
Colorful variation of tetraphenylethene derivatives in the solid state
Guangxi
Huang
 ,
Xinxiang
Du
,
Heng
Bo
and
Bing Shi
Li
*
,
Xinxiang
Du
,
Heng
Bo
and
Bing Shi
Li
*
Key Laboratory of New Lithium-Ion Battery and Mesoporous Material, College of Chemistry and Environmental Engineering, Shenzhen University, 1066 Xueyuan Avenue, Nanshan, Shenzhen 518055, China. E-mail: phbingsl@szu.edu.cn
First published on 26th September 2023
Abstract
Stimuli-responsive chromic materials respond sensitively to mechanical, thermal, chemical vapor, and light stimuli. These responses are exhibited as mechanochromism, mechanoluminesence, thermochromism, vapochromism, photochromism and excitation wavelength dependent (Ex-De) luminescence, etc. As one of the most favourable aggregation induced emission luminogens with a twisted molecular conformation, the tetraphenylethene (TPE) scaffold has been widely used in the design of stimuli-responsive materials. This review provides a summary of the stimuli-responsive molecular design strategy, photophysical properties and application of these TPE-based molecular systems. The correlation between stimuli-responsive properties and different molecular structures, conformations and crystal packing modes is also discussed. The progress of these TPE-based stimuli-responsive materials offers a more comprehensive understanding of stimuli-responsive material design and provides helpful suggestions for future research.
 Guangxi Huang | Guangxi Huang received his BS in 2008 from Wuhan University. He then obtained his PhD degree from Institute of Chemistry, Chinese Academy of Science under the supervision of Prof. Deqing Zhang in 2013. During November 2013 to December 2016, he carried out his postdoctoral work at Geneva University and Fudan University for one and two years, respectively. From April 2017 to June 2021, he worked in Shenzhen University as an associated researcher. His current research interests are focused on multi-stimuli responsive materials and nanoimprint lithography. |
 Bing Shi Li | Bing Shi Li is a professor in the Department of Chemistry, Shenzhen University. She obtained her PhD degree from the Institute of Chemistry, Chinese Academy of Sciences in 2004 under the supervision of Prof. Chunli Bai and Prof. Ben Zhong Tang. She did her postdoc research in The University of Toronto from 2004 to 2007. She joined Shenzhen University in 2009. Her current research includes circularly polarized luminescence and stimuli-responsive materials. |
Introduction
In nature, chameleons and mimic octopus can alter their skin colors to match the surrounding background when the environment varies in order to stay out of danger. This prompt stimuli-responsive ability has attracted a lot of research attention. With the aim of mimicking these extraordinary stimuli-responsive behaviors, scientists have devoted great efforts to develop a large number of stimuli-responsive materials.1–8 These materials are capable of changing their macro-/micro- physical or chemical properties, such as appearance color, luminescence, shape, conductivity, hydrophobicity, hydrophilicity, viscosity, etc, in response to diverse external stimuli.1 Among them, the variation of fluorescence properties and appearance color upon external stimuli is the most commonly used feature for the advantage of easy detection and low cost. According to external stimuli including light, temperature, force, electricity, magnet, and so on, stimuli-responsive chromic materials can be divided into the corresponding categories as follows: photochromism,9–12 thermochromism,13–15 mechanochromsim,16–26 electrochromism,27–30 and magnetochromism.31,32 These stimuli-responsive chromic materials have exhibited a wide range of promising applications in chemosensors,33–36 biosensors,37–39 photoelectric devices,40–42 displays,43–45 anti-counterfeiting,46,47etc.Designing desirable stimuli-responsive molecules that can respond to external stimuli in a controllable and predictable fashion is an appealing but challenging task.48–52 There are several key points to be considered in the design. First, the synthesis of molecules should be a facile and feasible process. Complicated molecular design and synthesis will restrict their applications. Second, the speed, amplitude, sensitivity and reversibility of the response determine the performance of the materials and their commercial potentiality, which need to be comprehensively taken into account. Third, though a structure–property relationship has been gradually presented, the construction of most stimuli-responsive materials is still lacking an efficient design theory and researchers usually make some decoration on the basis of previous reported molecular structures. Therefore, the development of novel stimuli-responsive chromic materials will remain a hot topic for many years.
In 2001, Tang et al. discovered the “aggregation-induced emission” (AIE) phenomenon of an organic molecule 1-methyl-1,2,3,4,5-pentaphenylsilole.53 Thereafter, the investigation of the photophysical properties of organic AIE luminogens in the powder state, film state, nanoparticle state and in rigid matrices becomes very prosperous, which greatly enriches the whole area of luminogens. To date, AIE materials have been widely applied in various fields, such as organic luminescence devices, chemo- and bio-sensors. Among them, tetraphenylethene (TPE) derivatives have gradually grown into a class of key and representative AIE molecules, which is most likely related to their advantages of simple molecular structure and synthetic route, convenient chemical modification and excellent AIE features. Moreover, thanks to their twisted molecular conformation, relatively weak intermolecular interactions, easy crystallinity and obvious luminescence in the solid state, TPE derivatives were found to display various fluorescence color changes from blue to green to yellow even to white upon the stimulation of multiple external stimuli in recent years. Thus, TPE exhibits great potential in the construction of stimuli-responsive chromic materials.
In consideration of the ever-growing interest and great progress in organic stimuli-responsive materials, this review will provide an overview of the published studies in the area of organic materials showing responses to mechanical, thermal, chemical, and light stimuli. Due to the huge database based on different structural types, we only focus on the TPE-based derivatives. In particular, according to the type of external stimuli, the mechanochromism, solvent guest responsibility, mechanoluminesence, photochromism, photo switch and excitation wavelength dependent (Ex-De) luminescence of the reported TPE derivatives are summarized (Fig. 1). The related mechanistic study and application of different stimuli-responsive properties of TPE derivatives will be given as well. Finally, challenges and prospects in this research area are also discussed. Given the limited length of this review, stimuli-responsive properties of TPE derivatives in solution,54–58 silicone oil,59–61 gel,62 polymer matrix,63–66 or stimuli-responsive TPE-based liquid crystals,67 and polymers68–71 are excluded. To obtain a more comprehensive understanding, other prevalent reviews on the topics of specific stimuli-responsive materials or stimuli-responsive AIEgens will be helpful for the readers.72–79
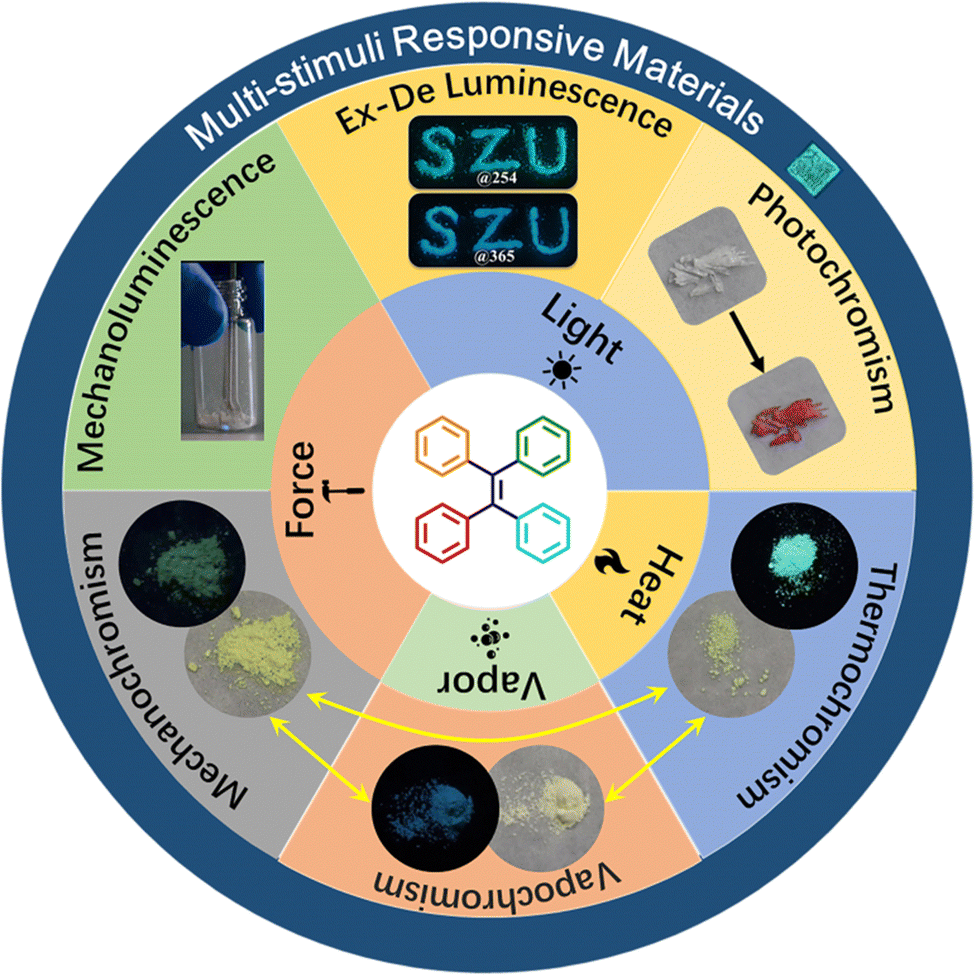 | ||
| Fig. 1 Schematic illustration of different external stimuli and stimuli-responsive properties exploited for TPE derivatives. | ||
Mechanochromism
The first TPE-based mechanochromic (or mechanofluorochromism, MFC) material, 1, can be traced back to 2011.80 This molecule was derived from the covalent connection of one anthracene and two TPE moieties (Fig. 2A), which was reported by Chi and Xu's group. Inherited from the AIE properties of TPE, compound 1 emitted observable fluorescence in the solid state. As shown in Fig. 2B and 2C, the yellow powder showed green emission at 506 nm; after grinding it turned into reddish-orange powder and emitted yellow fluorescence centered at 574 nm. Upon thermal annealing at 340 °C for 1 min and then grinding, both states could be reversibly transformed into each other without deterioration. They found the absorption spectra of 1 showed a significant change after grinding, and thus excluded excimer formation and believed planarization of molecular conformation should be more reasonable. The PXRD results demonstrated the change of fluorescence was attributed to phase transition between crystalline and amorphous states. DSC results revealed a cold-crystallization peak at approximately 336 °C for the ground sample, which suggested the amorphous state was metastable and could be converted into a more stable crystalline state through heating. All these results indicated that mechanochromism was related with the alteration of conformation and aggregation state. More experiments were carried out to press/grind over thirty new AIE compounds and from the results it was determined that almost all of them exhibited a piezofluorochromic effect. Hence these luminogens were named “piezofluorochromic aggregation-induced emission (PAIE) compounds” for their dual properties of MFC and AIE.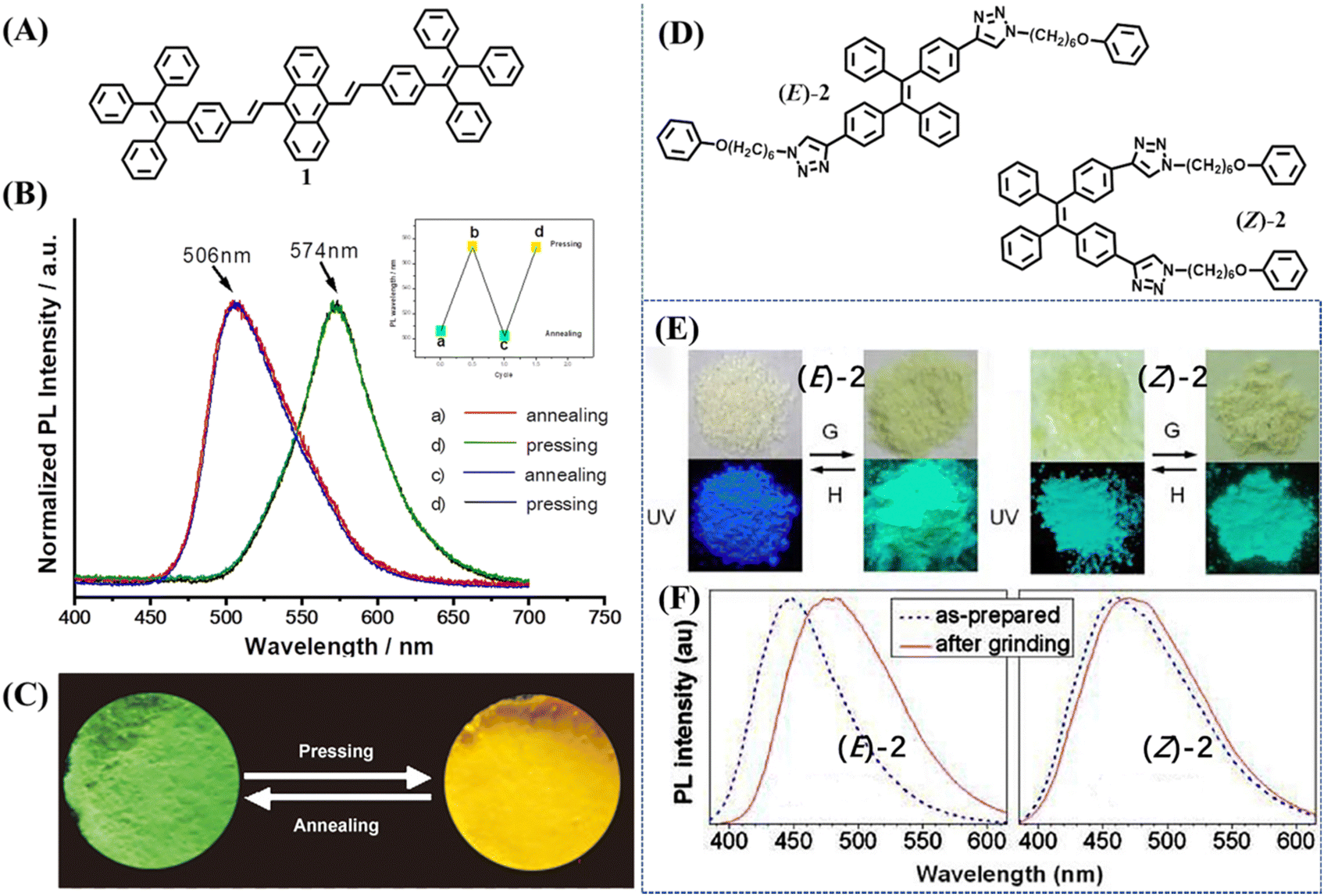 | ||
| Fig. 2 (A) The molecular structure of 1. (B) Normalized fluorescence spectra of 1 after annealing at 340 °C for 1 min and pressing. The inset shows the reversible recycle of fluorescence. (C) The images of annealed sample and pressed sample of 1 taken at room temperature. Reproduced with permission.80 Copyright 2011, Wiley-VCH. (D) The molecular structure of 2. (E) Photographs of the as-prepared (left) and ground (right) samples of (E)-2 and (Z)-2 taken under room lighting (upper) and UV illumination (lower); G = grinding; H = heating (at 120 °C for 1 min). (F) Fluorescence spectra of (E)-2 and (Z)-2 before and after grinding. Reproduced with permission.101 Copyright 2012, American Chemical Society. | ||
After that, PAIE compounds sprang up like mushrooms. As an important part of PAIE compounds, paramount TPE-based mechanochromic materials are involved, especially those with two fluorescence states.81–100 In recognition of this, here we only specify a few representative compounds/phenomena in this review.
In 2012, Sun et al. reported the mechanochromism of pure stereoisomers of a TPE derivative 2 (Fig. 2D).101 The as-prepared (E)-2 was an off-white solid with blue emission, whereas its isomer (Z)-2 was a pale-yellow solid with bluish-green emission (Fig. 2E). After mechanical grinding, (E)-2 showed a mechanochromic effect with fluorescence and appearance changing into bluish-green and pale-yellow, respectively. The mechanochromic activity of (Z)-2 was very weak and grinding merely caused its emission peak to redshift by 10 nm with a negligible change in the appearance color (Fig. 2F). The XRD patterns clearly indicated that E-isomer had intense and sharp reflection peaks and the Z-isomer only displayed a few blunt “peaks”, which revealed the different crystallization capability of the as-prepared powder, accounting for their distinct mechanochromic property. These results also suggested the correlation between mechanochromism and the modes of molecular packing.
Unlike most mechanochromic materials displaying two distinct emission colors, some special molecules showed intriguing turn-on luminescence in response to the mechanical grinding. The high contrast luminescence of mechanical stimuli-responsive materials was valuable in the practical applications of optical recording and sensing, which was also an important approach in exploring the mechanochromic mechanism. Tian et al. synthesized compound 3 through incorporating a twisted TPE skeleton and a rigid acridone moiety as an electron donor and acceptor (Fig. 3A), respectively.102 The as-prepared powder of 3 had very weak blue emission with the quantum yield of only 1% ignoring the presence of TPE. But its ground powder showed extremely bright cyan fluorescence with a 43-fold enhancement of the quantum yield to 43% (Fig. 3B and C). PXRD results confirmed that the two contrast states were related to the crystalline and amorphous state (Fig. 3D). Crystallographic analysis revealed that there were no face-to-face π–π interactions between adjacent molecules; large steric hindrance from the twisted TPE unit led to relatively loose packing of 3 crystals, which were submissive to transformation into the amorphous state with quenched emission under mechanical stimulus.
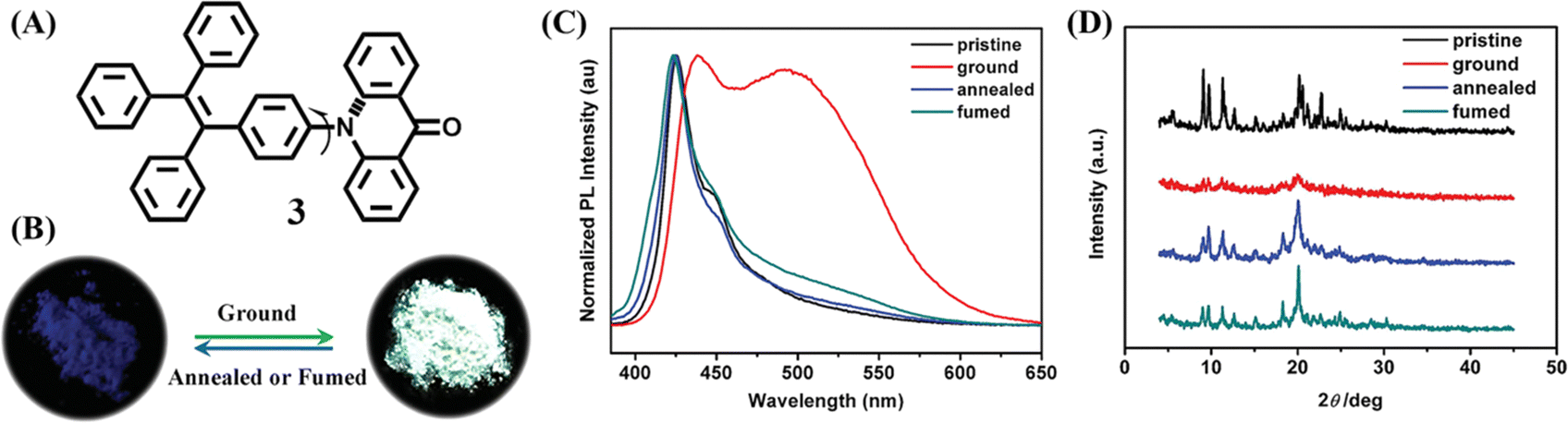 | ||
| Fig. 3 (A) Structural formula of 3. (B) Fluorescence images 3 under different treatments. (C) Fluorescence spectra and (D) PXRD patterns of powders of 3 under different treatments: pristine, ground and annealed sample (150 °C for 1 min) and fumed sample (fumed with ethyl acetate). Reproduced with permission.102 Copyright 2015, Wiley-VCH. | ||
A novel strategy for developing turn-on mechanochromic materials was explored by Chi's group103 and Tang's group.104 They introduced different numbers of nitro groups into the TPE skeleton (Fig. 4A). All the compounds 4–7 inherited the AIE property of the TPE skeleton, but their single crystals exhibited different emission intensities: crystals 4 and 5 showed intense emission with a high quantum yield of more than 20%, while 6 and 7 were nearly non-emissive. After grinding into amorphous powder, their emission intensified as the fluorescence on–off ratio increased from 1.2 to 1417 with the increase of nitro groups (Fig. 4B). The emission could be turned off upon heating or exposure to acetone/dichloromethane vapor. To gain insight into the mechanism of turn-on fluorescence, Chi et al. analyzed the molecular packings from the view of crystal structure. They considered the porous structure in 6 and 7 provided sufficient space for the intramolecular rotations of nitrophenyl groups (Fig. 4C), which ultimately quenched the emission. Tang et al. performed theoretical calculations on single molecules in both gas and crystalline state. The Nitro group could facilitate the formation of a non-radiative ISC channel, and more nitro groups would further boost this process, leading to the fluorescence quenching of 6 and 7. Once the crystal structure was destroyed, variations of molecular packing and conformation would block the efficient ISC process and switched on the fluorescence in the amorphous state. The high contrast and fast responsive fluorescence on–off performance of 6 and 7 were reversible and sensitive. On this basis, the fluorescence-off state of 7 was adopted to display fingerprints on aluminum, ceramic and wooden substrates upon pressing with fingers.104 At the same time, Tang and Zhang et al. used this ultra-sensitive fluorescence switch to visualize metal stress/strain distribution and fatigue crack propagation.105 As illustrated in Fig. 4D, an amorphous film of 7 was first coated on a metallic tensile specimen with bright green fluorescence and then annealed to quench its fluorescence. Due to the sensitivity of mechanical stimuli, the crystalline film of 7 cracked into fluorescent amorphous fragments upon stretching (Fig. 4E). Around the notch and hole in the metallic specimen, the strong fluorescence of 7 was turned on and gave the mapping of stress/strain concentrations with the merits of real-time, full-field, and on-site visualization. These results demonstrated the great potential of the fluorescence switch of the mechanochromic material for mechanical analysis on various metals and different application scenarios.
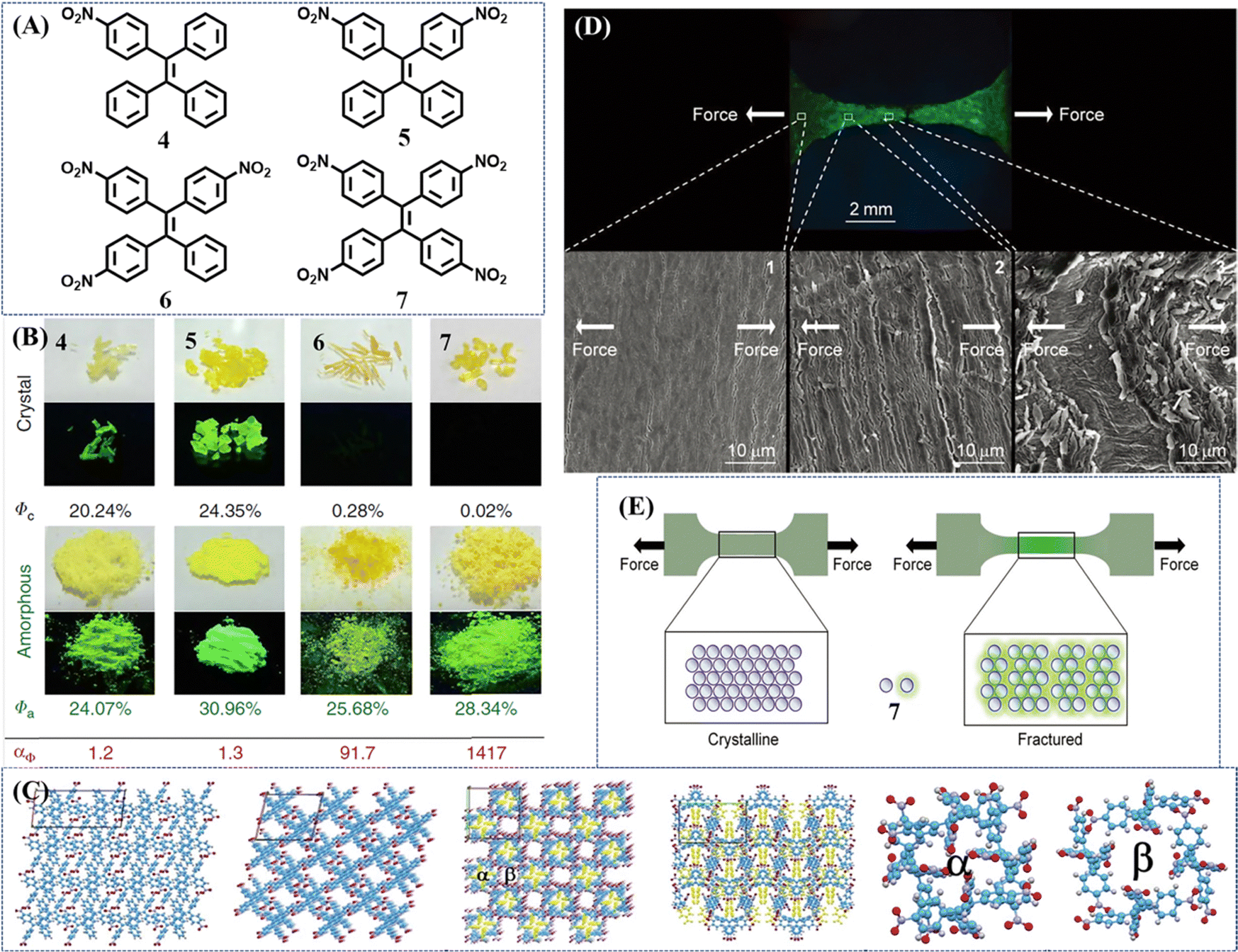 | ||
| Fig. 4 (A) Chemical structures of compounds 4–7. (B) Photos of compounds 4–7 in crystalline and amorphous states taken under room and UV light irradiation with their quantum yields (Φ). Luminescence contrast ratio: αΦ = Φa/Φc. Reproduced with permission.104 Copyright 2018, Nature Publishing Group. (C) From left to right: the single crystal structure of 5; single crystal structure of 6; single crystal structure of 7 viewed down the c axis; single crystal structure of 7 viewed down the b axis (the nitro substituted phenyls in pore α are labeled in yellow); structure of pore α in 7; structure of pore β in 7. Reproduced with permission.103 Copyright 2017, Royal Society of Chemistry. (D) Fluorescence image of fractured steel tensile specimen and SEM images of the selected areas for surface analysis. Direction of stretching force: horizon. The cracks were approximately vertical to the direction of force. (E) Working mechanism of 7-coating for stress/strain visualization. Reproduced with permission.105 Copyright 2018, Wiley-VCH. | ||
Although most mechanochromic TPE derivatives could achieve a reversible two-color transformation, only a limited number of multicolor switched materials were available due to a lack of universal design principles. One of these approaches is the facilitation of acid or base stimuli, which apparently enriches current molecular structures and their fluorescence states. Chi et al. developed a TPE derivative 8 with remarkable four-colored switching (Fig. 5A).106 The as-prepared powder 8B exhibited strong blue emission. After exposure to HCl vapor, benzothiazole was protonated accompanied by an enhanced electron-withdrawing ability; the push–pull effect of the whole molecule was consequently strengthened and resulted in the redshift of the HCl-fumed powder to yellow emission (8Y). After subsequent exposure to NH3 vapor, the yellow emission reverted to its initial blue emission. Meanwhile, grinding could transform the as-prepared powder into green emissive state 8G, which was recovered upon heating or organic CH2Cl2 fuming. The ground powder 8G could also undergo a HCl-NH3 vapor cycle with the alternation of orange (8O) and green (8G) fluorescence. Additional transition occurred from the yellow emissive 8Y to orange emissive 8O after grinding and both these deprotonated states could be restored to the as-prepared state 8B by heating.
