
An efficient and economical degradation strategy for epoxy thermosets based on a low-cost transesterification catalyst†
Zhijun
Yang
a,
Shuhan
Zhang
a,
Huan
Liang
a,
Enjian
He
a,
Yixuan
Wang
a,
Ting
Lei
b,
Zhicheng
Wu
c,
Qiulin
Chen
d,
Fusheng
Zhou
d,
Yen
Wei
 *a and
Yan
Ji
*a
*a and
Yan
Ji
*a
aThe Key Laboratory of Bioorganic Phosphorus Chemistry & Chemical Biology (Ministry of Education), Department of Chemistry, Tsinghua University, Beijing 100084, China
bDepartment of Electrical Engineering, Tsinghua University, Beijing 100084, China
cState Key Laboratory of Electrical Insulation and Power Equipment, Xi'an Jiaotong University, Xi'an 710049, China
dElectric Power Research Institute, China Southern Power Grid Co. Ltd, Guangzhou 510623, China
First published on 2nd November 2024
Abstract
Epoxy thermoset materials are widely used in the field of electrical engineering, including gas-insulated enclosed switches and drytype transformers. It is a challenge to degrade and recycle traditional epoxy thermosets, which can lead to serious resource wastage and solid waste pollution. Previously reported strategies have often required harsh chemical conditions or complex chemical syntheses, which cannot effectively recycle and degrade the large amount of existing anhydride-cured epoxy thermoset waste used in electrical engineering. Herein, we propose an efficient and economical degradation strategy for epoxy thermosets based on a commercial transesterification catalyst (1,8-diazabicyclo[5.4.0]undec-7-ene, DBU). DBU is much cheaper than the widely reported transesterification catalyst in the field of degrading and recycling epoxy thermosets (1,5,7-triazabicyclo[4.4.0]dec-5-ene, TBD). We demonstrated that an anhydride-cured epoxy thermoset could be completely depolymerized in DBU-ethylene glycol solution below 140 °C in 2 h (or 120 °C in 3 h). Our study offers an effective method to recycle anhydride-cured epoxy thermoset wastes.
1. Introduction
Epoxy thermosets are widely used in daily life and industrial fields (e.g., electrical engineering, wind-power generation, aerospace and transportation) owing to their outstanding properties (e.g., adhesion, insulation, and chemical resistance).1,2 In the field of electrical engineering, epoxy thermoset materials are widely used as gas-insulated enclosed switches and dry-type transformers. Epoxy thermosets widely used in electrical engineering are epoxy-anhydride resins, and exhibit outstanding mechanical performance and a high glass transition temperature.2 However, it is a challenge to degrade and recycle the wastes of these epoxy thermosets, which can lead to severe resource wastage and solid waste pollution.In fact, degrading and recycling epoxy thermoset has always been a research “hotpot”. In 2011, Leibler et al. were the first to report an epoxy dynamic crosslinked network based on transesterification, and named the dynamic crosslinked network as a “vitrimer”.3,4 Since then, epoxy-based vitrimers containing different dynamic bonds have been widely reported, including the ester bond,3–8 disulfide bond,9–19 imine bond,20–23 silyl ether equilibration,24,25 boronic ester bond,26 and the Diels–Alder reaction.27–31 The exchange of dynamic bonds in epoxy-based vitrimers is usually reliant on catalysts, whereas no transesterification catalyst exists in epoxy thermosets of electrical equipment wastes and, thus, they cannot be reprocessed or reshaped. Researchers have also designed degradable and recyclable hyperbranched epoxy resins enabled by reversible moieties, which can be digested at 90 °C in 2 h.32 However, the synthesis of hyperbranched epoxy resins is complex, and scaling up for mass production is challenging. In 2023, Ahrens et al. proposed a transition-metal-catalyzed approach for recovery of block bisphenol A and fiber in epoxy composites.33 However, the yield of degradation products is not satisfying and, thus, it is not suitable for the recycling and degradation of epoxy thermosets. In 2024, Barta et al. fabricated a recyclable epoxy-amine thermoset, which could be degraded by methanolysis at 70 °C.34 Epoxy thermosets in electrical engineering are mostly anhydride-cured epoxy resins. Thus, the epoxy-amine thermoset is not suitable in the field of electrical engineering. In recent years, biomass-derived epoxy thermosets have garnered attention, but their application in electrical engineering is rare because they cannot address the challenge of recycling and degradation of epoxy thermosets wastes.20,35–43
Although several strategies have been proposed to recycle epoxy thermosets, an effective approach to degrade and reuse epoxy thermoset wastes of electrical equipment is lacking. Many ester bonds are present in epoxy resins, so degrading ester bonds in the robust epoxy network is important. The commonly used degradation strategy of epoxy thermosets is based on alcoholysis: the transesterification reaction between alcohol and ester bonds in the epoxy network enables the chemical degradation of epoxy resins. Under supercritical methanol, carbon fiber-reinforced epoxy resin composites can be degraded efficiently, but harsh degradation conditions (270 °C, 8 MPa) are required, resulting in enormous energy consumption.44,45 In recent years, ethylene glycol has also been utilized to degrade epoxy resins. At 180 °C, ethylene glycol can effectively degrade epoxy resins containing transesterification catalysts such as 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD).46–51 However, the ethylene glycol-based degradation method for epoxy thermosets usually requires a relatively high temperature (∼180 °C). Besides, TBD is an expensive transesterification catalyst, which increases the cost of degrading epoxy thermoset wastes. Thus, there is an urgent need to degrade epoxy thermoset wastes using an efficient and economical method (Fig. 1).
