
Isomer-driven polymerization, depolymerization, and reconstruction†
Herbert
Wakefield IV
a,
Nicholas J.
Fromel
a,
Jennifer
Jiang
a,
Ilia
Kevlishvili
 b,
Yunxin
Yao
c,
Stephen L.
Craig
c,
Heather J.
Kulik
b and
Rebekka S.
Klausen
*a
b,
Yunxin
Yao
c,
Stephen L.
Craig
c,
Heather J.
Kulik
b and
Rebekka S.
Klausen
*a
aDepartment of Chemistry, Johns Hopkins University, 3400 N. Charles St, Baltimore, MD 21218, USA. E-mail: klausen@jhu.edu
bDepartment of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
cDepartment of Chemistry, Duke University, Durham, NC, USA
First published on 20th November 2024
Abstract
We report that differences in ring strain enthalpy between cis and trans isomers of sila-cycloheptene provide a driving force for both polymerization and depolymerization via olefin metathesis. A need for new methods to reintroduce the low-strain isomer into the plastic economy inspired the development of a polymerization based on ring-opening/cross-metathesis step polymerization, which afforded perfect sequence control for an alternating copolymer. The chemical principles are a platform for achieving both efficient polymerization and depolymerization with high mass recovery in functional polymers.
Introduction
Chemical solutions to end-of-life management of post-consumer plastic waste are an emerging and urgent area of research.1 Currently, less than 10% of plastic waste is mechanically recycled, instead ending in the landfill or incinerator (a linear plastic economy). In contrast to mechanical recycling, which can erode material properties due to changes in molecular weight characteristics arising from mechanical degradation, chemical recycling to monomer transforms plastic waste into feedstocks indistinguishable from petroleum-derived monomer, which can then reform pristine plastics (a more circular plastic economy).2 Approaches to chemical recycling to monomer include comonomers or end groups in poly(methyl methacrylate) (PMMA) that trigger β-scission,3–9 transesterification of poly(ethylene terephthalate),10–13 self-immolative polymers,14–16 and more.17Olefin metathesis-based approaches to polymer deconstruction have been recently reviewed.18 An attractive feature of this approach is that the polymer is stable until addition of the metathesis catalyst. Deconstruction can be a depolymerization process that returns the original starting monomer or a reaction yielding a different chemical building block.19 Polymerization thermodynamics are critical:20–23 if ring-opening metathesis polymerization (ROMP) is highly exothermic due to significant strain release, the reverse reaction (ring-closing metathesis (RCM) depolymerization) is challenging to effect. Chemical recycling to monomer by olefin metathesis was until recently limited to monomers with low-to-moderate ring strain,22 while polymeric materials derived from higher strain monomers like norbornene and cis-cyclooctene24 were not depolymerizable to monomer. Recently, structural modifications to cis-cyclooctene enabled chemical recycling to monomer by modulating ring-strain enthalpy (RSE) and polymerization–depolymerization thermodynamics.21,25
Exploiting the differences in RSE between cis and trans geometric isomers of the same cycloalkene has potential to broaden the scope of polymeric materials amenable to olefin metathesis polymerization–depolymerization, as shown by Wang et al. in a “closed-loop” photoisomerization-ROMP-RCM cycle of fused bicyclic cyclooctene monomers.26 However, olefin photoisomerization has limitations: high-energy light is typically needed (e.g., 254 nm) and the amount of cis isomer that can be converted to trans isomer is fundamentally limited by the photostationary trans![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :cis ratio. This means that not all the chemical matter recovered by depolymerization to the cis-isomer can reenter the cycle, highlighting the need for additional chemical methods to reintroduce the low-strain isomer into the plastic economy.
:cis ratio. This means that not all the chemical matter recovered by depolymerization to the cis-isomer can reenter the cycle, highlighting the need for additional chemical methods to reintroduce the low-strain isomer into the plastic economy.
Herein, we demonstrate isomer-driven polymerization, depolymerization, and reconstruction of sila-cycloheptene by multiple olefin metathesis mechanisms. We recently reported the geometrically-selective synthesis of both the cis and trans isomers of sila-cycloheptene 1 in which trans-1 was high strain (ca. 10 kcal mol−1) and readily underwent ROMP.27P1 is a novel example of a hybrid polymer incorporating both C and Si into the backbone. We showed via single molecule force spectroscopy that the Si incorporation into P1 reduced single chain elasticity relative to all-carbon polymer backbones, resulting in a softer, less stiff polymer strand, which was hypothesized to arise from force-induced changes in geometry and conformation at Si.28 While trans-1 was a good ROMP monomer, low strain cis-1 (ca. 1–2 kcal mol−1) did not polymerize. DFT calculations predicted that metathesis initiation is kinetically accessible for cis-1, but ROMP is endothermic.
We now report well-controlled ROMP of trans-1 to afford a high molecular weight (>300 kg mol−1) homopolymer P1 that is stable in the absence of a metathesis initiator but undergoes 100% depolymerization to low-strain cis-1 upon addition of an appropriate initiator (Fig. 1). To reuse the low-strain isomer, we hypothesized that ring-opened cis-1 could be captured in a selective cross-metathesis reaction, resulting in a novel tandem ring-opening/cross-metathesis (RO-CM) olefin metathesis polymerization that is sequence-controlled. We show that recovered cis-1 participates in RO-CM with butanediol diacrylate (BDA) to afford a perfectly alternating copolymer P2. This work demonstrates the ability of isomerism to drive novel polymer design, optimize reactivity, mass recovery, and reuse, as well as the role of Si for C replacement29 in modulating monomer ring strain, polymerization thermodynamics, and polymer properties.
