
Association of Zn- and Mg-based chain transfer agents in coordinative chain transfer polymerizations of olefins for enhanced control and activity†
Ariane Desgranges‡
ab,
Ludmilla Verrieux§
ac,
Victor Lancenetab,
Nicolas Baulu‡ab,
François Jean-Baptiste-dit-Dominiquead,
Robert Ngoad,
Franck D'Agosto *ab,
Marie-Eve L. Perrin*ac and
Christophe Boisson*ab
*ab,
Marie-Eve L. Perrin*ac and
Christophe Boisson*ab
aChemistLab, Michelin CP2M ICBMS joint Laboratory, 69616, Villeurbanne, France. E-mail: franck.dagosto@univ-lyon1.fr; Marie-Eve.Perrin@univ-lyon1.fr; christophe.boisson@univ-lyon1.fr
bUniversite Claude Bernard Lyon 1, CPE-Lyon, CNRS UMR 5128, Laboratoire CP2M, Equipe PCM, 69616 Villeurbanne, France
cUniversite Claude Bernard Lyon I, CNRS, CPE-Lyon, UMR 5246, ICBMS, 1 rue Victor Grignard, F-69622 Villeurbanne cedex, France
dManufacture Michelin, 23 place Carmes Déchaux, F-63000 Clermont-Ferrand, France
First published on 12th December 2024
Abstract
Based on DFT-level computational studies, ZnEt2 was implemented as a chain transfer agent (CTA) to promote the coordinative chain transfer (co)polymerization of ethylene for the first time with a metallocene complex of Nd. The use of ZnEt2 in combination with Mg(nBu)1.5(nOct)0.5 compared to the use of Mg(nBu)1.5(nOct)0.5 alone, improves several key aspects of the polymerization process. When ethylene is polymerized, an increase of catalytic activities is observed and narrower molar mass distributions are obtained due to reduced β-H transfer and faster reversible chain transfer reactions. In copolymerization of ethylene with butadiene, the presence of ZnEt2 has no impact on the polymerization process in terms of polymerization kinetics and microstructure of the final copolymer. Nevertheless, it acts as an excellent CTA. Using ZnEt2 in combination with MesMgBr rather than Mg(nBu)1.5(nOct)0.5 enables selective chain transfer between neodymium and zinc and promotes a nearly quantitative chain-end functionalization with acyl chloride.
Introduction
Polyolefins represent the most significant family of polymers in the industry. They are produced in large volumes and at low cost through radical polymerization or coordination catalysis and serve a broad spectrum of applications. A long-standing challenge in this field has been achieving the control of the chain growth to allow the formation of more complex architectures such as block copolymers. Both radical and coordination polymerizations can achieve this by utilizing systems leading to controlled and/or living polymerizations. For instance, recent advancements have applied reversible deactivation radical polymerizations and controlled coordination polymerizations to successfully control the polymerization of ethylene. Although not applied to the high-pressure and high-temperature gas-phase process used for producing low-density polyethylene (LDPE), recent studies have demonstrated that reversible deactivation radical polymerization of ethylene is achievable.1–4 Besides, since the early 1990s, the development of coordinative chain transfer polymerization (CCTP)5–8 has garnered increasing interest from both academia and industry. For example, Dow Chemical marketed olefin block copolymers (OBC) produced by this approach.9 CCTP is a polymerization catalysis based on a degenerative transfer mediated by an organometallic compound such as AlR3, MgR2, ZnR2 that acts as a reversible chain transfer agent (CTA).10 Polymer chains grow on the catalyst and are reversibly transferred to the CTA, where they become intermittently dormant. In the absence of β-H elimination and with an excess of CTA relative to the catalyst, the polymerization process becomes controlled—meaning the number of polymer chains is fixed and directly proportional to the amount of CTA. As a result, the degree of polymerization is predictable based on the ratio of monomer consumed to the initial amount of CTA used. Since the chains produced by CCTP remain connected to a metal center throughout the polymerization, this method not only allows for the production of precision polyolefins,11–13 but also facilitates the introduction of functional groups at their chain ends14 and gives access to diverse macromolecular architectures.15–20 Interestingly, the CTA itself can be active under polymerization conditions. In the case of (co)polymerization of 1,3-dienes and styrene mediated by lanthanide borohydride complexes, it is possible to adjust the microstructure of the (co)polymers by regulating the amount of MgR2.21–23 In the same works, Zinck et al. have also shown the beneficial use of ternary systems based on the association of MgR2 and AlR3 to control the stereospecificity of 1,3-diene polymerization by CCTP.Neodymium-based metallocenes enable coordinative chain transfer (co)polymerization (CCT(co)P) of ethylene and butadiene using MgR2 as the CTA such as Mg(nBu)1.5(nOct)0.5 (BOMAG) (Scheme 1).24–26 The unique properties of MgR2 in this CCTP process has been elucidated through a theoretical mechanistic study.25 ZnEt2 is commonly used in association with a large panel of olefin polymerization catalysts resulting from the combination of Fe-, Hf- or Zr-based complexes with borate activators or methylaluminoxane (MAO).7,8 In these systems, the underlying reaction mechanisms remain intricate. In addition, the group of Sita suggested that ZnEt2 can mediate the chain transfer between the active catalyst and the second co-catalyst.13
 | ||
| Scheme 1 Implementation of the CCTP of ethylene and CCTcoP of ethylene and butadiene mediated by binary or ternary Nd-based catalysts. [Nd] corresponds to the metal center and its ligands (Me2Si(C13H8)2), [Nd]X = ({(Me2Si(C13H8)2)Nd(μ-BH4)[(μ-BH4)Li(THF)]}2). | ||
In light of these results, developing more efficient and versatile conditions for Nd-based CCTP is of significant interest, especially when compared to those achieved with MgR2 as a lone CTA. However, a major challenge in using ZnEt2 as a CTA with Nd-based pre-catalysts is its inability to activate the Nd precursor.27
In this communication, we circumvent this issue by combining ZnEt2 and MgR2 or RMgX with ({(Me2Si(C13H8)2)Nd(μ-BH4)[(μ-BH4)Li(THF)]}2) Nd-based complex 1 (Scheme S1†). Building on our previous expertise with MgR2 as an efficient CTA for the CCTP of ethylene and the CCT(co)P of ethylene and butadiene using a Nd-based catalyst,28–30 and supported by preliminary DFT-level computational investigations, we evaluated the potential of ZnEt2 as a CTA in MgR2-mediated CCTP systems. The resulting ternary Nd/Mg/Zn catalysts were then investigated for the polymerization of ethylene and its copolymerization with butadiene.
