
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
RAFT step-growth polymerization via the Z-group approach and deconstruction by RAFT interchange†
Jiajia
Li
 ab,
Joji
Tanaka
*b,
Qing
Li
a,
Claire Jing
Jing Wang
b,
Sergei
Sheiko
b,
Samantha Marie
Clouthier
b,
Jian
Zhu
a and
Wei
You
*b
ab,
Joji
Tanaka
*b,
Qing
Li
a,
Claire Jing
Jing Wang
b,
Sergei
Sheiko
b,
Samantha Marie
Clouthier
b,
Jian
Zhu
a and
Wei
You
*b
aState and Local Joint Engineering Laboratory for Novel Functional Polymeric Materials, Jiangsu Key Laboratory of Advanced Functional Polymer Design and Application, Department of Polymer Science and Engineering, College of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou 215123, China
bDepartment of Chemistry, University of North Carolina at Chapel Hill, NC 27599, USA. E-mail: joji@email.unc.edu; wyou@unc.edu
First published on 23rd February 2024
Abstract
Recycling vinyl polymers is essential to mitigate the environmental impact of plastic waste. However, typical polymerization strategies to construct vinyl polymers lack the ability to incorporate degradable linkers throughout the main chain. We report a RAFT step-growth polymerization through the Z-group approach that is directly carried out by using a common class of symmetric trithiocarbonate based RAFT agents and commercially available bismaleimide monomers. Such synthesized RAFT step-growth polymers contain embedded RAFT agents in every structural unit, allowing chain expansion of the step-growth backbone via controlled chain growth to yield linear multiblock (co)polymers. These polymers can undergo deconstruction via the RAFT interchange process with exogeneous RAFT agents, generating smaller uniform species with narrow molecular weight distribution. In addition, the telechelic bifunctional RAFT agent nature after deconstruction allows repolymerization, showing a promising method for recycling common vinyl polymers.
Introduction
The C–C single bonds in vinyl polymers offer excellent stability for the intended applications, yet these polyethylene-like polymers are notoriously difficult to degrade, posing grand challenges in recycling these polymers and when degradation is desirable (e.g., drug delivery).1–3 Various strategies have been developed to overcome this challenge, including copolymerization with degradable monomers,4–18 introduction of degradable groups by combining with step-growth polymerization,19–24 depolymerization to monomers under special conditions,25–31 and degradation to oligomers catalyzed by special reactions.32–35 Among them, introducing labile carbon-heteroatom bonds (e.g., ester) into vinyl polymers via copolymerization of a second monomer containing such labile bonds is one of the most efficient methods.2,3 However, the insertion of these special monomers is usually random/statistical due to their different reactivities from the vinyl monomers; such synthesized “random” copolymers would thus result in uncontrolled degradation into a mixture of oligomeric species.3 For instance, Kiel et al. recently copolymerized styrene with a thionolactone (dibenzo[c,e]-oxepine-5(7H)-thione, DOT) to successfully impart degradation to polystyrene (PS);7 yet the degraded PS showed a molar mass dispersity around 1.9, due to the statistical nature of their copolymerization.To ensure a homogeneous degradation, it would require uniform insertion of these degradable labile carbon-heteroatom bonds into the main chain. One such method has been elegantly demonstrated by Uchiyama et al. where they applied controlled cationic copolymerization of vinyl ethers with a 7-membered cyclic thioacetal to achieve a rather uniform degradation of poly(vinyl ether)s.36 Furthermore, these thioacetals can serve as in-chain dormant species to enable the controlled growth of internal segments of poly(vinyl ether)s with cationic polymerization of different vinyl ethers via the degenerative chain-transfer (DT) mechanism; such multiblock copolymers can degrade into homogeneous diblock copolymers. However, this method is limited by the monomer scope of cationic polymerization; by contrast, radical polymerization is generally applicable to a wide scope of vinyl monomers and is much more tolerant to various functional groups and solvents. Thus it would be desirable to design similar strategies to achieve homogeneous degradation, in particular, for biological applications.3
Indeed, installation of in-chain dormant species in polymers has been explored via reversible addition–fragmentation chain transfer (RAFT) polymerization, one of the most versatile reversible deactivation radical polymerization (RDRP).37–39 Such polymers can be prepared via either chain-growth copolymerization with cyclic trithiocarbonates40–43 or step-growth polymerization of a bifunctional reagent tethered by a trithiocarbonate core.44–47 Subsequent RAFT controlled chain-growth polymerization of vinyl monomers would produce internal segments between in-chain dormant species, and such synthesized vinyl polymers could then degrade into uniform oligomers (i.e., “freed” internal segments). However, the syntheses of these cyclic chain transfer agents (CTAs) or RAFT agents with special functional groups (with some exceptions46,47) are rather complicated, limiting the scale-up and wide applications. In addition, there are limited strategies for repolymerization of degraded units following the degradation,7 yet such repolymerizations are required for closed loop recycling. We noticed that in-chain RAFT agents in polymer networks have been exploited to achieve self-healing materials via photoinduced covalent bond rearrangement through the RAFT process (or RAFT interchange);48–53 these intriguing results prompted us to explore the dynamic RAFT interchange for recycling polymers with in-chain CTAs; ideally, such polymers could be prepared in a simple and highly efficient manner.
Previously, we demonstrated RAFT step-growth polymerization (or polyaddition, suggested by IUPAC54,55) by exploiting bifunctional reagents bearing a monomer and CTA functionality that can generate high yields of single unit monomer inserted (SUMI)56 CTA adducts under stoichiometrically balanced conditions.57–62 In our earlier reports, the CTA was tethered with the monomer (i.e., for AB step-growth) or with the same CTA (i.e., for A2 + B2 step-growth) through the R-group, which yields a polymer backbone with thiocarbonylthio units appended as the side chains on each structural unit. This unique configuration allows subsequent graft polymerization from the step-growth backbone (Fig. 1). We envisioned that RAFT step-growth can also be achieved by tethering the CTA through the Z-group, or more simply by employing a symmetric trithiocarbonate that bears two R-groups as the B2 for A2 + B2 step-growth polymerization. It is important to clarify that, although the Z-group is not explicitly presented in Fig. 1, the implied Z-group is the –SR group opposite the polymerizing side of the RAFT agent. By taking advantage of this proposed RAFT step-growth via the Z-group approach, herein we report a simple approach with easy-to-prepare or commercially available monomers to construct a polymer with uniformly installed RAFT agents, which can be chemically recycled through RAFT interchange with exogenous RAFT agents. Additionally, in contrast to the former R-group approach, multi-segment vinyl polymers can be obtained via subsequent main chain expansion of the step-growth polymer with vinyl monomers through RAFT controlled chain-growth. Furthermore, deconstruction of such vinyl polymers through RAFT interchange with exogenous RAFT agents results in lower molecular weight vinyl polymer species with narrow molecular weight distribution; these species can be considered as a bifunctional macro-CTA for repolymerization through the RAFT step-growth via the Z-group approach, thereby closing the loop for chemically recycling vinyl polymers.