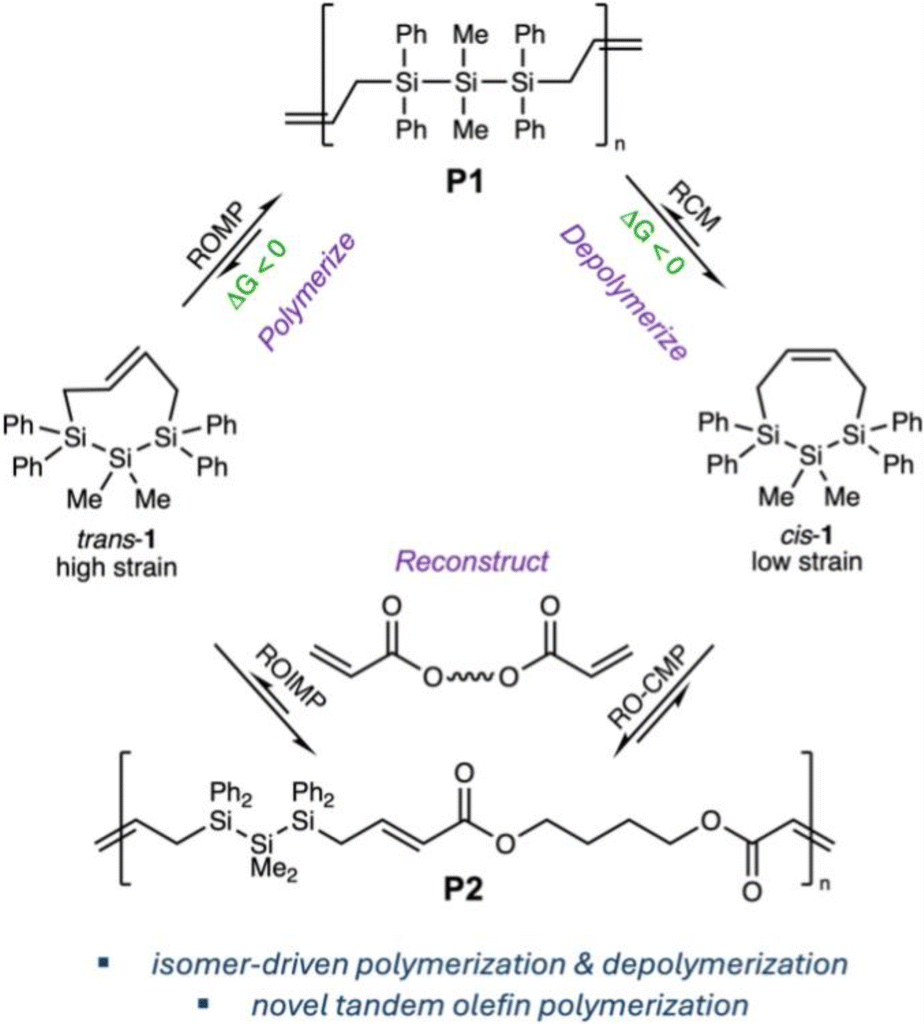 | ||
| Fig. 1 Intrinsic differences in ring strain of trans- and cis-1 enable polymerization, depolymerization, and reconstruction by multiple olefin metathesis mechanisms. ROMP = ring-opening metathesis polymerization; RCM = ring-closing metathesis; ROIMP = ring-opening-insertion metathesis polymerization; RO-CMP = ring-opening/cross-metathesis polymerization. | ||
Results and discussion
Our initial efforts focused on identifying if molecular weight characteristics are important in depolymerization of P1 as our initial publication reported a high molecular weight and high dispersity polymer (Mn = 62.6 kg mol−1, Mw/Mn = 3.50) reflecting a multimodal sample containing both high molecular weight strands and oligomeric fragments.27 The low molecular weight fraction included cyclic oligomers arising from ring-closing macrocyclization, which lack end groups and could complicate end-group selective initiation of depolymerization. To remove the lower molecular weight fraction, trans-1 was polymerized to P15.03 as a polymodal sample then purified via recycling size exclusion chromatography (RSEC) and reprecipitated in methanol to afford narrow dispersity P11.23 (Fig. 2).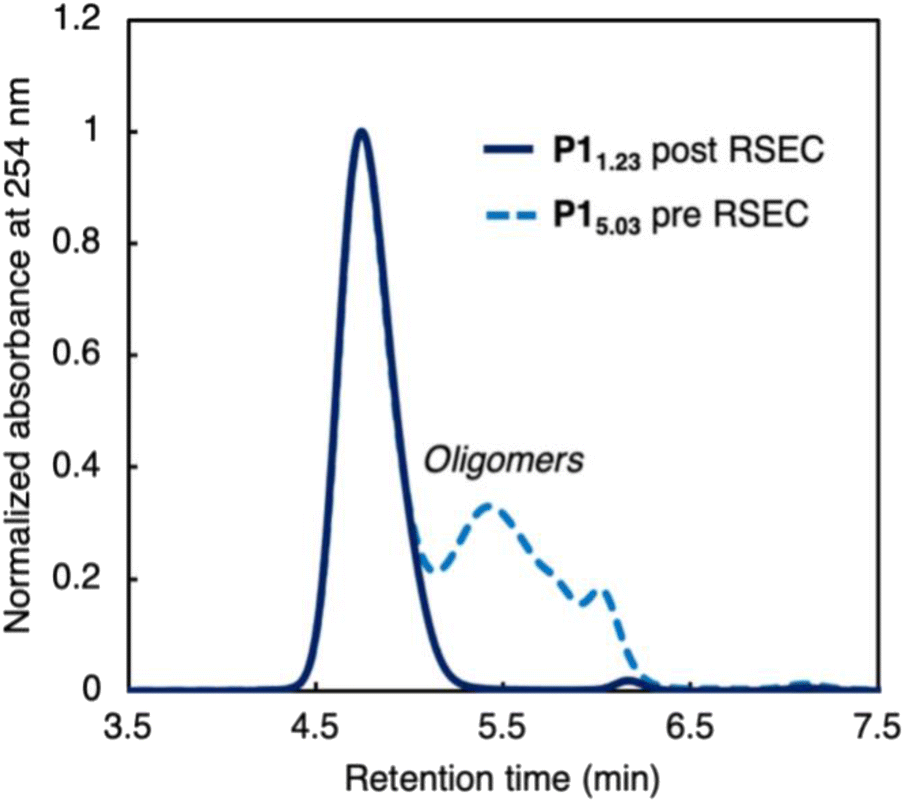 | ||
| Fig. 2 Size exclusion chromatography (SEC) elugram of P1 before (dashed) and after (solid) purification by recycling SEC, normalized to the largest molecular weight peak at 4.74 min ([P1] = 1.00 mg mL-1, THF, RT) relative to polystyrene standards. P1 (before RSEC) Mn = 2.03 kg mol−1, Mw/Mn = 5.03. P1 (after RSEC) Mn = 13.0 kg mol−1, Mw/Mn = 1.23. | ||
Unimodal P1 can also be synthesized directly, as we have now also identified improved ROMP conditions. Hypothesizing that the formation of lower molecular weight oligomers arose from secondary metathesis reactions, we sought to rigorously purify trans-1 by recrystallization to remove any olefinic contaminants from synthesis (e.g., trans-1,4-dichlorobutene). With highly purified starting material and a much shorter reaction time of 10 minutes, P1 was synthesized in high molecular weight and in a unimodal distribution (Mn = 92.9 kg mol−1, Mw/Mn = 1.14, Scheme 1a).