Results and discussion
Insights from molecular modelling
Before implementing the experimental design for the ternary catalytic system 1/BOMAG/ZnEt2, we first obtained structural and mechanistic insights through DFT-level molecular modelling. This study presents several challenges, including the large size of the system (>100 atoms), the presence of three different metals, and multiple polymer chains. To address these challenges, we relied on our previous theoretical modelling strategies for speciation of heteropolymetallic complexes and the exploration of chain exchange reactions.25,31 To reduce computational costs, BOMAG was modelled using Mg(nBu)2 clusters, with the rhombohedral tetramer being selected as the most stable configuration.25 Regarding ZnEt2, optimization of a dimer consistently leads to its dissociation into monomers. Since ZnEt2 is purchased and used in a hydrocarbon solution, the most stable form present in this context is considered to be the monomer. The fully detailed computational method is described in the ESI.†To validate the computational method, the thermodynamics of the formation of alkylated monometallic species [Nd]-R (R = nBu or Et, [Nd] = Me2Si(C13H8)2Nd) from the Nd precursor Me2Si(C13H8)2Nd(μ-Cl)2Li(OEt2)2 (NdClLiCl) was computed for both Zn and Mg alkylating agent (Scheme 2). This precursor has been taken as both as model and analog of 1. While the activation reaction of NdClLiCl is endergonic by 6 kcal mol−1 in the case of Mg(nBu)2, it is endergonic by 37 kcal mol−1 in the case of ZnEt2. This is in line with experimental results for this specific system,27 but also matches with the activation conditions reported for other polymerization catalytic systems.32,33
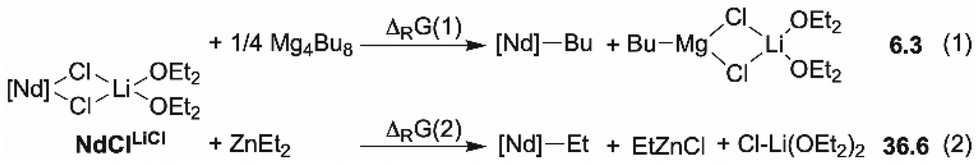 | ||
| Scheme 2 Thermodynamics (ΔRG, kcal mol−1) of [Nd]-R species formation (R = nBu or Et) as a function of the alkylating agent. [Nd] corresponds to the metal center and its Me2Si(C13H8)2 ligand. Gaussian 09, B3PW91/D3BJ(SP), SMD(TolH), 298 K, 1 atm. | ||
As suggested in the literature and mentioned above, ZnEt2 may mediate the chain transfer between the catalyst and a second CTA, namely AlR3.13 This mechanism hypothesis has been considered and will be compared to the chain transfer mechanism previously reported for the CCTP of ethylene mediated by BOMAG and [Nd]-nBu.25 Of note, the latter revealed that the chain exchange reaction proceeds via a two-step association/dissociation transmetalation in which the intermediate heterotrimetallic complex [Nd]-(μ-nBu)2-Mg2nBu3 (NdMg2) is the resting state of the polymerization catalyst.25
Considering now the addition of ZnEt2 as a third component of the catalytic system, the speciation of thermodynamically accessible complexes has been performed. Relative to NdMg2 and ZnEt2, a set of complexes can be formed with an endergonicity ranging from 1 to 10 kcal mol−1 (Scheme 3). As a general trend, relative to separated NdMg2 and ZnEt2, the formation of complexes in which zinc is the β position to neodymium is endergonic by at least 10 kcal mol−1. Interestingly, the formation the heterobimetallic dimer NdZn is almost as endergonic as the release of the NdBu complex from which monomer coordination–insertion mechanism takes place.25
