
Design of Bi-functional mixed oxide electrodes for selective oxidative C–C cleavage of glycerol to formate and synchronized green hydrogen production†
Arindam
Saha
 ab,
Vasantharadevi
Murugiah
a,
Ravi
Ranjan
ab,
Inderjeet
Chauhan
ab,
Kshirodra Kumar
Patra
a,
Himanshu
Bajpai
a,
Avisekh
Saha
ab and
Chinnakonda S.
Gopinath
*ab
ab,
Vasantharadevi
Murugiah
a,
Ravi
Ranjan
ab,
Inderjeet
Chauhan
ab,
Kshirodra Kumar
Patra
a,
Himanshu
Bajpai
a,
Avisekh
Saha
ab and
Chinnakonda S.
Gopinath
*ab
aCatalysis and Inorganic Chemistry Division, CSIR-National Chemical Laboratory, Dr Homi Bhabha Road, Pune 411 008, India. E-mail: cs.gopinath@ncl.res.in
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad 201002, India
First published on 22nd May 2024
Abstract
Alkaline water electrolysis is a mature method to produce green hydrogen; however, it suffers from significantly high cost as high overpotentials are required for the oxygen evolution reaction (OER). However, the OER could be avoided altogether by replacing it with kinetically favorable oxidation of abundantly available feedstock molecules at a significantly low potential to value-added product(s) together with green hydrogen generation. This is a potential method to address the high cost of green hydrogen production while converting waste to wealth. Herein, we report green, template-free hydrothermal synthesis of an electrochemically active NiCoMn mixed oxide (NCMO) electrocatalyst with multiple sites, porous structure, large surface area, and nanoneedle (NN) morphology deposited directly over Ni foam (NF). Sustainable electrocatalytic performance was demonstrated for 120 h in 0.2 M alkaline glycerol using chronoamperometry and chronopotentiometry. Highly selective formate production demonstrated an exclusive C–C cleavage with the present catalyst system. Oxides of individual metal-ions (Ni, Co, and Mn) and their bimetallic combination (NiCo, NiMn, and CoMn) exhibited lower activity and product selectivity than the trimetallic NCMO electrocatalyst. The membrane-free two-electrode electrolyzer setup with NCMO/NF at both the anode and cathode (NCMO/NF‖NCMO/NF) requires 1.63 V to accomplish 100 mA cm−2 with 0.2 M glycerol, which is 296 mV less than that of 1 M KOH solution. High faradaic efficiency was observed for hydrogen (98%) with highly selective formate (90%) production. Electrocatalytic formate generation from an alkaline glycerol solution with NCMO is an energy-efficient and promising approach that also supplies carbon-negative green H2.
Introduction
The global research community has long been aware of the incubating problem of climate change, mainly due to CO2 pollution.1 Owing to climate change, mankind has witnessed several climate catastrophes in the 21st century, such as soaring temperatures and record rainfall causing deadly floods, across the globe, thereby resulting in economic challenges.2 These challenges seeking possible solutions for clean-energy production without damaging the climate are looming large currently, and they need to be answered soon.3,4 Recently, Domen's group5 reported a proof of concept for the promising utilization of sunlight for water splitting; similarly, Salgaonkar et al.6 reported a proof of concept for artificial photosynthesis of methanol and formaldehyde from CO2 in direct sunlight. Utilizing water as a bulk and cheap feedstock with power generated through Si-PV has attracted attention to address the green hydrogen issue through water electrolysis. Although industrial electrolyzers have been proven to produce green H2 on a large scale, high operating expenses (opex), apart from employing noble metal catalysts and membranes, increase the capex and necessitate cheaper alternatives.Recently, glycerol-assisted water electrolysis using transition metal oxides7–9 has been highlighted as a preferred way towards concurrently achieving economically viable green H2 and value-added products (VAPs). Emerging bimetallic and multi-metallic materials have underscored the enormous scope with multiple cooperative oxidations sites10–13 for the selective oxidation of cheap building-block biomass component molecules to VAP and concurrent green H2 generation.8,14 NiCoMn mixed oxide (NCMO) has been recently employed for water splitting.15–17 Although the anode potential for NCMO (1.26 V for 10 mA cm−2 for OER) resides around that of a noble metal catalyst (RuO2@NF anode at 1.60 V for 10 mA cm−2),18 it offers a more cost-effective solution owing to the expensive nature of ruthenium. However, it is essential to decrease the opex, which is a critical challenge in water electrolysis. To answer this, many efforts have been made for the oxidation of cheaper biomass-derived molecules such as glycerol, glucose, and 5-hydroxymethylfurfural at low potentials.19–24 These carbon sources are selectively oxidized at the anode, and the OER is completely avoided. This strategy is expected to favorably lower the opex and generate VAP from the biomass components. However, the finer challenge is to make a catalyst that operates at low oxidation potentials with the highly selective formation of VAPs. Glycerol25 contains hydrophilic hydroxyl groups to ensure solubility in water. A suitable electrocatalyst with a highly hydrophilic surface is likely to work better while bringing down the anodic potential; the advantage of interacting hydroxyl ligands26 on the electrocatalyst surface contributes to faster redox reactions with transition metal catalysts.
Biodiesel has attracted the attention of several researchers. Crude glycerol is a byproduct (10% w/w) generated during the transesterification process in the biodiesel plant,27 and expected to grow annually by 4.5%.28 However, glycerol can be electrochemically oxidized to several products such as dihydroxyacetone, glyceric acid, glyceraldehyde, acetic acid, methanol, and formate.14,29 Hence, the challenge is to find an electrocatalyst that converts glycerol selectively to one product predominantly. Glycerol to single carbon-containing products such as formic acid and methanol require the electrocatalyst to primarily cleave both C–C bonds in glycerol.7,26,30 Indeed, an earlier report shows promising results for the conversion of glycerol to formate and hydrogen with a Ni–Mo–N/carbon cloth; however, it suffers from long-term stability due to the leaching of Mo from the anode.31 Industrial formic acid production by methyl formate hydrolysis is typically labor- and energy-intensive, and relies on raw materials such as CO from natural gas or coal gasification.32,33 While glycerol, a byproduct of the biodiesel industry, must be processed downstream to avoid unwanted pile up, converting this feedstock selectively via an electrochemical means could solve the existing problem and produce VAPs. Furthermore, the theoretical oxidation potential needed to oxidize one glycerol molecule to formic acid is 0.69 V (ESI note 1†),31 which is about 50% that of the theoretical water oxidation34 (1.23 V) under the same conditions; hence, it is prudent to deploy glycerol oxidation at the anode.