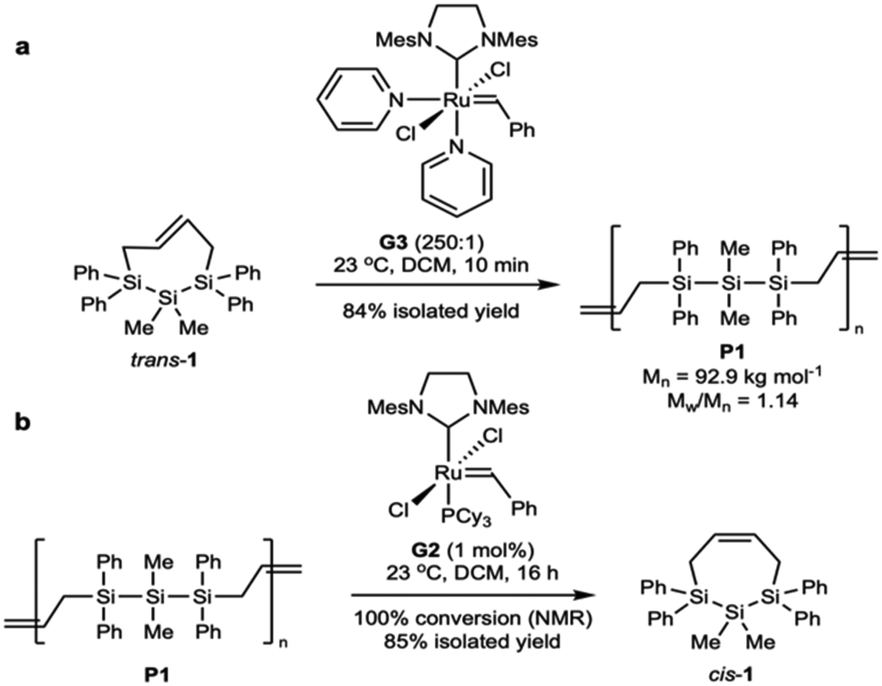 | ||
| Scheme 1 (a) Optimized polymerization of trans-1 to afford low dispersity P1 and (b) optimized depolymerization of P1 to afford cis-1 in 85% yield. | ||
Upon treatment of P11.23 with 1 mol% of the metathesis initiator G1, we observed after 16 hours by 1H NMR spectroscopy the growth of peaks assigned to cis-1 (55% conversion) with polymeric material remaining (Fig. 3). Consistent with chain transfer competing with depolymerization by RCM, multimodal P111.1 (containing residual oligomers) depolymerized in only 28% conversion under the same conditions (Fig. S2†).
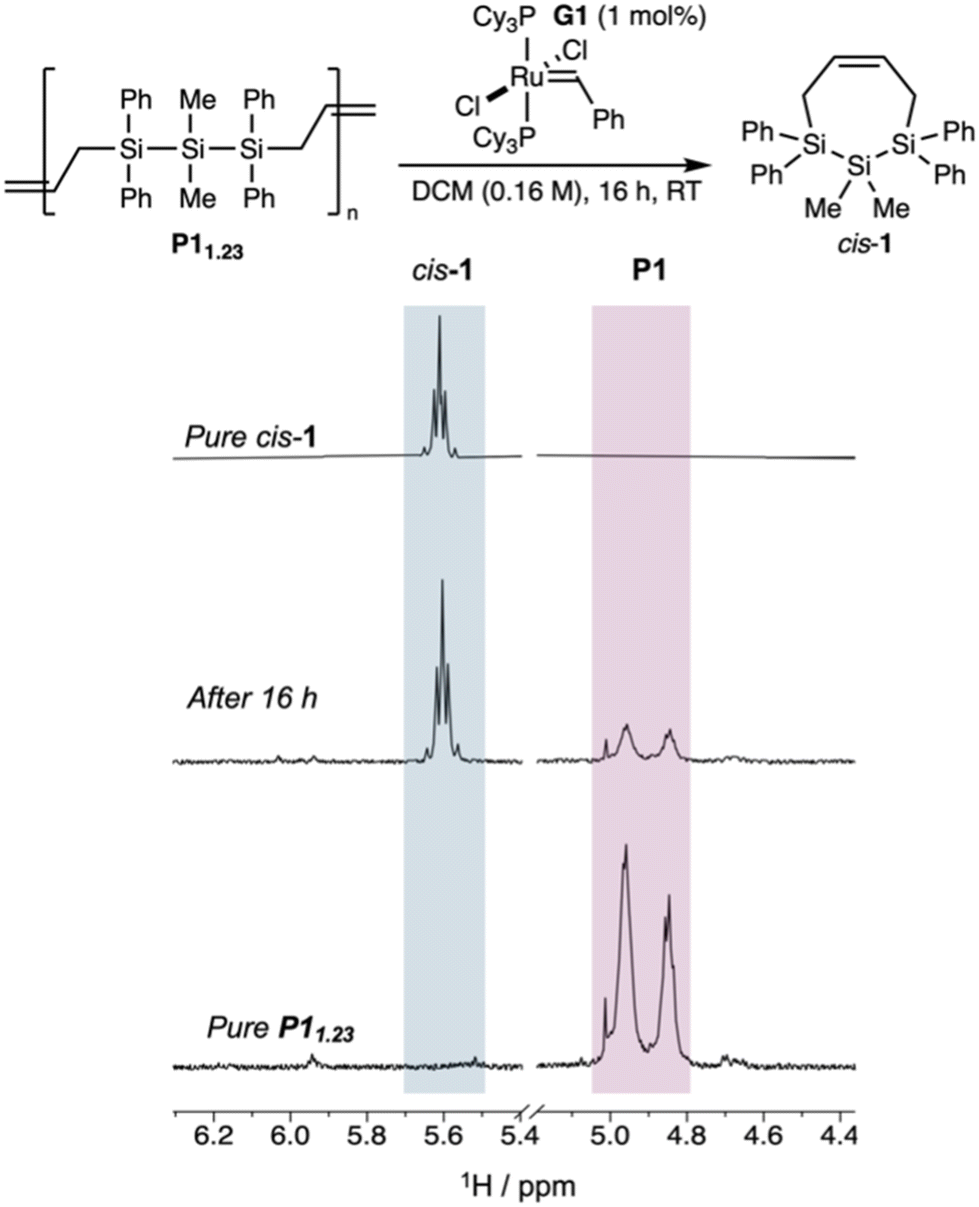 | ||
| Fig. 3 Cropped 1H NMR spectra of pure cis-1 (top), after 16 hours of depolymerization (middle), and pure P11.23 (bottom) showing 55% depolymerization to cis-1. | ||
Monitoring the depolymerization of P11.23 over time by SEC (Fig. 4 and Table 1), we observed a gradual decrease in the amount of P1 and an increase in cis-1. Reisolation of P1 at each time point showed that Mn decreased from 11.9 kg mol−1 to 6.92 kg mol−1 while dispersity increased modestly from 1.30 to 1.81 (Table 1). Deconstruction at random internal sites would result in many more short oligomers and a much broader Mw/Mn than we observed here, suggesting a possible end-to-end depolymerization. At the same time, the modest increase in dispersity suggested that chain transfer might also be occurring between strands, reducing the efficiency of depolymerization.