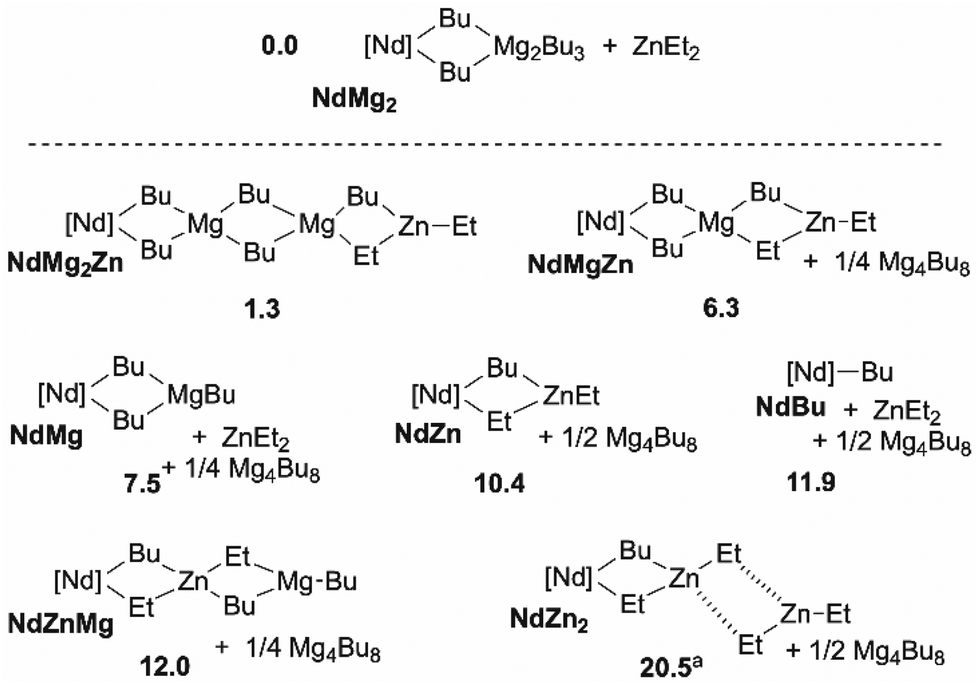 | ||
| Scheme 3 Speciation of the alkylated species yielded from [Nd]-nBu, Mg4nBu8 and ZnEt2. Only the most stable species are shown, the fully detailed speciation is provided in ESI.† ΔG in kcal mol−1. aIn NdZn2, interaction between the ZnEt2 fragments is weak, the endergonicity results from the entropy cost of the association of free and separated ZnEt2. Gaussian 09, B3PW91/D3BJ(SP), SMD(TolH), 298 K, 1 atm. | ||
From a kinetic point of view, the Gibbs energy barrier for ethylene insertion in the Nd–C bond of the NdBu complex is computed to 10.7 kcal mol−1. Concerning, the Nd/Mg binary system, in presence of Mg4Bu8, the overall Gibbs energy barrier is raised to 22.6 kcal mol−1 relative to NdMg2 and ethylene due to the formation of the NdMg2 dormant complex.25 In the Nd/Mg/Zn ternary system, the presence of zinc leads to the formation of the bimetallic species NdZn as an intermediate that can become the most abundant Nd-based complex along with NdMg if ZnEt2 is added in large excess relative to BOMAG, the amount of NdMg2 becoming small. NdZn and NdMg becoming the most abundant resting state of the catalyst, the overall Gibbs energy barrier for ethylene insertion is decreased from 22.6 kcal mol−1 in a window ranging from 12.2 to 15.1 kcal mol−1. Thereof, adding an excess of ZnEt2 to the catalytic mixture would lead to a significant boost in catalytic activity. Regarding the influence on the chain transfer, we were unable to adequately converge the geometry of the transition states(s) as it relies on association/dissociation processes, coupled with re-orientation of the chain end carbanion doublet by rotations.25,26 However, an energy barrier of less than 5 kcal mol−1 can be roughly estimated leading to an overall fast exchange between Nd and Zn, this barrier is thereof lower than the one computed between Nd and Mg by 8 kcal mol−1,25 thus enabling a much faster chain shuttling. Finally, as ZnR2 and presumably Zn(polymer)2, do not aggregate, a more straightforward chain transfer between Nd and Zn may be inferred.
Polymerization of ethylene and copolymerization of ethylene with butadiene
In a first blank experiment, ethylene was polymerized with the 1/BOMAG catalyst ([Nd]/[Mg] = 1/80, run 1, Table 1). In agreement with our previous works,25 the catalyst meets the requirement of CCTP but the chain exchange rate is low compared with the propagation rate as indicated by a dispersity value of 2.1. This has been attributed to a high-energy barrier for dissociation of the heterobimetallic species formed between the active species and BOMAG.25 When the ternary system 1/BOMAG/ZnEt2 ([Nd]/[Mg]/[Zn] = 1/5/75) was used (run 2, Table 1), complex 1 was first activated by BOMAG in a separate vial and the mixture was transferred in a solution containing ZnEt2 before starting the polymerization. A strong decrease of Đ from 2.1 to 1.1 (Table 1 and Fig. 1) is observed and the content of vinyl-terminated chains resulting from β-H elimination drops from 10 to 2%.