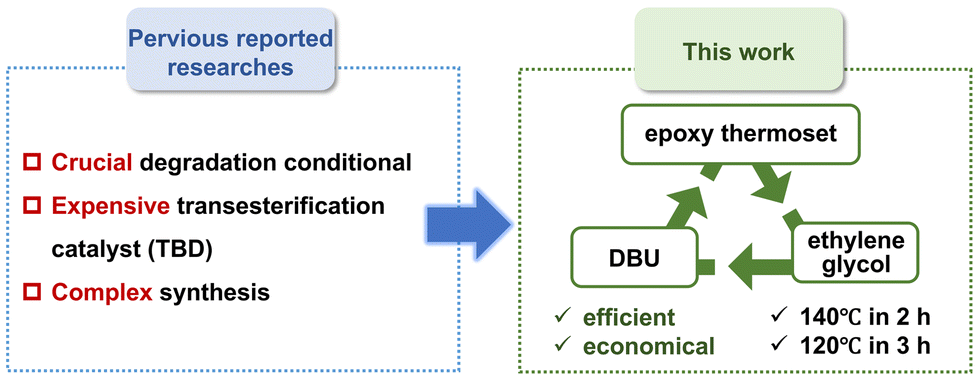 | ||
| Fig. 1 Comparison between previously reported studies and this work (schematic). | ||
We propose an efficient and economical degradation strategy for epoxy thermoset wastes based on a commercial low-cost transesterification catalyst (1,8-diazabicyclo[5.4.0]undec-7-ene, DBU). Compared with TBD, DBU is much more cost-effective. In previous works, DBU has usually been employed as an accelerator in the polymerization of epoxy resins, but not used as a transesterification catalyst in the degradation of epoxy thermoset.52–54 Herein, we utilized DBU as an efficient and economical catalyst to depolymerize epoxy resins, which has not been reported previously. We found that the content of DBU and the degradation temperature could influence the degradation rate. Specifically, an epoxy thermoset could be completely depolymerized in DBU-ethylene glycol solution below 140 °C in 2 h (or 120 °C in 3 h). By precipitation in water, the degradation product (DGEBA derivative tetra-alcohol) could be recycled simply.
2. Experimental
Materials
Bisphenol A diglycidyl ether (DGEBA, Energy Chemical, 85%), methyl tetrahydrophthalic anhydride (MeTHPA, Macklin, 95%), benzyl-N,N-dimethylamine (BDMA, J&K Scientific, 99%), 1,8-diazabicyclo[5.4.0]undec-7-ene, (DBU, Bidepharm, 99.62%), ethylene glycol (EG, Energy Chemical, 99%) epoxy composite (resin-impregnated paper bushing, Xian XD High Voltage Bushing) were used directly without further purification.Preparation of anhydride-cured epoxy thermoset
First, 4.004 g (10 mmol) of DGEBA, 3.498 g (20 mmol) of MeTHPA, and 75.03 mg of BDMA were stirred homogenously at 60 °C for 30 min. The mixture was transfered into a polydimethylsiloxane (PDMS) mold and cured at 140 °C for 12 h for full polymerization (Fig. 2).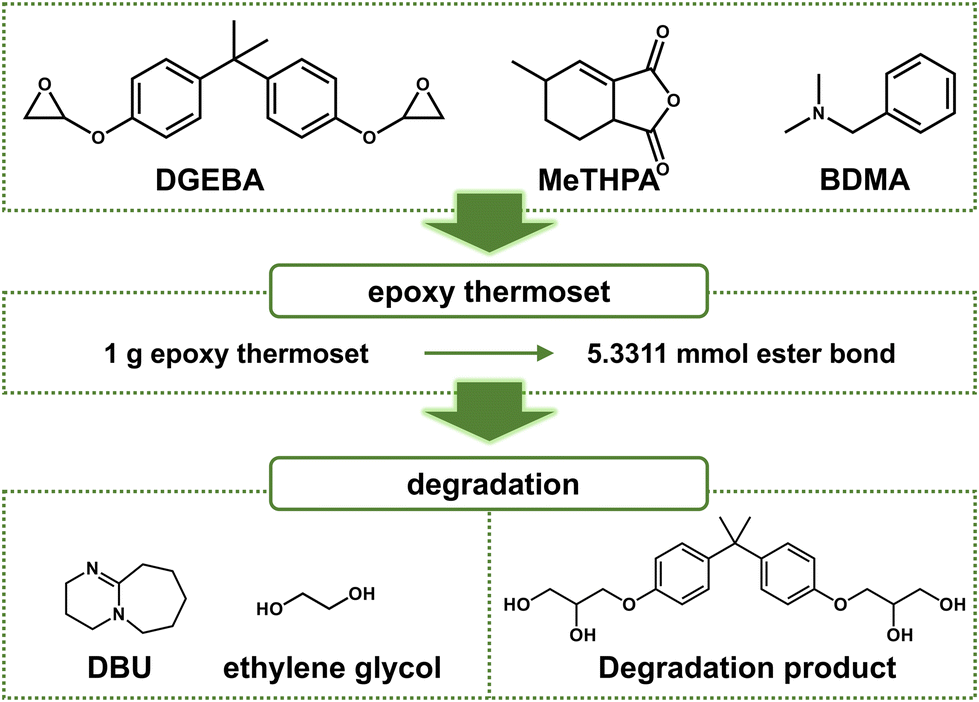 | ||
| Fig. 2 Chemical structure of reactants, catalysts, and recycled component of degradation products (schematic). | ||
General characterization
Differential scanning calorimetry (DSC) experiments were conducted on a Q2000 system (TA Instruments) with a scanning rate of 10 °C min−1 from −20 to 150 °C. 1H NMR spectroscopy was done using a JNM-ECA400 (400 MHz) spectrometer (JEOL, Tokyo, Japan) in CDCl3. 1H NMR (400 MHz, methanol-d4) δ 7.15–7.05 (m, 5H), 6.87–6.77 (m, 5H), 4.81 (d, J = 5.0 Hz, 7H), 4.55 (s, 1H), 4.23 (s, 1H), 4.14–4.01 (m, 1H), 4.00 (s, 2H), 3.93 (pt, J = 5.3, 3.1 Hz, 2H), 3.65 (dt, J = 9.0, 4.5 Hz, 1H), 3.61 (s, 1H), 3.33–3.26 (m, 2H), 1.63–1.55 (m, 7H), 1.29 (s, 1H).3. Results and discussion
Degradation properties of epoxy thermosets with different content of DBU
The content of DBU could obviously influence the degradation rate of epoxy thermosets, and we found that epoxy thermosets could be degraded more quickly as the temperature increased. Herein, to control the different content of DBU, we chose different mole ratios between DBU and ester bonds. As the epoxy thermosets were cured by 4.004 g (10 mmol) of DGEBA and 3.498 g (20 mmol) of MeTHPA, the total mass of epoxy thermosets was 7.503 g, and the total mole number of the ester bond was 40 mmol. Thus, 1 g of epoxy thermosets contained 5.331 mmol of ester bonds. The mole ratio between EG and ester bonds was 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, while the mole ratios between DBU and ester bonds was 0:1, 0.1:1, 0.3:1, and 0.5:1. Accordingly, we named the experimental groups at different content of DBU as group 0–1, group 0.1–1, group 0.3–1, and group 0.5–1, respectively. The degradation temperature was controlled at 120 °C.