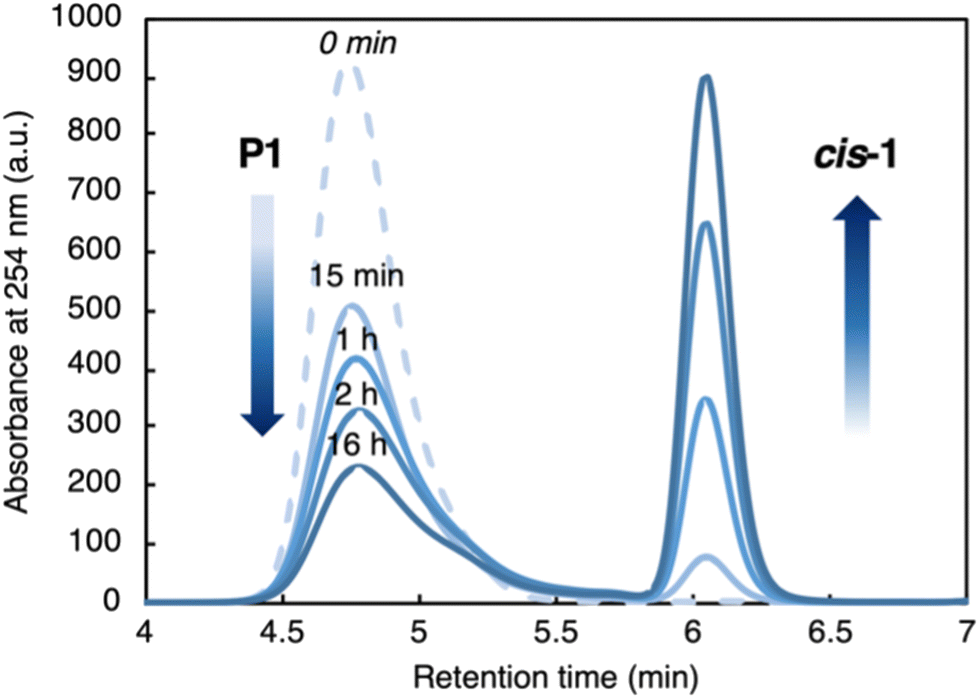 | ||
| Fig. 4 SEC elugrams of recovered P1 over time ([P1] = 0.75 mg mL−1, THF, RT). See Table 1 for molecular weight characteristics of recovered P1. | ||
| Reaction time |
M
nb (kg mol−1) |
M
w/Mnb |
|---|---|---|
| a Depolymerization of P1 performed at room temperature over 16 h with a concentration of 0.16 M in DCM and 1 mol% G1. b Determined by size exclusion chromatography relative to polystyrene standards at 254 nm (THF, [P1] = 0.75 mg mL−1, 40 °C, 0.35 mL min−1, 10 μL injection). | ||
| 0 | 11.9 | 1.30 |
| 15 min | 9.55 | 1.51 |
| 1 h | 8.0 | 1.68 |
| 2 h | 7.12 | 1.79 |
| 16 h | 6.92 | 1.81 |
We investigated other metathesis initiators. While G3 improved the depolymerization efficiency compared to G1, resulting in 86% depolymerization of P1 to cis-1, the best conditions employed 1 mol% of G2 and increased depolymerization efficiency to 100% conversion to cis-1 (Table 2, entries 2 and 3), with no remaining high molecular weight polymer. In general, higher loadings of metathesis promoter and higher temperatures led to higher quantities of recovered cis-1 (entries 5–7). Scaling up the best conditions, we were able to depolymerize 80 mg of P1 to 100% conversion by 1H NMR and with 85% isolated yield of cis-1 (Table 2, entry 3). We hypothesize that the higher depolymerization efficiency with G2 is related to its generally higher reactivity relative to G130 and employing G2 instead of G1 also increased the depolymerization efficiency of high dispersity material (compare entries 5 and 9).
| Entry | M w/Mn of starting P1 | Cat.a | mol% | Temp. (°C) | % NMR yieldbcis-1 |
|---|---|---|---|---|---|
| a Catalyst used for depolymerization. b Determined from the relative integration of the polymer olefinic protons and the olefinic protons of cis-1. | |||||
| 1 | 1.23 | G1 | 1 | 23 | 55 |
| 2 | 1.23 | G2 | 1 | 23 | 100 |
| 3 | 2.13 | G2 | 1 | 23 | 100 (85% isolate yield) |
| 4 | 1.23 | G3 | 1 | 23 | 86 |
| 5 | 11.1 | G1 | 1 | 23 | 28 |
| 6 | 11.1 | G1 | 3 | 23 | 38 |
| 7 | 11.1 | G1 | 5 | 23 | 37 |
| 8 | 11.1 | G1 | 1 | 50 | 36 |
| 9 | 8.61 | G2 | 1 | 23 | 84 |
Having identified the circumstances leading to complete depolymerization of P1 to cis-1, we considered potential reuse scenarios for this low-strain cycloalkene. Entropy-driven ROMP can be an effective method for polymerization of low-strain cycloalkenes but requires high concentrations31 and cis-1 is a crystalline solid that is poorly soluble in CH2Cl2, CHCl3 and other solvents (Table S1†). We considered alkene photoisomerization, but photochemical silylene extrusion32 is possible for cis-1. In addition, the amount of trans isomer available from the cis isomer is limited by the photostationary trans:cis ratio.33,34
For these reasons, we sought an alternative polymerization method for cis-1. Prior calculated free energy profiles for initiation of cis- and trans-1 ROMP with G227 indicated that cis-1 is kinetically able to form a ring-opened, Ru-terminated structure similar to A (Fig. S3†).35 While the reaction of A with cis-1 to form a homopolymer via ROMP (a chain polymerization) is endothermic and not observed, we hypothesized that intermediate A could instead be captured in cross-metathesis. Reaction of A with a bifunctional Type 2 olefin B2 would release the metathesis catalyst and difunctional molecule AB, which we anticipated would be a suitable step polymerization monomer36via selective cross-metathesis between terminal alkene and acrylate end groups.37,38 Formation of the low energy α,β-unsaturated ester provides the driving force for ring-opening/cross-metathesis (RO-CM). While RO-CM is a tandem reaction extensively developed for small molecule synthesis,39,40 it does not appear that RO-CM step polymerization has previously been reported. The polymerization by selective cross-metathesis of linear monomers with a terminal alkene and an acrylate supports our hypothesized mechanism.37,38 It is plausible that both RO-CM and the reverse order of fundamental steps (cross metathesis/ring-opening, CM-RO) operate simultaneously, although CM-RO would require G2 to react first with B2 and then ring open cis-141,42 while the reaction of G2 with acrylates is slower than with more activated olefins.
