certain organized assemblies. Presumably, domains with low affinity to water (containing isopropyl and/or butenyl substituents) form the interior of the self-assembled polymeric structure, which is surrounded by a shell consisting of more hydrophilic copolymer domains (containing methyl and amine-modified substituents). Observed self-organization of the studied copolymers in water could indicate a good availability of amino groups (in the outside part of the structure) to form complexes with negatively charged phosphates of DNA at certain concentrations. Our considerations were confirmed by measurements of the sizes of the copolymer structures in aqueous solutions, which were performed for a concentration of 7.5 mg mL−1 (above cac) at room temperature using the DLS technique (Fig. 3).
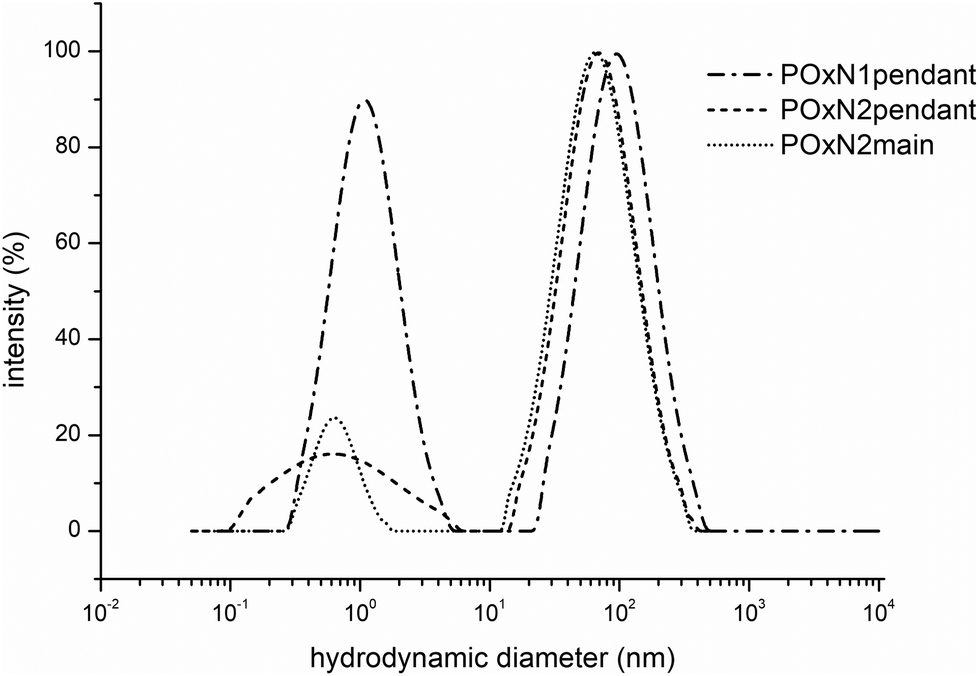 | ||
| Fig. 3 Distribution of the hydrodynamic diameter (intensity weighted distribution) for aqueous solutions of POxN1pendant, POxN2pendant and POxN2main (7.5 mg mL−1, 25 °C). | ||
For all the copolymer solutions, two populations of particles were observed: smaller particles with sizes of up to 10 nm and organized structures with sizes depending on the studied copolymer. The average hydrodynamic diameter of the organized structures (for a population of particles of larger sizes) was Dh = 153 nm for POxN1pendant, Dh = 67 nm for POxN2pendant and Dh = 69 nm for POxN2main. It should be noted that obtained sizes of self-assembled POxs particles do not seem to limit their uses in medicine and biology.
Differences in particle sizes originate probably from the ability of various amino groups to form hydrogen bonds with water molecules. Generally, the particles containing 1° amines can form more hydrogen bonds with water molecules than those containing 2 or 3° amines. Therefore, the particles of POxN1pendant are larger than those of copolymers with secondary amines, regardless of their location in the POx structure (in the substituent or in the main chain). This consistency could be also seen in the cloud point temperatures range, as POxN1pendant has the greatest affinity to water, forming the greatest number of hydrogen bonds among all studied copolymers which results in the highest value of TCP (Fig. S5A in the ESI†).
As a result of prolonged annealing of all aqueous copolymer solutions at temperatures above TCP, precipitation of macromolecules from the solution occurred, as confirmed by turbidimetric measurements (Fig. 4A). When the temperature was lowered, the polymer was redissolved in water. Thus, it can be concluded that macroscopically, no crystallization of copolymers in water occurred.
