
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUpconverting photons at the molecular scale with lanthanide complexes
Loïc J.
Charbonnière
 *a,
Aline M.
Nonat
a,
Richard C.
Knighton
ab and
Léna
Godec
a
*a,
Aline M.
Nonat
a,
Richard C.
Knighton
ab and
Léna
Godec
a
aEquipe de Synthèse Pour L'Analyse (SynPA), Institut Pluridisciplinaire Hubert Curien (IPHC), UMR7178, CNRS, Université de Strasbourg, ECPM, 67087 Strasbourg Cedex, France. E-mail: l.charbonn@unistra.fr
bSchool of Chemistry, University of Southampton, Southampton SO17 1BJ, UK
First published on 30th January 2024
Abstract
In this perspective, we summarise the major milestones to date in the field of molecular upconversion (UC) with lanthanide based coordination complexes. This begins from the leap firstly from solid-state to nanoparticular regimes, and further down the scale to the molecular domain. We explain the mechanistic intricacies of each differing way of generating upconverted photons, critiquing them and outlining our views on the benefits and limitations of each process, also offering our perspective and opinion on where these new molecular UC edifices will take us. This nascent area is already rapidly expanding and improving, having increased in luminance efficiency by more than four orders of magnitude in the last decade: we conclude that the future is bright for molecular UC.
 Loïc J. Charbonnière | Loïc Charbonnière got a diploma of engineer in chemistry (1991) and a master in supramolecular chemistry (1993) in Strasbourg, before joining the University of Geneva for a PhD thesis in coordination chemistry with Prof. Alan Williams (1996). He then entered the word of lanthanide chemistry with a first post-doctoral position in Lausanne with Prof. jean-Claude Bünzli and a second one in Strasbourg (1997) with Dr Françoise Arnaud-Neu. He was appointed as researcher at the French CNRS (1998) and then as research Director (2011). In 2009, he created the SynPA team dedicated to fundamental and applied aspects of coordination chemistry. |
 Aline M. Nonat | Aline Nonat is researcher at CNRS, IPHC/Strasbourg. She is currently in charge of the SynPA research team. She earned her degree from ENSC Montpellier and a PhD from CEA/Université Grenoble Alpes under Prof. M. Mazzanti's supervision in France. In 2007, she pursued a postdoctoral position at Trinity College Dublin, Ireland, with Prof. T. Gunnlaugsson. Returning to France in 2009, she took an ATER position at Burgundy University before joining IPHC in 2010 with Dr L. Charbonnière. Honored with the CNRS Bronze Medal in 2019, her research delves into applying coordination chemistry involving d-block metals and f-elements for energy conversion, as well as biomedical applications. |
 Richard C. Knighton | Dr Richard Knighton received his DPhil in 2015 at the University of Oxford under the supervision of Prof. Paul D. Beer working on mechanically interlocked molecules. This was followed by multiple appointments in the UK and France working on organometallic chemistry and lanthanide complexes, including with Dr Charbonnière in Strasbourg. He was appointed as Lecturer in Inorganic Chemistry at the University of Southampton in 2023. His research interests centre on photon upconverting materials utilising transition-metal and lanthanide assemblies. |
 Léna Godec | Léna Godec got a master’s degree in Chemistry and Interfaces with Living Organisms in 2020 at the University of Brest. Then, she obtained her PhD in 2023 under the supervision of Dr Loïc Charbonnière in the SynPA team. Her research mainly focuses on developing new supramolecular lanthanide assemblies for upconversion in water. |
1 Introduction
Upconversion (UC) is the process by which two or more photons are accumulated by a compound and restored as a photon of higher energy, with exclusion of the phenomenon associated to simple thermal population of the excited states.1 A full classification of the different mechanisms leading to UC and their relative efficiencies was given by Auzel in his seminal review of 2004.1This anti-Stokes process allows to convert near infrared (NIR) light into UV-Vis light and finds applications in various fields. In microscopy, the UC light drastically removes artifact associated to auto-fluorescence of the biological samples and of light scattering in the setups, affording an excellent signal to noise ratio,2 which can also be of interest to biological sensing and analysis.3 Bringing NIR photons in the visible domain is also a promising way to improve the efficiency of solar cells, which are up to now poorly efficient in the NIR domain, but have their best performances in the visible.4 Alternatively, UC has proved to be of interest for numerous other applications such as anticounterfeiting inks,5 photodynamic therapy,6 diagnosis,7… However, whatever the application and for reasons that will be described in details in this perspective article, UC is still restricted to the use of bulk solids for materials and nanoparticles for biological applications. Considering issues such as reproducibility, stability and toxicity of nanoparticles,8–10 there is a large interest in designing chemically controlled molecular upconverting devices, that could be covalently bonded to (bio)molecules of interest,11 with improved chemical stability.
Historically, the observation of the phenomenon was rendered possible in the 1960's by the development of high energy luminescence sources,12 but essentially on solids doped with d- or f-elements and lately on UC nanoparticles (UCNPs),13 a field which has exploded since then, with thousands of publications on the synthesis of UCNPs14 and their applications.15 It is only in 1996 that UC could be observed for the first time at the molecular level for ErCl3 salts dissolved in POCl3/SnCl4 liquid medium, with a green emission band observed at 550 nm upon laser excitation at 800 nm.16
At this stage of the discussion, it is important to note that UC was also reported as soon as 1962 for solutions containing purely organic luminescent compounds in cold (72 K) ethanol.17 The process was termed photosensitized triplet–triplet annihilation (TTA) and has been the subject of numerous research articles which can be found in different authoritative reviews.18,19 Although TTA is a topic of great interest for UC, with potential applications such as in solar energy conversion,20 the phenomenon is not purely molecular as it requests energy transfers between freely diffusing donors and acceptors in concentrated solution, with exceptions of very recent examples of supramolecular adducts.21 Hence, TTA will not be covered in this perspective article which is focused on lanthanide (Ln) based molecular UC devices.
The basic pre-requisites for the observation of UC are: firstly, the necessity to successively climb different energy levels, necessitating compounds with ladder like energy levels; and secondly long lived intermediate excited states enabling the ascent to the higher energy levels. This occurs by absorption of the next quantum of energy before it decays back to the ground state, either by simple luminescence or by non-radiative pathways. It is therefore unsurprising to find Ln elements as top players in the field of UC, whatever the physical state of the sample (solid, nanoparticulate, or molecules) as they possess both these characteristics. As evidenced in Fig. 1,22 the electronic configuration of Ln(III) cations ([Xe]4fn with n = 0 to 14) is at the origin of a large number of electronic transitions, spanning the UV to near-infrared (NIR) domain, a perfect example of ladder like energy level diagram. Additionally, the Laporte forbidden character of electric dipole f–f electronic transitions provides them with very long radiative lifetimes, sometimes reaching the millisecond (ms) regime, that are two to six orders of magnitude larger than those of d-element coordination complexes or organic molecules respectively.23 Unfortunately, a corollary of the forbidden character of the f–f electronic transition is that the corresponding absorption coefficients are very weak (a few units of M−1 cm−1), requiring high intensity excitation sources to populate the excited states.
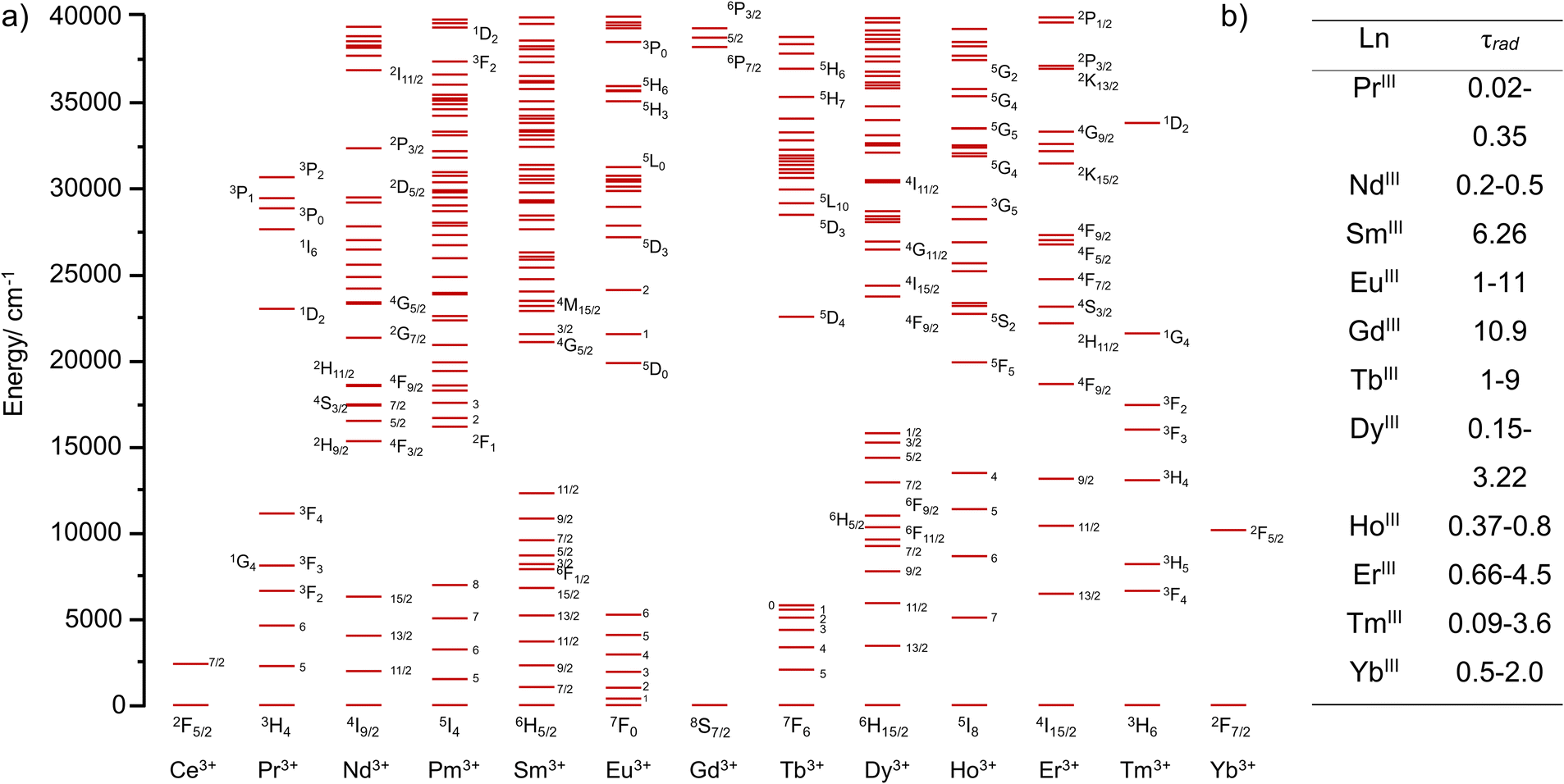 | ||
| Fig. 1 (a) Calculated energy levels of Ln(III)22 and (b) ranges of radiative lifetimes τrad for different Ln(III) ions (in ms).23 | ||
The radiative lifetime (Fig. 1b) is defined as the lifetime of a luminescent species in total absence of non-radiative contributions, that is for a 100% luminescence quantum yield. Unfortunately, luminescent compounds, including Ln compounds, are rarely perfect luminescent probes and an important part of the energy is lost by non-radiative pathways, among which is energy dissipation through the vibrations of the environment. In solids, these vibrations are called phonons and are defined as quasi-particles having the energy of a vibration of the lattice.24 In solution, the corresponding vibrations are those of the environment, the ligand for complexes and the solvent molecules for solvated ions. Part of the energy of the excited states can then be transferred to these vibrations or, more precisely, to their overtones.25 Despite some known deviations from the theory,26 it is generally assumed that the rate of the non-radiative processes, knr, is determined by the energy gap law,27 which states that for a transition at an energy ΔE, knr is inversely proportional to the exponential of ΔE/hν, hν being the energy of the vibration. In other words, the higher the energy of the vibration, the more efficient the non-radiative processes. Therefore, O–H, N–H and C–H vibrations, which can be found in the 3000–4000 cm−1 region of the infrared domain are far more efficient at quenching the intermediate excited states of Ln ions than phonons in solids, which rarely exceed 1200 cm−1,28 explaining why it is so difficult to observe UC at the molecular scale compared to solids or nanoparticles.