To test the hypothesized RO-CM reactivity, we carried out two small molecule studies in which we reacted cis-1 with 1-hexene (Type 1 olefin) or methyl acrylate (Type 2 olefin). With 1-hexene, we only observed the formation of the homodimerization product 4-decene and residual cis-1 (Fig. S4†). But in the presence of excess methyl acrylate, the ring-opened structure 2 was isolated in 61% yield after purification by silica gel chromatography (Scheme 2). The RO-CM product 2 has exclusively the trans olefin geometry, as determined by the coupling constants of the olefinic peaks δ 6.81 (J1H–1H = 15.5 Hz) and δ 5.39 (J1H–1H = 15.4 Hz) (Fig. S5†).
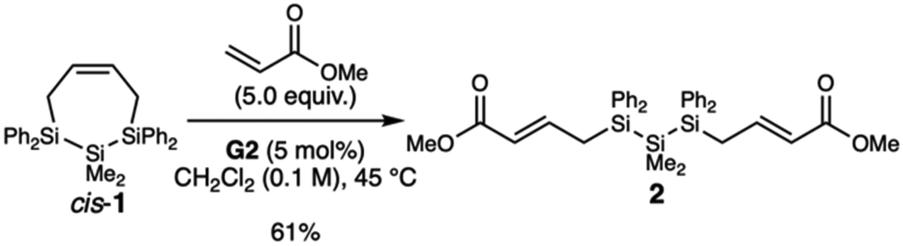 | ||
| Scheme 2 Model reaction for ring opening-cross metathesis (RO-CM) of cis-1 and methyl acrylate. | ||
Expanding from molecular RO-CM, we then investigated the RO-CM step polymerization of cis-1 and 1,4-butanediol diacrylate (BDA) (Fig. 5b). We observed 91% consumption of cis-1 within 4.5 hours. By precipitation from hexanes, we obtained polymer P2 (Mn = 3.31 kg mol−1, Mw/Mn = 1.87). Structural characterization supported incorporation of both monomers. ATR-IR spectroscopy (Fig. S5†) indicated the presence of an α,β-unsaturated carbonyl at 1706 cm−1, as well as a SiMe functional group at 1257 cm−1.
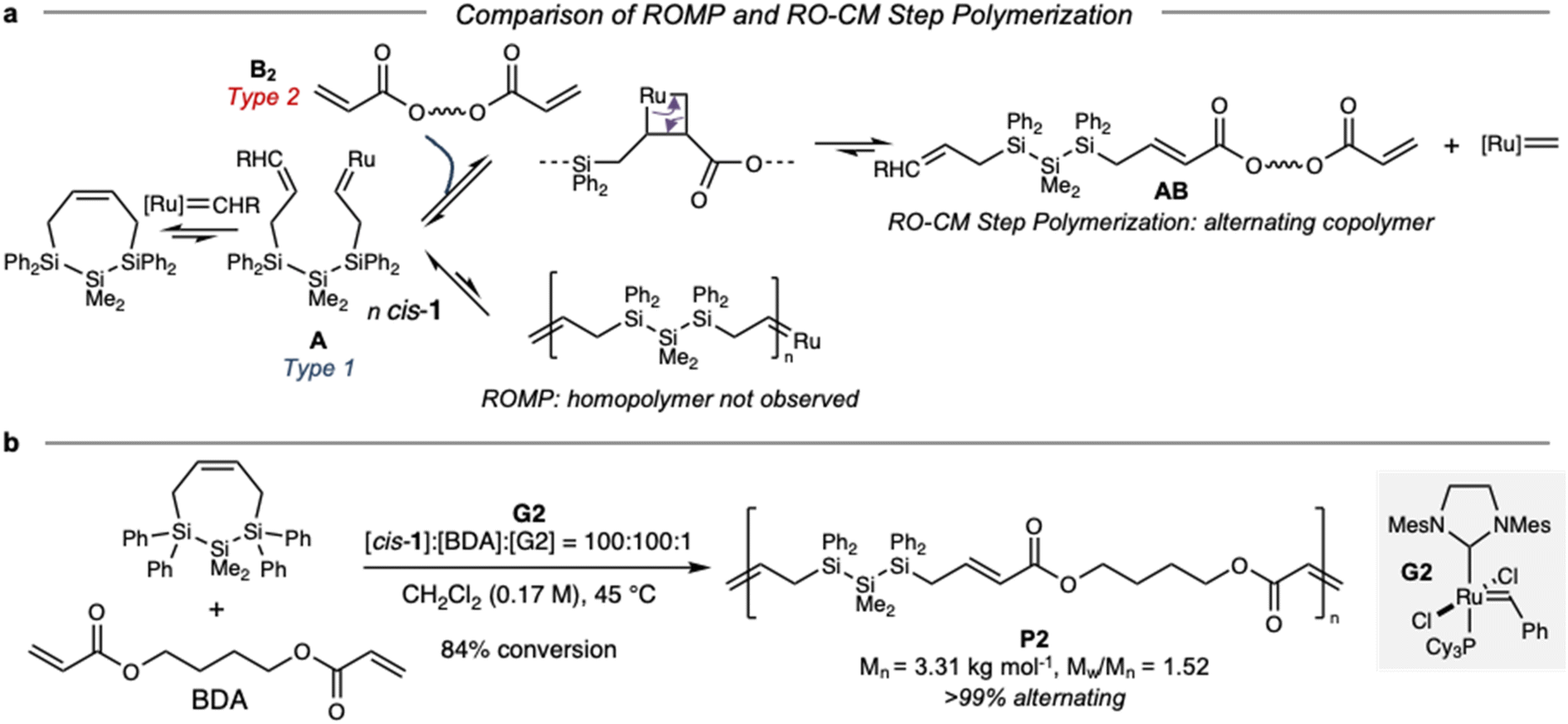 | ||
| Fig. 5 (a) Proposed mechanism for ring-opening/cross-metathesis step polymerization yielding highly alternating copolymers. (b) Synthesis of P2via RO-CM step polymerization. | ||
Since cis-1 is a poor ROMP monomer and BDA is a poor ADMET monomer, RO-CM predicts an alternating copolymer due to faster crosspolymerization than homopolymerization. Structural characterization supported assignment of P2 to a highly alternating polymer (>99% alteration) based on 1H NMR spectroscopy (Fig. 6) in which neither the singlet consistent with a fumarate resonance nor the lower field resonances of a diallyl silane were observed. The major peaks were assigned to the alternating copolymer and were consistent with an (E)-geometry. Resonances consistent with both acrylate and styrenic end groups were identified and full details of copolymer structural characterization are reported in the ESI.† We note that P2 can itself be deconstructed at end of life by ester hydrolysis.