:1, while the mole ratios between DBU and ester bonds was 0:1, 0.1:1, 0.3:1, and 0.5:1. Accordingly, we named the experimental groups at different content of DBU as group 0–1, group 0.1–1, group 0.3–1, and group 0.5–1, respectively. The degradation temperature was controlled at 120 °C.
Usually, DBU is employed as an accelerator in the polymerization of epoxy resins, and its content is relatively low. In this work we found that, as the content of DBU increased, it could act as an effective catalyst and efficiently degrade epoxy thermosets. Without DBU as the transesterification catalyst, the epoxy thermoset could barely be degraded (Fig. 3). Meanwhile, its weight increased slightly because it would swell at the EG solution. As the DBU/ester bond mole ratios increased, the epoxy thermoset degraded in DBU/EG solution more rapidly. In particular, the epoxy thermoset was totally dissolved in DBU/EG solution within 3 h as the DBU/ester bond mole ratio increased to 0.5 : 1.
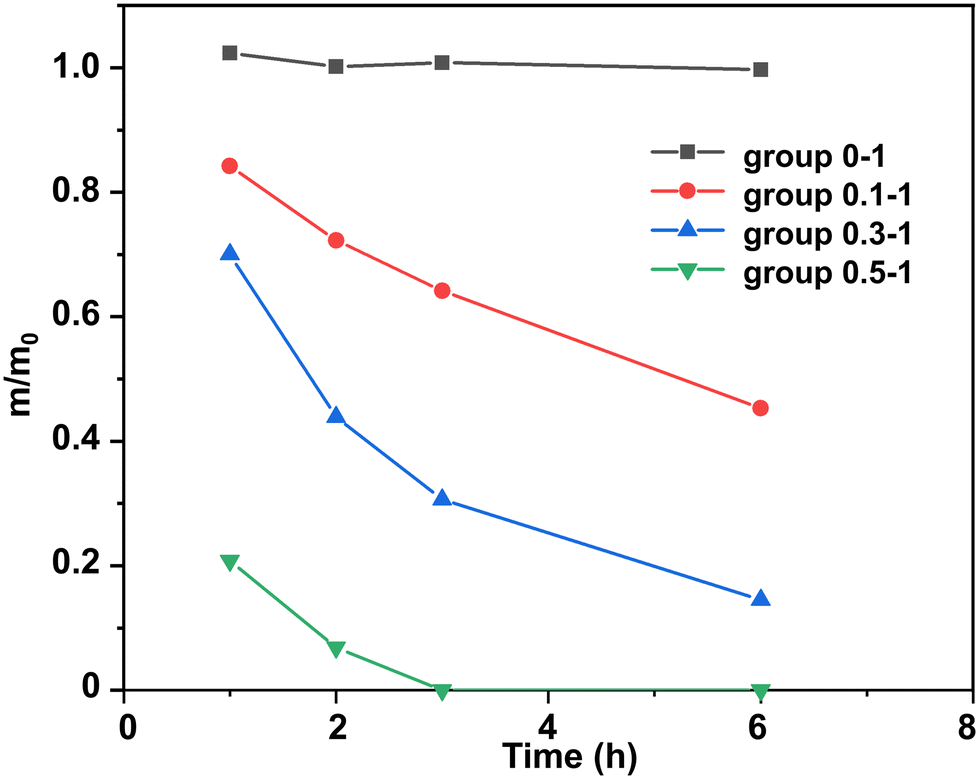 | ||
| Fig. 3 Normalized residual mass of epoxy in DBU/EG solution with different DBU/ester bond mole ratios as a function of time at 120 °C. | ||
We postulated a mechanism of degradation of epoxy resin catalyzed by DBU/EG solution (Fig. S1†). Owing to the hydrogen-bonding activation of DBU, the transesterification between EG and ester could be catalyzed.55 As a result, epoxy resin could be effectively degraded by DBU/EG solution.
