
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructure-aware dual-target drug design through collaborative learning of pharmacophore combination and molecular simulation†
Sheng
Chen
 ab,
Junjie
Xie
ab,
Renlong
Ye
b,
David Daqiang
Xu
b and
Yuedong
Yang
*a
ab,
Junjie
Xie
ab,
Renlong
Ye
b,
David Daqiang
Xu
b and
Yuedong
Yang
*a
aSchool of Computer Science and Engineering, Sun Yat-sen University, Guangzhou 510006, China
bAixplorerBio Inc., Jiaxing 314031, China. E-mail: yangyd25@mail.sysu.edu.cn
First published on 13th June 2024
Abstract
Dual-target drug design has gained significant attention in the treatment of complex diseases, such as cancers and autoimmune disorders. A widely employed design strategy is combining pharmacophores to leverage the knowledge of structure–activity relationships of both targets. Unfortunately, pharmacophore combination often struggles with long and expensive trial and error, because the protein pockets of the two targets impose complex structural constraints. In this study, we propose AIxFuse, a structure-aware dual-target drug design method that learns pharmacophore fusion patterns to satisfy the dual-target structural constraints simulated by molecular docking. AIxFuse employs two self-play reinforcement learning (RL) agents to learn pharmacophore selection and fusion by comprehensive feedback including dual-target molecular docking scores. Collaboratively, the molecular docking scores are learned by active learning (AL). Through collaborative RL and AL, AIxFuse learns to generate molecules with multiple desired properties. AIxFuse is shown to outperform state-of-the-art methods in generating dual-target drugs against glycogen synthase kinase-3 beta (GSK3β) and c-Jun N-terminal kinase 3 (JNK3). When applied to another task against retinoic acid receptor-related orphan receptor γ-t (RORγt) and dihydroorotate dehydrogenase (DHODH), AIxFuse exhibits consistent performance while compared methods suffer from performance drops, leading to a 5 times higher performance in success rate. Docking studies demonstrate that AIxFuse can generate molecules concurrently satisfying the binding mode required by both targets. Further free energy perturbation calculation indicates that the generated candidates have promising binding free energies against both targets.
The paradigm of targeted-drug design has been dominated by the “one target, one drug” concept.1 However, single-target drugs are prone to drug resistance. For complex diseases like cancers and autoimmune disorders, current single-target therapy struggles to achieve long-term therapeutic effects.2,3 To engage different therapeutic targets simultaneously, combination therapy has been widely employed to produce additive or synergistic effects by using multiple drugs that act on different targets.4–6 Nevertheless, using multiple drugs involves drug–drug interactions, dose-limiting toxicities, unpredictable pharmacokinetics, adverse off-target effects, and poor patient compliance.7 A promising strategy to overcome these limitations is the design of dual/multi-target drugs.8–10 In recent years, there has been an increasing trend towards dual/multi-target drugs in FDA-approved medications, particularly in the treatment of malignant tumors and nervous system diseases, as well as digestive system and metabolic diseases.11,12
Towards rational design of dual-target drugs, the pharmacophore combination strategy has been widely employed.10,13–17 It includes simple “linking” of two distinct pharmacophores with a linker, “merging” of pharmacophores with shared fragments, and “fusing” of pharmacophores. The “linking” approach often results in higher molecular weight and is prone to suffering from pharmacokinetic issues.10,13 The “merging” approach is limited to targets that share some binding modes.14,15 The “fusing” approach allows tight joining of active fragments and offers the opportunity to achieve favorable physicochemical properties in the resulting molecules.16,17 Designing dual-target drugs through pharmacophore combination allows for effective integration of structure–activity relationship (SAR) knowledge for both targets.2 However, finding correct pharmacophore combination patterns is often hindered by long and expensive trial and error, mainly due to the complex structural constraints imposed by the protein pockets of the dual targets. Therefore, it is valuable to develop dual-target drug design methods that improve the efficiency and success rate.
Computational drug design methods have been developed over several decades to generate molecules with desired properties.18,19 Traditional molecular generation primarily navigated the chemical-space exploration through population-based stochastic optimization procedures, such as evolutionary algorithms (EA)20 or swarm intelligence.21 In recent years, deep learning algorithms have been widely employed in molecular generation.22,23 Early studies such as those on ChemVAE,24 CVAE25 and Heteroencoder26 generated SMILES strings as molecular representation. On the other hand, Liu et al.27 and You et al.28 made exploratory attempts to generate molecules represented by the graph, whose node-edge structure naturally matches the atom-bond structure of molecules. However, most of them focused on non/single-objective generation and thus are not suitable for dual-target drug design. There are only a handful of multi-objective molecular generation algorithms.29–33 Li et al.33 trained machine learning (ML) models as approximate empirical measurements of activity against glycogen synthase kinase-3 beta (GSK3β) and c-Jun N-terminal kinase 3 (JNK3). Following their work, related methods (e.g. RationaleRL,31 MARS29) employed the same activity predictors in their methods and assessments. The utilization of ML predictors brought up two issues. Firstly, the generalizability of the ML model cannot be guaranteed when available activity data are limited. Secondly, the ML model based on molecular fingerprint representation is not structurally interpretable. Compared with the ML-based activity predictor, molecular-simulation-based computational tools such as molecular docking and free energy perturbation (FEP) are more generalizable and structurally interpretable. Since molecular simulation is less computationally efficient, recent efforts have been made on molecular docking34,35 and FEP36,37 by active learning (AL) to improve the efficiency of virtual screening. Therefore, integrating multi-objective molecular generation with active learning on molecular-simulation-based activity estimation is a promising way to improve generalizability and structural interpretability.
However, combining molecule generation with AL presents another challenge. In the context of virtual screening, the target domain for AL is well-defined, given that the compound library is known and readily available. In contrast, when it comes to molecule generation, the target domain for AL remains elusive because the molecules within this domain haven't been generated. This dilemma is akin to training an artificial intelligence algorithm for the GO game in the absence of any historical chess records. AlphaZero38 has masterfully navigated this very conundrum by self-play with reinforcement learning (RL) and Monte Carlo tree search (MCTS). Inspiringly, searching pharmacophore combination patterns to satisfy the dual-target structural constraints can also be modeled as two self-play MCTS agents against two targets. Through self-play pharmacophore combination, target domain molecules can be generated for AL training, and the trained models can subsequently provide feedback to navigate the molecular generation.
In this study, we propose AIxFuse, a structure-aware dual-target drug design method. To our knowledge, this is the first approach that learns pharmacophore fusion patterns to satisfy the dual-target structural constraints simulated by molecular docking. In AIxFuse, pharmacophores are extracted automatically through protein–ligand interaction analysis of active compounds and target proteins. The pharmacophore-fusion chemical space is represented by multi-level molecular sub-structure trees. Exploration in this chemical space towards multiple desired properties (i.e. binding affinity, drug-likeness, and synthetic accessibility) is navigated by an actor–critic-like39 RL framework. The RL framework consists of two self-play MCTS actors for molecule generation and a dual-target docking score critic trained by AL. After iterations of collaborative RL and AL, AIxFuse learns to generate molecules with multiple desired properties. AIxFuse was first evaluated on designing a dual-inhibitor for GSK3β and JNK3, where it showed outperformance (32.3% relative improvement in success rate) compared to other state-of-the-art (SOTA) methods. We also applied AIxFuse to another task that aims to design dual inhibitors against retinoic acid receptor-related orphan receptor γ-t (RORγt) and dihydroorotate dehydrogenase (DHODH).3 Consistently, AIxFuse achieves the best success rate of 23.96%, over 5 times higher than that of other methods. Docking studies demonstrated that AIxFuse can generate molecules that concurrently satisfy the binding mode required by both targets. Furthermore, as revealed by FEP, a generated candidate exhibits better/comparable binding free energy with known inhibitors on both targets, demonstrating its dual-target activity potential. This holds promising prospects for AIxFuse to accelerate dual-target drug design in real-world drug development scenarios.