Unlike costly noble metals, abundant first-row transition metal-based oxide catalysts with green methods of preparation, high activity, and chemical stability under alkaline conditions make them industrially relevant. Herein, we present a template-free green synthesis method for preparing a NCMO electrocatalyst directly bound to a nickel foam (NF), which is designated as NCMO/NF. The high surface area with a porous structure facilitates the diffusivity of reactants to multiple active sites simultaneously. Further, the catalyst shows bifunctionality (as both the anode and the cathode), with extended hours of operation and fast reaction kinetics to oxidize glycerol to formate, having high selectivity at ambient temperature. A minimal voltage of 1.21 V (vs. RHE) was needed to produce 10 mA cm−2 current density in a 3-electrode system, while a membrane-free two-electrode setup required 1.38 and 1.64 V to obtain current densities of 10 and 100 mA cm−2, respectively; this is in fact 251 mV (10 mA cm−2) and 296 mV (100 mA cm−2) lower than that of the water splitting in 1 M KOH. Utilizing earth-abundant metal oxide electrocatalysts to augment sustainable reactions in a membrane-free setup with selective formate and carbon negative green H2 is expected to be attractive from a green chemistry perspective.
Experimental
Materials and methods
Glycerol (C3H8O3, ≥99.0%, CAS# 56-81-5), nickel(II) nitrate hexahydrate (Ni(NO3)2·6H2O, ≥97.0%, CAS# 13478-00-7), cobalt(II) nitrate hexahydrate (Co(NO3)2·6H2O, ≥98.0%, CAS# 10026-22-9), manganese(II) nitrate tetrahydrate (Mn(NO3)2·4H2O, ≥98.0%, CAS# 13446-34-9), urea ((NH2)2CO, ≥99.5%, CAS# 57-13-6), and deuterium oxide (D2O, ≥99.5%, CAS# 7789-20-0) were purchased from Sigma-Aldrich. Potassium hydroxide (KOH, ≥85.0%, CAS# 1310-58-3) and isopropyl alcohol ((CH3)2CHOH, ≥98.5, CAS# 67-63-0) were procured from Merck. All the chemicals were used as received unless otherwise mentioned. The NF (1.6 mm width) was purchased from Global Nanotech (Mumbai, 400062) and was subjected to hydrophilic treatment (sonicated for 1 min in 3 M HCL, followed by sonication in deionized (DI) water for 1 min) to remove the surface oxide before use. KHP (potassium hydrogen phthalate) was employed as an internal standard for the quantification of all the products observed in 1H NMR. The test solution was directly extracted from the electrolyzer. Then 130 μL D2O and 20 μL KHP were added to 450 μL of solution extracted from the electrolyzer and thoroughly mixed and homogenized in an NMR tube. Post dilution, the KHP concentration reached 1 mM, and its integrated peak area was employed for the quantification of various products. The concentration of liquid products formed after the electrocatalytic reaction was calculated using the following equation:where nx represents the molar concentration of KHP, Ix represents the integral area in the 1H NMR spectra for KHP, and Nx is the number of nuclei (4 equivalent protons for KHP appearing at 7.25 ppm). Similarly, ny was calculated from the above-mentioned formula for the liquid product, Iy is the integral area of the product formed and Ny is the number of nuclei associated with the peak. The concentration of products obtained from the 1H NMR spectra was used to calculate the conversion, selectivity, and carbon balance (ESI note 2†).
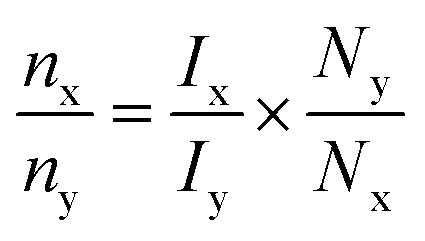
Direct synthesis of NiCoMn mixed oxide (NCMO) on the Ni-foam
A trimetallic NCMO catalyst was prepared using a template-free green synthesis method. Briefly, the catalyst was grown directly over a NF support by using 70 mM of Co(NO3)2·6H2O, 70 mM of Ni(NO3)2·6H2O, and 70 mM of Mn(NO3)2·4H2O; all three metal-ion precursors were added in a equimolar metal ratio, with 30 mM urea dissolved in 30 mL of Milli-Q water to form a pale reddish pink colour solution. This precursor mixture was kept under stirring for 30 min. The solution was then transferred into a 60 mL Teflon-lined stainless-steel autoclave. The 2 × 2 cm2 acid-treated NF was covered with Teflon tape from the top (1 × 2 cm2), and the remaining 1 × 2 cm2 was uncovered and submerged into the solution to obtain the selective deposition of the metal precursors on the uncovered NF. Then, the autoclave was put into a hot air convection oven; the oven was heated at a ramping rate of 2 °C min−1 to 140 °C and held at the same temperature for 1 h, followed by natural cooling. After the hydrothermal reaction, uniform deposition was observed over the uncovered NF (see Fig. S1†), resulting in NiCoMn-hydroxide-coated electrodes. The Teflon tape was removed from the NF, and the catalyst section was meticulously washed several times with DI water to remove any loosely held particles. The as-grown hydroxide-deposited catalyst was heated at a rate of 2 °C min−1 in a quartz tube furnace under an Ar flow to 300 °C and held at the same temperature for 2 h, followed by natural cooling to ambient temperature under the continuous Ar flow to obtain the final NCMO catalyst. The same procedure was also applied to make monometal and bimetal combinations of electrocatalysts such as NiCo, NiMn, MnCo, and Co. The deposited amount of catalyst, after the calcination, was found to be ∼3 ± 0.2 mg cm−2.Characterization
The morphological and compositional information was obtained by field emission scanning electron microscopy (FESEM; Nova Nano SEM 450). A Tecnai T-20 instrument at an accelerating voltage of 200 kV was used for recording transmission electron microscopic (TEM) images. High-resolution TEM (HRTEM) and HAADF-STEM mapping were performed using a JEOL JEM F-200 HRTEM electron microscope. The TEM and HRTEM samples were prepared by drop-casting the thoroughly dispersed catalysts in isopropanol on a carbon-coated 200 mesh lacey carbon grid followed by overnight drying. The crystallinity of the catalyst was investigated by X-ray diffraction (XRD) using a Rigaku SmartLab X-ray diffractometer with Cu Kα radiation (l = 1.5406 Å), at a scan rate of 2° min−1. The textural properties were investigated from N2 adsorption–desorption isotherms, measured using a Quantachrome quadrasorb instrument version 2.0; surface area and pore size distribution were calculated by adopting the BET (Bruner–Emmett–Teller) and BJH (Barrett–Joyner–Halenda) models, respectively. X-ray photoelectron spectroscopy (XPS) analysis was performed using a Thermo Scientific Kα, fully integrated, monochromatic small-spot XPS system with Al Kα as the X-ray source. The solid–liquid contact interface between the catalysts and water was characterized using an optical contact angle system (KRUS drop shape analyzer). A 500 MHz NMR (Bruker) spectrometer was employed to analyze the chemical composition of product molecules in the liquid phase with a water suppression sequence.Electrochemical studies
