
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA cyclic trinuclear silver complex for photosynthesis of hydrogen peroxide†
Ri-Qin
Xia
 ,
Zhen-Na
Liu
,
Yu-Ying
Tang
,
Xiao
Luo
,
Rong-Jia
Wei
,
Tao
Wu
,
Guo-Hong
Ning
* and
Dan
Li
*
,
Zhen-Na
Liu
,
Yu-Ying
Tang
,
Xiao
Luo
,
Rong-Jia
Wei
,
Tao
Wu
,
Guo-Hong
Ning
* and
Dan
Li
*
College of Chemistry and Materials Science, Guangdong Provincial Key Laboratory of Supramolecular Coordination Chemistry, Jinan University, Guangzhou 510632, People's Republic of China. E-mail: guohongning@jnu.edu.cn; danli@jnu.edu.cn
First published on 8th August 2024
Abstract
The development of metal complexes for photosynthesis of hydrogen peroxide (H2O2) from pure water and oxygen using solar energy, especially in the absence of any additives (e.g., acid, co-catalysts, and sacrificial agents), is a worthwhile pursuit, yet still remains highly challenging. More importantly, the O2 evolution from the water oxidation reaction has been impeded by the classic bottleneck, the photon-flux-density problem of sunlight that could be attributed to rarefied solar radiation for a long time. Herein, we reported synthesis of boron dipyrromethene (BODIPY)-based cyclic trinuclear silver complexes (Ag-CTC), and they exhibited strong visible-light absorption ability, a suitable energy bandgap, excellent photochemical properties and efficient charge separation ability. The integration of BODIPY motifs as oxygen reduction reaction sites and silver ions as water oxidation reaction sites allows Ag-CTC to photosynthesize H2O2 either from pure water or from sea water in the absence of any additives with a high H2O2 production rate of 183.7 and 192.3 μM h−1, which is higher than that of other reported metal-based photocatalysts. The photocatalytic mechanism was systematically and ambiguously investigated by various experimental analyses and density functional theory (DFT) calculations. Our work represents an important breakthrough in developing a new Ag photocatalyst for the transformation of O2 into H2O2 and H2O into H2O2.
Introduction
Hydrogen peroxide (H2O2), an environment-friendly oxidant and a clean fuel is widely used in medical disinfection, water treatment and chemical synthesis,1–10 with an annual demand of 5.7 million tons by 2027.11 So far, the anthraquinone oxidation process is still the most used approach for H2O2 production in industry; however, it requires expensive palladium catalysts and a large amount of harmful organic solvent and consumes lots of energy.12 Therefore, it is a worthwhile pursuit to develop green and efficient catalysts, especially solar photocatalysts, for H2O2 production from water and oxygen due to its potential for solving global energy shortage and ecological issues.Artificial photosynthesis of H2O2 using metal complexes (MCs) requires several processes including light absorption, charge-separation, the water oxidation reaction (WOR) and the oxygen reduction reaction (ORR). Currently, the ORR half-reaction has been predominantly investigated for photosynthesis of H2O2; however, the WOR half-reaction is not sufficiently exploited,13–17 which severely hindered practical application. This because the WOR process suffers from sluggish kinetics, the so called “photo-flux-density problem of sunlight”,18,19 especially for the photocatalytic four-electron (4e−) oxygen evolution reaction (OER). Typically, the generation of O2 photo-catalysis by MCs takes seconds, which is far longer than the timescale for hole generation and migration.18,19 During this long period, the photocatalyst would undergo undesired decomposition or transformation, leading to a loss of catalytic activity. Therefore, it is a worthwhile pursuit to develop MCs that are capable of photo-catalyzing both the ORR and WOR, yet it still remains highly challenging.
Conventionally, to bypass the WOR issues, hybrid catalytic systems involving two or more different components are designed (Scheme 1a).11,12,20–22 For instance, Ru or Cu complexes as photocatalysts or photosensitizers for the 2e− ORR, and Co/Ir complexes or semiconductors (i.e., WO3 or BiVO4) as water oxidation catalysts (WOCs) are combined for producing H2O2.11,23–28 Moreover, the addition of sacrificial agents, a superoxide anion radical (O2˙−) stabilizer (i.e., Sc(NO3)3) and a proton source (e.g., H2SO4 or HClO4) is required in these homogeneous catalytic systems. Thus, these processes not only produce by-products and harmful waste, but also require additional purification steps. Furthermore, low visible-light absorption ability of reported MCs, decomposition of H2O2 triggered by MCs, and the unsatisfactory H2O2 production performance also remains unresolved problems in this field. More importantly, these photocatalytic systems did not address the WOR bottleneck.
 | ||
| Scheme 1 Schematic illustration of (a) conventional metal-based hybrid catalytic systems and (b) a Ag-CTC complex in this work for photosynthesis of H2O2. (ORC and WOC mean the oxygen reduction catalyst and water oxidation catalyst). | ||
Recently, great efforts have been put into developing transition MCs with strong visible-light absorption, which can be used in photocatalytic oxidation, and hydrogen evolution from water.29–32 For instance, the incorporation of boron dipyrromethene (BODIPY) into a cyclic trinuclear copper(I) complex (Cu-CTC) has significantly improved its visible-light absorption, leading to light-induced O2˙− generation from molecular oxygen (O2). Since O2˙− is involved in one of the reaction pathways for photosynthesis of H2O2 (see eqn (1)–(5)), we hypothesize that BODIPY-based CTCs might be promising photocatalysts for H2O2 production and address the above-mentioned issues.
Herein, we rationally designed BODIPY-based Ag-CTC (Ag3L3, 1) and Cu-CTC (Cu3L3, 2) by complexation between a BODIPY-based pyrazolyl ligand (HL) and silver benzoate (PhCOOAg) or Cu(NO3)2, respectively (Scheme 1b). For comparison, Ag3L13 (3, HL1 represents 3,5-bis(trifluoromethyl)-1H-pyrazole) as a reference compound was also synthesized.33 The incorporation of the BODIPY motif remarkably improved the photophysical properties of 1 and 2, and they both featured high molar absorptivity (ε = ∼7.4 and 7.9 × 104 M−1 cm−1), and good photo-induced charge-separation efficiency in the visible-light range. Under light irradiation, Ag-CTC 1 as a heterogenous photocatalyst delivered a high H2O2 production rate of 183.7 μM h−1 in the absence of a co-catalyst, sacrificial agents and acid, and it performed better than HL, Cu-CTC 2 and reference compound 3. Notably, to the best of our knowledge, Ag-CTC 1 is the best metal complex-based photocatalyst for H2O2 production to date. The mechanistic studies further revealed that Ag-CTC 1 can generate H2O2 through a stepwise e− ORR pathway (i.e., O2 → O2˙− → H2O2), and we unexpectedly found that the holes generated after visible-light irradiation can be used to perform the oxygen evolution reaction (OER). The 4e− WOR occurred at the silver center. Our work, for the first time, has demonstrated that molecular metal complexes composed of oxygen reduction and oxidation centers can be used in the full reaction photosynthesis of H2O2 without sacrificial agents.