Results
Fig. 1 displays the overall pipeline of AIxFuse. First, the targeting protein structures and known active compounds are collected for both targets. Subsequently, their protein–ligand complex structures are simulated using molecular docking by Glide.40 From the complex structures, protein–ligand interactions (PLIs) are extracted using the PLIP41,42 program. The extracted PLIs are then combined to define the pharmacophores (Fig. 1B), which are ranked based on interaction scores, frequency scores, etc. The top-scored pharmacophores split each compound into core and side chain fragments. These fragments can be rearranged by two searching trees (Fig. 1C), where the leaf nodes of two trees will be fused to generate the pharmacophore-fused molecules. An actor–critic-like reinforcement learning (RL) framework is employed to learn the optimal pharmacophore fusion patterns. It consists of two self-play Monte Carlo Tree Searching (MCTS) actors and a dual-target docking score critic. The MCTS actors aim to maximize the upper confidence bounds (UCB) of the reward for the generated molecules. More specifically, the reward function encompasses various properties, including drug-likeness, synthetic accessibility, and dual-target docking scores. As a dual-target docking score critic, a multi-task AttentiveFP43 model is iteratively trained by the active learning (AL). After several iterations of collaborative RL and AL, the final models will be used to generate molecules.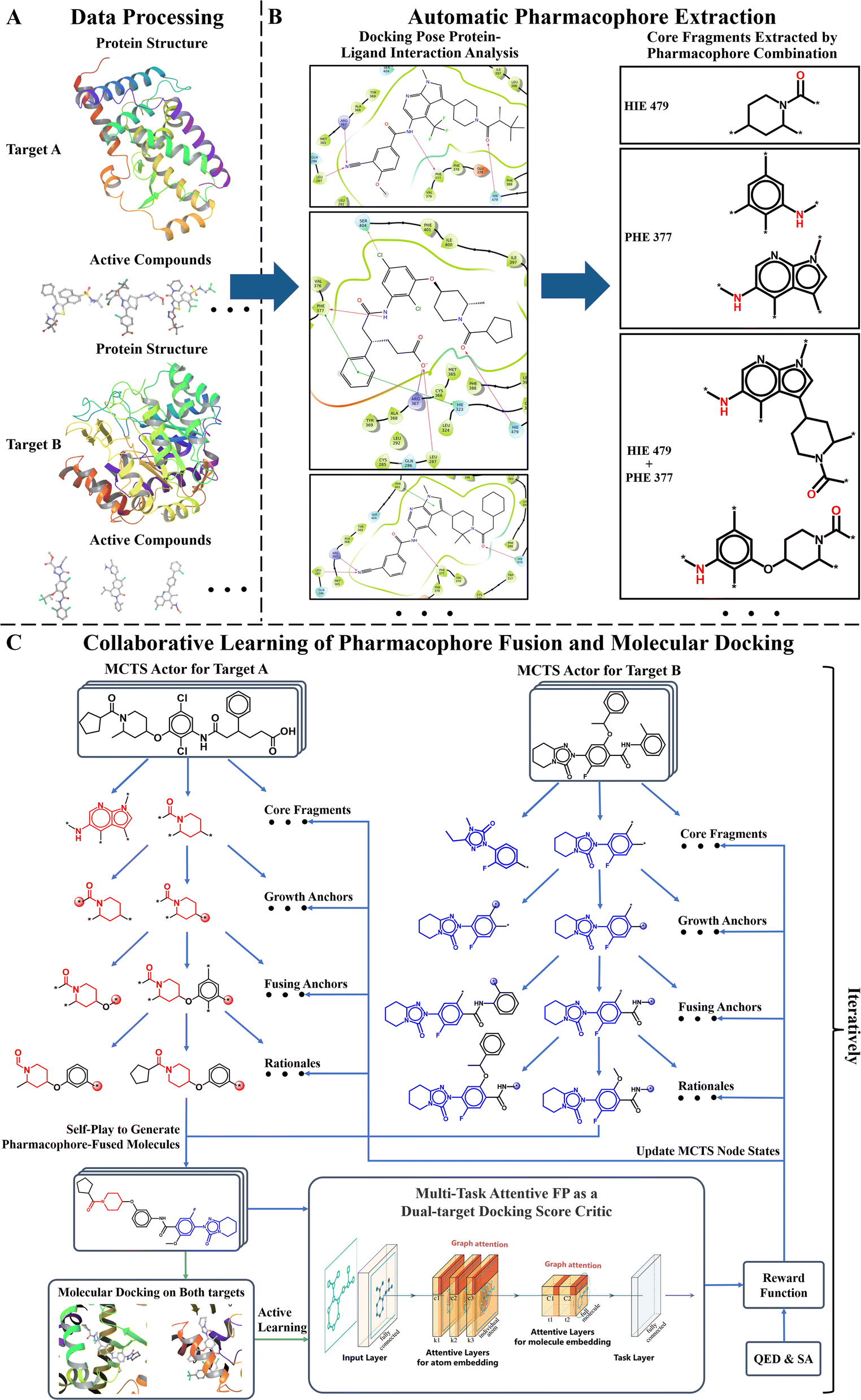 | ||
| Fig. 1 The AIxFuse pipeline encompasses three core stages: (A) data processing (example shown by RORγt and DHODH), entailing the preprocessing of protein structures and active compounds for both targets; (B) automatic pharmacophore extraction (example shown by RORγt), accomplished through automatic scrutiny of protein–ligand interactions within the docking poses of active compounds to isolate core fragments that preserve key pharmacophores; and (C) collaborative learning of pharmacophore fusion and molecular docking. The actor–critic-like reinforcement learning on pharmacophore fusion and the active learning on molecular docking are run collaboratively and iteratively. It employs two self-play MCTS actors for molecule generation and a dual-target docking score critic to navigate generation. The critic is a multi-task AttentiveFP (schematic diagram depicted by Xiong et al.43) and is trained by active learning. The green arrow lines represent the training procedure. | ||
AIxFuse outperformed previous SOTA methods on GSK3β|JNK3 dual-inhibitor design
AIxFuse was compared to state-of-the-art (SOTA) methods: RationaleRL,31 MARS,29 and REINVENT2.0.32 We ran each method to generate 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 molecules for designing dual inhibitors against glycogen synthase kinase-3 beta (GSK3β) and c-Jun N-terminal kinase 3 (JNK3), a classical benchmark for dual-target drug design.29,31,33
000 molecules for designing dual inhibitors against glycogen synthase kinase-3 beta (GSK3β) and c-Jun N-terminal kinase 3 (JNK3), a classical benchmark for dual-target drug design.29,31,33
All methods were first assessed in terms of three general metrics for molecular generation: validity, uniqueness, and diversity. As shown in Table 1, all methods achieved a validity rate exceeding 98%, indicating their ability to generate molecules that conform to essential chemical constraints. Notably, significant differences were observed in the uniqueness metric. AIxFuse demonstrated the highest uniqueness at 89.7%, surpassing other previous methods, including REINVENT2.0 (82.6%), RationaleRL (53.3%), and MARS (24.1%). While fragment-based methods often struggle with diversity, AIxFuse obtained a score of 0.719, outperforming RationaleRL (0.656) and MARS (0.597), and matching the performance of REINVENT2.0 (0.722). This unexpected success may be attributed to the upper confidence bounds (UCB) of reward expectations in MCTS, which aims to strike a balance between sampling frequency and reward expectations.
| Metrics | REINVENT2.0 | RationaleRL | MARS | AIxFuse |
|---|---|---|---|---|
| a Success rate is defined as the proportion of generated molecules that obtain better multiple-property values than the average of active compounds. b USR (unique success rate) denotes the proportion of non-repeating molecules that obtain better single-property values than the average of active compounds. | ||||
| Validity (%) | 98.9 | 99.9 | 100 | 100 |
| Uniqueness (%) | 82.6 | 53.3 | 24.1 | 89.7 |
| Diversity | 0.722 | 0.656 | 0.597 | 0.719 |
| Success ratea (%) | 1.75 | 11.95 | 17.83 | 23.59 |
| USR QEDb (%) | 14.77 | 46.84 | 24.05 | 37.64 |
| USR SA (%) | 55.45 | 16.39 | 17.02 | 55.66 |
| USR dockingGSK3β (%) | 19.75 | 38.47 | 11.90 | 81.74 |
| USR dockingJNK3 (%) | 16.73 | 23.94 | 6.63 | 78.20 |
| FCD (GSK3β|JNK3) | 29.5|30.8 | 28.2|27.8 | 53.8|46.8 | 16.7|24.9 |
| SNN (GSK3β|JNK3) | 0.35|0.41 | 0.38|0.44 | 0.36|0.45 | 0.47|0.45 |
We then checked the ability of each method to generate molecules with multiple desired properties. The success rate is defined as the proportion of generated molecules that outperform the mean values of known active compounds in multiple properties. These properties include dual-target docking scores, Quantitative Estimate of Drug-likeness (QED), and synthetic accessibility (SA). As shown in Table 1, AIxFuse achieved the highest success rate at 23.59%, outperforming the second-place method MARS (17.83%) with a relative improvement of 32.3%. This result demonstrated AIxFuse's capability to generate molecules that simultaneously satisfy multiple property constraints. To evaluate the performance of each individual property, we calculated the unique success rate (USR), which represents the proportion of non-repeating molecules satisfying a specific property constraint. The baseline model AIxFuse(w/o RLAL) was constructed by replacing RL and AL with random sampling on both trees. As shown in Fig. 2, baseline model AIxFuse(w/o RLAL) exhibited a promising USR in docking scores, but its USRs in SA and QED were not optimal. By incorporating RL and AL, AIxFuse achieved the best USR in SA (55.66%) and the second-highest USR in QED (37.64%), demonstrating significant improvements to AIxFuse(w/o RLAL). Furthermore, AIxFuse achieved the highest USR on the docking scores of GSK3β (81.74%) and JNK3 (78.20%), surpassing other methods by a significant margin.
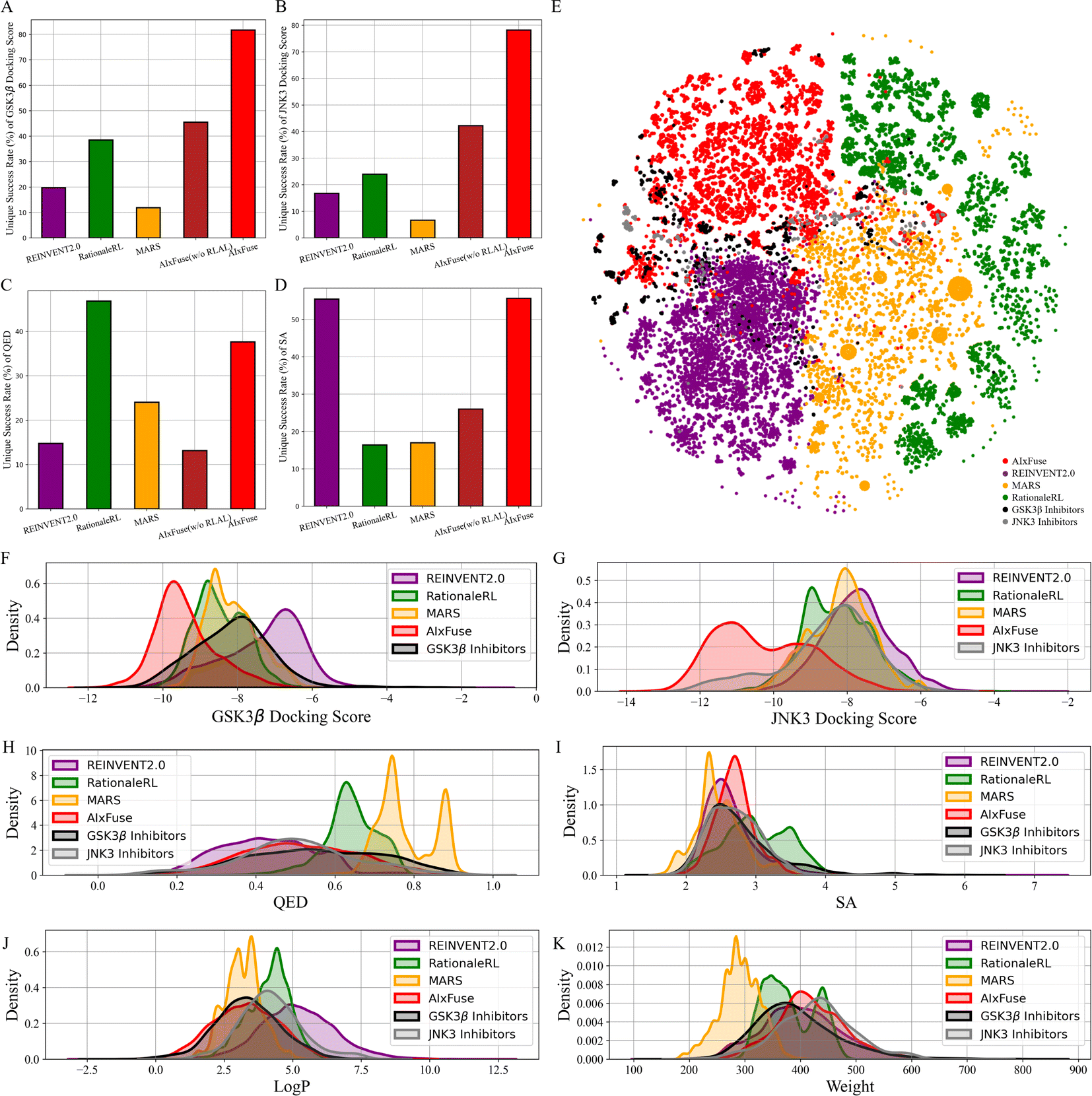 | ||
| Fig. 2 Statistics and visualization of molecules generated for GSK3β|JNK3 dual-inhibitor design: the unique success rate of (A) GSK3β docking score, (B) JNK3 docking score, (C) QED, and (D) SA; (E) the t-SNE visualization of Morgan fingerprints of known inhibitors and generated molecules; the property distributions of (F) GSK3β docking score, (G) JNK3 docking score, (H) QED, (I) SA, (J) logP, and (K) molecular weight. | ||
We were interested in studying the similarity between generated molecules and active compounds. As shown in Table 1, AIxFuse consistently achieved the lowest Fréchet ChemNet Distance (FCD) and the highest Similarity to Nearest Neighbor (SNN) against active compounds of both targets. This is as expected since AIxFuse preserved key pharmacophores of active compounds to incorporate the SARs of both targets. To gain an intuitive understanding of similarity, Fig. 2E visualizes the chemical spaces of generated molecules and active compounds. The chemical space of GSK3β inhibitors closely resembled that of JNK3 inhibitors. Notably, AIxFuse explored the gaps between the main clusters of known inhibitors, while other methods didn't. Molecules generated within the chemical space gaps between the inhibitors of the two targets are likely to exhibit high similarity to both ends. Looking deeper into the molecular property similarity between generated molecules and active compounds, we plotted the property distributions in Fig. 2F–K. It can be found that the QED, SA, logP, and weight distribution of AIxFuse-generated molecules closely aligned with those of GSK3β inhibitors and JNK3 inhibitors. However, for both GSK3β and JNK3 docking scores, AIxFuse-generated molecules were distributed towards the lower end of the spectrum compared to the corresponding known inhibitors. It demonstrated that AIxFuse-generated molecules obtained better estimated binding affinities against both targets while keeping other properties similar to those of the known inhibitors.