Electrochemical experiments were performed using a Gamry 3000 potentiostat in a 1 M KOH electrolyte at ambient temperature (25 °C). Hydrogen evolved as a single gaseous product, which was confirmed by GC (gas chromatograph, Agilent 7890A) measurements. The activity and durability of the electrocatalysts were measured by cyclic voltammetry (CV), linear sweep voltammetry (LSV), chronopotentiometry, and chronoamperometry using a three-electrode setup. The NCMO/NF was employed as the working electrode with an active area of 1 cm2, Hg/Hg2Cl2 as the reference electrode, and graphite rod as the counter electrode. To evaluate the HER performance of the samples, LSV was recorded from 0.20 to −0.60 V vs. RHE at a scan rate of 5 mV s−1. To estimate the OER activity of the materials, LSVs were recorded from 0.4 to 1.5 V vs. RHE at a scan rate of 5 mV s−1. Chronoamperometric performance was evaluated at 1.4 V vs. RHE. Chronopotentiometry performance was evaluated at current densities of 10 and 50 mA cm−2 in 3 electrodes (up to 8 h) and 2 electrodes (up to 120 h), respectively. All the potentials mentioned in the work are based on the RHE scale by calibrating Hg/Hg2Cl2 in a KCl-saturated solution using the following eqn (1):| E(vs. RHE) = E(vs. Hg/Hg2Cl2) + 0.241 V + (0.059 × pH) V | (1) |
The faradaic efficiency (FE) of hydrogen production was evaluated using the following equation:
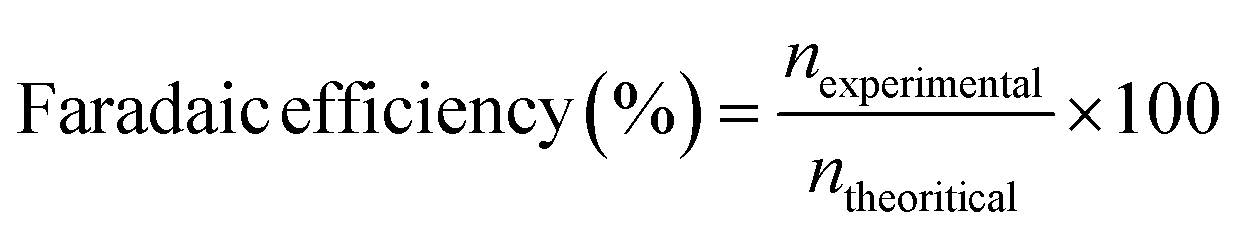
where Q is the transferred charge, n is the number of electrons transferred to form each H2 molecule and F is Faraday's constant (96
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1).
485 C mol−1).
The electrochemical impedance spectroscopy (EIS) measurements were performed at the open-circuit voltage, while the frequency limits ranged from 100 Hz to 200 kHz with a voltage amplitude of 10 mV. The electrochemical active surface area (ECSA) of each electrode was evaluated via the electric double-layer capacitance (Cdl), which was determined by the CV measurements between 0.85 and 0.95 V vs. RHE with different scan rates (2–14 mV s−1) in N2-saturated 1 M KOH with 0.2 M glycerol, and the ECSA was calculated using the following equation (eqn (2)):
 | (2) |
The specific capacitance was noted as 40 μF cm−2 for oxides.7,13
Results and discussion
Structural and textural analysis
Powder XRD is a crucial technique to characterize the crystalline structure and related details of the material under consideration. The XRD results obtained from NCMO/NF are shown, along with NF, in Fig. 1a. Apart from the three narrow features of NF, a few other broad and low-intensity features are observed in NCMO. This suggests the nanocrystalline nature of NCMO grown over the NF support, with a crystallite size of 1.92 nm, calculated using the Scherrer equation. The metal oxide has a homogenous and nanocrystalline distribution of three metal oxides together. The inset in Fig. 1a shows the FESEM of NCMO, and the same demonstrates 1D NCMO nanoneedles (NCMO NNs). The characteristic diffraction peaks observed in Fig. 1a indicate that NCMO NNs exhibit a multi-phase nature. A good correlation is observed between the observed XRD features with the standard JCPDS patterns of NiO (JCPDS no. 96-153-4271), Co3O4 (JCPDS no. 96-900-5890), and Mn3O4 (JCPDS no. 96-154-1154) distributed evenly over each NN. It should be noted that the broad XRD peaks with low intensity are ascribed to the typical overall nanocrystalline characteristics of the resulting NCMO NNs grown over NF. Fig. 1b shows the N2 adsorption–desorption isotherm measured at 77 K. The result implies a type-IV adsorption isotherm, and the hysteresis indicates H3 type, possibly with narrow and open mesopores. The inset in Fig. 1b shows the pore size distribution; 4.5 nm diameter pore size seems to contribute to the maximum. The surface area, measured by the BET model, was found to be 109.8 m2 g−1. | ||
| Fig. 1 (a) Wide-angle XRD pattern of NCMO/NF and NF, and (b) N2 adsorption–desorption isotherm recorded at 77 K and pore size distribution of NCMO. | ||
The chemical composition and oxidation state of NCMO/NF were characterized by XPS measurements, and the corresponding results are shown in Fig. 2. The survey spectrum (Fig. S2†) shows the presence of all three cations, namely, Ni, Co, and Mn present on the surface; however, not in equal proportions which is in contrast to the nominal input ratio (1:1:1). Charge calibration was performed by using a C 1s peak, which appears on metallic surfaces at 284.6 eV. The cobalt 2p core-level spectra (Fig. 2a) show the presence of spin–orbit doublets with strong satellite features. Deconvolution was carried out using the Casa XPS software, and the Shirley background removal procedure was applied. The Co 2p spectrum was deconvoluted, and it displays characteristics peaks of Co2+ oxidation state and hydroxide (Co(OH)2) at 780.0 and 780.5 eV, respectively, along with subpeaks at 782.3 eV and two satellite peaks at high binding energy (BE).35 XPS analysis confirms the presence of both oxide and hydroxide on the NCMO/NF surface, which is consistent with the XRD results (Fig. 1a), indicating their presence in the bulk of the material as well. Ni 2p3/2 and 2p1/2 spin–orbit doublet shows multiple splitting and satellites, as depicted in Fig. 2b; indeed, the results are consistent with the literature data reported for the NiO phase.36 The XPS spectra of Ni show the characteristic peaks of Ni2+ oxidation state, expected for NiO, and hydroxide Ni(OH)2 at 854.0 and 855.8 eV, respectively, along with broad satellite peaks around 861 eV.37 The red peak (853.3 eV) fitted originates from the NF, and the same was confirmed from the XPS analysis of the NCMO prepared on the Cu-foam; the red colour peak is not observed on the Cu-foam (Fig. S3†), which reiterates that the catalyst preparation might have led to Ni-oxidation.