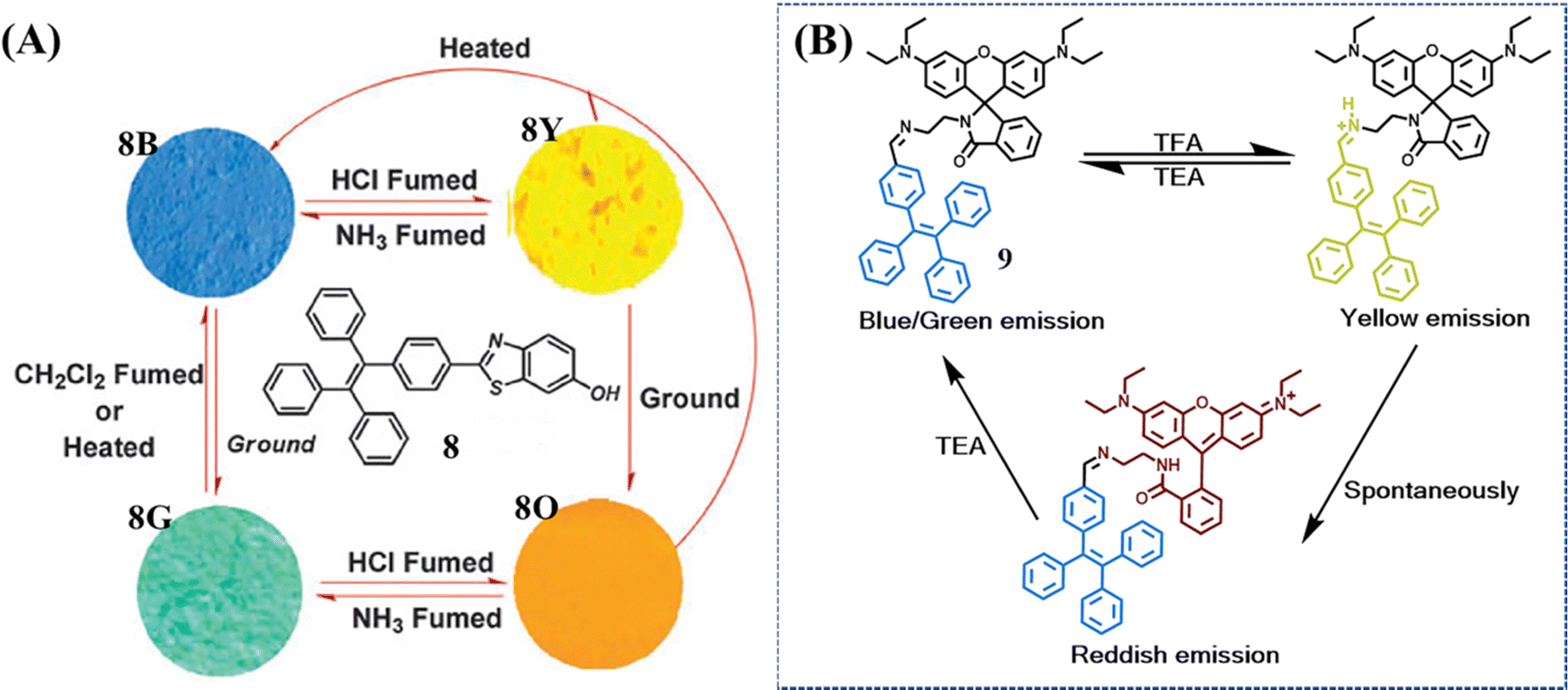 | ||
| Fig. 5 (A) Molecular structure of compound 8 and fluorescence images of the powders. 8B: as-prepared 8; 8G: ground sample; 8Y: 8B in the vapor of HCl for 10 min; 8O: 8G in the vapor of HCl for 10 min (excitation wavelength: 365 nm). Reproduced with permission.106 Copyright 2014, Royal Society of Chemistry. (B) The multicolored acidichromic property of 9. Reproduced with permission.107 Copyright 2019, Royal Society of Chemistry. | ||
Another approach is to merge a reactive moiety with a TPE skeleton. Ma et al. took advantage of the acid-induced ring-open reaction of rhodamine to construct a multicolor switched molecule 9 (Fig. 5B).107 The as-prepared powder and ground powder achieved a color transition from blue/green to yellow when fuming with trifluoroacetic acid (TFA). After removing TFA vapor, the yellow emissive powder gradually changed to red emissive powder. The yellow state was mainly ascribed to the protonation of the imine group, which was not stable enough thus spontaneously transferred to the ring-open form of rhodamine lactam (red state). When TFA was neutralized by trimethylamine (TEA), both the yellow and red state returned to the as-prepared blue state.
Fuming with acid or amine vapor though can produce multicolor mechanochromic materials, it is not an optimal method compared to thermal or optical stimuli because of using harmful and irritant vapor as the stimuli. A novel design strategy thus needs to be developed.
In 2016, Dong et al. synthesized a tricolored mechanochromic compound 10 by combining a TPE unit and rhodamine B with the presence of the boron atom (Fig. 6A).108 Without the boron atom, compound 11 could not be cultured into a single crystal and only displayed two-color mechanochromism. The as-prepared powder of 10 was in a crystalline state with blue emission, which was transformed to blue-green emission by gently grinding (Fig. 6B and C). Further intensively in situ grinding induced the emergence of reddish fluorescence. Heating the reddish state induced the recovery of a blue-green state, but the original blue state could not be restored by either thermal or solvent treatment. The tricolored switch behavior was explained as follows: the blue-green state was the result of disorderly packing and conformation planarization; the reddish state originated from the ring-open reaction of the rhodamine B moiety due to the greater energy input of intensively grinding. PXRD revealed that the ground states of 10 and 11 were amorphous, so BH3 was considered to play a vital role in the formation of the blue crystalline state.
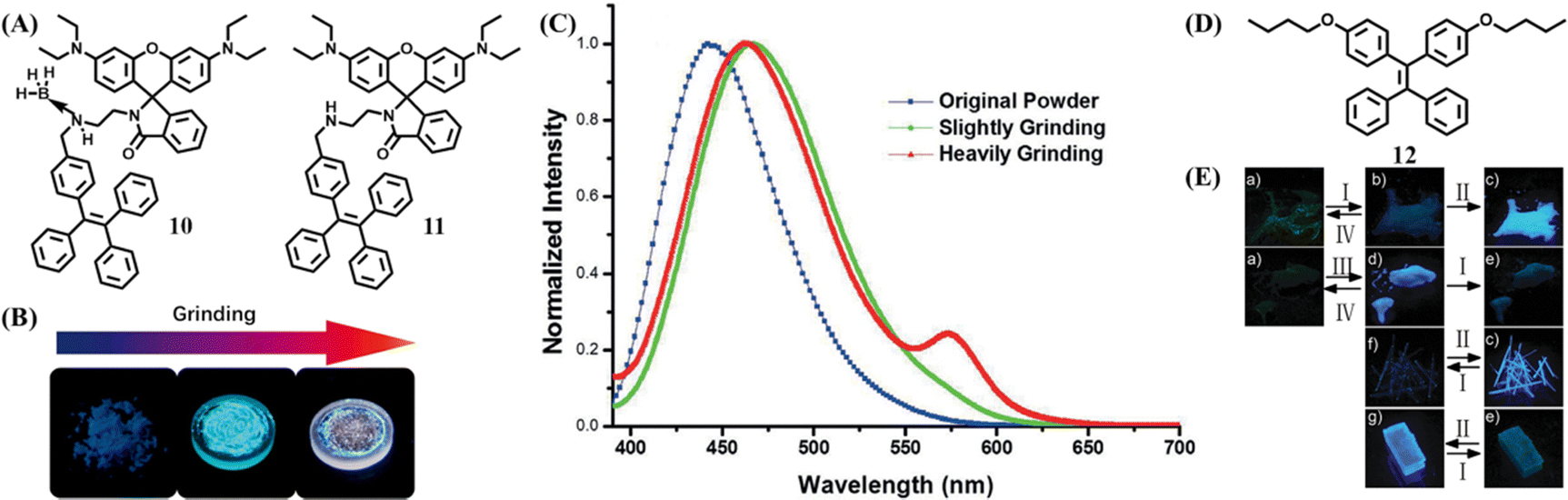 | ||
| Fig. 6 (A) The chemical structures of 10 and 11. (B) Optical images of the original powder of 10 and powder 10 after slight and continuous grinding. (C) Fluorescent spectra of 11. Reproduced with permission.108 Copyright 2016, Wiley-VCH. (D) The chemical structures of 12. (E) Photos of 12Am (a), 12CB (f) and 12CA (g) after different treatments. (I, 90 °C; II, fuming with diethyl ether vapor; III, fuming with diethyl ether vapor; IV, heating to melt and cooling with liquid nitrogen). Reproduced with permission.109 Copyright 2017, Royal Society of Chemistry. | ||
The TPE derivative could still exhibit multistate mechanochromism without the assistance of the reactive group. Dong et al. reported a butoxyl group decorated TPE derivative 12 (Fig. 6D), which displayed three fluorescence states with high contrast in both emission color and efficiency.109 The wo crystalline states of 12 (12CA and 12CB) had obvious distinct quantum yield (Fig. 6E): 12CA was deep blue emissive (λem = 420 nm, Φfl = 50.4%) and 12CB was sky blue emissive (λem = 460 nm, Φfl = 8.3%). Its amorphous powder 12Am showed green emission (λem = 496 nm, Φfl = 7.6%), which could be converted to 12CB by heating at 90 °C for 30 min or to 12CA by fuming with diethyl ether vapor for 5 min. Heating 12CA or 12CB to melt and then cooling with liquid nitrogen led to the recovery of 12Am. Moreover, 12CA and 12CB could be switched into each other by annealing at 90 °C for 30 min and exposure to diethyl ether vapor for 15 min. The regeneration of 12Am was slightly troublesome, as it required heating 12CA or 12CB to melt and then cooling with liquid nitrogen. These results indicated the transformation among the three states of 12 was completely reversible.
Compound 13 was the first example to realize an intact three-state cycle through a simple grinding/fuming/annealing process, which is shown in Fig. 7A.110 The molecule was obtained by incorporating a TPE skeleton with carboxyl and methoxyl groups. The yellow ground powder 13p-g emitted yellow fluorescence (λem = 496 nm, Φfl = 10.5%), which could be prepared through easy grinding from other states and also ascribed to the amorphous state (Fig. 7B). When 13p-g was fumed with acetone (or other solvents) for 3 min, it transformed into a yellowish-white fumed state 13p-f with blue emission (λem = 462 nm, Φfl = 7.4%). Three single crystals of 13 (13c-a, 13c-c and 13c-d) displayed a porous hydrogen-bonded organic framework (HOF) with or without solvent molecules, but their photophysical properties and PXRD patterns resembled those of 13p-f (Fig. 7C), indicating their close identity. Heating the above powder or crystalline states at 140 °C for 3 min produced the light-yellow heated state 13p-h, which exhibited very strong cyan fluorescence (λem = 482 nm, Φfl = 82.3%). According to the single crystal structure analysis and PXRD spectra, the porous HOF structure was considered a key factor for the weak blue emission. When the HOF structure was destroyed by heating, the molecular packing became closer and the non-radiative energy loss was inhibited, leading to the intense increase of quantum yield. Thanks to the reversible transformation among three states, a tricolored Chinese seal pattern was successfully drawn with the powder of molecule 13 (Fig. 7D).
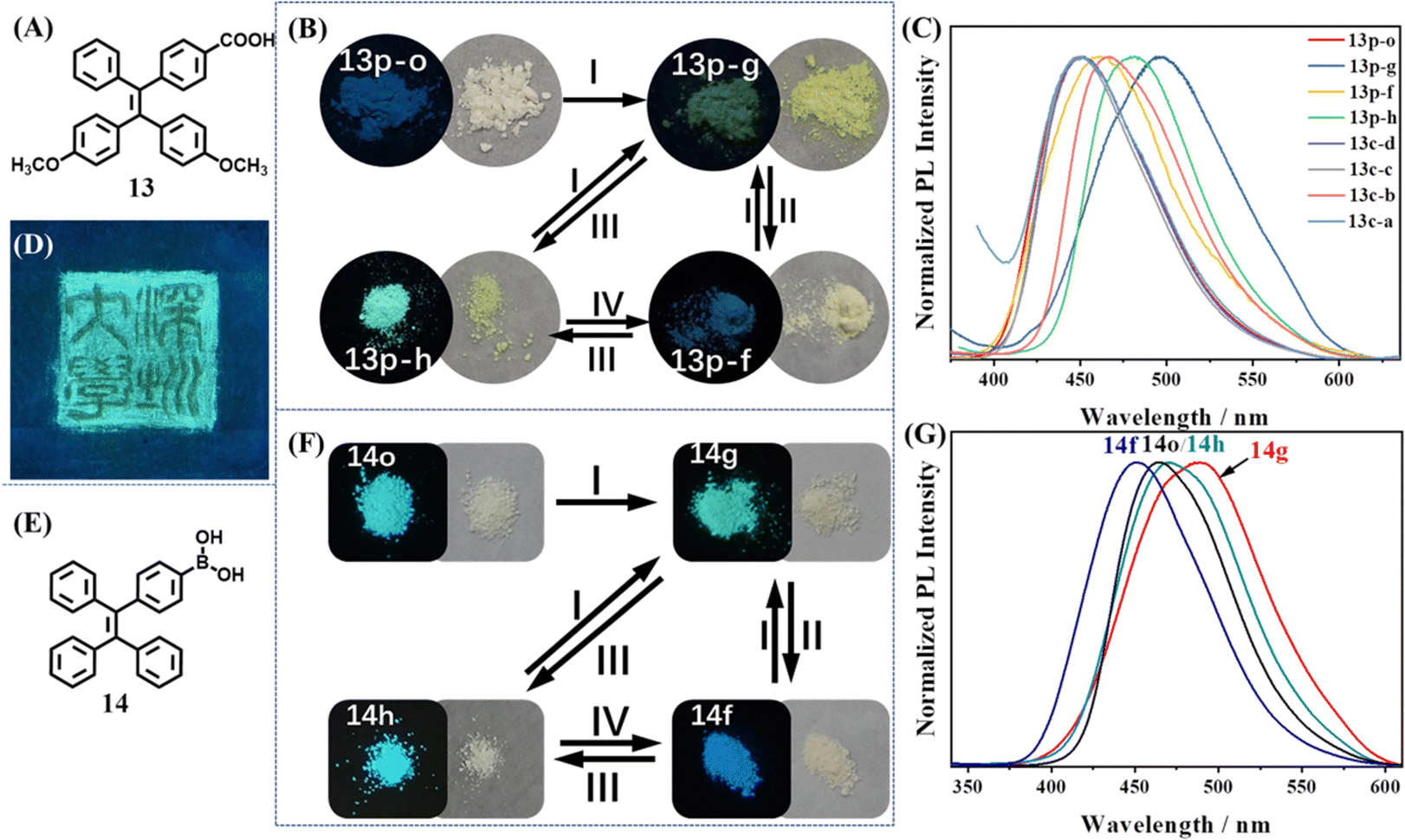 | ||
| Fig. 7 (A) The chemical structures of 13. (B) Photos of 13 upon different treatments (I, grinding; II, fuming with solvent vapor; III, annealing at 140 °C; IV, fuming with THF or acetone vapor). (C) Normalized PL intensity of different states of 13. (D) The tricolored Chinese seal pattern drawn with the powder of molecule 13 on a weighing paper under UV illumination at 365 nm. Reproduced with permission.110 Copyright 2019, Wiley-VCH. (E) The chemical structures of 14. (F) Photos of 14 in different powder states after different treatments (I, grinding; II, fuming with methanol; III, annealing at 145 °C; IV, fuming with methanol). (G) Normalized PL intensity of the four powder states of 14. Reproduced with permission.111 Copyright 2022, Royal Society of Chemistry. | ||
Recently, our group discovered an excellent tricolored mechanochromic property of a commonly used TPE intermediate 14 with simple chemical structures (Fig. 7E).111 As shown in Fig. 7F, the amorphous ground state 14g showed strong green emission (λem = 490 nm, Φfl = 50.9%). After fuming with methanol for 1 min or heating at 145 °C for 5 min, 14g was converted into the blue emissive crystalline state 14f (λem = 449 nm, Φfl = 48.1%) or cyan emissive crystalline state 14h (λem = 465 nm, Φfl = 52%). 14h had similar PXRD and photophysical properties with those of the as-prepared powder 14o. 14h and 14f could be transformed into each other by fuming with methanol for 10 min and heating at 145 °C for 5 min (Fig. 7G). All three states had high fluorescence quantum yields and therefore their color could be distinguished by naked eyes. The interconversion cycles among 14g, 14f and 14h could be repeatedly realized through alternate grinding/fuming/annealing processes without any deterioration. The potential application of 14 in optical recording and anticounterfeiting was also explored.
Although the above three molecules exhibited a three-state mechanochromic property, the time of their transformation among different states was still too long, which would restrain their applications. Our group developed two three-state mechanochromic TPE derivatives 15 and 16 with fast responsiveness,112,113 which was a pair of isomers (Fig. 8A and D). They were synthesized through a one-step reaction between a TPE moiety and different substitution positioned cholesterol moiety in high yields. The TPE and cholesterol moiety had no strong hydrogen bond donor group or receptor group, determining their weak intermolecular interactions. The single crystal of 15 was successfully cultivated, but the single crystal of 16 failed to be obtained due to its highly twisted conformation. The crystal of 15 had loose packing because no intermolecular π–π stacking and few C–H⋯O intermolecular interactions were formed (Fig. 8C). This loose packing mode probably made its crystalline state susceptible to external stimuli. Conversely, the crystalline state should be easily and quickly recovered from other states, resulting in excellent multistimuli-responsive properties. All states of both molecules showed a white appearance but different fluorescence color. The reversible three states of 15 were as follows (Fig. 8B): cyan emissive ground state 15g (λem = 465 nm, Φfl = 22.0%), blue emissive fumed state 15f (λem = 450 nm, Φfl = 24.8%) and dim heated state 15h (λem = 457nm, Φfl = 1.1%). The response time for interconversion among the three states of 15 was quick, for example, annealing of 15g or 15f at 120 °C for 10 s almost quenched their fluorescence, corresponding to the generation of 15h. The emissions of three states of 16 under 365 nm UV light irradiation were relatively weak compared to those of 15. As shown in Fig. 8D, the color changing of 16 was sky-blue to deep-blue to dim, corresponding to its ground state 16g (λem = 460 nm, Φfl = 10.53%), fumed state 16f-dh (with CH2Cl2 and hexane, λem = 451 nm, Φfl = 5.52%) and heated state 16h (λem = 460 nm, Φfl = 2.39%), respectively. The fluorescence spectrum of 16g resembled that of 16h, though one was amorphous and the other was in the crystalline state (Fig. 8E). The three states of both molecules could interconvert repeatedly without any deterioration, revealing excellent reversibility.
 | ||
| Fig. 8 (A) The chemical structures of 15. (B) Photographs of 1 upon the treatments of grinding (I), fuming with solvent vapor (II), annealing at 120 °C (III) and fuming with DCM vapor (IV), respectively. (C) Intermolecular interactions between molecules of crystal 15c viewed along the b-axis. Reproduced with permission.112 Copyright 2019, Wiley-VCH. (D) The molecular structure and photographs of 16 upon the treatment of grinding (I), annealing at 130 °C (II), fuming with dichloromethane and hexane vapor (III), fuming with dichloromethane and hexane vapor (IV), fuming with hexane vapor (V), heating to molten state and cooling, then annealing at 130 °C (VI) and crystallizing from acetone/H2O solution (VII), respectively. Photos were taken under available UV lamps (@365/@302/@254), respectively. (E) PXRD patterns of all the corresponding states of 16. Reproduced with permission.113 Copyright 2023, CCS Chemistry. | ||
Solvent guest regulated fluorescence
In common vapochromism phenomenon, solvent molecules only played a role in accelerating the ordered arrangement of host molecules. When the crystalline state of host molecules recovered, solvent molecules escaped and only the host remained. But for acid-stimuli-responsive or base-stimuli-responsive molecules, acid (HCl, AcOH, TFA etc.) or amine (NH3, TEA, etc) vapor did not leave the host molecules; instead they reacted with them and changed their chemical structures. Recently, several porous materials, HOFs, which were constructed from pure organic building blocks through hydrogen-bonding interactions, displayed a different vapochromism phenomenon. The solvent molecules acted as guests to coexist with host molecules and regulate the emissive property of HOFs.Chi et al. decorated the TPE skeleton with four nitrobenzene moieties (Fig. 9A).114 The phenyl rings rendered the new molecule 17 with more flexibility, while the nitro groups could help to form HOF through hydrogen bonds. The rotation of phenyl rings was expected to provide enough spatial tolerance through conformational changes, achieving accommodation of solvent molecules with different sizes. The frameworks of most reported HOFs collapsed upon removal of the solvent molecules from the pores. However, flexible HOFs constructed from 17 displayed unexpected permanent porosity. Single crystal to single crystal transformation could take place during the removal of the ethyl acetate (EA) guest from 17-EA (EA fumed state) to 17-Heated. During this transformation, the crystalline framework was firmly retained. When the solvent-free 17-Heated was exposed to the corresponding solvent vapors, structural transformations of 17 into host–guest co-crystals were realized, accompanied by changes in emission color, lifetime and brightness (Fig. 9B). Moreover, different states of 17 with or without solvent molecules could be converted into each other by multiple external stimuli including heating, grinding and solvent fuming. This powerful multi-stimuli responsive HOF enriched the application fields of existing porous materials. When the nitro group of 17 was replaced with a hydroxyl group, the new molecule 18 could also form HOF (Fig. 9C).115 In this HOF, X-shaped molecules 18 arranged into 1D strands via intermolecular O–H⋯O hydrogen bonds (Fig. 9D). Then the interlocking of one-dimensional (1D) strands was woven into a dynamic two-dimensional (2D) topological structure. This type of 2D structure possessed exceptional flexibility by virtue of flexible molecules 18 and much softer O–H⋯O hydrogen bonds. When a protic solvent MeOH molecule was introduced into the 2D woven structure, the intermolecular O–H⋯O hydrogen bonds of molecules 18 were disturbed, but the MeOH molecule could act as a bridge to link two original molecules 18 and maintained the whole 2D woven structure (Fig. 9E). Therefore, the green fluorescence of amorphous HOF was transformed into cyan after the fuming with MeOH. Similar results were observed when amorphous 18 was exposed to EA.