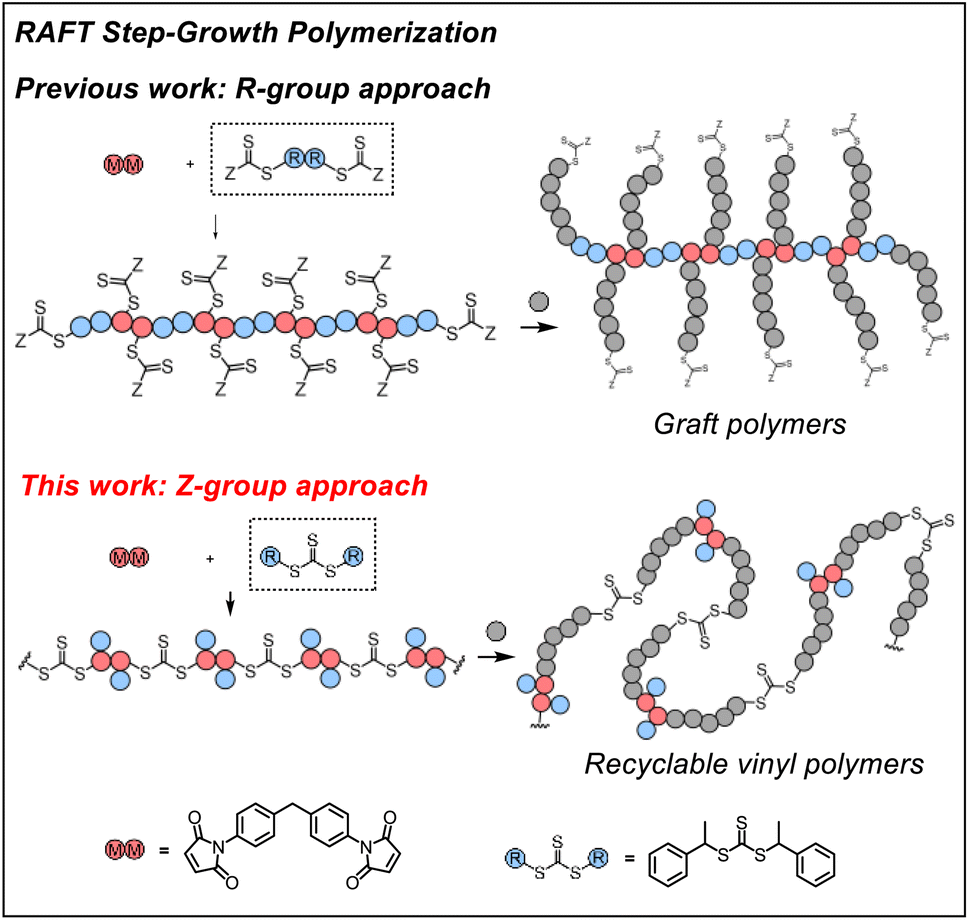 | ||
| Fig. 1 RAFT step-growth polymerization via the R-group approach or the Z-group approach. | ||
Results and discussion
Mechanistic difference between the ‘R-group approach’ and ‘Z-group approach’ for RAFT step-growth polymerization
Though RAFT step-growth polymerizations via the R-group approach and the Z-group approach proceed through an identical SUMI process, we highlight some key mechanistic differences in Fig. 2. For clarity, the thiocarbonyl thiol present for every repeat unit is not drawn in the illustration. In addition, specific to this work where a symmetric trithiocarbonate is used, the Z-group is a sulfur atom bearing a fragmentable group (polymer backbone or another R-group). With other classes of dithiocarbonyl thio based CTAs, the Z-group would be required to be tethered to another Z-group bearing a dithiocarbonyl thio unit via a non-fragmentable linker. The simplification of treating two groups of dithiocarbonyl thio units to a single core is unique to this class of symmetric trithiocarbonates.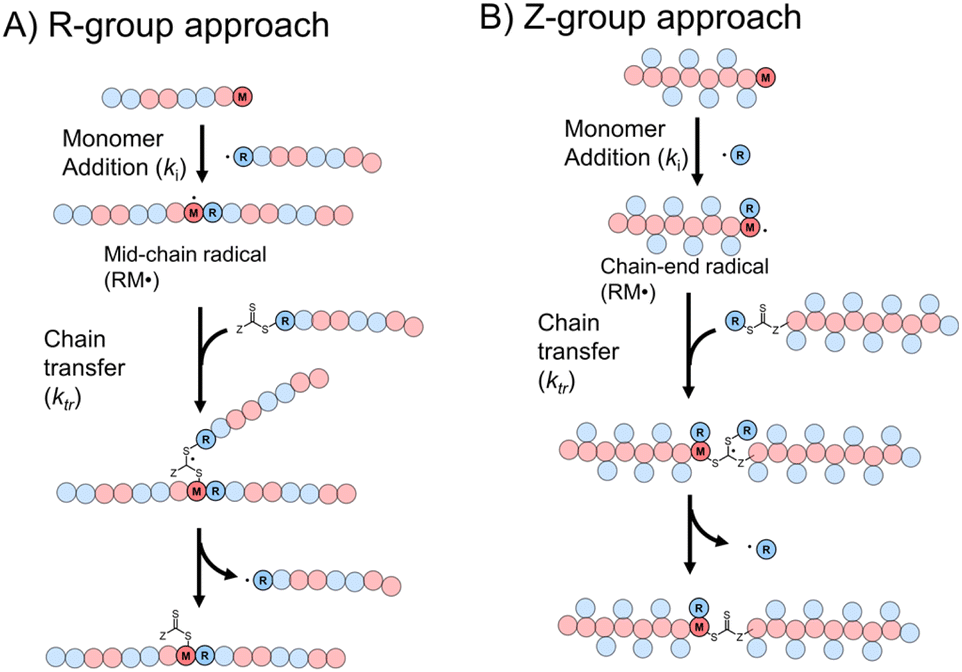 | ||
| Fig. 2 The difference between RAFT step-growth polymerization via (A) the R-group approach and (B) the Z-group approach. | ||
While with both R-group and Z-group approaches, monomer addition (ki) and chain transfer (ktr) are key steps to complete the RAFT step-growth cycle, the difference between how two CTAs are tethered (R vs. Z) has implications in the radical intermediate species and the way the polymeric chains are formed. In the R-group approach, the R˙ species exists as a (macro)radical end-group that adds to the monomer end-group, forming the R–M˙ adduct as a mid-chain radical species (Fig. 2A). However, in the Z-group approach, the “free” R˙ species adds to a monomer end group, forming an R–M˙ adduct as a chain-end radical species (Fig. 2B). The R–M˙ adduct will then be added to a chain end CTA to form a 3-arm star like polymer as a chain transfer intermediate adduct in the R-group approach (Fig. 2A), while in the Z-group approach this intermediate resembles a linear chain (Fig. 2B). Finally, following fragmentation from the chain transfer intermediate adduct, the respective R˙ species is regenerated, concurrently forming the CTA backbone. We emphasize that a ‘stable’ main chain is formed upon an irreversible monomer addition step in the R-group approach (Fig. 2A), whereas a ‘dynamic’ mainchain is formed upon the reversible chain transfer step in the Z-group approach (Fig. 2B).
RAFT SUMI model reaction
We chose a trithiocarbonate based CTA, bis(1-phenylethyl)trithiocarbonate (CTA2 in Fig. 3), to experimentally validate the RAFT step-growth polymerization via the Z-group approach, since this R-group has been previously reported to undergo an efficient RAFT-SUMI process with maleimides.60,62 In addition, we found this particular bifunctional CTA can be synthesized with high yield under mild conditions.63N-Ethyl maleimide was chosen as the monomer to attempt the model RAFT-SUMI reaction, to confirm if symmetrical trithiocarbonate based CTAs can efficiently yield SUMI-CTA adducts.62 In theory, one equivalent of CTA2 would consume two equivalents of monofunctional maleimide (Fig. 3). As shown in Fig. 3, S1 and Table S1,† near quantitative yield of the SUMI-CTA adduct was observed within 4 hours using a thermal initiator, 2,2-azobisisobutyronitrile (AIBN) at 70 °C, indicating the feasibility of this monomer/CTA pair for the RAFT step-growth polymerization.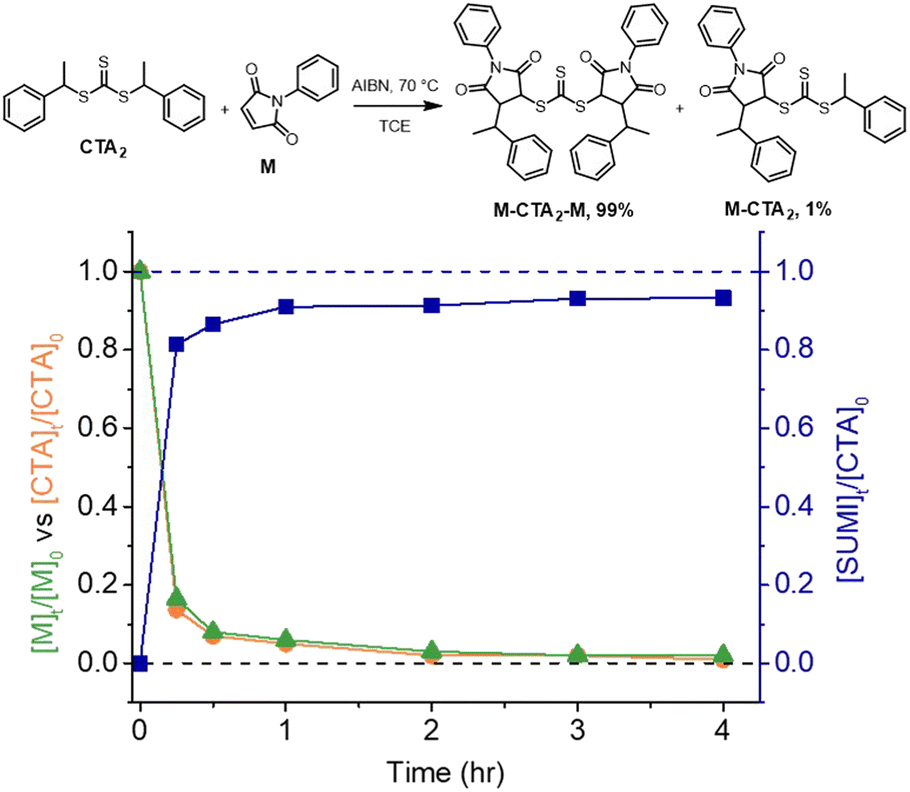 | ||
Fig. 3 Kinetic analysis of the RAFT-SUMI model reaction between the RAFT agent and maleimide with [CTA2]0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[M]0:[AIBN]0 = 0.5:1:0.05 M in TCE at 70 °C. The RAFT-SUMI-CTA2 adduct (M-CTA2-M) yields (blue line) include both the dual SUMI-CTA2 adduct and mono-SUMI adduct (M-CTA2); the relative fraction of each species after 4 hours is included in the reaction scheme. :[M]0:[AIBN]0 = 0.5:1:0.05 M in TCE at 70 °C. The RAFT-SUMI-CTA2 adduct (M-CTA2-M) yields (blue line) include both the dual SUMI-CTA2 adduct and mono-SUMI adduct (M-CTA2); the relative fraction of each species after 4 hours is included in the reaction scheme. | ||