What contributed to the outperformance of AIxFuse? We first explored the contribution of running multiple iterations of AL. As shown in Fig. 3A and B, as AL iterated, the overall MSE decreased while R2 increased. This trend demonstrated that multiple iterations of AL contribute to the accuracy of the dual-target docking score critic. We then presented the accuracy of the final dual-target docking score critic in Fig. 3C and D. The final Pearson correlation coefficients indicated strong positive correlations between the predicted docking scores and the ground truth (0.691 and 0.704 for GSK3β and JNK3, respectively). This affirmed the models' robust ranking ability, which is an essential factor for molecular generation to improve molecular properties. We then investigated the contribution of RL optimization. As is shown in Fig. 3E–H, the docking score distribution of AIxFuse(w/o RLAL) closely resembles that of known active molecules. This suggests that our automatically extracted pharmacophores can effectively preserve the essential SARs of both targets. However, AIxFuse(w/o RLAL)'s QED distribution and SA distribution are not ideal. After iterations of collaborative RL and AL, AIxFuse achieves improvements in all four properties. These phenomena underscore the synergistic effect of collaborative RL and AL: AL trained dual-target docking score critic for RL optimization, while RL provided molecules with higher quality for AL training.
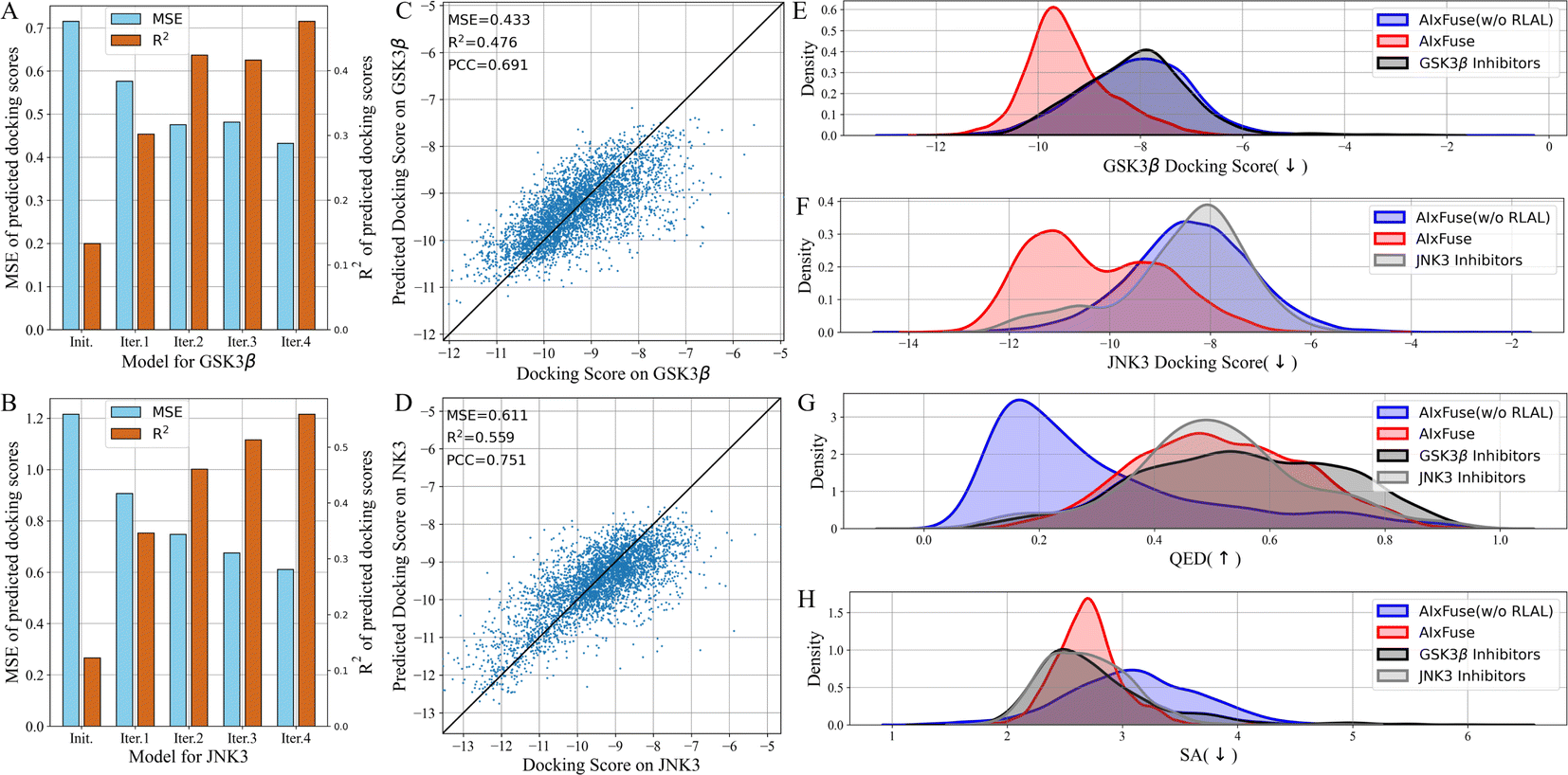 | ||
| Fig. 3 The ablation study of multiple iterations of AL: the MSE and R2 of docking scores on (A) GSK3β and (B) JNK3 predicted by models at various iteration steps; the scatter plot illustrating the correlation between (C) GSK3β and (D) JNK3 docking scores and predicted docking scores at final iteration; the ablation study of collaborative RL and AL: the property distribution of (E) GSK3β docking scores, (F) JNK3 docking scores, (G) QED, and (H) SA of molecules generated by AIxFuse(w/o RLAL), AIxFuse, and active compounds of GSK3β and JNK3. | ||
AIxFuse achieved consistent outperformance on RORγt|DHODH dual-inhibitor design
We then applied AIxFuse to another dual-target design task, where there is a precedent for rational design. Chen et al. previously designed a potential dual-target inhibitor, (R)-14d,3 achieving IC50 values of 0.110 μM for RORγt (retinoic acid receptor-related orphan receptor γ-t) and 0.297 μM for DHODH (dihydroorotate dehydrogenase). Following the evaluation setting of the GSK3β|JNK3 benchmark task, we ran AIxFuse, RationaleRL, MARS, and REINVENT2.0 to generate 10000 molecules for RORγt|DHODH dual-inhibitor design.
Compared with other methods, AIxFuse demonstrated superior performance in various evaluation metrics, as depicted in Table 2. Specifically, AIxFuse achieved the highest validity, uniqueness, success rate, USR QED, USR SA, USR dockingRORγt, USR dockingDHODH, USR 3D SNNRORγt, and USR 3D SNNDHODH. In terms of diversity, that achieved by AIxFuse is lower than that of REINVENT2.0, comparable with that of RationaleRL, and far higher than that of MARS. When compared across benchmark tasks, AIxFuse exhibited consistent results, whereas other methods displayed fluctuations. Fig. 4A highlights that AIxFuse achieved a success rate of 23.96%, consistent with its performance in the GSK3β|JNK3 task (23.59%, Fig. 4B). Conversely, MARS failed to generate any successful molecules against RORγt|DHODH, resulting in a success rate drop (17.83% → 0%). A similar decrease in success rate occurred in RationaleRL (11.95% → 1.16%). Both MARS and RationaleRL rely on machine learning (ML) activity predictors. However, due to the limited availability of public non-active data for DHODH, training an ML predictor posed challenges (detailed in ESI Section 1†). In contrast, AIxFuse replaced ML-based activity predictors with molecular docking, which should contribute to its consistent performance.
| Metrics | REINVENT2.0 | RationaleRL | MARS | AIxFuse |
|---|---|---|---|---|
| a 3D SNN measures the maximum 3D similarity to known active compounds, while USR 3D SNN is the proportion of non-repeating compounds that obtained higher 3D SNN than dual-target inhibitor (R)-14d.3 | ||||
| Validity (%) | 100 | 99.9 | 100 | 100 |
| Uniqueness (%) | 33.5 | 33.7 | 9.9 | 94.1 |
| Diversity | 0.708 | 0.672 | 0.530 | 0.661 |
| Success rate (%) | 4.65 | 1.16 | 0.00 | 23.96 |
| USR QED (%) | 25.94 | 1.39 | 9.83 | 41.53 |
| USR SA (%) | 31.39 | 33.58 | 9.77 | 82.41 |
| USR dockingRORγt (%) | 21.60 | 23.56 | 1.67 | 85.04 |
| USR dockingDHODH (%) | 4.94 | 16.54 | 0.02 | 72.74 |
| USR 3D SNNRORγta (%) | 20.33 | 12.83 | 3.17 | 58.18 |
| USR 3D SNNDHODH (%) | 22.81 | 10.71 | 6.95 | 83.87 |
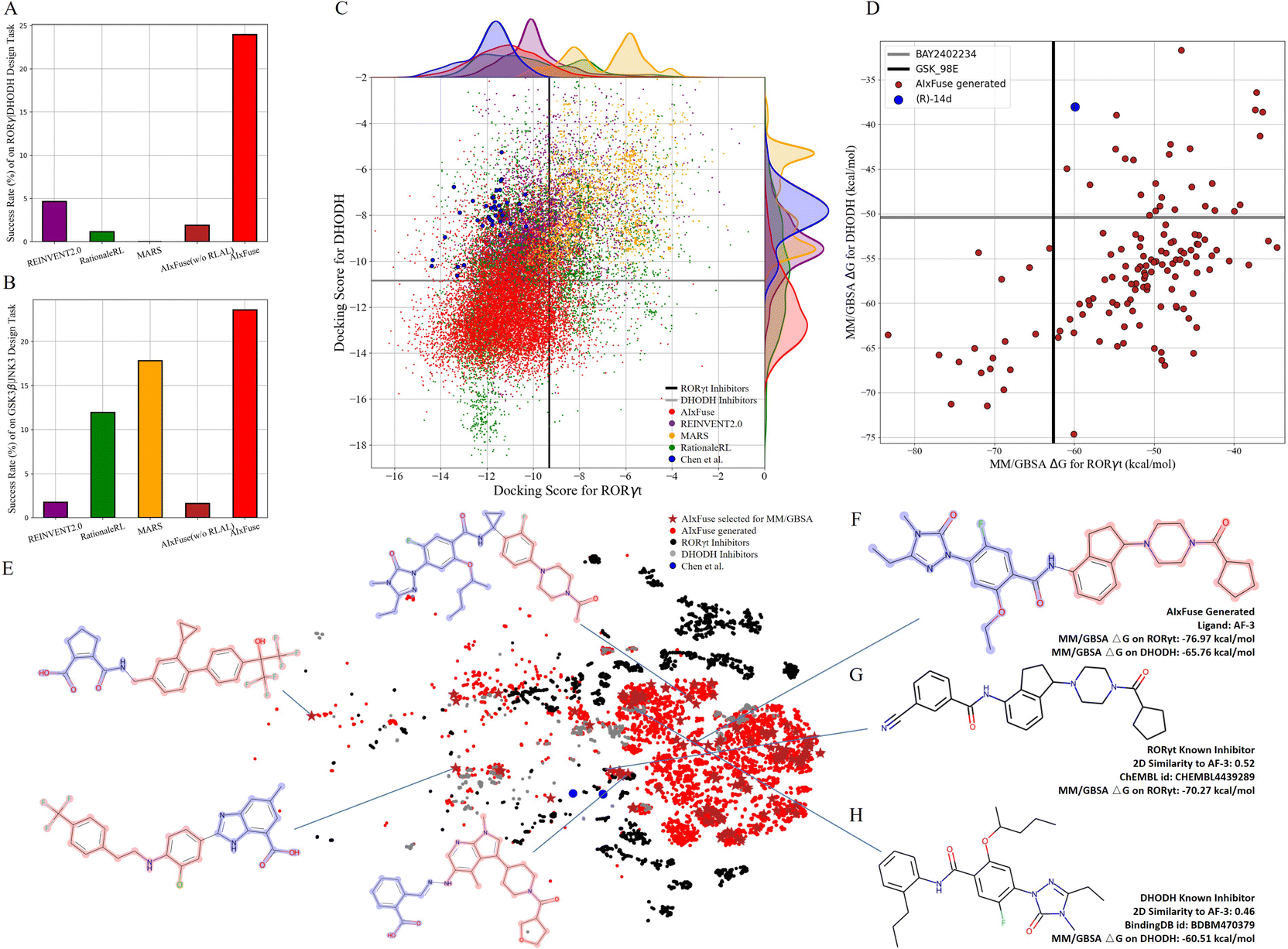 | ||
| Fig. 4 The performance of AIxFuse on RORγt|DHODH dual-inhibitor design: the success rates of AIxFuse and compared methods on (A) RORγt|DHODH dual-inhibitor design with (B) GSK3β|JNK3 task as a reference; the scatter plot of dual-target docking scores (C) obtained by AIxFuse (red), REINVENT2.0 (purple), MARS (yellow), and RationaleRL (green) with compounds designed by Chen et al. as references (blue); (D) the dual-target MM/GBSA binding free energies of 134 AIxFuse-generated molecules (brick red) with dual-target inhibitor (R)-14d (blue) as a reference; the t-SNE visualization (E) of AIxFuse-generated molecules that are selected for MM/GBSA calculation with molecular structure of some examples; (F) the molecular structure and MM/GBSA binding free energies of AIxFuse-generated drug candidate AF-3 with its most similar RORγt inhibitor (G) CHEMBL4438289 and its most similar DHODH inhibitor (H) BDBM470379. | ||