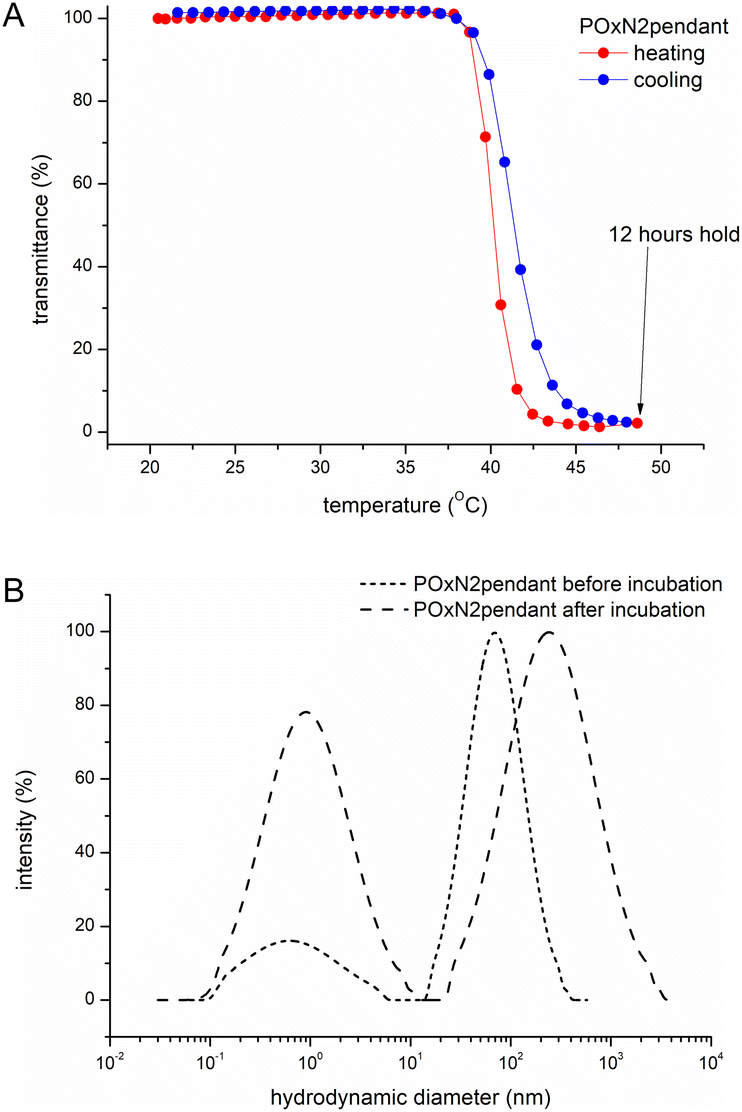 | ||
| Fig. 4 Transmittance as a function of temperature for an exemplary aqueous solution of POxN2pendant (c = 2 mg mL−1) incubated at 50 °C for 12 hours and cooled to room temperature (A); distribution of the hydrodynamic diameter (intensity weighted distribution) for aqueous solution of POxN2pendant before and after incubation at 50 °C for 12 hours (7.5 mg mL−1, 25 °C) (B). | ||
The DLS measurements of the size of particles formed in an aqueous solution of exemplary POxN2pendant, incubated for 12 hours at a temperature above TCP and cooled to room temperature, revealed the presence of two populations (Fig. 4B). The hydrodynamic diameters of formed aggregates were larger than before incubation and increased more than threefold from Dh = 67 nm to Dh = 216 nm. The observation indicates that although macroscopically no crystallization was observed (Fig. 4A), at the molecular level the particles in solution do not return to their initial sizes after prolonged annealing. However, the nanoparticles that remained in the solution are within the size range that allows for their potential use in biology and medicine.
To determine whether the copolymers could potentially be used for complexation with nucleic acids, we analyzed their zeta potential. In most works devoted to gene therapy conducted with the use of polymer carriers, attention is paid to the fact that the surface charge of the created polyplexes has a positive value. This facilitates the internalization of polyplexes in cells through electrostatic interaction with the negatively charged cell membranes and then may promote the polyplex release from endosomal compartments to the cytosol.60 Therefore, it was important to determine whether the starting polymers would have the appropriate zeta potential to form polyplexes that could efficiently enter cells.
At 25 °C, for the aqueous solutions (7.5 mg mL−1) of the POxN1pendant polymer, a positive value of zeta potential ζ = +10 mV was observed, and it was essentially neutral for POxN2pendant and POxN2main. At a temperature of 50 °C, the ζ value increased to +17 mV for POxN1pendant, +7 mV for POxN2pendant and +12 mV for POxN2main. This could indicate that self-assembly of the copolymers in water to larger structures at increased temperature, confirmed by DLS measurements (Fig. 4), might cause the relocation of the positive charges to the outermost area of the formed particles, resulting in the increase of the surface charge. The similar results were obtained by Hoogenboom and Rangelov40 who observed such an increase for nanoparticles based on partially hydrolyzed poly(2-n-propyl-2-oxazoline).
Polyplexes – formation and physicochemical characterization
It should be noted that in numerous studies, the information on the structure and size of complexes formed from polymers and various nucleic acids is omitted. Here, the ability to condense deoxyribonucleic acid from salmon sperm (DNA) by the POxN1pendant, POxN2pendant and POxN2main polymers into polyplexes was studied, and DLS, zeta potential measurements and cryo-TEM imaging were used to characterize the obtained complexes.The size of the pure (non-complexed) DNA in aqueous solution was measured at a concentration of 5 mg mL−1 at 25 °C, using the DLS technique. A few populations of particles were observed: the smallest structures of Dh = 3 nm, larger structures of Dh = ∼100 nm and aggregates greater than 1 μm in diameter. The sizes of DNA measured by DLS depend on many factors and are the subject of many studies. They may depend on the type and number of the base pairs in DNA, on the conformation it assumes in solution (open circle, supercoiled, or linear) and also on the type of interaction (electrostatic, stacking, strand-strand etc.) between the structural elements of the nucleic acid. The presence of the salt in water and the ionic strength of the solution have also an influence on the DNA sizes. Many authors observe a multimodal size distribution and explain it with the above factors.61–63
Based on our previous results on the transfection study using the gold standard polyethyleneimine, which is generally a derivative of polyoxazoline, we have established values of applicable N/P ratios of 3, 7 and 10. This is due to the fact that at higher N/P ratios, despite the satisfactory transfection efficiency, a low viability of HT-1080 cells was observed.7
For POxN1pendant polyplexes with DNA (DNA-POxN1pendant) at 25 °C, two populations of particles were observed, with the size dependent on the N/P ratio used: particles with a Dh of up to 300 nm and aggregates greater than 1 μm in diameter (Fig. S6A in the ESI†). It has to be however noted that with the highest amount of DNA (N/P = 3) at 25 °C, a partial clouding of the polyplex solution was observed, and TCP of the POxN1pendant decreased from 62 to 27 °C. This indicates that the addition of DNA to polyoxazolines containing primary amino groups in the substituents strongly affects their interactions with water via weakening the hydration sphere and increasing the contribution of the hydrophobic interactions. Thus, in this case, the size of the DNA-POxN1pendant was also studied at 10 °C, but two populations of structures were still observed in the solution (Fig. S6B in the ESI†).
