DOI:
10.1039/D4PY00239C
(Paper)
Polym. Chem., 2024,
15, 2055-2062
Unsaturated sulfur-containing polymers from modular alcohol click chemistry†
Received
2nd March 2024
, Accepted 15th April 2024
First published on 16th April 2024
Abstract
The traditional polymerization methods utilizing thiol–yne Michael addition have made significant progress in the past decades, despite suffering from the poor availability of dithiols. Here, a modular and efficient click reaction for connecting primary and secondary alcohols with activated alkynes via carbonyl sulfide (COS) was developed. The click reaction is successfully applied to synthesize different kinds of polythiocarbonates by the step-wise polyaddition of diols, dipropiolates, and COS. Diols are a diverse and abundant class of compounds that can be obtained from biomass and COS is released as industrial waste, while dipropiolates can be obtained through a one-step condensation reaction of diols with propiolic acid. In addition to the ease of obtaining the monomers, this method is atom-economical, metal-free, and involves mild conditions. Overall, the polymers have broad prospects for green materials given their facile synthesis, readily available feedstocks, desirable performance.
Introduction
An effective method for synthesizing sulfur-containing polymers is through the Michael addition of thiols and alkynes.1–12 For example, functional hyperbranched polymers of prop-2-ynyl-3-mercaptopropanoate have been prepared by photo-initiated thiol–yne click polymerization under the assistance of the initiator of 2,2-dimethoxy-2-phenylacetophenone (DMPA),13 and linear main chain polymers can be prepared by the UV-initiated click polymerizations of terminal monoynes with dithiols.14 With applications in high performance polymer,15–17 optical,18,19 luminescent,20 opto-electronic,21 self-healing,2,22 metal adsorption23 and other fields, these polymers are in high demand. However, the utilization of thiols is unpleasant since they are expensive with a persistent irritating odor and they have lower ignition points and flash points compared to alcohols.
Conversely, diols represent readily available and sustainable building blocks with an extremely rich variety, and many of them can be synthesized from renewable biomass resources.24 In general, diols are typically condensed with diacids to synthesize polymers, but due to the need for high temperature and vacuum conditions to remove water, the energy consumption of this process is relatively high, and the atom-economy is poor.25 Therefore, there is a high demand for the atom-economic utilization of diols in polymerization reactions under mild conditions.
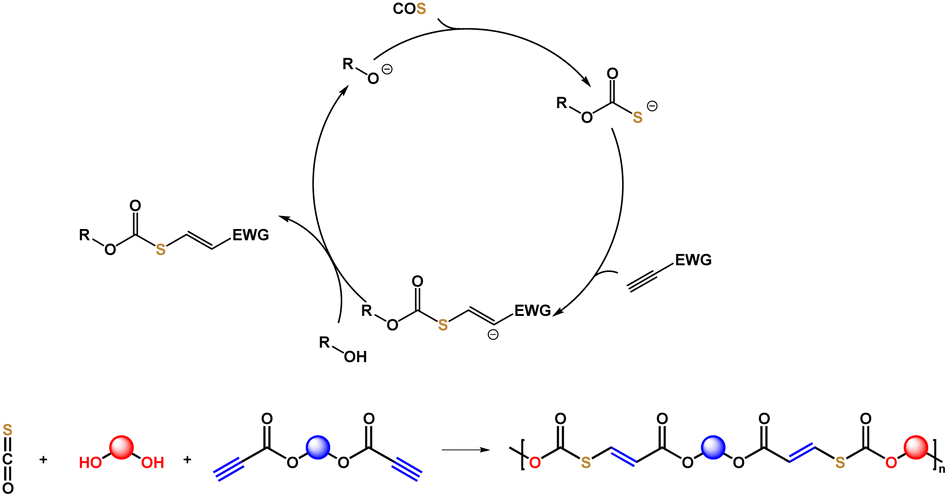
Carbonyl sulfide (COS) is a waste product from mining of fossil resources, which can cause acid rain and destruction of the ozone layer.26,27 COS can be separated by selective adsorption to obtain a high purity gas. Additionally, COS is industrially mass produced from CO and sulfur, and has been widely used as a fumigant for grain storage.28 In very recent years, the synthesis and applications of sulfur-containing polymers using COS29–46 were investigated by our group and others.47,48 Using metal or organic catalysts, different kinds of well-defined polythioethers and polythiocarbonates have been obtained. During the polymerization process using COS and epoxides, we realized that there might be an intermediate with a thiol group formed. This intermediate could potentially undergo a click reaction with olefins or alkynes.
Next, we discovered that the carbonothioate intermediate formed with a base as catalyst using COS and an alcohol can react with electron-deficient C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bonds (Fig. 1) to generate thiocarbonates. Based on the newly developed click reaction, this study discloses the step-wise polyaddition of diols, dipropiolates, and COS to synthesize a series of poly(thiocarbonate ester)s with C
C bonds (Fig. 1) to generate thiocarbonates. Based on the newly developed click reaction, this study discloses the step-wise polyaddition of diols, dipropiolates, and COS to synthesize a series of poly(thiocarbonate ester)s with C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C remaining in the main chain. Dipropiolates are accessible by the esterification of diols and propiolic acid, which serve to incorporate in-chain ester groups as potentially biodegradable and chemically breakable points. Compared with the well-known polyaddition of dithiols and monoynes, the strategy avoids the use of unpleasant and expensive thiols. Utilization of COS for manufacturing valuable sulfur-containing polymers is also of significance for both polymer chemistry and environmental science. Our method is atom-economical, performed under mild conditions, and uses common organic bases as catalysts.
C remaining in the main chain. Dipropiolates are accessible by the esterification of diols and propiolic acid, which serve to incorporate in-chain ester groups as potentially biodegradable and chemically breakable points. Compared with the well-known polyaddition of dithiols and monoynes, the strategy avoids the use of unpleasant and expensive thiols. Utilization of COS for manufacturing valuable sulfur-containing polymers is also of significance for both polymer chemistry and environmental science. Our method is atom-economical, performed under mild conditions, and uses common organic bases as catalysts.