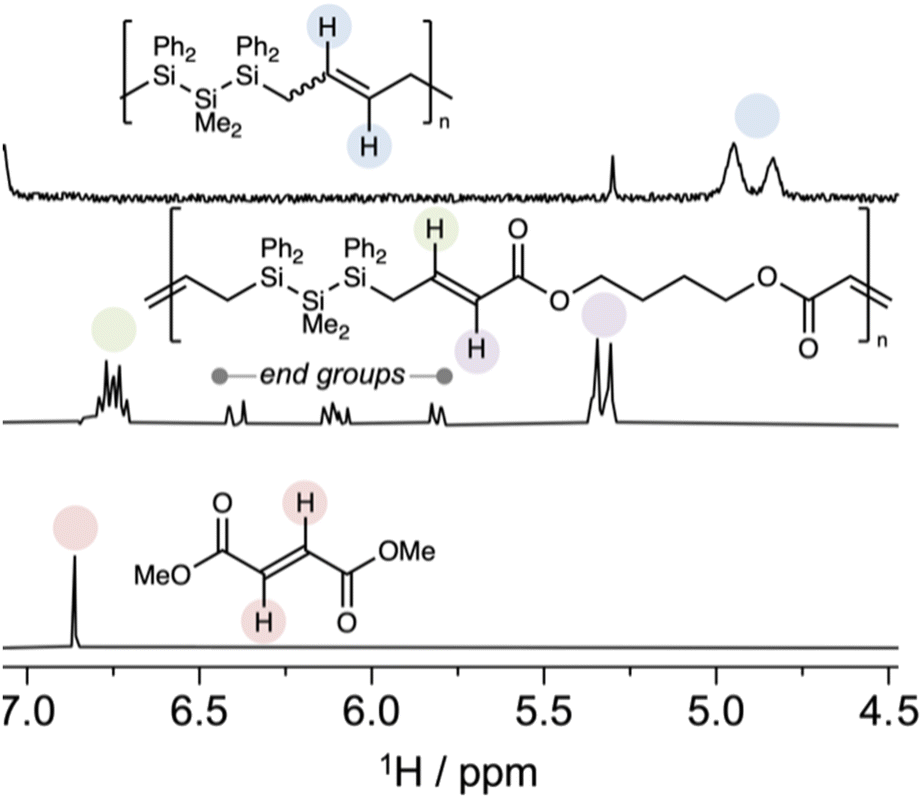 | ||
| Fig. 6 Cropped 1H NMR spectra (CDCl3, 400 MHz) comparing the olefinic regions of P1 (top), P2 (middle), and dimethyl fumarate (bottom). There is no evidence of signals consistent with consecutive homopolymerization repeat units, supporting a highly alternant copolymer microstructure. | ||
To understand the impact of BDA incorporation onto the properties of a hybrid carbosilane polymer, we measured the glass transition temperature (Tg) of P2. While the homopolymer P1 exhibits a glass transition temperature a little above room temperature (ca. 35 °C), the alternating copolymer P2 had a Tgca. 13 °C. The lower Tg may reflect the additional incorporation of the flexible butane chain and a lower overall aromatic side chain content. In recent years, the role of sequence control has emerged as a tactic for modulating the glass transition temperature of a copolymer.43 The onset of thermal decomposition occurred at ca. 300 °C, consistent with other Si–Si containing polymers.27,44
This tandem ring-opening and selective cross-metathesis polymerization has the advantage of very high sequence control, a grand challenge in polymer synthesis.45,46 The copolymerization of cycloalkenes and α,ω-diene monomers related to Type 1 olefins is known, but not sequence controlled.47,48 Alternating copolymerization by olefin metathesis has been reported with cyclohexene and electron-poor strained cycloalkenes (e.g., cyclobutenecarboxamide).49 The most similar reaction to ours is ring-opening-insertion metathesis polymerization (ROIMP). ROIMP is the copolymerization of cycloalkenes and diacrylates to yield highly alternant copolymers,50–53 but ROIMP proceeds by a distinctly different mechanism involving first rapid cycloalkene ROMP (e.g., cis-cyclooctene), followed by insertion of BDA or other diacrylate into the homopolymer by a slower secondary metathesis process.
In the current work, the lack of cis-1 homopolymerization and the well-precedented slow rate of acrylate dimerization are not consistent with a ROIMP mechanism involving insertion of a second monomer into a homopolymer. A series of control experiments suggested that cis-1/BDA RO-CM copolymerization proceeds by a mechanism that is distinct from ROIMP and that these differences result in meaningful impacts on polymer microstructure e.g., sequence control.
First, we evaluated the reactivity of trans-1 and BDA under the same conditions employed for cis-1. We observed a mixture that consisted predominantly of P1 and unreacted BDA, with evidence of smaller amounts of dimeric BDA and P2 (Fig. S6†). This is distinctly different from the outcome with cis-1, where no homopolymers were observed, and is consistent with high strain trans-1 being suitable for ROIMP while low strain cis-1 reacts via RO-CM step polymerization.
Second, we noted that Choi and Grubbs reported that polycyclooctene could be used directly as a substrate for ROIMP, supporting the hypothesis that the homopolymer is an intermediate. If ROIMP was operative for the copolymerization of cis-1/BDA, P1 should react with BDA to yield P2. However, the reaction between homopolymer P1 and BDA afforded only residual P1 and small quantities of the BDA dimer/oligomer, but no spectroscopic evidence of either cis-1 or P2 (Fig. S10†).