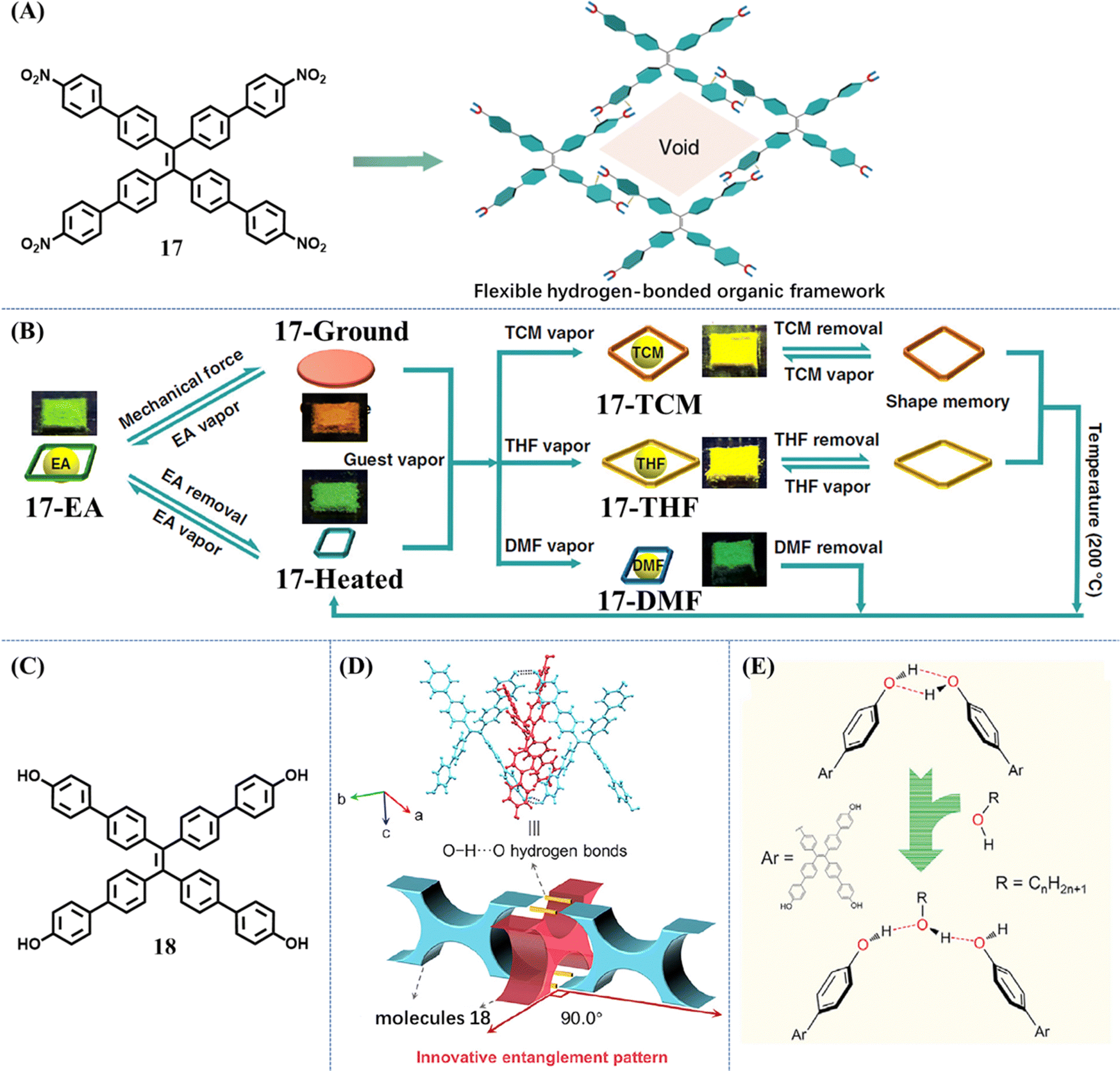 | ||
| Fig. 9 (A) The flexible framework is constructed from molecule 17 through hydrogen-bonding interactions. (B) Illustration of the multimode reversible structural transformations realized in 17 including transitions between various porous phases as well as transitions between nonporous and porous states in response to different kinds of external stimuli. Reproduced with permission.114 Copyright 2019, Nature Publishing Group. (C) The chemical structures of 18. (D) The interlocking mode at the entanglement location in the XRD structure of crystal 18 (top) and the corresponding schematic diagram (bottom). (E) Illustration of the influence of alcohols on the formation of O–H⋯O hydrogen bonds in the molecular woven framework. Reproduced with permission.115 Copyright 2021, Elsevier. | ||
Recently, Wei et al. developed a special TPE-based HOF based on molecule 19.116 It had diversified kinetic-stable porous and thermo-stable nonporous crystals. Due to dynamic intermolecular interactions and flexible molecular conformation, the transformations among different crystalline phases under the stimulation of different solvent vapor and temperature could be readily achieved (Fig. 10A), accompanying a fluorescence change. It is worth mentioning that when flexible long-chain alkane guests were entrapped in the cavity of this HOF, the corresponding five crystals emitted near pure white-light (Fig. 10B). The shorter-wavelength emission centered at 450–470 nm can be reasonably classified as the intrinsic emission. The yellow emission centered at 550–570 nm was deduced to be relaxed from the intrinsic blue emission. This is because disordered alkane molecules in the crystals offered the benzonitrile groups adequate room of motion, resulting in the relaxation of the excited state. The dual emissions generated from the same molecular skeleton through flexible environment induction should be an alternative strategy to obtain white-light emission materials.
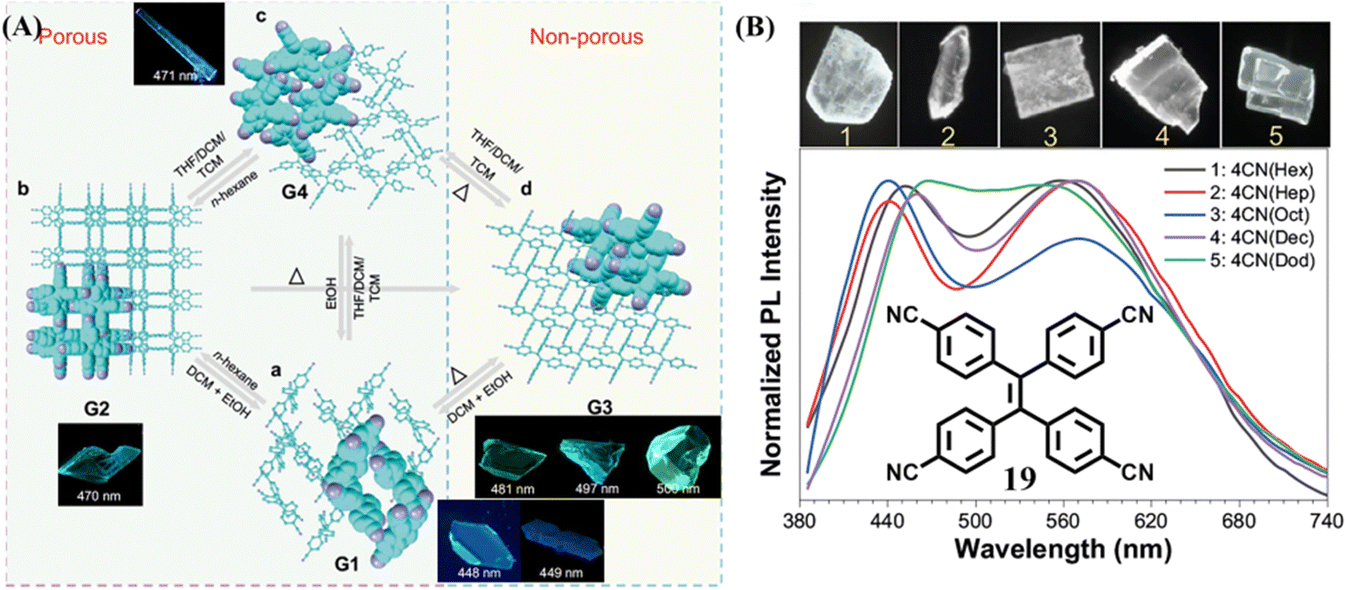 | ||
| Fig. 10 (A) Multimode reversible transformations of 19. (B) Normalized fluorescent emission spectra and corresponding fluorescent images of five white-light crystals of 19. Reproduced with permission.116 Copyright 2022, Nature Publishing Group. | ||
Mechanoluminescence
TPE derivatives have achieved great success in designing mechanochromic materials. It is not exaggerative to say, TPE derivatives have become the main stream of mechanochromic materials for their simple synthesis. As a category of multiple functional molecules, the TPE derivative still has a place in the field of mechanoluminescence materials. However, the characteristic mechanoluminescence feature of TPE-based mechanoluminescence materials remained to be revealed compared to other organic skeletons, such as triphenylamine, carbazole, pyrene, phenothiazine, and phthalimide, etc.79 TPE-based derivatives only generate blue or green mechanoluminescence but do not exhibit mechanical induced phosphorescence,117 tri-color mechanoluminescence,118 color-changeable mechanoluminescence,119,120 or solar-renewable mechanoluminescence,121etc., thus the exploration of TPE derivatives was relatively unattractive and slow.In 2015, Chi and Xu et al. reported the first TPE-based mechanoluminescence molecule 20 (Fig. 11A),122 whose two crystalline polymorphs displayed distinct luminescent properties. The block-like polymorph (Cb phase) showed blue emission (λem = 476 nm) but no mechanoluminescence, while the prism-like polymorph (Cg phase) showed green emission (λem = 498 nm) with bright green mechanoluminescence (λem = 517 nm). Interestingly, the Cb phase could be converted into the Cbf(Cg) phase under the fuming of dichloromethane and the mechanoluminescence occurred again while the samples were crushed (Fig. 11B). Both crystals belonged to the non-centrosymmetric polar space group of P21. Theoretical calculations indicated that the Cg phase had a stronger dipole moment and smaller energy gap than those of the Cb phase. It was believed that non-centrosymmetric molecular packing and a dipolar structure are favorable for a mechanoluminescence property through the piezoelectric effect at the early study of organic mechanoluminescence materials. Crystal structure analysis revealed that the Cg phase possessed more intermolecular interactions such as C–H⋯π and C–H⋯S aside from the C–H⋯O hydrogen bonding. Thus, the rigidization of molecular conformations and suppression of intramolecular rotation may also be responsible for the mechanoluminescence activity.
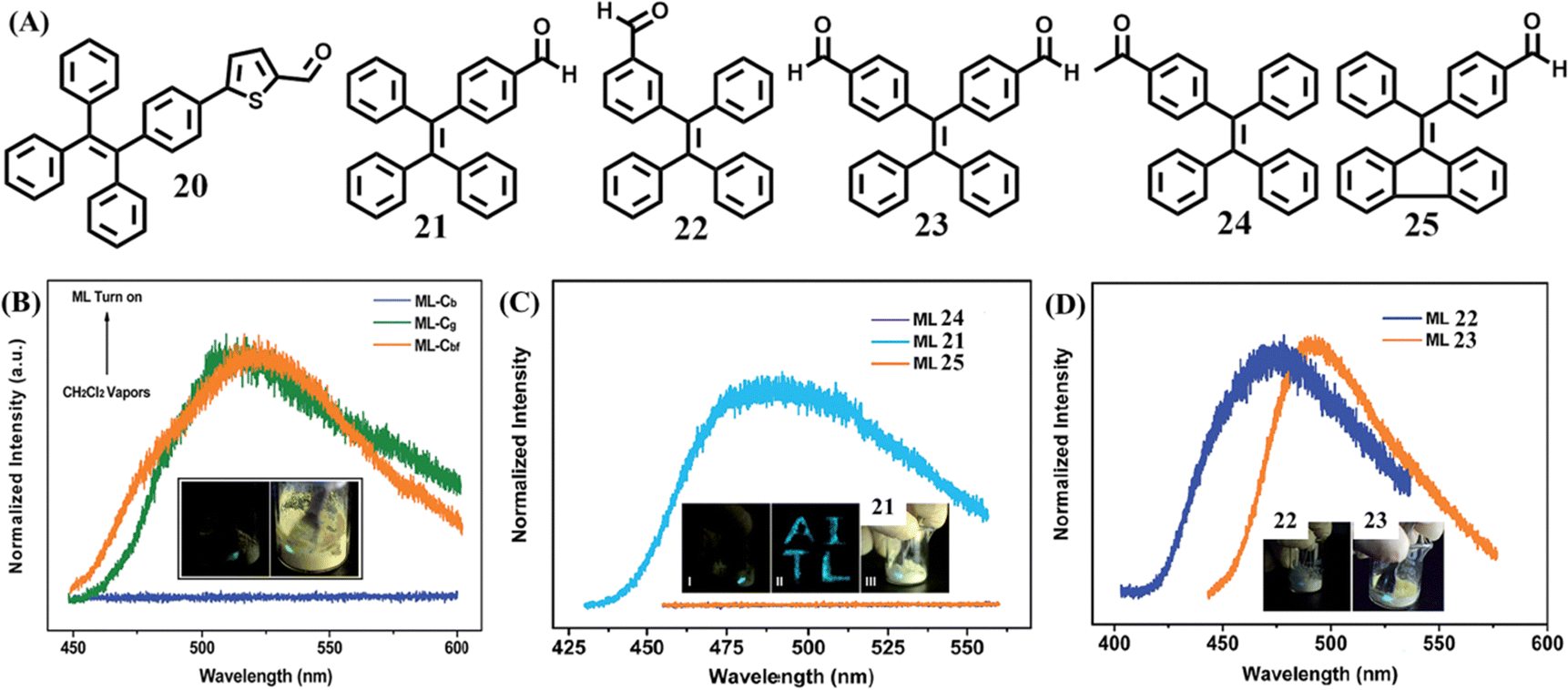 | ||
| Fig. 11 (A) Molecular structures of 20–25. (B) The mechanoluminescence spectra of 20 in the Cb, Cg and Cbf phase (fumed by dichloromethane); the inset shows the mechanoluminescence photos. Reproduced with permission.122 Copyright 2015, Royal Society of Chemistry. (C) The mechanoluminescence spectra of 21, 24 and 25; the inset shows the mechanoluminescence photos of 21. (D) The mechanoluminescence spectra and photos of 21 and 23. Reproduced with permission.123 Copyright 2016, Royal Society of Chemistry. | ||
Chi's group also reported a series of mechanoluminescence-active TPE derivatives 21–23 with an aldehyde group (Fig. 11A).123 Two reference compounds 24 and 25 were derived from a subtle structural change of 21, and somehow lost their mechanoluminescence (Fig. 11C). By shearing the crystals with a spatula, 21–23 emitted mechanoluminescence with obvious distinct intensity of 21 > 23 > 22 (Fig. 11C and D). The three single crystals of 22, 23 and 21 belonged to the non-centrosymmetric polar space groups of P21, P21 and Pna21, respectively, which was different to the centrosymmetric non-polar space groups of mechanoluminescence-inactive reference compounds. Their dipole moments suggested that the two conformations of 21 (4.79 and 5.42 Debye, respectively), were both larger than that of 23 and 22 (4.78 Debye and 3.83 Debye, respectively). The solid powders of 21 and 23 possessed impressive fluorescence quantum yields of 0.28 and 0.21, respectively, which are much higher than that of 22 (0.10). Thus, the weak mechanoluminescence of 22 was considered to be caused by its small dipole moment and low quantum yield.
A pure hydrocarbon TPE derivative 26 was then reported by Li et al. (Fig. 12A)124 Crystal 26 emitted redshifted mechanoluminescence compared to its blue fluorescence. The PL spectra of 26 under different conditions showed that the mechanoluminescence matched well with the fluorescence of 26 absorbed on thin-layer chromatography or in a 95% water/THF solution (Fig. 12B). So it was proposed that the mechanoluminescence more likely originated from the excited state of unimolecular 26 at the surface of crystal, which was produced while the crystal was cracking, rather than aggregated molecules 26 in the inner of the crystal. Crystal 26 belonged to the monoclinic crystal system with a chiral C2 space group. Molecule 26 was a non-polar molecule for its dipole moment was as extremely small as 0.007 Debye. The aforementioned mechanoluminescence-active molecules all contained an aldehyde group, which can form multiple C–H⋯O intermolecular interactions. In crystal 26, the molecules were very close to each other but common hydrogen bonds were absent due to the lack of a hydrogen bond donor. In consideration of the abundant electrons of the ethynyl group, it was believed that strong intermolecular interactions should occur among the ethynyl group and acetylene/aromatic hydrogens, which was probably responsible for the generation of mechanoluminescence.
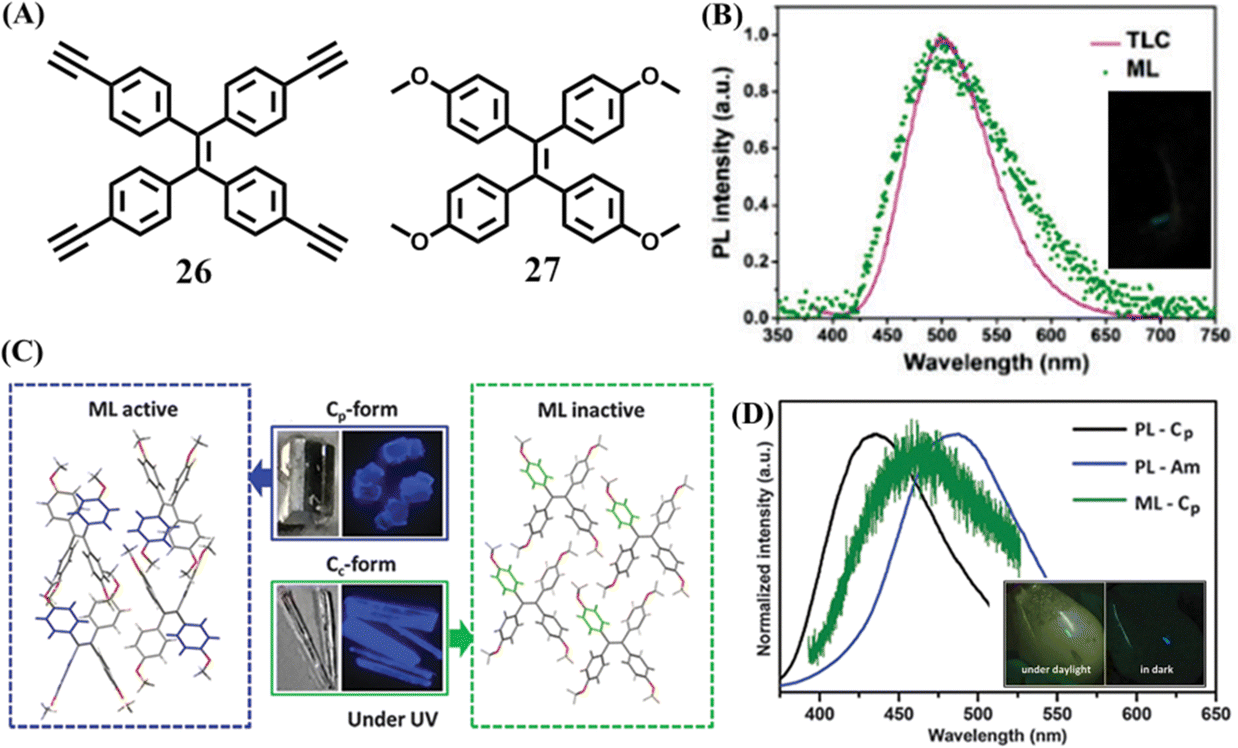 | ||
| Fig. 12 (A) The molecular structures of 26 and 27. (B) The fluorescence spectrum of 26 absorbed by a TLC plate, and the mechanoluminescence spectrum; the inset shows a photograph of the mechanoluminescence. Reproduced with permission.124 Copyright 2017, Royal Society of Chemistry. (C) Stacking mode and images of active Cp-form crystal and inactive Cc-form crystal. (D) The fluorescence spectra of 27 in Cp crystal form and amorphous state, and the mechanoluminescence spectrum of the Cp crystal form; the inset shows mechanoluminescence photos of 27 under daylight and dark conditions. Reproduced with permission.125 Copyright 2016, Royal Society of Chemistry. | ||
The piezoelectric effect is frequently used to explain the mechanoluminescence phenomenon of inorganic mechanoluminescence materials, though it is not omnipotent. For organic mechanoluminescence materials, there is a lack of feasible explanation about the mechanoluminescence mechanism. The photophysical properties of organic molecules in the aggregated state have a close relationship with molecular structure and packing. Therefore, researchers undertook studies to expand the library of organic mechanoluminescence molecules and obtain clues about the influence of the molecule packing in the crystalline state on the mechanoluminescence properties, which was not limited to TPE-based mechanoluminescence materials.
Li et al. obtained two polymorphs with different mechanoluminescence activities from 27 (Fig. 12A).125 The block-like polymorph (Cp phase) had a centrosymmetric non-polar P21/c space group, while the prism-like polymorph (Cc phase) belonged to a chiral polar C2 space group. Different to those of 20, the two polymorphs Cp and Cc phase of 27 emitted similar blue fluorescence peaking at 420 and 429 nm (Fig. 12C), respectively. After grinding, only the Cp phase exhibited bright deep-blue mechanoluminescence (Fig. 12D), which contradicted with previous criterion about the space group. From the view of the crystal structure, the Cp phase had more C–H⋯π and C–H⋯O intermolecular interactions with a shorter distance than those in the Cc phase. The greater number and stronger intermolecular interactions in the Cp phase could help to rigidify the molecular conformation and resist the crush of crystal. In contrast, the Cc phase was fragile and could be easily ground into amorphous powder compared to the Cp phase. Once the crystal was crushed, the slippage of molecular layers in the Cc phase might exhaust the mechanical energy through non-radiative pathways. However, the non-radiative transition was probably impeded by the rigid conformation and compact packing in the Cp phase. Thus mechanical energy could be transformed into mechanoluminescence through a radiative channel.
In 2018, Li's group investigated the effect of different linkage modes on the mechanoluminescence property based on two TPE derivatives.126 When two bulky phenanthro[9,10-d]imidazole groups decorated the meta- and para-position of the TPE skeleton, mm-28 and pp-28 were obtained (Fig. 13A and C). In spite of their similar molecular structure, mm-28 was mechanoluminescence-active (Fig. 13A), while pp-28 exhibited no mechanoluminescence property. The unit cell of pp-28 and mm-28 was centrosymmetric with a non-polar space group of P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) and P21/n, respectively. The dipole moment of the two main molecular configurations in mm-28 was 4.90 and 13.80 Debye, respectively, much larger than those in pp-28 (0.01 Debye). Crystal structure analysis revealed that pp-28 showed weaker intermolecular interactions and looser molecular packing (Fig. 13B and D), so the energy loss via non-radiative relaxation channels might be enhanced, causing its inactive mechanoluminescence.
and P21/n, respectively. The dipole moment of the two main molecular configurations in mm-28 was 4.90 and 13.80 Debye, respectively, much larger than those in pp-28 (0.01 Debye). Crystal structure analysis revealed that pp-28 showed weaker intermolecular interactions and looser molecular packing (Fig. 13B and D), so the energy loss via non-radiative relaxation channels might be enhanced, causing its inactive mechanoluminescence.