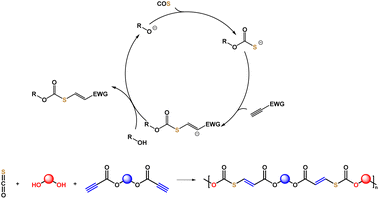 |
| | Fig. 1 Connecting alcohols with activated alkynes via a bridge molecule of carbonyl sulfide (COS). | |
Results and discussion
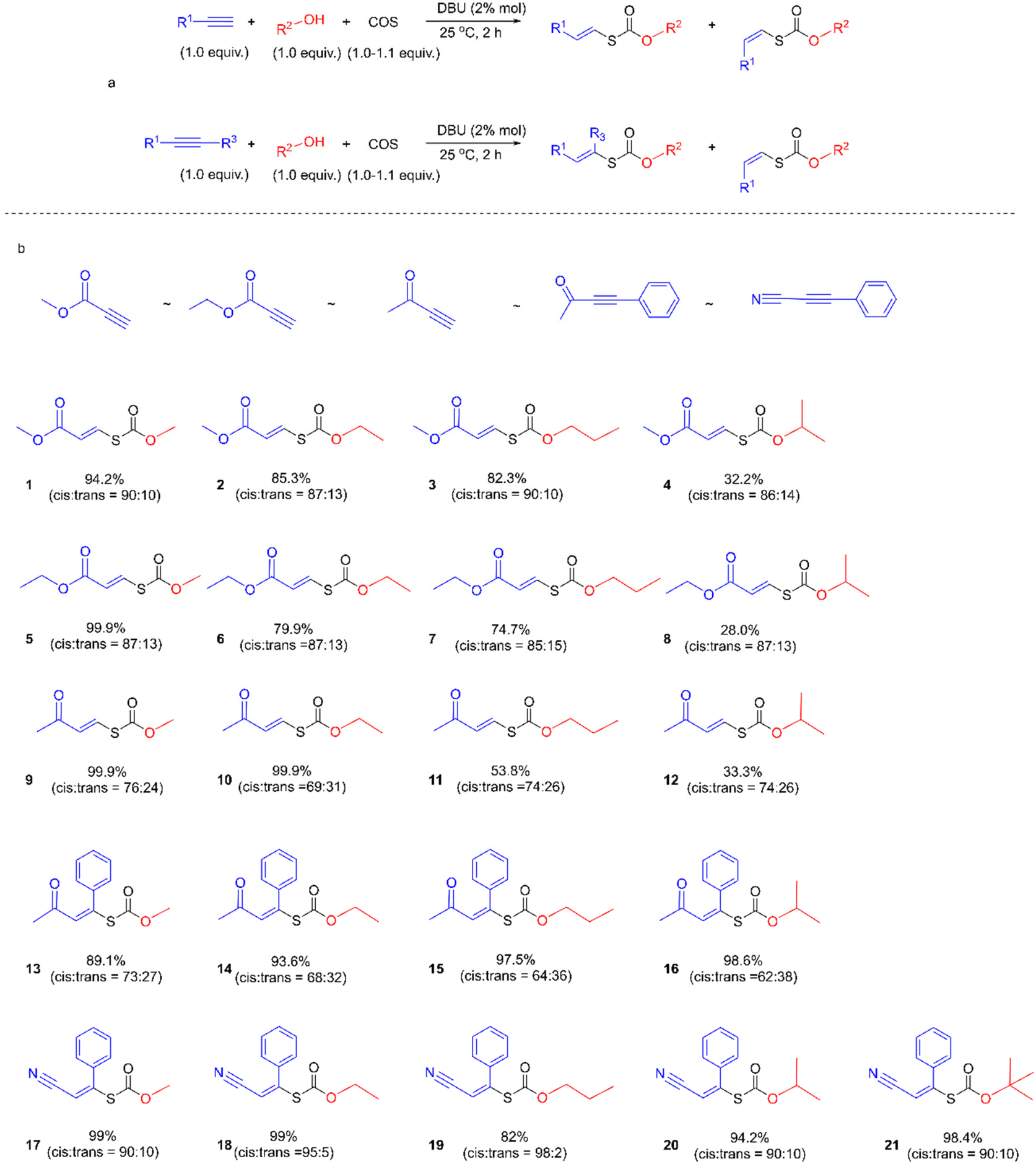
We first examined the alcohol–COS–alkyne coupling as the model reaction using 5 common activated alkynes (methyl propiolate (MP), ethyl propiolate, 3-butyn-2-one, 4-phenyl-3-butyn-2-one, and 3-phenyl-2-propynenitrile) and 5 representative alcohols (methanol, ethanol (EtOH), 1-propanol, isopropanol, and tert-butanol) (Fig. 2). All of these chemicals were used directly after purchase without purification. Since alkoxides formed from the alcohol and the base catalyst could attack the triple bonds of the alkyne units, the sequence of the feedstocks is quite important for the control of selectivity. With COS added before the methanol and DBU (see more details in the ESI†), the nucleophilic attack of the alkynes by alkoxide is avoided. The desired coupling was achieved with high efficiency under mild and robust conditions. The conversion of activated alkynes increased in the order methyl propiolate ∼ ethyl propiolate ∼ 3-butyn-2-one < 4-phenyl-3-butyn-2-one < 3-phenyl-2-propynenitrile due to the degree of electron deficiency of the CC bond and the benzene ring. The resonance of the vinylic carbanion with the benzene ring would help stabilize intermediates, resulting in increased conversion of alkynes with a benzene ring (4-phenyl-3-butyn-2-one and 3-phenyl-2-propynenitrile). A range of commercially available organobases are effective catalysts for this method (Table 1). For instance, under the same reaction conditions (Scheme 1), 1,5-diazabicyclo(5,4,0)undec-5-ene (DBU), 1,5,7-triazabicyclo(4,4,0)dec-5-ene (TBD), 7-methyl-1,5,7-triazabicyclo(4,4,0)dec-5-ene (MTBD), 4-dimethylaminopyridine (DMAP), potassium tert-butoxide (t-BuOK), N,N,N′,N′-tetraethyl ethylenediamine (TEEDA), tri-tert-butylphosphine (t-Bu3P) and tributylphosphine (n-Bu3P) afforded the product 1 in 99.9%, 99.9%, 99.9%, 99.9%, 76.2%, 88.6%, 97.4%, and 25.2% yields (entries 1–8, Table 1), respectively. A simple silica gel column process yielded purified 1 by removing excess methanol and catalysts (1H-NMR, Fig. 3). TBACl and PPNCl did not present catalytic activity (entries 9 and 10, Table 1), even in solvents like DMF and DMSO. Besides the mechanism of typical basic catalysts that we provide (Fig. 5), there may be another reaction route for the nucleophilic catalysts. Nucleophilic catalysts first attack the carbon atom of COS, and then the ethanol inserts to gain an intermediate similar to S6, thus the sterically hindered catalysts would benefit the insertion of ethanol. Higher temperatures led to increased efficiency. By elevating the reaction temperature to 60 °C and using 2% DBU, we obtained 1 in 98% yield within 5 min.
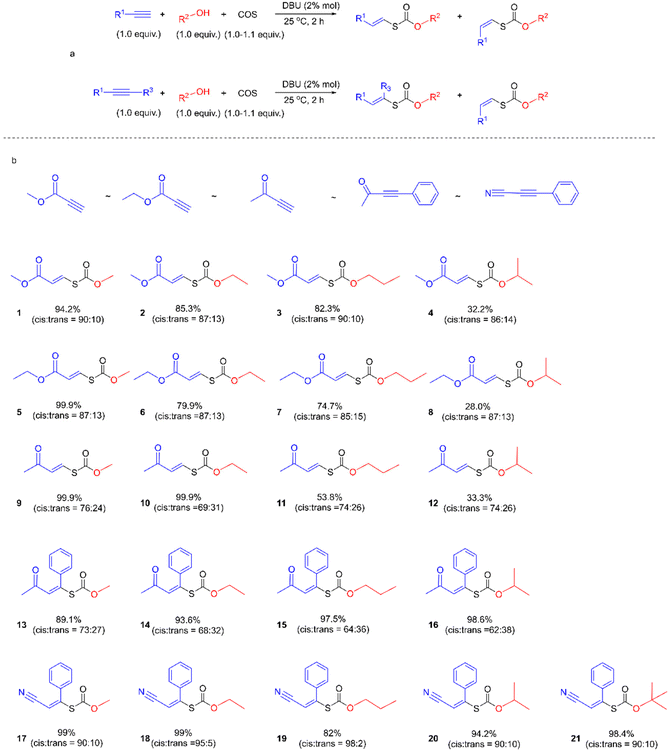 |
| | Fig. 2 Substrate scope for the coupling of alcohols, COS, and activated alkynes. | |
 |
| | Scheme 1 The model reaction of MP/EtOH/COS in Table 1. | |
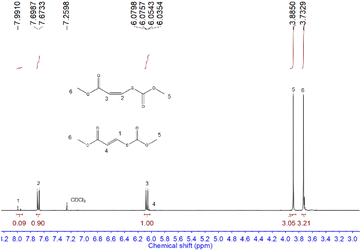 |
| | Fig. 3
1H-NMR spectrum for the product 1 from COS, MP, and methanol. | |
Table 1 The isomer content obtained under different reaction conditionsa
| Entry |
Cat. |
Solvent |
C
trans![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)
|
C
cis
c |
Conv.d |
|
The reactions were conducted at 25 °C in 8 h.