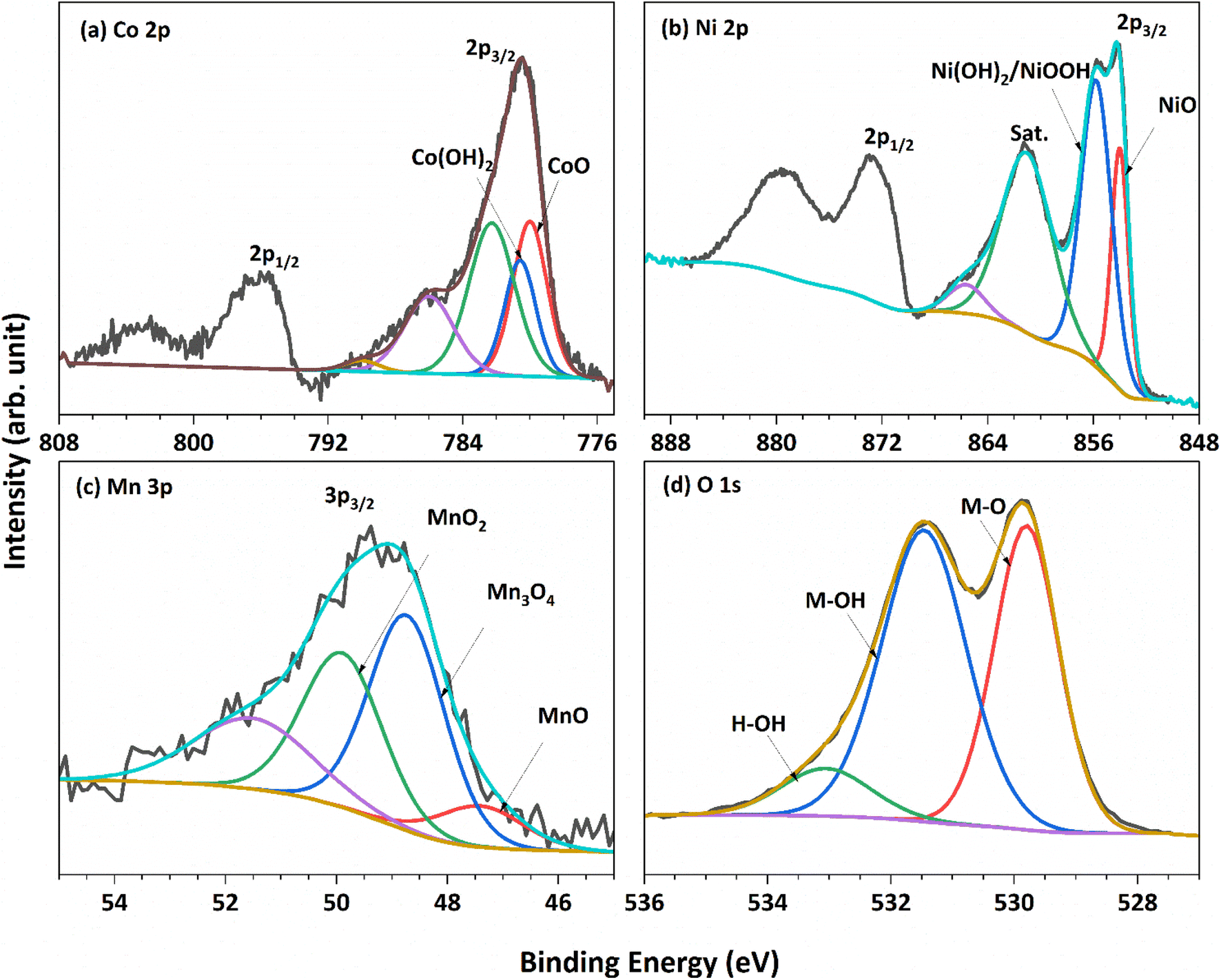 | ||
| Fig. 2 XPS spectra recorded for NCMO for core levels of all constituent elements. (a) Co 2p, (b) Ni 2p, (c) Mn 3p, and (d) O 1s. | ||
The Mn 2p core level overlaps with Auger peaks from Ni-ions, and hence, the Mn 3p core level was explored. The Mn 3p (Fig. 2c) shallow core-level spectrum was fitted with Gaussian–Lorentzian combination peaks at 47.4, 48.7, and 49.9 eV, corresponding to Mn2+, Mn3+ and Mn4+ respectively,38 as reported by Shaju et al.39 The presence of variable Ni3+ and Mn3+ is attributed to electronic redox transformation between Ni2+ and Mn4+ represented as Ni4+ + Mn2+ = Ni3+ + Mn3+. The core-level spectra of the O 1s core level (Fig. 2d) present three distinct features, and they are deconvoluted for clarity. The features at 529.8 and 530.4 eV correspond to metal–oxygen bonds and metal hydroxides, respectively. The peak at 533.0 eV is attributed to adsorbed water and/or carbonate.37
Electron microscopy was employed to observe the morphology and elemental mapping to understand the heterogeneity of the distribution of various elements. Fig. 3a shows the FESEM images of a collection of the NNs, having a cactus-like morphology with vertical 1D NNs, projected out perpendicularly from the center of the cactus. A similar NN-like structure was also observed emanating from the top of the NF (see Fig. S4†) throughout. Although the cactus size is observed to be between few hundred nanometers to 2 μm, 1D NNs attached to it make the morphology attractive, with the width of few nanometers and a length of few hundred nanometers (Fig. 3b and c). The NN makes the entire surface area available for the reaction with high surface area and porosity. The TEM and HRTEM images were recorded after prolonged sonication of NCMO in isopropanol. The NN morphology shows the integrity that kept the structure intact, even after breaking from the initial cactus-like morphology. A careful analysis of TEM results reveals the following information about the NNs. The needle-like NCMO with tip diameters ranging from a few nanometers to several tens of nanometers can be seen in Fig. 3c, which agrees with the previous FESEM results. Notably, the high-resolution TEM image in Fig. 3d reveals a porous nature of NNs (see also Fig. S4c and d†). Such a porous structure is beneficial in facilitating electrolyte diffusion to the inner surface of electrodes and increasing the electrode/electrolyte contact area; this also enhances the possibility of effective utilization of charges from every active electrocatalyst site, consequently enhancing the electrochemical performance. The enlarged version of NNs shown in Fig. 3c and d (shown in Fig. S3c and d†) displays a highly porous nanoneedle structure. The HRTEM elemental maps in Fig. 3e confirm the homogenous distribution of Ni, Co, and Mn elements in the nanoneedle structure.