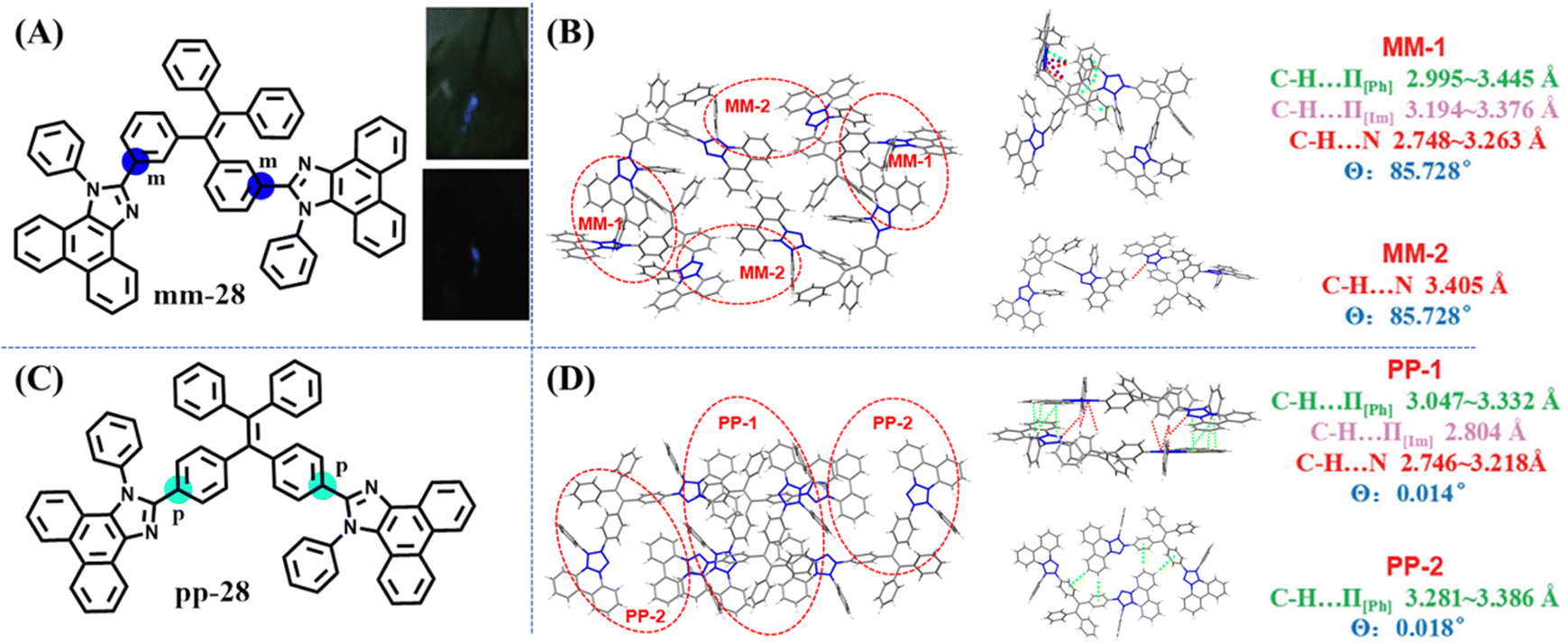 | ||
| Fig. 13 (A) The molecular structure and mechanoluminescence photos of mm-28. (C) The molecular structure of pp-28. Unit cell (left) and relevant molecular configurations (right) of (B) mm-28 and (D) pp-28. Reproduced with permission.126 Copyright 2018, Royal Society of Chemistry. | ||
Recently, Chi et al. reported the first example of a flexible HOF with mechanoluminescence behavior. Molecule 29 was constructed from a TPE skeleton and four acetophenone moieties (Fig. 14A).127 Due to the rotations between phenyl rings in TPE and phenyl rings in acetophenone moieties around the C–C single bond, the relative locations of the methyl groups and oxygen atoms in the acetophenone moieties were different when distinct solvent molecules were included in the pores of crystals of 29. Only the 29-DCM crystal (included dichloromethane) was mechanoluminescence-active (Fig. 14B). Among these host–guest single crystals, the large supramolecular dipole moments of 29-DCM were determined to contribute significantly to its mechanoluminescence activity. When dichloromethane molecules in the voids of 29-DCM were released upon heating, a new crystal 29-H with narrow pores was formed accompanying the loss of mechanoluminescence activity (Fig. 14C). Moreover, the mechanoluminescence of the ground powder of 29-DCM could be regenerated facilely by exposure to dichloromethane vapor or simple recrystallization. This example revealed that the mechanoluminescence property could be realized through supramolecular dipole moment regulation.
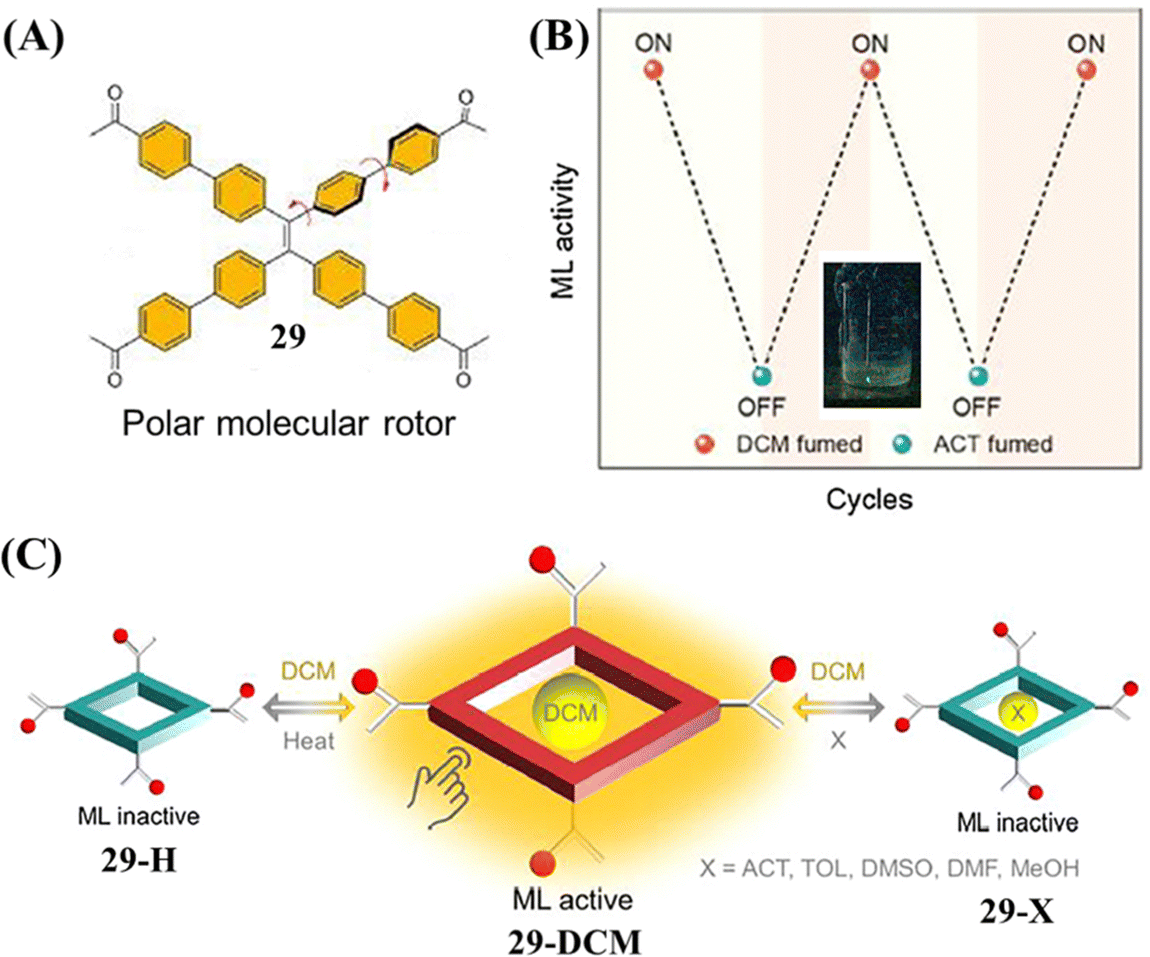 | ||
| Fig. 14 (A) The molecular structure of 29. (B) Diagram of on–off mechanoluminescence switching between 29-DCM and 29-ACT in response to dichloromethane and acetone vapor, respectively. The inset shows the mechanoluminescence photo of 29-DCM. (C) Diagram of the guest-induced breathing behaviors with controlled ML activity. Reproduced with permission.127 Copyright 2022, CCS Chemistry. | ||
From 2019, our group started to investigate mechanoluminescence based on meta- and para-substituted TPE derivatives (14, 30–43) due to their easy synthesis process and relatively simple intermolecular interactions.111,128–131 These substituted groups included –OH, –NH2, –NO2, –N(CH3)2, –CN, –COOH, –COOCH3, –B(OH)2. –CHO (21, 22) and –COCH3 (24) substituted TPE and were reported by Chi et al.123 The mechanoluminescence activity of –OH, –NO2, –N(CH3)2 substituted TPE have not been organized into articles for publication, but here we have summarized them in Fig. 15. Some of the compounds could not grow into single crystals so we used the as-prepared powder to test the mechanoluminescence activity. It can be found that most of the mono-substituted TPE derivative exhibited obvious mechanoluminescence activity. Some potentially interesting clues and phenomena were also explored during the investigation.
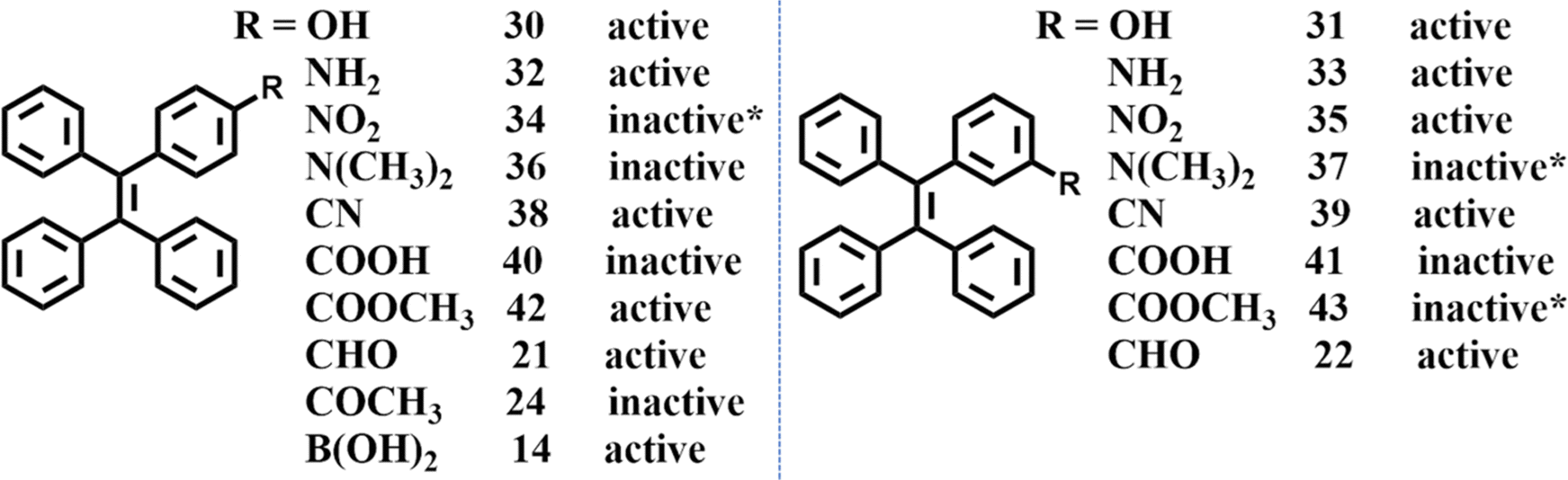 | ||
| Fig. 15 Summary of the mechanoluminescence activity of mono-substituted TPE compounds. Compounds 34, 37 and 43 were measured using their polycrystal samples because single crystals could not be obtained. | ||
Mechanoluminescence active 32 and 33 were TPE derivatives with an amino group substituted at the para- or meta-position, respectively.12832 had two polymorphs, 32-c1 exhibited obvious blue mechanoluminescence when it was ground, while the mechanoluminescence color of 32-c2 was cyan (Fig. 16A). The quantum yield of crystal 33 was as low as 1.6%, however, both its as-prepared powder 33-o and single crystal 33-c still possessed characteristic mechanoluminescence properties (Fig. 16C); it thus suggested that the origin paths of fluorescence and mechanoluminescence were not totally coincident. The mechanoluminescence spectra of 32 and 33 were closer to the fluorescence of their amorphous state in 99% water/THF solution rather than in single crystal (Fig. 16B and D), which was consistent with the result of 26. Molecule 32-c1 adopted a centrosymmetric P space group, whereas 32-c2 and 33-c belonged to the centrosymmetric P21/c and chiral P21 space group, respectively. The amino group in the three crystals was in a disordered state, but abundant intermolecular interactions could still be assured, helping to realize mechanoluminescence properties. The hydroxyl group substituted TPE derivative 44 also displayed mechanoluminescence activity (Fig. 16E and F).129 Crystal 44 belongs to a monoclinic system with a centrosymmetric P21/n space group. By virtue of three oxygen containing groups, each molecule in the crystal had various C–H⋯O and O–H⋯O intermolecular interactions with neighbouring molecules.
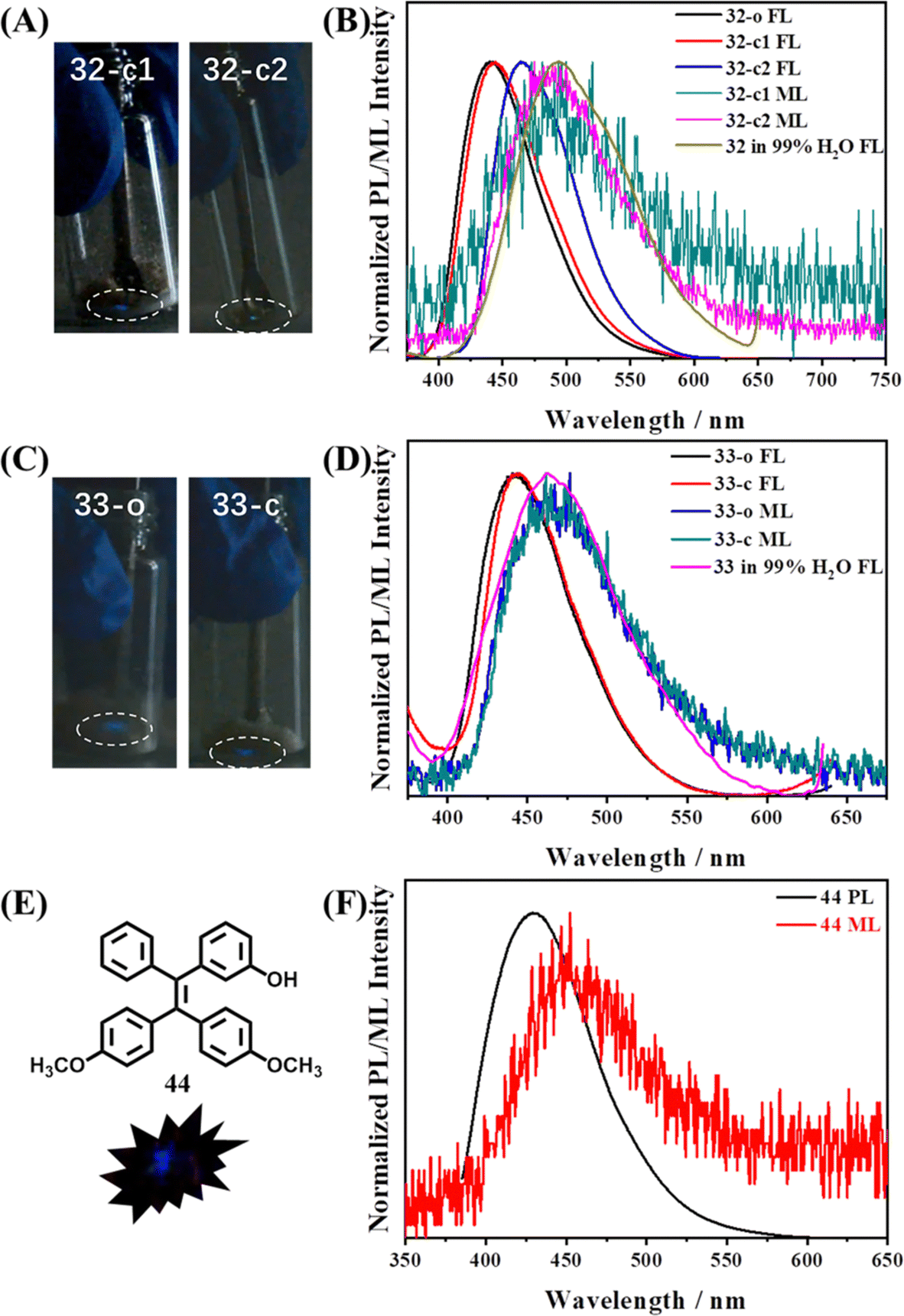 | ||
| Fig. 16 Mechanoluminescence images of (A) 32-c1 and 32-c2, and (C) 33-o and 33-c in the low light situation at room temperature. The normalized photoluminescence and mechanoluminescence spectra of (B) 32 and (D) 33 in different states. Reproduced with permission.128 Copyright 2019, Royal Society of Chemistry. (E) Molecular structure and mechanoluminescence image of crystal 44. (F) The normalized photoluminescence and mechanoluminescence spectra of crystal 44. Reproduced with permission.129 Copyright 2022, Elsevier. | ||
When the TPE skeleton was decorated with cyano groups, 38 and 39 were produced and the mechanoluminescence property was still active.130 As shown in Fig. 17A and C, bright mechanoluminescence could be clearly observed by naked eyes when crushing all the crystals or as-prepared powders of 38 and 39. The mechanoluminescence intensity was obviously stronger than that of other TPE derivatives, which can be judged by the non-black background of the mechanoluminescence photos. Molecule 39 had two polymorphs, while 39-c1 belonged to a tetragonal system with a centrosymmetric P4/ncc space group and 39-c2 belonged to a monoclinic system with a chiral P21 space group. Crystal 38c adopted a chiral P31 space group belonging to the trigonal system. Due to the abundant electrons of the cyano group and the existence of nitrogen atoms, many C–H⋯N intermolecular interactions with short distances were found in three crystals. For organic mechanoluminescence molecules, the emission inevitably became weak and then disappeared under continuous mechanical grinding. Interestingly, the mechanoluminescence signals of ground powders of 38 and 39 could be recovered after annealing for 6 min (Fig. 17B and D). The unchanged PXRD patterns of 38 demonstrated that the recycling process was only related to the variation in the particle size of the crystalline powder. For 39, the phenomenon was somewhat different. Annealing induced a one-time crystalline transformation from phase 39-c1 to phase 39-c2 (Fig. 17F), accompanying the irreversible switch of mechanoluminescene color from sky-blue to cyan (Fig. 17E). When the TPE skeleton was decorated with a boronic acid group at the para position (Fig. 15), the resultant compound 14 displayed not only tricolored mechanochromism (in the above section) but also a recyclable mechanoluminescene property.111 The reborn temperature of mechanoluminescene was lowered to 100 °C, though the signal intensity was relatively weak.
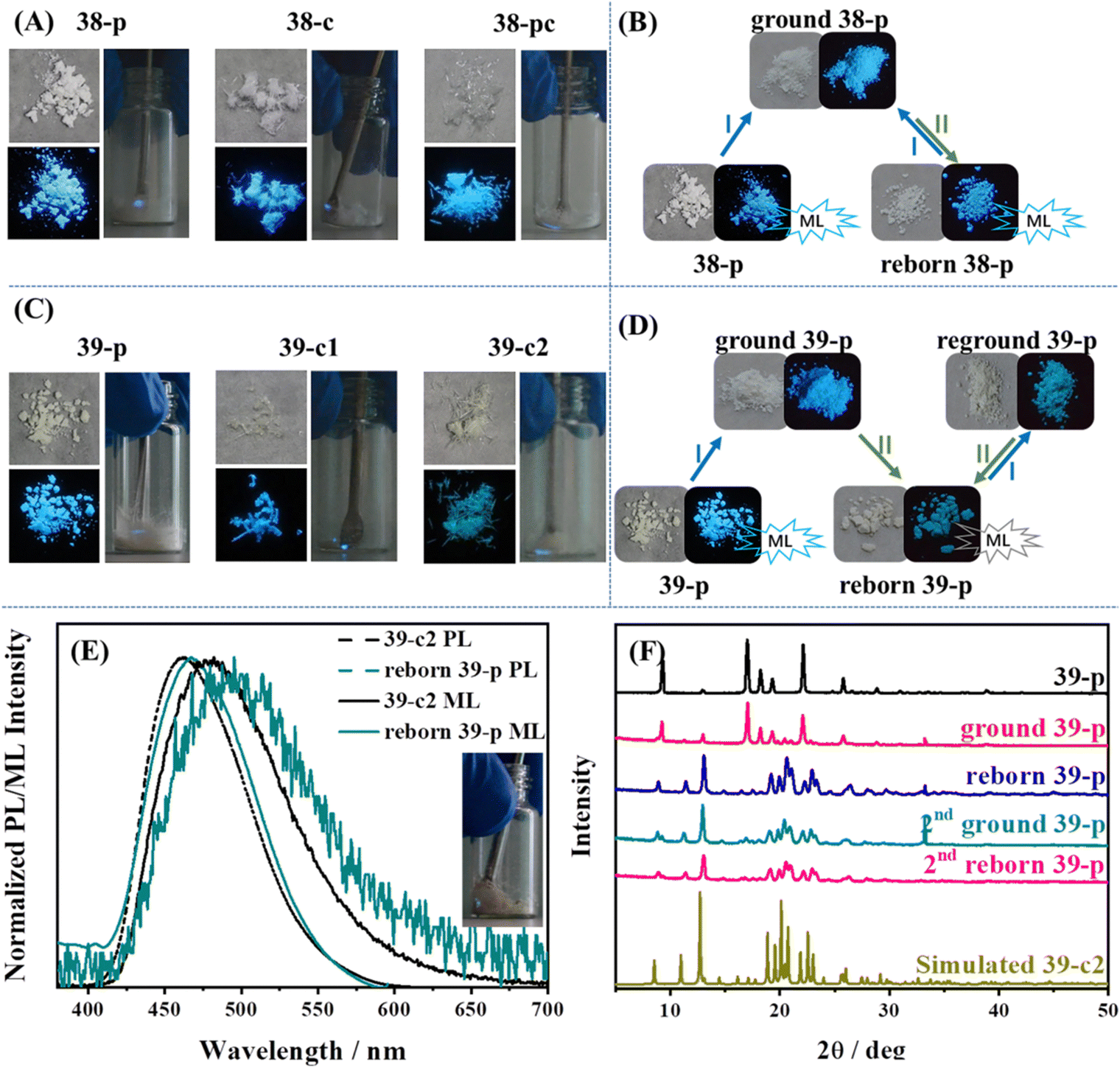 | ||
| Fig. 17 Photos of (A) 38 and (C) 39 in different states; those in the left panel correspond to natural light (upper) and UV illumination (down) and those in the right panel correspond to the mechanoluminescence images under daylight at room temperature. Recyclable utilization process of (B) 38-p and (D) 39-p. (E) Fluorescence and mechanoluminescence spectra of 39-c2 and reborn 39-p. The inset shows the mechanoluminescence image of reborn 39-p. (F) PXRD patterns of different states of 39-p during the reborn ML process. Conditions: I, grinding; and II, heating at 138 °C for 6 min. Reproduced with permission.130 Copyright 2021, Royal Society of Chemistry. | ||
To further probe the underlying mechanism, we synthesized TPE derivatives 40–43 (Fig. 15).131 Apart from 43, the single crystals of the other three compounds were successfully obtained. Only crystal 42 emitted bright blue light after grinding. Compared with –COOCH3, –COOH which can act as a stronger hydrogen bond donor/acceptor unit, mechanoluminescene was unexpectedly absent in 40 and 41. Crystal 40 and 41 belonged to the monoclinic system with a P21/c space group, whereas 42 belonged to the monoclinic system with a P21 space group. The molecular packings suggested that strong interactions existed between carboxyl groups and outstretched conformation and molecules 40 formed a three-dimensional porous structure with a low packing density (ρ = 1.061 g cm−3, Mw = 376.43 g mol−1) (Fig. 18A). The stacking of molecule 41 was very compact (ρ = 1.208 g cm−3, Mw = 376.43 g mol−1), however, intermolecular interactions had an unbalanced distribution in the crystal. The unsubstituted phenyl rings of 41 had very weak intermolecular interactions, resulting in a defective three-dimensional hydrogen-bonded network (Fig. 18B). The packing density of 42 (ρ = 1.187 g cm−3, Mw = 390.46 g mol−1) was between the two former crystals, and the balanced distribution of multiple intermolecular interactions gave crystal 42 with an intact three-dimensional hydrogen-bonded network (Fig. 18C). The energy of the excited state in 40 was likely dissipated by the intramolecular motions of phenyl rings around the porous region. For 41, the fracture of the crystal probably happened along the row of unsubstituted phenyl rings with weak intermolecular interactions. Then dislocation or squeezing probably caused these phenyl rings closer to form new/strong interactions, leading to the consumption of excited energy. In contrast, the moderate compact packing with an intact three-dimensional hydrogen-bonded network in crystal 42 avoided the energy loss in crystal 40 and 41 so as to realize bright mechanoluminescene (Fig. 18D). These views partially overlap with previous results explored by Li et al.132 Their results of a series of halogen-substituted triphenylamine derivatives suggested that loose packing inclines to collapse and too tight packing resists the breaking of crystals, which are not beneficial for the mechanoluminescence effect by increasing energy loss through non-radiative relaxation channels under mechanical stimulation. Only tight packing with moderate compactness helped to induce mechanoluminescence (Fig. 18E). It should be noted that the above mechanisms are only rather rough descriptions, more details need to be clarified in the future.