Degradation properties of epoxy thermosets at different temperatures
The degradation rate of epoxy thermosets increased as the degradation temperature increased, and they could be totally depolymerized at 140 °C for 2 h. As aforementioned, epoxy thermosets could be efficiently dissolved in DBU/EG solution at 120 °C when the mole ratio between DBU and ester bonds was 0.5:1. Thus, we kept the DBU/ester bond mole ratio as 0.5:1 and investigated the influence of temperature on the degradation rate of epoxy thermosets.
The glass transition temperature (Tg) is a vital thermal property of polymers. DSC indicated that Tg of the epoxy thermoset was ∼103 °C (Fig. 4). At temperatures lower than the Tg, the chain segments of epoxy crosslinked networks are almost “frozen” and can hardly move. Accordingly, it would be difficult to degrade an epoxy thermoset at a temperature lower than its Tg. As the degradation temperature increases, EG would be more likely to enter the interior of an epoxy crosslinked network, thereby accelerating the degradation of epoxy thermosets. In addition, the rate of transesterification could increase at a higher temperature.
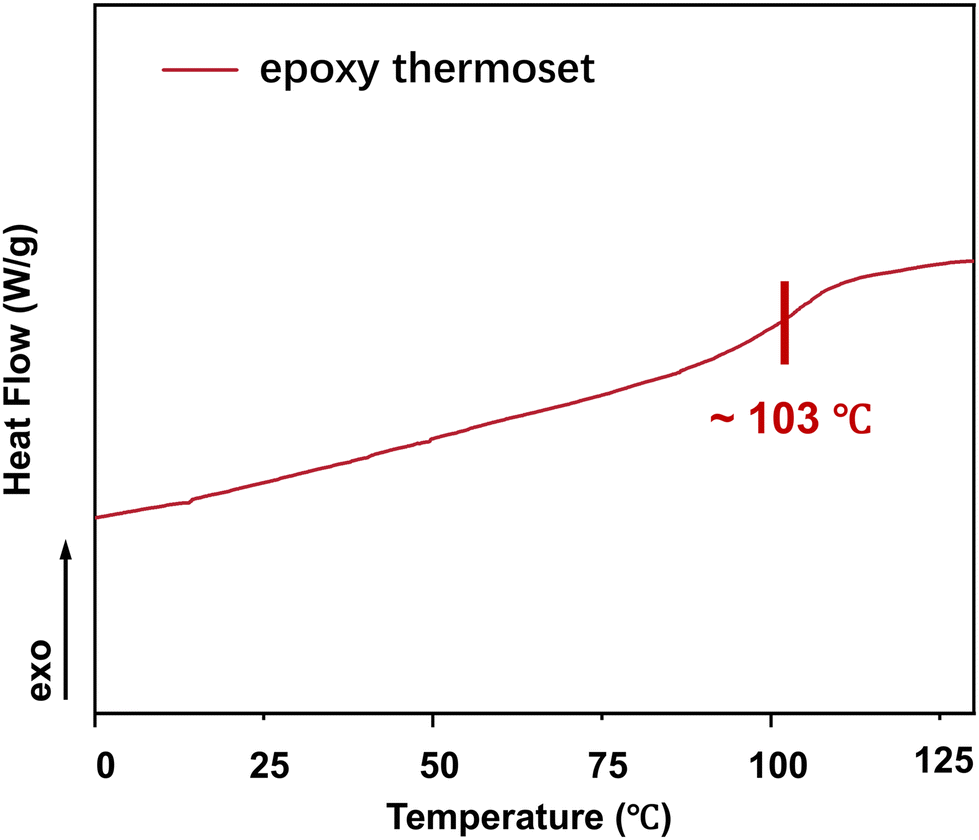 | ||
| Fig. 4 DSC curve of an epoxy thermoset. | ||
At a temperature lower than the Tg of epoxy thermosets (80 °C and 100 °C), the epoxy thermosets were dissolved at a much lower degradation rate and they could be hardly dissolved at 80 °C (Fig. 5). When the degradation temperature was higher than the Tg (120 °C and 140 °C), the epoxy thermosets were quickly degraded in 3 h. Notably, they were dissolved at 140 °C in <2 h, indicating that an increased temperature could effectively promote the degradation performance of epoxy thermosets.