 | ||
| Fig. 3 (a) FESEM image of as-grown NCMO/NF. (b and c) TEM image of a collection of NNs. (d) HRTEM image identified with lattice fringes (inset shows single NN). (e) STEM-EDX mapping of NCMO. | ||
The EDX data (see Fig. S5†) confirms the presence of all three cations (Ni, Mn and Co) in the nanoneedle structure. The amount of the metal components present in the oxide prepared are estimated from the EDX mapping and shown in the inset of Fig. S5a.† From the large-area analysis, the atomic ratio of Ni/Co/Mn in the NCMO nanoneedles was estimated to be 5/5/1. It should also be noted that the chemical mapping displayed in Fig. S5b† shows 5/2/1 for Ni/Co/Mn, and hence, there are local variations; nonetheless, the composition is always dominated by Ni, and Mn appears to be minimum. ICP-OES (inductively coupled plasma-optical emission spectroscopy) analysis was also performed to find out the bulk composition of NCMO. The atomic percentage of Ni (51.7%), Co (32.8%), and Mn (15.5%) leads to 3.4:2:1 ratio of Ni:Co:Mn. As noted above, local variation occurs in the chemical composition data obtained from EDX; nonetheless, the results are in agreement with each other. The bulk composition of NCMO was estimated to be Ni0.52Co0.33Mn0.16Ox. The length and width of the NN range between 700–900 nm and 15–25 nm, respectively. This leads to a high aspect ratio of 30–60, which helps faster diffusion of reactants and products.
Electrocatalytic performance with OERs/GORs
To investigate the oxidative electrocatalytic performance of NCMO for the glycerol oxidation reaction (GOR), a series of electrochemical measurements were performed in a three-electrode setup. Fig. 4a presents the LSV curves of NCMO/NF and its bimetallic and monometallic oxide counterparts in 1.0 M KOH with 0.2 M glycerol, recorded in a three-electrode setup. NCMO/NF was evaluated for water splitting with and without glycerol, and LSV results are given in the inset in Fig. 4a. The effect of glycerol concentration in the electrolyte on the GOR performance was also investigated; the concentration of glycerol has been optimized in a separate study, and the results are shown in Fig. S6.† The highest GOR activity at the lowest potential was achieved with 0.2 M glycerol in a 1 M KOH solution. In the absence of glycerol, the electrode shows moderate OER activity, which reaches the anodic current densities of 10 and 100 mA cm−2 at potentials of 1.26 and 1.56 V (vs. RHE), respectively (see Fig. S7†). The peak centered at 1.4 V (vs. RHE) can be ascribed to the oxidation peak of Ni2+/Ni3+ by forming NiOOH,40 and often Ni4+ species26 could be observed during the GOR. After introducing 0.2 M glycerol, the current density increased markedly, and the anodic potential decreased to 1.21 V (vs. RHE) to achieve 10 mA cm−2. In terms of the performance of the NCMO catalyst, 100 and 200 mA cm−2 current densities were achieved at an applied potential of 1.36 and 1.43 V (vs. RHE), respectively (see Fig. S6†), which is far lower than the potential required without glycerol for the OER. A direct comparison of electrocatalytic performance with and without glycerol is evident from the results shown in the inset of Fig. 4a; a reduction in anodic potential by 200 and 180 mV for 100 and 200 mA cm−2 current densities was observed with glycerol, compared to without glycerol, underlining the superior performance of NCMO. Indeed, this performance is significantly higher than that of reported systems with glycerol.41–44 The bimetallic combinations (NiCo, NiMn, and CoMn), as well as monometallic component (Ni, Co, and Mn) electrodes, were also prepared and tested (Fig. S8†). It is directly evident that the trimetallic NCMO catalyst exhibits superior performance over any of the bimetallic and monometallic oxides, both in 1 M KOH (Fig. S8†) and 1 M KOH mixed with 0.2 M glycerol (Fig. 4a).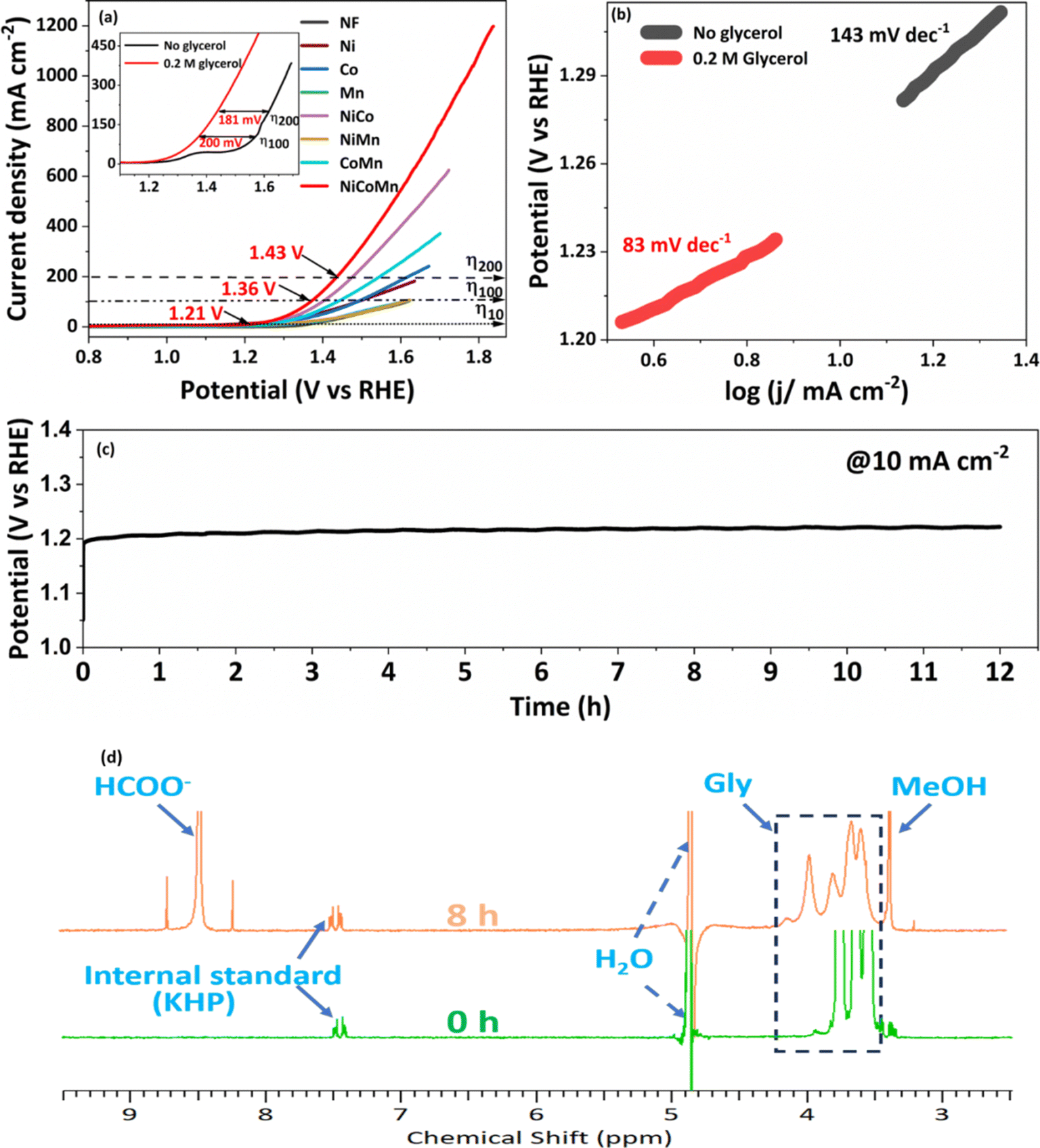 | ||