The content of trans-isomers determined by 1H-NMR spectroscopy.
The content of cis-isomers determined by 1H-NMR spectroscopy.
The conversion of MP determined by 1H-NMR spectroscopy.
The temperature was 40 °C.
The temperature was 60 °C.
|
| 1 |
DBU |
DCM |
11.5 |
88.5 |
99.9 |
| 2 |
TBD |
DCM |
11.5 |
88.5 |
99.9 |
| 3 |
MTBD |
DCM |
11.5 |
88.5 |
99.9 |
| 4 |
DMAP |
DCM |
9.9 |
90.1 |
99.9 |
| 5 |
t-BuOK |
DCM |
11.5 |
88.5 |
76.2 |
| 6 |
TEEDA |
DCM |
13.8 |
86.2 |
88.6 |
| 7 |
t-Bu3P |
DCM |
9.9 |
90.1 |
97.4 |
| 8 |
n-Bu3P |
DCM |
9.1 |
90.9 |
25.2 |
| 9 |
TBACl |
DCM |
0 |
0 |
0 |
| 10 |
PPNCl |
DCM |
0 |
0 |
0 |
| 11 |
DBU |
— |
11.5 |
88.5 |
99.9 |
| 12e |
DBU |
— |
11.5 |
88.5 |
99.9 |
| 13f |
DBU |
— |
11.5 |
88.5 |
99.9 |
| 14 |
DBU |
MeCN |
10.7 |
89.3 |
99.9 |
| 15 |
DBU |
Acetone |
11.5 |
88.5 |
99.9 |
| 16 |
DBU |
n-Hexane |
12.3 |
87.7 |
99.9 |
| 17 |
DBU |
DMF |
13.0 |
87.0 |
99.9 |
| 18 |
DBU |
DMSO |
13.8 |
86.2 |
99.9 |
| 19 |
DBU |
THF |
18.0 |
82.0 |
99.9 |
| 20 |
DBU |
Ethyl acetate |
18.0 |
82.0 |
99.9 |
Since the double bonds in the generated products do not undergo further Michael addition reactions, the products are a mixture of cis–trans isomers coexisting simultaneously. Thus, we investigated the control of the content of cis–trans isomers in the products under different conditions, including varying temperatures, catalysts, and solvents. The content of isomers makes no difference at 25, 40, and 60 °C (entries 7–9, Table 1), indicating that the Gibbs free energy of the isomers are nearly the same. The results indicate that the catalysts and the solvents have a weak relationship with the cis–trans isomer content of the reaction product. With different catalysts such as n-Bu3P and TEEDA, the content of trans isomers changed slightly from 9.1% (entry 8, Table 1) to 13.8% (entry 6, Table 1). With different solvents such as MeCN and ethyl acetate, the content of trans isomers changed slightly from 10.7% (entry 14, Table 1) to 18.0% (entry 20, Table 1).
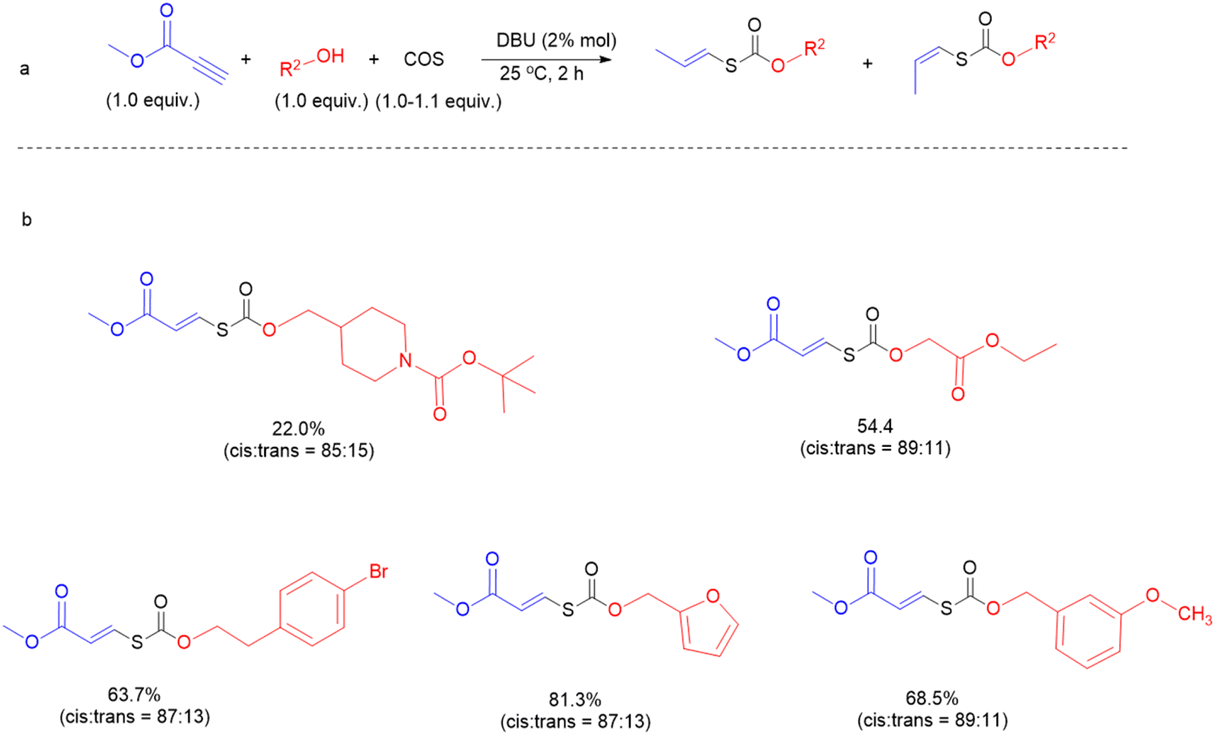