RAFT step-growth polymerization via the Z-group approach
We chose a low-cost and commercially available bismaleimide, 1,1′-(methylenedi-4,1-phenylene)bismaleimide, as the bifunctional monomer (M2) to demonstrate A2 + B2 type RAFT step-growth polymerization via the Z-group approach. Tetrachloroethane (TCE) was used as a solvent for its good solubility with maleimide monomers.60 Pleasingly, the evolution of number-average (Mn), weight-average (Mw), and Z-average (Mz) molecular weight with the extent of reaction (p) (determined from 1H NMR, Fig. S2†) tracked very well with the theoretical values (Fig. 4A), validating the polymerization to proceed through linear step-growth molecular weight evolution. The lower experimental Mn values can be attributed to the formation of cyclic species observed in the SEC traces (Fig. 4B). The deviation of experimental Mw from theoretical values for balanced stoichiometry ([CTA2]0:[M2]0 = 1:1) can be accounted for by the stoichiometric imbalance caused by external initiation (Table S2†).62,64 It is worth noting that irreversible radical termination events would also contribute to imbalanced stoichiometry.
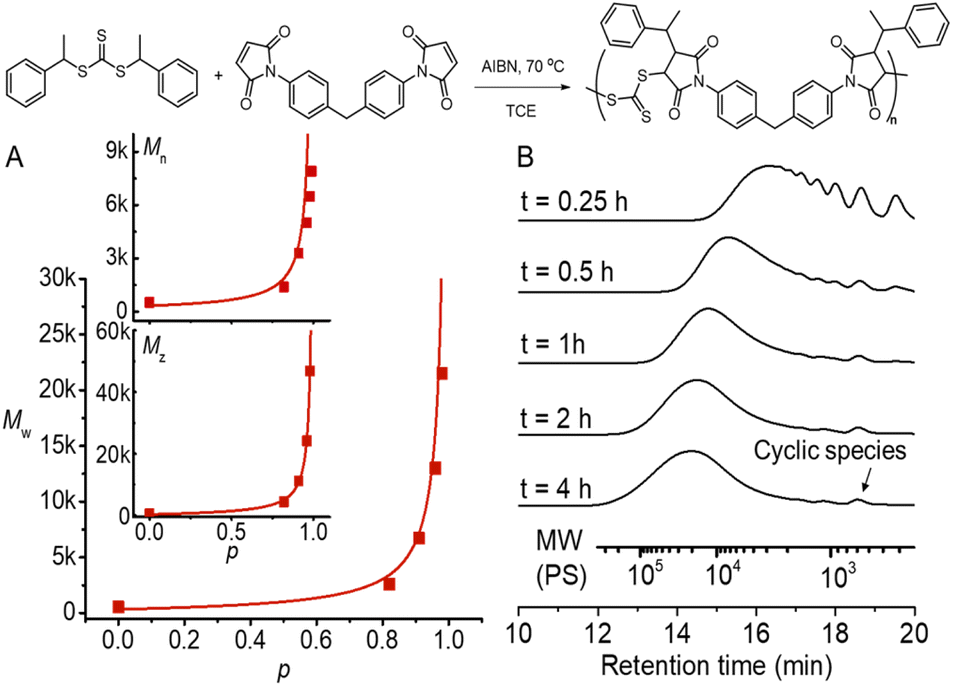 | ||
| Fig. 4 RAFT step-growth polymerization results using CTA2 and M2 with [CTA2]0:[M2]0:[AIBN]0 = 0.5:0.5:0.05 M in TCE at 70 °C. (A) Evolution of experimental Mn, Mw, and Mz determined by SEC analysis using polystyrene as a standard and extent of reaction (p) calculated from 1H NMR, plotted with theoretical molecular weight averages predicted without considering cyclization (see the ESI† for more information); (B) SEC traces of the obtained polymers at different time points. | ||
Recycling the RAFT step-growth polymers
The presence of the in-chain trithiocarbonate (TTC) in the alternating copolymers prepared from RAFT step-growth polymerization via the Z-group approach would allow for both deconstruction through RAFT interchange and chain expansion through RAFT chain-growth polymerization. Before attempting the chain expansion from this polymer, we first examined its recyclability through RAFT interchange in the presence of exogenous RAFT agents. Mechanistically, as shown in Fig. 5, the exogenous CTA2 can undergo direct photolysis to generate a reactive carbon-centered radical, R˙, which can react with the in-chain TTC group through RAFT interchange, leading to fragmentation of the alternating copolymers. It is worth noting that this process is reversible and controlled by equilibrium; thus increasing the amount of the exogenous CTA2 will result in more complete deconstruction. On the other hand, owing to the dynamic nature of the in-chain TTC, reshuffling reactions of the polymer chain could also happen during the polymerization. Moreover, the direct photolysis of the carbon–sulfur bond in the backbone would promote the RAFT interchange process, which is independent of the structure difference between the backbone and exogenous CTA2 (Fig. S3†). The deconstruction process could also be considered as RAFT step-growth polymerization under imbalanced stoichiometry, which should in theory lead to a low degree of polymerization (DP).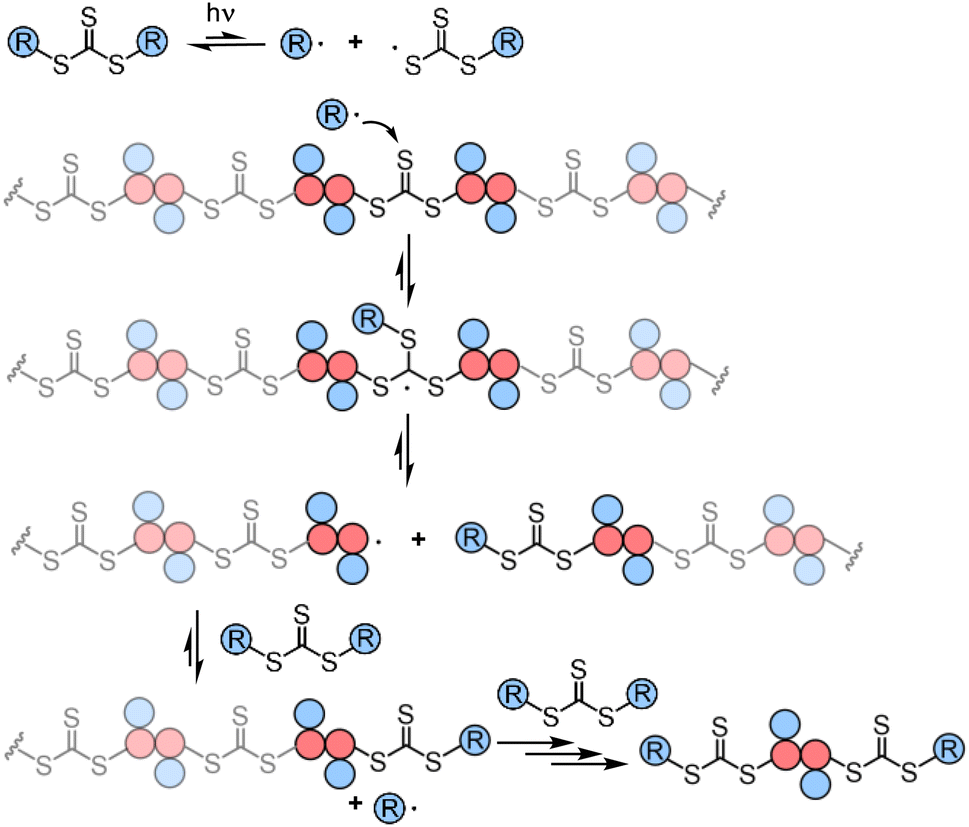 | ||
| Fig. 5 Proposed mechanism for the deconstruction of the backbone. | ||
As shown in Fig. 6, deconstruction of a backbone polymer (Mw = 11900, Đ = 2.29) was conducted with CTA2 ([TTC]0:[CTA2]0 = 1:5) under irradiation of a 405 nm LED. After 2 hours, the Mw decreased from 11900 to 1000, consistent with the theoretical value of 994.2 for 3 structural units (2 × CTA2 + 1 × M2, Fig. 5), indicating successful deconstruction of this alternating copolymer. Moreover, it can be observed from 1H NMR that the signal of the benzylic CH (peak b, Fig. S4†) of the benzyl R group at the RAFT terminal increased and the signal of the CH (peak c, Fig. S4†) of the maleimide adjacent to trithiocarbonate progressed to a more defined splitting pattern after photoirradiation of the backbone with CTA2, further supporting the successful deconstruction. The minor peak in the higher molecular weight side in the SEC curve (Fig. 6) after deconstruction indicated incomplete deconstruction of the backbone. Decreasing the amount of CTA2 (e.g. [TTC]0:[CTA2]0 = 1:1 or 1:2) resulted in more incomplete deconstruction, where more oligomers were observed (Fig. S5 and S6†). After the deconstruction, since the mixture consists of extra CTA2 and a newly generated bifunctional RAFT agent, adding M2 into this mixture would allow RAFT step-growth (re)polymerization. The exact amount of M2 can be calculated based on the amount of previously added CTA2 during the deconstruction. Indeed, successful repolymerization was achieved through both a photoiniferter method (Fig. 6 and S7–S9†) and thermally-induced method (Fig. S10†).59