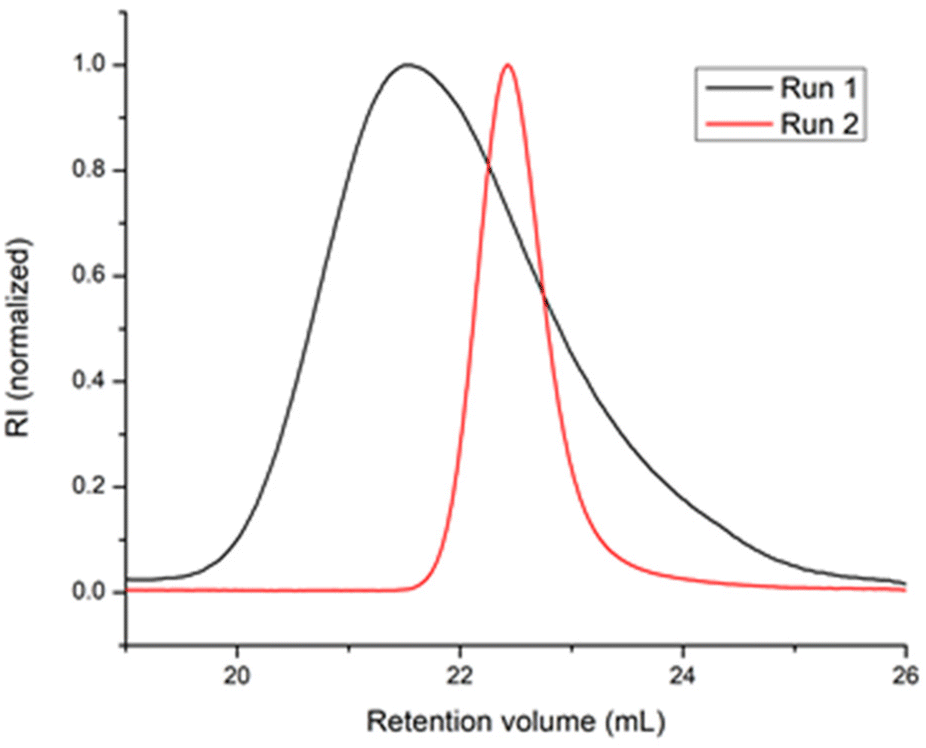 | ||
| Fig. 1 SEC traces of polyethylene obtained with 1/BOMAG (run 1, Table 1) and 1/BOMAG/ZnEt2 (run 2, Table 1) catalysts. | ||
We also observe a good agreement between the theoretical number-average molar mass (Mtheon) calculated considering that ZnEt2 is concomitantly acting as chain transfer agent with BOMAG and the experimental Mn confirming that ZnEt2 definitively acts as a CTA in this CCTP.
In addition, a sharp increase in activity from 200 to 3700 kg mol−1 h−1 is obtained, in good agreement with the modelling study. Indeed, the formation of a heterobimetallic NdZn (Scheme 3) formed between the active species Me2Si(C13H8)2NdR and ZnEt2 is exergonic by only 1.5 kcal mol−1, whereas the heterobimetallic NdMg2 (Scheme 3) species formed with BOMAG is exergonic by 11.9 kcal mol−1. Consequently, the overall energy barrier of the ethylene insertion is lowered in the presence of ZnEt2 and chain exchange is much faster between Nd and zinc than between Nd and magnesium. However, at this stage of our study, chain exchange between magnesium and zinc is not demonstrated. To evaluate a potential chain transfer between magnesium and zinc, a model reaction was performed in an NMR tube. The 1H NMR analysis of BOMAG (Mg(nBu)1.5(nOct)0.5) and ZnEt2 was carried out (Fig. S1 and S2†), followed by that of a BOMAG/ZnEt2 mixture ([Mg]/[Zn] = 0.8, Fig. S3–S7†). Three new signals are observed: two triplets at 0.23 and 0.27 ppm respectively, and a quadruplet at 0.13 ppm (Fig. S4†). An assignment of the two triplets is better performed when the NMR analysis is carried with a spectrometer operating at 500 MHz (Fig. S5†). As the signals are as well defined as those of ZnEt2, it is reasonable to assume the formation of ZnEtR (R = nBu and nOct respectively) and to confirm an exchange of alkyl groups between zinc and magnesium under these conditions.