In the case of DNA-POxN2pendant and DNA-POxN2main, containing secondary amines, stable polyplexes much smaller in diameter were formed at 25 °C. Fig. 5A shows the influence of the N/P ratio on the hydrodynamic diameter of the polyplexes based on POxN2pendant and POxN2main. In general, with increasing polymer content, that is, increasing N/P, the size of the complexes decreased. Dh of DNA-POxN2pendant was equal to 911 nm at N/P = 3, while it dropped to Dh = 120 nm at N/P = 10. For DNA-POxN2main, Dh = 710 nm at N/P = 3 and it decreased to 140 nm at N/P = 10. Fig. 5B shows the hydrodynamic diameter for aqueous solutions of pure DNA, the exemplary copolymer POxN2pendant, and their complexes at different N/P ratios at 25 °C.
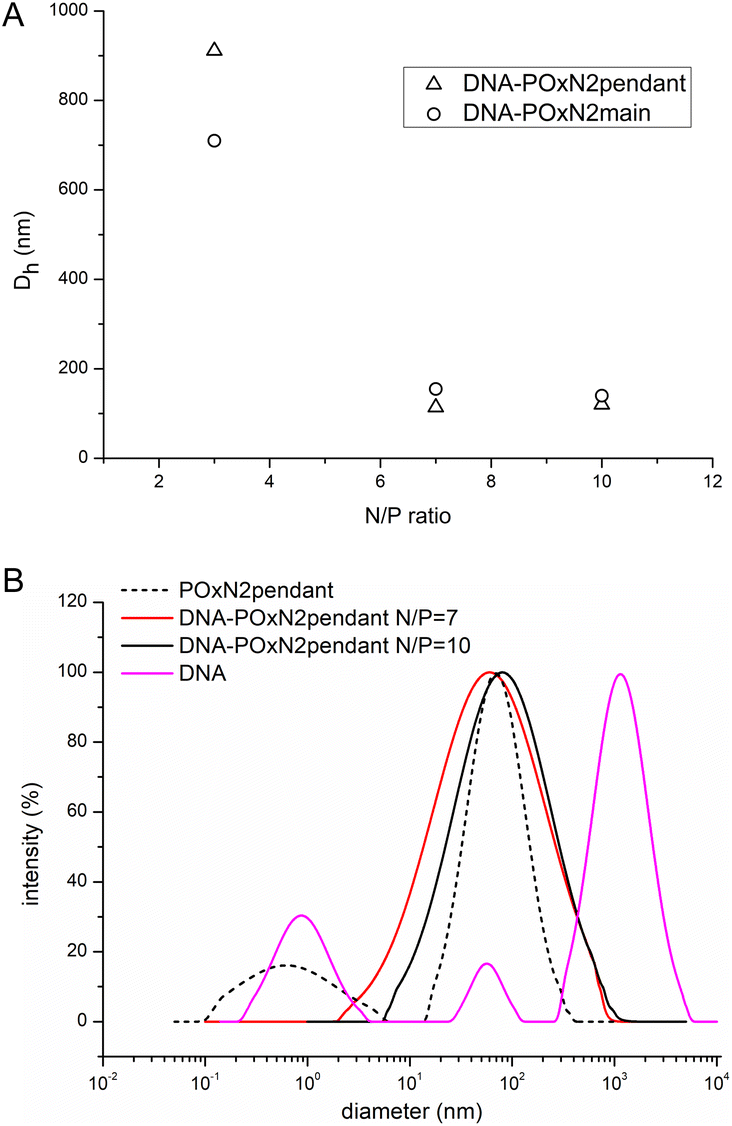 | ||
| Fig. 5 D h of the DNA-POxN2pendant and DNA-POxN2main as a function of N/P (A); distribution of the hydrodynamic diameter (intensity weighted distribution) for aqueous solutions of pure DNA, pure POxN2pendant, and their polyplexes at different N/P (7.5 mg mL−1, 25 °C) (B). | ||
Copolymers containing secondary amino groups exhibited good ability to condense the bulky structure of DNA at N/P ratios of 7 and 10. Polyplexes formed more unified structures (one population of particles) than the naked DNA (three populations of particles).
Importantly, copolymers of 2-oxazolines containing secondary amino groups exhibited a better ability to condense DNA, compared to POxs with primary amines. The composition and structure of these two copolymers (POxN2pendant and POxN2main) afford a good hydrophilic–hydrophobic balance, and hence their solution conformation as well as the occurrence of electrostatic and hydrophobic interactions may result in stronger complexation with DNA than that in the case of POxN1pendant. As was shown, the POxN1pendant copolymer was able to form bigger and looser structures in aqueous solution, compared to POxN2pendant and POxN2main (Fig. 3). Based on this research, a possible conclusion can be drawn that the hydrogen bonds in the case of POxN1pendant interfere or compete with DNA condensation.
Besides the sizes of polyplexes, their zeta potential and morphology were also studied (Fig. 6).