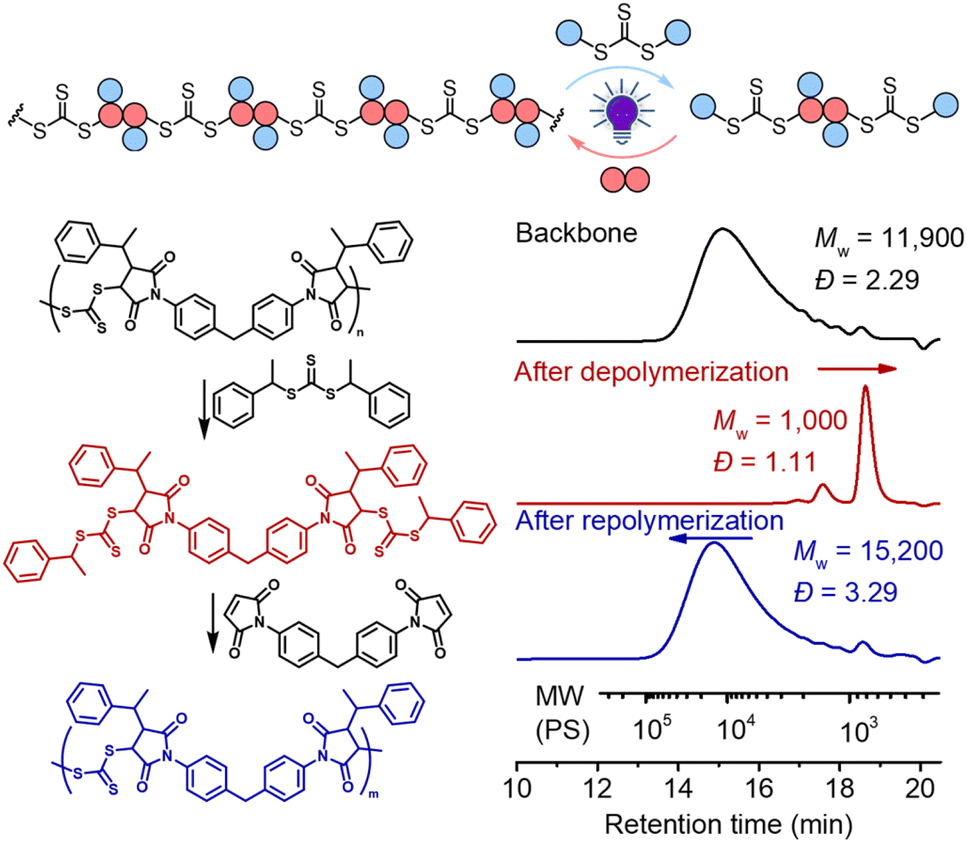 | ||
| Fig. 6 Deconstruction of the backbone with [TTC]0:[CTA2]0 = 1:5 for 2 hours under irradiation of a 405 nm LED and (re)polymerization of the formed mixture with [CTA2]0:[M2]0 = 1:1 for 96 hours under irradiation of a 405 nm LED. | ||
Deconstruction of (multiblock) vinyl polymers
We next subjected this alternating copolymer (i.e., the backbone) to chain expansion experiments with methyl acrylate (MA) and styrene (St), respectively, via photoiniferter RAFT polymerization under UV-irradiation of 405 nm.65 To avoid copolymerization of the possible terminal maleimide from the precursor step-growth polymer during the chain expansion, we applied a step-growth polymerization with an imbalanced stoichiometry at [CTA2]0:[M2]0 = 1:0.9, which in theory should consume all bismaleimides during the step-growth. 1H NMR analysis of the obtained polymer (Mw = 12100, Đ = 2.46) (Fig. S11†) disclosed that the signal assigned to the double bond in maleimide (peak d) at around 6.8 ppm nearly completely disappeared and the benzylic CH proton (peak i) was clearly visible at the chain end, supporting the anticipated alternating copolymer terminated by the CTA end group (structure in Fig. S11†). The targeted DP of the vinyl polymer was controlled by the molar ratio between the monomer and the in-chain TTC and the reaction time. As depicted in Fig. 7A, the SEC trace showed a clear shift after chain expansion with MA (the blue line, monomer conversion = 18.3% from 1H NMR), with Mw increasing from 12100 to 140100. The low molecular weight peak (though minor) is likely caused by the chain expansion of the cyclic species. Triple detection SEC (dRI, LS, VS) analysis was further employed for characterization of the polymers before and after chain expansion (Fig. S12 and S13†). The Mark–Houwink plots showed an α value of 0.66 for the backbone polymer and 0.677 for the PMA, consistent with the hydrodynamic volume as a function of molecular weight distribution for linear polymers.66 This contrasts to the case of subsequent RAFT chain-growth polymerization from RAFT step-growth polymers prepared by the R-group approach, where the α value decreases as the obtained graft copolymers behave more compact in solution.62 Furthermore, following the deconstruction via RAFT interchange with CTA2, a more symmetrical SEC trace was observed with narrow molecular weight distribution (the red line in Fig. 7A). Pleasingly, the experimentally determined Mn of 11100 (from conventional SEC analysis with polystyrene calibration in THF, see the ESI† for SEC information) for the PMA segments after deconstruction was consistent with the theoretical value of 8900 (Mn,th = ([MA]0/[CTA]0) × MMA × conv.% + MM2 + 2 × MCTA2). These results demonstrated the successful chain expansion of the backbone and its subsequent deconstruction into well-defined PMA segments. The chain expansion of the backbone was also carried out with St to prepare degradable polystyrene (PS). As depicted in Fig. 7B, successful chain expansion was observed with molecular weight increasing from 12100 to 120100 (blue line, monomer conversion = 26.4%). Meanwhile, the Mark–Houwink plots showed an α value of 0.655 (Fig. S14†), indicative of a linear PS as expected. The molecular weight of PS segments (Mn = 13200, Đ = 1.29) after deconstruction was also consistent with the theoretical value of 14700 (Mn,th = ([St]0/[CTA]0) × MSt × conv.% + MM2 + 2 × MCTA2). We have also performed the controlled experiment of PS in the absence of CTA2, and the reshuffling of backbone trithiocarbonate resulted in minor cyclization (Fig. S15†).