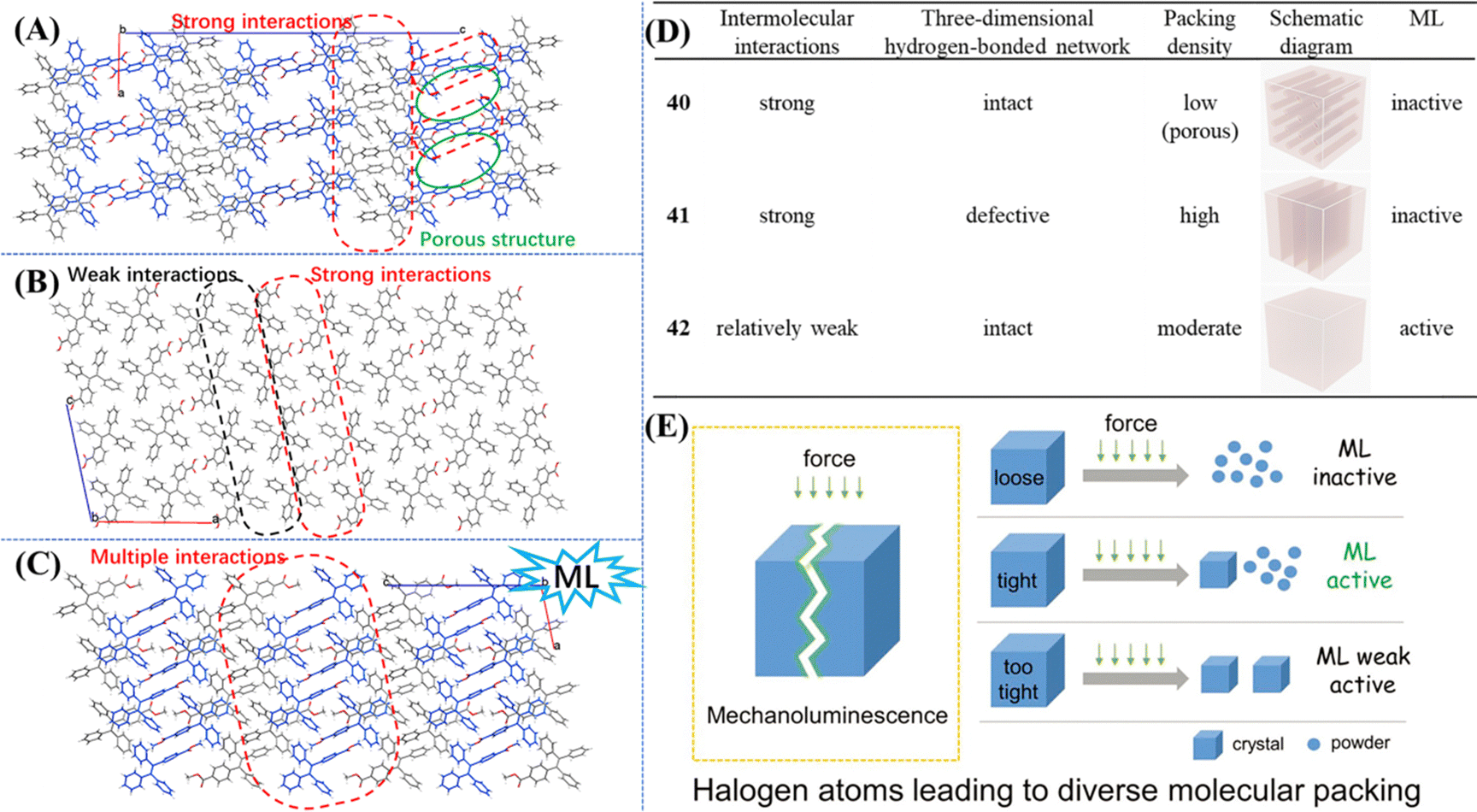 | ||
| Fig. 18 The detailed molecular packing modes in (A) 40, (B) 41 and (C) 42 viewed along the b-axis. (D) Summary of single crystal information and the mechanoluminescence property of 40–42. Reproduced with permission.131 Copyright 2019, Royal Society of Chemistry. (E) Molecular packing mode of crystals with different mechanoluminescence properties. Reproduced with permission.132 Copyright 2019, Wiley-VCH. | ||
Photochromism
Harnessing light to manipulate the appearance color or/and fluorescence properties of organic molecules is of great importance for their wide applications in molecular switches, anti-counterfeiting, optical storing and super-resolution imaging and so on. Non-contact stimulus and facile control/detection are the advantages of photochromic materials. Azobenzene, spiropyran, bisthienylethene, and phenoxyquinone derivatives are the most commonly explored photochromic materials to date. However, the photochromic property of these materials is always inactive in the aggregation state. Photochromism occurring only in the solution state would apparently restrict their application scenarios. Thus, constructing effective solid-state photochromic switches requires smart design. The major obstacle for the photoswitching of common photochromic molecules in the solid state is the strong molecular packing or not sufficient space for the large change in size/geometry. Taking steps along this path, bulky substituents could be used to enable the photoswitching in the solid state. Therefore, a direct method to design TPE-based photochromic switches is to merge the TPE moiety with a traditional photochromic moiety.133–138 The TPE moiety is mainly responsible for fluorescence emission, while the geometrical and electronic changes of the photochromic moiety on the molecular level are harnessed to modulate the optical properties of the material.In 2018, Yin et al. designed a novel solid-state photo-switch 45 by integrating two spiropyran moieties with a TPE skeleton (Fig. 19A).137 Powder 45 changed its appearance color from light yellow to green and further to deep blue with the irradiation of UV light (Fig. 19B). After visible light irradiation (400–750 nm) for 10 min, the deep blue color of the ring-open form would revert to light yellow color. The reversible photochromic behavior of 45 could be repeated multiple times without obvious decay. PXRD revealed that 45 was an amorphous powder with loose molecular packing, thus efficient photoswitching between 45 and its ring-open form is believed to be achieved with the aid of large free volumes caused by the nonplanar molecular structures of TPE moieties.
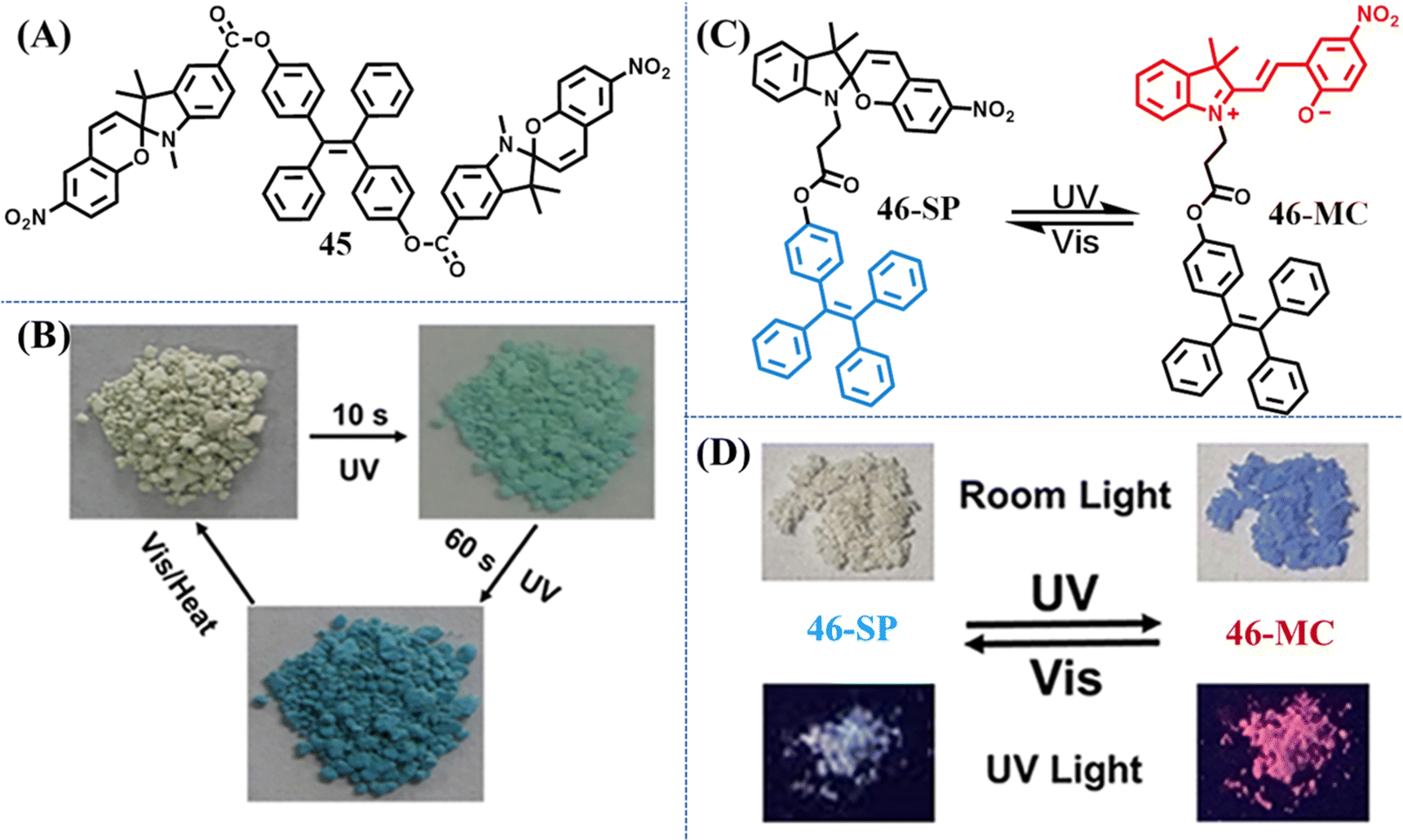 | ||
| Fig. 19 (A) Chemical structure of compound 45. (B) Apparent colors of powder 45 before and after UV irradiation. Reproduced with permission.137 Copyright 2018, American Chemical Society. (C) Reversible structural isomerization of 46-SP and 46-MC. (D) Fluorescent and visible images of powders 46-SP and 46-MC under the alternating UV and visible light treatment. Reproduced with permission.138 Copyright 2021, American Chemical Society. | ||
When spiropyran moieties were reduced to one, Tian et al. reported a reversible dual fluorescent photoswitching TPE derivative 46 (Fig. 19C).138 The colorless powder 46-SP changes to dark purple 46-MC after 365 nm UV light irradiation for 5 min, accompanying its fluorescence altered from bright cyan to red (Fig. 19D). Visible light irradiation could prompt the occurrence of a contrary process. Detailed spectroscopic and theoretical studies also suggested the nonplanar conformation of TPE moieties provided large free volumes, facilitating the reversible photoswitching of the spiropyran moiety. The reversibility and high-contrast dual-photoswitches of fluorescence and appearance color enabled its great application prospect in anticounterfeiting marks and multimodality optical memory.
Compared with usual photochromic molecules, salicylaldehyde hydrazone is also a good candidate for a reversible photochromic group with good fatigue resistance.139 The salicylaldehyde hydrazone moiety has a reversible tautomerism between its enol-form and keto-forms, as shown in Fig. 20A. The former is an “on” state due to the excited-state intramolecular proton transfer (ESIPT) process, while the latter is an “off” state by virtue of a strong electron-withdrawing ability. With rational molecular design, they could be used to exhibit excellent photochromic property.
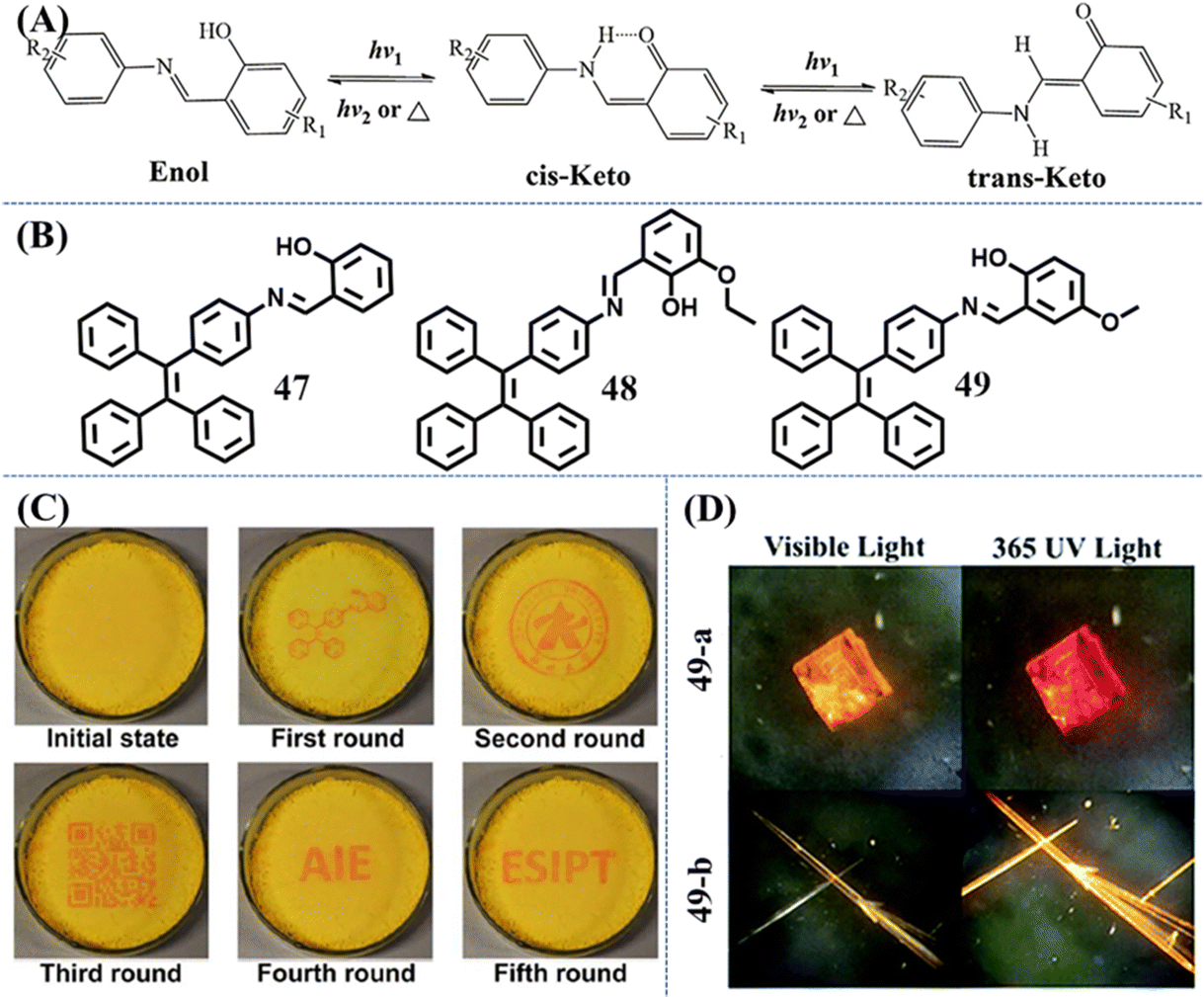 | ||
| Fig. 20 (A) Photochromic mechanism of salicylaldehyde hydrazone upon UV irradiation. (B) Chemical structures of 47–49. (C) Generating different patterns on the same film of 47. Reproduced with permission.140 Copyright 2016, Royal Society of Chemistry. (D) Photos of 49-a and 49-b under 365 UV and visible light. Reproduced with permission.142 Copyright 2019, Royal Society of Chemistry. | ||
Li et al. developed a reversible photochromic AIE molecule, 4-(1,2,2-triphenylvinyl)aniline salicylaldehyde hydrazine 47, by simply connecting salicylaldehyde hydrazone with TPE (Fig. 20B).140 The appearance and fluorescence of the as-prepared powder was yellow, corresponding to the enol-form. After UV irradiation, the powder was transformed into red keto-form with weakened fluorescence (Fig. 20C). According to experiments, the main conformation in the aggregation state was the trans-keto form for its relative high stability. Thus, the color of UV-irradiated powder can persist for hours at RT in the dark. Visible light or thermal stimulus could accelerate the recovery of the red keto-form to yellow enol-form. The switchable appearance and fluorescence color with good fatigue resistance was then demonstrated to be a promising solid material for erasable photo-patterning. A similar TPE derivative with an additional ethoxyl group was reported by Zhang et al.141 Molecule 48 also exhibited reversible photochromic behavior and excellent fatigue resistance (Fig. 20B). Due to the steric hindrance of the ethoxyl group, the transformation between the cis-keto form and the trans-keto form was impeded, leading to more than 15 days recovery time of irradiated powder in the dark at RT. So 410 nm light served as a pen and 420–590 nm white light served as an eraser to construct rewritable photochromic paper based on 48.
Ni et al. investigated the relationship between the crystal packing styles and photochromism property of Schiff base-TPE derivative 49 (Fig. 20B).142 Slow evaporation of different concentrations of solutions gave two different polymorphs: the square-block crystal 49-a exhibited reversible photochromic behavior and very weak emission, while the needle-like crystal 49-b emitted relatively stronger orange fluorescence but was photochromism-inactive (Fig. 20D). Crystal structure analysis revealed that molecules in the photochromic crystal had local unconsolidated stacking around the salicylaldimine moiety, which provided enough space for the photo-reaction. Photochromism-inactive crystal was in a close packing style, whose photoisomerization process of the salicylaldimine moiety should be impeded and responsible for the absence of photochromic behavior.
The above TPE-based photochromic materials focused on the native photochromic moieties. Actually, the diphenylethylene unit of the TPE/triphenylethylene skeleton could undergo reversible photocyclization or irreversible dehydrogenation reaction, leading to a large variation in conformation, structure, electronic state and photophysical property in solution.143–150 In 2019, our group found that the photocyclization reaction of TPE derivatives could also take place in solid powder. Several novel TPE-based photochromic materials were then explored.112,113,129,151 Compound 15 with multi-state mechanochromism has been mentioned in the above section, and only its single crystal and heated state exhibited special self-recovery photochromism (Fig. 21A).112 After UV irradiation, the white heated powder 15h or transparent crystal 15c became deep red and gradually faded within 1 min, forming strong visual contrast. Accordingly, their absorption spectra contained a strong absorption band centered at 514 nm, indicating the conjugation of the molecule was elongated, which was consistent with previous reports about the formation of the diphenylethylene unit.148–150 The self-recovery photochromism process could be repeated many times with good fatigue resistance. According to the crystal structure, weak intermolecular interactions between adjacent phenyl rings would be favorable for the photocyclization of the TPE moiety in the solid state under UV-light irradiation. DFT calculations indicated that the reactive site of the photocyclization reaction was probably near the substitute group. In combination with reversible multi-state mechanochromism, the application potential of 15 in multidimensional anticounterfeiting was demonstrated (Fig. 21B). A rose with cyan leaves, dim petals and blue background was drawn by the ground, heated and fumed state of 15, respectively. After UV irradiation, a picture of red rose was present and gradually faded. These dynamic patterns or paintings could be designed in light of specific requirements. Similarly, the agglomerated state of compound 16m prepared using a thermal-melting and annealing process also displayed a self-recovery photochromic property (Fig. 21C).113
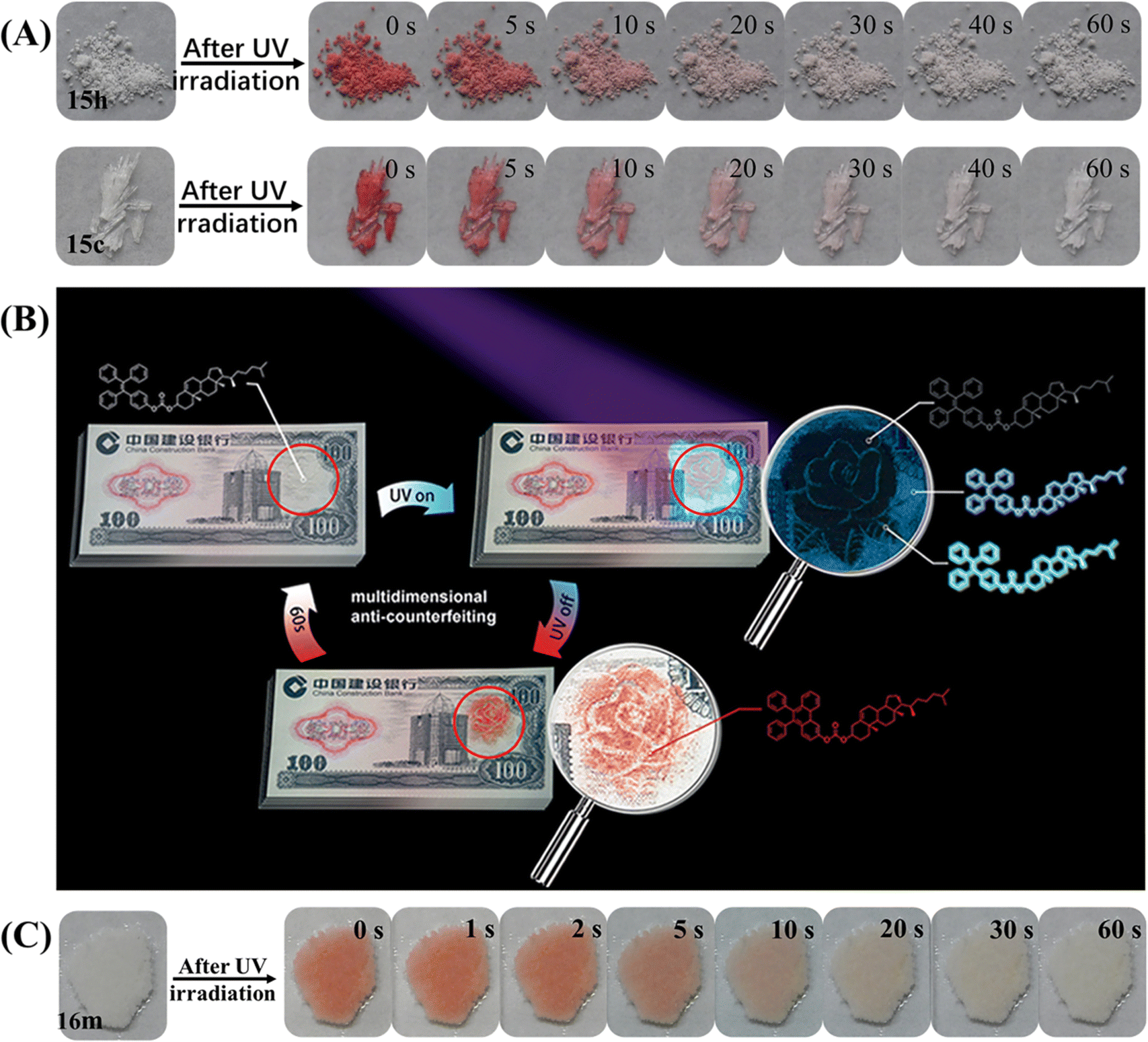 | ||
| Fig. 21 (A) Photochromic bleaching process of 15h and 15c at room temperature after for 0–60 s after irradiation with UV light. (B) Illustration of a model banknote as a prototype for multidimensional anti-counterfeiting design based on 15. Reproduced with permission.112 Copyright 2019, Wiley-VCH. (C) Photochromic bleaching process of 16m at room temperature after for 0–60 s after irradiation with UV light. Reproduced with permission.113 Copyright 2022, CCS Chemistry. | ||
In contrast to 15 and 16, the photochromism phenomenon of compound 44 took place on its amorphous state.129 The self-recovery photochromic bleaching time of irradiated amorphous powder was elongated to about 1 h. The TPE-based molecular cage 50 can also display self-recovery photochromism (Fig. 22A), which was recently reported by Cao et al.152 The color of 50 turned red after being irradiated by 365 nm UV light and the red color faded within 60 s after switching off the UV light both in the amorphous solid state and solution (Fig. 22B). Apart from self-recovery photochromism, we also explored an irreversible photochromic TPE derivative 51 (Fig. 22C), which was facilely synthesized by aldimine condensation reaction through the corresponding TPE derivative and p-toluenesulfonyl hydrazide.151 With the UV light irradiation time elapsing, the blue emission of the as-prepared powder 51p could be transformed into cyan, yellowish green, yellow and orange emission, accompanying their appearance change from white to orange (Fig. 22D). Based on 1H NMR, fluorescence spectrum and high-resolution mass spectrometry, the isomerization of the hydrazine group was excluded as the main reason for the photochromism. The photocyclization and dehydrogenation reaction of partial TPE moieties in the external layer of powder was believed to induce the photochromism of compound 51. We considered that with the appropriate selection of a modified moiety, the photochromic property of the TPE skeleton through the occurrence of photocyclization reaction could be turned on.