We then checked the dual-target activity potential of AIxFuse-generated molecules. As shown in Fig. 4C, AIxFuse's docking score distributions on both targets consistently favored the lower (better) end of the spectrum. A majority of AIxFuse-generated molecules exhibited lower RORγt and DHODH docking scores than the average score of known RORγt inhibitors and DHODH inhibitors respectively. When referring to compounds designed by Chen et al., AIxFuse also successfully generated numerous molecules with comparable RORγt docking scores and lower DHODH docking scores. This observation indicates promising dual-target activity potential. For a more accurate estimation of protein–ligand binding free energies, we employed the Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) calculation.44Fig. 4D illustrates the computation results. When referring to the GSK_98E (active ligand in the co-crystal structure of RORγt, PDB id: 5NTP45), 17 out of 134 AIxFuse-generated molecules obtained lower binding free energies. All these 17 candidates achieved lower binding free energies than BAY2402234 (active ligand in the co-crystal structure of DHODH, PDB id: 6QU7 (ref. 46)). Additionally, we performed a t-SNE visualization of the chemical space of these generated molecules in Fig. 4E. We also displayed the structures of 5 representative molecules, showcasing their notable structural diversity. Looking into a detailed case, AF-3 (structure shown in Fig. 4F) exhibited dual-target activity potential with MM/GBSA binding free energies of −76.97 kcal mol−1 on RORγt and −65.76 kcal mol−1 on DHODH. We also displayed structures of AF-3's most similar RORγt inhibitor (ChEMBL id: CHEMBL4438289) and most similar DHODH inhibitor (BindingDB id: BDBM470379) in Fig. 4G and H, respectively. Notably, the left half structure (LHS) of CHEMBL4438289 showed a similar pattern to the LHS of BDBM470379. AF-3 successfully merged this pattern by fusing the RORγt rationale (red in Fig. 4F) and the DHODH rationale (blue in Fig. 4F). This fusion likely contributes to its lower MM/GBSA binding free energies on both targets.
Binding mode and affinity study on the AIxFuse-generated dual-target drug candidate against RORγt|DHODH
Absolute protein–ligand binding free-energy calculation by free energy perturbation (ABFEP)47 is one of the most reliable in silico approaches. ABFEP calculation has achieved a remarkable root mean square error of 1.1 kcal mol−1 after zero-point shifting to match the experimental values in a previous study.48 Therefore we employed ABFEP calculation as the last process of molecular screening. As shown in Fig. 5A, we selected 17 molecules with balanced dual-target MM/GBSA binding free energies for ABFEP calculations. Among them, we found AF-5, AF-20, AF-3, and AF-16 with binding free energies comparable to those of the active molecules for both targets (detailed in ESI Table S6†). Finally, AF-5 was chosen as the top dual-target drug candidate as it exhibited the most promising dual-target activity potential.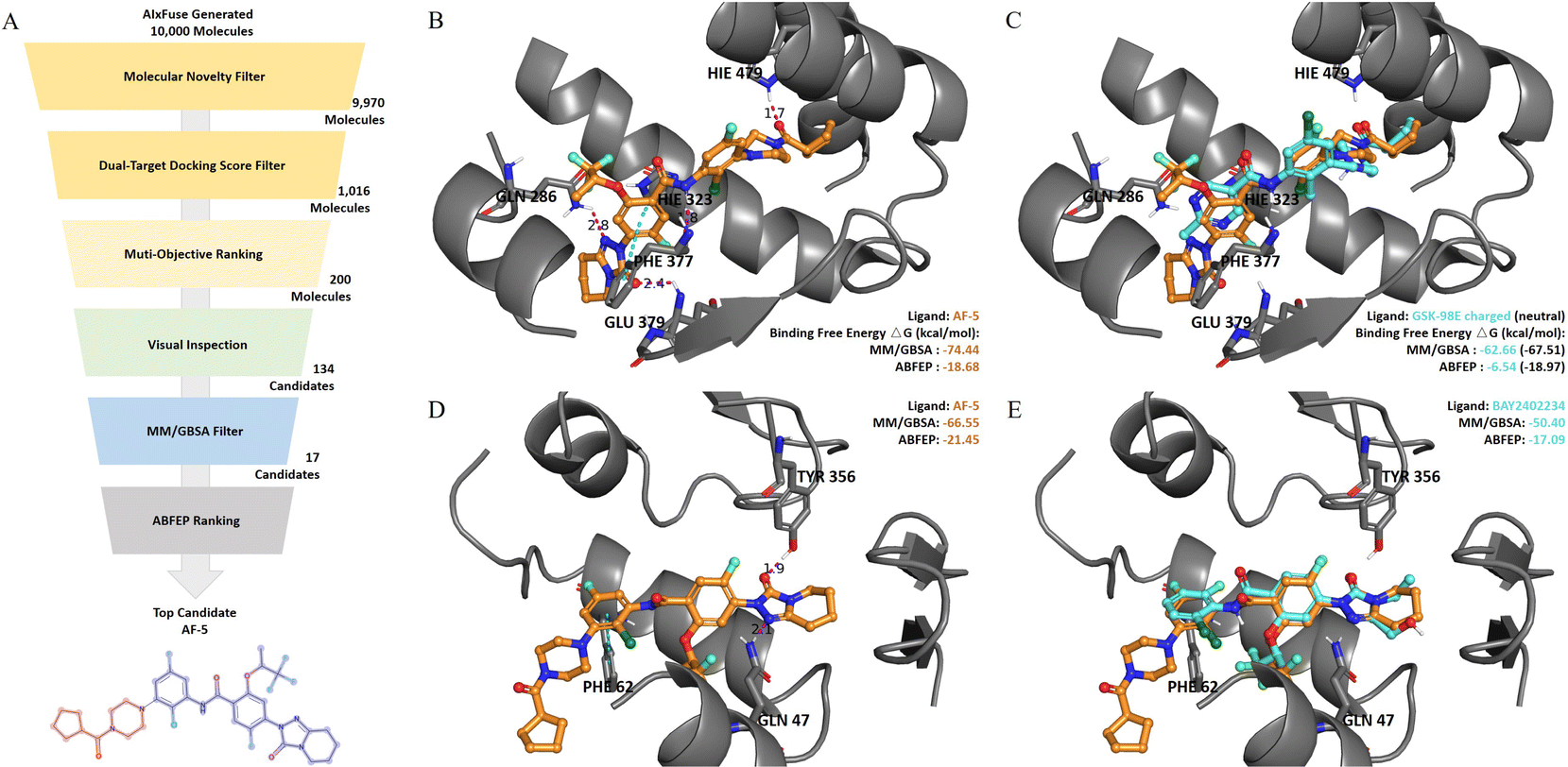 | ||
| Fig. 5 (A) The virtual screening process on AIxFuse-generated molecules. (B) The binding mode of AIxFuse generated compound AF-5 (orange stick) with RORγt-LBD revealed by the docking study. (C) Overlay of AF-5 (orange stick) with RORγt-LBD co-crystal ligand GSK-98E (cyan stick). (D) Binding mode of AF-5 (orange stick) with DHODH. (E) Overlay of AF-5 (orange stick) with DHODH co-crystal ligand BAY2402234 (cyan stick). The pink dotted lines represent hydrogen bonds, and the cyan dotted lines represent π–π stacking. | ||
We first studied the binding mode of AF-5 on both targets by molecular docking. As shown in Fig. 5B, when docking into RORγt (PDB id: 5NTP), AF-5 established hydrogen bonds and π–π stacking interactions with key residues, including HIE479, PHE377, GLU379, HIE323, and GLN 286. When docking into DHODH (PDB id: 6QU7, Fig. 5D), AF-5 exhibited hydrogen bonds and π–π stacking between AF-5 and key residues GLN47, TYR356, and PHE62. AF-5's binding modes on both targets matched previous studies on corresponding known inhibitors.3,45,46Fig. 5C and E illustrated that the docking pose of AF-5 (colored in orange) on both targets closely resembled those of known inhibitors in the co-crystal structure (RORγt inhibitor GSK-98E45 and DHODH inhibitor BAY2402234,46 both colored in cyan).
We proceeded to analyze the dual-target activity potential of AF-5 using ABFEP. AF-5 was compared with GSK-98E and BAY2402234 on their binding free energies against corresponding targets. According to its optimal protonation state at pH 7.0 ± 2.0, GSK-98E had a +1 net charge, while AF-5 was electrically neutral. The disparity in net charge would cause systematic errors. To account for this, we also calculated the binding free energy of the GSK-98E in the electrically neutral state. As depicted in Fig. 5C, AF-5 exhibited a lower RORγt binding free energy (−18.68 kcal mol−1) compared to the charged GSK-98E (−6.54 kcal mol−1) which was comparable to that of the neutral GSK-98E (−18.97 kcal mol−1). Regarding the other target, DHODH, AF-5's ABFEP binding free energy (−21.45 kcal mol−1) was significantly lower than that of BAY2402234 (−17.09 kcal mol−1). We also included additional references by supplementing the ABFEP calculation results of several other active molecules. As shown in ESI Table S6,† AF-5's RORγt binding free energy was comparable to those of known RORγt inhibitors, while its DHODH binding free energy was lower than those of known DHODH inhibitors. In summary, the in silico analysis demonstrated the potential of AF-5 as a dual-target inhibitor against RORγt and DHODH.