Third, a consequence of the ROIMP mechanism is that extended homopolycycloalkene segments were found at early time points (e.g., 20 min).50 For cis-1/BDA copolymerization, high alternation of 82.0% was observed after 15 minutes (Fig. S11†). This points to the very high selectivity for cross-metathesis rather than homopolymerization.
Conclusions
The isomer-driven metathesis reactivity reported herein highlights how subtle differences in structure, thermodynamics, and kinetics can be exploited in the well-controlled, serial polymerization/depolymerization/reconstruction pathways. Only the trans isomer of sila-cycloheptene 1 undergoes ROMP by chain polymerization, driven by strain release. The reverse ring-closing metathesis reaction yields only the low-strain cis isomer of 1, driven by the entropic benefit of depolymerization, and leading to 100% selectivity for a single product and 100% conversion in chemical recycling. The use of cycloalkene geometric isomerism to affect the position of polymer thermodynamics is recently emerging, building onto the use of cis/trans relative configuration at tetrahedral stereoegenic centers.54,55 We reintroduce cis-1 into polymeric materials by a novel tandem olefin metathesis pathway involving ring-opening/cross-metathesis step polymerization. Tandem olefin metathesis reactions remain rare in macromolecular synthesis due to poor control,51 but herein we achieved perfect selectivity for the cross-metathesis reaction and obtained a highly alternant copolymer from cis-1 and the diacrylate BDA. The resulting polymer was fully characterized by ATR-IR, 1H, 13C, 29Si, COSY, and HSQC NMR spectroscopy to determine polymer microstructure.That a 7-membered ring can switch from a high to a low-strain reactivity manifold is enabled not only by controlling olefin geometry, but also by the ability of Si for C replacement to modulate ring conformation and geometry (trans-cycloheptene is unstable >−40 °C).56 The work described herein showcases that subtle structural modifications can have dramatic effects on chemical reactivity relevant to polymer end-of-life management.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NSF Center for the Chemistry of Molecularly Optimized Networks (MONET; Award CHE-2116298). This work used Expanse at San Diego Supercomputing Center through allocation CHE140073 from the Advanced Cyberinfrastructure Coordination Ecosystem: Services and Support (ACCESS) program, which is supported by National Science Foundation Grants #2138259, #2138286, #2138307, #2137603, and #2138296. J. J. thanks Johns Hopkins University for a Provost's Undergraduate Research Award (PURA).References
- D. E. Fagnani, J. L. Tami, G. Copley, M. N. Clemons, Y. D. Y. L. Getzler and A. J. McNeil, ACS Macro Lett., 2021, 10, 41–53 CrossRef CAS PubMed.
- G. W. Coates and Y. D. Y. L. Getzler, Nat. Rev. Mater., 2020, 5, 501–516 CrossRef CAS.
- J. B. Young, R. W. Hughes, A. M. Tamura, L. S. Bailey, K. A. Stewart and B. S. Sumerlin, Chem, 2023, 9, 2669–2682 CAS.
- R. W. Hughes, M. E. Lott, I. S. Zastrow, J. B. Young, T. Maity and B. S. Sumerlin, J. Am. Chem. Soc., 2024, 146, 6217–6224 CrossRef CAS PubMed.
- M. R. Martinez, S. Dadashi-Silab, F. Lorandi, Y. Zhao and K. Matyjaszewski, Macromolecules, 2021, 54, 5526–5538 CrossRef CAS.
- H. S. Wang, N. P. Truong, Z. Pei, M. L. Coote and A. Anastasaki, J. Am. Chem. Soc., 2022, 144, 4678–4684 CrossRef CAS PubMed.
- M. T. Chin, T. Yang, K. P. Quirion, C. Lian, P. Liu, J. He and T. Diao, J. Am. Chem. Soc., 2024, 146, 5786–5792 CrossRef CAS PubMed.
- R. Whitfield, G. R. Jones, N. P. Truong, L. E. Manring and A. Anastasaki, Angew. Chem., Int. Ed., 2023, 62, e202309116 CrossRef CAS PubMed.
- V. Bellotti, K. Parkatzidis, H. S. Wang, N. De Alwis Watuthanthrige, M. Orfano, A. Monguzzi, N. P. Truong, R. Simonutti and A. Anastasaki, Polym. Chem., 2023, 14, 253–258 RSC.
- N. George and T. Kurian, Ind. Eng. Chem. Res., 2014, 53, 14185–14198 CrossRef CAS.
- H. Tang, N. Li, G. Li, A. Wang, Y. Cong, G. Xu, X. Wang and T. Zhang, Green Chem., 2019, 21, 2709–2719 RSC.
- M. Wang, Y. Li, L. Zheng, T. Hu, M. Yan and C. Wu, Polym. Chem., 2024, 15, 585–608 RSC.
- K. R. Delle Chiaie, F. R. McMahon, E. J. Williams, M. J. Price and A. P. Dove, Polym. Chem., 2020, 11, 1450–1453 RSC.
- J. P. Lutz, O. Davydovich, M. D. Hannigan, J. S. Moore, P. M. Zimmerman and A. J. McNeil, J. Am. Chem. Soc., 2019, 141, 14544–14548 CrossRef CAS PubMed.
- J. Yuan, G. J. Giardino and J. Niu, Angew. Chem., Int. Ed., 2021, 60, 24800–24805 CrossRef CAS PubMed.
- M. Hansen-Felby, A. Sommerfeldt, M. L. Henriksen, S. U. Pedersen and K. Daasbjerg, Polym. Chem., 2022, 13, 85–90 RSC.
- C. F. Gallin, W. Lee and J. A. Byers, Angew. Chem., Int. Ed., 2023, 62, e202303762 CrossRef CAS PubMed.
- D. Sathe, S. Yoon, Z. Wang, H. Chen and J. Wang, Chem. Rev., 2024, 124, 7007–7044 CrossRef CAS PubMed.
- R. J. Conk, S. Hanna, J. X. Shi, J. Yang, N. R. Ciccia, L. Qi, B. J. Bloomer, S. Heuvel, T. Wills, J. Su, A. T. Bell and J. F. Hartwig, Science, 2022, 377, 1561–1566 CrossRef CAS PubMed.
- P. Olsén, K. Odelius and A.-C. Albertsson, Biomacromolecules, 2016, 17, 699–709 CrossRef PubMed.