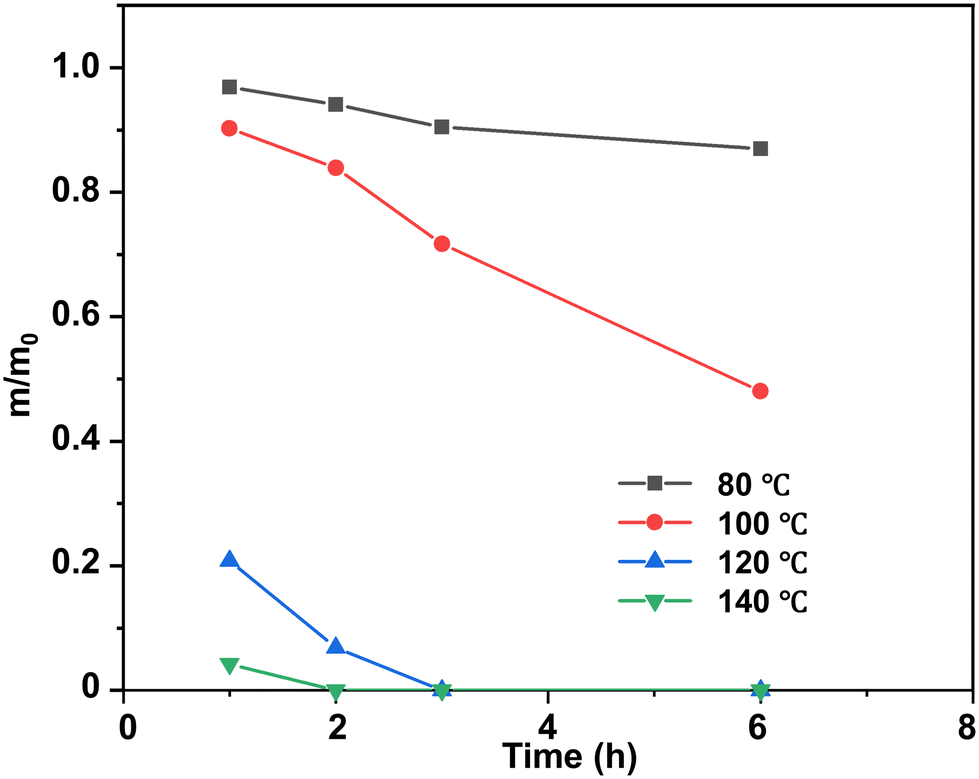 | ||
| Fig. 5 Normalized residual mass of epoxy in DBU/EG solution at different temperatures as a function of time at 120 °C. | ||
Recycling of depolymerized products
We further recycled the depolymerized epoxy products (mainly the DGEBA derivative tetra-alcohol). According to previous reports, the DGEBA derivative tetra-alcohol is not, but the anhydride derivative diester is, soluble in water.51,56 Thus, we could separate the recycled products in dissolution solution by precipitation in water (Fig. 6). After precipitation in water, the mixture became a suspension. By centrifugation, the DGEBA derivative tetra-alcohol could be precipitated. After drying at 100 °C for 12 h, the yield of DGEBA derivative tetra-alcohol was calculated to be ∼62%. | ||
| Fig. 6 Separation of DGEBA derivative tetra-alcohol. | ||
The chemical structure of the DGEBA derivative tetra-alcohol was analysed by NMR spectroscopy. Fig. 7 demonstrated that the calculated hydrogen ratio between BA (bisphenol A) and GI (glycidyl) was consistent with the structure of the DGEBA derivative tetra-alcohol, indicating that the DGEBA derivative tetra-alcohol could be recycled by simple precipitation in water. Moreover, a signal of anhydride (HP) linkages56 was not detected, nor were the signals of EG, BDMA or DBU (Fig. S2, S3 and S4†), thereby demonstrating that the DGEBA derivative tetra-alcohol could also be purified by precipitation in water.
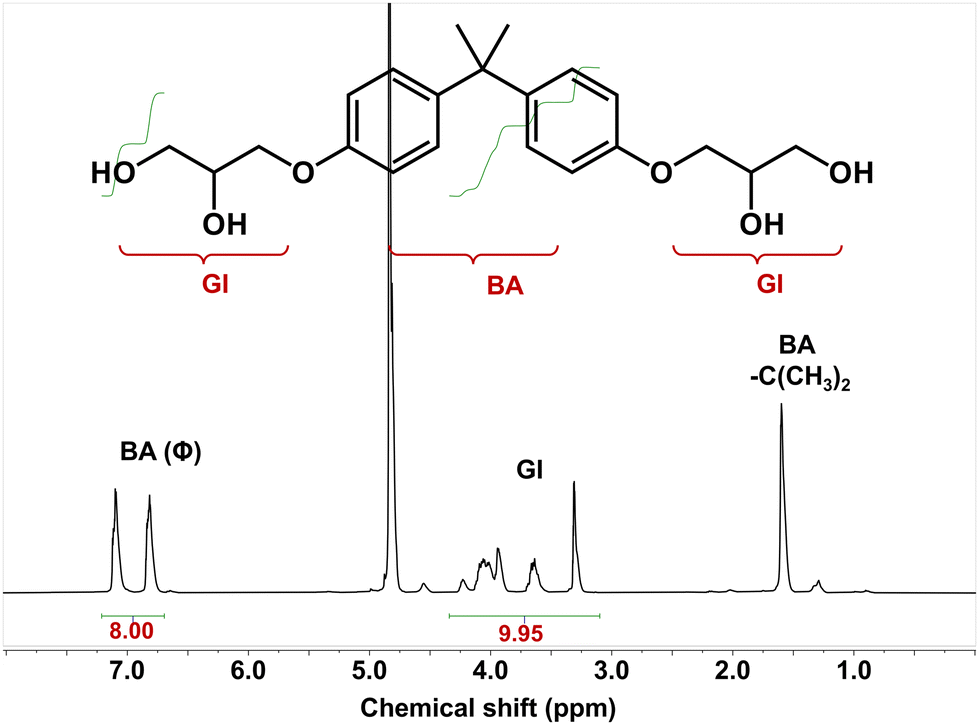 | ||
| Fig. 7 1H NMR spectrum of recycled DGEBA (BA: bisphenol A; GI: glycidyl). | ||
Degradation of commercial epoxy composites
Commercially available epoxy resin cured materials were also degraded using the method stated above. Resin-impregnated paper bushing is a key component to maintain the safe operation of dc transmission lines.57 Resin-impregnated paper bushing is mainly composed of epoxy resin and impregnated paper. Herein, we investigated whether the DBU/EG solution could degrade the epoxy composite efficiently.Results indicated that that epoxy resin of the resin-impregnated paper bushing could be almost degraded in DBU/EG solution at 140 °C in ∼3 h (Fig. 8). The other insoluble components of the epoxy composites remained, indicating that this strategy could be applied to separate and recycle epoxy resin of commercial epoxy composites, as well as other non-resin components.