Discussion
Dual-target drug design is attractive but still challenging. Traditional pharmacophore-combination-based manual design often struggles with long and expensive trial and error. This is because the protein pockets of the two targets impose complex structural constraints on the pharmacophore combination. Therefore, it is important to develop computational methods that learn pharmacophore fusion patterns to satisfy the dual-target structural constraints. AIxFuse, as an innovative computational dual-target drug design method, has made solid progress on the challenging problem of improving the generalizability and structural interpretability of dual-target molecular generation. By utilizing molecular docking rather than ML-based activity prediction, AIxFuse exhibited enhanced generalizability (independent of the size of the training dataset). AIxFuse was also structurally interpretable, as its pharmacophores were extracted based on protein–ligand interaction and it optimized the target-specific binding affinities by molecular docking. AIxFuse makes pharmacophore fusion and molecular docking learnable through iterations of collaborative reinforcement learning and active learning. To our knowledge, AIxFuse is the first structure-aware dual-target drug design method that learns pharmacophore fusion patterns to satisfy the dual-target structural constraints simulated by molecular docking.AIxFuse was benchmarked on two dual-target drug design tasks and compared with previous SOTA methods. In the dual-target drug design task against GSK3β and JNK3, AIxFuse exhibited 32.3% relative improvement in success rate compared with the best of other methods. In the RORγt and DHODH dual-target inhibitor design task with limited activity data, AIxFuse achieves a success rate of 23.96% while compared methods suffered from performance drops, leading to a 5 times higher performance in success rate. Further in silico binding free-energy calculation results highlighted the dual-target activity potential of AIxFuse-generated drug candidates.
As a structure-aware dual-target drug design method, AIxFuse can generate potential dual-target hit compounds for subsequent lead optimization. For example, our latest structure-based scaffold decoration method DiffDec can be used for lead optimization on AIxFuse-generated molecules. In the future, active learning on FEP36,37 can also be introduced into AIxFuse to enhance the accuracy of binding affinity estimation. As the demand for dual-target drugs becomes significant in the treatment of complex diseases, AIxFuse can provide rapid and effective starting points for various dual-target design tasks.
Materials and methods
Benchmark curation
To assess the efficacy of our method, we established two dual-target inhibitor design benchmark tasks. The first benchmark involved the development of dual-target inhibitors against glycogen synthase kinase-3 beta (GSK3β) and c-Jun N-terminal kinase 3 (JNK3), denominated as the GSK3β|JNK3 benchmark. Notably, GSK3β and JNK3 have been associated with the pathogenesis of Alzheimer's disease (AD) and other disorders.49,50 The concept of designing dual inhibitors concurrently against GSK3β and JNK3 holds the potential to introduce a novel multi-target therapy for AD.33The second benchmark task aims to design dual-target inhibitors against retinoic acid receptor-related orphan receptor γ-t (RORγt) and dihydroorotate dehydrogenase (DHODH), named as the RORγt–DHODH benchmark. RORγt and DHODH have both emerged as attractive targets for the treatment of autoimmune diseases.51,52 Chen et al.3 advocated for the development of the RORγt|DHODH dual inhibitor as a promising strategy to enhance therapeutic efficacy, reduce toxicity, and mitigate drug resistance in the context of inflammatory bowel disease (IBD) therapy. They designed a series of compounds and identified one candidate with both in vitro and in vivo activities, along with favorable mouse pharmacokinetic profiles.
Evaluation settings
On the above two benchmarks, we compared AIxFuse with the following methods, and their implementation details are available in ESI Section 4:†• RationaleRL:31 RationaleRL begins by extracting rationales from active compounds via MCTS to retain essential activity-contributing elements. Subsequently, it employs the pharmacophore merge strategy to combine rationales, preserving the maximum common substructure. Finally, a molecular generation model is fine-tuned to produce side chain groups.
• MARS:29 MARS selects the top 1000 fragments from the ChEMBL dataset, ensuring that each fragment contains no more than 10 heavy atoms and appears most frequently. It employs Graph Neural Networks (GNN) and Markov Chain Monte Carlo (MCMC) sampling to manipulate these fragments and generate molecules with enhanced properties.
• REINVENT2.0:32 This method generates SMILES representations of molecules using a Recurrent Neural Network (RNN). Reinforcement learning is then applied to optimize the properties of the generated molecules, facilitating the generation of multi-target molecules through the application of multiple rewards.
We employed a diverse set of metrics to assess the performance of each method. Initially, we computed general metrics such as validity, uniqueness, and diversity using Moses.53 Additionally, we scrutinized properties including drug-likeness and synthesizability of the generated molecules and dual-target activity. Quantitative Estimate of Drug-likeness (QED) and synthetic accessibility (SA) were calculated using RDKit.54 Notably, for dual-target activity estimation, no public ML-based active predictor is available for the RORγt|DHODH task. Therefore, we opted for the molecular docking score as the approximation of binding affinity in both tasks. As a molecular-simulation-based method, molecular docking offers structural interpretability and has been widely used in drug design.35,55–58 Following the approach of Chen et al.,3 Glide40 software was employed to estimate binding modes and affinities. The co-crystal structures of RORγt LBD (PDB: 5NTP45), DHODH (PDB: 6QU7 (ref. 46)), GSK3β (PDB: 6Y9S59), and JNK3 (PDB: 4WHZ60) were selected and processed for docking preparation. The generated molecules were designed to concurrently satisfy multiple property constraints, prompting us to calculate the success rate, denoting the percentage of generated molecules meeting multiple property constraints concurrently. Diverging from prior works29,31 which set the thresholds of success as fixed values, we adopted the average value of active compounds as the threshold for each property. Those active compounds were collected from ChEMBL and BindingDB (details available in ESI Section 4†). For the GSK3β|JNK3 benchmark, the thresholds for QED, SA, and dual-target docking scores were set at 0.538, 2.76, −8.12, and −8.53 according to the average value of active compounds. For the RORγt|DHODH benchmark, the corresponding thresholds were 0.430, 3.49, −9.31, and −10.8. To mitigate the contribution of repeated molecules, we also computed the “unique success rate” for each property, which accounts for the proportion of non-repeating compounds that satisfied the constraint. Fréchet ChemNet Distance (FCD)61 and Similarity to Nearest Neighbor (SNN) scores were calculated using Moses53 to assess the similarity between the generated compounds and known inhibitors. Since designing molecules with higher 3D similarity to lead compounds is desirable in drug design,62 for the RORγt|DHODH benchmark, we additionally calculated the 3D SNN of the generated molecules by ShapeTanimotoDist in RDKit,54 measuring the maximum 3D similarity to known active compounds.
Extracting core fragments with key pharmacophores
The pharmacophore fusing strategy emphasizes the preservation of crucial pharmacophores across diverse targets.2 Here the pharmacophore can be defined as the combination of protein–ligand interactions (PLIs). We devised a dedicated module for the extraction of core fragments that retain the essential pharmacophores present in active compounds. This module was designed to facilitate both automatic extraction and guided selection, where expert-recommended key residues can influence the process. In this study, with reference to previous studies,3,45,46 we selected specific key residues for RORγt and DHODH. For RORγt, HIE479 was set as the key residue. For DHODH, GLN47 and TYR356 were identified as key residues. Regarding GSK3β|JNK3, we referred to the binding mode analysis in the co-crystal structures,59,60 which emphasized the contribution of PLIs associated with VAL135 in GSK3β59 and MET149 in JNK3.60To analyze PLIs, we employed the PLIP tool41,42 on the molecular docking complex structures due to the unavailability of co-crystal structures for most active compounds. For each pharmacophore, we systematically extracted its minimal connected substructure, termed core fragments, as illustrated in Fig. 1B. Our interest lies in core fragments that possess the following characteristics: ① interaction or contact with key residues, ② prevalence across multiple active compounds, and ③ appropriate size. To achieve this, we have reward fragments using scoring functions of interactions, distance, and heavy atom number, with details shown in ESI Section 2.† Furthermore, for the core fragment shared by two or more compounds, we aggregated its score so that fragments favored by medchemists rank higher. Ultimately, the top-scoring core fragments were chosen to construct trees of the two targets. The overall pharmacophore extraction and selection process is visualized in Fig. 1B and detailed in ESI Algorithm 1.†
Tree structure for dual-target pharmacophore fusion
Drawing inspiration from the Monte Carlo Tree Search (MCTS) employed in AlphaZero, we were motivated to explore the utilization of tree structures as a representation of the pharmacophore-fusion chemical space. While previous studies have made forays into using tree structures as representations of molecules,63,64 our approach stands apart in that it introduced a well-defined starting point. This starting point, denoted as core fragments in Fig. 1C, was derived from the pharmacophore extraction module. Consequently, the initial tier of our tree structure was dedicated to the selection of cores, which exerted control over the preservation of specific key pharmacophores.Once two cores have been selected from two trees, the challenge at hand transforms into how to modify and fuse them. Given that the byproduct of extracting cores from active compounds is the side chain fragments, it is natural to consider using side chains from two trees to build linkers. The linker construction process unfolds through three stages. First, two anchor atoms were chosen from two core fragments as the linker growth anchors, named as Growth Anchor in Fig. 1C. Second, for each growth anchor, we extracted a side chain that grew from this anchor. Third, from each side chain, we selected an anchor atom to fuse them, denoted as Fusing Anchor in Fig. 1C. By connecting two fusing anchors from distinct trees, core fragments from distinct targets can be consequently linked together. For growth anchors that have not been selected, decorating R-groups was considered to help improve the diversity of generated molecules. Side chains growing from these anchors can be sliced at different atoms to construct R-groups. Ultimately, as illustrated in Fig. 1C, the leaf node rationale was constructed by decorating valid R-groups (R-groups that are chemically valid and do not disrupt the ring within the original side chain) on unselected growth anchors. The final molecule was generated by fusing two rationales at their fusing anchors.
The sizes of the rationale libraries for GSK3β, JNK3, RORγt, and DHODH surpassed 130k, 60k, 150k, and 13k, respectively. It can be estimated that the chemical space accessible to AIxFuse ranges between 109 and 1010 on both GSK3β|JNK3 and RORγt|DHODH benchmarks. Virtual screening within chemical spaces of such magnitude demands an overwhelming amount of computational resources, often beyond reach. This challenge mirrors that encountered by AlphaZero when evaluating the value of numerous states within the game space. Therefore, we did not assess the entire chemical space but instead employed iterative simulation and exploration by Monte Carlo tree search (MCTS). To guide the exploration, we also trained a multi-task graph neural network (GNN) to predict the docking scores against distinct targets, which is described in the next subsection.
Graph neural network for dual-target docking score prediction
We trained a multi-task GNN as a dual-target docking score prediction. Prior to the graph encoding by the GNN, the extraction of atomic features and bond features is imperative. We utilized a combination of nine atomic features and four bond features (refer to ESI Table S3†), which were adopted by Xiong et al.43Graff et al. demonstrated that in the realm of molecular docking active learning, message-passing neural networks (MPNN) outperformed both random forest and traditional neural network approaches.35 Nonetheless, MPNN, when applied to the processing of molecular graphs, may encounter the issue of diluted effects. This arises from its uniform treatment of all nodes regardless of their distance to a target node, leading to weakened impacts of topologically adjacent nodes and functional groups.43 To address this limitation, attention mechanisms have been introduced, offering improved neighbor aggregation in the form of the Graph Attention Network (GAT).65 More recently, the Attentive Fingerprints (AttentiveFP) framework43 was proposed as an approach capable of capturing both local atomic interactions through node information propagation and non-local effects within a molecule through a graph attention mechanism. It was tailored for molecular feature extractions and outperformed other architectures (including MPNN) across comprehensive benchmarks.