Even if the first evidence of molecular UC dates back to the end of the last century,16 it was only in 2011 that UC could be observed on a chemically elaborated discrete molecule, a hetero-trinuclear triple stranded helicate of Cr(III) and Er(III) designed by Piguet and co-workers.29 At that time, these premises were obtained in organic solvents at very low temperatures in solid solutions, but they opened the door to this burgeoning field. Even if still rare, examples of molecular UC are now generally observed at room temperature and has even been observed once in non-deuterated water.30
The purpose of this perspective article is not to review in depth the field of molecular UC with Ln complexes, this has been thoroughly done in different recent review articles,31–34 but rather to expound the different mechanisms affording UC at the molecular scale, their interest or drawbacks and possible directions to improve molecular UC probes.
2 Excited state absorption (ESA) based molecular UC
Excited-state absorption (ESA) is the simplest and least demanding mechanism for molecular UC in terms of molecular design. Many lanthanide-based molecular species, ranging from mononuclear complexes to extended supramolecular edifices, have been shown to exhibit UC properties via this mechanism.35 In the ESA mechanism, the lanthanide complex is excited to a higher energy state via the absorption of a first low-energy photon, bringing the ground state to an intermediate excited state, followed by the absorption of a second photon by the excited state to reach the doubly excited state, which finally emits light (Fig. 2). In this mechanism, the primary requirement arises from the lanthanide itself, which should possess multiple energy levels with a ladder-like energy structure. The most straightforward example is the Er(III) ion. In its ground state, the electronic structure of erbium(III) is defined by its 4f electron shell, which is incompletely filled with 11 electrons ([Xe]4f11). Like other Ln(III) ions, the 4f electrons possess a series of energy levels, forming a ladder-like pattern due to the complex interactions between the electrons.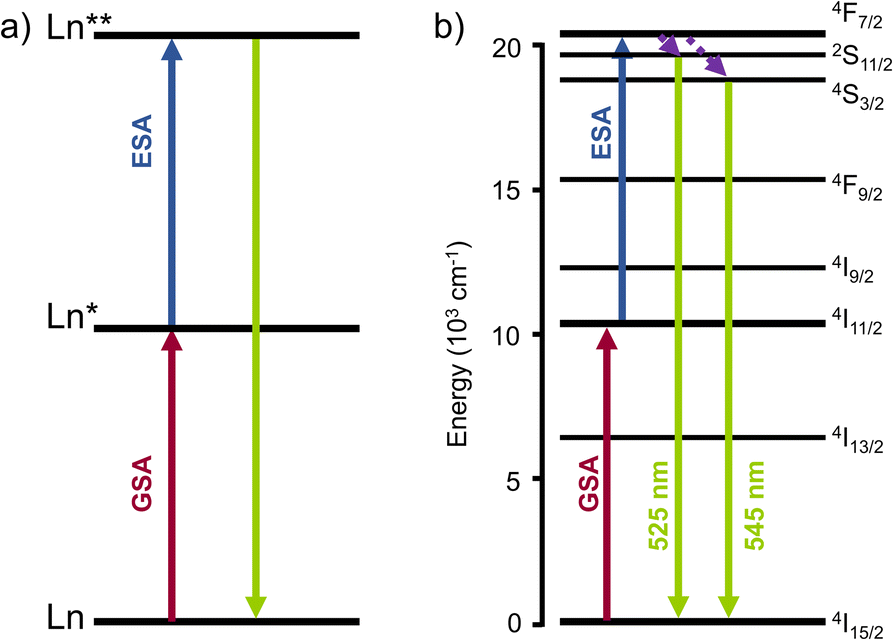 | ||
| Fig. 2 (a) Schematic representation of the ESA mechanism for Ln luminescence UC. (b) ESA mechanism for Er(III). | ||
These energy levels are conventionally described by the spectroscopic terms in the form (2S+1LJ) where S, L, and J stands for the usual quantum numbers: S, the spin quantum number defines the number of unpaired electrons; L is the total orbital momentum resulting from the electrostatic interaction within the orbital and J = L + S is the total angular momentum as a result of the spin–orbit coupling.36
As shown in Fig. 1, Er(III) is one of the lanthanide ions displaying transitions in both the near-infrared and visible regions. This characteristic is crucial in achieving ESA UC from NIR to visible light. In addition, Er(III) has a particularly strong absorption cross-section at 980 nm, which is a commonly used excitation wavelength for UC studies and application, establishing erbium as a prototypical species for ESA and for UC in general.35
There are two important electronic transitions for Er(III) in view of ESA UC. The first absorption band at ca. 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1 (around 1000 nm), corresponds to the transition from the 4I15/2 ground state to the 4I11/2 excited state (in red, Fig. 2b). Further excitation at the same energy allows to reach the higher 4F7/2 excited state (in blue, Fig. 2b). The second important transition is at ca. 12500 cm−1 and is responsible for the ascendance from the ground state to the 4I9/2 excited state. From there, decays to the 4I11/2 or 4I13/2 excited states, followed by absorption of the second photon allows the climbing to higher excited states emitting in the green region at ca. 550 nm (in green, Fig. 2b).
000 cm−1 (around 1000 nm), corresponds to the transition from the 4I15/2 ground state to the 4I11/2 excited state (in red, Fig. 2b). Further excitation at the same energy allows to reach the higher 4F7/2 excited state (in blue, Fig. 2b). The second important transition is at ca. 12500 cm−1 and is responsible for the ascendance from the ground state to the 4I9/2 excited state. From there, decays to the 4I11/2 or 4I13/2 excited states, followed by absorption of the second photon allows the climbing to higher excited states emitting in the green region at ca. 550 nm (in green, Fig. 2b).
In that context, basic ErCl3 salts dissolved in low phonon liquids composed of a mixture of POCl3 and SnCl4 were shown to display ESA UC upon excitation at 800 nm with a modest 500 mW excitation power.16 The clever use of low phonon liquids is unfortunately not compatible with Er(III) based coordination complexes composed of organic ligands which might react chemically with reactive species such as POCl3. For coordination compounds, it is then mandatory to use benign solvents, generally displaying high energy vibrations such as O–H, N–H or C–H oscillators, with the concomitant deleterious effect on the lifetime and quantum efficiencies of the excited states (vide supra). A way to bypass this inconvenience is then to use high intensity excitation sources. But even in this case, the studies of Reinhard and Güdel on Er(III) tris-dipicolinate DPA (DPA = 2,6-pyridine-dicarboxylate, Fig. 3) complexes in water failed to show any UC.37 Similar experiments were also conducted with isostructural Tm(III) and Yb(III) complexes, yielding the same outcome.