| O2 + 2H+ + 2e− → H2O2, (Eθ = +0.68 V vs. NHE) | (1) |
| O2 + e− → O2˙−, (Eθ = −0.33 V vs. NHE) | (2) |
| O2˙− + 2H+ + e− → H2O2, (Eθ = +1.44 V vs. NHE) | (3) |
| 2H2O + 4h+ → O2 + 4H+, (Eθ = +1.23 V vs. NHE) | (4) |
| 2H2O + 2h+ → H2O2 + 2H+, (Eθ = +1.76 V vs. NHE) | (5) |
Results and discussion
Synthesis and characterization
The BODIPY-based pyrazolyl ligand (HL) was prepared according to our previous work.29 The mixture of HL and PhCOOAg in anhydrous THF solution was stirred for 24 hours in the dark at room temperature (rt) to give compound 1 as an orange powder (Scheme S1†). The structure of 1 was characterized by using 1H, 19F, and 13C NMR spectra (Fig. S1–S3†). The disappearance of the N–H signal of ligand HL at 13.81 ppm suggested the deprotonation of HL and formation of Ag–N bonds. The 19F NMR spectra of 1 and HL revealed that they both display one 19F peak located at −141.89 and −141.66 ppm, respectively, further proving the purity of 1. The X-ray photoelectron spectroscopy (XPS) measurement of 1 showed intensely sharp and symmetrical Ag(I) 2d3/2 and 2d5/2 peaks at 374.09 and 368.10 eV, respectively (Fig. S5†). Energy-dispersive X-ray spectroscopy (EDS) elemental mapping confirmed that Ag, C, N, B, and F elements were evenly distributed in 1 (Fig. S6†). The orange crystals suitable for single-crystal X-ray diffraction (SXRD) analysis can be obtained with a yield of 13.7% by the solvothermal synthesis method.34As shown in Fig. 1, 1 was crystalized in the Ia![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) cubic space group, exhibiting nine-membered Ag3N6 units with a distorted planar structure (Fig. 1a). The dihedral angle between the Ag3N6 plane and the BODIPY unit is ∼31°, leading to a propeller-shaped structure with three blades (Fig. 1a). In addition, two Ag-CTCs tightly stacked with each other to form a dimer with an interdimer Ag⋯Ag distance of 3.75 Å (Fig. 1b), revealing strong Ag–Ag interactions. Reference compound Cu-CTC 2 (ref. 29) and Ag3L133 (ref. 35 and 36) were synthesized for comparison and their crystal structures are shown in Fig. 1c and d, respectively. They both exhibited similar nine-membered planar configurations. Furthermore, thermo-gravimetric analysis (TGA) of 1 revealed that it started to decompose at 260 °C (Fig. S9†). The phase purity of 1 was confirmed by powder X-ray diffraction (PXRD) (Fig. S10†). The water contact angles of HL, 1, 2 and 3 are all larger than 110°, suggesting that they are all hydrophobic (Fig. S13†).
cubic space group, exhibiting nine-membered Ag3N6 units with a distorted planar structure (Fig. 1a). The dihedral angle between the Ag3N6 plane and the BODIPY unit is ∼31°, leading to a propeller-shaped structure with three blades (Fig. 1a). In addition, two Ag-CTCs tightly stacked with each other to form a dimer with an interdimer Ag⋯Ag distance of 3.75 Å (Fig. 1b), revealing strong Ag–Ag interactions. Reference compound Cu-CTC 2 (ref. 29) and Ag3L133 (ref. 35 and 36) were synthesized for comparison and their crystal structures are shown in Fig. 1c and d, respectively. They both exhibited similar nine-membered planar configurations. Furthermore, thermo-gravimetric analysis (TGA) of 1 revealed that it started to decompose at 260 °C (Fig. S9†). The phase purity of 1 was confirmed by powder X-ray diffraction (PXRD) (Fig. S10†). The water contact angles of HL, 1, 2 and 3 are all larger than 110°, suggesting that they are all hydrophobic (Fig. S13†).
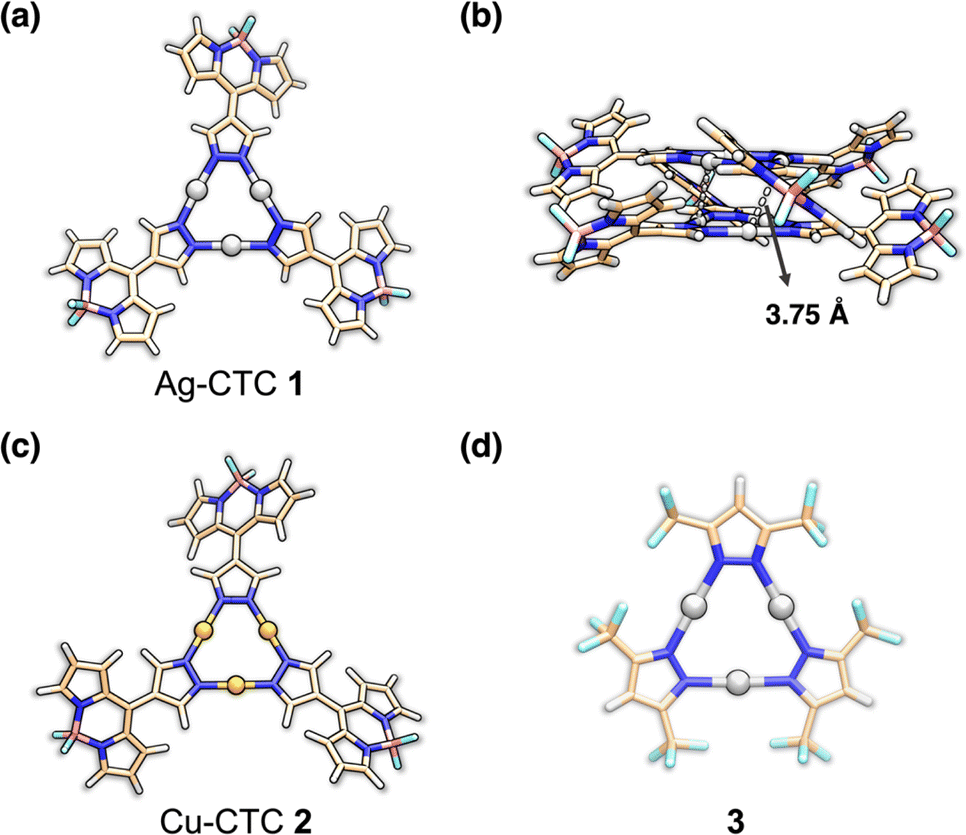 | ||
| Fig. 1 Crystal structures of (a) Ag-CTC 1, (c) Cu-CTC 2 and (d) Ag3L133, exhibiting one CTC unit structure. (b) Side view of tightly packed 1 showing an interdimer Ag⋯Ag distance of 3.75 Å (ball-stick model) (color codes: C, wheat; N, blue; Cu, orange; Ag, silver; B, light pink; and F, light green). | ||
Optics and electrochemistry