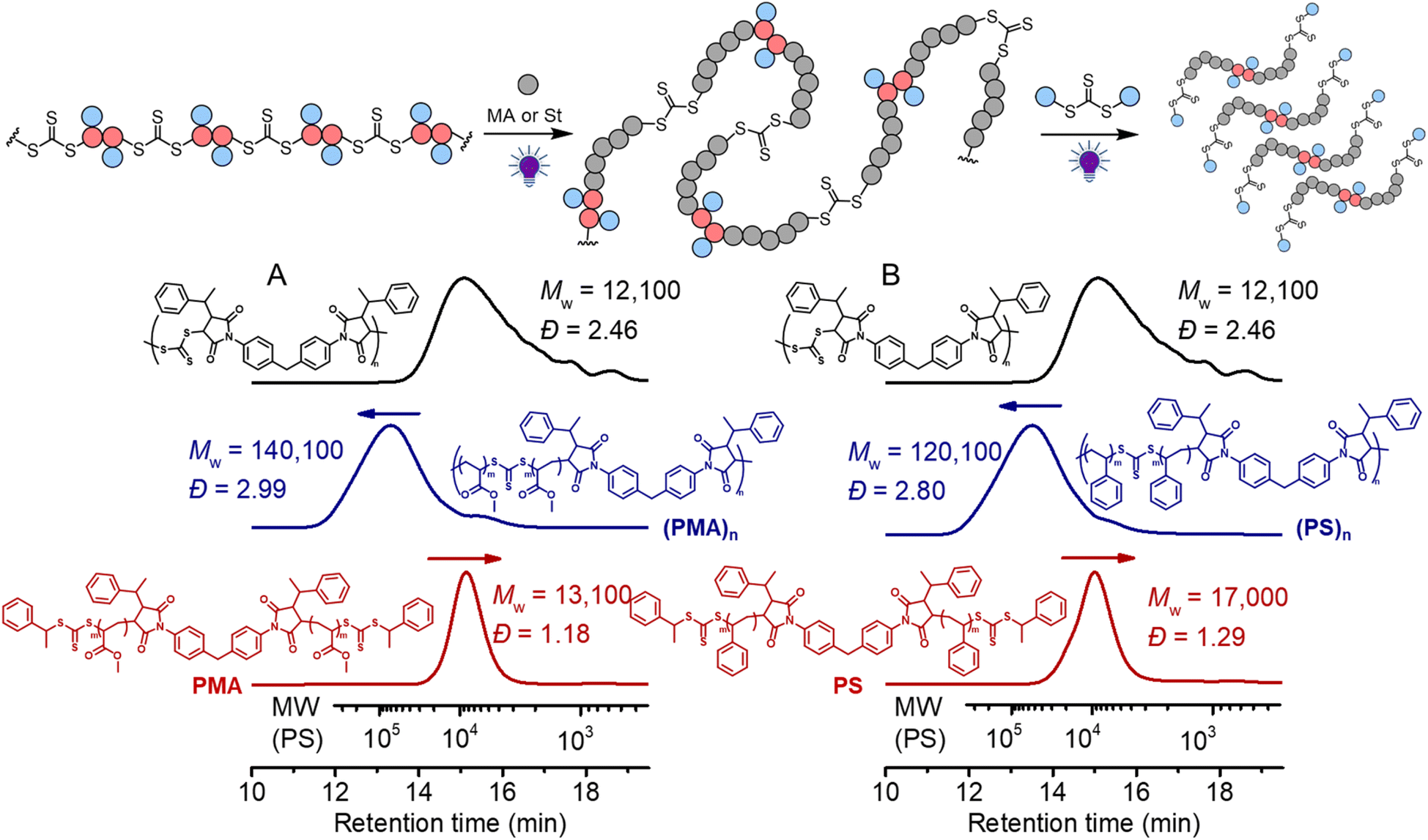 | ||
| Fig. 7 Black line: the backbone (Mw = 11800); blue line: chain expansion under 405 nm light with [monomer]0:[TTC]0 = 500:1; red line: after deconstruction of the obtained polymers with [TTC]0:[CTA2]0 = 1:10 for 24 hours; (A) using MA as a monomer at conv.% = 18.3%; (B) using St as a monomer at conv.% = 26.4%. | ||
In traditional RAFT chain-growth polymerization, multiple chain extensions are required to prepare multiblock copolymers (typically generating one block per chain extension).67 By exploiting the in-chain RAFT agent, multiblock copolymers can be easily prepared by simple chain expansions from the step-growth backbone. As shown in Fig. 8, we demonstrate this by first chain expansion of a backbone (Mw = 12100) with St (Mw = 59400), followed by the second chain expansion with MA (Mw = 182600), which results in multiblock copolymer of P(PS-b-PMA)n. The Mark–Houwink plots of the PS and multiblock showed α values of 0.69 and 0.617, respectively (Fig. S16†), confirming the formation of linear polymers. After deconstruction of the multiblock copolymer, a decreased Mw of 13800 (from 182600 of the multiblock copolymer) was observed with a narrow molecular weight distribution (Đ = 1.23), indicating a successful deconstruction of the multiblock copolymer to segments of controlled lengths (consisting of a triblock copolymer PMA-b-PS-b-PMA). Furthermore, the experimental number average molecular weight (Mn = 11200, Ð = 1.23) matched well with the theoretical value of 12900 predicted by the monomer conversion (Mn,th = ([St]0/[CTA]0) × MSt × conv.% (St) + ([MA]0/[CTA]0) × MMA × conv.% (MA) + MM2 + 2 × MCTA2).
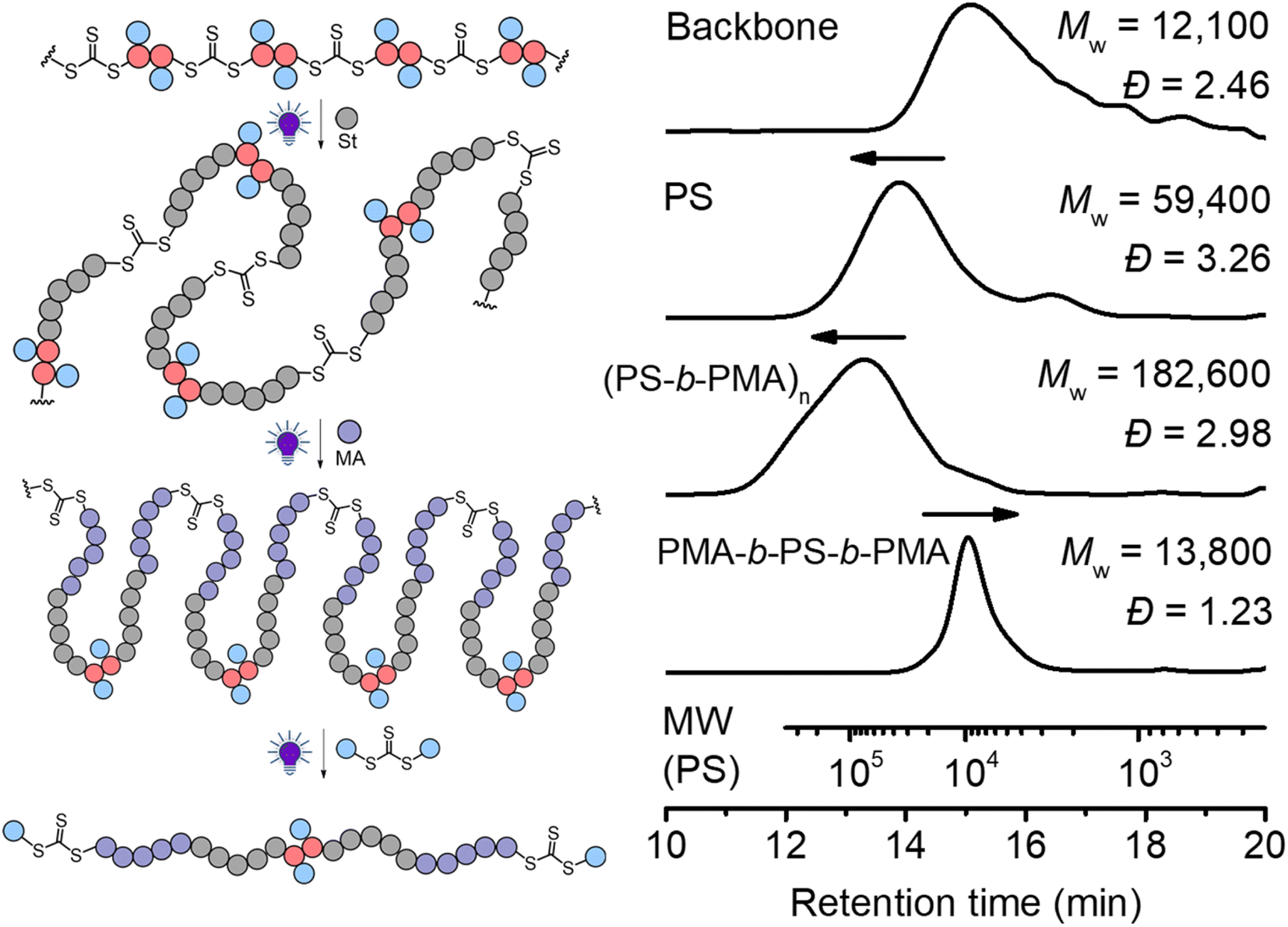 | ||
| Fig. 8 Chain expansion with St was carried out under 405 nm light with [St]0:[CTA]0 = 330:1, conv.% = 12.7%; chain expansion with MA was carried out under 405 nm light with [MA]0:[CTA]0 = 330:1, conv.% = 23.2%; deconstruction of the obtained polymer was carried out with [TTC]0:[CTA2]0 = 1:10 under 405 nm light for 24 hours. | ||
Recycling vinyl polymers
Finally, we explored the iterative cycles of repolymerization and deconstruction to recycle vinyl polymers. We first examined (PMA)n using the same bifunctional CTA (CTA2) for the deconstruction and bis-maleimide (M2) for repolymerization. We chose a polymer (after chain expansion with MA) having relatively low molecular weight PMA segments (Fig. 9, purple line, Mw = 26400, Đ = 3.65), to allow repolymerization of the degradants (after deconstruction) at sufficiently high molar concentrations, because a high concentration is required to achieve high molecular weight.62 It is worth noting that 1H NMR reveals shifts in the CH protons next to the trithiocarbonate following chain expansion of the backbone with MA: the first two sets of signals (end-group – benzyl CH, peak a at 5.25 ppm vs. in-chain maleimidic CH, peak b at 4.70 ppm, Fig. S17†) of the step-growth backbone converted to single species of CH in-chain termini of PMA segments at 4.82 ppm (peak c, Fig. S17†) after chain expansion of MA. These observable differences in 1H NMR became convenient in analyzing each step of the recycling process (vide infra).