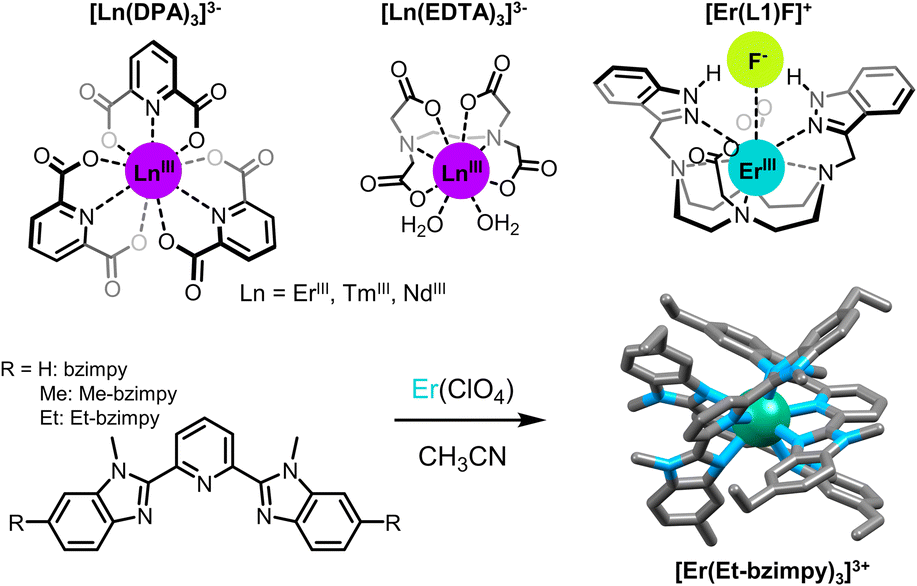 | ||
| Fig. 3 Examples of Er(III) complexes showing ESA (ion colors: Er = teal).37,38,44,46 | ||
Nevertheless, UC was achieved with the same molecules ([Er(DPA)3] and [Er(EDTA)]−, Fig. 3) three years later by Xiao and co-workers in D2O.38 By combining two highly focused high intensity (ca. 100 kW, P > 109 W cm−2) lasers, corresponding to the two electronic transitions from the ground state to the first excited state and from the latter to the second excited state, and by reducing the losses by non-radiative deactivation using a deuterated solvent, visible UC emission from Er(III) was finally observed. With a similar two-laser setup with high pump power, the energy ladder of Nd(III) and Tm(III) could also be climbed and ESA UC emission was observed in the UV (386 nm) and visible (480 nm), respectively. However, very weak UC signals were observed for the solutions in H2O.
The intensity of the UC signal arising from an ESA mechanism depends on various factors such as the lanthanide ion of interest and its absorption cross-section (or extinction coefficient in solution studies), the experimental conditions (phonon energy of the matrix, solvent, temperature), the concentration of the lanthanide ions in the sample, the excitation wavelength and the laser power. Generally, the efficiency of UC expressed by its UC quantum yield (ΦUC, eqn (1)), typically falls within the range of 10−10 to 10−7 in solution.
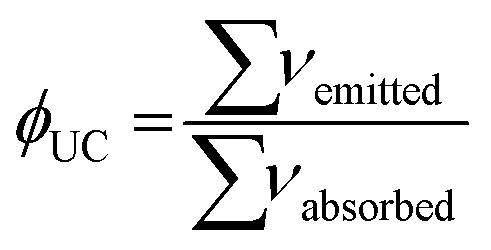 | (1) |
For more details, these parameters have been recently reviewed by Yao and co-workers.32 As demonstrated in the study by Xiao and co-workers discussed above,38 vibrational quenching is the principal enemy of ESA-based UC systems but it is also a parameter that can be limited by a careful design of the ligands. Several strategies have been investigated over the last ten years in the quest of limiting high-energy oscillators from the ligand and solvent molecules.
The simplest one is basically to use deuterated solvents as the impact of deuteration was recognized to improve the luminescence properties of Ln ions.39 Introducing X–D in place of X–H bonds (where X = O, N or C atoms) increases the reduced mass of the bond within the frame of an harmonic oscillation, thereby decreasing the energy of the corresponding oscillation, while requiring more of these oscillators to quench the luminescence.31 For coordination compounds, the impact of deuteration of the solvent is essentially on the second coordination sphere. Thus, a logical step forward is the deuteration of the ligands themselves, which now requires far more synthetic work.26,40,41 Some authors even went a step further by fluorination of the ligands, which appears particularly efficient for Yb complexes.42 In order to repel solvent molecules as far as possible from the Ln atoms, another strategy consists in using bulky aromatic substituents, possibly fluorinated ones.43
In our very first study on UC, we measured ESA on a mononuclear [ErL(H2O)] complex in D2O (Fig. 4a) and demonstrated that the signal was substantially increased while replacing the inner sphere water molecule by a fluoride ion.44 Furthermore, the signal was further enhanced when the structure was rigidified through the formation of a supramolecular dimer [F⊂(ErL)2]+, stabilized by π–π interactions between the aromatic indazolyl moieties and augmented by a network of hydrogen bonds.45 The UC efficiency, as reflected by the emission intensity at 541 nm upon excitation at 980 nm (Fig. 4b), was very well correlated with the concentration of the dimeric species in solution. For a [NaF]/[ErL(H2O)] ratio of 0.5 (corresponding to approximately 90% dimer formation), UC emission increased by a factor of 7.7. Under these conditions, the UC process was confirmed by a slope of two in the logI/logP plot (Fig. 4c).
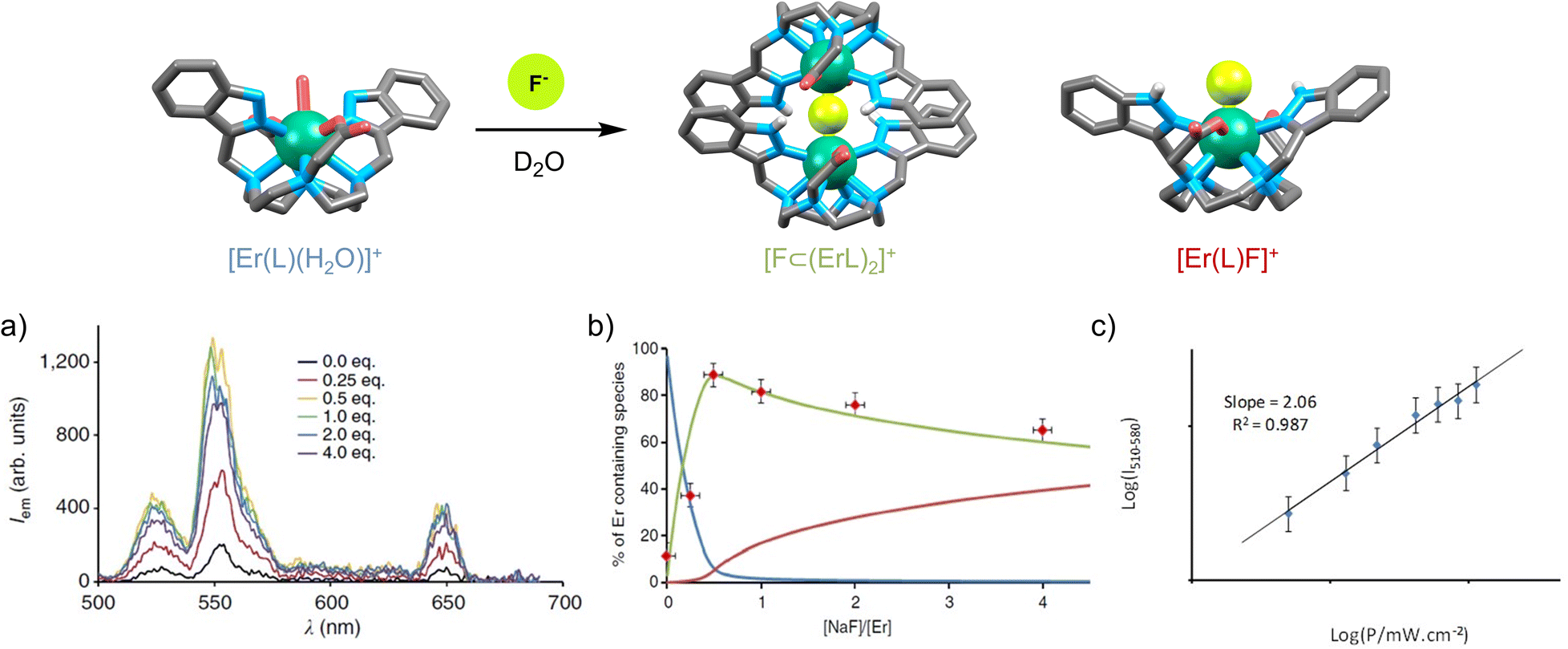 | ||
| Fig. 4 (a) Increase of ESA efficiency of [ErL(H2O)] upon addition of NaF (D2O, c = 200 μM, r.t., λex = 980 nm, P = 5 W). (b) UC intensity at 541 nm as a function of the [NaF]/[ErL(H2O)] ratio in correlation with the concentration of [ErL(H2O)] (in blue), [F⊂(ErL)2]+ (in green) and [ErLF] (in red). (c) Emitted intensity at 541 nm as a function of LASER power for a solution of [ErL(H2O)] containing 0.5 eq. of NaF in D2O (c = 200 μM, r.t.).45 | ||
Subsequently, Piguet and his group designed a series of nonadentate triple–helicate complexes with polyaromatic tridentate ligands derived from terpyridine and bis(benzimidazole)pyridine (Fig. 3).46,47 The absence of metal-bound solvent molecules is favorable for long-lived excited states (τEr,obs = 2–6 μs) and efficient UC properties, with a noticeable advantage for bis(benzimidazole)pyridine derivatives. Replacing terpyridine by benzimidazole substituents nonetheless enhances the UC intensity by extending the Er(III) excite state lifetime (from τEr,obs = 1.88 μs for [Er(Et-tpy)3]3+ to τEr,obs = 5.56 for [Er(Et-bzimpy)3]3+), but it also significantly raises the Er(III) molar absorption at 980 nm (increasing from 0.7 M−1 cm−1 for [Er(Et-tpy)3]3+ to 26 M−1 cm−1 for [Er(Et-bzimpy)3]3+). Within this series, quantum yield values up to 1.7 × 10−9 at P = 25 W cm−2 have been measured in CH3CN at room temperature upon NIR excitation of the 4I9/2 ← 4I15/2 absorption band of Er(III) at 801 nm. Although low, these values reflect a clear trend in adjusting the UC capabilities through structural modifications of the ligands.47
As observed in the pioneering work of by Xiao and co-workers on Tm(III) and Nd(III) complexes with DPA and EDTA (Fig. 2), these two metal ions are also capable of ESA UC.38 So far, molecular ESA UC emission with these two ions has been limited to simple complexes or, simple solvates (such as [Tm(CF3SO3)3] in DMSO-d6), as reported by Sorensen, Beeby, and Faulkner, necessitating the use of high-power NIR lasers.48 It can be anticipated that UC with Nd(III) and Tm(III) is less efficient compared to Er(III) due to the shorter excited state lifetimes exhibited by Nd(III) and Tm(III), which are unfavorable for UC. As examples, τR (4F3/2,Nd(NO3)3) = 0.42 ms;26τR (4I13/2 Er(NO3)3) = 0.66 ms;36τR (4F3/2, Y2O3:Nd3+) = 0.27 ms;34τR (4I13/2, Y2O3:Er3+) = 7.75 ms;49τR (1G4, Y2O3:Tm3+) = 0.29 ms.49 In addition, Nd(III) exhibits absorption cross-sections that are usually lower than that of Er(III) and Tm(III).36
UC is a valuable process, particularly in bioimaging, where the conversion of low-energy near-infrared light to visible light is advantageous for deep-tissue imaging.50 Despite its potential, it is important to highlight that the efficiency of UC via ESA may currently be insufficient, leading to a preference for other mechanisms. As a consequence, several research groups have turned towards mixed heterometallic coordination complexes or assemblies with the idea of taking advantage of the long-lived excited state and/or increased molar absorption coefficients of donors such as Cr(III)51 or Yb(III)52 and to favor UC emission processes other than ESA, such as energy transfer up-conversion (ETU) which will be detailed in the following section. Also, when ETU takes place, it is important to note that ESA UC is still present. This was indeed the case for the 3:2 sandwich-type complexes with naphthylsalophen ligands developed by A. de Bettencourt-Dias et al.52 and for the [CrErCr(bpb-bzimpy)3]9+ triple-stranded helicates synthesized by Piguet et al.51 In this very particular example were the experimental UC quantum yields was determined to be φUC,tot = 1.7(2) × 10−9 (λexc = 801 nm and P = 25 W cm−2), state-of-the art photophysical and theoretical studies have shown that ESA remains significantly more efficient than ETU, with a theoretical contribution of ESA exceeding 98.8%, while the theoretical contribution of ETU is limited to a maximum of 1.2%.