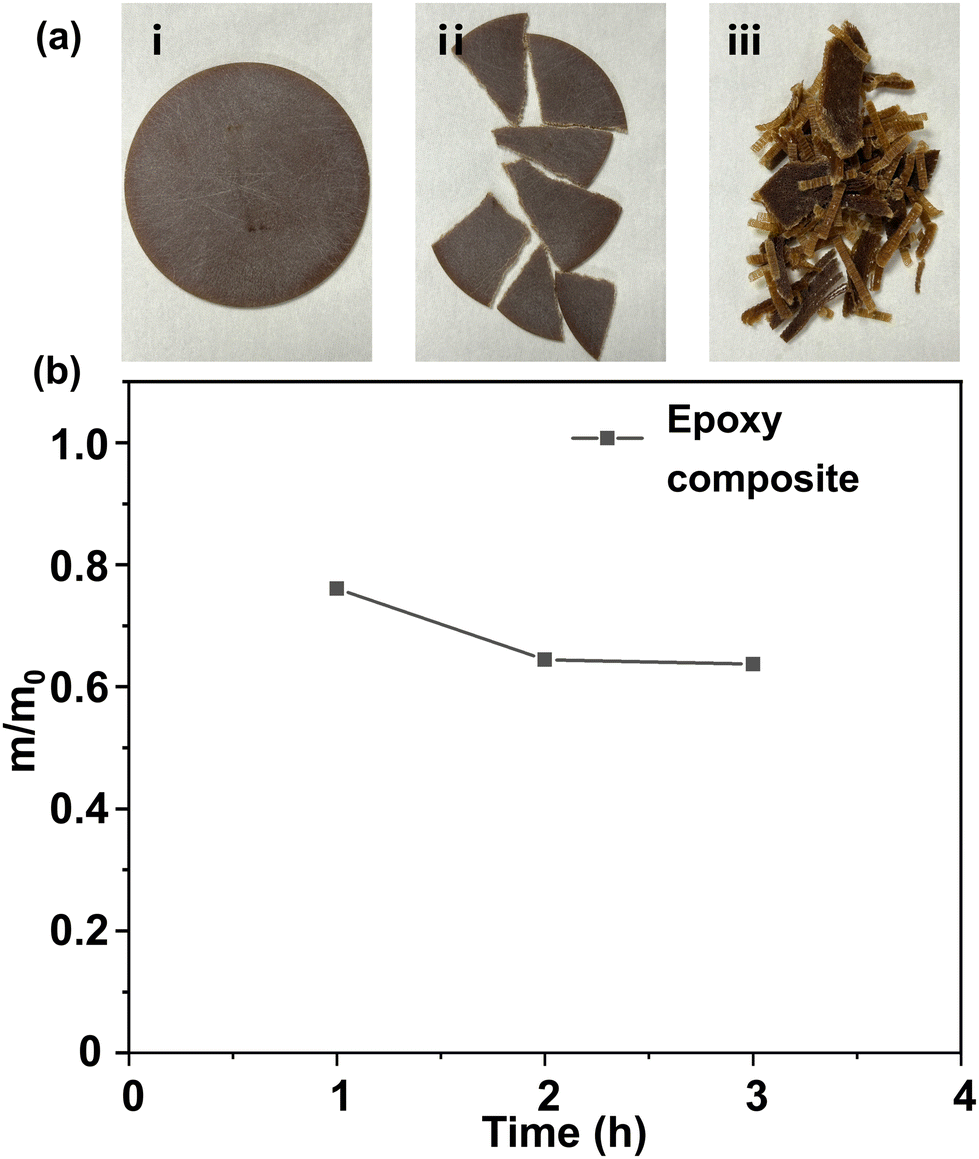 | ||
| Fig. 8 (a) Commercial epoxy composite sample before and after degradation. (i) Complete epoxy composite sample before degradation. (ii) Epoxy composite sample in pieces; (iii) epoxy composite sample after degradation. (b) Normalized residual mass of epoxy composite in DBU/EG solution as a function of time at 140 °C. | ||
4. Conclusions
The degradation and recycling of epoxy thermosets have been a great challenge in the field of polymer study. Even though many studies have been reported, some problems remain. For example, epoxy-based vitrimers can be recycled and reprocessed by hot-pressing, but transesterification catalysts (zinc acetate or TBD) are required, which do not exist in current epoxy thermoset wastes in the field of electrical engineering. Besides, many reported recyclable epoxy thermosets are amine-cured epoxy thermosets, while epoxy thermosets in electrical engineering are usually cured based on anhydrides. Moreover, crucial degradation conditions, an expensive transesterification catalyst, and complex synthesis are required for effective degradation, which increases the cost of recycling epoxy thermosets. Thus, it is vital to develop an efficient and economical degradation strategy.In this work, we proposed an efficient and economical degradation strategy based on DBU. By controlling the content of DBU and the degradation temperature, epoxy thermosets could be effectively dissolved in DBU/EG solution at 140 °C in <2 h (or at 120 °C in <3 h). The degradation product (DGEBA derivative tetra-alcohol) could be simply recycled by precipitation in water, which might be re-cured with DGEBA as the recycled epoxy resin or upcycled as polyurethane by reacting with isocyanate.58
However, to realize the closed-loop recycling of an anhydride-cured epoxy resin, two main problems must be resolved. First, DBU in the water/EG solution should also be recycled, which might be separated by a basic ion-exchange resin.59 Second, the MeTHPA derivative di-alcohol should also be reused. One possible solution to upcycle the MeTHPA derivative di-alcohol is reacting it with isocyanate to produce polyurethane.60
Herein, an anhydride-cured epoxy resin demonstrated an effective approach to degrade an epoxy resin in the field of electrical engineering. This strategy is applicable to other epoxy resin systems, and provides a general approach to recycling epoxy resin wastes.