- J. Zhou, D. Sathe and J. Wang, J. Am. Chem. Soc., 2022, 144, 928–934 CrossRef CAS PubMed.
- W. J. Neary and J. G. Kennemur, ACS Macro Lett., 2019, 8, 46–56 CrossRef CAS PubMed.
- D. Zhang, X. Wang, Z. Zhang and N. Hadjichristidis, Angew. Chem., Int. Ed., 2024, 63, e202402233 CrossRef CAS PubMed.
- H. Martinez, N. Ren, M. E. Matta and M. A. Hillmyer, Polym. Chem., 2014, 5, 3507 RSC.
- D. Sathe, J. Zhou, H. Chen, H.-W. Su, W. Xie, T.-G. Hsu, B. R. Schrage, T. Smith, C. J. Ziegler and J. Wang, Nat. Chem., 2021, 13, 743–750 CrossRef CAS PubMed.
- H. Chen, Z. Shi, T. Hsu and J. Wang, Angew. Chem., Int. Ed., 2021, 60, 25493–25498 CrossRef CAS PubMed.
- H. Wakefield, I. Kevlishvili, K. E. Wentz, Y. Yao, T. B. Kouznetsova, S. J. Melvin, E. G. Ambrosius, A. Herzog-Arbeitman, M. A. Siegler, J. A. Johnson, S. L. Craig, H. J. Kulik and R. S. Klausen, J. Am. Chem. Soc., 2023, 145, 10187–10196 CrossRef PubMed.
- K. E. Wentz, Y. Yao, I. Kevlishvili, T. B. Kouznetsova, B. A. Mediavilla, H. J. Kulik, S. L. Craig and R. S. Klausen, Macromolecules, 2023, 56, 6776–6782 CrossRef CAS.
- H. Wakefield, S. J. Melvin, J. Jiang, I. Kevlishvili, M. A. Siegler, S. L. Craig, H. J. Kulik and R. S. Klausen, Chem. Commun., 2024, 60, 4842–4845 RSC.
- M. Scholl, S. Ding, C. W. Lee and R. H. Grubbs, Org. Lett., 1999, 1, 953–956 CrossRef CAS PubMed.
- A. K. Pearce, J. C. Foster and R. K. O'Reilly, J. Polym. Sci., Part A:Polym. Chem., 2019, 57, 1621–1634 CrossRef CAS.
- A. G. Moiseev and W. J. Leigh, J. Am. Chem. Soc., 2006, 128, 14442–14443 CrossRef CAS PubMed.
- G. S. Hammond and J. Saltiel, J. Am. Chem. Soc., 1962, 84, 4983–4984 CrossRef CAS.
- Y. Inoue, T. Kobata and T. Hakushi, J. Phys. Chem., 1985, 89, 1973–1976 CrossRef CAS.
- A. K. Chatterjee, T.-L. Choi, D. P. Sanders and R. H. Grubbs, J. Am. Chem. Soc., 2003, 125, 11360–11370 CrossRef CAS PubMed.
- G. Odian, Principles of polymerization, J. Wiley & sons, Hoboken (N.J.), 4th edn, 2004 Search PubMed.
- L. Montero De Espinosa and M. A. R. Meier, Chem. Commun., 2011, 47, 1908–1910 RSC.
- I. A. Gorodetskaya, T.-L. Choi and R. H. Grubbs, J. Am. Chem. Soc., 2007, 129, 12672–12673 CrossRef CAS PubMed.
- J. P. Morgan, C. Morrill and R. H. Grubbs, Org. Lett., 2002, 4, 67–70 CrossRef CAS PubMed.
- M. L. Randall, J. A. Tallarico and M. L. Snapper, J. Am. Chem. Soc., 1995, 117, 9610–9611 CrossRef CAS.
- M. Ulman, T. R. Belderrain and R. H. Grubbs, Tetrahedron Lett., 2000, 41, 4689–4693 CrossRef CAS.
- T.-L. Choi, C. W. Lee, A. K. Chatterjee and R. H. Grubbs, J. Am. Chem. Soc., 2001, 123, 10417–10418 CrossRef CAS PubMed.
- W. F. Drayer and D. S. Simmons, Macromolecules, 2022, 55, 5926–5937 CrossRef CAS.
- Q. Jiang, S. Wong and R. S. Klausen, Polym. Chem., 2021, 12, 4785–4794 RSC.
- J.-F. Lutz, M. Ouchi, D. R. Liu and M. Sawamoto, Science, 2013, 341, 1238149 CrossRef PubMed.
- Z. Li and Z. Li, in Sequence–Controlled Polymers, ed. J. Lutz, Wiley, 1st edn, 2018, pp. 349–377 Search PubMed.
- G. Si and C. Chen, Nat. Synth., 2022, 1, 956–966 CrossRef.
- G. Si, Z. Wang, C. Zou and C. Chen, CCS Chem., 2024, 1–10 Search PubMed.
- K. A. Parker and N. S. Sampson, Acc. Chem. Res., 2016, 49, 408–417 CrossRef CAS PubMed.
- T.-L. Choi, I. M. Rutenberg and R. H. Grubbs, Angew. Chem., 2002, 114, 3995–3997 CrossRef.
- H.-K. Lee, K.-T. Bang, A. Hess, R. H. Grubbs and T.-L. Choi, J. Am. Chem. Soc., 2015, 137, 9262–9265 CrossRef CAS PubMed.
- C. Chauveau, S. Fouquay, G. Michaud, F. Simon, J.-F. Carpentier and S. M. Guillaume, ACS Appl. Polym. Mater., 2019, 1, 1540–1546 CrossRef CAS.
- C. Chauveau, S. Fouquay, G. Michaud, F. Simon, J.-F. Carpentier and S. M. Guillaume, ACS Appl. Polym. Mater., 2020, 2, 5135–5146 CrossRef CAS.
- S.-Y. Shan, W. Zhang, Q. Cao, Y.-C. Ye, Z. Cai and J.-B. Zhu, Polym. Chem., 2024, 15, 1070–1076 RSC.
- M. L. Smith, T. M. McGuire, A. Buchard and C. K. Williams, ACS Catal., 2023, 13, 15770–15778 CrossRef CAS PubMed.
- M. E. Squillacote, J. DeFellipis and Q. Shu, J. Am. Chem. Soc., 2005, 127, 15983–15988 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplemental figures, experimental procedures, NMR spectra, SEC elugrams. See DOI: https://doi.org/10.1039/d4py01281j |
| This journal is © The Royal Society of Chemistry 2024 |