We next sought to investigate the general applicability of this method to various alcohols using methyl propiolate as a Michael acceptor (Fig. 4). Under mild conditions (2% DBU, at 25 °C, for 2 h), diverse and densely functionalized alcohols (N-Boc-4-piperidinemethanol, ethyl glycolate, 4-bromophenethyl alcohol, furfuryl alcohol, and m-anisyl alcohol (with DCM as solvent)) produced several thiocarbonates with CC bonds in 22.0%, 54.4%, 63.7%, 68.5%, and 81.3% yields and the ratio of Ctrans to Ccis is about 1:7 (Fig. S39–S43†), indicating that this reaction is tolerant of many primary alcohols with reactive functions such as halide, ester, pyridyl and furyl groups. Notably, click chemistry has emerged as one of the most powerful tools for constructing carbon–heteroatom bonds in organic chemistry, materials science, bioconjugation, etc.49–51
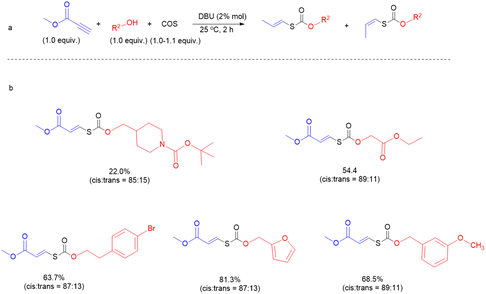 |
| | Fig. 4 Substrate scope for the coupling of alcohols, COS, and activated alkynes. | |
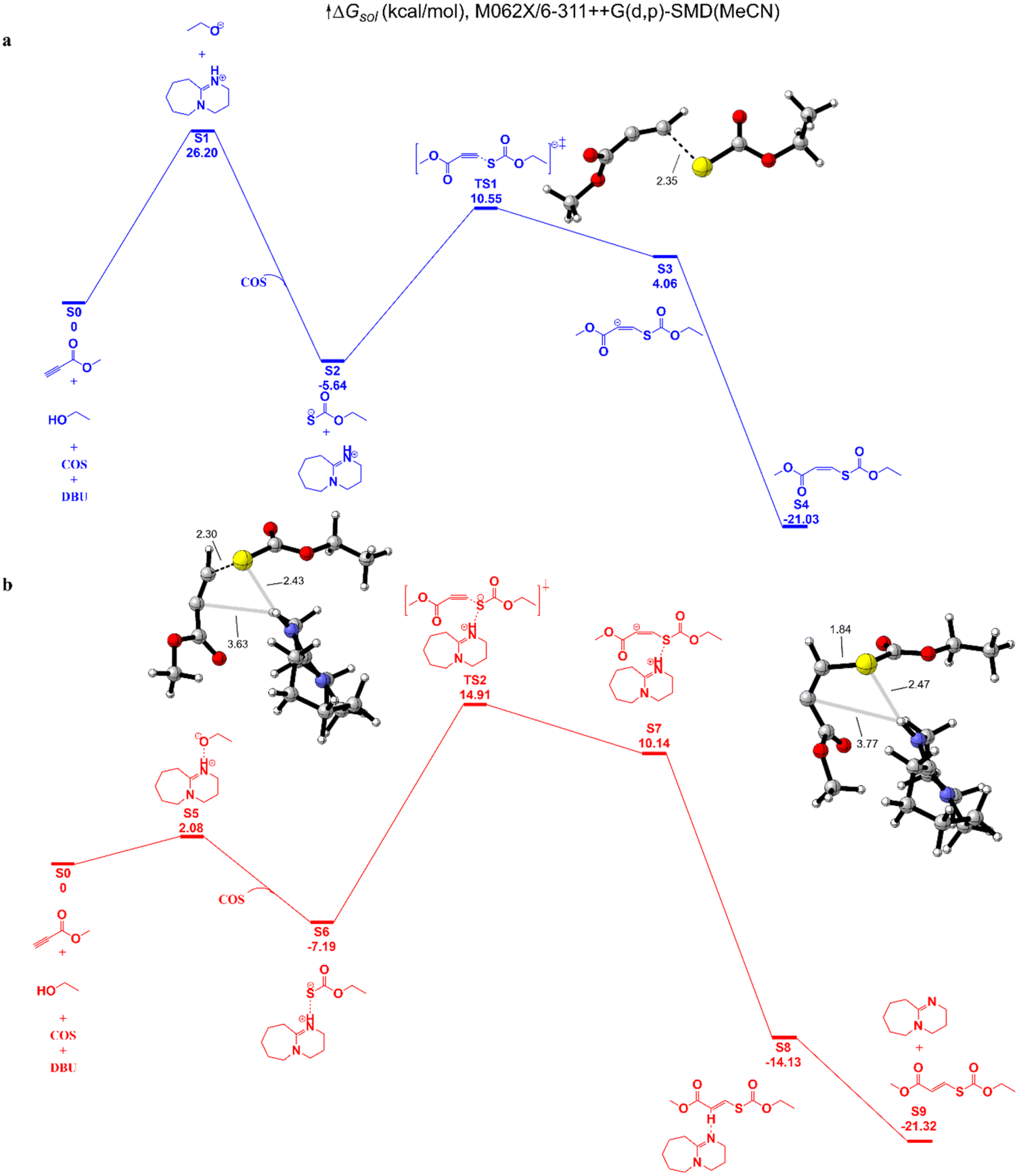
We then explored the hypothetical mechanism through density functional theory (DFT) calculations (Fig. 5). With DBU as the catalyst, MP and ethanol are used as representative reactants. Under the deprotonation of DBU, the hydroxyl group selectively attacks the CS bond of COS to generate the carbonothionate. Insertion of MP via the transition state determines the reaction rate in route b, with a barrier of 22.1 kcal mol−1, while the barrier for the hydroxyl group and DBU in route b is 26.2 kcal mol−1. In both routes the cis conformations are dominant, therefore the cis-isomers are preferentially formed. The Gibbs free energies of cis/trans isomers are nearly the same with a 0.29 kcal mol−1 difference, so under the influence of a base catalyst, the cis–trans isomers could transform reversibly.
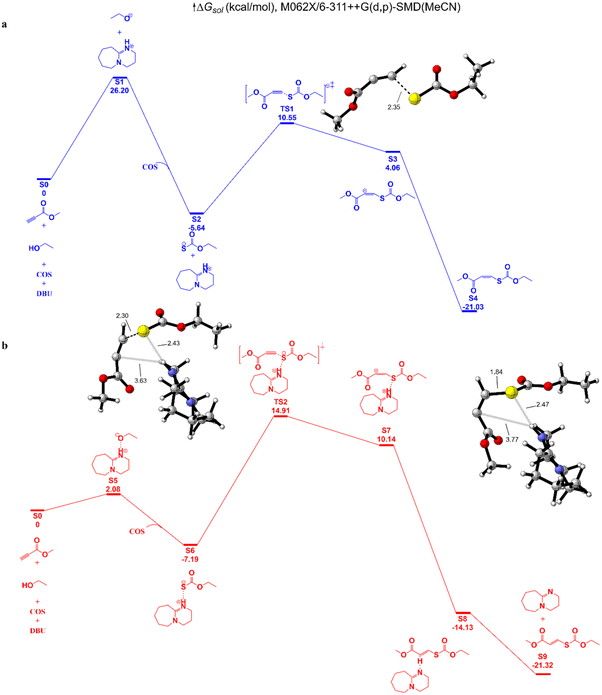 |
| | Fig. 5 Free energies associated with DBU-catalyzed coupling of COS, MP, and ethanol. In route (a), the proton of the hydroxy group is abstracted by DBU; in route (b), the hydroxy group forms a hydrogen bond with the nitrogen atom of DBU. | |
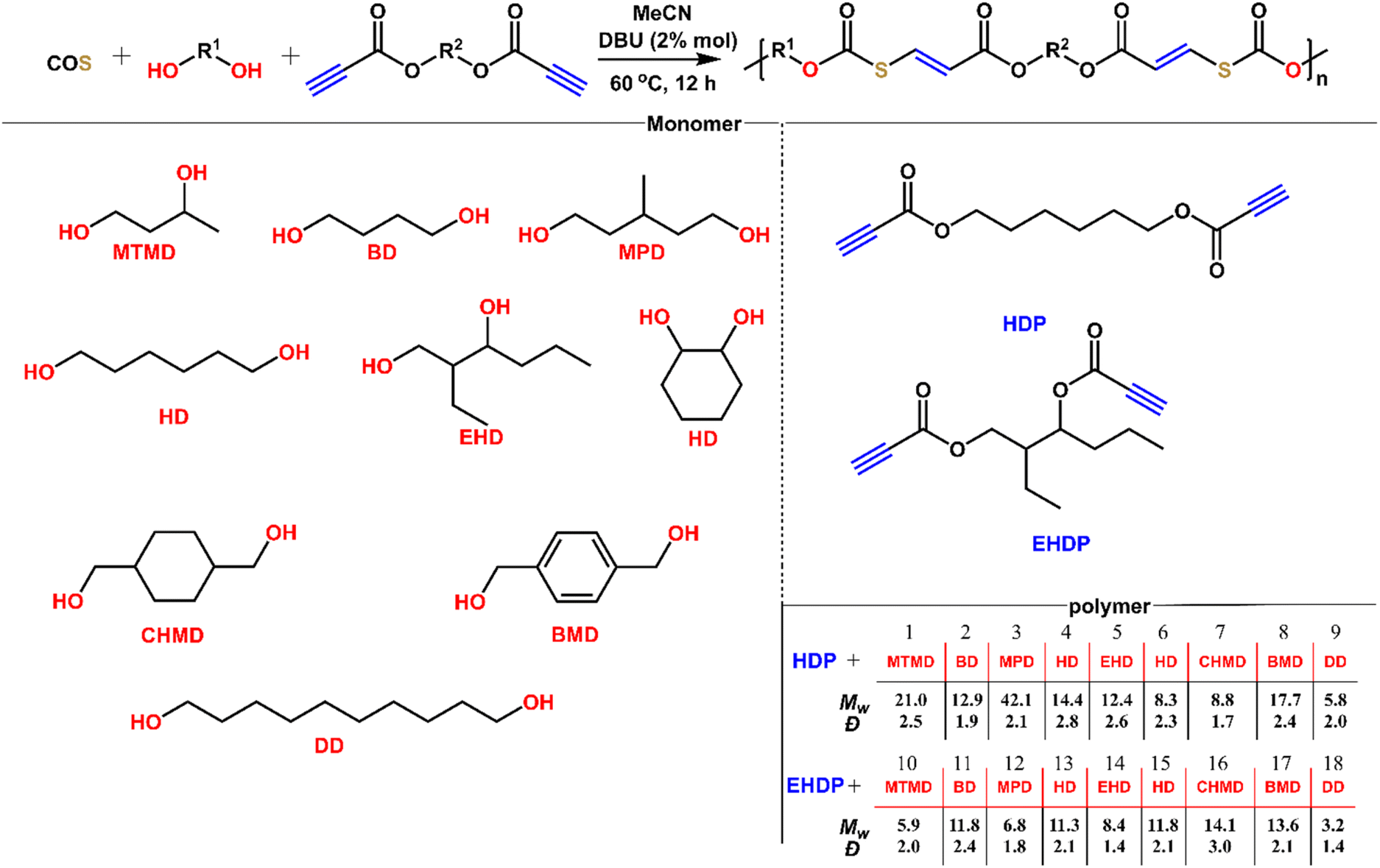