In addition, in the context of exploratory research, there remain unexplored possibilities for enhancing ESA efficiency. These possibilities involve exploring the use of pulse-shaping53 or laser pumping54 techniques to enhance ESA by optimizing the absorption of excited states, as well as further exploiting the temperature-dependent ligand field effect to encourage hyperfine transitions, with the goal of increasing their absorption coefficient.55–57
3 Energy transfer upconversion (ETU) based molecular UC
The energy transfer UC (ETU) mechanism (Fig. 5) is distinguishable by its core principle: unlike ESA, it requires two ions in a neighboring environment.1 In this process, the first ion, termed the sensitizer (S), is excited from the ground state to a higher excited level by absorption of a photon. Energy is then transferred to the second ion, termed the activator (A), which is itself in an excited state, enabling it to reach a doubly excited state, from which light is emitted.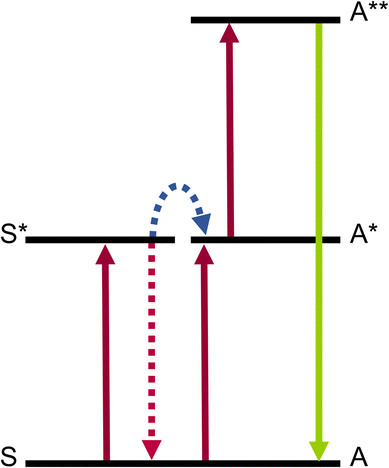 | ||
| Fig. 5 Illustration of the ETU mechanism. | ||
This breakthrough finds its roots in the pioneering work of Auzel reported in 1966, who introduced the concept of ETU to address the limitations associated with the direct excitation of parity-forbidden intra-f transitions within trivalent lanthanide cations.58,59 This concept revolutionized the landscape by proposing the use of sensitizers possessing heightened absorption cross sections, initiating sequential energy transfer processes that lead to the multistep excitation of the activator. Thus, the ETU mechanism offers an efficient pathway to circumvent the challenges posed by activators with short-lived intermediate excited states, by using sensitizers with long-lived excited states.
Harnessing this mechanism, lanthanide-doped solids or nanoparticulate materials have emerged as promising platforms. In particular, the unique long-lived excited state of ytterbium ions, exploited as a relay in ETU, has enabled the effective UC of ions including erbium, thulium, and praseodymium.1
Building upon this fundamental understanding of the ETU mechanism, we now investigate its intricate dynamics at the molecular scale. Through an analysis of established molecular examples, a comprehensive perspective on how ETU operates at the molecular level is attained, providing indispensable insights to optimize this phenomenon for diverse applications.
At the heart of molecular-scale ETU studies lies the trinuclear complex [CrErCr(L1)3]9+ (Fig. 6a), developed by Piguet and co-workers, where chromium (Cr) acts as the sensitizer (S) and erbium (Er) as the activator (A), being the first isolated molecular system to exhibit near-infrared to visible UC.29 Several factors, especially the high concentration of sensitizers, contribute to detectable molecular UC in frozen acetonitrile solution at 77 K. The quadratic dependence of upconverted emission on incident intensity and the crucial role of Cr → Er energy transfer processes supported the presence of the ETU pathway in these supramolecular complexes. The system's main limitation lies in its relatively low efficiency in energy transfer, potentially attributed to a significant Cr–Er intermetallic distance which averages 8.8 Å.31 The significance of the number of sensitizers becomes apparent when comparing with a comparable dinuclear complex with a singular sensitizer (Cr3+) per erbium activator, forming a SA pair.29,33,60 The resulting upconverted emission from NIR to green displayed a noticeable reduction in intensity by more than one order of magnitude compared to CrErCr.
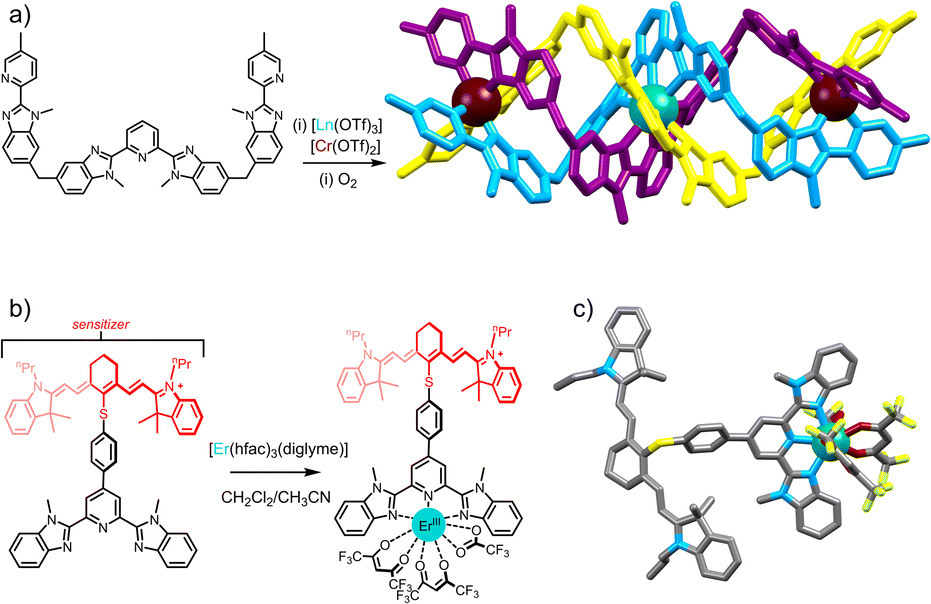 | ||
| Fig. 6 (a) Heterotritopic ligand L1 used in the preparation of the heterotrinuclear [Cr2Er(L1)3]9+ complex displaying NIR to visible ETU UC.29 (b) NIR dye containing tridentate ligand L2 displaying ligand to Er ETU UC.72 with (c) single-crystal X-ray structure (ion colors: Cr = brown, Er = teal). | ||
These examples were followed by the first evidence of molecular photon UC achieved by ETU through an organic antenna-sensitized erbium complex in organic solution at room temperature.61 Efficiency gains were achieved through the utilization of the IR-806 dye with a large infrared absorption band, contributing to the notable improvement in the UC process compared to f–f transitions. Continuing from the progress achieved through organic dye antennas, another notable advancement emerged,62 involving a NIR dye antenna linked to a tridentate binding unit, effectively capturing NIR photons and transferring them efficiently thanks to the spatial proximity. The resulting complex, [L2Er(hfac)3] (hfac = hexafluoroacetyl acetonate) (Fig. 6b), exhibited a remarkable UC brightness in nondeuterated acetonitrile at room temperature.62
Employing a co-doping strategy with Yb3+ ions has emerged as a powerful tool to improve the NIR absorption lanthanide ions used for UC. In particular, the focus shifts to the well-explored Er3+–Yb3+ pair, a prominent and extensively investigated duo in the field of UC in solids.1
Thus, Zeng et al. developed heteronuclear lanthanide complexes strategically composed of Yb/Er pairs, harnessing the ability of Yb3+ to trigger notable Er3+ UC emission through an ETU mechanism in CH2Cl2 solution.63 Remarkably, this strategic assembly of complexes, carefully balanced the sensitizer-to-activator (S/A) ratio and incorporating fluoride for reduced vibrational quenching, yielded an UC quantum efficiency that surpassed previous examples by a factor of 20.
The abundance of Yb3+ significantly enhanced the probability of Yb → Er energy transfer, emphasizing the critical role of the Yb/Er ratio in augmenting UC efficiency.64 This phenomenon has been particularly noticeable in the nanosized [Er2Yb13] complex. In light of their comparable chemical characteristics, the substitution of one lanthanide with another in these molecular entities is a viable option. This reveals the first example of Ho3+ complex exhibiting UC at the molecular scale in deuterated methanol solution with a [Gd8Ho2Yb10] cluster.65
The quest for efficient UC luminescence in molecular systems by ETU mechanism steers towards careful ligand selection, aiming to combine multiple lanthanides into cohesive polynuclear assemblies. In this context, molecular lanthanide clusters,66,67 also called molecular cluster aggregates (MCA)68,69 offer distinct advantages. Their spherical topology confers structural rigidity, ensuring a stable framework. The high nuclearity and the possibility to interchange lanthanides within these molecular structures provide a versatile platform for tailoring properties and transitions. Moreover, these structures exhibit stability in various solvents, and their structural rigidity helps dampen molecular vibrations, leading to notably high UC quantum yields. These combined features position clusters as promising candidates for efficient and customizable UC applications.