The UV-vis absorption spectra of 1 exhibited strong adsorption at 497 nm with an exceptionally high molar extinction coefficient (ε) of 74![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 353 M−1 cm−1 (Fig. S16 and S17†). This value is similar to that of compound 2 (ε = 78515 M−1 cm−1) and approximately 5.17 times higher than that of HL (ε = 14379 M−1 cm−1).13 In addition, the solid-state UV-vis diffuse reflection spectra of HL, 1, 2 and 3 were obtained to study light absorption ability. Specifically, 2 exhibited the widest absorption range (i.e., 200–800 nm), while 3 showed the narrowest absorption range (i.e. 200–400 nm) (Fig. 2a). HL and 1 delivered similar absorption edges at ∼600 nm. These results suggested that the incorporation of BODIPY units remarkably improved visible-light harvesting ability of CTCs. The optical bandgaps (Eg) of HL, 1, 2 and 3 were estimated to be 1.97, 2.16, 1.76 and 2.94 eV, respectively by using the Tauc plot (Fig. 2b). The flat band potentials of HL, 1, 2 and 3 were determined to be −1.38, −0.80, −1.33 and −1.47 eV vs. NHE at pH = 7 through the Mott–Schottky experiments,37 respectively (Fig. S18–S21†), which were equal to their conduction band (CB) potentials. Combining the Mott–Schottky experiments and optical bandgap data, valence band (VB) potentials of HL, 1, 2 and 3 were calculated to be 0.59, 1.36, 0.43 and 1.47 eV vs. NHE at pH = 7 (Fig. 2c). Since the reduction potentials of O2(g)/O2˙− and O2(g)/H2O2 are known to be −0.35 and 0.28 V vs. NHE at pH = 7,38 they are thermodynamically suitable for photocatalytic reduction of O2 to give H2O2. In addition, the oxidation potential of H2O/O2 is 0.82 V vs. NHE at pH = 7;39 thus, compounds 1 and 3 are thermodynamically suitable for photocatalytic oxidation of water to produce O2 and protons.
353 M−1 cm−1 (Fig. S16 and S17†). This value is similar to that of compound 2 (ε = 78515 M−1 cm−1) and approximately 5.17 times higher than that of HL (ε = 14379 M−1 cm−1).13 In addition, the solid-state UV-vis diffuse reflection spectra of HL, 1, 2 and 3 were obtained to study light absorption ability. Specifically, 2 exhibited the widest absorption range (i.e., 200–800 nm), while 3 showed the narrowest absorption range (i.e. 200–400 nm) (Fig. 2a). HL and 1 delivered similar absorption edges at ∼600 nm. These results suggested that the incorporation of BODIPY units remarkably improved visible-light harvesting ability of CTCs. The optical bandgaps (Eg) of HL, 1, 2 and 3 were estimated to be 1.97, 2.16, 1.76 and 2.94 eV, respectively by using the Tauc plot (Fig. 2b). The flat band potentials of HL, 1, 2 and 3 were determined to be −1.38, −0.80, −1.33 and −1.47 eV vs. NHE at pH = 7 through the Mott–Schottky experiments,37 respectively (Fig. S18–S21†), which were equal to their conduction band (CB) potentials. Combining the Mott–Schottky experiments and optical bandgap data, valence band (VB) potentials of HL, 1, 2 and 3 were calculated to be 0.59, 1.36, 0.43 and 1.47 eV vs. NHE at pH = 7 (Fig. 2c). Since the reduction potentials of O2(g)/O2˙− and O2(g)/H2O2 are known to be −0.35 and 0.28 V vs. NHE at pH = 7,38 they are thermodynamically suitable for photocatalytic reduction of O2 to give H2O2. In addition, the oxidation potential of H2O/O2 is 0.82 V vs. NHE at pH = 7;39 thus, compounds 1 and 3 are thermodynamically suitable for photocatalytic oxidation of water to produce O2 and protons.
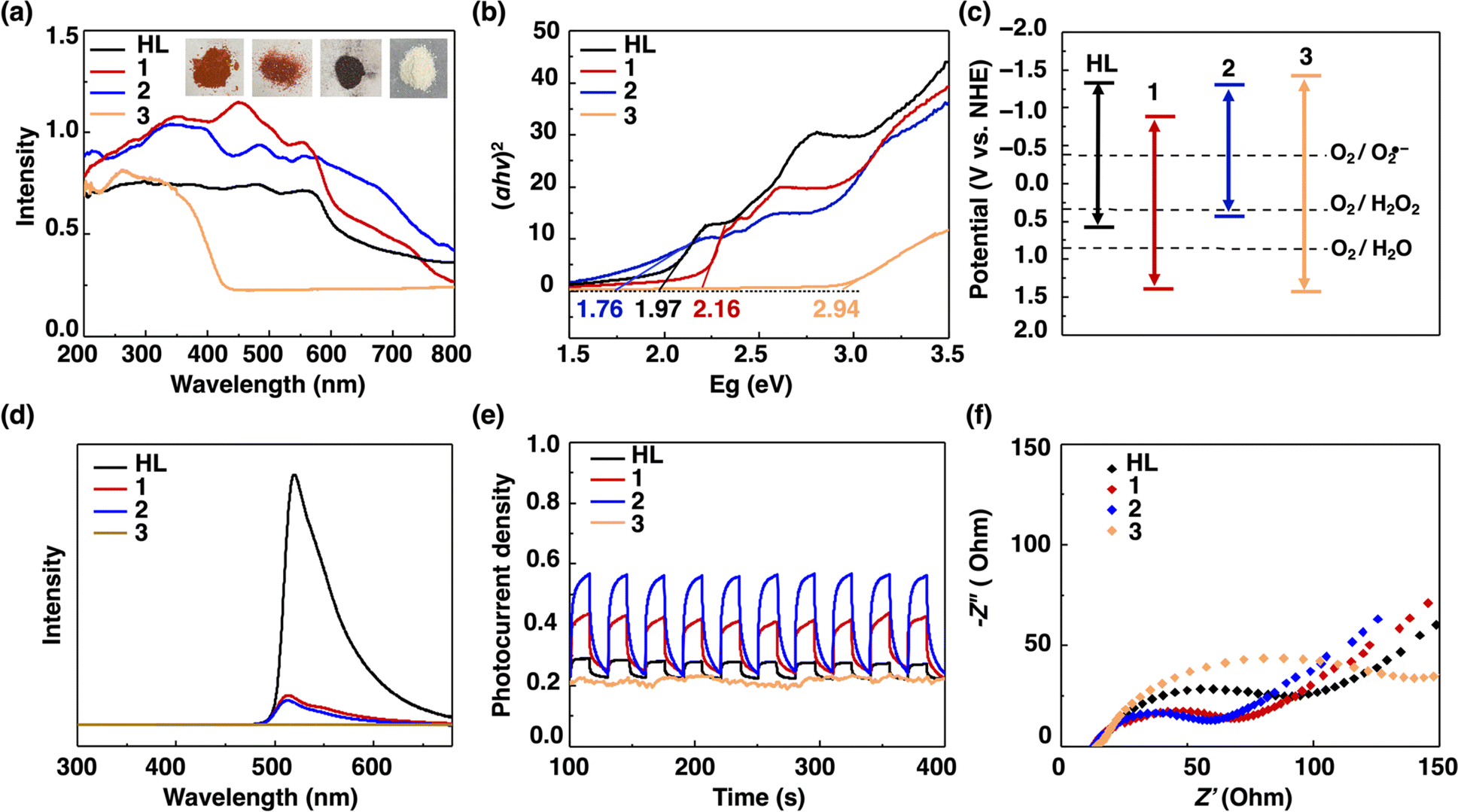 | ||
| Fig. 2 (a) The solid-state UV-vis diffuse reflectance spectra of HL, 1, 2 and 3. The inset shows the photographs of powder samples of HL, 1, 2 and 3. (b) The Tauc plot curves of HL, 1, 2 and 3. (c) Energy level diagrams of HL, 1, 2 and 3. (d) PL spectra of HL, 1, 2 and 3. (e) Photocurrent response curves of HL, 1, 2 and 3 (the unit is μA cm−1). (f) Nyquist plots of HL, 1, 2 and 3. | ||