| Fig. 4 Electrocatalytic performance of NCMO/NF evaluated with aqueous glycerol. (a) LSV result observed from NCMO/NF, bimetallic combinations (NiCo, CoMn, and NiMn), and all three monometal catalysts coated on NF as the anode in 1.0 M KOH with 0.2 M glycerol (Gly) solution. The same scan rate of 5 mV s−1 was employed for all measurements. The inset shows the LSV curve of the NCMO/NF catalyst with and without glycerol (0.2 M). (b) Tafel plots of the anodic GOR and OER derived from the inset of (a). (c) Sustainable chronopotentiometry test for the anodic GOR with NCMO/NF at a current density of 10 mA cm−2. (d) 1H NMR spectra of the reactant at 0 h and product + reactant after 8 h GOR on the NCMO/NF electrode. This demonstrates the predominant conversion of glycerol to formate; KHP was used as an internal standard for quantification. | ||
The Tafel slope analysis provides insights into the activation energy and the rate of reaction. The NCMO shows the smallest Tafel slope (33 mV dec−1, Fig. 4b) for the GOR, which is far lower than that of the OER (113 mV dec−1), indicating a faster kinetics with the GOR. Noticeably, NCMO performs significantly better than the NF, Ni, Co, Mn, and bimetallic combinations loaded over NF. Compared to the lowest Tafel slope (33 mV dec−1) observed with NCMO, all other catalysts show higher values: NF (84 mV dec−1), Ni/NF (75 mV dec−1), Co/NF (41 mV dec−1), Mn/NF (74 mV dec−1), NiCo/NF (49 mV dec−1), NiMn/NF (64 mV dec−1), and CoMn/NF (61 mV dec−1), as shown in Fig. S9.† The lowest Tafel slope with NCMO indicates the possible synergetic effects45 of all three metal ions towards the GOR. According to the EIS results shown in Fig. S10† and the corresponding fitting results, NCMO/NF electrode possesses a much smaller charge transfer resistance (Rct = 4.6 Ω cm2) than that of NF (45.9 Ω cm2), monometals (21.2 Ω cm2) and bimetals (16.6 Ω cm2). This observation suggests a low electric impedance and a fast charge transfer rate (or low resistance) with the trimetal combination catalyst and the electrolyte. With the increase in calcination temperature, the possibility of oxidation of core metallic Ni in NF to its oxides can increase the resistivity and decrease the efficiency of charge transfer and, eventually the GOR rate. The electrochemically active surface areas (ECSAs) of NCMO/NF, NiCo/NF, NiMn/NF, CoMn/NF and Ni foam electrodes were determined by measuring double-layer capacitances (Cdl) via CV tests, as can be seen in Fig. S11a.† The Cdl values derived from the CV curves, recorded in a voltage range without any faradaic current, form the basis for calculating ECSA. Among them, NCMO/NF shows a much higher electrochemical double-layer capacitance (Cdl) value (254.3 mF cm−2) than that of NiCo (136.2 mF cm−2), NiMn (142.2 mF cm−2), and CoMn (121 mF cm−2) in Fig. S11b.† Very low scan rates were employed, as mentioned in Fig. S11a.† The ECSA values of various catalysts were observed as follows: NCMO/NF (6333 cm2), NiCo (2216 cm2), NiMn (1626 cm2), and CoMn (1216 cm2). This indicates a very high ECSA, and most exposed active sites are present with trimetallic NCMO/NF. The ECSA normalized current output in Fig. S12† suggests the superior contribution of higher surface-mediated activation and reactions for the GOR. This is also supported from the high BET surface area. The FE of the formate produced at different potentials is shown in Fig. S13.† This shows the highest FE at 1.4 V (vs. RHE), and decreased on either side of 1.4 V.
In view of the superior performance of NCMO, the same is explored in detail, and further studies are restricted to this composition only. The catalyst was explored to oxidize glycerol under different conditions to increase the selectivity of formate and generate green hydrogen on the counter electrode. Moreover, the electrochemical stability of the NCMO/NF electrode for the GOR was evaluated by recording time-dependent chronopotentiometry performance at 10 mA cm−2 (Fig. 4c). No significant increase in potential was observed during the 12 h stability test, elucidating the robust durability of the anode. The 1H NMR spectrum of the products reveals the presence of formate predominantly; however, a small amount of methanol and some unreacted glycerol was also found (Fig. 4d). Product quantification was carried out by employing KHP as an internal standard6,46 in the test solution.