In our study, we applied the AttentiveFP framework to the domain of molecular docking active learning. Two convolutional layers were constructed to extract atomic features, and a readout layer was utilized for generating molecular embeddings. Subsequently, docking scores were predicted by feeding molecular embeddings into a multi-task fully connected layer. The network architecture can be represented as eqn (1)–(3):
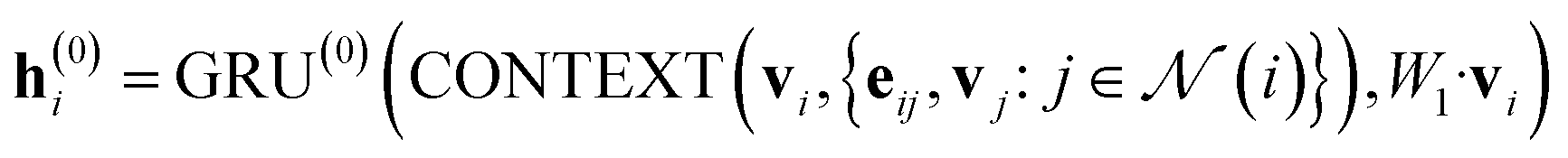 | (1) |
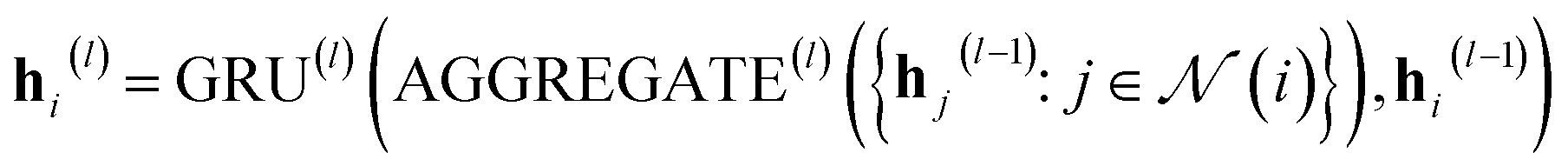 | (2) |
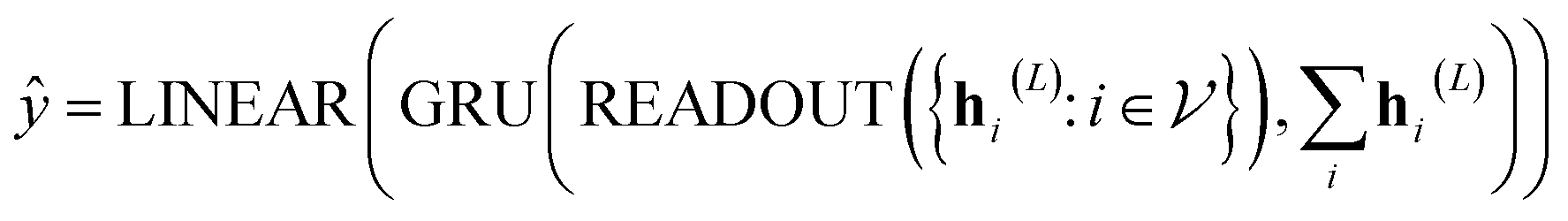 | (3) |
The attention mechanism utilized in CONTEXT, AGGREGATE, and READOUT functions is described by eqn (4), (6) and (8):
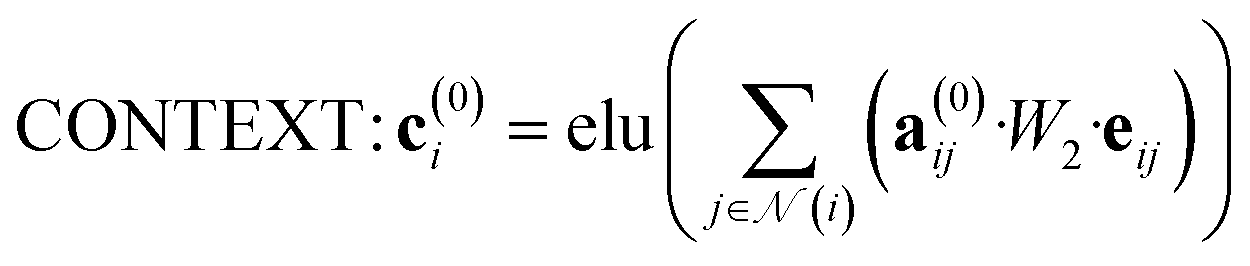 | (4) |
| a(0)ij = softmax(leaky_relu(W3[W4·vi, leaky_relu(W5[eij, vj])])) | (5) |
![[Doublestruck R]](https://www.rsc.org/images/entities/i_char_e175.gif) N denotes the context messages activated by the elu function.
N denotes the context messages activated by the elu function.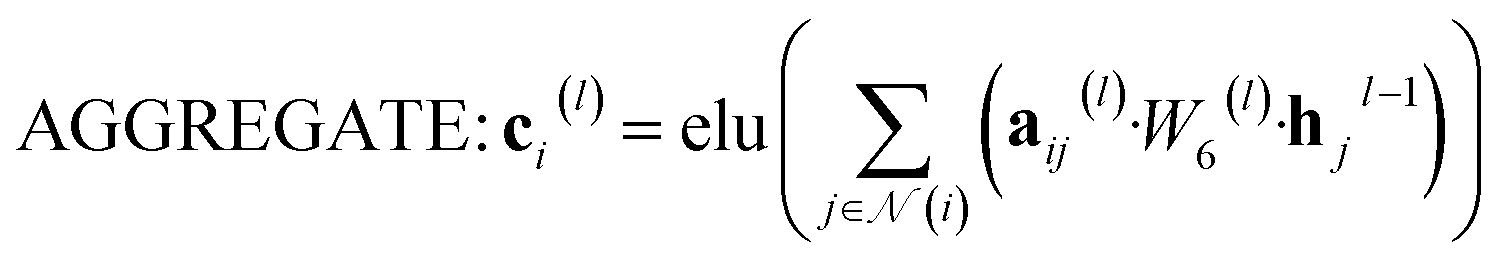 | (6) |
| aij(l) = softmax(leaky_relu(W7(l)[hi(l−1), hj(l−1)])) | (7) |
N is to be processed by GRU and then fed into the next layer.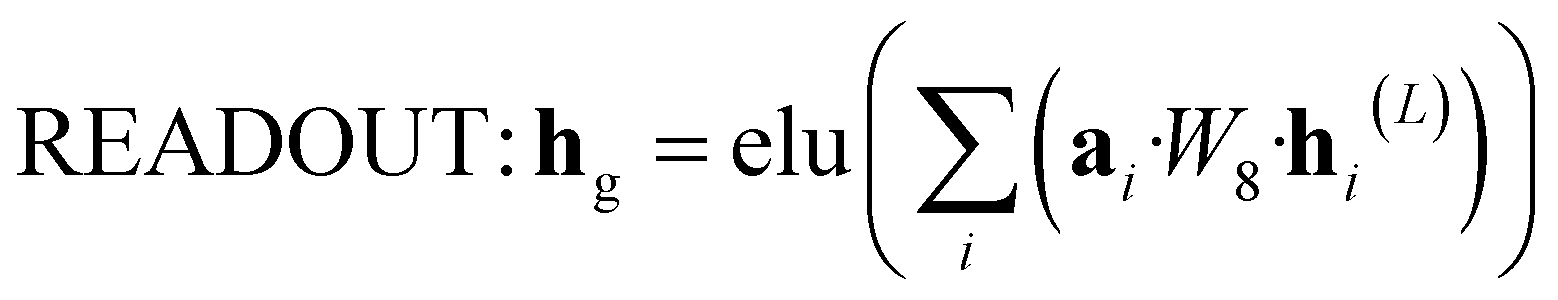 | (8) |
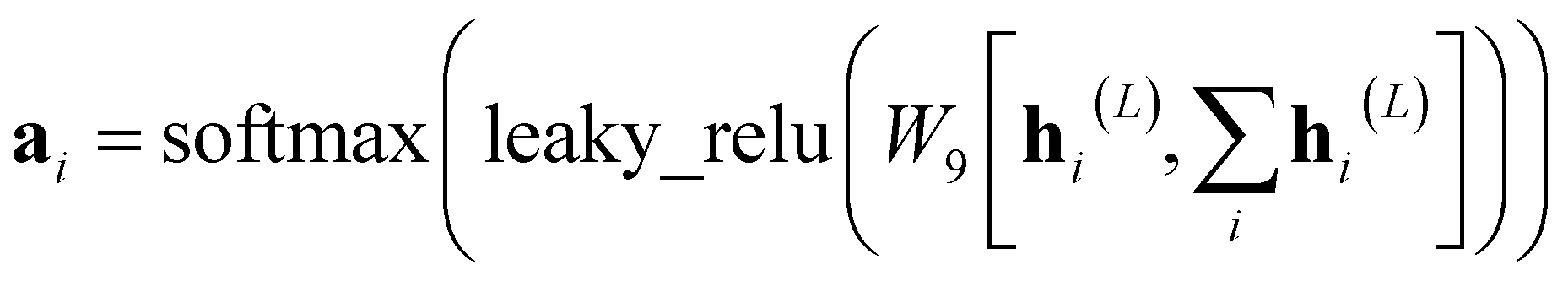 | (9) |
N is the final representation at the molecular level.
We used MSELoss to measure the mean-squared error of our docking score regression model, which can be mathematically represented as:
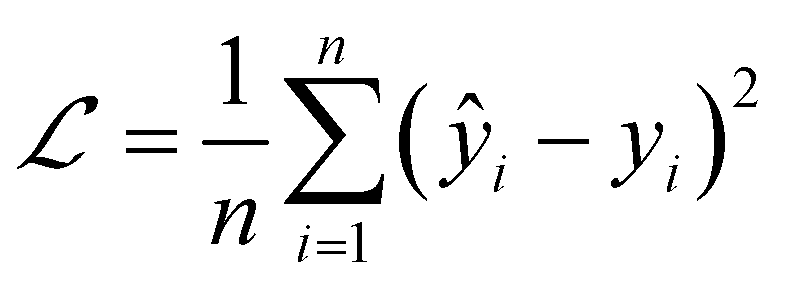 | (10) |
The entire network was trained in a multi-target fashion, yielding two predicted docking scores for a given molecule on the two targets. During each training iteration, the dataset was randomly divided into a training dataset and a validation dataset in a 9:1 ratio. To mitigate the risk of overfitting, early stopping techniques were implemented. Specifically, we monitored each model's performance on the validation set and continued training until no improvement was observed over 20 epochs or the maximum epochs of 1000 was reached.
Our models were trained on the Pytorch71 framework using the Adam72 optimizer for gradient descent optimization. The Deep Graph Library (DGL) package73 and the DGLLifeSci74 extension were employed for the implementation of AttentiveFP. All experiments were conducted within a uniform computational environment, consisting of 4 GPUs of Quadro RTX 5000, 96 CPU cores of Intel(R) Xeon(R) Gold 6248R, and the Ubuntu 20.04.6 LTS operating system. ESI Section 5† describes the details of training and evaluation.
Collaborative learning of pharmacophore fusion and molecular docking
AIxFuse employs collaborative RL and AL to learn pharmacophore fusion patterns that satisfy the dual-target structural constraints simulated by molecular docking. As is shown in Fig. 1C, the RL framework is actor–critic-like, involving two MCTS actors to generate pharmacophore-fused molecules and a dual-target docking score critic trained by active learning. Algorithm 1 describes the collaborative learning procedure, where the INIT_GEN function (i.e. AIxFuse(w/o RLAL)) generates the pharmacophore-fused molecules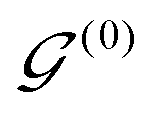 by random sampling and fusing rationales from distinct trees, the DOCKING function dock generates molecules into target binding pockets to estimate their dual-target binding affinities, the TRAIN function represents active learning of the dual-target docking scores by a multi-task AttentiveFP model
by random sampling and fusing rationales from distinct trees, the DOCKING function dock generates molecules into target binding pockets to estimate their dual-target binding affinities, the TRAIN function represents active learning of the dual-target docking scores by a multi-task AttentiveFP model 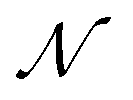 , and the trained parameters
, and the trained parameters 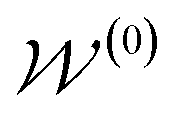 of
of 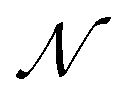 are then used in the SELF_PLAY function to generate a new batch of pharmacophore-fused molecules
are then used in the SELF_PLAY function to generate a new batch of pharmacophore-fused molecules 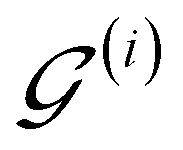 .