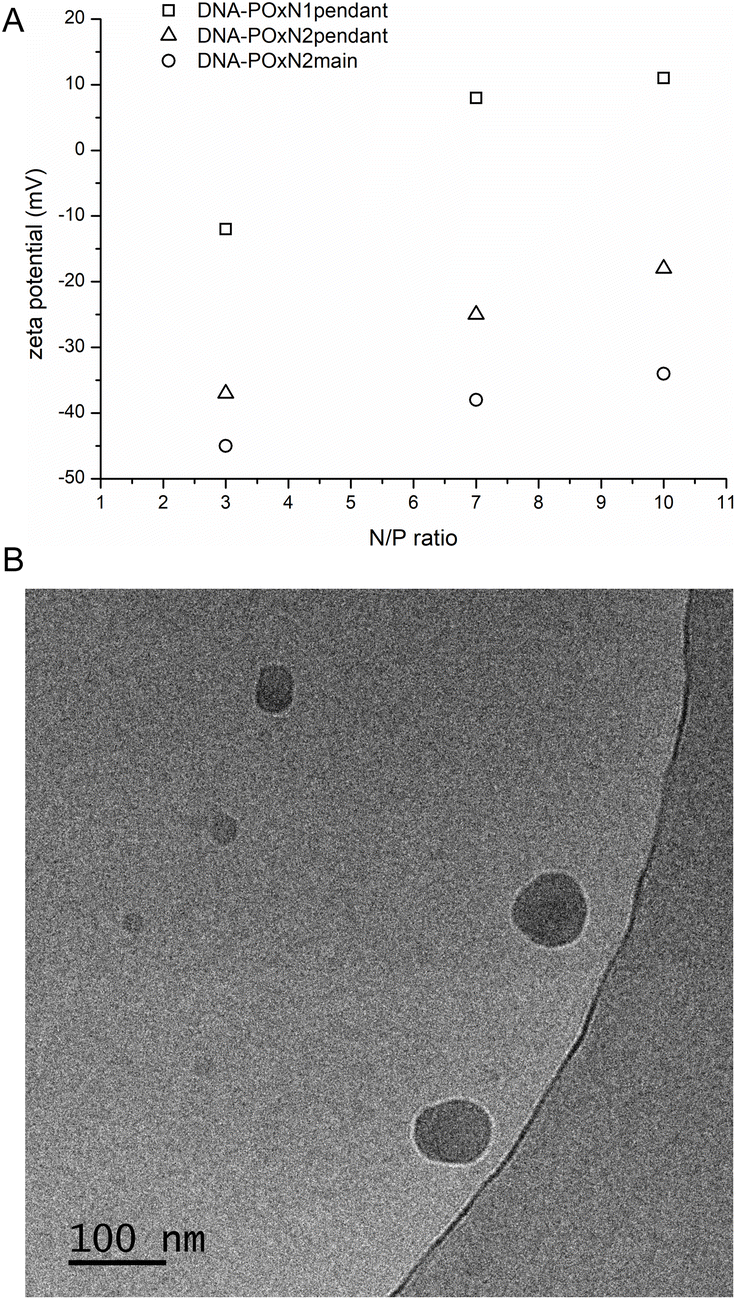 | ||
| Fig. 6 ζ of DNA-POxN1pendant, DNA-POxN2pendant and DNA-POxN2main as a function of N/P (7.5 mg mL−1, 25 °C) (A); cryo-TEM images of DNA-POxN2pendant at N/P = 7 (B). | ||
The zeta potential of bare DNA in aqueous solution was −140 mV at 25 °C. With the increase of the N/P ratio, the substantial increase of the zeta potential of polyplexes was observed compared to that of the bare nucleic acid. Only for polyplexes of POxN1pendant, the positive values of zeta potential were obtained (Fig. 6), which is probably due to the fact that this polymer had a positive ζ value before DNA complexation (+10 mV). It should be mentioned that small positive or even almost neutral zeta potential values were also obtained for DNA complexes with other 2-oxazoline copolymers, in contrast to positively charged linear and branched PEI.36,38,40
The polyplexes formed compact nanoparticles with spherical morphology at 25 °C. Fig. 6B shows the exemplary cryo-TEM image of DNA-POxN2pendant at N/P = 7. The sizes of the polyplexes estimated from cryo-TEM analysis were 50–100 nm, which is in the same range as the hydrodynamic diameters obtained by DLS measurements.
The stability of polyplexes is an important issue, from the point of view of their potential applications. We assessed the sizes of polyplexes in aqueous solutions at N/P ratios of 3, 7 and 10, upon their storage at 4 °C for 14 days. Fig. 7 shows changes in Dh of exemplary POxN2pendant and POxN2main polyplexes, as a function of time.
 | ||
| Fig. 7 D h as a function of time for polyplexes based on POxN2pendant (A) and POxN2main (B) (measured at 25 °C in aqueous solution at a concentration of 7.5 mg mL−1). | ||
During 14 days, we observed no considerable changes of the polyplexes sizes, or even slight reduction in particle diameter in the case of polyplexes based on both the studied copolymers, obtained at N/P = 3. This indicates the stability of polyplexes and their suitability for further research.
Analysis of the sizes of the polyplexes along with the change in their zeta potential, morphology and stability gave us certainty of the DNA-complexing ability of the studied copolymers and thus their appropriateness to undertake biological testing. It was shown that copolymers containing secondary amines exhibited a better ability to condense the bulky structure of DNA, as compared to POxs with primary pendant amino groups, as polyplexes much smaller in diameter were formed. Therefore, for preliminary biological studies we have chosen the polymer POxN2pendant.