After understanding the alcohol–COS–activated alkyne coupling reaction, we sought to perform the step-wise polyaddition of diols, dipropiolates, and COS (Fig. 6). We chose MeCN as the solvent for polymerization because of its good performance in the polymerization (monomer solubility, boiling point, moisture content, etc.). All of these diols are commercially available and used as received. 1,6-Hexanediol dipropiolates (HDP) and 2-ethyl-1,3-hexanediol dipropiolates (EHDP) are synthesized from 1,6-hexanediol, 2-ethyl-1,3-hexanediol and propiolic acid. Under mild conditions (2% DBU, 60 °C, 12 h), we obtained the title polymers P1 to P18 in 99% yields. These polymers have weight-average molecular weights of 3.2 to 42.1 kDa and dispersities of 1.4 to 3.0. The low Mw polymer of linear primary diol DD maybe due to the relative insolubility of DD in MeCN, while in a good solvent for DD such as DMSO and DMF, the resulting polymer tends to crosslink. The polymerization proceeding for 12 h was long enough for the secondary diols to react with COS and alkynes (the low conversion of alkynes in 4 was because the reaction of isopropanol, COS and methyl propiolate was conducted at 25 °C in only 2 h). Thus, the difference in the Mw also depends on the solubility of the diols. Because of the poor solubility of diols with long carbon chains in solvents like MeCN and DCM in which polymers have better solubility, it is quite hard to gain polymers with higher molecular weight from such diols, thus we didn't make many attempts for the polymerization of long carbon chain diols, COS and dipropiolates. But it's suggested that those polymers may have stronger crystallinity from previous work. At low temperature (e.g., 25 °C), the Mw of P4 gained in 12 h would decrease to about 8.8 kDa (with Đ = 1.9). Reducing the reaction time is also not conducive to the increase of Mw, (e.g., 4 h), and the Mw of P4 gained under the same conditions is about 11.3 kDa (with Đ = 2.4). All of these polymers possess well-defined and predesigned chain structures according to NMR analysis (Fig. S47–S66†), in which each repeating unit contains thiocarbonate and ester groups and CC bonds. The controlled experiment confirmed that the polymerization did not occur without a catalyst. Accordingly, the readily accessible monomers, mild polymerization conditions, and high efficiency reveal a facile and versatile method for polymer synthesis.