To further probe the optical and photo-electrochemical properties of HL, 1, 2 and 3, photoluminescence (PL) spectra were obtained, and electrochemical impedance spectroscopy (EIS) and transient photocurrent measurements were performed. The steady-state PL spectra of HL, 1, 2 and 3 in the suspended-state were obtained (Fig. 2d). HL, 1 and 2 showed bright green emission with maximum emission peak (λem) positions at 514 nm, while the emission intensity of 3 was very weak (Fig. S24–S28†). Compounds 1 and 2 exhibited similar emission intensity, much lower than that of HL, suggesting that 1 and 2 possessed the highest separation efficiency of photoinduced electron–hole pairs and the presence of electron transfer.40 Moreover, 2 possessed the highest photocurrent density, while the transient photocurrent density of 1 was larger than that of HL and 3 under visible-light irradiation, implying an effective spatial separation of photogenerated charge carriers in 1 and 2 (Fig. 2e). The EIS spectra of HL, 1, 2 and 3 exhibited semicircles; meanwhile 1 and 2 delivered the lowest charge transfer resistance (Fig. 2f), indicating the highest charge-separation efficiency in 1 and 2.
Photosynthesis of H2O2
The visible-light photosynthesis of H2O2 was initially tried in pure water under an O2 atmosphere and the yield of H2O2 was determined by iodometric titration.41 The production rate of H2O2 with different amounts of photocatalyst 1 (i.e. 1, 2, 3, 4 and 5 mg) in 5 mL pure water was measured. It was found that 4 mg of 1 furnished the highest H2O2 production rate of 229.7 μmol g−1 h−1 (Fig. S33†). Fig. 3a shows the photocatalytic performances of HL, 1, 2 and 3, and a linear relationship between H2O2 production and irradiation time was observed under optimal conditions. With compound 1, the concentration of H2O2 gradually increased to 918.5 μM after 5 hours, and the production rate was estimated to be 183.7 μM h−1 (Fig. 3a). In sharp contrast, when HL, 2 and 3 were employed as photocatalysts, no H2O2 was detected (Fig. 3a). Notably, although Cu-CTC 2 exhibited better photoinduced charge separation efficiency than Ag-CTC 1, 2 was not able to produce H2O2 under light irradiation. This is because H2O2 decomposition is catalyzed by 2 (Fig. S35†), which has also observed for other reported Cu-CTC complexs.42 In addition, the unsuitable bandgaps of HL, and 2 for the 4e− WOR also hampered the photosynthesis of H2O2. Moreover, although Ag-CTC 3 had a suitable optic bandgap, the low light absorption ability impeded the photo-production of H2O2.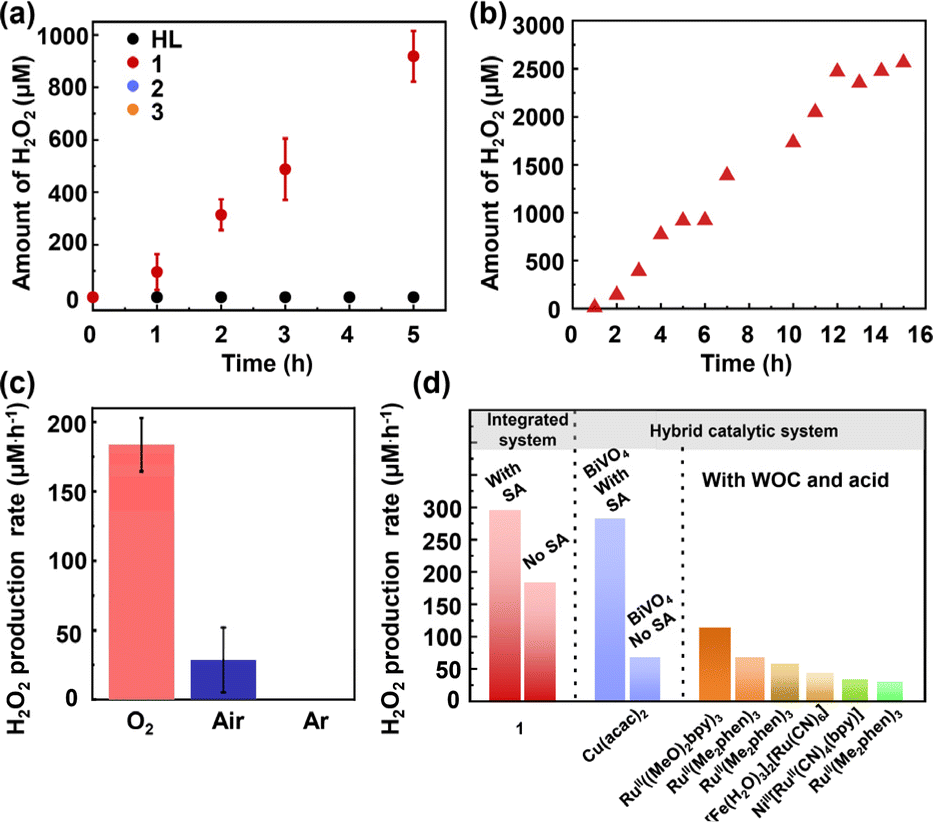 | ||
| Fig. 3 (a) Time course of photocatalytic H2O2 production for HL, 1, 2 and 3 irradiated with a 300 W xenon lamp fitted with a λ > 420 nm filter. (b) Long-term photocatalytic H2O2 production of 1. (c) Photocatalytic H2O2 production activities of 1 under different gas atmospheres. (d) Comparison of the H2O2 production rate with representative metal-based photocatalysts. The error bars denote ±s.d. of the mean for three independent experiments in (a) and (c). | ||
The long-term photostability of catalysts is practically important; thus, the continuous photosynthesis of H2O2 was conducted. In a continuous experiment (Fig. 3b), the H2O2 production rate leveled off after about 15 h using 1. The amount of H2O2 produced under pure O2 (99%) was 6.7 times greater than that in air, and no H2O2 was detected under a continuous Ar atmosphere, suggesting that O2 is essential for H2O2 production (Fig. 3c and see the ESI† for details). The apparent quantum yield (AQY) of 1 was measured determined to be 0.039%, 0.043%, 0.035%, 0.040% and 0.042% at 420, 450, 485, 520 and 535 nm, respectively (Fig. S38†), suggesting a visible-light promoted H2O2 production process. Compared to pure water, sea water is the most earth-abundant resource. Thus, photosynthesis of H2O2 from sea water is highly desired, yet is a significant challenge due to the complex composition and high salt concentration of seawater. Interestingly, 1 demonstrated an even higher H2O2 photo-production rate of 192.3 μM h−1 using seawater (Fig. S40 and S41†). Notably, 1 delivered the highest H2O2 production rate among reported metal-based photocatalysts (Fig. 3d).23,25,43–47 These results indicate that 1 is a promising photocatalyst for H2O2 production. However, with the increase in photo-generated holes and concentration of H2O2, the photo-catalytic performance decreased after 15 h of photo-irradiation, and the crystallinity of 1 declined as confirmed by PXRD analysis (Fig. S42†). These results suggested the moderate stability and durability of complex 1 during photosynthesis of H2O2. Nevertheless, our work presented a rare example of a Ag complex for photo-catalyzing both the ORR and OER.