Biological testing of copolymers and polyplexes
Prior to polyplex transfection activity assays, the cytotoxicity tests of the chosen POxN2pendant copolymer were performed. The HT-1080 cells originated from a human fibrosarcoma, a malignant tumor of fibrous connective tissue, were used for tests. They reproduce rapidly and are highly invasive, and therefore are often used as models in transfection studies with various gene delivery systems.64–66 After 24 hours of culture, the cells were supplemented with the POxN2pendant copolymer solution at various concentrations in DMEM. The viability of the HT-1080 cells was assessed using an alamarBlue reduction test assay. The results are plotted in Fig. 8.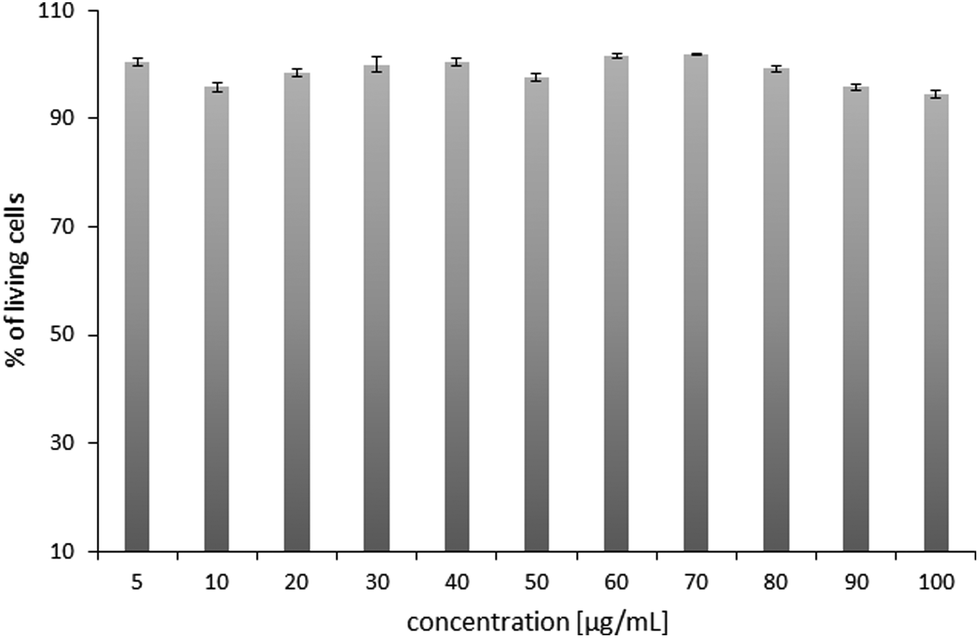 | ||
| Fig. 8 The cytotoxicity assay of POxN2pendant at different concentrations after 24 hours of cell culture. The results are shown as a percentage of the control, where untreated cells constituted 100%. | ||
It can be seen that the copolymer is non-toxic to the cells in an entire range of studied concentrations, which provided the premise for further research.
In preliminary studies, to confirm whether polyplexes based on the obtained polyoxazolines are capable of transfecting HT-1080 cells, an in vitro model experiment was performed. Polyplexes were formed using POxN2pendant and plasmid DNA with secreted Metridia luciferase as a reporter gene driven by the CMV promoter, which is often applied to quickly and accurately assess transfection efficiency. The gene transfection efficiency was estimated by the overall proper protein production (luciferase) by transfected cells. It was assumed that only living cells manufactured proteins at the time of measurement. The transfection efficiency, visible as luciferase activity, was evaluated indirectly as an average of three luminescence measurements. The obtained results were analyzed together with the cytotoxicity of polyplexes.
The cytotoxicity assay was used to estimate the viability of cells during their transfection. The results after 24 hours of incubation are shown as a percentage of the control, which is the culture of cells without polyplexes (Fig. 9A). The same N/P values were used as for polyplex characterization in aqueous solutions (3, 7 and 10) (Fig. 5). Additional tests at higher N/P values (16 and 32) were carried out to preliminarily assess the correlation between HT-1080 cell viability and transfection efficiency, as it is known that this can drop significantly, due to, among others, the presence of a high concentration of polymer-derived amino groups in the polyplex.7
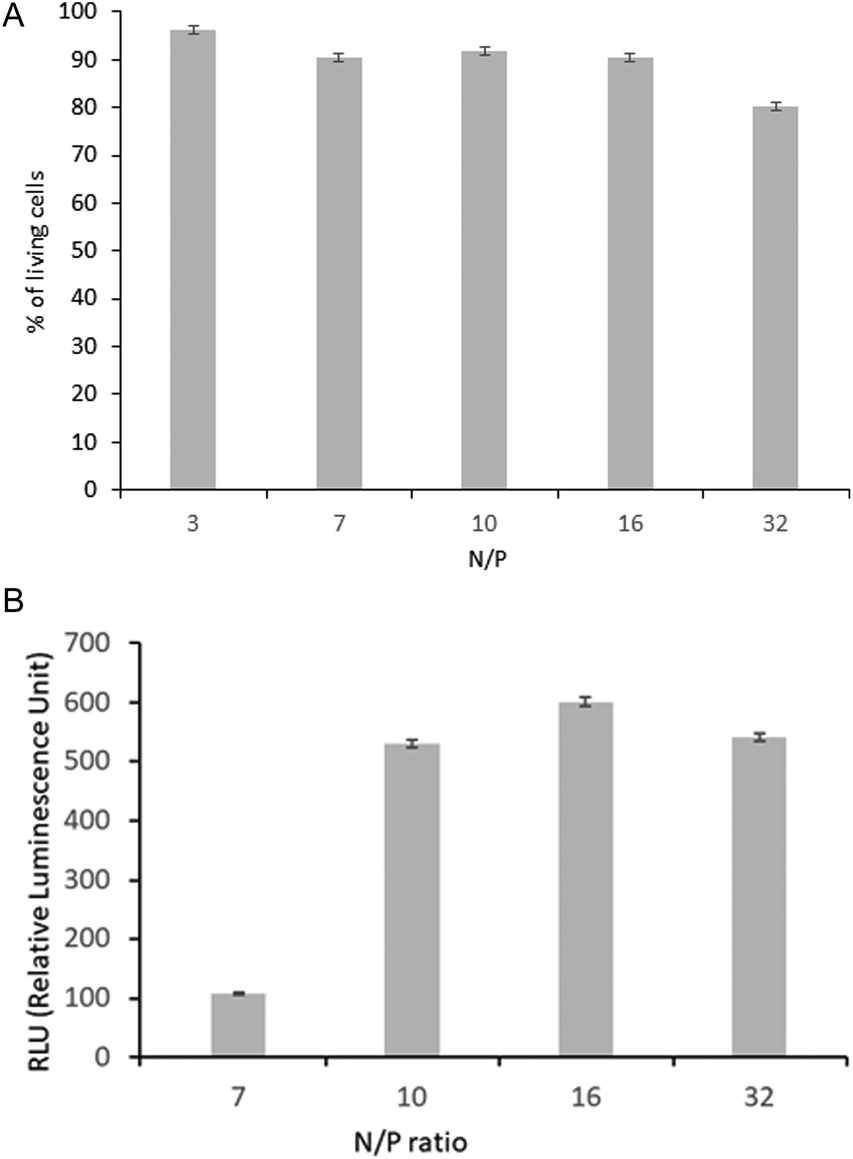 | ||
| Fig. 9 Viability of HT-1080 in the presence of DNA-POxN2pendant at increasing N/P, shown as a percentage of the control, where untreated cells constituted 100% (A); efficiency of HT-1080 cells transfection with DNA-POxN2pendant at different N/P ratios, shown as overall luminescence values (B). | ||
Luciferase expression mediated by the analyzed polyplexes was found to be dependent on the N/P ratio (Fig. 9B). For polyplexes of the studied copolymer, the luciferase activity significantly increased at N/P = 10. The DNA-POxN2pendant with a stable size revealed the most efficient transfection efficiency in the N/P range from 10 to 32, simultaneously preserving 100% cell viability. It means that the pDNA binding efficiency with the use of the polyoxazoline copolymer containing the secondary amino groups in the substituents provided the best protection for the nucleic acid, which resulted in the highest transfection efficiency.