Author contributions
Y. J. and Z. Y. conceived the central idea. Z. Y. performed the main experiments. S. Z. contributed to in the experiment. Z. Y. and Y. J. wrote the draft version of the manuscript. All authors participated in the discussion of the results. Y. W. reviewed and edited the manuscript. Y. J. arranged the funding and infrastructure for the project.Data availability
All the data needed to evaluate the conclusions reached from our study are present in this manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by National Key R&D Program of China (2023YFB2407100).References
- Y. Yang, Y. Xu, Y. Ji and Y. Wei, Prog. Mater. Sci., 2021, 120, 100710 CrossRef CAS.
- X. Zhao, Y. Long, S. Xu, X. Liu, L. Chen and Y.-Z. Wang, Mater. Today, 2023, 64, 72–97 CrossRef CAS.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS PubMed.
- M. Capelot, D. Montarnal, F. Tournilhac and L. Leibler, J. Am. Chem. Soc., 2012, 134, 7664–7667 CrossRef CAS PubMed.
- C. Li, B. Ju and S. Zhang, Cellulose, 2021, 28, 2879–2888 CrossRef CAS.
- Y. Yang, Z. Pei, Z. Li, Y. Wei and Y. Ji, J. Am. Chem. Soc., 2016, 138, 2118–2121 CrossRef CAS PubMed.
- Y. Yang, E. M. Terentjev, Y. Wei and Y. Ji, Nat. Commun., 2018, 9, 1906 CrossRef PubMed.
- K. Yu, P. Taynton, W. Zhang, M. L. Dunn and H. J. Qi, RSC Adv., 2014, 4, 10108–10117 RSC.
- I. Azcune and I. Odriozola, Eur. Polym. J., 2016, 84, 147–160 CrossRef CAS.
- J. Canadell, H. Goossens and B. Klumperman, Macromolecules, 2011, 44, 2536–2541 CrossRef CAS.
- M. Chen, L. Zhou, Y. Wu, X. Zhao and Y. Zhang, ACS Macro Lett., 2019, 8, 255–260 CrossRef CAS PubMed.
- Z. Huang, Y. Wang, J. Zhu, J. Yu and Z. Hu, Compos. Sci. Technol., 2018, 154, 18–27 CrossRef CAS.
- L. M. Johnson, E. Ledet, N. D. Huffman, S. L. Swarner, S. D. Shepherd, P. G. Durham and G. D. Rothrock, Polymer, 2015, 64, 84–92 CrossRef CAS.
- M. Pepels, I. Filot, B. Klumperman and H. Goossens, Polym. Chem., 2013, 4, 4955–4965 RSC.
- A. Ruiz de Luzuriaga, R. Martin, N. Markaide, A. Rekondo, G. Cabañero, J. Rodríguez and I. Odriozola, Mater. Horiz., 2016, 3, 241–247 RSC.
- A. Ruiz de Luzuriaga, J. M. Matxain, F. Ruipérez, R. Martin, J. M. Asua, G. Cabañero and I. Odriozola, J. Mater. Chem. C, 2016, 4, 6220–6223 RSC.
- A. Takahashi, T. Ohishi, R. Goseki and H. Otsuka, Polymer, 2016, 82, 319–326 CrossRef CAS.
- Y. Zhang, L. Yuan, G. Liang and A. Gu, Ind. Eng. Chem. Res., 2018, 57, 12397–12406 CrossRef CAS.
- F. Zhou, Z. Guo, W. Wang, X. Lei, B. Zhang, H. Zhang and Q. Zhang, Compos. Sci. Technol., 2018, 167, 79–85 CrossRef CAS.
- S. Wang, S. Ma, Q. Li, X. Xu, B. Wang, W. Yuan, S. Zhou, S. You and J. Zhu, Green Chem., 2019, 21, 1484–1497 RSC.
- X. Xu, S. Ma, J. Wu, J. Yang, B. Wang, S. Wang, Q. Li, J. Feng, S. You and J. Zhu, J. Mater. Chem. A, 2019, 7, 15420–15431 RSC.
- S. Zhao and M. M. Abu-Omar, Macromolecules, 2018, 51, 9816–9824 CrossRef CAS.
- H. Liu, H. Zhang, H. Wang, X. Huang, G. Huang and J. Wu, Chem. Eng. J., 2019, 368, 61–70 CrossRef CAS.
- Q. Liu, L. Jiang, Y. Zhao, Y. Wang and J. Lei, Macromol. Chem. Phys., 2019, 220, 1900149 CrossRef.
- X. Wu, X. Yang, R. Yu, X.-J. Zhao, Y. Zhang and W. Huang, J. Mater. Chem. A, 2018, 6, 10184–10188 RSC.
- W. Wu, H. Feng, L. Xie, A. Zhang, F. Liu, Z. Liu, N. Zheng and T. Xie, Nat. Sustain., 2024, 7, 804–811 CrossRef.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1702 CrossRef CAS PubMed.
- H. Hou, J. Yin and X. Jiang, Adv. Mater., 2016, 28, 9126–9132 CrossRef CAS PubMed.
- X. Kuang, G. Liu, X. Dong, X. Liu, J. Xu and D. Wang, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 2094–2103 CrossRef CAS.
- Q. Tian, Y. C. Yuan, M. Z. Rong and M. Q. Zhang, J. Mater. Chem., 2009, 19, 1289–1296 RSC.
- D. H. Turkenburg and H. R. Fischer, Polymer, 2015, 79, 187–194 CrossRef CAS.
- Z. Xu, Y. Liang, X. Ma, S. Chen, C. Yu, Y. Wang, D. Zhang and M. Miao, Nat. Sustain., 2020, 3, 29–34 CrossRef.