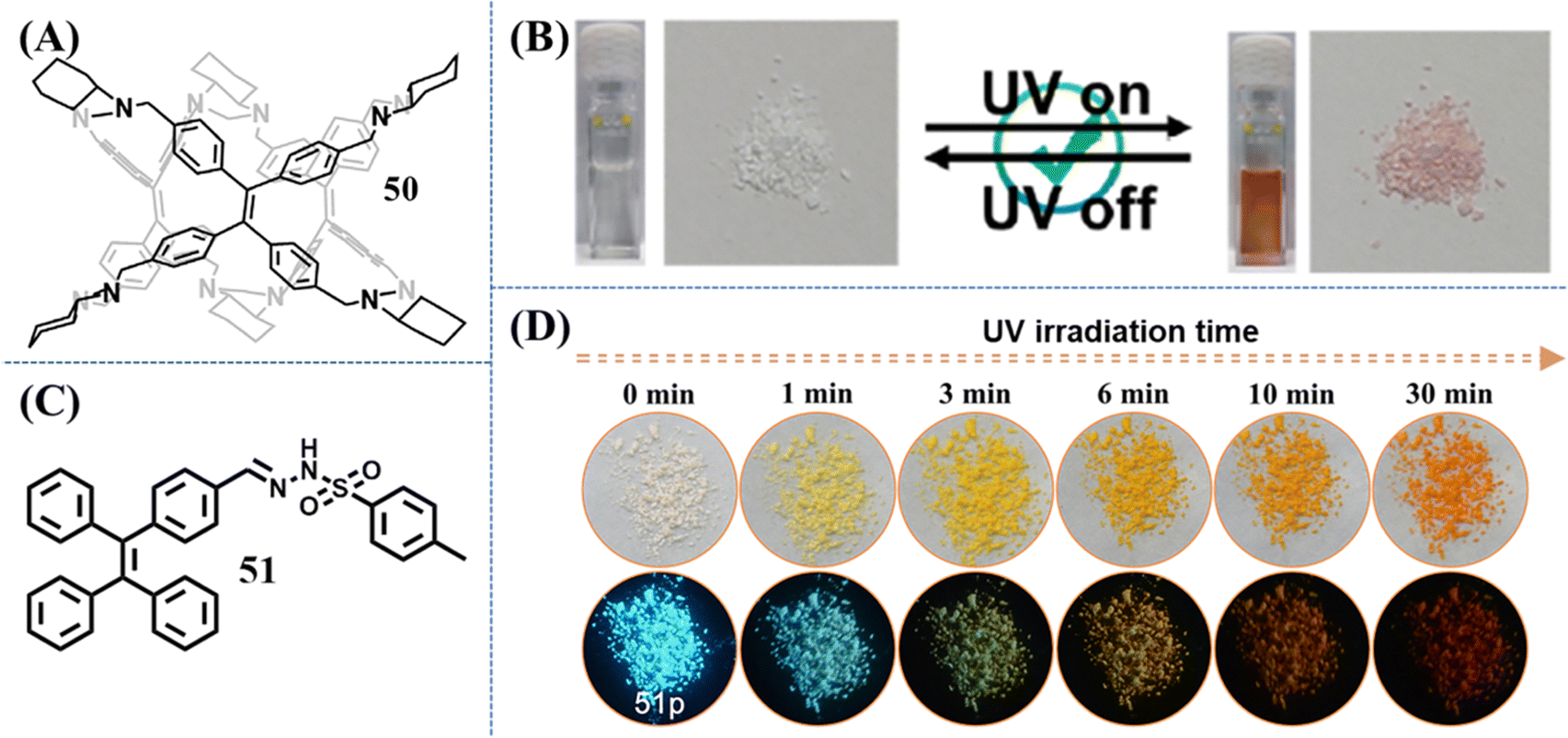 | ||
| Fig. 22 Chemical structure of compound (A) 50 and (C) 51. (B) Photochromic behaviors of 50 in solution and solid states. Reproduced with permission.152 Copyright 2023, American Chemical Society. (D) Images of 51p after 365 nm UV irradiation with different irradiation time, which were taken under ambient light (top) and 365 nm UV light (bottom). Reproduced with permission.151 Copyright 2021, Elsevier. | ||
Photoswitch
Photoswitch was another type of photoresponsive TPE material, which showed apparent changes in the fluorescence intensity or fluorescence/appearance color with the irradiation of UV light. As shown in Fig. 23A, Chi et al. synthesized a mechanical and photo induced fluorescence turn-on TPE-modified diaminomaleonitrile derivative, 52.153 Amino groups and the strongly electron-withdrawing cyan groups of diaminomaleonitrile moieties can help to form strong intermolecular hydrogen bonds. As a result, 52 displays a sandwich-type crystal structure with tightly packed areas (TPE moieties) and loosely packed areas (diaminomaleonitrile moieties) through the assistance of the above hydrogen bonding. The loosely packed crystal structure can provide enough space for significant molecular rotations and vibrations, resulting in the almost non-emissive property of crystal 52 (the quantum yield is less than 0.1%). Once the loose packing was broken, the emission band of 52 with the maximum at ca. 562 nm was switched on. Interestingly, the non-emissive crystal became emissive after UV irradiation (Fig. 23B), while its emissive amorphous sample did not show obvious changes. Combing the data of PXRD, NMR, reflectance and absorption spectra, photocyclization or photooxidation was precluded in this system. While after UV light irradiation, there was an increased amount of the cis-isomer related to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond. In the crystal state, the trans- to cis-isomer transition of diaminomaleonitrile moieties might destroy the well-aligned loose packing of the soft-layer, while the main crystal structure is maintained after irradiation. Without the orderly packing in the soft-layer, the intermolecular motions became active to some extent and the emission was turned on. Although the effects of grinding and UV irradiation were similar, their internal mechanism are likely different.
N bond. In the crystal state, the trans- to cis-isomer transition of diaminomaleonitrile moieties might destroy the well-aligned loose packing of the soft-layer, while the main crystal structure is maintained after irradiation. Without the orderly packing in the soft-layer, the intermolecular motions became active to some extent and the emission was turned on. Although the effects of grinding and UV irradiation were similar, their internal mechanism are likely different.
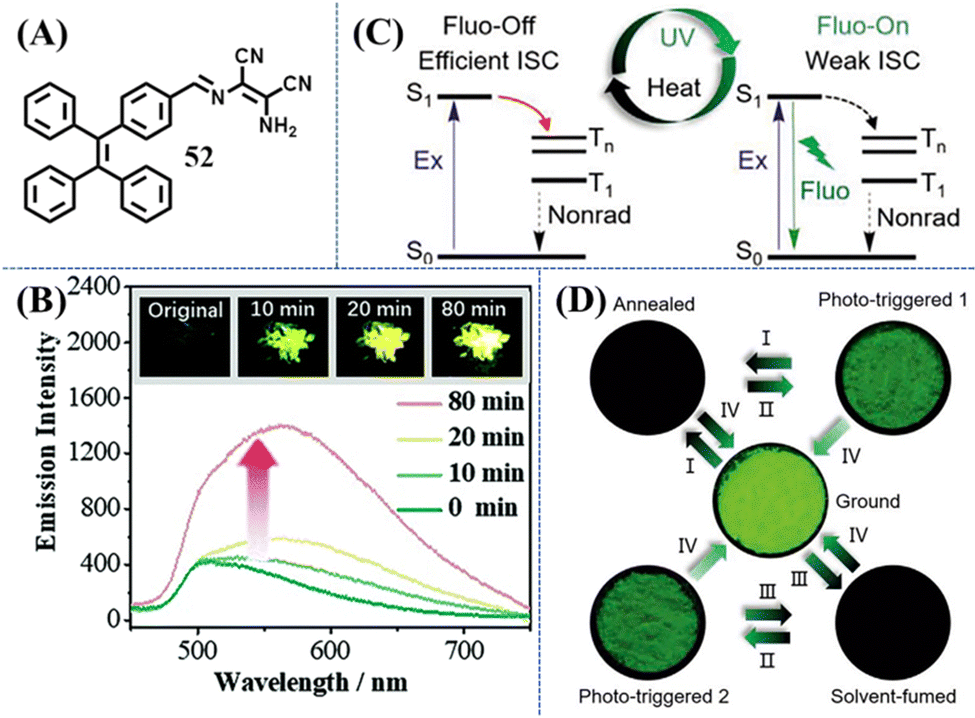 | ||
| Fig. 23 (A) Chemical structure of compound 52. (B) Emission spectra of crystal 52 before and after UV-light irradiation at 365 nm, excitation wavelength = 365 nm. The inset shows the fluorescence photographs (under UV-light) of the original and irradiated samples. Reproduced with permission.153 Copyright 2017, Royal Society of Chemistry. (C) Illustration of the photo- and thermoresponsive mechanism of 7. (D) Switching the luminescence of powder 7 under different stimulus: (I) heating at 150 °C for 5 min; (II) UV irradiation for 5 min; (III) fuming with acetone for 30 s; (IV) grinding; the fluorescence photos were taken by a handheld camera under 365 nm UV lamp. Reproduced with permission.154 Copyright 2020, Wiley-VCH. | ||
Another phototriggered fluorescence turn-on example was reported by Tang et al.154 Compound 7 was found to have high contrasts (on–off), ultrasensitive, and reversible mechanochromic property (Fig. 23C and D). Its crystalline state possesses a non-emissive molecular conformation because the efficient ISC was activated and excitons were exhausted by non-radiation decay. The ISC channel can not only be manipulated by the mechano-stimulated morphology mentioned in previous content, but also be closed by photo-stimulus. This is a rather confusing phenomenon since the possibility of photoreaction was already excluded based on the unchanged NMR and HPLC spectra of the powder and film of 7 before and after phototriggering. Quantum chemical calculation on both crystalline and gas states suggested that they possessed distinct twisted torsion angles of phenyl/nitro groups and excitation energy of S1 state, which agrees well with the redshift absorption from the crystalline film to amorphous film. Further evidence came from the ultra-surface-sensitive Raman spectra and related DFT calculations. The Raman peak at 1616 cm−1 is correlated with the characteristic vibrations of phenyl groups, which showed reversible changes between fluorescence “on” states and fluorescence “off” states. Photoirradiation was thus considered to cause slight morphology changes of crystalline molecules 7 on the surface, leading to the amorphous state by releasing molecular conformations. Subsequently, ISC channels were blocked and the fluorescence “on” state was formed. Utilizing this non-destructive photoresponsive luminescent behavior with a high on–off contrast (>102), good light transmittance (>72.3%), good durability and reversibility, the film of 7 was explored as invisible anti-counterfeiting and dynamic optical data storage with micro-resolution.
Ex-De luminescence
Ex-De luminescence is a special non-invasive/destructive property related to adjustable emission color. Different emission colors of Ex-De luminescent materials can be realized by merely tuning the excitation wavelength. This dynamic control of luminescence is convenient and promising for bioimaging, sensors and anti-counterfeiting applications. Ex-De luminescence is commonly observed in nanoparticles (nanocrystals and carbon/silica dots) or metal complexes due to their composite consisting of multiple aggregated states.155–158 In contrast, purely organic Ex-De luminescent molecules are still rarely observed to show Ex-De luminescence. For a single organic molecule, the excited state relaxation pathways should obey Kasha's rule: The emitting level of a given multiplicity is the lowest excited level of that multiplicity,159 and thus the excitation wavelength is independent.Organic Ex-De Luminescence molecules actually bypassed Kasha's rule and the underlying mechanism has been exploited with several strategies. Zhang et al. utilized the excited state intramolecular proton transfer (ESIPT) property of a specific molecular structure to produce two relaxation pathways of the excited states,160 just like incorporating different structures into a single molecule, leading to green and yellow emission upon irradiation with low-energy and high-energy light, respectively. Another strategy was derived from the exploration of mechanochromism phenomenon of several TPE derivatives. Zhang et al. explored a series of single-arm extension TPE derivatives 53–58 (Fig. 24A), which displayed efficient solid-state emission and mechanochromic activity.161 Unexpectedly, these derivatives showed Ex-De emission. Taking 53 as an example, the ground powder of 53 could emit cyan and green fluorescence under the excitation of 365 and 460 nm light (Fig. 24B), respectively. However, the as-prepared, annealed and fumed powder of 53 could only emit observable fluorescence by 365 nm UV light irradiation. Besides 53, TPE and other TPE derivatives 54–58 all exhibited different extents of Ex-De emission in the ground, annealed and solvent fumed states. However, many of them were probably difficult to distinguish with the naked eye, probably due to the extra interference of excited light when the excitation wavelength is longer than 400 nm and the insignificant emission wavelength shifts. The Ex-De luminescence property of 53 unambiguously could provide application potential in dual-channel anti-counterfeiting (Fig. 24C). 53 was printed on a practice banknote and then annealed to form a crystalline state, which could not be observed under ambient light. When the two characters “5 yuan” of TPE-VB were rubbed, the dual variation of fluorescence intensity (off-to-on or weak-to-strong) under 460 nm visible light and 365 nm UV-light could be thought of as encryption for banknotes.
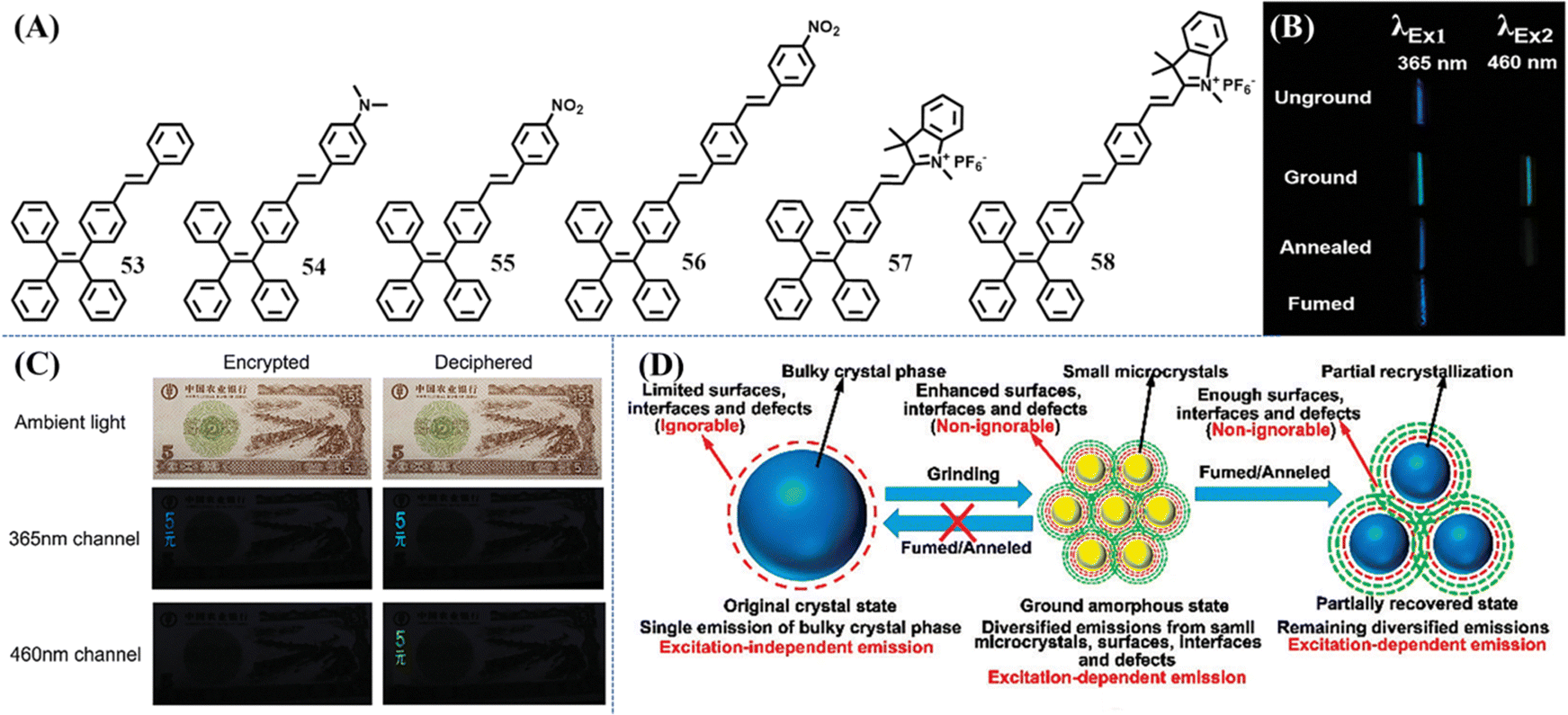 | ||
| Fig. 24 (A) Chemical structures of compounds 53–58. (B) Optical images of 53 in different states with the excitation light at 365 nm and 460 nm, respectively (the optical images excited by the light of 460 nm were taken under a light filter with the cut-off wavelength at 500 nm). (C) Illustration of the application potential of 53 in dual-channel anti-counterfeiting (the optical images in the 460 nm channel were taken under a light filter with the cut-off wavelength at 500 nm). (D) Illustration of the irreversible influence of mechanical stress on mechanochromic materials. Reproduced with permission.161 Copyright 2015, Royal Society of Chemistry. | ||
Our group also reported a single-arm extension TPE derivative 16 with a long cholesterol moiety (Fig. 8D).113 The ground and fumed powder (with dichloromethane and hexane) of 16 did not exhibit an Ex-De luminescence property. Its heated powder 16h, fumed powder (with hexane) 16f–h and polycrystal 16c emitted greenish fluorescence under 254 nm UV light, which was much brighter than the dim blueish fluorescence under 365 nm UV light. Its agglomerated state 16m showed a slightly enhanced emission with its fluorescence color transitioning from bluish into greenish. Contrary to the bathochromic shift phenomena of the aforementioned Ex-De TPE derivatives 53–58,161 our molecule 16 showed a differentiated Ex-De property that is the higher energy excitations resulting in enhanced emissions at longer wavelengths. More importantly, most of the transformation among Ex-De luminescence active states and inactive states was reversible, which greatly facilitated its multi-channel anti-counterfeiting application. Three emissive color patterns under 254 nm UV light could be easily drawn with the dichloromethane solution of 16 after the solvent evaporating, fuming and heating process (Fig. 25A). When the excited wavelength was switched to 365 nm, the green fluorescence “birds” formed by the heated state almost disappeared due to the low quantum yield, while the cyan deep-blue bamboo trunks and sky-blue bamboo leaves remain unchanged. Practically, different patterns could be devised according to specific requirements in the personalized anticounterfeiting field. Besides, the agglomerated state of 16m gathered in integral whole with photochromism and Ex-De luminescence behaviors (Fig. 25B), resulting in the non-destructive switching of fluorescence and appearance color. The combination of Ex-De luminescence and other stimuli-responsive properties undoubtedly could elevate anti-counterfeiting to a new level.
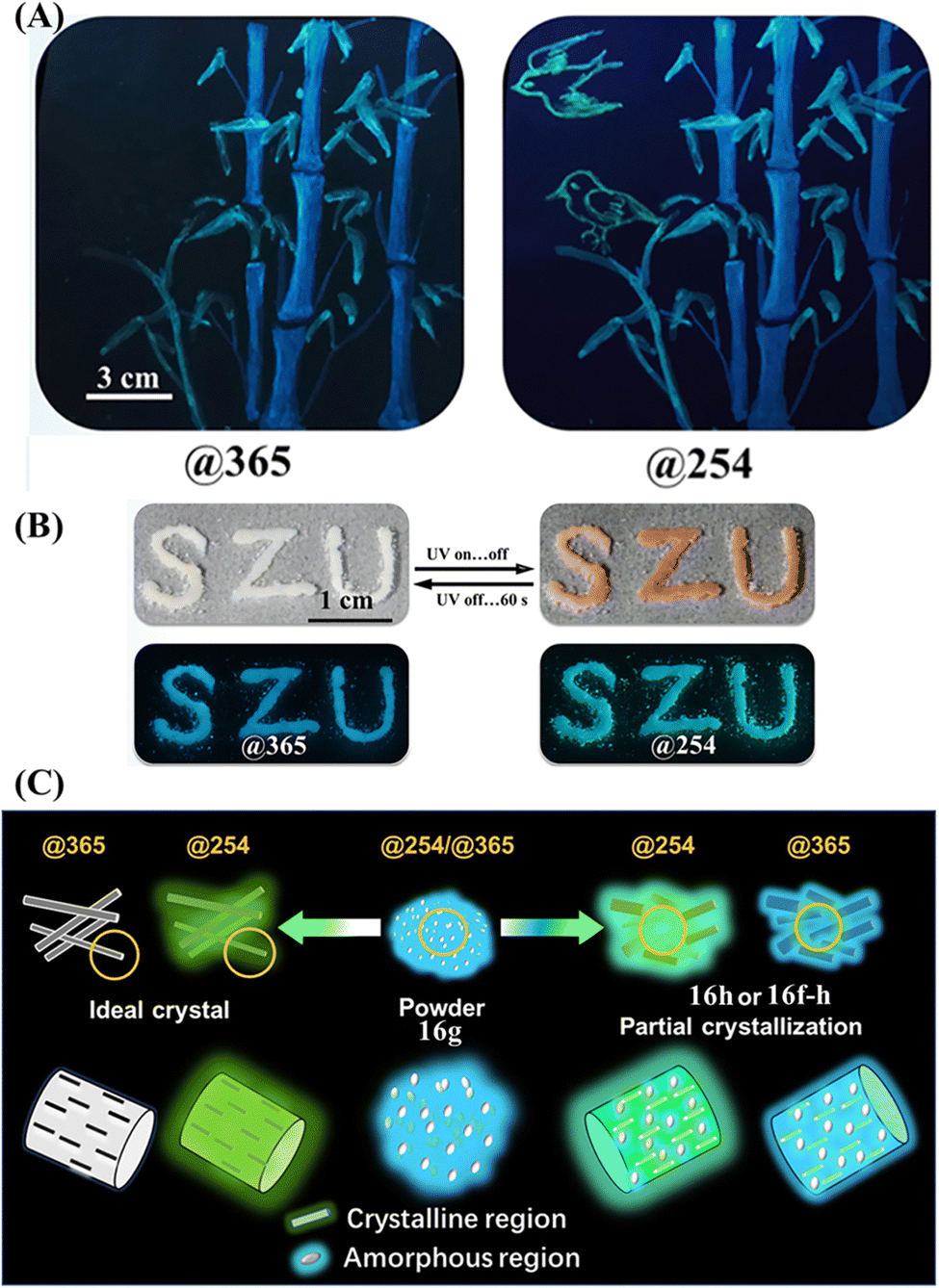 | ||
| Fig. 25 (A) Illustration of the multi-channel anti-counterfeiting application of 16. (B) Photochromism and Ex-De luminescence behaviors of 16. (C) Schematic illustration of the Ex-De mechanism of 16. The amorphous state 16g can be excited @254 and @365 with blueish emission while the ideal crystal can only be excited @254 with greenish fluorescence. State 16f-h, 16h or 16m with the coexistence of crystalline and amorphous regions showed combinational greenish and blueish fluorescence. Reproduced with permission.113 Copyright 2022, CCS Chemistry. | ||
Now a question arises: why do these TPE derivatives display Ex-De luminescence? Zhang et al. found that the Ex-De behavior of the TPE derivatives with adjustable conformations can be activated in PMMA films, while planar molecules with fixed conformation (Anthracene and Rhodamine 6G) showed independence of the excitation wavelength both in solutions and in PMMA films. Combined with the excitation spectra and excitation-dependent emission spectra during the mechanochromic process, they deduced that through manual grinding it is very difficult to destroy all the crystals into the amorphous state; a number of small microcrystals were still covered by multiple-layers of amorphous molecules (Fig. 24D). Therefore various surfaces, interfaces and defects of the microcrystals/amorphous mixture are non-ignorable, thus giving the unique Ex-De luminescence. Meanwhile, the annealing and fuming treatments could not completely induce recrystallization of the amorphous state either due to the low mobility of molecules in the solid state. For molecule 16, its relatively twisted conformation is unfavorable for the formation of perfect crystal packing when comparing to the stretched conformation of its isomer 15. Similarly, annealing, fuming or even slow solvent evaporation could not produce a single ordered state of 16, leading to the coexistence of crystalline and amorphous regions. The ratio of blueish emissive amorphous state and green emissive crystalline state in heated powder, fumed powder and agglomerated state collaboratively determined the remarkable degree of their Ex-De property (Fig. 25C).
The above examples imply that new Ex-De molecules might be explored based on TPE derivatives with an irregular/twisted molecular structure. Twisted conformation may help to reduce intermolecular interactions and facilitate the transformation between different states, while irregular molecular structure may also help to form mixtures of different states after grinding, annealing or fuming treatments. However, the above explanations about Ex-De luminescence dominated by conjecture, more examples and research methods are still in urgent need.
Summary and outlook
In summary, various TPE derivatives in the solid state have achieved breakthrough developments in the past decade. We have highlighted the representative progress of stimuli-responsive TPE-based compounds. External stimuli could be a mechanical force, heat, light, solvent vapor, etc. Many interesting features have been gradually discovered with the in-depth study of TPE and more novel discoveries will be made in the future. Based on what have been achieved now, we have the following comments on the development trend of stimuli-responsive materials (not limited to TPE derivatives) probably including the following respects:(1) Exploration of more multi-stimuli responsive materials will be an important approach. Quite a number of different types of compounds only respond to a single external stimulus. Integrating different stimuli-responsive functions to a single molecule is still challenging work. For example, two-state mechanochromic materials are relatively easy to realize for TPE derivatives now, but three colored materials are still lacking in design strategy. Multi-stimuli responsive materials can undoubtedly simplify the synthesis and combination of multiple raw materials, which single functional materials have to confront with. Moreover, investigation of multi-stimuli responsive materials is obviously favorable for an understanding of the underlying mechanism of single function and the relationship among different stimuli-responsive properties. TPE has demonstrated that it is a potential building block for multi-stimuli responsive materials, which as a matter of course is worth further exploration.