The promising results of this model experiment open the way to our further research, where we plan to investigate the correlations between the size and structure of the labeled DNA polyplexes with the cell viability and transfection efficiency in order to obtain the most efficient vectors based on functional 2-oxazoline polymers.
Conclusions
iPrOx-based copolymers containing amino groups in an amount not exceeding 15 mol% were obtained. Copolymers with primary and secondary amino groups in the substituents were obtained via a thio-click reaction between the appropriate aminothiols and vinyl double bonds of 2-(3-butenyl)-2-oxazoline present in the copolymer. For the first time, a polyoxazoline containing secondary amino groups in substituents was obtained by post-polymerization modification. A copolymer with secondary amino groups in the main chain was obtained via hydrolysis of the amide bonds of 2-methyl-2-oxazoline units present in the copolymer. Macromolecules were characterized in terms of their ability to crystallize and it was verified how long, rigid, aliphatic substituents derived from ButEnOx affected the thermal and crystalline properties of the copolymers. Results have shown that the obtained copolymers did not contain a crystalline fraction, even upon prolonged annealing, which made them possible to apply as nucleic acid carriers. All the copolymers assembled in the aqueous solutions at certain concentrations. Two populations of particles were observed: smaller particles with a hydrodynamic diameter of up to 10 nm and organized structures of Dh = 153 nm for POxN1pendant, Dh = 67 nm for POxN2pendant and Dh = 69 nm for POxN2main. POxN1pendant due to the presence of 1° amines was able to form more hydrogen bonds with water molecules than copolymers containing 2° amines, and thus the particles were significantly larger. Upon addition of DNA to POxN1pendant, its condensation into polyplexes of scales appropriate for effective cellular internalization was not satisfactory and aggregates greater than 1 μm in diameter were formed. In the case of DNA-POxN2pendant and DNA-POxN2main, containing secondary amines, stable polyplexes much smaller in diameter were formed at N/P ratios of 7 and 10. Three populations of particles were observed for naked DNA, while one population of particles was observed in the case of polyplexes based on these copolymers. The studies with the use of exemplary POxN2pendant revealed that it was non-toxic towards HT-1080 cells from a human fibrosarcoma in a wide range of concentrations, and it was able to transfect those cells.Data availability
Repository for Open Data: https://doi.org/10.18150/G9HIHO.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Synthesis of modified 2-oxazoline copolymers was supported by the National Science Centre, Project 2021/43/B/ST4/01493. The study of complexation of copolymers with nucleic acids and characterization of the obtained polyplexes were financed within the framework of the Polish-Bulgarian Research Project under the agreement on scientific cooperation between the Polish Academy of Sciences and the Bulgarian Academy of Sciences “Polyoxazoline-derived nanobiomaterials” (IC-PL/07/2022-2023). We thank Dr. Aleksander Forys for cryo-TEM images.References
- A. S. Piotrowski-Daspit, A. C. Kauffman, L. G. Bracaglia and W. M. Saltzman, Adv. Drug Delivery Rev., 2020, 156, 119–132 CrossRef CAS PubMed.
- U. Lächelt and E. Wagner, Chem. Rev., 2015, 115, 11043–11078 CrossRef PubMed.
- L. Zhou, M. Emenuga, S. Kumar, Z. Lamantia, M. Figueiredo and T. Emrick, Biomacromolecules, 2022, 23, 4029–4040 CrossRef CAS PubMed.
- S. Y. Wong, J. M. Pelet and D. Putnam, Prog. Polym. Sci., 2007, 32, 799–837 CrossRef CAS.
- E. Junquera and E. Aicart, Adv. Colloid Interface Sci., 2016, 233, 161–175 CrossRef CAS.
- A. Fus-Kujawa, P. Teper, M. Botor, K. Klarzyńska, Ł. Sieroń, B. Verbelen, M. Smet, A. L. Sieroń, B. Mendrek and A. Kowalczuk, Int. J. Polym. Mater. Polym. Biomater., 2021, 70, 356–370 CrossRef CAS.
- B. Mendrek, L. Sieroń, M. Libera, M. Smet, B. Trzebicka, A. L. Sieroń, A. Dworak and A. Kowalczuk, Polymer, 2014, 55, 4551–4562 CrossRef CAS.
- K. C. R. Bahadur and H. Uludağ, in Polymers and Nanomaterials for Gene Therapy, Woodhead Publishing, 2016, pp. 29–54 Search PubMed.