| Run | [Nd]/[Mg]/[Zn] | Yield | Time | Activity | Mtheon | Mn′![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
Đ | PE-vinylb |
|---|---|---|---|---|---|---|---|---|
| g | min | kg mol−1 h−1 | g mol−1 | g mol−1 | % | |||
| [Nd] = 20 μM (runs 3 and 4), 62.5 μM (runs 5 to 9), 200 mL toluene, 4 bar, 90 °C (run 8 at 80 °C), Mtheon = yield/2*(nMg + nZn).a Determined by HT-SEC with PE standards.b Determined by 1H NMR.c Total deactivation of the catalytic system at the end of polymerization. | ||||||||
| 3 | 1/255/0 | 3.9 | 207 | 290 | 1950 | 2000 | 2.2 | 17 |
| 4 | 1/15/240 | 4.8 | 8 | 9200 | 2400 | 1900 | 1.2 | 5 |
| 5 | 1/60/20 | 4.1 | 48 | 410 | 2050 | 1400 | 1.6 | 29 |
| 6 | 1/40/40 | 4.9 | 14 | 1700 | 2450 | 2000 | 1.4 | 17 |
| 7 | 1/20/60 | 5.3 | 6 | 4600 | 2650 | 1800 | 1.4 | 5 |
| 8 | 1/20/60 | 9.7 | 12 | 3100 | 4850 | 4900 | 1.2 | 11 |
| 9 | 1/5/75 | 3.6 | 7 | 2500c | 1800 | 1400 | 1.2 | 5 |
In the present case, ZnEt2 increases the quantity of active neodymium as shown by the computational investigation. The good agreement between theoretical and experimental Mn indicates that polymerizations are controlled according to CCTP. The final PE show narrow molar mass distributions and the content of PE-vinyl chains decreases when the amount of ZnEt2 increases. Decreasing the [Mg]/[Zn] ratio (runs 5–7, Table 2) leads to an increase in activity. When the lowest [Mg] is used (run 9, Table 2), a sudden deactivation of the catalyst is observed at the end of polymerization (Fig. S8†). The deactivation process appears to be linked to the increase in [Nd]-P active species in the presence of ZnEt2. This deactivation might occur from inter- or intra-molecular reactions between the active species, reactions that are not identified yet even by molecular modelling. In summary, the dialkylmagnesium compound not only acts as an alkylating agent for 1 but also brings stability to the catalytic system via a mechanism that still not understood at the molecular level. Building on these promising results on the PE chain transfer ability when additional ZnEt2 is employed, the combination of 1 and mixed CTA based on Mg and Zn was further evaluated in the coordination chain transfer copolymerization (CCT(co)P) of ethylene and butadiene for the synthesis of ethylene butadiene rubber (EBR).
Copolymerizations of ethylene and butadiene were then carried out with a ([Mg] + [Zn])/[Nd] ratio fixed to 10 and targeting 15 g of polymer, corresponding under these conditions to a theoretical Mtheon of 15000 g mol−1 (Table 3). Very similar molar masses (between 12500 and 14200 g mol−1) consistent with the targeted value were obtained for various [Mg]/[Zn] ratios showing again here that both BOMAG and ZnEt2 are acting as CTAs.
| Run | [Nd]/[Mg]/[Zn] | Yield | Time | Activity | Mna | Đ |
|---|---|---|---|---|---|---|
| g | min | kg mol−1 h−1 | g mol−1 | |||
| [Nd] = 250 μM, 4 bar (ethylene/butadiene = 80/20), 90 °C, 200 mL toluene.a Determined by SEC in THF using an universal calibration.b Low activity then total deactivation of the catalytic system after 35 minutes of polymerization. | ||||||
| 10 | 1/10/0 | 14.9 | 81 | 220 | 13800 |
1.4 |
| 11 | 1/8/2 | 15.4 | 87 | 210 | 14200 |
1.5 |
| 12 | 1/5/5 | 14.8 | 94 | 190 | 13800 |
1.5 |
| 13 | 1/4/6 | 14.8 | 76 | 230 | 12500 |
1.4 |
| 14b | 1/2/8 | 1.6 | 35 | 60b | — | — |
For CCTcoP of ethylene and butadiene, the number of polymer chains is fixed by the number of magnesium-alkyl and zinc-alkyl bonds. For both binary and ternary catalytic systems, similar molar mass distributions are obtained (Fig. S9†), but contrary to ethylene homopolymerization, the use of ZnEt2 as co-CTA in CCTcoP of ethylene and butadiene does not raise the catalytic activity nor narrow the molar mass distribution that respectively remain around 200 kg mol−1 h−1 and 1.4 (Table 3). The microstructure of the EBR formed in the presence of ZnEt2 is similar to the one of the EBR obtained with BOMAG alone (Table 4). This suggest that the same active species are formed in both systems. Indeed, whereas the overall energy barrier for ethylene insertion includes the dissociation of dormant heterobimetallic species into the active species, the kinetics of the copolymerization of ethylene and butadiene is controlled by monomer insertions into the Nd-allyl and the Nd-(vinylcyclohexyl)methylene active chain ends, as demonstrated by joint experimental and computational mechanism investigation.26,31 The nature of the CTA has thus very little impact on the kinetics.
| Run | [Nd]/[Mg]/[Zn] | E/B | Vinyl (%) | trans-1,4% | 1,2-Cyclohexyl (%) | Ethylenea (%) |
|---|---|---|---|---|---|---|
| a Except those included in cyclic units. | ||||||
| 10 | 1/10/0 | 80.0/20.0 | 7.7 | 6.0 | 10.4 | 75.9 |
| 11 | 1/8/2 | 81.2/18.8 | 6.3 | 5.9 | 10.7 | 77.2 |
| 12 | 1/5/5 | 80.7/19.3 | 6.6 | 6.2 | 10.7 | 76.6 |
| 13 | 1/4/6 | 81.0/19.0 | 6.4 | 6.0 | 10.7 | 76.9 |
| 17 | 1/16/10 | 79.7/20.3 | 8.6 | 5.8 | 9.9 | 75.8 |
As in the case of ethylene polymerization (run 9, Table 2), the catalytic system deactivates at low [Mg]/[Nd] ratios (run 14, Table 3). In CCTcoP case, since the initial activity for this run is low (60 kg mol−1 h−1, Table 3), the deactivation of the catalytic system may result from the inefficient activation when using only 2 equivalents of magnesium relative to neodymium.