To reveal the photocatalytic mechanism, several control experiments were conducted. Firstly, the H2O2 production rate increased to 237.8 μM h−1 with tert-butanol (TBA) as the sacrificial agent. The enhancement of the H2O2 production rate in the presence of TBA as electron donors indicated that ˙OH did not participate in the H2O2 production reaction (Fig. 4a).48 Secondly, the addition of AgNO3 as electron acceptors significantly decreased the H2O2 production rate to 30.5 μM h−1, indicating that photogenerated electrons played a vital role in the photocatalytic ORR. Thirdly, in the presence of La2O3, NaIO3 and 1 in pure water, O2 was detected and the production rate was estimated to be 81.8 μmol g−1 h−1 after 5 h of photo-irradiation (Table S3 and Fig. S44, S45†). This observation suggests that Ag-CTC 1 is able to directly photo-produce O2 from water through the 4e− OER. To further prove that O2 was photo-generated from water, the H218O isotopic labeling experiment was conducted.49,50 As shown in Fig. S46,† in the presence of 16O2 and H218O, the reaction mixture was irradiated after 12 hours. Afterward, the resulting solution was decomposed by MnO2, and 18O2 was observed by mass spectroscopy, indicating that H218O participated in the OER to generate 18O2, and the generated 18O2 was further captured in the ORR to convert H218O2. Finally, the addition of p-benzoquinone (BQ) as an O2˙− scavenger51 completely inhibited H2O2 production (Fig. 4a), suggesting that O2˙− was required during the photosynthesis of H2O2. These results are consistent with the observed optical properties of 1. For instance, the CB and VB of 1 also suggested that the 2e− ORR (i.e., O2 to H2O2) and 4e− WOR (i.e., H2O to O2) are thermodynamically possible, while e− (i.e., H2O to ˙OH) and 2e− WOR (i.e., H2O to H2O2) are thermodynamically prohibited.
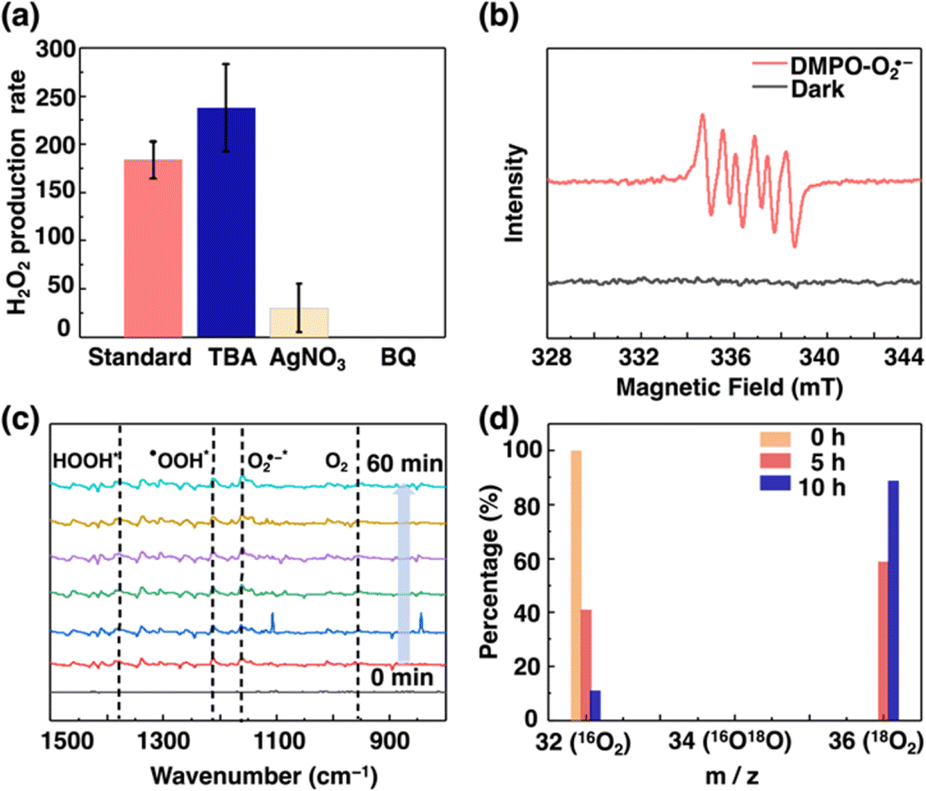 | ||
| Fig. 4 (a) Photocatalytic H2O2 production activities of 1 with the addition of different sacrificial agents (the unit is μM h−1). The error bars denote ±s.d. of the mean for three independent experiments. (b) EPR trapping experiments in the presence of DMPO before (gray line) and after (light red line) light irradiation. (c) In situ DRIFT spectra of 1 during H2O2 photosynthesis (the time interval is 10 min). (d) Isotopic 18O2 labeling experiments. | ||
To further study the reaction pathway of H2O2 production by Ag-CTC 1, the average electron transfer number of Ag-CTC 1 was determined to be 2.00 based on the results of rotating disc electrode (RDE) studies conducted on O2 reduction processes (Fig. S47†), which showed that Ag-CTC 1 could reduce O2 to H2O2 using the direct two-electron reduction pathway. Meanwhile, 5,5-dimethyl-1-pyrroline N-oxide (DMPO) was employed as the radical trapping agent for detecting O2˙− in electron paramagnetic resonance (EPR) measurements. No noticeable signals can be found under dark conditions, while the characteristic signals of DMPO-O2˙− appeared after light irradiation (Fig. 4b).52 Such results further supported the generation of O2˙− though the one-electron reduction of O2. In addition, the adsorbed intermediates on 1 during the H2O2 production process were revealed by in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS). As shown in Fig. 4c, the O–O stretching signals for O2 as well as O2˙−*, ˙OOH*, and HOOH* (* denoted as surface-adsorbed) were found at 954, 1160, 1211 and 1379 cm−1, respectively.53–55 Moreover, isotopic labeling experiments using 18O2 were also performed. The percentage of 18O2 detected by mass spectroscopy increased from 0% to 88.2% over 5 h and further decreased to 75.3% after 10 h (Fig. 4d), suggesting that the 16O2 generated by the OER would be captured in the ORR and converted into H216O2. Such results are consistent with the H218O isotopic labeling experiment, and reveal that the photosynthesis of H2O2 using Ag-CTC 1 involves stepwise ORR (i.e., O2 → O2˙− → H2O2)56 and the 4e− OER process (i.e., H2O → O2).