- A. Skandalis, M. Uchman, M. Štěpánek, S. Kereïche and S. Pispas, Macromolecules, 2020, 53, 5747–5755 CrossRef CAS.
- E. Haladjova, V. Chrysostomou, M. Petrova, I. Ugrinova, S. Pispas and S. Rangelov, Macromol. Biosci., 2021, 21, 1–13 CrossRef PubMed.
- P. D. Petrov, N. I. Ivanova, M. D. Apostolova and C. B. Tsvetanov, RSC Adv., 2013, 3, 3508–3511 RSC.
- E. Haladjova, S. Halacheva, D. Momekova, V. Moskova-Doumanova, T. Topouzova-Hristova, K. Mladenova, J. Doumanov, M. Petrova and S. Rangelov, Macromol. Biosci., 2018, 18, 1–12 CrossRef.
- E. Haladjova, G. Mountrichas, S. Pispas and S. Rangelov, Macromol. Chem. Phys., 2018, 219, 1–10 CrossRef.
- E. Haladjova, S. Rangelov, C. B. Tsvetanov and S. Pispas, Soft Matter, 2012, 8, 2884–2889 RSC.
- R. Kalinova, J. A. Doumanov, K. Mladenova, D. Janevska, M. Georgieva, G. Miloshev, T. Topouzova-Hristova and I. Dimitrov, ChemistrySelect, 2017, 2, 12006–12013 CrossRef CAS.
- M. Licciardi, M. Campisi, G. Cavallaro, B. Carlisi and G. Giammona, Eur. Polym. J., 2006, 42, 823–834 CrossRef CAS.
- A. Chroni, A. Forys, B. Trzebicka, A. Alemayehu, V. Tyrpekl and S. Pispas, Polymers, 2020, 12, 1283 CrossRef CAS PubMed.
- T. G. Park, J. H. Jeong and S. W. Kim, Adv. Drug Delivery Rev., 2006, 58, 467–486 CrossRef CAS PubMed.
- S. Ganta, H. Devalapally, A. Shahiwala and M. Amiji, J. Controlled Release, 2008, 126, 187–204 CrossRef CAS PubMed.
- B. Mendrek, L. Sieroń, I. Zymełka-Miara, P. Binkiewicz, M. Libera, M. Smet, B. Trzebicka, A. L. Sieroń, A. Kowalczuk and A. Dworak, Biomacromolecules, 2015, 16, 3275–3285 CrossRef CAS PubMed.
- E. Ivanova, I. Dimitrov, R. Kozarova, S. Turmanova and M. Apostolova, J. Nanopart. Res., 2013, 15, 1358 CrossRef.
- R. Hoogenboom, Eur. Polym. J., 2022, 179, 111521 CrossRef CAS.
- N. Adams and U. S. Schubert, Adv. Drug Delivery Rev., 2007, 59, 1504–1520 CrossRef CAS.
- T. Lorson, M. M. Lübtow, E. Wegener, M. S. Haider, S. Borova, D. Nahm, R. Jordan, M. Sokolski-Papkov, A. V. Kabanov and R. Luxenhofer, Biomaterials, 2018, 178, 204–280 CrossRef CAS PubMed.
- N. Oleszko-Torbus, Polym. Rev., 2022, 62, 529–548 CrossRef CAS.
- M. A. Mees and R. Hoogenboom, Polym. Chem., 2018, 9, 4968–4978 RSC.
- H. M. L. Lambermont-Thijs, J. P. A. Heuts, S. Hoeppener, R. Hoogenboom and U. S. Schubert, Polym. Chem., 2011, 2, 313–322 RSC.
- V. R. De La Rosa, E. Bauwens, B. D. Monnery, B. G. De Geest and R. Hoogenboom, Polym. Chem., 2014, 5, 4957–4964 RSC.
- H. M. L. Lambermont-Thijs, F. S. Van Der Woerdt, A. Baumgaertel, L. Bonami, F. E. Du Prez, U. S. Schubert and R. Hoogenboom, Macromolecules, 2010, 43, 927–933 CrossRef CAS.
- H. P. C. Van Kuringen, V. R. De La Rosa, M. W. M. Fijten, J. P. A. Heuts and R. Hoogenboom, Macromol. Rapid Commun., 2012, 33, 827–832 CrossRef CAS PubMed.
- H. P. C. Van Kuringen, J. Lenoir, E. Adriaens, J. Bender, B. G. De Geest and R. Hoogenboom, Macromol. Biosci., 2012, 12, 1114–1123 CrossRef CAS PubMed.
- E. Vlassi, A. Papagiannopoulos and S. Pispas, Macromol. Chem. Phys., 2018, 219, 1–9 CrossRef.
- M. H. Litt and C. S. Lin, J. Polym. Sci., Part A: Polym. Chem., 1992, 30, 779–786 CrossRef CAS.
- S. Cesana, J. Auernheimer, R. Jordan, H. Kessler and O. Nuyken, Macromol. Chem. Phys., 2006, 207, 183–192 CrossRef CAS.
- M. Hartlieb, D. Pretzel, K. Kempe, C. Fritzsche, R. M. Paulus, M. Gottschaldt and U. S. Schubert, Soft Matter, 2013, 9, 4693–4704 RSC.
- Z. He, L. Miao, R. Jordan, D. S-Manickam, R. Luxenhofer and A. V. Kabanov, Macromol. Biosci., 2015, 15, 1004–1020 CrossRef CAS PubMed.
- A. C. Rinkenauer, L. Tauhardt, F. Wendler, K. Kempe, M. Gottschaldt, A. Traeger and U. S. Schubert, Macromol. Biosci., 2015, 15, 414–425 CrossRef CAS PubMed.
- A. Zahoranová and R. Luxenhofer, Adv. Healthcare Mater., 2021, 10, 2001382 CrossRef.