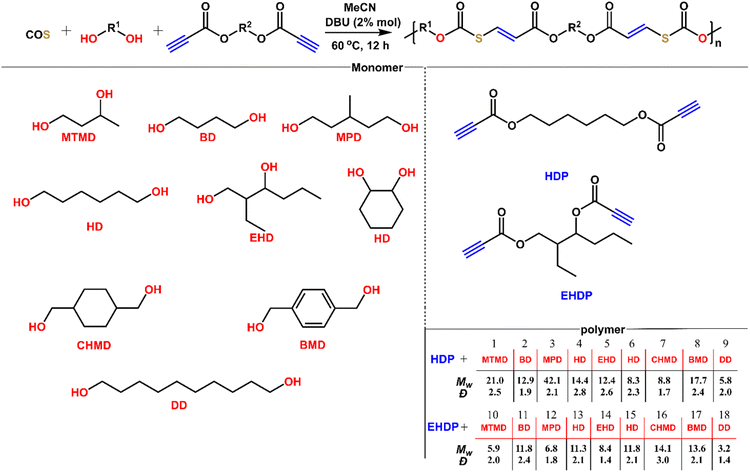 |
| | Fig. 6 Synthesis of a series of polymers from diols, dipropiolates, and COS. | |
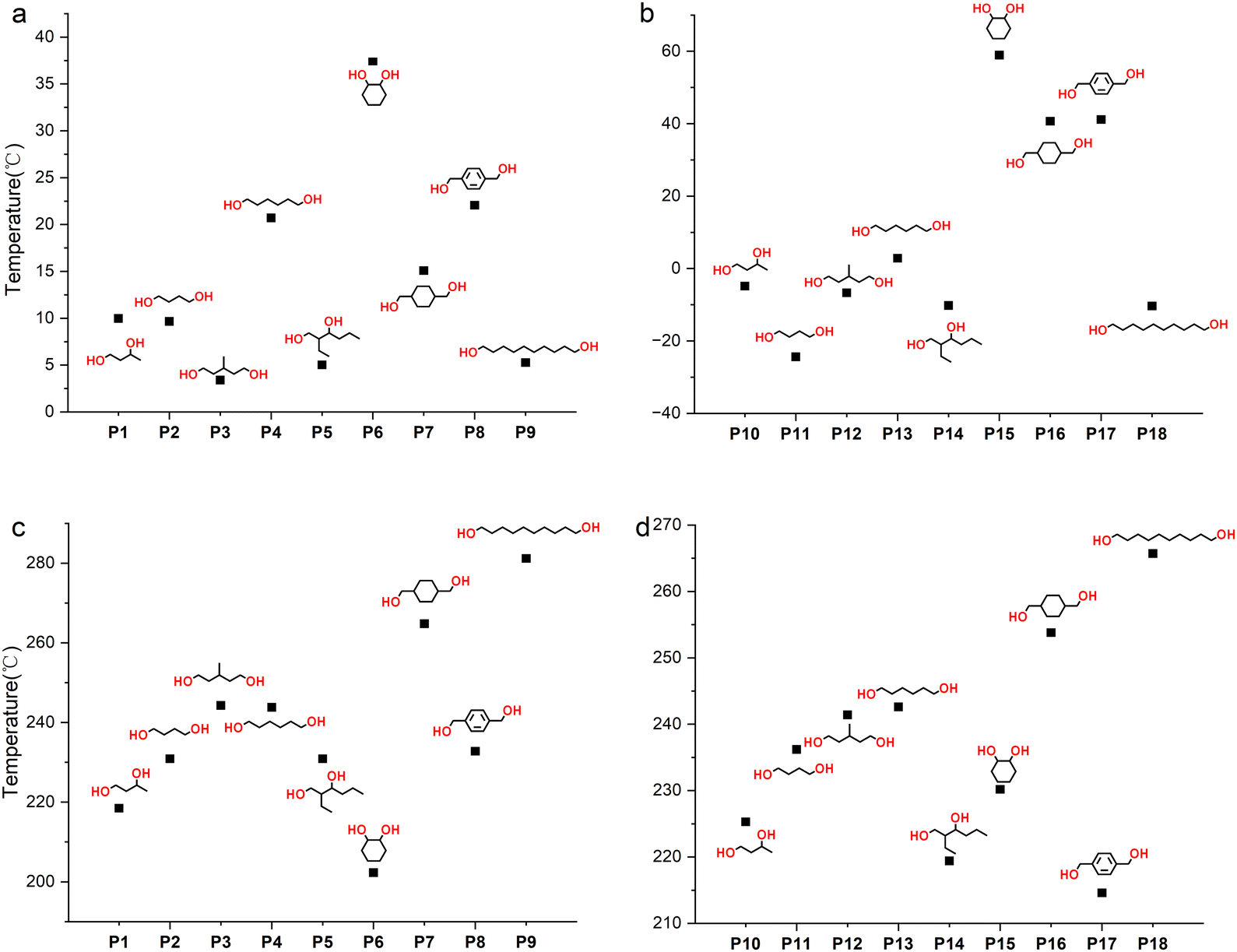
The thermodynamic properties of polymers were tested using differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA), and the results are shown in Table 2 and Fig. 7. The Tg of the polymer is mainly affected by the functional groups of the polymer and the number of methylene –CH2– units in the polymer main chain. The stronger the rigidity of the polymer chain and side groups, the higher the Tg. For example, for P4 and P7, cyclohexane is more rigid than –(CH2)4–, so the Tg rises from 20.7 to 37.4 °C. The possible reason for Tg first increasing and then decreasing with the number of –CH2– units in the main chain (entries 1, 4 and 9, Table 2) is that the chain regularity increases while the density of the rigid groups (ester groups and thiocarbonate groups) decreases. The improvement of chain regularity will also increase the crystallization ability of the polymer. For instance, for linear primary diols such as HD, no obvious melting peak is observed in the DSC curve (Fig. S70†), indicating its low crystallization ability, while DD, a linear primary diol containing ten –CH2– groups, shows obvious crystallization ability (Fig. S75†), with a crystallization temperature (Tc) of 93.3 °C. At the same time, the thermal stability of the polymer increases with the length of the main chain (entries 2, 4 and 9, Table 2), indicating that the polymer's stability is mainly affected by the thiocarbonate groups. The greater the density of thiocarbonate groups, the easier it is for the polymer to decompose at high temperatures.
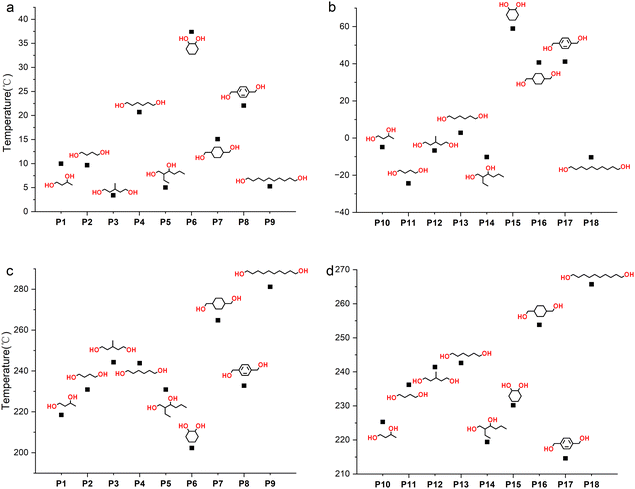 |
| | Fig. 7 (a) Tg values of P1 to P9. (b) Tg values of P10 to P18. (c) Td values of P1 to P9. (d) Td values of P10 to P18. | |
Table 2 Polymerizations of HDP&EHDP/COS/diols catalysed by DBU at 60 °Ca
| Entry |
Diols |
No. |
M
wb |
Đ
|
T
gc |
T
d,5%d |
|
The reactions were conducted in 12 h with [HDP]:[diol]:[COS]:[DBU] = 50:50:55:1 with MeCN as solvent.
The molecular weight and distribution of polymers were determined by GPC with polystyrene standards.
The glass transition temperature was determined by DSC.
The decomposition (5 wt% polymer) temperature was determined by TGA.