Theoretical studies
Density functional theory (DFT) calculations were conducted by Gaussian 09E (ref. 57) software to further study the reaction mechanism. The electrostatic potential (ESP)58,59 distribution is used to describe the charge distribution on the surface of molecule 1 (Fig. S53†). The electrostatic potential value surrounding the BODIPY moiety has a negative value, while the nine-membered Ag3N6 unit exhibits a positive value, suggesting that the BODIPY region has a higher affinity for electrons and is more electrophilic compared to the nine-membered Ag3N6 unit.60 Thus, the electrons prefer to be transferred from the Ag3N6 unit to the BODIPY motifs.1 In addition, the cyclic voltammetry (CV) test was performed to study their redox potentials. The CV curve of 1 exhibited two reversible redox processes with the peak potential at approximately −1.65 and −1.26 V (vs. Fc+/0), respectively, which can be assigned to the redox couple of AgII/AgI, and the BODIPY ligands (Fig. S52†). The more negative reduction potential of the Ag center compared to that of BODIPY ligands further suggests that the electron transfer process from the Ag to BODIPY unit is thermodynamic feasible.61 Moreover, as shown in Fig. 5a, the electrons were primarily localized at the BODIPY units either in the highest occupied or lowest unoccupied molecular orbital (HOMO or LUMO). According to the time-dependent density-functional theory (TDDFT) calculation results, it is indicated that BODIPY units contribute more in the transition from the ground state (S0) to the first 10 Sn states with an f larger than 0.01 (Table S4†). This further confirms that for 1, the electrons are mainly localized on the BODIPY units. Based on the above discussion, we propose that the mechanism for H2O2 production involves the ORR occurring on BODIPY, while the WOR takes place on the Ag3N6 unit. | ||
| Fig. 5 (a) HOMO and (b) LUMO of 1 in the optimized structure (isovalue = 0.05 a.u.). (c) Calculated free energy for H2O2 formation from O2 at the DFT-D3 (BJ) level of theory. (d) The proposed photocatalytic mechanism for H2O2 synthesis in the presence of photocatalyst 1 (* denoted as surface-adsorbed). | ||
For the ORR process, a plausible reaction mechanism is proposed in Fig. 5b. The adsorption energy (ΔEad) of O2 in the BODIPY region of 1 and 2 was calculated to be 0.2469 and 0.2709 eV, respectively (Fig. S55–S58 and Table S5†), suggesting that the adsorption of O2 either on 1 or 2 is thermodynamically infeasible. Nevertheless, the more negative ΔEad value of 1 compared to 2 indicates that O2 prefers to be absorbed on 1 (Fig. 5c). It is well known that the formation of the ˙OOH intermediate is a crucial step during the synthesis of H2O2;55,62,63 thus the Gibbs free energy of the ˙OOH intermediate (ΔGOOH*) adsorbed in the BODIPY region for 1 (i.e., 0.1502 eV) and 2 (i.e., 0.2664 eV) is assessed and compared (Fig. 5c). The more negative value of ΔGOOH* for 1 suggests that 1 is more active for ˙OOH intermediate production. Furthermore, the negative value of ΔGH2O2 (−0.8719 eV) produced on the BODIPY unit for 1 (Fig. 5c) implies that the production of H2O2 from the ˙OOH intermediate is thermodynamically favored.
For the 4e− WOR process, two H2O molecules are required to be adsorbed on the surface of the catalysts to produce one O2. Based on the above-mentioned results, the WOR possibly takes place on the Ag3N6 unit of 1.64 The ΔEad of the two H2O molecules on the Ag3N6 active site of 1 was also calculated to be −0.3806 eV (Fig. S59 and S60†), further evidencing that the adsorption of H2O on the Ag3N6 unit of 1 is thermodynamically favored. We calculated the energy path for water oxidation into oxygen. Based on the experimental results and calculation, a possible mechanism is proposed (Fig. 5d): initially, O2 is adsorbed on the BODIPY sites. After light irradiation, photoinduced electrons are produced at the BODIPY site, and then O2 obtains the electron and proton to generate the ˙OOH intermediate. Afterward, ˙OOH further obtains the electron and proton to produce H2O2. Meanwhile, two H2O molecules can be absorbed on the Ag3N6 site, and then photoinduced holes obtain electrons from H2O to produce O2 and protons.