The main difficulty to develop efficient ETU based complexes of Ln rely on the choice of the activator. Apart from the rare case of Ho, most of these systems are based on Er, where Tm or Pr should also be envisaged. But all of these potential activators possess NIR electronic transitions (Fig. 1), which are subject to efficient non-radiative quenching from the vibrations of the surrounding environment. The current trends to improve molecular ETU are therefore focused on the enhancements of the population of the first excited state of the activator, either by NIR dyes, or through the rigidification of the ligands and/or the decrease of the symmetry around the activator, increasing the transition probabilities.70 In that respect, one could imagine that the combination of the NIR absorbing properties of bulky and hydrophobic derivatives of porphyrins may be an interesting scaffold to adopt, as has been demonstrated for Yb complexes.71 Considering that the conventional downshifted luminescence is increased by ca. one order of magnitude in these systems, an impact of two orders of magnitude would be expected for the UC quantum yields, with an even larger impact on the overall brightnesses of such probes possibly reaching thousands of M−1 cm−1.72
Concerning the cluster approach, whilst it appears well suited for new ETU systems, but the chemical similarity of Ln cations in the series preclude complete synthetic control of their compositions and a statistical approach has to be considered. However, combining these UC properties with properties brought by ligands in the supramolecular structures, one can envisage hierarchical arrangements leading to usage as liquid crystalline UC materials.
4 Cooperative photosensitization (CP) and cooperative luminescence (CL)
4.1 Cooperative photosensitisation
Cooperative photosensitization (CP) (also called cooperative sensitization; Fig. 7a) is a special case of cooperative energy transfer and differs from the mechanisms discussed above. Unlike ESA and ETU mechanisms – which rely on utilizing lanthanides with ladder-like energy levels – CP uses energy donors and acceptors which do not utilize intermediate excited states. Commonly employed strategies are using donor–acceptor pairs where the energy-gap between the ground and excited-state of the acceptor are half that of the acceptor.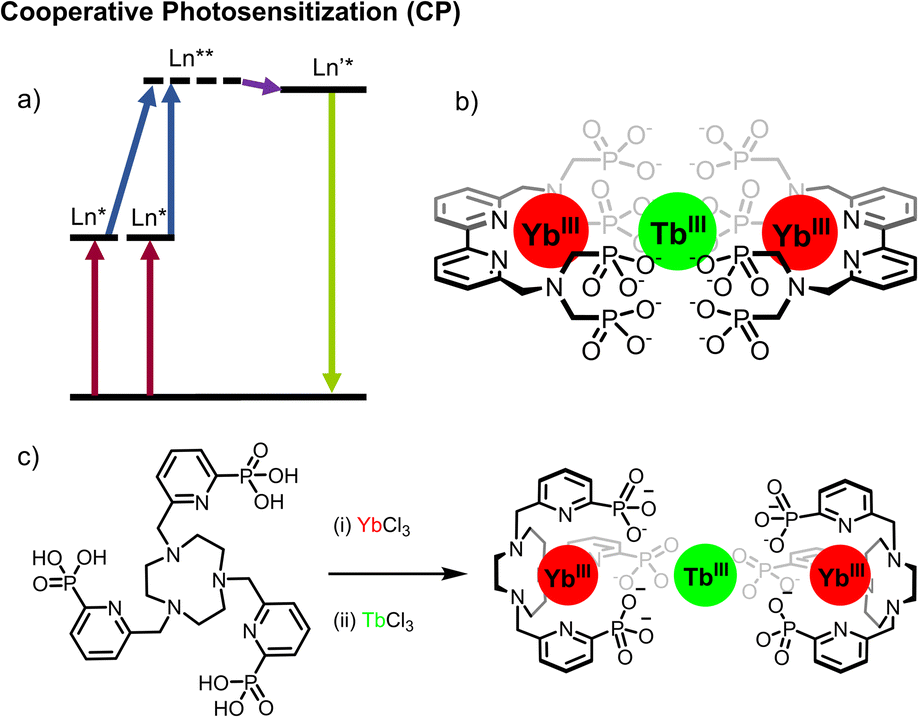 | ||
| Fig. 7 (a) Cooperative photosensitization (CP) mechanism. (b) bipyridine-based Yb2Tb assembly.41 (c) Stepwise formation of TACN-based Yb2Tb assembly.30 | ||
Thus, by using two donors the combined energy gap is in resonance with that of a single acceptor. The prototypical example is that of the (ytterbium)2-terbium triad, for which the 2F5/2 → 2F7/2 transition of Yb at 980 nm is at half the energy of the 5D4 → 7F6 transition of Tb, and which was first reported in 2017 (Fig. 7b).41 In this example a bipyridine ligand substituted with a bis-phosphonated amine was able to coordinatively saturate a Yb complex (τYb = 35 μs in D2O). The dodecadeuterated congener was prepared, minimizing quenching by ligand CH oscillators, with a corresponding increase in the excited state lifetime in D2O (τYb = 65 μs for YbLD). This increase in lifetime permitted the first observation of solution-state CP UC, although this was only operative in D2O, the significantly decreased lifetime of Yb in H2O precluded CP UC in non-deuterated water. Irradiation of the NIR absorption band of Yb (2F5/2 ← 2F7/2, λex = 980 nm) gave rise to the characteristic emission of Tb (5D4 → 7FJ with J = 6 to 3, λem = 475–630 nm).
Subsequent work in the group built on this, using a trifunctionalized pyridylphosphonate triazacyclonane (TACN) scaffold.30 In this case, UC was visible in D2O, but also importantly in H2O. This case highlights the combined importance of both quantum yield and lifetime – and will be an important factor in the design of future upconverting molecular edifices. The TACN complex displays a comparable excited-state lifetime in H2O (2.8 μs vs. 3.3 μs for YbLD), but exhibits quantum yield of 0.75% (cf. 0.2% for YbLD). These results are encouraging for future iterations73 but clearly one prospective challenge is enabling high stability of all metal ions – not just the sensitizers. We envisage supramolecular multitopic ligands to satisfy this criterion.
While UC systems operative in aqueous solvents are highly desirable, they also represent the biggest challenge due to the appreciable quenching effect of outer-sphere OH and, to a lesser extent, OD oscillators. This is currently impinged by the fact that the molecular ligand toolbox for strong Ln chelators in water rarely includes bridging modalities for multimetallic assemblies. Indeed, we have already stated that short internuclear Ln–Ln distances are crucial for the energy-transfer step (vide supra). In the area of CP UC, molecular clusters have again been promising UC candidates. While these systems are not amenable to aqueous environments, they are a miniaturized congeners of the well-known Ln-nanoparticle based emitters, displaying short Ln–Ln distances of ca. 3.5 Å. The first example reported was that of an Ln9 (Ln = Tb/Yb) cluster66 the central Ln9 core being isostructural with hexagonal doped NaYF4 UC nanoparticles. In this case the ligands were present as bridging deuteroxide (μ3-OD or μ4-OD), rather than fluoride in nanoparticles, which represents the primary mechanism of excited-state deactivation. Studies revealed that, contrary to the mechanistic interpretation, sensitizer to activator ratios approaching parity gave the most efficient UC platforms (ΦUC = 2.8 × 10−6 for Tb4Yb5 in CD3OD).67 The commercial availability of these ligands and ease of synthesis has allowed rapid diversification of the acceptor. Murugesu and co-workers have worked extensively on an Ln20 cluster compound and have reported a mixed Yb/Tb/Eu system (Fig. 8a) by doping a Gd host cluster matrix.68 Excitation of the Yb ions at λex = 980 nm gives rise to both Tb (5D4 → 7FJ with J = 6 to 3) and Eu (5D0 → 7FJ with J = 4 to 0) emission which is modulated by the temperature. The authors ascribe the emission of Eu* to arise from energy transfer from the UC generated Tb* excited state, although a phonon-assisted process directly from the diexcited Yb (Yb** → Eu*) cannot be ruled out.
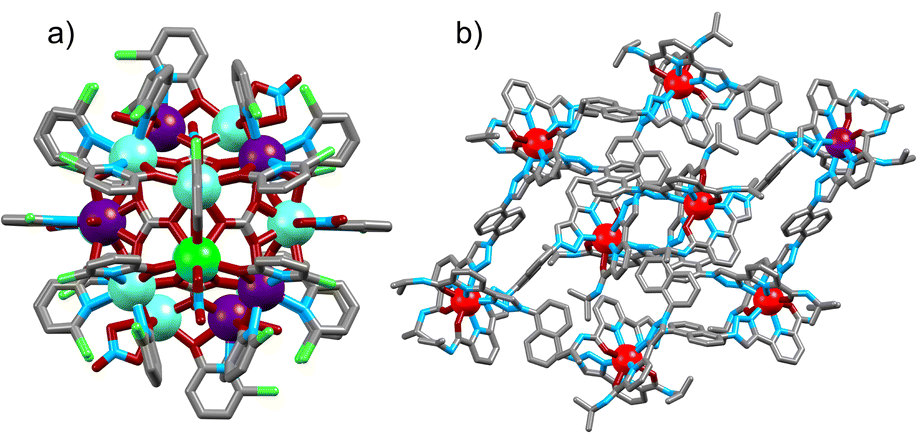 | ||
| Fig. 8 Single-crystal X-ray structure of (a) [Gd7Tb2Eu11] cluster,68 (b) Yb7Eu1 metal–organic cage (ion colors: Eu = purple, Tb = green, Gd = teal, Yb = red).74 | ||
Indeed, an example of Yb/Eu UC has recently been reported using a Ln8 metal–organic cage containing a mixture of Eu and Yb ions.74 By employing a statistical mixture of Yb and Eu, in analogy to the strategy used for cluster-based materials, cooperative photosensitization of Eu emission was observed in the solid-state upon irradiation of the Yb absorption band at 980 nm, with a QY of ca. 2 × 10−9. The Ln–Ln distances were approximately 14.3 Å, demonstrating the ability for UC over significantly longer distances than previously thought (Fig. 8b).