(2) The internal mechanism of stimuli responsive properties is still compelling. It is clear that the mechanism of the mechanochromic property is related to the phase transformation between crystalline state and amorphous state or different crystalline states. In contrast, exploration of organic mechanoluminescence materials is still in its infant stage. Therefore, the design of organic mechanoluminescence molecules still has a long way to go. Multiple challenging but critical questions need to be answered. How to characterize the sensitivity of mechanoluminescence? How to quantitatively measure the emissive intensity? How to clarify the photophysical process of mechanoluminescence? How to correctly describe the structure–property relationship (the aforementioned investigations on polymorphs, isomers or different substituted groups are not very reasonable)? These questions are hard to address in a short time. Reversible TPE-based photochromic or organic Ex-De molecules are also in their initial stage. It is extremely difficult to derive a structure–property relationship from quite limited reported examples. Therefore, enlarging the library of these stimuli-responsive molecules and exploring their internal mechanism are urgently in demand.
(3) Photochromism, photo switch and Ex-De luminescence materials are more promising towards practical application. Currently, the mechanochromic property of small organic molecules can hardly satisfy practical applications for the risk of organic powder leaking and organic solvent pollution/harm are always involved. While compared to mechanochromic polymers, they also have the drawback of failing to form a free-standing film. The application of organic mechanoluminescence materials is also challenging in several aspects. Their signal intensity is obviously weaker than that of inorganic mechanoluminescence materials. In addition, a recovery step is necessary for repeated use. These drawbacks are great obstacles for displaying, sensing and other applications. At this point, non-invasive stimuli-responsive properties, such as photochromism, photoswitching and Ex-De luminescence, are more promising for practical applications, which might be an important approach in the future.
We hope that this review can help researchers to construct a cognition framework and give some clues for further investigation towards the rational design and synthesis of novel multi-stimuli responsive materials with the potential to be used in a variety of practical applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Nature Science Foundation of China (21905177), the Natural Science Foundation of Guangdong Province (2019KZDXM008 and 2021A1515010192) and the Fundamental Foundation of Shenzhen (JCYJ20210324094607021). In addition, Dr Huang dedicates this manuscript to his parents Mr Huang Jinchuan and Mrs Song Meiyu on the occasion of their 60th birthday.References
- S. K. Mellerup and S. Wang, Boron-based stimuli responsive materials, Chem. Soc. Rev., 2019, 48, 3537–3549 RSC.
- P. She, Y. Qin, X. Wang and Q. Zhang, Recent Progress in External-Stimulus-Responsive 2D Covalent Organic Frameworks, Adv. Mater., 2022, 34, 2101175 CrossRef CAS.
- X. Lin, X. Wang, L. Zeng, Z. L. Wu, H. Guo and D. Hourdet, Stimuli-Responsive Toughening of Hydrogels, Chem. Mater., 2021, 33, 7633–7656 CrossRef CAS.
- R. Kumar, A. Sharma, H. Singh, P. Suating, H. S. Kim, K. Sunwoo, I. Shim, B. C. Gibb and J. S. Kim, Revisiting Fluorescent Calixarenes: From Molecular Sensors to Smart Materials, Chem. Rev., 2019, 119, 9657–9721 CrossRef CAS PubMed.
- P. Mahalingavelar and S. Kanvah, α-Cyanostilbene: a multifunctional spectral engineering motif, Phys. Chem. Chem. Phys., 2022, 24, 23049–23075 RSC.
- P. Theato, B. S. Sumerlin, R. K. O'Reilly and I. I. I. T. H. Epps, Stimuli responsive materials, Chem. Soc. Rev., 2013, 42, 7055–7056 RSC.
- P. Naumov, S. Chizhik, M. K. Panda, N. K. Nath and E. Boldyreva, Mechanically Responsive Molecular Crystals, Chem. Rev., 2015, 115, 12440–12490 CrossRef CAS PubMed.
- Y. Dong, J. Wang, X. Guo, S. Yang, M. O. Ozen, P. Chen, X. Liu, W. Du, F. Xiao, U. Demirci and B.-F. Liu, Multi-stimuli-responsive programmable biomimetic actuator, Nat. Commun., 2019, 10, 4087 CrossRef PubMed.
- M. Li and W.-H. Zhu, Sterically Hindered Diarylethenes with a Benzobis(thiadiazole) Bridge: Enantiospecific Transformation and Reversible Photosuperstructures, Acc. Chem. Res., 2022, 55, 3136–3149 CrossRef CAS PubMed.
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Photochromism of Diarylethene Molecules and Crystals: Memories, Switches, and Actuators, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS PubMed.
- L. Kortekaas and W. R. Browne, The evolution of spiropyran: fundamentals and progress of an extraordinarily versatile photochrome, Chem. Soc. Rev., 2019, 48, 3406–3424 RSC.
- A. B. A. Kayani, S. Kuriakose, M. Monshipouri, F. A. Khalid, S. Walia, S. Sriram and M. Bhaskaran, UV Photochromism in Transition Metal Oxides and Hybrid Materials, Small, 2021, 17, 2100621 CrossRef CAS PubMed.
- L. M. C. Luong, M. A. Malwitz, V. Moshayedi, M. M. Olmstead and A. L. Balch, Role of Anions and Mixtures of Anions on the Thermochromism, Vapochromism, and Polymorph Formation of Luminescent Crystals of a Single Cation, [(C6H11NC)2Au]+, J. Am. Chem. Soc., 2020, 142, 5689–5701 CrossRef CAS PubMed.
- J. Feng, K. Tian, D. Hu, S. Wang, S. Li, Y. Zeng, Y. Li and G. Yang, A Triarylboron-Based Fluorescent Thermometer: Sensitive Over a Wide Temperature Range, Angew. Chem., Int. Ed., 2011, 50, 8072–8076 CrossRef CAS.
- D. Hu, T. Zhang, S. Li, T. Yu, X. Zhang, R. Hu, J. Feng, S. Wang, T. Liang, J. Chen, L. N. Sobenina, B. A. Trofimov, Y. Li, J. Ma and G. Yang, Ultrasensitive reversible chromophore reaction of BODIPY functions as high ratio double turn on probe, Nat. Commun., 2018, 9, 362 CrossRef PubMed.
- G. Chen and W. Hong, Mechanochromism of Structural-Colored Materials, Adv. Opt. Mater., 2020, 8, 2000984 CrossRef CAS.
- T. Han, L. Liu, D. Wang, J. Yang and B. Z. Tang, Mechanochromic Fluorescent Polymers Enabled by AIE Processes, Macromol. Rapid Commun., 2021, 42, 2000311 CrossRef CAS.
- K. Seshimo, H. Sakai, T. Watabe, D. Aoki, H. Sugita, K. Mikami, Y. Mao, A. Ishigami, S. Nishitsuji, T. Kurose, H. Ito and H. Otsuka, Segmented Polyurethane Elastomers with Mechanochromic and Self-Strengthening Functions, Angew. Chem., Int. Ed., 2021, 60, 8406–8409 CrossRef.
- Q. Qi, G. Sekhon, R. Chandradat, N. M. Ofodum, T. Shen, J. Scrimgeour, M. Joy, M. Wriedt, M. Jayathirtha, C. C. Darie, D. A. Shipp, X. Liu and X. Lu, Force-Induced Near-Infrared Chromism of Mechanophore-Linked Polymers, J. Am. Chem. Soc., 2021, 143, 17337–17343 CrossRef CAS PubMed.
- H. Hu, X. Cheng, Z. Ma, R. P. Sijbesma and Z. Ma, Polymer Mechanochromism from Force-Tuned Excited-State Intramolecular Proton Transfer, J. Am. Chem. Soc., 2022, 144, 9971–9979 CrossRef CAS PubMed.
- T. Jadhav, B. Dhokale and R. Misra, Effect of the cyano group on solid state photophysical behavior of tetraphenylethene substituted benzothiadiazoles, J. Mater. Chem. C, 2015, 3, 9063–9068 RSC.
- A. Ekbote, S. H. Han, T. Jadhav, S. M. Mobin, J. Y. Lee and R. Misra, Stimuli responsive AIE active positional isomers of phenanthroimidazole as non-doped emitters in OLEDs, J. Mater. Chem. C, 2018, 6, 2077–2087 RSC.
- T. Jadhav, B. Dhokale, Y. Patil and R. Misra, Aggregation induced emission and mechanochromism in tetraphenylethene substituted pyrazabole, RSC Adv., 2015, 5, 68187–68191 RSC.
- T. Jadhav, B. Dhokale, S. M. Mobin and R. Misra, Mechanochromism and aggregation induced emission in benzothiazole substituted tetraphenylethylenes: a structure function correlation, RSC Adv., 2015, 5, 29878–29884 RSC.
- F. Khan, A. Ekbote and R. Misra, Reversible mechanochromism and aggregation induced enhanced emission in phenothiazine substituted tetraphenylethylene, New J. Chem., 2019, 43, 16156–16163 RSC.
- A. Ekbote, T. Jadhav and R. Misra, T-Shaped donor–acceptor–donor type tetraphenylethylene substituted quinoxaline derivatives: aggregation-induced emission and mechanochromism, New J. Chem., 2017, 41, 9346–9353 RSC.
- G. Yang, Y.-M. Zhang, Y. Cai, B. Yang, C. Gu and S. X.-A. Zhang, Advances in nanomaterials for electrochromic devices, Chem. Soc. Rev., 2020, 49, 8687–8720 RSC.
- H. Fu, L. Zhang, Y. Dong, C. Zhang and W. Li, Recent advances in electrochromic materials and devices for camouflage applications, Mater. Chem. Front., 2023, 7, 2337–2358 RSC.
- Y. Zhai, J. Li, S. Shen, Z. Zhu, S. Mao, X. Xiao, C. Zhu, J. Tang, X. Lu and J. Chen, Recent Advances on Dual-Band Electrochromic Materials and Devices, Adv. Funct. Mater., 2022, 32, 2109848 CrossRef CAS.
- V. Rai, R. S. Singh, D. J. Blackwood and D. Zhili, A Review on Recent Advances in Electrochromic Devices: A Material Approach, Adv. Eng. Mater., 2020, 22, 2000082 CrossRef CAS.
- X. Chen, L. Li, X. Sun, Y. Liu, B. Luo, C. Wang, Y. Bao, H. Xu and H. Peng, Magnetochromatic Polydiacetylene by Incorporation of Fe3O4 Nanoparticles, Angew. Chem., Int. Ed., 2011, 50, 5486–5489 CrossRef CAS PubMed.
- Z. Huang, T. Lan, L. Dai, X. Zhao, Z. Wang, Z. Zhang, B. Li, J. Li, J. Liu, B. Ding, A. K. Geim, H.-M. Cheng and B. Liu, 2D Functional Minerals as Sustainable Materials for Magneto-Optics, Adv. Mater., 2022, 34, 2110464 CrossRef CAS PubMed.
- V. Kumar, H. Kim, B. Pandey, T. D. James, J. Yoon and E. V. Anslyn, Recent advances in fluorescent and colorimetric chemosensors for the detection of chemical warfare agents: a legacy of the 21st century, Chem. Soc. Rev., 2023, 52, 663–704 RSC.
- Z. Meng and K. A. Mirica, Covalent organic frameworks as multifunctional materials for chemical detection, Chem. Soc. Rev., 2021, 50, 13498–13558 RSC.
- D. Cao, Z. Liu, P. Verwilst, S. Koo, P. Jangjili, J. S. Kim and W. Lin, Coumarin-Based Small-Molecule Fluorescent Chemosensors, Chem. Rev., 2019, 119, 10403–10519 CrossRef CAS PubMed.
- T. L. Mako, J. M. Racicot and M. Levine, Supramolecular Luminescent Sensors, Chem. Rev., 2019, 119, 322–477 CrossRef CAS PubMed.
- D. Mao and B. Liu, Biology-Oriented Design Strategies of AIE Theranostic Probes, Matter, 2021, 4, 350–376 CrossRef CAS.
- J. Yin, L. Huang, L. Wu, J. Li, T. D. James and W. Lin, Small molecule based fluorescent chemosensors for imaging the microenvironment within specific cellular regions, Chem. Soc. Rev., 2021, 50, 12098–12150 RSC.
- Y.-M. Zhang, Y.-H. Liu and Y. Liu, Cyclodextrin-Based Multistimuli-Responsive Supramolecular Assemblies and Their Biological Functions, Adv. Mater., 2020, 32, 1806158 CrossRef CAS PubMed.
- Y. Wakayama, R. Hayakawa, K. Higashiguchi and K. Matsuda, Photochromism for optically functionalized organic field-effect transistors: a comprehensive review, J. Mater. Chem. C, 2020, 8, 10956–10974 RSC.
- M. H. Barbee, K. Mondal, J. Z. Deng, V. Bharambe, T. V. Neumann, J. J. Adams, N. Boechler, M. D. Dickey and S. L. Craig, Mechanochromic Stretchable Electronics, ACS Appl. Mater. Interfaces, 2018, 10, 29918–29924 CrossRef CAS PubMed.
- H. Zhang, H. Chen, J.-H. Lee, E. Kim, K.-Y. Chan, H. Venkatesan, X. Shen, J. Yang and J.-K. Kim, Mechanochromic Optical/Electrical Skin for Ultrasensitive Dual-Signal Sensing, ACS Nano, 2023, 17, 5921–5934 CrossRef CAS PubMed.
- Y. Wang, Y.-M. Zhang and S. X.-A. Zhang, Stimuli-Induced Reversible Proton Transfer for Stimuli-Responsive Materials and Devices, Acc. Chem. Res., 2021, 54, 2216–2226 CrossRef CAS PubMed.
- C. Gu, A.-B. Jia, Y.-M. Zhang and S. X.-A. Zhang, Emerging Electrochromic Materials and Devices for Future Displays, Chem. Rev., 2022, 122, 14679–14721 CrossRef CAS PubMed.
- Z. Wang, X. Wang, S. Cong, F. Geng and Z. Zhao, Fusing electrochromic technology with other advanced technologies: A new roadmap for future development, Mater. Sci. Eng., R, 2020, 140, 100524 CrossRef.
- X. Yu, H. Zhang and J. Yu, Luminescence anti-counterfeiting: From elementary to advanced, Aggregate, 2021, 2, 20–34 CrossRef CAS.
- Y. Sun, X. Le, S. Zhou and T. Chen, Recent Progress in Smart Polymeric Gel-Based Information Storage for Anti-Counterfeiting, Adv. Mater., 2022, 34, 2201262 CrossRef CAS PubMed.
- Q. Li and Z. Li, Molecular Packing: Another Key Point for the Performance of Organic and Polymeric Optoelectronic Materials, Acc. Chem. Res., 2020, 53, 962–973 CrossRef CAS PubMed.
- M. Fang, J. Yang and Z. Li, Light emission of organic luminogens: Generation, mechanism and application, Prog. Mater. Sci., 2022, 125, 100914 CrossRef CAS.
- C. Wang and Z. Li, Molecular conformation and packing: their critical roles in the emission performance of mechanochromic fluorescence materials, Mater. Chem. Front., 2017, 1, 2174–2194 RSC.
- H. Zhang, Z. Zhao, A. T. Turley, L. Wang, P. R. McGonigal, Y. Tu, Y. Li, Z. Wang, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Aggregate Science: From Structures to Properties, Adv. Mater., 2020, 32, 2001457 CrossRef CAS.
- Y. Fan, Q. Li and Z. Li, Organic luminogens bearing alkyl substituents: design flexibility, adjustable molecular packing, and optimized performance, Mater. Chem. Front., 2021, 5, 1525–1540 RSC.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Aggregation-induced emission of 1-methyl-1,2,3,4,5-pentaphenylsilole, Chem. Commun., 2001, 1740–1741 RSC.
- Y. Chen, W. Zhang, Y. Cai, R. T. K. Kwok, Y. Hu, J. W. Y. Lam, X. Gu, Z. He, Z. Zhao, X. Zheng, B. Chen, C. Gui and B. Z. Tang, AIEgens for dark through-bond energy transfer: design, synthesis, theoretical study and application in ratiometric Hg2+ sensing, Chem. Sci., 2017, 8, 2047–2055 RSC.
- S. Chen, J. Liu, Y. Liu, H. Su, Y. Hong, C. K. W. Jim, R. T. K. Kwok, N. Zhao, W. Qin, J. W. Y. Lam, K. S. Wong and B. Z. Tang, An AIE-active hemicyanine fluorogen with stimuli-responsive red/blue emission: extending the pH sensing range by “switch + knob” effect, Chem. Sci., 2012, 3, 1804–1809 RSC.
- L. Cheng, P. Tian, H. Duan, Q. Li, X. Song, A. Li and L. Cao, Chiral adaptive recognition with sequence specificity of aromatic dipeptides in aqueous solution by an achiral cage, Chem. Sci., 2023, 14, 833–842 RSC.
- Y. Zhang, D. Li, Y. Li and J. Yu, Solvatochromic AIE luminogens as supersensitive water detectors in organic solvents and highly efficient cyanide chemosensors in water, Chem. Sci., 2014, 5, 2710–2716 RSC.
- B. Yuan, D.-X. Wang, L.-N. Zhu, Y.-L. Lan, M. Cheng, L.-M. Zhang, J.-Q. Chu, X.-Z. Li and D.-M. Kong, Dinuclear HgII tetracarbene complex-triggered aggregation-induced emission for rapid and selective sensing of Hg2+ and organomercury species, Chem. Sci., 2019, 10, 4220–4226 RSC.
- H. Liu, Y. Gu, Y. Dai, K. Wang, S. Zhang, G. Chen, B. Zou and B. Yang, Pressure-Induced Blue-Shifted and Enhanced Emission: A Cooperative Effect between Aggregation-Induced Emission and Energy-Transfer Suppression, J. Am. Chem. Soc., 2020, 142, 1153–1158 CrossRef CAS PubMed.
- X. Wang, C. Qi, Z. Fu, H. Zhang, J. Wang, H.-T. Feng, K. Wang, B. Zou, J. W. Y. Lam and B. Z. Tang, A synergy between the push–pull electronic effect and twisted conformation for high-contrast mechanochromic AIEgens, Mater. Horiz., 2021, 8, 630–638 RSC.
- H. Yuan, K. Wang, K. Yang, B. Liu and B. Zou, Luminescence Properties of Compressed Tetraphenylethene: The Role of Intermolecular Interactions, J. Phys. Chem. Lett., 2014, 5, 2968–2973 CrossRef CAS.
- Z. Li, P. Liu, X. Ji, J. Gong, Y. Hu, W. Wu, X. Wang, H.-Q. Peng, R. T. K. Kwok, J. W. Y. Lam, J. Lu and B. Z. Tang, Bioinspired Simultaneous Changes in Fluorescence Color, Brightness, and Shape of Hydrogels Enabled by AIEgens, Adv. Mater., 2020, 32, 1906493 CrossRef CAS.
- Y. Cheng, J. Wang, Z. Qiu, X. Zheng, N. L. C. Leung, J. W. Y. Lam and B. Z. Tang, Multiscale Humidity Visualization by Environmentally Sensitive Fluorescent Molecular Rotors, Adv. Mater., 2017, 29, 1703900 CrossRef PubMed.
- C. Li, K. Xiong, Y. Chen, C. Fan, Y.-L. Wang, H. Ye and M.-Q. Zhu, Visible-Light-Driven Photoswitching of Aggregated-Induced Emission-Active Diarylethenes for Super-Resolution Imaging, ACS Appl. Mater. Interfaces, 2020, 12, 27651–27662 CrossRef CAS.
- B. Wu, T. Xue, W. Wang, S. Li, J. Shen and Y. He, Visible light triggered aggregation-induced emission switching with a donor–acceptor Stenhouse adduct, J. Mater. Chem. C, 2018, 6, 8538–8545 RSC.
- S. Liu, Y. Cheng, Y. Li, M. Chen, J. W. Y. Lam and B. Z. Tang, Manipulating Solid-State Intramolecular Motion toward Controlled Fluorescence Patterns, ACS Nano, 2020, 14, 2090–2098 CrossRef CAS PubMed.
- J. Wang, Q. Jiang, S. Cao, C. Sun, Y. Zhang, Y. Qiu, H. Wang, G. Yin, Y. Liao and X. Xie, Z/E Effect on Phase Behavior of Main-Chain Liquid Crystalline Polymers Bearing AIEgens, Macromolecules, 2021, 54, 10740–10749 CrossRef CAS.
- S.-W. Cheng, T. Han, T.-Y. Huang, B.-Z. Tang and G.-S. Liou, High-performance electrofluorochromic devices based on aromatic polyamides with AIE-active tetraphenylethene and electro-active triphenylamine moieties, Polym. Chem., 2018, 9, 4364–4373 RSC.
- S. Mi, J. Wu, J. Liu, Z. Xu, X. Wu, G. Luo, J. Zheng and C. Xu, AIEE-Active and Electrochromic Bifunctional Polymer and a Device Composed thereof Synchronously Achieve Electrochemical Fluorescence Switching and Electrochromic Switching, ACS Appl. Mater. Interfaces, 2015, 7, 27511–27517 CrossRef CAS.
- N. Sun, K. Su, Z. Zhou, Y. Yu, X. Tian, D. Wang, X. Zhao, H. Zhou and C. Chen, AIE-Active Polyamide Containing Diphenylamine-TPE Moiety with Superior Electrofluorochromic Performance, ACS Appl. Mater. Interfaces, 2018, 10, 16105–16112 CrossRef CAS.
- R. Singh, H.-Y. Wu, A. Kumar Dwivedi, A. Singh, C.-M. Lin, P. Raghunath, M.-C. Lin, T.-K. Wu, K.-H. Wei and H.-C. Lin, Monomeric and aggregation emissions of tetraphenylethene in a photo-switchable polymer controlled by cyclization of diarylethene and solvent conditions, J. Mater. Chem. C, 2017, 5, 9952–9962 RSC.
- Z. Yang, Z. Chi, Z. Mao, Y. Zhang, S. Liu, J. Zhao, M. P. Aldred and Z. Chi, Recent advances in mechano-responsive luminescence of tetraphenylethylene derivatives with aggregation-induced emission properties, Mater. Chem. Front., 2018, 2, 861–890 RSC.
- F. Khan, A. Ekbote, G. Singh and R. Misra, Mechanochromic luminogens with hypsochromically shifted emission switching property: recent advances and perspectives, J. Mater. Chem. C, 2022, 10, 5024–5064 RSC.
- Q. Yan and S. Wang, Fusion of aggregation-induced emission and photochromics for promising photoresponsive smart materials, Mater. Chem. Front., 2020, 4, 3153–3175 RSC.
- J. Zhang, B. He, Y. Hu, P. Alam, H. Zhang, J. W. Y. Lam and B. Z. Tang, Stimuli-Responsive AIEgens, Adv. Mater., 2021, 33, 2008071 CrossRef CAS.
- Y. Sun, Z. Lei and H. Ma, Twisted aggregation-induced emission luminogens (AIEgens) contribute to mechanochromism materials: a review, J. Mater. Chem. C, 2022, 10, 14834–14867 RSC.
- J. Yang, M. Fang and Z. Li, Organic luminescent materials: The concentration on aggregates from aggregation-induced emission, Aggregate, 2020, 1, 6–18 CrossRef.
- P. Zhou and K. Han, ESIPT-based AIE luminogens: Design strategies, applications, and mechanisms, Aggregate, 2022, 3, e160 CrossRef CAS.
- Y. Xie and Z. Li, The development of mechanoluminescence from organic compounds: breakthrough and deep insight, Mater. Chem. Front., 2020, 4, 317–331 RSC.
- X. Zhang, Z. Chi, H. Li, B. Xu, X. Li, W. Zhou, S. Liu, Y. Zhang and J. Xu, Piezofluorochromism of an Aggregation-Induced Emission Compound Derived from Tetraphenylethylene, Chem. – Asian J., 2011, 6, 808–811 CrossRef CAS PubMed.
- X. Tian, H. Wang, S. Cao, Y. Liu, F. Meng, X. Zheng and G. Niu, Cationic tetraphenylethylene-based AIE-active acrylonitriles: investigating the regioisomeric effect, mechanochromism, and wash-free bioimaging, J. Mater. Chem. C, 2023, 11, 5987–5994 RSC.
- W. Qiao, P. Yao, Y. Chen, Q. Xiao, L. Zhang and Z. A. Li, Squaraine-based AIEgens for reversible mechanochromism, sensitive and selective hypochlorite detection and photostable far-red fluorescence cell imaging, Mater. Chem. Front., 2020, 4, 2688–2696 RSC.
- X. Yang, Q. Wang, P. Hu, C. Xu, W. Guo, Z. Wang, Z. Mao, Z. Yang, C. Liu, G. Shi, L. Chen, B. Xu and Z. Chi, Achieving remarkable and reversible mechanochromism from a bright ionic AIEgen with high specificity for mitochondrial imaging and secondary aggregation emission enhancement for long-term tracking of tumors, Mater. Chem. Front., 2020, 4, 941–949 RSC.
- P. Xu, Q. Qiu, X. Ye, M. Wei, W. Xi, H. Feng and Z. Qian, Halogenated tetraphenylethene with enhanced aggregation-induced emission: an anomalous anti-heavy-atom effect and self-reversible mechanochromism, Chem. Commun., 2019, 55, 14938–14941 RSC.