| Run | [Nd]/[Mg]/[Zn] | Yield | Time | Activity | Mtheon | Mnc |
Đ | PE-vinyld |
PE(EBR)-C(O)C6H4X |
|---|---|---|---|---|---|---|---|---|---|
| g | min | kg mol−1 h−1 | g mol−1 | g mol−1 | % | % | |||
| [Nd] = 62.5 μM, 4 bar, 80 °C, 200 mL toluene.a Reference test with BOMAG.b [Nd] = 250 μM, 4 bar (ethylene/butadiene feed ration: 80/20), 80 °C, 200 mL toluene.c Determined by HT-SEC with PE standards for PE samples and by SEC-THF with universal calibration for the EBR sample.d Determined by 1H NMR.e Partial discoloration of the solution, loss of exotherm and decrease of ethylene consumption.f Functionalization yield = MSECn/MRMNn. | |||||||||
| 1a | 1/80/0 | 3.6 | 82 | 200 | 1800 | 2400 | 2.1 | 10 | — |
| 15 | 1/20/80 | 4.2 | 31 | 690 | 2010 | 2000 | 1.1 | 3 | 79d |
| 16 | 1/8/80 | 3.5 | 35 | 500e | 1750 | 1700 | 1.1 | 4 | 86d |
| 17b | 1/16/10 | 10.3 | 174 | 70 | 10000 |
16400 |
1.3 | — | 34f |
Here again, 4 g of PE was targeted, corresponding to a theoretical Mn of 2000 g mol−1 considering that ZnEt2 governs the number of chains formed, i.e. all chains were initiated by an ethyl group (Table 5). Whatever the reaction conditions used, polymerization takes place. Considering that ZnEt2 does not alkylate 1 and that the Nd-Mes was previously shown not to initiate ethylene polymerization,40 initiation of the polymerization presumably proceeds via an arylation of the Nd center by MesMgBr followed by a transmetalation between Nd-Mes and Zn–Et bonds. This was indeed confirmed by a good agreement between experimental and theoretical Mn. The reversibility of the chain transfer between Nd and Zn centers is also confirmed by lower the dispersity values obtained (Đ < 1.3) compared to the one obtained when the experiment was conducted with BOMAG alone (Đ = 2.1, run 1, Table 5).
Concerning chain-end functionalization, preliminary results gathered on the reactivity of MgPE2 prepared with (C5Me5)2NdCl2Li(OEt)2/BOMAG binary catalyst reveals that functionalization upon the reaction with two equivalents of p-toluoyl chloride does not provide the expected ketone functionalized PE chains. Instead, a mixture of secondary and tertiary alcohols (PE-CH(OH)-(C6H4Me): 25%, PE2-C(OH)-(C6H4Me): 27%), PE-vinyl (30%) et PE-CH3 (16%) (Scheme S2 and Fig. S10†) is obtained. Using 1/MesMgBr/ZnEt2 ternary system, chain-end functionalization with two equivalents of p-toluoyl chloride relative to Zn was performed (runs 15 and 16, Table 1). A representative 1H NMR spectrum of the final PE obtained is shown on Fig. 2 for run 16 (Table 5). The assignment of the signals performed on Fig. 2 strongly supports the selective formation of the targeted p-methylphenone-PE and shows the absence of any other signals including those related to chains initiated by a mesityl moiety. High functionalization yields from 79 to 86% were obtained using only one equivalent of acyl chloride per Zn–alkyl bond. This selectivity outcome reinforces the role played by Zn in mostly supporting PE chains and eludes an efficient chain transfer reactions between Nd or Zn with Mg. Eventually, run 16 (Table 5), with [Mg]/[Nd] = 8, shows a slowdown in polymerization rate over time. As already mentioned in the case of BOMAG (Table 2), an excess of magnesium compound compared to neodymium is required to achieve stable activity.
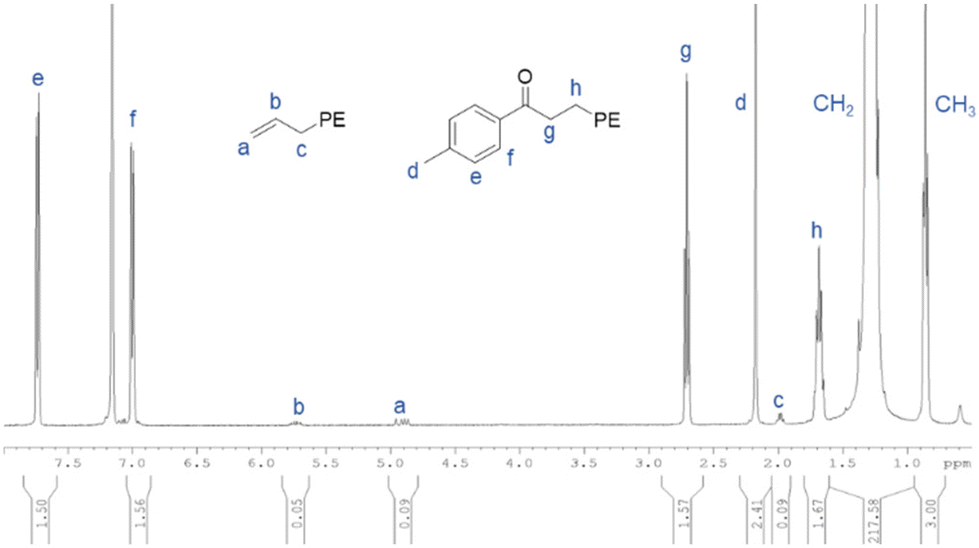 | ||
| Fig. 2 1H NMR spectrum of the p-methylphenone-PE obtained with the ternary catalytic system 1/MesMgBr/ZnEt2 after deactivation with p-toluoyl chloride (run 16, Table 5). | ||