Conclusions
In summary, we have prepared BODIPY decorated Ag-CTC 1, and it features a suitable energy bandgap, strong visible-light absorption, excellent photochemical properties and efficient charge separation ability. Owing to these merits, complex 1 can be used as a photocatalyst for producing H2O2 either from pure water or from sea water with high photocatalytic activities (183.7 and 192.3 μM h−1) in the absence of any additives (e.g., acid, co-catalysts, and sacrificial agents). The photocatalytic performance is attributed to the prominently enhanced two-electron ORR by forming endoperoxide at the BODIPY unit and highly concentrated holes at the Ag3N6 site. The generated O2 from the OER is subsequently consumed by the ORR, thereby boosting overall reaction kinetics. The photocatalytic mechanism was systematically investigated by various experimental analyses and DFT calculations. Our studies demonstrated that the integration of organic chromophores as ORR sites and silver centers as OER sites is a new approach for design of efficient metal-based photocatalysts for H2O2 production using solar energy.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for complex 1 has been deposited at the CCDC (DOI: https://doi.org/10.5517/ccdc.csd.cc2gqh2s) and can be obtained from https://www.ccdc.cam.ac.uk.Author contributions
G.-H. N., and D. L. designed the research; R.-Q. X. conducted the experiments and data analysis; R.-Q. X. contributed to data analysis and theoretical calculation; R.-Q. X., G.-H. N., and D. L. co-wrote the manuscript. All authors read and commented on the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
G.-H. N. is thankful for the financial support from the Guangzhou Science and Technology Project (202201020038). This project was supported financially by the National Natural Science Foundation of China (No. 22371091, 21975104, 21731002, 22150004 and 92261205) and the Guangdong Major Project of Basic and Applied Research (No. 2019B030302009). R.-J. W is thankful to the Open Fund of Guangdong Provincial Key Laboratory of Functional Supramolecular Coordination Materials and Applications (No. 2020B121201005).Notes and references
- J. N. Chang, J. W. Shi, Q. Li, S. Li, Y. R. Wang, Y. Chen, F. Yu, S. L. Li and Y. Q. Lan, Angew. Chem., Int. Ed., 2023, 135, e202303606 CrossRef.
- J.-N. Lu, L. J.-J. Liu, L.-Z. Dong, J.-M. Lin, F. Yu, J. Liu and Y.-Q. Lan, Angew. Chem., Int. Ed., 2023, 62, e202308505 CrossRef CAS PubMed.
- J.-Y. Yue, L.-P. Song, Y.-F. Fan, Z.-X. Pan, P. Yang, Y. Ma, Q. Xu and B. Tang, Angew. Chem., Int. Ed., 2023, 62, e202309624 CrossRef CAS.
- Y. Zhang, C. Pan, G. Bian, J. Xu, Y. Dong, Y. Zhang, Y. Lou, W. Liu and Y. Zhu, Nat. Energy, 2023, 8, 361–371 CrossRef CAS.
- W. Zhao, P. Yan, B. Li, M. Bahri, L. Liu, X. Zhou, R. Clowes, N. D. Browning, Y. Wu and J. W. Ward, J. Am. Chem. Soc., 2022, 144, 9902–9909 CrossRef CAS PubMed.
- J. Liu, Y. Zou, B. Jin, K. Zhang and J. H. Park, ACS Energy Lett., 2019, 4, 3018–3027 CrossRef CAS.
- K. Sato, M. Aoki and R. Noyori, Science, 1998, 281, 1646–1647 CrossRef CAS PubMed.
- R. J. Lewis, K. Ueura, X. Liu, Y. Fukuta, T. E. Davies, D. J. Morgan, L. Chen, J. Qi, J. Singleton and J. K. Edwards, Science, 2022, 376, 615–620 CrossRef CAS.
- Y. Sun, L. Han and P. Strasser, Chem. Soc. Rev., 2020, 49, 6605–6631 RSC.
- Y. Kondo, Y. Kuwahara, K. Mori and H. Yamashita, Chem, 2022, 8, 2924–2938 CAS.
- X. Zeng, Y. Liu, X. Hu and X. Zhang, Green Chem., 2021, 23, 1466–1494 RSC.
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. G. Fierro, Angew. Chem., Int. Ed., 2006, 45, 6962–6984 CrossRef CAS.
- F. Kuttassery, Y. Ohsaki, A. Thomas, R. Kamata, Y. Ebato, H. Kumagai, R. Nakazato, A. Sebastian, S. Mathew, H. Tachibana, O. Ishitani and H. Inoue, Angew. Chem., Int. Ed., 2023, 135, e202308956 CrossRef.
- T. J. Meyer, M. V. Sheridan and B. D. Sherman, Chem. Soc. Rev., 2017, 46, 6148–6169 RSC.
- Y. Kou, Y. Nabetani, D. Masui, T. Shimada, S. Takagi, H. Tachibana and H. Inoue, J. Am. Chem. Soc., 2014, 136, 6021–6030 CrossRef CAS PubMed.
- L. Sun, Science, 2015, 348, 635–636 CrossRef CAS PubMed.
- F. Zhou, C. McDonnell-Worth, H. Li, J. Li, L. Spiccia and D. R. Macfarlane, J. Mater. Chem. A, 2015, 3, 16642–16652 RSC.
- H. Inoue, T. Shimada, Y. Kou, Y. Nabetani, D. Masui, S. Takagi and H. Tachibana, ChemSusChem, 2011, 4, 173–179 CrossRef CAS PubMed.
- F. Kuttassery, S. Mathew, S. N. Remello, A. Thomas, K. Sano, Y. Ohsaki, Y. Nabetani, H. Tachibana and H. Inoue, Coord. Chem. Rev., 2018, 377, 64–72 CrossRef CAS.
- S. Fukuzumi, Y. M. Lee and W. Nam, ChemCatChem, 2018, 10, 9–28 CrossRef CAS.
- C. Qin, X. Wu and L. Tang, Angew. Chem., Int. Ed., 2024, 136, e202402297 CrossRef.
- H. Yu, F. Zhang and Q. Chen, Nat. Commun., 2023, 14, 5238–5250 CrossRef.
- S. Kato, J. Jung, T. Suenobu and S. Fukuzumi, Energy Environ. Sci., 2013, 6, 3756–3764 RSC.
- K. Mase, M. Yoneda, Y. Yamada and S. Fukuzumi, Nat. Commun., 2016, 7, 11470 CrossRef CAS PubMed.
- T. Suenobu, S. Shibata and S. Fukuzumi, Inorg. Chem., 2016, 55, 7747–7754 CrossRef CAS.
- Y. Wang, Y. Wang, J. Zhao, M. Chen, X. Huang and Y. Xu, Appl. Catal. B, 2021, 284, 119691 CrossRef CAS.
- L. Wang, J. Zhang, Y. Zhang, H. Yu, Y. Qu and J. Yu, Small, 2022, 18, 2104561 CrossRef CAS.
- H. Hou, X. Zeng and X. Zhang, Angew. Chem., Int. Ed., 2020, 59, 17356–17376 CrossRef CAS.
- K. C. Chong, C. Li and B. Liu, CCS Chem., 2023, 5, 2436–2447 CrossRef CAS.
- J. Wang, J. Zhang, S. B. Peh, G. Liu, T. Kundu, J. Dong, Y. Ying, Y. Qian and D. Zhao, Sci. China: Chem., 2020, 63, 192–197 CrossRef CAS.
- R.-Q. Xia, J. Zheng, R.-J. Wei, J. He, D.-Q. Ye, G.-H. Ning and D. Li, Inorg. Chem. Front., 2022, 9, 2928–2937 RSC.