- D. N. Yamaleyeva, N. Makita, D. Hwang, M. J. Haney, R. Jordan and A. V. Kabanov, Macromol. Biosci., 2023, 2300177, 1–12 Search PubMed.
- M. Mees, E. Haladjova, D. Momekova, G. Momekov, P. S. Shestakova, C. B. Tsvetanov, R. Hoogenboom and S. Rangelov, Biomacromolecules, 2016, 17, 3580–3590 CrossRef CAS PubMed.
- E. Vlassi and S. Pispas, Macromol. Chem. Phys., 2015, 216, 873–883 CrossRef CAS.
- T. Ivanova, E. Haladjova, M. Mees, D. Momekova, S. Rangelov, G. Momekov and R. Hoogenboom, Pharmacia, 2016, 63, 3–8 Search PubMed.
- N. Toncheva-Moncheva, E. Veleva-Kostadinova, C. Tsvetanov, D. Momekova and S. Rangelov, Polymer, 2017, 111, 156–167 CrossRef CAS.
- E. Haladjova, M. Smolíček, I. Ugrinova, D. Momekova, P. Shestakova, Z. Kroneková, J. Kronek and S. Rangelov, J. Appl. Polym. Sci., 2020, 137, e494400 CrossRef.
- A. K. Blakney, G. Yilmaz, P. F. McKay, C. R. Becer and R. J. Shattock, Biomacromolecules, 2018, 19, 2870–2879 CrossRef CAS.
- N. Oleszko-Torbus, B. Mendrek, A. Kowalczuk, A. Utrata-Wesołek, A. Dworak and W. Wałach, Materials, 2020, 13, 1–18 Search PubMed.
- H. Witte and W. Seeliger, Justus Liebigs Ann. Chem., 1974, 1974, 996–1009 CrossRef.
- A. Gress, A. Völkel and H. Schlaad, Macromolecules, 2007, 40, 7928–7933 CrossRef CAS.
- N. Oleszko-Torbus, M. Bochenek, A. Utrata-Wesołek, A. Kowalczuk, A. Marcinkowski, A. Dworak, A. Fus-Kujawa, A. L. Sieroń and W. Wałach, Materials, 2020, 13, 2702 CrossRef CAS PubMed.
- W. Wałach, N. Oleszko-Torbus, A. Utrata-Wesołek, M. Bochenek, E. Kijeńska-Gawrońska, Ż. Górecka, W. Święszkowski and A. Dworak, Polymers, 2020, 12, 295 CrossRef PubMed.
- W. Wałach, A. Klama-baryła, A. Sitkowska, A. Kowalczuk and N. Oleszko-Torbus, Int. J. Mol. Sci., 2021, 22, 1–11 Search PubMed.
- N. Oleszko-Torbus, A. Utrata-Wesołek, W. Wałach and A. Dworak, Eur. Polym. J., 2017, 88, 613–622 CrossRef CAS.
- N. Oleszko-Torbus, W. Wałach, A. Utrata-Wesołek and A. Dworak, Macromolecules, 2017, 50, 7636–7645 CrossRef CAS.
- N. Oleszko, W. Wałach, A. Utrata-Wesołek, A. Kowalczuk, B. Trzebicka, A. Klama-Baryła, D. Hoff-Lenczewska, M. Kawecki, M. Lesiak, A. L. Sieroń and A. Dworak, Biomacromolecules, 2015, 16, 2805–2813 CrossRef CAS.
- M. Meyer, M. Antonietti and H. Schlaad, Soft Matter, 2007, 3, 430–431 RSC.
- A. L. Demirel, M. Meyer and H. Schlaad, Angew. Chem., Int. Ed., 2007, 46, 8622–8624 CrossRef CAS PubMed.
- C. Diehl, I. Dambowsky, R. Hoogenboom and H. Schlaad, Macromol. Rapid Commun., 2011, 32, 1753–1758 CrossRef CAS PubMed.
- N. Oleszko-Torbus, A. Utrata-Wesołek, M. Bochenek, D. Lipowska-Kur, A. Dworak and W. Wałach, Polym. Chem., 2020, 11, 15–33 RSC.
- N. Oleszko, A. Utrata-Wesołek, W. Wałach, M. Libera, A. Hercog, U. Szeluga, M. Domański, B. Trzebicka and A. Dworak, Macromolecules, 2015, 48, 1852–1859 CrossRef CAS.
- D. W. Pack, A. S. Hoffman, S. Pun and P. S. Stayton, Nat. Rev. Drug Discovery, 2005, 4, 581–593 CrossRef CAS PubMed.
- G. Yang and Y. Wang, Methods Mol. Biol., 2018, 1837, 161–176 CrossRef CAS PubMed.
- T. Ivanova, E. Haladjova, M. Mees, D. Momekova, S. Rangelov, G. Momekov and R. Hoogenboom, Pharmacia, 2016, 63, 3–8 Search PubMed.
- S. K. Filippov, R. Khusnutdinov, A. Murmiliuk, W. Inam, L. Y. Zakharova, H. Zhang and V. V. Khutoryanskiy, Mater. Horiz., 2023, 10, 5354–5370 RSC.
- J. A. Shepard, P. J. Wesson, C. E. Wang, A. C. Stevans, S. J. Holland, A. Shikanov, B. A. Grzybowski and L. D. Shea, Biomaterials, 2011, 32, 5092–5099 CrossRef CAS PubMed.
- M. Sugiyama, M. Matsuura, Y. Takeuchi, J. Kosaka, M. Nango and N. Oku, Biochim. Biophys. Acta, Biomembr., 2004, 1660, 24–30 CrossRef CAS PubMed.
- M. I. Patrício, A. R. Barnard, C. I. Cox, C. Blue and R. E. MacLaren, Mol. Ther. – Methods Clin. Dev., 2018, 9, 288–295 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py01313h |
| This journal is © The Royal Society of Chemistry 2024 |