Thus far, all systems have employed lanthanide sensitizers and activators. One advantage of the molecular approach – diverging from nanoparticle-based systems – is the potential for changing the identity of the accepting chromophore. Indeed, the case of cooperative phostosensitization is enigmatic in that the activator does not require intermediate energy levels to ascend the energy ladder (viz. Tb). This was exploited elegantly by Heinze, Resch-Genger, and Seitz through co-crystallisation of a mixture of a tricationic chromium complex, mer-[CrIII(dppd)3]3+ (dppd = N,N′-di-methyl-N,N′-dipyridine-2-ylpyridine-2,6-diamine) and a trianionic ytterbium complex, [Yb(DPA)3]3− (Fig. 9a).75
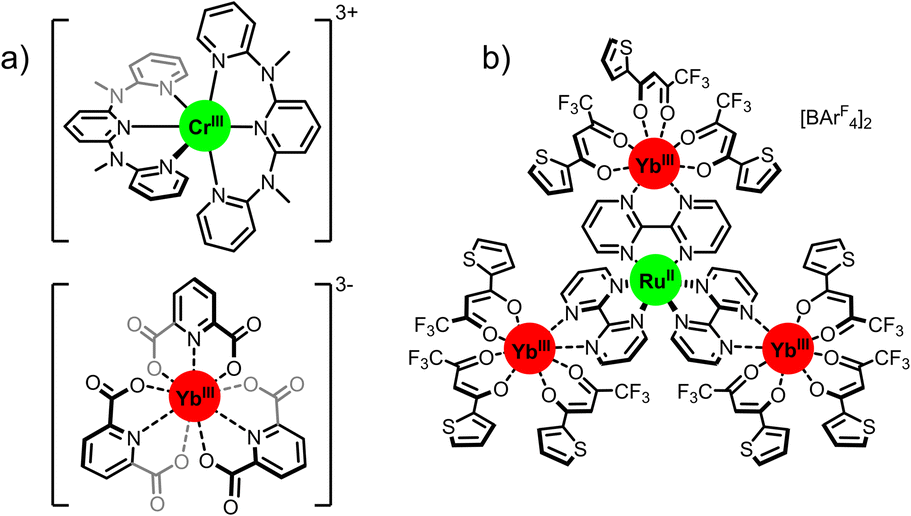 | ||
| Fig. 9 CP with d–f systems: (a) solid-state crystals of CrIII/YbIII.73 (b) supramolecularly assembled metastable RuYb3.76 | ||
Upon excitation into the Yb manifold at 980 nm, concomitant energy transfer occurred, populating first the 4T2 state, followed by intersystem crossing (ISC) to the 2E/2T1 excited state of the Cr ion, resulting in emission at 775 nm arising from the 2E/2T1 → 4A2 transition. As with other systems use of deuterium was key to observing the phenomenon, the included CH3OH/CD3OD solvates providing a significant quenching pathway. This work highlights the potential advantages of transition-metal acceptors; the additional excited state ISC relaxation mechanism permits both NIR-excitation and NIR-emission.
In a similar approach, we reported a related example which exhibits solution-state UC in a tetrametallic RuYb3 assembly (Fig. 9b).76 Through combination of [Ru(bpm)3][BarF4] (bpm = 2,2-bipyrimidine, BArF4 = tetrakis[3,5-bis(trifluoromethyl)phenyl]borate), and [Yb(ttfa)3(OH)2] (ttfa = 2-thenoyltrifluoroacetonate) in anhydrous acetonitrile we were able to form an assembled RuYb3 array. Irradiation of the peripheral Yb ions (λex = 980 nm) produced CP which sensitized the 1MLCT state of [Ru(bpm)]2+, and ISC led to the emission from the 3MLCT state at 636 nm. Importantly the rate of kUC was found to be 25 times higher than for Yb–Tb CP systems, demonstrating the advantage of using a spin-allowed absorption of d-metals vs. spin-forbidden Ln transitions. The quantum yield was determined to be 2.1 × 10−8 which compares favorably with Ln only systems, particularly given that these results were obtained in non-deuterated media. A unique case of CP UC was reported by Pérez-Preto, González-Béjar and co-workers using a fluorescein analogue. The authors mixed Yb(OTf)3 and Eosin Y to produce a material with the general constitution [Eosin Y]x[Yb]x (x = integer).77 This compound exhibits UC via sensitization of the Eosin YS1 state by the Yb2** state after NIR excitation (λex = 980 nm). Emission from the S1 → S0 transition is observed at 600 nm, with an impressive calculated quantum yield of 7.2 × 10−5. Both this example and the case using ruthenium demonstrates that the use of spin-allowed absorption can have a strong effect on the overall quantum yield by enhancing the efficiency of the energy-transfer step, and this area should be a strong focus of UC research in the future.
4.2 Cooperative luminescence
Cooperative luminescence (CL) is related to CP in that both use two Yb ions as energy relays for UC. Where the two differ is that in CP two Yb sensitizers transfer their energy to a single acceptor ion, while in CL there is no central visible-emitting acceptor. Rather – conversely to all other mechanisms considered here – CL can be considered a second order process whereby a Yb*Yb* state recombines to emit from a doubly Yb** excited state (Fig. 10a). The phenomenon of CL had, until recently, only been reported in solid-state materials.78,79 The absence of reports of CL in nanoparticles owe to the fact that such materials display a high propensity for energy migration and as such either exhibit conventional Yb-based NIR downshifting photoluminescence or non-radiative decay of the Yb excited state before CL can occur. In this regard, molecular level systems have proved invaluable due to their lower nuclearity and hence reduced susceptibility for quenching via successive energy migration steps.80 Despite this, such molecular CL systems are still relatively rare. The first such molecular example was reported in a powdered sample of a monometallic [Yb(N,N)(ttfa)3] complex in 2019.50 Cooperative luminescence occurred through an intermolecular process between neighbor Yb complex in the solid-state, and exhibited emission at λem = 490 nm upon excitation at 950 nm. In terms of discrete single-molecule UC in solution we first noticed an anomalous signal present in CP UC in molecular hetero-nonanuclear Yb/Tb clusters.67 This signal was not present in the downshifted photoluminescent spectra indicating the presence of an alternative UC process. Irradiation (λex = 980 nm) of the homonuclear [Yb9(acac)16(OD)10][OD] complex (Fig. 10b) in a CD3OD solution gave rise to a highly symmetrical structureless emission band at 503 nm. Importantly, the log/log plot of integrated emission intensity vs. incident laser power gave a slope of 1.8, confirming the two-photon process. The lifetime of the CL emission (8 μs) is approximately half that of the Yb-centred lifetime (15 μs), in good agreement with theory.79 Although this occurs within a Yb9 cluster, in theory this requires only two proximate Yb ions. Unequivocal proof of this bimetallic mechanism was demonstrated in through simplifiction into the mechanistically simplest components, molecular Yb2 dimers. This was observed in an homo-dimetallic ytterbium cryptate (Fig. 11a) and a 2,2′-bipyrimidine bridged complex.81 The CL UC signal was significantly more intense for the cryptate complex owing to the much shorter intermetallic distance (3.445 Å vs. 6.639 Å). Importantly the quantum efficiency of the CL phenomenon (4.7 × 10−8 in (CD3)2SO) far exceeded that of the higher nuclearity Yb9 cluster (cf. 3 × 10−9 in CD3OD), informing that CL does not show an improvement with increasing nuclearity, the additional absorption cross-section of high Yb concentrations notwithstanding. Time-resolved measurements and kinetic modelling reveals a rate-determining kUC step to the (YbYb)** state. Interestingly the CL emission band profile at ca. 500 nm was shown to directly relate to the corresponding downshifted Yb photoluminescence profile due to the Stark-split sublevels of the 2F5/2 → 2F7/2 transition between 980–1050 nm (Fig. 11b), indicating the emission profile has some ‘memory’ of the excited state of each individual Yb* state from which it is derived.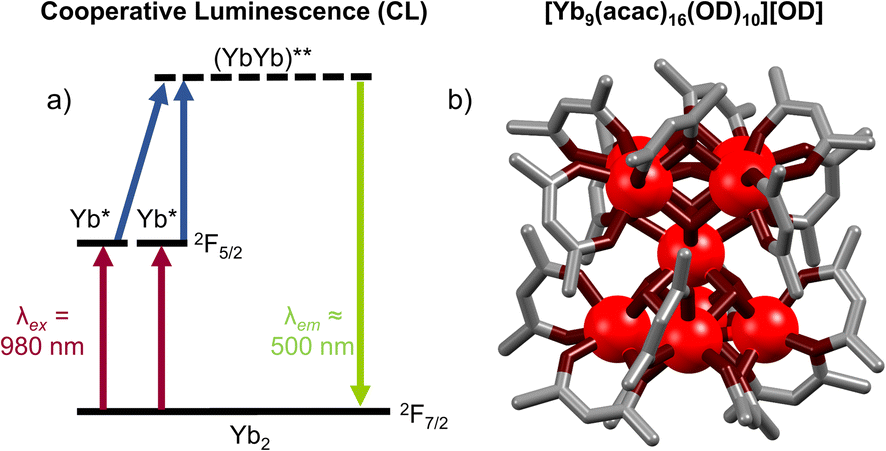 | ||
| Fig. 10 (a) Energy-level diagram for cooperative luminescence of Yb ions. (b) A [Yb9(acac)16(OD)10][OD] system exhibiting CL (ion colors: Yb = red)67 | ||
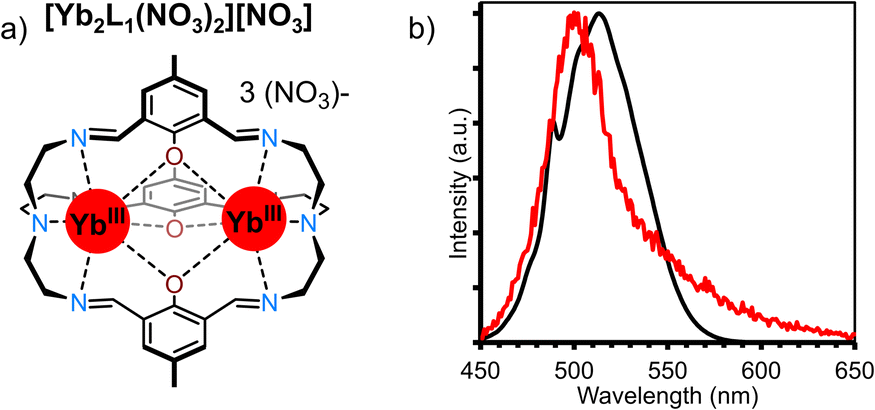 | ||
| Fig. 11 (a) Structure of [Yb2L1(NO3)][NO3]. (b) CL spectra in red (λex = 980 nm, p = 6.9 W cm−1, (CD3)2SO), convoluted downshifted spectra in black.81 | ||
One can conclude that the observation of molecular scale CL holds many possibilities for future research; circumventing the need for heterometallic CP systems, which can be hard to prepare, while not suffering from the main limitation of homometallic ETU phosphors based upon Er: significant quenching due to the presence of very low energy states (>1500 nm). We hope that these first few examples will serve as inspiration to more diverse and efficient systems.