1/MesMgBr/ZnEt2 catalyst system was further tested for the copolymerization of ethylene and butadiene (run 17, Table 5). An EBR (20.3% of inserted butadiene) with a similar microstructure (Table 4) to that obtained when BOMAG is used (run 10, Table 3) was isolated however with lower activity. The polymerization medium was deactivated by addition of 4-dimethylaminobenzoyl chloride. The 1H NMR spectrum shows the characteristic signal of methyl groups from –NMe2 group in agreement with the formation of p-dimethylaminophenone chain end (Fig. S11†). A functionalization rate of 34% was measured. This relatively low functionalization efficiency in comparison to functionalization reaction conducted on PE chains can be ascribed to the different nature of the chain ends in the two systems26 and their respective reactivity but also to the higher molar mass obtained for EBR.41
Conclusions
In this work, we have demonstrated that ZnEt2 is an effective CTA when used in combination with 1 for CCTP of ethylene or CCTcoP of ethylene and butadiene to produce EBR. However, being unable to alkylate 1 when used alone, the effectiveness ZnEt2 as CTA depends on the additional presence of an organomagnesium compound that additionally stabilize the resulting catalyst. Compared to magnesium, the storage of the polymer chains on zinc modifies the reactivity of the chain ends, enabling a selective terminal ketone formation through deactivation reactions of the polymerization medium with acyl chlorides. Despite this, the functionalization rate remains low for EBR, making the synthesis of highly functionalized EBRs with high molar masses a future challenge. Additionally, combining ZnR2 and MgR2 CTA could open new avenues for integrating Nd metallocenes in original catalyst combinations for the production of original OBC. This approach holds the potential to combine the properties of polyolefins exclusively accessible with Nd metallocene with other polyolefins.Author contributions
The original ideas were conceived by the corresponding authors FdA, MEP, and CB in collaboration with industrial partners FJBdD and RN. Experimental work was carried out by AD, VL and NB, while the computational studies were conducted by LV. The manuscript was written collectively by all the authors.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Manufacture Michelin is acknowledged for scientific and financial support of this project. The CCIR of ICBMS is acknowledged for a generous allocation of computational resources and providing technical support.References
- C. Dommanget, F. D'Agosto and V. Monteil, Angew. Chem., Int. Ed., 2014, 53, 6683–6686 CrossRef PubMed.
- Y. Nakamura, B. Ebeling, A. Wolpers, V. Monteil, F. D'Agosto and S. Yamago, Angew. Chem., Int. Ed., 2018, 57, 305–309 CrossRef PubMed.
- A. Wolpers, C. Bergerbit, B. Ebeling, F. D'Agosto and V. Monteil, Angew. Chem., Int. Ed., 2019, 58, 14295–14302 CrossRef PubMed.
- A. Wolpers, F. Baffie, L. Verrieux, L. Perrin, V. Monteil and F. D'Agosto, Angew. Chem., Int. Ed., 2020, 59, 19304–19310 CrossRef PubMed.
- R. Kempe, Chem. – Eur. J., 2007, 13, 2764–2773 CrossRef PubMed.
- L. R. Sita, Angew. Chem., Int. Ed., 2009, 48, 2464–2472 CrossRef PubMed.
- A. Valente, A. Mortreux, M. Visseaux and P. Zinck, Chem. Rev., 2013, 113, 3836–3857 CrossRef PubMed.
- R. Mundil, C. Bravo, N. Merle and P. Zinck, Chem. Rev., 2024, 124, 210–244 CrossRef PubMed.
- P. D. Hustad, Science, 2009, 325, 704–707 CrossRef PubMed.
- F. D'Agosto and C. Boisson, Aust. J. Chem., 2010, 63, 1155–1158 CrossRef.
- W. Zhang and L. R. Sita, J. Am. Chem. Soc., 2008, 130, 442–443 CrossRef PubMed.
- W. Zhang, J. Wei and L. R. Sita, Macromolecules, 2008, 41, 7829–7833 CrossRef.
- J. Wei, W. Zhang and L. R. Sita, Angew. Chem., Int. Ed., 2010, 49, 1768–1772 CrossRef PubMed.
- J. Mazzolini, E. Espinosa, F. D'Agosto and C. Boisson, Polym. Chem., 2010, 1, 793–800 RSC.
- D. J. Arriola, E. M. Carnahan, P. D. Hustad, R. L. Kuhlman and T. T. Wenzel, Science, 2006, 312, 714–719 CrossRef.
- P. D. Hustad, G. R. Marchand, E. I. Garcia-Meitin, P. L. Roberts and J. D. Weinhold, Macromolecules, 2009, 42, 3788–3794 CrossRef.
- S. S. Park, C. S. Kim, S. D. Kim, S. J. Kwon, H. M. Lee, T. H. Kim, J. Y. Jeon and B. Y. Lee, Macromolecules, 2017, 50, 6606–6616 CrossRef.