To explore the sustainability of the catalyst for the GOR, an extended chronoamperometry test was carried out with NCMO/NF at 1.4 V (vs. RHE) for 8 h, and the result is shown in Fig. 5a. High-performance liquid chromatography (HPLC) analysis was also conducted to confirm the NMR results. An applied potential of 1.4 V (vs. RHE) was selected to study chronoamperometry, as this potential completely avoids the OER, while the GOR is allowed. The results indicate that formate is the main oxidation product of glycerol, demonstrating a very high selectivity (90%). The chronoamperometry at the same potential with bimetallic combination has been carried out, and the results are shown in Fig. S14.† The chronoamperometry pattern, expectedly, shows the highest current density at the starting point of the profile, around 350 mA cm−2 for the first few minutes (Fig. 5a), suggesting a rapid rate of glycerol conversion; as the concentration of glycerol decreases, predictably, the reaction profile shows a decreasing current density as the reaction progresses. Within the first 30 min., current density reaches ∼100 mA cm−2. A very low current density was observed after a reaction time of five hours, indicating the depletion of glycerol content. Time-dependent product analysis was performed using the NMR spectral method, and the product distribution pattern is shown in Fig. S15.† The product distribution strongly implies that the reaction goes through a glycolic acid intermediate. Various products observed in NMR results have been confirmed either by the established values of NMR chemical shift or by spiking with a small amount of pure compounds to the test solution, increasing the intensity of the respective NMR features. To support the fast conversion of glycerol as the reactant, a small amount of 0.2 M glycerol (46 mg dissolved in 2 mL) was added at the end of the reaction to observe the augmentation of current density or an increase in reaction rate; the result is shown in Fig. S16.† The above-mentioned addition of glycerol was carried out at 5 h 35 min, which increases the current density instantly, followed by a quick decrease, underscoring its fast consumption. The product distribution observed by NMR is shown in Fig. 5b and S15.† From the first hour of the reaction, formate dominates the products, followed by methanol and glycolic acid as secondary products. Minor amounts of lactic and acetic acids are also observed. Major amounts of C1-products (formate and methanol; selectivity >90%) along with glycolic acid directly indicate the C–C cleavage from either end of glycerol followed by oxidation to acids. Glycerol probably gets oxidized and forms glyceraldehyde in the first step; further, oxidative C–C cleavage of glyceraldehyde yields glycolic acid and results in very high selectivity of formate. In addition, a significant amount of methanol and formate are present in the products. All these observations lead to the path of glycerol to glycolaldehyde followed by Cannizzaro's reaction with formaldehyde to form methanol and formate. Indeed, this accounts for high formate selectivity. This also implies that glycolic