 | ||
| Fig. 9 Recycling PMA through RAFT step-growth polymerization and deconstruction. Black line: the backbone; purple line: chain expansion with MA by RAFT chain-growth under 405 nm light with [MA]0:[TTC]0 = 50:1, conv.% = 18.0%; red line: deconstruction of the obtained polymer under 405 nm for 24 h with [TTC]0:[CTA2]0 = 1:10; blue line: repolymerization of PMA under 405 nm for 96 h with [PMA]0:[M2]0 = 1:1. | ||
As depicted in Fig. 9, (PMA)n with relatively low molecular weight (purple line, Mw = 26400, Đ = 3.65) was prepared by chain expansion of the backbone (black, Mw = 11800, Đ = 2.79); after deconstruction with CTA2, this (PMA)n degraded into PMA with low molecular weight and narrow dispersity (red line, Mw = 1,800, Đ = 1.14). After deconstruction, it should be pointed out that each terminal RAFT agent of the PMA has two different fragmentations, including a benzyl R-group and a secondary ester macro-R-group, which can be observed by 1H NMR (approximately 50:50 distribution of Hd and He, Fig. S17†). For (re)polymerization of the PMA via RAFT step-growth, a selective reaction between only one of the two fragmentations at each chain end is required to avoid the crosslinking reaction. In theory, selective (re)polymerization through the macro-R-group would lead to the RAFT step-growth via the R-group approach, whilst to achieve step-growth via the Z-group approach, selective repolymerization must occur through the other R-group. In our case, we chose the bismaleimide (M2) for the repolymerization because of its preferable RAFT-SUMI reaction with the benzyl R-group than the secondary ester R-group.62 As shown in Fig. 9 (blue line), the Mw increased from 1800 to 31900 after (re)polymerization with M2, demonstrating the success of this approach. Furthermore, 1H NMR analysis revealed the expected disappearance of the CH peak of the benzyl R-group next to the trithiocarbonate after repolymerization (Fig. S17†). Moreover, this polymer can be deconstructed again by adding CTA2 through the RAFT interchange (red line, Fig. S18†); interestingly, a peak at the low molecular weight side after deconstruction was observed, which was assigned to the adduct of M2 and CTA2 generated during the RAFT interchange (Fig. S19†). Unfortunately, separating this adduct from the degraded low molecular weight PMA was found to be challenging as it was more insoluble than PMA during purification by precipitation. Nonetheless, successful (re)polymerization can still be achieved by adding M2 to this mixture (blue line, Fig. S18†), though likely leading to a random copolymer containing segments from the original backbone and from the (PMA)n.
We also applied the same strategy for recycling PS. However, we noticed after deconstruction with CTA2 that each terminal RAFT agent of the PS has two phenyl fragmentations (Fig. 7B), which would lead to crosslinking during the (re)polymerization, due to the lack of selectivity. Therefore, a RAFT agent with a tertiary ester R group (CTA2E) was synthesized and used (instead of CTA2) for the deconstruction, aiming to subsequently achieve selective repolymerization. As shown in
Fig. 10, deconstruction of the (PS)n (grey line, Mw = 98000, Đ = 3.03) with CTA2E resulted in PS with lower molecular weight and narrow MWD (Mw = 10200, Đ = 1.28), indicating the high efficacy of CTA2E for the deconstruction (step A). Divinylbenzene (DVB) was then chosen as the bifunctional vinyl monomer for RAFT step-growth (re)polymerization of these PS macro-CTAs (step B), because of the highly efficient RAFT-SUMI process between the styrene monomer and trithiocarbonate bearing tertiary alkyl carboxyl fragmentation (Fig. S20 and S21†). Successful repolymerization with DVB was observed by the shift of the SEC curve towards higher molecular weight distribution (Mw = 50000, Đ = 1.99). Similar to the case of recycling PMA, the adduct of DVB and CTA2E was generated during the RAFT interchange for the second deconstruction (step C); however, unlike the case of recycling PMA, this adduct can be easily removed by precipitation in methanol whilst retaining the PS macro-CTA (Mw = 10900, Đ = 1.29). With the purified PS macro-CTA, we conducted the second repolymerization and third deconstruction through the same methods. The yield of PS obtained after each deconstruction and repolymerization is around 93–98%, showing that it is a promising method towards circular recycling of PS. The thermal decomposition temperature of the multiblock polystyrene was assessed by thermogravimetric analyses (TGA). The thermal decomposition temperature at 5% mass loss (Td, 5%) was slightly lower (371.6 °C) than that of non-degradable high molecular weight PS (reported value of 408 °C) (Fig. S22†).7 In theory, the thermal decomposition of the trithiocarbonate core is expected to occur between 210 and 250 °C;68 however, such transition is not observable due to the relatively low abundance of the trithiocarbonate by weight (∼1.52 wt%). Furthermore, Differential Scanning Calorimetry (DSC) revealed a glass transition temperature (Tg) of 106.7 °C which matched well with expected values for high molecular weight PS (Fig. S23†).7 Furthermore, thermomechanical properties were investigated with temperature sweep experiments of Dynamic Mechanical Analysis (DMA); at temperatures below the Tg, the elastic modulus was found to be within a similar order of magnitude to that of commercial PS (Fig. S24†).69
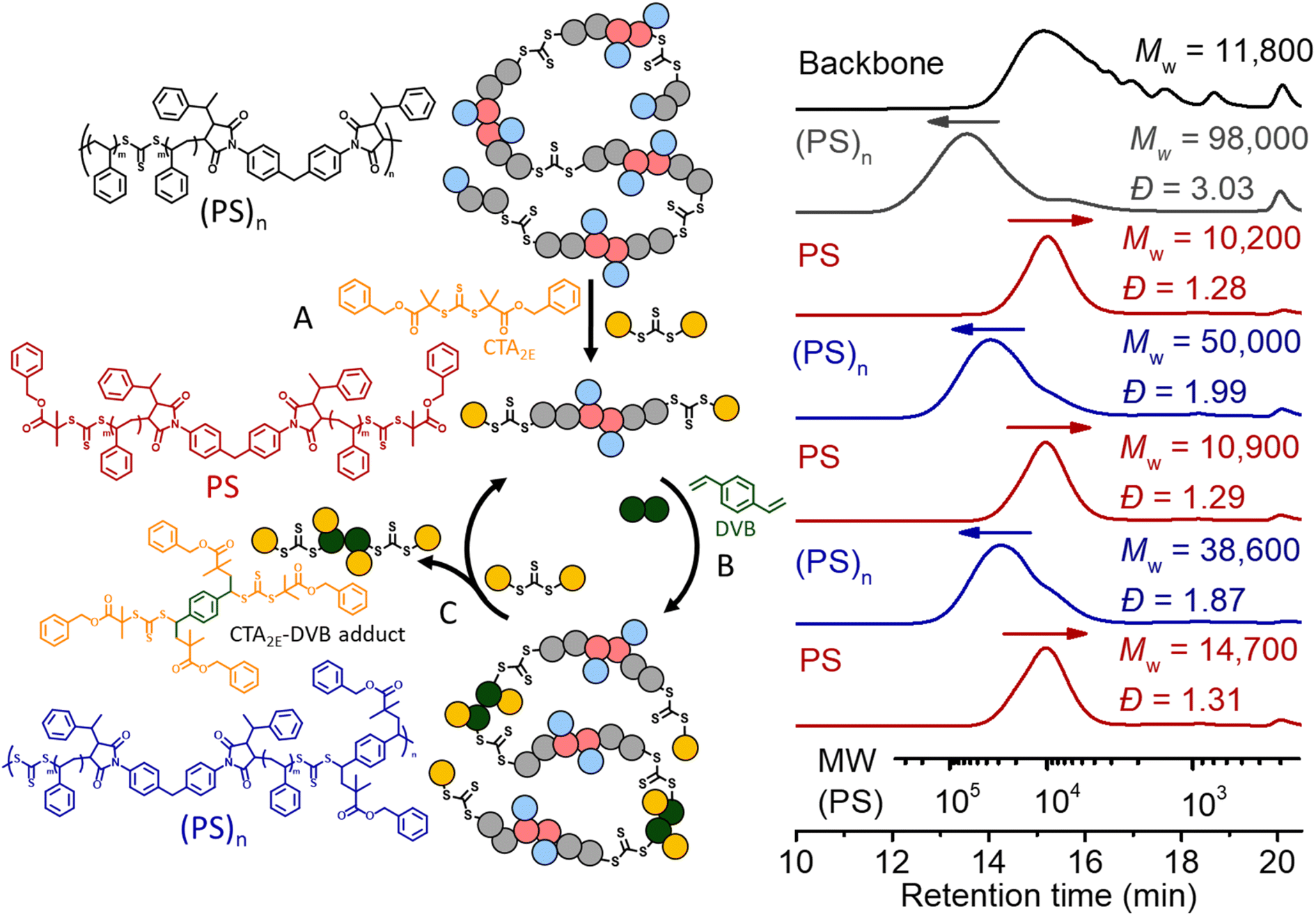 | ||
| Fig. 10 Recycling PS through RAFT step-growth polymerization and deconstruction. Black line: the backbone; grey line: chain expansion with St by RAFT chain-growth under 405 nm light with [St]0:[TTC]0 = 500:1, conv.% = 14.1%; red line: deconstruction of the obtained polymer under 405 nm for 24 h with [TTC]0:[CTA2]0 = 1:10; blue line: repolymerization of PS under 405 nm for 24 h with [PS]0:[DVB]0 = 1:1. | ||
Conclusions
In summary, RAFT step-growth polymerization via the Z-group approach was developed with a symmetrical trithiocarbonate as the CTA2 and a bismaleimide as the M2, resulting in an easy and efficient preparation of poly(trithiocarbonate)s. The in-chain dormant trithiocarbonate CTAs can be further utilized for chain expansion and as dynamic covalent bonds for deconstruction. Degradable PMA, PS and their multiblock block copolymers were successfully prepared by one step or two step chain expansion under 405 nm light. Segments generated after deconstruction showed controlled molecular weights and narrow molecular weight distributions. Moreover, these segments can be further repolymerized through the Z-group approach. We anticipate further expansion of the monomer scope for this new archetype of RAFT step-growth polymerization, considering many monomer and CTA pairings have already been successfully demonstrated through the former R-group approach. This will subsequently allow greater diversity of the step-growth backbone containing the in-chain CTAs and greater selection of vinyl monomers for chain-expansion. Thus, this new polymerization methodology can be considered as a general platform for preparing both degradable and recyclable yet functional vinyl polymers.Data availability
All experimental data is available in the ESI.†Author contributions
The manuscript was written through contributions of all authors.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was financially supported by the National Science Foundation (NSF) under Award CHE-2108670. The Bruker AVANCE III Nanobay 400 MHz NMR spectrometer was supported by the National Science Foundation under Grant No. CHE-0922858. Authors thank Dr Marc A. ter Horst and Dr Andrew Camp from the University of North Carolina's Department of Chemistry NMR Core Laboratory for the use of their NMR spectrometers.Notes and references
- J. Pan, X. Ai, C. Ma and G. Zhang, Acc. Chem. Res., 2022, 55, 1586–1598 CrossRef CAS
.
- M. R. Martinez and K. Matyjaszewski, CCS Chem., 2022, 4, 2176–2211 CrossRef CAS
- V. Delplace and J. Nicolas, Nat. Chem., 2015, 7, 771–784 CrossRef CAS PubMed
- W. Wang, Z. Zhou, D. Sathe, X. Tang, S. Moran, J. Jin, F. Haeffner, J. Wang and J. Niu, Angew. Chem., Int. Ed., 2022, 61, e202113302 CrossRef CAS PubMed
- H. Huang, B. Sun, Y. Huang and J. Niu, J. Am. Chem. Soc., 2018, 140, 10402–10406 CrossRef CAS PubMed
- R. J. A. Jos, M. J. Paulusse, R. A. Evans and C. J. Hawker, J. Am. Chem. Soc., 2009, 131, 9805–9812 CrossRef
- G. R. Kiel, D. J. Lundberg, E. Prince, K. E. L. Husted, A. M. Johnson, V. Lensch, S. Li, P. Shieh and J. A. Johnson, J. Am. Chem. Soc., 2022, 144, 12979–12988 CrossRef CAS PubMed
- N. Gil, B. Caron, D. Siri, J. Roche, S. Hadiouch, D. Khedaioui, S. Ranque, C. Cassagne, D. Montarnal, D. Gigmes, C. Lefay and Y. Guillaneuf, Macromolecules, 2022, 55, 6680–6694 CrossRef CAS
- P. Galanopoulo, N. Gil, D. Gigmes, C. Lefay, Y. Guillaneuf, M. Lages, J. Nicolas, M. Lansalot and F. D'Agosto, Angew. Chem., Int. Ed., 2022, 61, e202117498 CrossRef CAS PubMed
- R. A. Smith, G. Fu, O. McAteer, M. Xu and W. R. Gutekunst, J. Am. Chem. Soc., 2019, 141, 1446–1451 CrossRef CAS PubMed
- N. M. Bingham and P. J. Roth, Chem. Commun., 2018, 55, 55–58 RSC
- S. Tang, F. W. Seidel and K. Nozaki, Angew. Chem., Int. Ed., 2021, 60, 26506–26510 (
Angew. Chem.