- S. D. Kim, T. J. Kim, S. J. Kwon, T. H. Kim, J. W. Baek, H. S. Park, H. J. Lee and B. Y. Lee, Macromolecules, 2018, 51, 4821–4828 CrossRef.
- N. Baulu, M. Langlais, R. Ngo, J. Thuilliez, F. Jean-Baptiste-dit-Dominique, F. D'Agosto and C. Boisson, Angew. Chem., Int. Ed., 2022, 61, e202204249 CrossRef PubMed.
- M. Langlais, N. Baulu, S. Dronet, C. Dire, F. Jean-Baptiste-dit-Dominique, D. Albertini, F. D'Agosto, D. Montarnal and C. Boisson, Angew. Chem., Int. Ed., 2023, 62, e202310437 CrossRef PubMed.
- A. Valente, P. Zinck, M. J. Vitorino, A. Mortreux and M. Visseaux, J. Polym. Sci., Part A:Polym. Chem., 2010, 48, 4640–4647 CrossRef.
- A. Valente, P. Zinck, A. Mortreux and M. Visseaux, J. Polym. Sci., Part A:Polym. Chem., 2011, 49, 1615–1620 CrossRef.
- S. Georges, A. O. Touré, M. Visseaux and P. Zinck, Macromolecules, 2014, 47, 4538–4547 CrossRef.
- J.-F. Pelletier, A. Mortreux, X. Olonde and K. Bujadoux, Angew. Chem., Int. Ed. Engl., 1996, 35, 1854–1856 CrossRef.
- R. Ribeiro, R. Ruivo, H. Nsiri, S. Norsic, F. D'Agosto, L. Perrin and C. Boisson, ACS Catal., 2016, 6, 851–860 CrossRef.
- I. Belaid, B. Macqueron, M.-N. Poradowski, S. Bouaouli, J. Thuilliez, F. D. Cruz-Boisson, V. Monteil, F. D'Agosto, L. Perrin and C. Boisson, ACS Catal., 2019, 9, 9298–9309 CrossRef CAS.
- X. Olonde, A. Mortreux, F. Petit and K. Bujadoux, J. Mol. Catal., 1993, 82, 75–82 CrossRef.
- M. F. Llauro, C. Monnet, F. Barbotin, V. Monteil, R. Spitz and C. Boisson, Macromolecules, 2001, 34, 6304–6311 CrossRef.
- V. Monteil, R. Spitz, F. Barbotin and C. Boisson, Macromol. Chem. Phys., 2004, 205, 737–742 CrossRef.
- J. Thuilliez, L. Ricard, F. Nief, F. Boisson and C. Boisson, Macromolecules, 2009, 42, 3774–3779 CrossRef.
- H. Nsiri, I. Belaid, P. Larini, J. Thuilliez, C. Boisson and L. Perrin, ACS Catal., 2016, 6, 1028–1036 CrossRef.
- G. J. P. Britovsek, S. A. Cohen, V. C. Gibson and M. Van Meurs, J. Am. Chem. Soc., 2004, 126, 10701–10712 CrossRef.
- J. Wei, W. Zhang, R. Wickham and L. R. Sita, Angew. Chem., Int. Ed., 2010, 49, 9140–9144 CrossRef PubMed.
- G. J. P. Britovsek, V. C. Gibson and D. F. Wass, Angew. Chem., Int. Ed., 1999, 38, 428–447 CrossRef.
- V. Grignard and L. Tissier, C. R. Acad. Sci., 1901, 683–685 Search PubMed.
- H. Gilman, R. E. Fothergill and H. H. Parker, Recl. Trav. Chim. Pays-Bas, 1929, 48, 748–751 CrossRef.
- H. Gilman and M. L. Mayhue, Recl. Trav. Chim. Pays-Bas, 1932, 51, 47–50 CrossRef.
- X. Wang, L. Zhang, X. Sun, Y. Xu, D. Krishnamurthy and C. H. Senanayake, Org. Lett., 2005, 7, 5593–5595 CrossRef PubMed.
- H. Makio, T. Ochiai, J. Mohri, K. Takeda, T. Shimazaki, Y. Usui, S. Matsuura and T. Fujita, J. Am. Chem. Soc., 2013, 135, 8177–8180 CrossRef PubMed.
- N. Baulu, M.-N. Poradowski, L. Verrieux, J. Thuilliez, F. Jean-Baptiste-dit-Dominique, L. Perrin, F. D'Agosto and C. Boisson, Polym. Chem., 2022, 13, 1970–1977 RSC.
- A. P. Gies, Z. Zhou, L. Sun, E. Szuromi, T. Huang, A. Krasovskiy, S. Mukhopadhyay, E. Herceg, I. Kobylianskii, Z. Shi and J. C. P. Reyes, Macromolecules, 2022, 55, 2542–2556 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental protocols, computational details, addition NMR spectra, microstructure of EBRs. See DOI: https://doi.org/10.1039/d4py01220h |
| ‡ Current address: Manufacture Michelin, 23 place Carmes Déchaux, F-63000 Clermont-Ferrand, France. |
| § Current address: Univ Rennes, École Nationale Supérieure de Chimie de Rennes, CNRS, ISCR (Institut des Sciences Chimiques de Rennes) – UMR6226, F-35000, Rennes, France. |
| This journal is © The Royal Society of Chemistry 2024 |