|
| 1 |
MTMD |
P1
|
21.0 |
2.5 |
10.0 |
218.5 |
| 2 |
BD |
P2
|
12.9 |
1.9 |
9.7 |
230.9 |
| 3 |
MPD |
P3
|
42.1 |
2.1 |
3.4 |
244.3 |
| 4 |
HD |
P4
|
14.4 |
2.8 |
20.7 |
243.8 |
| 5 |
EHD |
P5
|
12.4 |
2.6 |
8.4 |
230.9 |
| 6 |
CHD |
P6
|
8.3 |
2.3 |
37.4 |
202.3 |
| 7 |
CHMD |
P7
|
8.8 |
1.7 |
15.1 |
264.8 |
| 8 |
BMD |
P8
|
17.7 |
2.4 |
22.1 |
232.8 |
| 9 |
DD |
P9
|
5.8 |
2.0 |
5.3 |
281.2 |
| 10 |
MTMD |
P10
|
5.9 |
2.0 |
−4.9 |
225.3 |
| 11 |
BD |
P11
|
11.8 |
2.4 |
−24.4 |
236.2 |
| 12 |
MPD |
P12
|
6.8 |
1.8 |
−6.7 |
215.9 |
| 13 |
HD |
P13
|
11.3 |
2.1 |
2.8 |
244.6 |
| 14 |
EHD |
P14
|
8.4 |
1.4 |
−10.2 |
219.4 |
| 15 |
CHD |
P15
|
11.8 |
2.1 |
58.9 |
230.2 |
| 16 |
CHMD |
P16
|
14.1 |
3.0 |
40.7 |
253.8 |
| 17 |
BMD |
P17
|
13.6 |
2.1 |
41.2 |
214.6 |
| 18 |
DD |
P18
|
3.2 |
1.4 |
−10.3 |
265.7 |
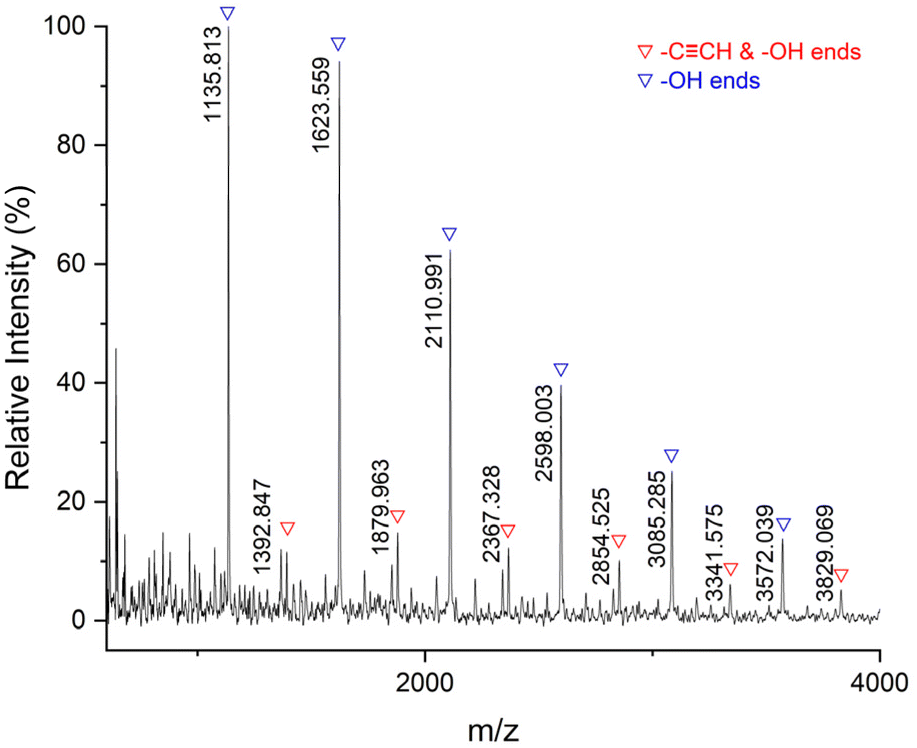
Subsequently, we tested the MALDI-TOF-MS of P7 (Fig. 8). There are mainly double –OH terminal species [(C22H30O8S2)n + C8H16O2 + Na+] = [486.14 × n + 144.12 + 22.99], accompanied by a small amount of –CCH and –OH terminal species [(C22H30O8S2)n + C21H30O7S + H+] = [486.14 × n + 426.17 + 1.01]. The thiocarbonate groups are not included at the ends of the polymer chains because the thiocarbonate groups are unstable and dissociate more easily.
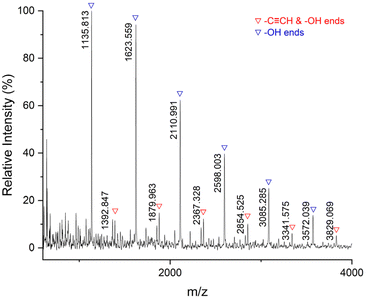 |
| | Fig. 8 MALDI-TOF-MS spectrum for P7 (entry 7, Table 2). | |
Conclusions
Through the coupling reaction of hydroxyl/COS/activated CC bonds, which is modular and efficient, we have obtained several small molecules (1 to 21) and macromolecules (P1 to P18) in high yield. The significance of this method lies in its avoidance of unpleasant and expensive thiols, utilizing diols as readily available and sustainable building blocks. The process is performed under mild conditions, utilizing common organic bases as catalysts, ensuring atom-economical and environmentally conscious polymerization. The produced polymers possess in-chain esters, thiocarbonate units and CC bonds. These polymers are expected to become green materials in practice given their widely available raw materials and facile synthesis.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
We acknowledge the financial support of the National Science Foundation of China (52373014, 52203129, U23A2083).
References
- Z. Geng, J. J. Shin, Y. Xi and C. J. Hawker, J. Polym. Sci., 2021, 59, 963–1042 CrossRef CAS.
- B. Yao, J. Sun, A. Qin and B. Z. Tang, Chin. Sci. Bull., 2013, 58, 2711–2718 CrossRef CAS.
- X. Yu, Y. Wang, M. Li, Y. Zhang, Y. Huang, Q. Qian, Y. Zheng, Q. Hou and X. Fan, ACS Appl. Polym. Mater., 2023, 5, 2750–2759 CrossRef CAS.
- X. Fu, A. Qin and B. Z. Tang, J. Polym. Sci., 2024, 62(5), 787–798 CrossRef CAS.
- B. J. Curole, W. J. Broussard, A. Nadeem and S. M. Grayson, ACS Polym. Au, 2023, 3, 70–81 CrossRef CAS.
- J. C. Worch and A. P. Dove, Acc. Chem. Res., 2022, 55, 2355–2369 CrossRef CAS PubMed.
- H. Mutlu, E. B. Ceper, X. Li, J. Yang, W. Dong, M. M. Ozmen and P. Theato, Macromol. Rapid Commun., 2019, 40, 1800650 CrossRef PubMed.
- E. Blasco, M. B. Sims, A. S. Goldmann, B. S. Sumerlin and C. Barner-Kowollik, Macromolecules, 2017, 50, 5215–5252 CrossRef CAS.
- D. A. Boyd, Angew. Chem., Int. Ed., 2016, 55, 15486–15502 CrossRef CAS PubMed.
- Y. Zheng, S. Li, Z. Weng and C. Gao, Chem. Soc. Rev., 2015, 44, 4091–4130 RSC.
- B. Yao, J. Mei, J. Li, J. Wang, H. Wu, J. Z. Sun, A. Qin and B. Z. Tang, Macromolecules, 2014, 47, 1325–1333 CrossRef CAS.
- G. Sagdic, O. Daglar, E. Cakmakci, V. Findik, S. S. Erdem, U. Tunca, U. S. Gunay and H. Durmaz, Macromolecules, 2023, 56, 7006–7022 CrossRef CAS.
- D. Konkolewicz, A. Gray-Weale and S. Perrier, J. Am. Chem. Soc., 2009, 131, 18075–18077 CrossRef CAS PubMed.
- O. Türünç and M. A. R. Meier, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1689–1695 CrossRef.
- V. X. Truong, M. P. Ablett, S. M. Richardson, J. A. Hoyland and A. P. Dove, J. Am. Chem. Soc., 2015, 137, 1618–1622 CrossRef CAS PubMed.
- L. J. Macdougall, V. X. Truong and A. P. Dove, ACS Macro Lett., 2017, 6, 93–97 CrossRef CAS PubMed.
- M. M. Pérez-Madrigal, J. E. Shaw, M. C. Arno, J. A. Hoyland, S. M. Richardson and A. P. Dove, Biomater. Sci., 2019, 8, 405–412 RSC.
- Q. Wei, R. Pötzsch, X. Liu, H. Komber, A. Kiriy, B. Voit, P.-A. Will, S. Lenk and S. Reineke, Adv. Funct. Mater., 2016, 26, 2545–2553 CrossRef CAS.