5 Future outlook
Since the very first examples of molecular UC with Ln complexes in solution16 to now, the UC quantum yield and the brightness of these objects have been boosted by more than four orders of magnitude, allowing for their easy observation with traditional spectroscopic setups. However, there is still room in this recent topic for improvements, either for the efficiency of the devices, or for their implementation into applied systems. Within the frame of the ESA mechanism, the Achilles heel stands in the very weak absorption of the Ln elements and further progress might arise from the design of new compounds of very low symmetry, to improve the Ln based transition probabilities.70 Alternatively, another way to improve UC is also to reduce non-radiative relaxation pathways. If large efforts have been devoted to the use of per-deuterated compounds, organic solvents may open the door to the replacement of hydroxide anions by the isoelectronic fluoride anions in Ln based architectures.In contrast, the use of molecular UC devices for photovoltaic applications might be regarded as a very unlikely outlook. The narrow and weak absorption bands of Ln atoms are here the bottlenecks limiting the photon collection in the NIR domain. The alternative would then be the development of Ln based complexes with broad band NIR absorption of associated ligands. Considering the efficiency spectra of common solar cells based on silicon or CIGS thin films, this absorption should be below 10000 cm−1. Then, the UC photons must match the efficiency window of the cell, that is an emission above 10000 cm−1, which, for the piling of two photons, fix the energy of the intermediate Ln excited state above 5000 cm−1. If one could potentially consider Ln elements such as Er (with its 4I13/2 → 4I15/2 transition at 1550 nm, i.e. 6450 cm−1) or Tm (3F4 → 3H6 transition at ca. 1900 nm, i.e. 5200 cm−1) for such UC process, the very low energy of these NIR transitions and their propensity to be quenched by the vibrations of the medium, are hardly compatible with a high quantum efficiency and an interesting UC process at the molecular scale.
Concerning applications, the holy grail would clearly be to reach efficient molecular UC in water for biolabeling and biosensing applications. The plethora of examples obtained with UC nanoparticles82 would be available with molecular precision and reproducibility, while removing the potential toxicity and stability aspects associated to nanoparticles83 (providing the molecular devices are themselves stable complexes in biological media). Examples of molecular UC in water are still rare,30 but have merit and can pave the way to improved performances. In that respect, a combination of ligand-based photosensitization with a convenient composition of Ln atoms in heteropolynuclear assemblies based on ETU or CL UC should be a convenient way to tackle this issue.
Author contributions
L. J. C. conceptualized, wrote and supervised the manuscript and A. M. N., R. C. K. and L. G. wrote and edited the perspective.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The French National Research Agency is gratefully acknowledged for financial support (ANR LUCAS ANR-19-CE29-0014-01 and POPUP project ANR-22-CE29-0005-01).Notes and references
- F. Auzel, Chem. Rev., 2004, 104, 139 CrossRef CAS PubMed.
- E. M. Mettenbrink, W. Yang and S. Wilhelm, Adv. Photonics Res., 2022, 3, 2200098 CrossRef CAS PubMed.
- W. Zheng, P. Huang, D. Tu, E. Ma, H. Zhuab and X. Chen, Chem. Soc. Rev., 2015, 44, 1379 RSC.
- P. Ramasamy, P. Manivasakan and J. Kim, RSC Adv., 2014, 4, 34873 RSC.
- H. Liu, J. Xu, H. Wang, Y. Liu, Q. Ruan, Y. Wu, X. Liu and J. K. W. Yang, Adv. Mater., 2019, 1807900 CrossRef PubMed.
- A. Garaikoetxea Arguinzoniz, E. Ruggiero, A. Habtemariam, J. Hernández-Gil, L. Salassa and J. C. Mareque-Rivas, Part. Part. Syst. Char., 2014, 31, 46 CrossRef.
- W. Li, J. S. Wang, J. S. Ren and X. G. Qu, J. Am. Chem. Soc., 2014, 136, 2248 CrossRef CAS PubMed.
- O. Dukhno, F. Przybilla, V. Muhr, M. Buchner, T. Hirsch and Y. Mély, Nanoscale, 2018, 10, 15904 RSC.
- S. Lahtinen, A. Lyytikainen, H. Pakkilä, E. Hömppi, N. Peralä, M. Lastusaari and T. Soukka, J. Phys. Chem. C, 2017, 121, 656 CrossRef CAS.
- A. Gnach, T. Lipinski, A. Bednarkiewicz, J. Rybkaab and J. A. Capobianco, Chem. Soc. Rev., 2015, 44, 1561 RSC.
- L. Francés-Soriano, N. Hildebrandt and L. J. Charbonnière, Lanthanides as luminescence imaging reagents, in Comprehensive Inorganic Chemistry III, ed. J. Reedijk and K. R. Poeppelmeier, Elsevier, 3rd edn, 2023, pp. 486–510 Search PubMed.
- T. H. Maiman, Nature, 1960, 187, 493 CrossRef.
- H. J. M. A. A. Zijlmans, J. Bonnet, J. Burton, K. Kardos, T. Vail, R. S. Niedbala and H. J. Tanke, Anal. Biochem., 1999, 267, 30 CrossRef CAS.
- C. Duan, L. Liang, L. Li, R. Zhang and Z. P. Xu, J. Mater. Chem. B, 2018, 6, 192 RSC.
- E. M. Mettenbrink, W. Yang and S. Wilhelm, Adv. Photonics Res., 2022, 3, 2200098 CrossRef CAS PubMed.
- C. Koeppen, G. Jiang, G. Zheng and A. F. Garito, Opt. Lett., 1996, 21, 653 CrossRef CAS PubMed.
- C. A. Parker and C. G. Hatchard, Proc. R. Soc. London, Ser. A, 1962, 386 CAS.
- P. Bharmoria, H. Bildirir and K. Moth-Poulsen, Chem. Soc. Rev., 2020, 49, 6529 RSC.
- T. N. Singh-Rachford and F. N. Castellano, Coord. Chem. Rev., 2010, 254, 2560 CrossRef CAS.
- A. J. Carrod, V. Gray and K. Börjesson, Energy Environ. Sci., 2022, 15, 4982 RSC.
- H. Chen, I. Roy, M. S. Myong, J. S. W. Seale, K. Cai, Y. Jiao, W. Liu, B. Song, L. Zhang, X. Zhao, Y. Feng, F. Liu, R. M. Young, M. R. Wasielewski and J. F. Stoddart, J. Am. Chem. Soc., 2023, 145, 10061 CrossRef CAS PubMed.
- C.-G. Ma, M. G. Brik, D.-X. Liu, B. Feng, Y. Tian and A. Suchocki, J. Lumin., 2016, 170, 369 CrossRef CAS.
- J. C. G. Bünzli, Chem. Rev., 2010, 110, 2729 CrossRef PubMed.
- I. Bertini and C. Lucchinat, in NMR of Paramagnetic Molecules in Biological Systems, The Benjamin/Cummings Publishing Company, California, 1986 Search PubMed.
- A. Beeby, I. M. Clarkson, R. S. Dickins, S. Faulkner, D. Parker, L. Royle, A. S. de Sousa, J. A. G. Williams and M. Woods, J. Chem. Soc., Perkin Trans. 2, 1999, 493 RSC.
- C. Doffek, J. Wahsner, E. Kreidt and M. Seitz, Inorg. Chem., 2014, 53, 3263 CrossRef CAS PubMed.
- E. B. Sveshnikova and V. L. Ermolaev, Opt. Spectrosc., 2011, 111, 34 CrossRef CAS.
- M. Haase and H. Schäfer, Angew. Chem., Int. Ed., 2011, 50, 5808 CrossRef CAS PubMed.
- L. Aboshyan-Sorgho, C. Besnard, P. Pattison, K. R. Kittilstved, A. Aebischer, J.-C. G. Bünzli, A. Hauser and C. Piguet, Angew. Chem., Int. Ed., 2011, 50, 4108 CrossRef CAS.
- A. M. Nonat, S. Bahamyrou, A. Lecointre, F. Przybilla, Y. Mély, C. Platas-Iglesias, F. Camerel, O. Jeannin and L. J. Charbonnière, J. Am. Chem. Soc., 2019, 141, 1568 CrossRef CAS PubMed.
- A. M. Nonat and L. J. Charbonnière, Coord. Chem. Rev., 2020, 409, 213192 CrossRef CAS.
- H.-J. Yin, Z.-G. Xiao, Y. Feng and C.-J. Yao, Materials, 2023, 16, 5642 CrossRef CAS PubMed.
- H. Bolvin, A. Fürstenberg, B. Golesorkhi, H. Nozary, I. Taarit and C. Piguet, Acc. Chem. Res., 2022, 55, 442 CrossRef CAS PubMed.
- L. J. Charbonnière, Dalton Trans., 2018, 47, 8566 RSC.