- H.-X. Yu, J. Zhi, T. Shen, W. Ding, X. Zhang and J.-L. Wang, Donor–acceptor type aggregation-induced emission luminophores based on the 1,1-dicyanomethylene-3-indanone unit for bridge-dependent reversible mechanochromism and light-up biosensing of hypochlorites, J. Mater. Chem. C, 2019, 7, 8888–8897 RSC.
- L. Huang, Y. Qiu, C. Wu, Z. Ma, Z. Shen and X. Jia, A multi-state fluorescent switch with multifunction of AIE, methanol-responsiveness, photochromism and mechanochromism, J. Mater. Chem. C, 2018, 6, 10250–10255 RSC.
- T. Jadhav, J. M. Choi, J. Shinde, J. Y. Lee and R. Misra, Mechanochromism and electroluminescence in positional isomers of tetraphenylethylene substituted phenanthroimidazoles, J. Mater. Chem. C, 2017, 5, 6014–6020 RSC.
- T. Jadhav, B. Dhokale, S. M. Mobin and R. Misra, Aggregation induced emission and mechanochromism in pyrenoimidazoles, J. Mater. Chem. C, 2015, 3, 9981–9988 RSC.
- R. Misra, T. Jadhav, B. Dhokale and S. M. Mobin, Reversible mechanochromism and enhanced AIE in tetraphenylethene substituted phenanthroimidazoles, Chem. Commun., 2014, 50, 9076–9078 RSC.
- C. Y. K. Chan, J. W. Y. Lam, Z. Zhao, S. Chen, P. Lu, H. H. Y. Sung, H. S. Kwok, Y. Ma, I. D. Williams and B. Z. Tang, Aggregation-induced emission, mechanochromism and blue electroluminescence of carbazole and triphenylamine-substituted ethenes, J. Mater. Chem. C, 2014, 2, 4320–4327 RSC.
- F. Ye, Y. Liu, J. Chen, S. H. Liu, W. Zhao and J. Yin, Tetraphenylene-Coated Near-Infrared Benzoselenodiazole Dye: AIE Behavior, Mechanochromism, and Bioimaging, Org. Lett., 2019, 21, 7213–7217 CrossRef CAS.
- T. Hu, B. Yao, X. Chen, W. Li, Z. Song, A. Qin, J. Z. Sun and B. Z. Tang, Effect of ionic interaction on the mechanochromic properties of pyridinium modified tetraphenylethene, Chem. Commun., 2015, 51, 8849–8852 RSC.
- P. Xue, Z. Yang and P. Chen, Hiding and revealing information using the mechanochromic system of a 2,5-dicarbazole-substituted terephthalate derivative, J. Mater. Chem. C, 2018, 6, 4994–5000 RSC.
- Z. Ruan, Y. Shan, Y. Gong, C. Wang, F. Ye, Y. Qiu, Z. Liang and Z. Li, Novel AIE-active ratiometric fluorescent probes for mercury(ii) based on the Hg2 + -promoted deprotection of thioketal, and good mechanochromic properties, J. Mater. Chem. C, 2018, 6, 773–780 RSC.
- H. Zhang, H. Zhang, G. Huang and B. S. Li, Two tetraphenylethene-pyrene isomers: Distinct fluorescence and mechanochromic properties, Dyes Pigm., 2021, 185, 108947 CrossRef CAS.
- A. Ekbote, S. M. Mobin and R. Misra, Stimuli-responsive phenothiazine-based donor–acceptor isomers: AIE, mechanochromism and polymorphism, J. Mater. Chem. C, 2020, 8, 3589–3602 RSC.
- F. Khan, E. Urbonas, D. Volyniuk, J. V. Grazulevicius, S. M. Mobin and R. Misra, White hyperelectrofluorescence from solution-processable OLEDs based on phenothiazine substituted tetraphenylethylene derivatives, J. Mater. Chem. C, 2020, 8, 13375–13388 RSC.
- F. Khan, A. Ekbote, S. M. Mobin and R. Misra, Mechanochromism and Aggregation-Induced Emission in Phenanthroimidazole Derivatives: Role of Positional Change of Different Donors in a Multichromophoric Assembly, J. Org. Chem., 2021, 86, 1560–1574 CrossRef CAS.
- F. Khan, M. Mahmoudi, D. Volyniuk, J. V. Grazulevicius and R. Misra, Stimuli-Responsive Phenothiazine-S,S-dioxide-Based Nondoped OLEDs with Color-Changeable Electroluminescence, J. Phys. Chem. C, 2022, 126, 15573–15586 CrossRef CAS.
- F. Khan, M. Mahmoudi, P. K. Gupta, D. Volyniuk, J. V. Grazulevicius and R. Misra, Mechanochromic Materials Based on Tetraphenylethylene-Substituted Phenothiazines: Substituent-Dependent Hypsochromic and Bathochromic Switching of Emission, J. Phys. Chem. C, 2023, 127, 1643–1654 CrossRef CAS.
- J. Wang, J. Mei, R. Hu, J. Z. Sun, A. Qin and B. Z. Tang, Click Synthesis, Aggregation-Induced Emission, E/Z Isomerization, Self-Organization, and Multiple Chromisms of Pure Stereoisomers of a Tetraphenylethene-Cored Luminogen, J. Am. Chem. Soc., 2012, 134, 9956–9966 CrossRef CAS PubMed.
- Q. Qi, J. Qian, X. Tan, J. Zhang, L. Wang, B. Xu, B. Zou and W. Tian, Remarkable Turn-On and Color-Tuned Piezochromic Luminescence: Mechanically Switching Intramolecular Charge Transfer in Molecular Crystals, Adv. Funct. Mater., 2015, 25, 4005–4010 CrossRef CAS.
- T. Yu, D. Ou, Z. Yang, Q. Huang, Z. Mao, J. Chen, Y. Zhang, S. Liu, J. Xu, M. R. Bryce and Z. Chi, The HOF structures of nitrotetraphenylethene derivatives provide new insights into the nature of AIE and a way to design mechanoluminescent materials, Chem. Sci., 2017, 8, 1163–1168 RSC.
- W. Zhao, Z. He, Q. Peng, J. W. Y. Lam, H. Ma, Z. Qiu, Y. Chen, Z. Zhao, Z. Shuai, Y. Dong and B. Z. Tang, Highly sensitive switching of solid-state luminescence by controlling intersystem crossing, Nat. Commun., 2018, 9, 3044 CrossRef PubMed.
- Z. Qiu, W. Zhao, M. Cao, Y. Wang, J. W. Y. Lam, Z. Zhang, X. Chen and B. Z. Tang, Dynamic Visualization of Stress/Strain Distribution and Fatigue Crack Propagation by an Organic Mechanoresponsive AIE Luminogen, Adv. Mater., 2018, 30, 1803924 CrossRef.
- C. Ma, B. Xu, G. Xie, J. He, X. Zhou, B. Peng, L. Jiang, B. Xu, W. Tian, Z. Chi, S. Liu, Y. Zhang and J. Xu, An AIE-active luminophore with tunable and remarkable fluorescence switching based on the piezo and protonation–deprotonation control, Chem. Commun., 2014, 50, 7374–7377 RSC.
- X. Liu, C. Ma, A. Li, W. Xu, Z. Ma and X. Jia, Schiff base-bridged TPE-rhodamine dyad: facile synthesis, distinct response to shearing and hydrostatic pressure, and sequential multicolored acidichromism, J. Mater. Chem. C, 2019, 7, 8398–8403 RSC.
- Z. Ma, Z. Wang, X. Meng, Z. Ma, Z. Xu, Y. Ma and X. Jia, A Mechanochromic Single Crystal: Turning Two Color Changes into a Tricolored Switch, Angew. Chem., Int. Ed., 2016, 55, 519–522 CrossRef CAS PubMed.
- H. Tian, P. Wang, J. Liu, Y. Duan and Y. Q. Dong, Construction of a tetraphenylethene derivative exhibiting high contrast and multicolored emission switching, J. Mater. Chem. C, 2017, 5, 12785–12791 RSC.
- G. Huang, Y. Jiang, S. Yang, B. S. Li and B. Z. Tang, Multistimuli Response and Polymorphism of a Novel Tetraphenylethylene Derivative, Adv. Funct. Mater., 2019, 29, 1900516 CrossRef.
- H. Zhang, B. Ma, F. Lin, Z. Yang, G. Huang, B. S. Li and B. Z. Tang, New shoots from old roots: multiple stimuli-responsive properties of a common tetraphenylethene derivative, Mater. Chem. Front., 2022, 6, 176–181 RSC.
- G. Huang, Q. Xia, W. Huang, J. Tian, Z. He, B. S. Li and B. Z. Tang, Multiple Anti-Counterfeiting Guarantees from a Simple Tetraphenylethylene Derivative – High-Contrasted and Multi-State Mechanochromism and Photochromism, Angew. Chem., Int. Ed., 2019, 58, 17814–17819 CrossRef CAS.
- Q. Xia, W. Xie, T. He, H. Zhang, Z. Zhao, G. Huang, B. S. Li and B. Z. Tang, A Versatile Tetraphenylethene Derivative Bearing Excitation Wavelength Dependent Emission, Multistate Mechanochromism, Reversible Photochromism, and Circularly Polarized Luminescence and Its Applications in Multimodal Anticounterfeiting, CCS Chem., 2023, 2, 1663–1673 CrossRef.
- Q. Huang, W. Li, Z. Mao, L. Qu, Y. Li, H. Zhang, T. Yu, Z. Yang, J. Zhao, Y. Zhang, M. P. Aldred and Z. Chi, An exceptionally flexible hydrogen-bonded organic framework with large-scale void regulation and adaptive guest accommodation abilities, Nat. Commun., 2019, 10, 3074 CrossRef.
- Q. Huang, W. Li, Z. Mao, H. Zhang, Y. Li, D. Ma, H. Wu, J. Zhao, Z. Yang, Y. Zhang, L. Gong, M. P. Aldred and Z. Chi, Dynamic molecular weaving in a two-dimensional hydrogen-bonded organic framework, Chem, 2021, 7, 1321–1332 CAS.
- Y. Shi, S. Wang, W. Tao, J. Guo, S. Xie, Y. Ding, G. Xu, C. Chen, X. Sun, Z. Zhang, Z. He, P. Wei and B. Z. Tang, Multiple yet switchable hydrogen-bonded organic frameworks with white-light emission, Nat. Commun., 2022, 13, 1882 CrossRef CAS PubMed.
- J.-A. Li, J. Zhou, Z. Mao, Z. Xie, Z. Yang, B. Xu, C. Liu, X. Chen, D. Ren, H. Pan, G. Shi, Y. Zhang and Z. Chi, Transient and Persistent Room-Temperature Mechanoluminescence from a White-Light-Emitting AIEgen with Tricolor Emission Switching Triggered by Light, Angew. Chem., Int. Ed., 2018, 57, 6449–6453 CrossRef CAS PubMed.
- J. Wang, C. Wang, Y. Gong, Q. Liao, M. Han, T. Jiang, Q. Dang, Y. Li, Q. Li and Z. Li, Bromine-Substituted Fluorene: Molecular Structure, Br–Br Interactions, Room-Temperature Phosphorescence, and Tricolor Triboluminescence, Angew. Chem., Int. Ed., 2018, 57, 16821–16826 CrossRef CAS PubMed.
- Z. Xie, T. Yu, J. Chen, E. Ubba, L. Wang, Z. Mao, T. Su, Y. Zhang, M. P. Aldred and Z. Chi, Weak interactions but potent effect: tunable mechanoluminescence by adjusting intermolecular C–H⋯π interactions, Chem. Sci., 2018, 9, 5787–5794 RSC.
- J. Yang, J. Qin, P. Geng, J. Wang, M. Fang and Z. Li, Molecular Conformation-Dependent Mechanoluminescence: Same Mechanical Stimulus but Different Emissive Color over Time, Angew. Chem., Int. Ed., 2018, 57, 14174–14178 CrossRef CAS PubMed.
- W. Li, Q. Huang, Z. Mao, Q. Li, L. Jiang, Z. Xie, R. Xu, Z. Yang, J. Zhao, T. Yu, Y. Zhang, M. P. Aldred and Z. Chi, Alkyl Chain Introduction: In Situ Solar-Renewable Colorful Organic Mechanoluminescence Materials, Angew. Chem., Int. Ed., 2018, 57, 12727–12732 CrossRef CAS PubMed.
- B. Xu, J. He, Y. Mu, Q. Zhu, S. Wu, Y. Wang, Y. Zhang, C. Jin, C. Lo, Z. Chi, A. Lien, S. Liu and J. Xu, Very bright mechanoluminescence and remarkable mechanochromism using a tetraphenylethene derivative with aggregation-induced emission, Chem. Sci., 2015, 6, 3236–3241 RSC.
- B. Xu, W. Li, J. He, S. Wu, Q. Zhu, Z. Yang, Y.-C. Wu, Y. Zhang, C. Jin, P.-Y. Lu, Z. Chi, S. Liu, J. Xu and M. R. Bryce, Achieving very bright mechanoluminescence from purely organic luminophores with aggregation-induced emission by crystal design, Chem. Sci., 2016, 7, 5307–5312 RSC.
- Y. Xie, J. Tu, T. Zhang, J. Wang, Z. Xie, Z. Chi, Q. Peng and Z. Li, Mechanoluminescence from pure hydrocarbon AIEgen, Chem. Commun., 2017, 53, 11330–11333 RSC.
- C. Wang, B. Xu, M. Li, Z. Chi, Y. Xie, Q. Li and Z. Li, A stable tetraphenylethene derivative: aggregation-induced emission, different crystalline polymorphs, and totally different mechanoluminescence properties, Mater. Horiz., 2016, 3, 220–225 RSC.
- F. Liu, J. Tu, X. Wang, J. Wang, Y. Gong, M. Han, X. Dang, Q. Liao, Q. Peng, Q. Li and Z. Li, Opposite mechanoluminescence behavior of two isomers with different linkage positions, Chem. Commun., 2018, 54, 5598–5601 RSC.
- Q. Huang, W. Li, Z. Yang, J. Zhao, Y. Li, Z. Mao, Z. Yang, S. Liu, Y. Zhang and Z. Chi, Achieving Bright Mechanoluminescence in a Hydrogen-Bonded Organic Framework by Polar Molecular Rotor Incorporation, CCS Chem., 2022, 4, 1643–1653 CrossRef CAS.
- Y. Jiang, J. Wang, G. Huang, Z. Li, B. S. Li and B. Z. Tang, Insight from the old: mechanochromism and mechanoluminescence of two amine-containing tetraphenylethylene isomers, J. Mater. Chem. C, 2019, 7, 11790–11796 RSC.
- H. Zhang, W. Li, Q. Huang, G. Huang, Z. Chi and B. S. Li, A multi-stimuli responsive tetraphenylethene derivative: Self-reversible mechanochromism, mechanoluminescence, switchable photochromism, Dyes Pigm., 2021, 187, 109128 CrossRef CAS.
- Y. Jiang, X. Chang, W. Xie, G. Huang and B. S. Li, Cyano-containing tetraphenylethene isomers: similar bright mechanoluminescence, but diverse recoverable processes, Mater. Chem. Front., 2021, 5, 885–892 RSC.
- G. Huang, Y. Jiang, J. Wang, Z. Li, B. S. Li and B. Z. Tang, The influence of intermolecular interactions and molecular packings on mechanochromism and mechanoluminescence – a tetraphenylethylene derivative case, J. Mater. Chem. C, 2019, 7, 12709–12716 RSC.
- Y. Yu, C. Wang, Y. Wei, Y. Fan, J. Yang, J. Wang, M. Han, Q. Li and Z. Li, Halogen-Containing TPA-Based Luminogens: Different Molecular Packing and Different Mechanoluminescence, Adv. Opt. Mater., 2019, 7, 1900505 CrossRef.
- S. Peng, J. Wen, M. Hai, Z. Yang, X. Yuan, D. Wang, H. Cao and W. He, Synthesis and application of reversible fluorescent photochromic molecules based on tetraphenylethylene and photochromic groups, New J. Chem., 2019, 43, 617–621 RSC.
- H. Dong, M. Luo, S. Wang and X. Ma, Synthesis and properties of tetraphenylethylene derivatived diarylethene with photochromism and aggregation-induced emission, Dyes Pigm., 2017, 139, 118–128 CrossRef CAS.
- L. Ma, C. Li, Q. Yan, S. Wang, W. Miao and D. Cao, Unsymmetrical photochromic bithienylethene-bridge tetraphenylethene molecular switches: Synthesis, aggregation-induced emission and information storage, Chin. Chem. Lett., 2020, 31, 361–364 CrossRef CAS.
- Q. Qi, J. Qian, S. Ma, B. Xu, S. X.-A. Zhang and W. Tian, Reversible Multistimuli-Response Fluorescent Switch Based on Tetraphenylethene–Spiropyran Molecules, Chem. – Eur. J., 2015, 21, 1149–1155 CrossRef CAS PubMed.
- Z. Wu, K. Pan, S. Mo, B. Wang, X. Zhao and M. Yin, Tetraphenylethene-Induced Free Volumes for the Isomerization of Spiropyran toward Multifunctional Materials in the Solid State, ACS Appl. Mater. Interfaces, 2018, 10, 30879–30886 CrossRef CAS PubMed.
- R. Yang, Y. Jiao, B. Wang, B. Xu and W. Tian, Solid-State Reversible Dual Fluorescent Switches for Multimodality Optical Memory, J. Phys. Chem. Lett., 2021, 12, 1290–1294 CrossRef CAS PubMed.
- E. Hadjoudis and I. M. Mavridis, Photochromism and thermochromism of Schiff bases in the solid state: structural aspects, Chem. Soc. Rev., 2004, 33, 579–588 CAS.
- L. Wang, Y. Li, X. You, K. Xu, Q. Feng, J. Wang, Y. Liu, K. Li and H. Hou, An erasable photo-patterning material based on a specially designed 4-(1,2,2-triphenylvinyl)aniline salicylaldehyde hydrazone aggregation-induced emission (AIE) molecule, J. Mater. Chem. C, 2017, 5, 65–72 RSC.
- H. Sun, J.-Y. Li, F.-F. Han, R. Zhang, Y. Zhao, B.-X. Miao and Z.-H. Ni, Reversible photochromic tetraphenylethene-based Schiff base: Design, synthesis, crystal structure and applications as visible light driven rewritable paper and UV sensor, Dyes Pigm., 2019, 167, 143–150 CrossRef CAS.
- H. Sun, S.-S. Sun, F.-F. Han, Z.-H. Ni, R. Zhang and M.-D. Li, A new tetraphenylethene-based Schiff base: two crystalline polymorphs exhibiting totally different photochromic and fluorescence properties, J. Mater. Chem. C, 2019, 7, 7053–7060 RSC.
- M. P. Aldred, C. Li and M.-Q. Zhu, Optical Properties and Photo-Oxidation of Tetraphenylethene-Based Fluorophores, Chem. – Eur. J., 2012, 18, 16037–16045 CrossRef CAS PubMed.
- G. Huang, B. Ma, J. Chen, Q. Peng, G. Zhang, Q. Fan and D. Zhang, Dendron-Containing Tetraphenylethylene Compounds: Dependence of Fluorescence and Photocyclization Reactivity on the Dendron Generation, Chem. – Eur. J., 2012, 18, 3886–3892 CrossRef CAS PubMed.
- T. Yu, D. Ou, L. Wang, S. Zheng, Z. Yang, Y. Zhang, Z. Chi, S. Liu, J. Xu and M. P. Aldred, A new approach to switchable photochromic materials by combining photochromism and piezochromism together in an AIE-active molecule, Mater. Chem. Front., 2017, 1, 1900–1904 RSC.
- Z. Xiong, X. Zhang, L. Liu, Q. Zhu, Z. Wang, H. Feng and Z. Qian, Achieving highly efficient aggregation-induced emission, reversible and irreversible photochromism by heavy halogen-regulated photophysics and D–A molecular pattern-controlled photochemistry of through-space conjugated luminogens, Chem. Sci., 2021, 12, 10710–10723 RSC.
- Y. Fan, M. Han, A. Huang, Q. Liao, J. Tu, X. Liu, B. Huang, Q. Li and Z. Li, Multi-photoresponsive triphenylethylene derivatives with photochromism, photodeformation and room temperature phosphorescence, Mater. Horiz., 2022, 9, 368–375 RSC.
- Z. He, L. Shan, J. Mei, H. Wang, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams, X. Gu, Q. Miao and B. Z. Tang, Aggregation-induced emission and aggregation-promoted photochromism of bis(diphenylmethylene)dihydroacenes, Chem. Sci., 2015, 6, 3538–3543 RSC.
- D. Ou, T. Yu, Z. Yang, T. Luan, Z. Mao, Y. Zhang, S. Liu, J. Xu, Z. Chi and M. R. Bryce, Combined aggregation induced emission (AIE), photochromism and photoresponsive wettability in simple dichloro-substituted triphenylethylene derivatives, Chem. Sci., 2016, 7, 5302–5306 RSC.
- L. Wang, T. Yu, Z. Xie, X. Chen, Z. Yang, Y. Zhang, M. P. Aldred and Z. Chi, Design, synthesis and photochromism studies of thienyl containing triarylethylene derivatives and their applications in real-time photoresponsive surfaces, J. Mater. Chem. C, 2018, 6, 8832–8838 RSC.
- W. Xie, X. Chang, L. Chen, L. Liu, G. Huang and B. S. Li, Construction of a multicolored emission tetraphenylethene derivative in response to multiple stimuli, Dyes Pigm., 2021, 195, 109723 CrossRef CAS.
- D. Zou, Z. Li, D. Long, X. Dong, H. Qu, L. Yang and X. Cao, Molecular Cage with Dual Outputs of Photochromism and Luminescence Both in Solution and the Solid State, ACS Appl. Mater. Interfaces, 2023, 15, 13545–13553 CrossRef CAS PubMed.
- Q. Huang, T. Yu, Z. Xie, W. Li, L. Wang, S. Liu, Y. Zhang, Z. Chi, J. Xu and M. P. Aldred, Hydrogen bonding-assisted loosely packed crystals of a diaminomaleonitrile-modified tetraphenylethene compound and their photo- and mechano-responsive properties, J. Mater. Chem. C, 2017, 5, 11867–11872 RSC.
- W. Zhao, Z. Liu, J. Yu, X. Lu, J. W. Y. Lam, J. Sun, Z. He, H. Ma and B. Z. Tang, Turning On Solid-State Luminescence by Phototriggered Subtle Molecular Conformation Variations, Adv. Mater., 2021, 33, 2006844 CrossRef CAS PubMed.
- J.-G. Yang, K. Li, J. Wang, S. Sun, W. Chi, C. Wang, X. Chang, C. Zou, W.-P. To, M.-D. Li, X. Liu, W. Lu, H.-X. Zhang, C.-M. Che and Y. Chen, Controlling Metallophilic Interactions in Chiral Gold(I) Double Salts towards Excitation Wavelength-Tunable Circularly Polarized Luminescence, Angew. Chem., Int. Ed., 2020, 59, 6915–6922 CrossRef CAS PubMed.
- H. Yang, Y. Liu, Z. Guo, B. Lei, J. Zhuang, X. Zhang, Z. Liu and C. Hu, Hydrophobic carbon dots with blue dispersed emission and red aggregation-induced emission, Nat. Commun., 2019, 10, 1789 CrossRef PubMed.
- B. Song, Y. Zhong, S. Wu, B. Chu, Y. Su and Y. He, One-Dimensional Fluorescent Silicon Nanorods Featuring Ultrahigh Photostability, Favorable Biocompatibility, and Excitation Wavelength-Dependent Emission Spectra, J. Am. Chem. Soc., 2016, 138, 4824–4831 CrossRef CAS PubMed.
- Y.-K. Jeong, Y. M. Lee, J. Yun, T. Mazur, M. Kim, Y. J. Kim, M. Dygas, S. H. Choi, K. S. Kim, O.-H. Kwon, S. M. Yoon and B. A. Grzybowski, Tunable Photoluminescence across the Visible Spectrum and Photocatalytic Activity of Mixed-Valence Rhenium Oxide Nanoparticles, J. Am. Chem. Soc., 2017, 139, 15088–15093 CrossRef CAS PubMed.
- G. N. Lewis and M. Kasha, Phosphorescence and the Triplet State, J. Am. Chem. Soc., 1944, 66, 2100–2116 CrossRef CAS.
- Y. Zhang, H. Yang, H. Ma, G. Bian, Q. Zang, J. Sun, C. Zhang, Z. An and W.-Y. Wong, Excitation Wavelength Dependent Fluorescence of an ESIPT Triazole Derivative for Amine Sensing and Anti-Counterfeiting Applications, Angew. Chem., Int. Ed., 2019, 58, 8773–8778 CrossRef CAS PubMed.
- Y. Wang, I. Zhang, B. Yu, X. Fang, X. Su, Y.-M. Zhang, T. Zhang, B. Yang, M. Li and S. X.-A. Zhang, Full-color tunable mechanofluorochromism and excitation-dependent emissions of single-arm extended tetraphenylethylenes, J. Mater. Chem. C, 2015, 3, 12328–12334 RSC.
| This journal is © the Partner Organisations 2024 |