, 133
, 26710–26714
) CrossRef CAS PubMed
- R. Hada, A. Kanazawa and S. Aoshima, Macromolecules, 2022, 55, 5474–5484 CrossRef CAS
- A. Kanazawa and S. Aoshima, Macromolecules, 2020, 53, 5255–5265 CrossRef CAS
- A. E. Neitzel, L. Barreda, J. T. Trotta, G. W. Fahnhorst, T. J. Haversang, T. R. Hoye, B. P. Fors and M. A. Hillmyer, Polym. Chem., 2019, 10, 4573–4583 RSC
- T. Shirouchi, A. Kanazawa, S. Kanaoka and S. Aoshima, Macromolecules, 2016, 49, 7184–7195 CrossRef CAS
- A. Kanazawa, S. Kanaoka and S. Aoshima, J. Am. Chem. Soc., 2013, 135, 9330–9333 CrossRef CAS PubMed
- Y. Ishido, R. Aburaki, S. Kanaoka and S. Aoshima, Macromolecules, 2010, 43, 3141–3144 CrossRef CAS
- C. Jehanno, J. Demarteau, D. Mantione, M. C. Arno, F. Ruiperez, J. L. Hedrick, A. P. Dove and H. Sardon, Angew. Chem., Int. Ed., 2021, 60, 6710–6717 CrossRef CAS PubMed
- M. Haussler, M. Eck, D. Rothauer and S. Mecking, Nature, 2021, 590, 423–427 CrossRef PubMed
- M. Mizutani, E. F. Palermo, L. M. Thoma, K. Satoh, M. Kamigaito and K. Kuroda, Biomacromolecules, 2012, 13, 1554–1563 CrossRef CAS PubMed
- M. Mizuntani, K. Satoh and M. Kamigaito, Macromolecules, 2011, 44, 2382–2386 CrossRef CAS
- K. S. Masato Mizutani and M. Kamigaito, J. Am. Chem. Soc., 2010, 132, 7498–7507 CrossRef
- M. Kamigaito, Polym. J., 2022, 54, 1391–1405 CrossRef CAS
- V. Bellotti, K. Parkatzidis, H. S. Wang, N. De Alwis Watuthanthrige, M. Orfano, A. Monguzzi, N. P. Truong, R. Simonutti and A. Anastasaki, Polym. Chem., 2023, 14, 253–258 RSC
- J. B. Young, J. I. Bowman, C. B. Eades, A. J. Wong and B. S. Sumerlin, ACS Macro Lett., 2022, 11, 1390–1395 CrossRef CAS PubMed
- H. S. Wang, N. P. Truong, Z. Pei, M. L. Coote and A. Anastasaki, J. Am. Chem. Soc., 2022, 144, 4678–4684 CrossRef CAS PubMed
- M. R. Martinez, F. De Luca Bossa, M. Olszewski and K. Matyjaszewski, Macromolecules, 2021, 55, 78–87 CrossRef
- Y. Sano, T. Konishi, M. Sawamoto and M. Ouchi, Eur. Polym. J., 2019, 120, 109181 CrossRef CAS
- M. J. Flanders and W. M. Gramlich, Polym. Chem., 2018, 9, 2328–2335 RSC
- L. Li, X. Shu and J. Zhu, Polymer, 2012, 53, 5010–5015 CrossRef CAS
- S. Yamamoto, T. Kubo and K. Satoh, J. Polym. Sci., 2022, 60, 3435–3446 CrossRef CAS
- J. B. Garrison, R. W. Hughes and B. S. Sumerlin, ACS Macro Lett., 2022, 11, 441–446 CrossRef CAS PubMed
- A. Adili, A. B. Korpusik, D. Seidel and B. S. Sumerlin, Angew. Chem., Int. Ed., 2022, 61, e202209085 CrossRef CAS PubMed
- T. Kimura, K. Kuroda, H. Kubota and M. Ouchi, ACS Macro Lett., 2021, 10, 1535–1539 CrossRef CAS PubMed
- M. Uchiyama, Y. Murakami, K. Satoh and M. Kamigaito, Angew. Chem., Int. Ed., 2022, e202215021 Search PubMed
- K. Parkatzidis, H. S. Wang, N. P. Truong and A. Anastasaki, Chem, 2020, 6, 1–14 Search PubMed
- M. D. Nothling, Q. Fu, A. Reyhani, S. Allison-Logan, K. Jung, J. Zhu, M. Kamigaito, C. Boyer and G. G. Qiao, Adv. Sci., 2020, 2001656 CrossRef CAS PubMed
- N. Corrigan, K. Jung, G. Moad, C. J. Hawker, K. Matyjaszewski and C. Boyer, Prog. Polym. Sci., 2020, 111, 101311 CrossRef CAS
- L. Zhang, Q. Wang, P. Lei, X. Wang, C. Wang and L. Cai, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2617–2623 CrossRef CAS
- P. Lei, Q. Wang, J. Hong and Y. Li, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 6600–6606 CrossRef CAS
- J. Hong, Q. Wang and Z. Fan, Macromol. Rapid Commun., 2006, 27, 57–62 CrossRef CAS
- J. Hong, Q. Wang, Y. Lin and Z. Fan, Macromolecules, 2005, 38, 2691–2695 CrossRef CAS
- T. Gegenhuber, A. M. Schenzel, A. S. Goldmann, P. B. Zetterlund and C. Barner-Kowollik, Chem. Commun., 2017, 53, 10648–10651 RSC
- T. Gegenhuber, L. De Keer, A. S. Goldmann, P. H. M. Van Steenberge, J. O. Mueller, M.-F. Reyniers, J. P. Menzel, D. R. D’hooge and C. Barner-Kowollik, Macromolecules, 2017, 50, 6451–6467 CrossRef CAS
- Q. Li, X. Hu and R. Bai, Macromol. Rapid Commun., 2015, 36, 1810–1815 CrossRef CAS PubMed
- Y. Liu and K. A. Cavicchi, Macromol. Chem. Phys., 2009, 210, 1647–1653 CrossRef CAS
- Z. Zhang, N. Corrigan and C. Boyer, Angew. Chem., Int. Ed., 2022, 61, e202114111 (
Angew. Chem.
, 2021
, 134
, e202114111
) CrossRef CAS PubMed
- S. Häkkinen, B. Dyer, A. Kerr and S. Perrier, Polym. Chem., 2022, 13, 479–484 RSC
- M. Chen, Y. Gu, A. Singh, M. Zhong, A. M. Jordan, S. Biswas, L. T. Korley, A. C. Balazs and J. A. Johnson, ACS Cent. Sci., 2017, 3, 124–134 CrossRef CAS PubMed
- M. B. Gordon, J. M. French, N. J. Wagner and C. J. Kloxin, Adv. Mater., 2015, 27, 8007–8010 CrossRef CAS PubMed
- Y. Amamoto, J. Kamada, H. Otsuka, A. Takahara and K. Matyjaszewski, Angew. Chem., Int. Ed., 2011, 50, 1660–1663 CrossRef CAS
- R. Nicolaÿ, J. Kamada, A. Van Wassen and K. Matyjaszewski, Macromolecules, 2010, 43, 4355–4361 CrossRef
- C. H. Chan, J.-T. Chen, W. S. Farrell, C. M. Fellows, D. J. Keddie, C. K. Luscombe, J. B. Matson, J. Merna, G. Moad, G. T. Russell, P. Théato, P. D. Topham and L. Sosa Vargas, Polym. Chem., 2022, 13, 2262–2270 RSC
- C. Boyer, M. Kamigaito, K. Satoh and G. Moad, Prog. Polym. Sci., 2023, 101648 Search PubMed
- J. Xu, Macromolecules, 2019, 52, 9068–9093 CrossRef CAS
- Z. Li, J. Li, B. Zhao, X. Pan, X. Pan and J. Zhu, Chin. J. Chem., 2023, 41, 503–508 CrossRef CAS
- Z. Li, J. Li, X. Pan, Z. Zhang and J. Zhu, ACS Macro Lett., 2022, 11, 230–235 CrossRef CAS PubMed
- S. M. Clouthier, J. Tanaka and W. You, Polym. Chem., 2022, 13, 6114–6119 RSC
- P. Boeck, N. Archer, J. Tanaka and W. You, Polym. Chem., 2022, 13, 2589–2594 RSC
- N. E. Archer, P. T. Boeck, Y. Ajirniar, J. Tanaka and W. You, ACS Macro Lett., 2022, 11, 1079–1084 CrossRef CAS
- J. Tanaka, N. E. Archer, M. J. Grant and W. You, J. Am. Chem. Soc., 2021, 143, 15918–15923 CrossRef CAS
- T. Endo, N. Aoyagi, B. Ochiai and H. Mori, Synlett, 2006, 636–638 CrossRef
- P. J. Flory, J. Am. Chem. Soc., 1936, 58, 1877–1885 CrossRef CAS
- M. Hartlieb, Macromol. Rapid Commun., 2022, 43, e2100514 CrossRef
- Y. Lu, L. An and Z.-G. Wang, Macromolecules, 2013, 46, 5731–5740 CrossRef CAS
- G. Gody, T. Maschmeyer, P. B. Zetterlund and S. Perrier, Nat. Commun., 2013, 4, 2505 CrossRef PubMed
- A. Postma, T. P. Davis, G. Moad and M. S. O'Shea, Macromolecules, 2005, 38, 5371–5374 CrossRef CAS
- https://www.matweb.com/search/DataSheet.aspx?MatGUID=df6b1ef50ce84e7995bdd1f6fd1b04c9%26ckck=1 .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc06736j |
| This journal is © The Royal Society of Chemistry 2024 |