- K. K. Chen, S. Guo, H. Liu, X. Li, Z. M. Zhang and T. B. Lu, Angew. Chem., Int. Ed., 2020, 59, 12951–12957 CrossRef CAS PubMed.
- H. R. Dias, S. A. Polach and Z. Wang, J. Fluor. Chem, 2000, 103, 163–169 CrossRef CAS.
- R.-Q. Xia, Z.-N. Liu, Y.-Y. Tang, X. Luo, R.-J. Wei, T. Wu, G.-H. Ning and D. Li, Experimental Crystal Structure Determination, 2024, DOI:10.5517/ccdc.csd.cc2gqh2s.
- O. Renn, L. M. Venanzi, A. Marteletti and V. Gramlich, Helv. Chim. Acta, 1995, 78, 993–1000 CrossRef CAS.
- C. Zhu, H. Zeng, C. Liu, Y. Cai, X. Fang and H. Jiang, Org. Lett., 2020, 22, 809–813 CrossRef CAS.
- J.-K. Jin, K. Wu, X.-Y. Liu, G.-Q. Huang, Y.-L. Huang, D. Luo, M. Xie, Y. Zhao, W. Lu and X.-P. Zhou, J. Am. Chem. Soc., 2021, 143, 21340–21349 CrossRef CAS PubMed.
- W. H. Koppenol, D. M. Stanbury and P. L. Bounds, Free Radical Biol. Med., 2010, 49, 317–322 CrossRef CAS.
- L. Liu, M.-Y. Gao, H. Yang, X. Wang, X. Li and A. I. Cooper, J. Am. Chem. Soc., 2021, 143, 19287–19293 CrossRef CAS PubMed.
- H. Lin, Y. Liu, Z. Wang, L. Ling, H. Huang, Q. Li, L. Cheng, Y. Li, J. Zhou and K. Wu, Angew. Chem., Int. Ed., 2022, 61, e202214142 CrossRef CAS.
- Z. Wei, M. Liu, Z. Zhang, W. Yao, H. Tan and Y. Zhu, Energy Environ. Sci., 2018, 11, 2581–2589 RSC.
- X. Li, J. Wang, F. Xue, Y. Wu, H. Xu, T. Yi and Q. Li, Angew. Chem., Int. Ed., 2021, 60, 2534–2540 CrossRef CAS PubMed.
- M. Teranishi, T. Kunimoto, S.-i. Naya, H. Kobayashi and H. Tada, J. Phys. Chem. C, 2020, 124, 3715–3721 CrossRef CAS.
- Y. Isaka, Y. Yamada, T. Suenobu, T. Nakagawa and S. Fukuzumi, RSC Adv., 2016, 6, 42041–42044 RSC.
- Y. Isaka, K. Oyama, Y. Yamada, T. Suenobu and S. Fukuzumi, Catal. Sci. Technol., 2016, 6, 681–684 RSC.
- Y. Isaka, S. Kato, D. Hong, T. Suenobu, Y. Yamada and S. Fukuzumi, J. Mater. Chem. A, 2015, 3, 12404–12412 RSC.
- Y. Aratani, T. Suenobu, K. Ohkubo, Y. Yamada and S. Fukuzumi, Chem. Commun., 2017, 53, 3473–3476 RSC.
- J. Yang, B. Pan, H. Li, S. Liao, D. Zhang, M. Wu and B. Xing, Environ. Sci. Technol., 2016, 50, 694–700 CrossRef CAS PubMed.
- D. Chen, W. Chen, Y. Wu, L. Wang, X. Wu, H. Xu and L. Chen, Angew. Chem., Int. Ed., 2023, 135, e202217479 CrossRef.
- Z. Teng, Q. Zhang, H. Yang, K. Kato, W. Yang, Y.-R. Lu, S. Liu, C. Wang, A. Yamakata and C. Su, Nat. Catal., 2021, 4, 374–384 CrossRef CAS.
- Y. Wang, Y. Xie, H. Sun, J. Xiao, H. Cao and S. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 9710–9720 CrossRef CAS.
- J.-L. Clément, N. Ferré, D. Siri, H. Karoui, A. Rockenbauer and P. Tordo, J. Org. Chem., 2005, 70, 1198–1203 CrossRef.
- J. N. Chang, Q. Li, J. W. Shi, M. Zhang, L. Zhang, S. Li, Y. Chen, S. L. Li and Y. Q. Lan, Angew. Chem., Int. Ed., 2023, 62, e202218868 CrossRef CAS.
- C. Zhao, X. Wang, Y. Yin, W. Tian, G. Zeng, H. Li, S. Ye, L. Wu and J. Liu, Angew. Chem., Int. Ed., 2023, 62, e202218318 CrossRef CAS PubMed.
- M. Kou, Y. Wang, Y. Xu, L. Ye, Y. Huang, B. Jia, H. Li, J. Ren, Y. Deng and J. Chen, Angew. Chem., Int. Ed., 2022, 61, e202200413 CrossRef CAS PubMed.
- H. Yang, C. Li, T. Liu, T. Fellowes, S. Y. Chong, L. Catalano, M. Bahri, W. Zhang, Y. Xu and L. Liu, Nat. Nanotechnol., 2023, 18, 307–315 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji and D. J. Fox, Gaussian 09 (Revision E . p. 01), Gaussian. Inc., Wallingford, CT, 2009 Search PubMed.
- H. Yao, Y. Cui, D. Qian, C. S. Ponseca Jr., A. Honarfar, Y. Xu, J. Xin, Z. Chen, L. Hong, B. Gao, R. Yu, Y. Zu, W. Ma, P. Chabera, T. Pullerits, A. Yartsev, F. Gao and J. Hou, J. Am. Chem. Soc., 2019, 141, 7743–7750 CrossRef CAS.
- J. S. Murray and P. Politzer, Rev.: Comput. Mol. Sci., 2017, 7, e1326 Search PubMed.
- J. Zheng, Z. Lu, K. Wu, G.-H. Ning and D. Li, Chem. Rev., 2020, 120, 9675–9742 CrossRef CAS PubMed.
- G.-Y. Wang, S. Guo, P. Wang, Z.-M. Zhang and T.-B. Lu, Appl. Catal. B, 2022, 316, 121655 CrossRef CAS.
- G. Han, X. Zhang, W. Liu, Q. Zhang, Z. Wang, J. Cheng, T. Yao, L. Gu, C. Du and Y. Gao, Nat. Commun., 2021, 12, 6335 CrossRef CAS PubMed.
- R. B. Rankin and J. Greeley, ACS Catal., 2012, 2, 2664–2672 CrossRef CAS.
- Y.-Y. Tang, X. Luo, R.-Q. Xia, J. Luo, S.-K. Peng, Z.-N. Liu, Q. Gao, M. Xie, R.-J. Wei, G.-H. Ning and D. Li, Angew. Chem., Int. Ed., 2024, e202408186 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04098h |
| This journal is © The Royal Society of Chemistry 2024 |