.
The INIT_GEN function in Algorithm 1 is described in ESI Section 3.† In short, AIxFuse initializes the weight of each node when building trees, and randomly samples child nodes according to their weights. The random-sampled rationales from the two trees were fused to generate molecules.
The SELF_PLAY function in Algorithm 1 is detailed in ESI Algorithm 3.† Two MCTS actors run in a self-play manner, where we run simulations of pharmacophore fusion to reward each exploration. The reward function of a given molecule m can be formulated as:
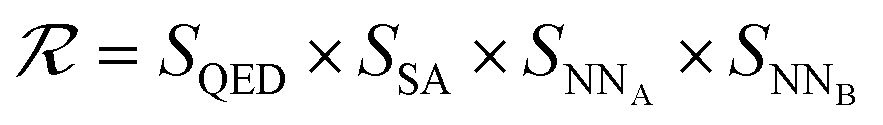 | (11) |
| SQED = max(min(10 × QED(m), 8) − 6, 0.5) | (12) |
| SSA = max(min(5.5 − SA(m)), 0.5) | (13) |
| SNN = max(−NN(m) − 6, 0.1) | (14) |
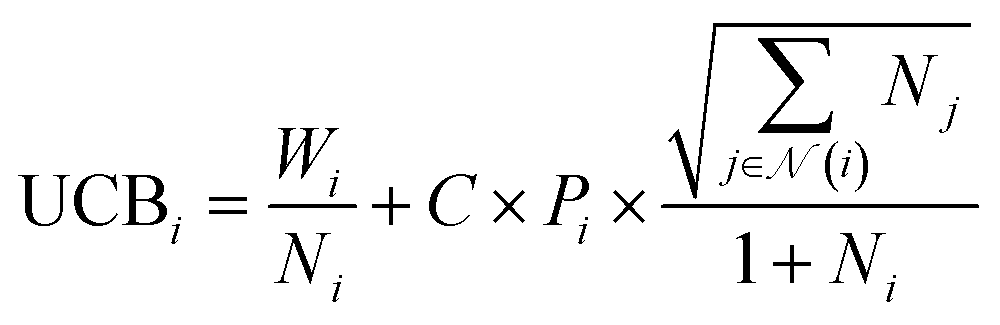 | (15) |
Computational methods of MM/GBSA and ABFEP calculations
Considering the huge computational consumption of molecular dynamics simulations, we conducted MM/GBSA binding free energy calculations for a subset of generated molecules. We initially selected the top 200 ranking molecules considering novelty, dual-target docking scores, 2D SNN, and 3D SNN. Subsequently, we filtered them by visual inspection, resulting in the retention of 134 molecules for MM/GBSA calculation. Among them, 17 representative molecules were selected for ABFEP calculation against RORγt and 7 out of 17 molecules were selected for ABFEP calculation against DHODH.Molecular dynamics simulations were performed by GROMACS (version 2020.6).75 The AMBER99SB-ILDN76 force field was used for proteins. The General Amber Force Field (GAFF)77 with AM1-BCC78,79 partial charges was used for ligands. The protein–ligand complex was solvated in the dodecahedral box of TIP3P80 water with the minimum distance between the solute and the box of 10 Å. The system was minimized using the steepest descent algorithm with max steps of 10000, followed by 100 ps NVT equilibration at 300 K and then 100 ps NPT equilibration at 300 K and 1 bar. Subsequently, 2 ns NPT production was performed at 300 K and 1 bar and the frames were saved every 10 ps for further analysis and MMGBSA calculations. The distance cutoff of short-range interactions was set to 10 Å. Long-range electrostatic interactions were treated with PME algorithm.81 LINCS82 algorithm was applied to constrain the hydrogen-involved bonds and all the MD simulations were performed with a timestep of 2 fs. The MM/GBSA calculations were performed with AMBER83 with a time interval of 20 ps for extracted frames via the single-trajectory scheme, where the structures of the separate protein and ligand were extracted from the trajectory of the complex. ABFEP calculations were performed following the schemes employed by Aldeghi et al.84,85 More specifically, 42 lambda windows and 31 lambda windows were used for the complex leg and solvent leg, respectively. The relative position of the ligand with respect to the protein was restrained by applying harmonic potentials to one distance, two angles, and three dihedrals as proposed by Boresch et al.86 The free energy difference for adding these restraints to the dummy ligand in solvent was calculated analytically.86 The perturbation of van der Waals interactions between the ligand and the environment was performed with soft-core potential.87 After the relaxation and equilibration of each lambda window, 5 ns NPT production runs were performed using Hamiltonian-exchange Langevin dynamics for enhanced sampling.
Data availability
Data and code will be soon available at https://github.com/biomed-AI/AIxFuse.Author contributions
S. C., Y. Y., and D. D. X. designed the study; S. C., J. X., and R. Y. performed the research; S. C. and R. Y. contributed new reagents/analytic tools; S. C., J. X., and R. Y. analyzed the data; S. C., Y. Y., D. D. X., J. X., and R. Y. wrote the paper.Conflicts of interest
The authors declare that they have no conflicts of interest related to this research.Acknowledgements
This study has been supported by the National Key R&D Program of China (2023YFF1204900), National Natural Science Foundation of China (T2394502), and Guangdong Key Field R&D Plan (2023B1111030002).References
-
M. L. Bolognesi and A. Cavalli, Multitarget drug discovery and polypharmacology, 2016 Search PubMed
.
- D. Sun,
et al., Dual-target kinase drug design: current strategies and future directions in cancer therapy, Eur. J. Med. Chem., 2020, 188, 112025 CrossRef CAS PubMed
- J. A. Chen,
et al., Discovery of orally available retinoic acid receptor-related orphan receptor γ-t/dihydroorotate dehydrogenase dual inhibitors for the treatment of refractory inflammatory bowel disease, J. Med. Chem., 2021, 65, 592–615 CrossRef PubMed
- G. V. Long,
et al., Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma, N. Engl. J. Med., 2014, 371, 1877–1888 CrossRef
- C. Robert,
et al., Improved overall survival in melanoma with combined dabrafenib and trametinib, N. Engl. J. Med., 2015, 372, 30–39 CrossRef PubMed
- R. B. Mokhtari,
et al., Combination therapy in combating cancer, Oncotarget, 2017, 8, 38022 CrossRef
- J. Ye, J. Wu and B. Liu, Therapeutic strategies of dual-target small molecules to overcome drug resistance in cancer therapy, Biochim. Biophys. Acta, Rev. Cancer, 2023, 188866 CrossRef CAS PubMed
- M. F. S. A. Ribeiro,
et al., Alectinib activity in chemotherapy-refractory metastatic RET-rearranged non-small cell lung carcinomas: a case series, Lung Cancer, 2020, 139, 9–12 CrossRef PubMed
- M. E. Van Dort,
et al., Discovery of bifunctional oncogenic target inhibitors against allosteric mitogen-activated protein kinase (MEK1) and phosphatidylinositol 3-kinase (PI3K), J. Med. Chem., 2016, 59, 2512–2522 CrossRef CAS
- K. Liu,
et al., 3,5-Disubstituted-thiazolidine-2,4-dione analogs as anticancer agents: design, synthesis and biological characterization, Eur. J. Med. Chem., 2012, 47, 125–137 CrossRef CAS PubMed
- H. H. Lin, L. L. Zhang, R. Yan, J. J. Lu and Y. Hu, Network analysis of drug–target interactions: a study on FDA-approved new molecular entities between 2000 to 2015, Sci. Rep., 2017, 7, 12230 CrossRef PubMed
- R. R. Ramsay, M. R. Popovic-Nikolic, K. Nikolic, E. Uliassi and M. L. Bolognesi, A perspective on multi-target drug discovery and design for complex diseases, Clin. Transl. Med., 2018, 7, 1–14 CrossRef
- M. Zhan,
et al., Design, synthesis, and biological evaluation of dimorpholine substituted thienopyrimidines as potential class I PI3K/mTOR dual inhibitors, J. Med. Chem., 2017, 60, 4023–4035 CrossRef CAS PubMed
- Q. Wang,
et al., Discovery of 4-methyl-n-(4-((4-methylpiperazin-1-yl)methyl)-3-(trifluoromethyl)phenyl)-3-((1-nicotinoylpiperidin-4-yl)oxy)benzamide (CHMFL-ABL/KIT-155) as a novel highly potent type II ABL/KIT dual kinase inhibitor with a distinct hinge binding, J. Med. Chem., 2017, 60, 273–289 CrossRef CAS
- Y. Tanaka,
et al., Discovery of potent Mcl-1/Bcl-xL dual inhibitors by using a hybridization strategy based on structural analysis of target proteins, J. Med. Chem., 2013, 56, 9635–9645 CrossRef CAS
- A. Hauschild,
et al., Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial, Lancet, 2012, 380, 358–365 CrossRef CAS
- Y. Chen,
et al., Integrated bioinformatics, computational and experimental methods to discover novel Raf/extracellular-signal regulated kinase (ERK) dual inhibitors against breast cancer cells, Eur. J. Med. Chem., 2017, 127, 997–1011 CrossRef CAS PubMed
- J. Meyers, B. Fabian and N. Brown, De novo molecular design and generative models, Drug Discovery Today, 2021, 26, 2707–2715 CrossRef CAS PubMed
-
M. Hartenfeller and G. Schneider, De novo drug design, in Chemoinformatics and computational chemical biology, 2011, pp. 299–323 Search PubMed
- N. Brown, B. McKay, F. Gilardoni and J. Gasteiger, A graph-based genetic algorithm and its application to the multiobjective evolution of median molecules, J. Chem. Inf. Comput. Sci., 2004, 44, 1079–1087 CrossRef CAS PubMed
- M. Reutlinger, T. Rodrigues, P. Schneider and G. Schneider, Multi-objective molecular de novo design by adaptive fragment prioritization, Angew. Chem., Int. Ed., 2014, 53, 4244–4248 CrossRef CAS PubMed
- M. Wang,
et al., Deep learning approaches for de novo drug design: an overview, Curr. Opin. Struct. Biol., 2022, 72, 135–144 CrossRef CAS PubMed
- V. D. Mouchlis,
et al., Advances in de novo drug design: from conventional to machine learning methods, Int. J. Mol. Sci., 2021, 22, 1676 CrossRef
- R. Gómez-Bombarelli,
et al., Automatic chemical design using a data-driven continuous representation
of molecules, ACS Cent. Sci., 2018, 4, 268–276 CrossRef PubMed
-
M. J. Kusner, B. Paige and J. M. Hernández-Lobato, Grammar variational autoencoder, in International conference on machine learning, PMLR, 2017, pp. 1945–1954 Search PubMed
- E. J. Bjerrum and B. Sattarov, Improving chemical autoencoder latent space and molecular de novo generation diversity with heteroencoders, Biomolecules, 2018, 8, 131 CrossRef PubMed
-
Q. Liu, M. Allamanis, M. Brockschmidt and A. L. Gaunt, Constrained graph variational autoencoders for molecule design in Proceedings of the 32nd International Conference on Neural Information Processing Systems, 2018, pp. 7806–7815 Search PubMed
-
J. You, B. Liu, R. Ying, V. Pande and J. Leskovec, Graph convolutional policy network for goal-directed molecular graph generation, in Proceedings of the 32nd International Conference on Neural Information Processing Systems, 2018, pp. 6412–6422 Search PubMed
- Y. Xie, et al., Mars: Markov molecular sampling for multi-objective drug discovery, arXiv, 2021, preprint, arXiv:2103.10432, DOI:10.48550/arXiv.2103.10432.