- A. Ahrens, A. Bonde, H. Sun, N. K. Wittig, H. C. D. Hammershøj, G. M. F. Batista, A. Sommerfeldt, S. Frølich, H. Birkedal and T. Skrydstrup, Nature, 2023, 617, 730–737 CrossRef CAS PubMed.
- X. Wu, P. Hartmann, D. Berne, M. De bruyn, F. Cuminet, Z. Wang, J. M. Zechner, A. D. Boese, V. Placet, S. Caillol and K. Barta, Science, 2024, 384, eadj9989 CrossRef CAS PubMed.
- T. Türel and Ž. Tomović, ACS Sustainable Chem. Eng., 2023, 11, 8308–8316 CrossRef.
- Q. Yu, X. Peng, Y. Wang, H. Geng, A. Xu, X. Zhang, W. Xu and D. Ye, Eur. Polym. J., 2019, 117, 55–63 CrossRef CAS.
- X. Liang, G. Chen, I. M. Lei, P. Zhang, Z. Wang, X. Chen, M. Lu, J. Zhang, Z. Wang, T. Sun, Y. Lan and J. Liu, Adv. Mater., 2022, 35, 2107106 CrossRef PubMed.
- X. Liu, L. Liang, M. Lu, X. Song, H. Liu and G. Chen, Polymer, 2020, 210, 123030 CrossRef CAS.
- H. Memon, H. Liu, M. A. Rashid, L. Chen, Q. Jiang, L. Zhang, Y. Wei, W. Liu and Y. Qiu, Macromolecules, 2020, 53, 621–630 CrossRef CAS.
- Z. Fang, S. Nikafshar, E. L. Hegg and M. Nejad, ACS Sustainable Chem. Eng., 2020, 8, 9095–9103 CrossRef CAS.
- V.-D. Mai, S.-R. Shin, D.-S. Lee and I. Kang, Polymers, 2019, 11, 293 CrossRef PubMed.
- X. Su, Z. Zhou, J. Liu, J. Luo and R. Liu, Eur. Polym. J., 2020, 140, 110053 CrossRef CAS.
- W. Xie, S. Huang, S. Liu and J. Zhao, Chem. Eng. J., 2021, 404, 126598 CrossRef CAS.
- K. Li and Z. Xu, Environ. Sci. Technol., 2015, 49, 1761–1767 CrossRef CAS PubMed.
- I. Okajima, M. Hiramatsu, Y. Shimamura, T. Awaya and T. Sako, J. Supercrit. Fluids, 2014, 91, 68–76 CrossRef CAS.
- C. Chung, H. Jiang, C. Luo and K. Yu, Mater. Today Sustain., 2024, 26, 100792 Search PubMed.
- Q. Shi, K. Yu, M. L. Dunn, T. Wang and H. J. Qi, Macromolecules, 2016, 49, 5527–5537 CrossRef CAS.
- Q. Shi, K. Yu, X. Kuang, X. Mu, C. K. Dunn, M. L. Dunn, T. Wang and H. Jerry Qi, Mater. Horiz., 2017, 4, 598–607 RSC.
- K. Yu, Q. Shi, M. L. Dunn, T. Wang and H. J. Qi, Adv. Funct. Mater., 2016, 26, 6098–6106 CrossRef CAS.
- Q. Mu, L. An, Z. Hu and X. Kuang, Polym. Degrad. Stab., 2022, 199, 109895 CrossRef CAS.
- X. Kuang, Q. Shi, Y. Zhou, Z. Zhao, T. Wang and H. J. Qi, RSC Adv., 2018, 8, 1493–1502 RSC.
- J. Tan, C. Li, K. De Bruycker, G. Zhang, J. Gu and Q. Zhang, RSC Adv., 2017, 7, 51763–51772 RSC.
- A. Bifulco, D. Parida, K. A. Salmeia, R. Nazir, S. Lehner, R. Stämpfli, H. Markus, G. Malucelli, F. Branda and S. Gaan, Mater. Des., 2020, 193, 108862 CrossRef CAS.
- W. G. Kim, H. G. Yoon and J. Y. Lee, J. Appl. Polym. Sci., 2001, 81, 2711–2720 CrossRef CAS.
- N. J. Sherck, H. C. Kim and Y.-Y. Won, Macromolecules, 2016, 49, 4699–4713 CrossRef CAS PubMed.
- X. Kuang, Y. Zhou, Q. Shi, T. Wang and H. J. Qi, ACS Sustainable Chem. Eng., 2018, 6, 9189–9197 CrossRef CAS.
- S. Shi, H. Tian, G. Xie, Q. Wang, T. Xie, X. Pang, G. Yang, P. Liu and Z. Peng, IEEE Trans. Dielectr. Electr. Insul., 2024, 31, 2776–2784 Search PubMed.
- R. W. Clarke, E. G. Rognerud, A. Puente-Urbina, D. Barnes, P. Murdy, M. L. McGraw, J. M. Newkirk, R. Beach, J. A. Wrubel, L. J. Hamernik, K. A. Chism, A. L. Baer, G. T. Beckham, R. E. Murray and N. A. Rorrer, Science, 2024, 385, 854–860 CrossRef CAS PubMed.
- P. R. Christensen, A. M. Scheuermann, K. E. Loeffler and B. A. Helms, Nat. Chem., 2019, 11, 442–448 CrossRef CAS PubMed.
- X. Zhao, R. Du, X. Chen, X. Wei, Y. Long, X. Liu, L. Chen, S. Xu and Y.-Z. Wang, J. Environ. Chem. Eng., 2023, 11, 109363 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py01137f |
| This journal is © The Royal Society of Chemistry 2024 |