- B. Golesorkhi, H. Nozary, A. Fürstenberg and C. Piguet, Mater. Horiz., 2020, 7, 1279 RSC.
- W. T. Carnall, in Handb. Phys. Chem. Rare Earths, Elsevier, 1979, p. 171 Search PubMed.
- C. Reinhard and H. Güdel, Inorg. Chem., 2002, 41, 1048 CrossRef CAS PubMed.
- X. Xiao, J. P. Haushalter and G. W. Faris, Opt. Lett., 2005, 30, 1674 CrossRef CAS PubMed.
- W. D. W. Horrocks and D. R. Sudnick, J. Am. Chem. Soc., 1979, 101, 334 CrossRef CAS.
- N. Souri, P. Tian, A. Lecointre, Z. Lemaire, S. Chafaa, J.-M. Strub, S. Cianférani-Sanglier, M. Elhabiri, C. Platas-Iglesias and L. J. Charbonnière, Inorg. Chem., 2016, 55, 12962 CrossRef CAS PubMed.
- N. Souri, P. Tian, C. Platas-Iglesias, K.-L. Wong, A. Nonat and L. J. Charbonnière, J. Am. Chem. Soc., 2017, 139, 1456 CrossRef CAS PubMed.
- Y. Ning, J. Tang, Y.-W. Liu, J. Jing, Y. Sun and J.-L. Zhang, Chem. Sci., 2018, 9, 3742 RSC.
- P. B. Glover, A. P. Bassett, P. Nockemann, B. M. Kariuki, R. Van Deun and Z. Pikramenou, Chem.–Eur. J., 2007, 13, 6308 CrossRef CAS PubMed.
- A. Nonat, C. F. Chan, T. Liu, C. Platas-Iglesias, Z. Liu, W.-T. Wong, W.-K. Wong, K.-L. Wong and L. J. Charbonnière, Nat. Commun., 2016, 7, 11978 CrossRef CAS PubMed.
- T. Liu, A. Nonat, M. Beyler, M. Regueiro-Figueroa, K. Nchimi Nono, O. Jeannin, F. Camerel, F. Debaene, S. Cianférani-Sanglier, R. Tripier, C. Platas-Iglesias and L. J. Charbonnière, Angew. Chem., Int. Ed., 2014, 53, 7259 CrossRef CAS PubMed.
- B. Golesorkhi, A. Furstenberg, H. Nozary and C. Piguet, Chem. Sci., 2019, 10, 6876 RSC.
- B. Golesorkhi, H. Nozary, L. Guénée, A. Fürstenberg and C. Piguet, Angew. Chem., Int. Ed., 2018, 57, 15172 CrossRef CAS PubMed.
- O. A. Blackburn, M. Tropiano, T. J. Sorensen, J. Thom, A. Beeby, L. M. Bushby, D. Parker, L. S. Natrajan and S. Faulkner, Phys. Chem. Chem. Phys., 2012, 14, 13378 RSC.
- M. J. Weber, Phys. Rev., 1968, 171, 283 CrossRef CAS.
- S. Dasari, S. Singh, P. Kumar, S. Sivakumar and A. K. Patra, Eur. J. Med. Chem., 2019, 163, 546 CrossRef CAS PubMed.
- B. Golesorkhi, I. Taarit, H. Bolvin, H. Nozary, J.-R. Jimenez, C. Besnard, L. Guenee, A. Furstenberg and C. Piguet, Dalton Trans., 2021, 50, 7955 RSC.
- J. H. S. K. Monteiro, E. A. Hiti, E. E. Hardy, G. R. Wilkinson, J. D. Gorden, A. E. V. Gorden and A. de Bettencourt-Dias, Chem. Commun., 2021, 57, 2551 RSC.
- X. Xie, C. Jin, Z. Pan, J. Wang, L. Deng, Y. Yao, D. Qi, Z. Sun, J. Qiu and S. Zhang, J. Lumin., 2023, 255, 119567 CrossRef CAS.
- F. Moglia, S. Mueller, F. Reichert, P. W. Metz, T. Calmano, C. Kraenkel, E. Heumann and G. Huber, Opt. Mater., 2015, 42, 167 CrossRef CAS.
- K. Driesen, P. Lenaerts, K. Binnemans and C. Görller-Walrand, Phys. Chem. Chem. Phys., 2002, 4, 552 RSC.
- N. V. Nikonorov, A. K. Przhevuskii and A. V. Chukharev, in Rare-Earth Doped Materials and Devices IV, ed. S. Jiang, Spie-Int. Soc. Optical Engineering, Bellingham, 2000, vol. 3942, pp. 183–191 Search PubMed.
- M. Iwamuro, Y. Hasegawa, Y. Wada, K. Murakoshi, N. Nakashima, T. Yamanaka and S. Yanagida, J. Lumin., 1998, 79, 29 CrossRef CAS.
- F. Auzel, C. R. Acad. Sci., 1966, 1016–1019 CAS.
- F. Auzel, C. R. Acad. Sci., 1966, 819 Search PubMed.
- D. Zare, Y. Suffren, L. Guénée, S. V. Eliseeva, H. Nozary, L. Aboshyan-Sorgho, S. Petoud, A. Hauser and C. Piguet, Dalton Trans., 2015, 44, 2529 RSC.
- I. Hyppänen, S. Lahtinen, T. Ääritalo, J. Mäkelä, J. Kankare and T. Soukka, ACS Photonics, 2014, 1, 394–397 CrossRef.
- B. Golesorkhi, S. Naseri, L. Guénée, I. Taarit, F. Alves, H. Nozary and C. Piguet, J. Am. Chem. Soc., 2021, 143, 15326 CrossRef CAS PubMed.
- J. Wang, Y. Jiang, J.-Y. Liu, H.-B. Xu, Y.-X. Zhang, X. Peng, M. Kurmoo, S. W. Ng and M.-H. Zeng, Angew. Chem., Int. Ed., 2021, 60, 22368 CrossRef CAS PubMed.
- D. A. Galico, J. S. Ovens, F. A. Sigoli and M. Murugesu, ACS Nano, 2021, 15, 5580 CrossRef CAS PubMed.
- D. A. Gálico, R. Ramdani and M. Murugesu, Nanoscale, 2022, 14, 9675 RSC.
- R. C. Knighton, L. K. Soro, A. Lecointre, G. Pilet, A. Fateeva, L. Pontille, L. Francés-Soriano, N. Hildebrandt and L. J. Charbonnière, Chem. Commun., 2021, 57, 53 RSC.
- R. C. Knighton, L. K. Soro, L. Francés-Soriano, A. Rodríguez-Rodríguez, G. Pilet, M. Lenertz, C. Platas-Iglesias, N. Hildebrandt and L. J. Charbonnière, Angew. Chem., Int. Ed., 2022, 61, e202113114 CrossRef CAS PubMed.
- D. A. Gálico and M. Murugesu, Angew. Chem., Int. Ed., 2022, 61, e202204839 CrossRef PubMed.
- D. A. Gálico, C. M. S. Calado and M. Murugesu, Chem. Sci., 2023, 14, 5827 RSC.
- S. Naseri, I. Taarit, H. Bolvin, J.-C. Bünzli, A. Fürstenberg, L. Guénée, G. Le-Hoang, M. Mirzakhani, H. Nozary, A. Rosspeintner and C. Piguet, Angew. Chem., Int. Ed., 2023 DOI:10.1002/anie.202314503.
- Y. Ning, J. Tang, Y.-W. Liu, J. Jing, Y. Sun and J.-L. Zhang, Chem. Sci., 2018, 9, 3742 RSC.
- I. Taarit, F. Alves, A. Benchohra, L. Guenee, B. Golesorkhi, A. Rosspeintner, A. Furstenberg and C. Piguet, J. Am. Chem. Soc., 2023, 145, 8621 CAS.
- L. K. Soro, C. Charpentier, F. Przybilla, Y. Mély, A. M. Nonat and L. J. Charbonnière, Chemistry, 2021, 3, 1037 CrossRef CAS.
- X.-F. Duan, L.-P. Zhou, H.-R. Li, S.-J. Hu, W. Zheng, X. Xu, R. Zhang, X. Chen, X.-Q. Guo and Q.-F. Sun, J. Am. Chem. Soc., 2023, 145, 23121 CrossRef CAS PubMed.
- J. Kalmbach, C. Wang, Y. You, C. Förster, H. Schubert, K. Heinze, U. Resch-Genger and M. Seitz, Angew. Chem., Int. Ed., 2020, 59, 18804 CrossRef CAS PubMed.
- R. C. Knighton, L. K. Soro, W. Thor, J.-M. Strub, S. Cianférani, Y. Mély, M. Lenertz, K.-L. Wong, C. Platas-Iglesias, F. Przybilla and L. J. Charbonnière, J. Am. Chem. Soc., 2022, 144, 13356 CrossRef CAS PubMed.
- R. B. Cevallos-Toledo, D. Bellezza, J. Ferrera-González, A. Giussani, E. Ortí, M. González-Béjar and J. Pérez-Prieto, ChemPhotoChem, 2023, 7, e202300156 CrossRef CAS.
- F. Auzel, D. Meichenin, F. Pellé and P. Goldner, Opt. Mater., 1994, 4, 35 CrossRef CAS.
- B. Chen and F. Wang, Acc. Chem. Res., 2020, 53, 358 CrossRef CAS PubMed.
- E. Nakazawa and S. Shionoya, Phys. Rev. Lett., 1970, 25, 1710 CrossRef CAS.
- K. L. Soro, R. C. Knighton, F. Avecilla, W. Thor, F. Przybilla, O. Jeannin, D. Esteban-Gomez, C. Platas-Iglesias and L. J. Charbonnière, Adv. Opt. Mater., 2022, 2202307 Search PubMed.
- A. Gnach, T. Lipinski, A. Bednarkiewicz, J. Rybka and J. A. Capobianco, Chem. Soc. Rev., 2015, 44, 1561–1584 RSC.
- E. Fröhlich, J. Nanobiotechnol., 2017, 15, 84 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2024 |