- F. Lu, M. Li, X. Min, C. Li and X. Zeng, De novo generation of dual-target ligands using adversarial training and reinforcement learning, Briefings Bioinf., 2021, 22, bbab333 CrossRef PubMed
-
W. Jin, R. Barzilay and T. Jaakkola, Multi-objective molecule generation using interpretable substructures, in International conference on machine learning, PMLR, 2020, pp. 4849–4859 Search PubMed
- T. Blaschke,
et al., Reinvent 2.0: an AI tool for de novo drug design, J. Chem. Inf. Model., 2020, 60, 5918–5922 CrossRef CAS
- Y. Li, L. Zhang and Z. Liu, Multi-objective de novo drug design with conditional graph generative model, J. Cheminf., 2018, 10, 1–24 Search PubMed
- Y. Yang,
et al., Efficient exploration of chemical space with docking and deep learning, J. Chem. Theory Comput., 2021, 17, 7106–7119 CrossRef CAS PubMed
- D. E. Graff, E. I. Shakhnovich and C. W. Coley, Accelerating high-throughput virtual screening through molecular pool-based active learning, Chem. Sci., 2021, 12, 7866–7881 CAS
- Y. Khalak, G. Tresadern, D. F. Hahn, B. L. de Groot and V. Gapsys, Chemical space exploration with active learning and alchemical free energies, J. Chem. Theory Comput., 2022, 18, 6259–6270 CrossRef CAS
- J. Thompson,
et al., Optimizing active learning for free energy calculations, Artif. Intell. Life Sci., 2022, 2, 100050 Search PubMed
- D. Silver,
et al., Mastering the game of go without human knowledge, nature, 2017, 550, 354–359 CAS
- V. Konda and J. Tsitsiklis, Actor-critic algorithms, Advances in Neural Information Processing Systems, 1999, 12, 1008–1014 Search PubMed
- R. A. Friesner,
et al., Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy, J. Med. Chem., 2004, 47, 1739–1749 CAS
- S. Salentin, S. Schreiber, V. J. Haupt, M. F. Adasme and M. Schroeder, PLIP: fully automated protein–ligand interaction profiler, Nucleic Acids Res., 2015, 43, W443–W447 CAS
- M. F. Adasme,
et al., PLIP 2021: expanding the scope of the protein–ligand interaction profiler to DNA and RNA, Nucleic Acids Res., 2021, 49, W530–W534 CrossRef CAS PubMed
- Z. Xiong,
et al., Pushing the boundaries of molecular representation for drug discovery with the graph attention mechanism, J. Med. Chem., 2019, 63, 8749–8760 CrossRef PubMed
- S. Genheden and U. Ryde, The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities, Expert Opin. Drug Discovery, 2015, 10, 449–461 CrossRef CAS
- J. Kallen,
et al., Structural states of RORγt: X-ray elucidation of molecular mechanisms and binding interactions for natural and synthetic compounds, ChemMedChem, 2017, 12, 1014–1021 CrossRef CAS
- S. Christian,
et al., The novel dihydroorotate dehydrogenase (DHODH) inhibitor bay 2402234 triggers differentiation and is effective in the treatment of myeloid malignancies, Leukemia, 2019, 33, 2403–2415 CrossRef CAS PubMed
- L. Wang,
et al., Accurate and reliable prediction of relative ligand binding potency in prospective drug discovery by way of a modern free-energy calculation protocol and force field, J. Am. Chem. Soc., 2015, 137, 2695–2703 CrossRef CAS PubMed
- W. Chen,
et al., Enhancing hit discovery in virtual screening through absolute protein–ligand binding free-energy calculations, J. Chem. Inf. Model., 2023, 10, 3171–3185 CrossRef
- J. A. McCubrey,
et al., Diverse roles of GSK-3: tumor promoter-tumor suppressor, target in cancer therapy, Adv. Biol. Regul., 2014, 54, 176–196 CrossRef CAS PubMed
- P. Koch, M. Gehringer and S. A. Laufer, Inhibitors of c-Jun N-terminal kinases: an update, J. Med. Chem., 2015, 58, 72–95 CrossRef CAS PubMed
- D. R. Withers,
et al., Transient inhibition of ROR-γt therapeutically limits intestinal inflammation by reducing TH17 cells and preserving group 3 innate lymphoid cells, Nat. Med., 2016, 22, 319–323 CrossRef CAS
- L. Klotz,
et al., Teriflunomide treatment for multiple sclerosis modulates T cell mitochondrial respiration with affinity-dependent effects, Sci. Transl. Med., 2019, 11, eaao5563 CrossRef CAS
- D. Polykovskiy,
et al., Molecular sets (MOSES): a benchmarking platform for molecular generation models, Front. Pharmacol., 2020, 11, 565644 CrossRef CAS
-
G. Landrum, RDKIT: open-source cheminformatics, 2006–2021 Search PubMed
- M. García-Ortegón,
et al., DOCKSTRING: easy molecular docking yields better benchmarks for ligand design, J. Chem. Inf. Model., 2022, 62, 3486–3502 CrossRef
- W. Jeon and D. Kim, Autonomous molecule generation using reinforcement learning and docking to develop potential novel inhibitors, Sci. Rep., 2020, 10, 22104 CrossRef CAS PubMed
- F. Gentile,
et al., Deep docking: a deep learning platform for augmentation of structure based drug discovery, ACS Cent. Sci., 2020, 6, 939–949 CrossRef CAS
- J. Lyu,
et al., Ultra-large library docking for discovering new chemotypes, Nature, 2019, 566, 224–229 CAS
- R. Buonfiglio,
et al., Discovery of novel imidazopyridine GSK-3β inhibitors supported by computational approaches, Molecules, 2020, 25, 2163 CrossRef CAS
- K. Zheng,
et al., Design and synthesis of highly potent and isoform selective JNK3 inhibitors: SAR studies on aminopyrazole derivatives, J. Med. Chem., 2014, 57, 10013–10030 CrossRef CAS PubMed
- K. Preuer, P. Renz, T. Unterthiner, S. Hochreiter and G. Klambauer, Fréchet ChemNet distance: a metric for generative models for molecules in drug discovery, J. Chem. Inf. Model., 2018, 58, 1736–1741 CrossRef CAS
- Y. Tan,
et al., DRlinker: deep reinforcement learning for optimization in fragment linking design, J. Chem. Inf. Model., 2022, 62, 5907–5917 CrossRef CAS PubMed
-
W. Jin, R. Barzilay and T. Jaakkola, Junction tree variational autoencoder for molecular graph generation, in International conference on machine learning, PMLR, 2018, pp. 2323–2332 Search PubMed
- T. Fu, et al., Differentiable scaffolding tree for molecular optimization, arXiv, 2021, preprint, arXiv:2109.10469 Search PubMed.
-
P. Veličković, et al., Graph attention networks, in International Conference on Learning Representations, 2018 Search PubMed
- S. Hochreiter and J. Schmidhuber, Long short-term memory, Neural Comput., 1997, 9, 1735–1780 CrossRef CAS PubMed
-
J. Chung, C. Gulcehre, K. Cho and Y. Bengio, Empirical evaluation of gated recurrent neural networks on sequence modeling, in NIPS 2014 Workshop on Deep Learning, December 2014, 2014 Search PubMed
- B. Xu, N. Wang, T. Chen and M. Li, Empirical evaluation of rectified activations in convolutional network, arXiv, 2015, preprint, arXiv:1505.00853, DOI:10.48550/arXiv.1505.00853.
- D. A. Clevert, T. Unterthiner and S. Hochreiter, Fast and accurate deep network learning by exponential linear units (ELUS), arXiv, 2015, preprint, arXiv:1511.07289, DOI:10.48550/arXiv.1511.07289.
-
X. Glorot, A. Bordes and Y. Bengio, Deep sparse rectifier neural networks, in Proceedings of the fourteenth international conference on artificial intelligence and statistics, JMLR Workshop and Conference Proceedings, 2011, pp. 315–323 Search PubMed
-
A. Paszke, et al., Automatic differentiation in pytorch, in NIPS 2017 Autodiff Workshop, December 2017, 2017 Search PubMed
- D. P. Kingma and J. Ba, Adam: a method for stochastic optimization, arXiv, 2014, preprint, arXiv:1412.6980, DOI:10.48550/arXiv.1412.6980.
- M. Wang, et al., Deep graph library: a graph-centric, highly-performant package for graph neural networks, arXiv, 2019, preprint, arXiv:1909.01315, DOI:10.48550/arXiv.1909.01315.
- M. Li,
et al., DGL-LifeSci: an open-source toolkit for deep learning on graphs in life science, ACS Omega, 2021, 6, 27233–27238 CrossRef CAS
- M. J. Abraham,
et al., Gromacs: high performance molecular simulations through multi-level parallelism from laptops to supercomputers, SoftwareX, 2015, 1, 19–25 CrossRef
- K. Lindorff-Larsen,
et al., Improved side-chain torsion potentials for the Amber ff99SB protein force field, Proteins: Struct., Funct., Bioinf., 2010, 78, 1950–1958 CrossRef CAS
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, Development and testing of a general AMBER force field, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS
- A. Jakalian, B. L. Bush, D. B. Jack and C. I. Bayly, Fast, efficient generation of high-quality atomic charges. AM1-BCC model: I. Method, J. Comput. Chem., 2000, 21, 132–146 CAS
- A. Jakalian, D. B. Jack and C. I. Bayly, Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation, J. Comput. Chem., 2002, 23, 1623–1641 CrossRef CAS PubMed
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, Comparison of simple potential functions for simulating liquid water, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS
- U. Essmann,
et al., A smooth particle mesh Ewald method, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS
- B. Hess, P-LINCS: a parallel linear constraint solver for molecular simulation, J. Chem. Theory Comput., 2008, 4, 116–122 CrossRef CAS PubMed
- B. R. Miller III,
et al., MMPBSA.py: an efficient program for end-state free energy calculations, J. Chem. Theory Comput., 2012, 8, 3314–3321 CrossRef PubMed
- M. Aldeghi, A. Heifetz, M. J. Bodkin, S. Knapp and P. C. Biggin, Accurate calculation of the absolute free energy of binding for drug molecules, Chem. Sci., 2016, 7, 207–218 RSC
- M. Aldeghi, A. Heifetz, M. J. Bodkin, S. Knapp and P. C. Biggin, Predictions of ligand selectivity from absolute binding free energy calculations, J. Am. Chem. Soc., 2017, 139, 946–957 CrossRef CAS
- S. Boresch, F. Tettinger, M. Leitgeb and M. Karplus, Absolute binding free energies: a quantitative approach for their calculation, J. Phys. Chem. B, 2003, 107, 9535–9551 CrossRef CAS
- T. C. Beutler, A. E. Mark, R. C. van Schaik, P. R. Gerber and W. F. Van Gunsteren, Avoiding singularities and numerical instabilities in free energy calculations based on molecular simulations, Chem. Phys. Lett., 1994, 222, 529–539 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc00094c |
| This journal is © The Royal Society of Chemistry 2024 |