- S. Mavila, J. Sinha, Y. Hu, M. Podgórski, P. K. Shah and C. N. Bowman, ACS Appl. Mater. Interfaces, 2021, 13, 15647–15658 CrossRef CAS PubMed.
- Q. Cao, R. Jiang, M. Liu, Q. Wan, D. Xu, J. Tian, H. Huang, Y. Wen, X. Zhang and Y. Wei, Mater. Sci. Eng., C, 2017, 80, 411–416 CrossRef CAS PubMed.
- A. Marrocchi, A. Facchetti, D. Lanari, S. Santoro and L. Vaccaro, Chem. Sci., 2016, 7, 6298–6308 RSC.
- L. J. Macdougall, M. M. Pérez-Madrigal, J. E. Shaw, M. Inam, J. A. Hoyland, R. O'Reilly, S. M. Richardson and A. P. Dove, Biomater. Sci., 2018, 6, 2932–2937 RSC.
- X.-M. He, G.-T. Zhu, Y.-Y. Zhu, X. Chen, Z. Zhang, S.-T. Wang, B.-F. Yuan and Y.-Q. Feng, ACS Appl. Mater. Interfaces, 2014, 6, 17857–17864 CrossRef CAS PubMed.
- F. M. Haque, J. S. A. Ishibashi, C. A. L. Lidston, H. Shao, F. S. Bates, A. B. Chang, G. W. Coates, C. J. Cramer, P. J. Dauenhauer, W. R. Dichtel, C. J. Ellison, E. A. Gormong, L. S. Hamachi, T. R. Hoye, M. Jin, J. A. Kalow, H. J. Kim, G. Kumar, C. J. LaSalle, S. Liffland, B. M. Lipinski, Y. Pang, R. Parveen, X. Peng, Y. Popowski, E. A. Prebihalo, Y. Reddi, T. M. Reineke, D. T. Sheppard, J. L. Swartz, W. B. Tolman, B. Vlaisavljevich, J. Wissinger, S. Xu and M. A. Hillmyer, Chem. Rev., 2022, 122, 6322–6373 CrossRef CAS PubMed.
- Q. Cai, T. Bai, H. Zhang, X. Yao, J. Ling and W. Zhu, Mater. Today, 2021, 51, 155–164 CrossRef CAS.
- J. Notholt, Z. Kuang, C. P. Rinsland, G. C. Toon, M. Rex, N. Jones, T. Albrecht, H. Deckelmann, J. Krieg, C. Weinzierl, H. Bingemer, R. Weller and O. Schrems, Science, 2003, 300, 307–310 CrossRef CAS PubMed.
- R. P. Turco, R. C. Whitten, O. B. Toon, J. B. Pollack and P. Hamill, Nature, 1980, 283, 283–285 CrossRef CAS.
- R. J. Ferm, Chem. Rev., 1957, 57, 621–640 CrossRef CAS.
- J. Kiriratnikom, X. Zhang, X. Cao, B. Chu, C. Zhang and X. Zhang, J. Polym. Sci., 2022, 60, 2262–2268 CrossRef CAS.
- Y. Wang, Y. Xia, Z. Hua, C. Zhang and X. Zhang, Polym. Chem., 2022, 13, 5397–5403 RSC.
- L.-F. Hu, Y. Li, B. Liu, Y.-Y. Zhang and X.-H. Zhang, RSC Adv., 2017, 7, 49490–49497 RSC.
- J.-L. Yang, H.-L. Wu, Y. Li, X.-H. Zhang and D. J. Darensbourg, Angew. Chem., Int. Ed., 2017, 56, 5774–5779 CrossRef CAS PubMed.
- J.-L. Yang, X.-H. Cao, C.-J. Zhang, H.-L. Wu and X.-H. Zhang, Molecules, 2018, 23, 298 CrossRef PubMed.
- C.-J. Zhang, H.-L. Wu, Y. Li, J.-L. Yang and X.-H. Zhang, Nat. Commun., 2018, 9, 2137 CrossRef PubMed.
- C.-J. Zhang, J.-L. Yang, L.-F. Hu and X.-H. Zhang, Chin. J. Chem., 2018, 36, 625–629 CrossRef CAS.
- J. Yang, H. Wang, L. Hu, X. Hong and X. Zhang, Polym. Chem., 2019, 10, 6555–6560 RSC.
- X. Cao, H. Wang, J. Yang, R. Wang, X. Hong, X. Zhang, J. Xu and H. Wang, Chin. Chem. Lett., 2022, 33, 1021–1024 CrossRef CAS.
- J. Kiriratnikom, J. Guo, X. Cao, M. U. Khan, C. Zhang and X. Zhang, J. Polym. Sci., 2022, 60, 3414–3419 CrossRef CAS.
- M. Ullah Khan, S. Ullah Khan, X. Cao, M. Usman, X. Yue, A. Ghaffar, M. Hassan, C. Zhang and X. Zhang, Chem. – Asian J., 2023, 18, e202201050 CrossRef CAS PubMed.
- Y. Xia, C. Zhang, Y. Wang, S. Liu and X. Zhang, Chin. Chem. Lett., 2023, 108860 Search PubMed.
- C. Zhang, X. Geng, X. Zhang, Y. Gnanou and X. Feng, Prog. Polym. Sci., 2023, 136, 101644 CrossRef CAS.
- Y. Li, H.-Y. Duan, M. Luo, Y.-Y. Zhang, X.-H. Zhang and D. J. Darensbourg, Macromolecules, 2017, 50, 8426–8437 CrossRef CAS.
- C.-J. Zhang, T.-C. Zhu, X.-H. Cao, X. Hong and X.-H. Zhang, J. Am. Chem. Soc., 2019, 141, 5490–5496 CrossRef CAS PubMed.
- Z. Zhang, Z. Xiong, B. Chu, Z. Zhang, Y. Xie, L. Wang, J. Z. Sun, H. Zhang, X.-H. Zhang and B. Z. Tang, Aggregate, 2022, 3, e278 CrossRef CAS.
- Y. Xia, X. Yue, Y. Sun, C. Zhang and X. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202219251 CrossRef CAS PubMed.
- Y. Xia, Y. Sun, Z. Liu, C. Zhang and X. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202306731 CrossRef CAS PubMed.
- D. K. Tran, A. N. Braaksma, A. M. Andras, S. K. Boopathi, D. J. Darensbourg and K. L. Wooley, J. Am. Chem. Soc., 2023, 145, 18560–18567 CrossRef CAS PubMed.
- Q. Wen, Q. Cai, P. Fu, D. Chang, X. Xu, T.-J. Wen, G.-P. Wu, W. Zhu, L.-S. Wan, C. Zhang, X.-H. Zhang, Q. Jin, Z.-L. Wu, C. Gao, H. Zhang, N. Huang, C.-Z. Li and H. Li, Chin. Chem. Lett., 2023, 34, 107592 CrossRef CAS.
- M. G. Finn, H. C. Kolb and K. B. Sharpless, Nat. Synth., 2022, 1, 8–10 CrossRef.
- G. Meng, T. Guo, T. Ma, J. Zhang, Y. Shen, K. B. Sharpless and J. Dong, Nature, 2019, 574, 86–89 CrossRef CAS PubMed.
- J. C. Worch, C. J. Stubbs, M. J. Price and A. P. Dove, Chem. Rev., 2021, 121, 6744–6776 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a and
Xinghong
Zhang
*a and
Xinghong
Zhang