acid is the reaction pathway through the oxidation of the α-OH group and cleavage of the terminal C–C bond. A plausible mechanism for the glycerol oxidation over NCMO is given in Scheme 1. It is also to be noted that a minimal amount of formate undergoes further oxidation to CO2. The 13C NMR results also do not show any carbonate or bicarbonate at potentials lower than 1.35 V. Very low carbonate formation was observed after 8 h of reaction at 1.4 V; the result is shown in Fig. S17.†
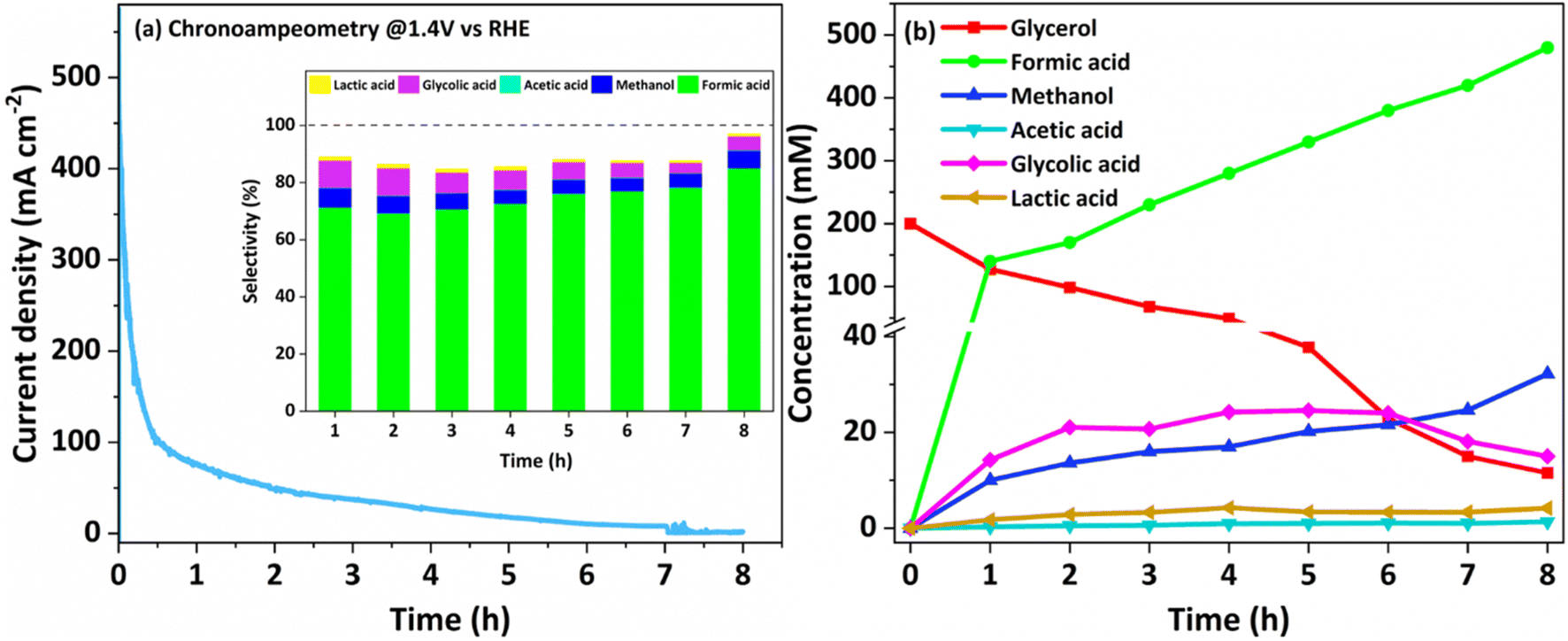 | ||
| Fig. 5 (a) Time-dependent chronoamperometry carried out for the GOR with NCMO/NF at 1.4 V (vs. RHE). Inset: percent selectivity of the products of the GOR. (b) Quantification of glycerol conversion and various products obtained from NMR analysis. Near 100% carbon balance was observed underscoring the maintenance of atom economy. | ||
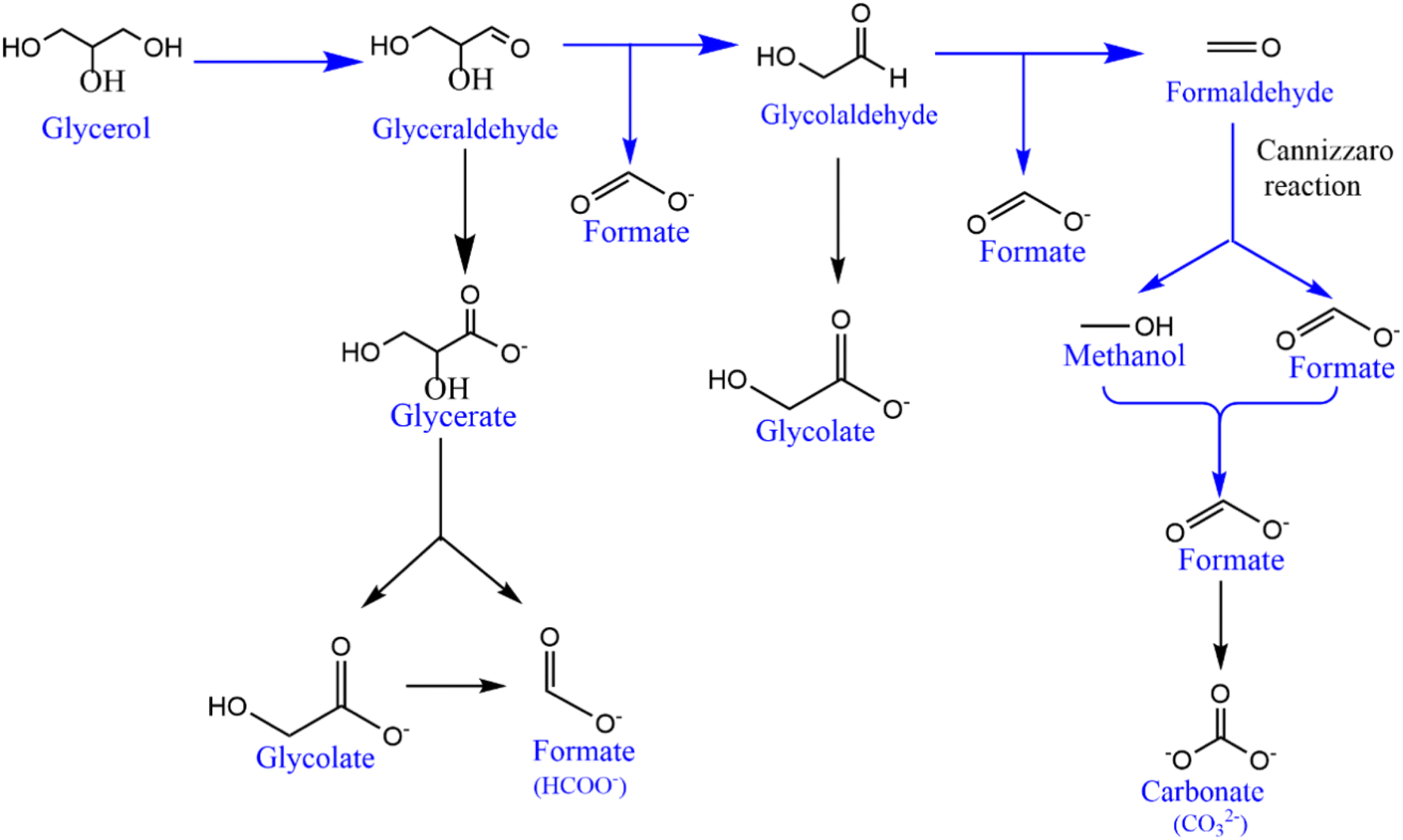 | ||
| Scheme 1 Major pathways in the suggested mechanistic framework of glycerol electrocatalytic oxidation to formate on NiCoMn oxide in an alkaline medium. | ||
Carbon-negative green hydrogen
NCMO/NF, other bimetallic combinations and monometallic contents, and Ni foam as cathodes were subjected to HER evaluations in a three-electrode system in 1 M KOH with 0.2 M glycerol. To explore the HER and to prove the bifunctionality of the NCMO/NF catalyst, LSV and chronopotentiometry were performed similarly to the evaluation for GORs and OERs. The NCMO/NF electrode shows the best performance for HERs at a lower potential with significantly high current density (10 and 100 mA cm−2 at −0.01 and −0.27 V vs. RHE, respectively), as shown in Fig. S18.† Nonetheless, NCMO/NF shows similar performance in alkaline and alkaline glycerol solutions, as shown in Fig. 6a. However, differences emerge at high current densities owing to the lesser proximal availability of hydroxides in a glycerol solution than with glycerol. Fig. 6b shows the Tafel slope of NCMO/NF (85 mV dec−1), which is in between that of precious metal catalyst Pt/C (58 mV dec−1) and other combinations (110 mV dec−1) and NF (191 mV dec−1), suggesting the favorable HER reaction kinetics with NCMO/NF (Fig. S17†). Meanwhile, EIS analyses show the lowest charge transfer resistance (Rct) of NCMO/NF owing to the presence of Ni, which directly contributes to its high catalytic activity (Fig. S8†). To gain deeper insights into the HER activity, the ECSA values of different catalysts were obtained by extracting the corresponding Cdl, 186.2 mF cm−2, and it is clear that NCMO/NF shows a larger ECSA than that of others (6333 cm2), and a little lower for other bimetallic combinations. The ECSA-normalized LSV results, shown in Fig. S19,† underscore the importance of the high surface area of NiCoMn for HERs. Again this is due to the large surface area and high porosity of the material to offer more reactive sites. As shown in Fig. 6b, the Tafel slope shows significant changes after the addition of 0.2 M glycerol, owing to the best performance without any non-conductive organic moiety leading to significant charge transfer resistance (here glycerol). Moreover, after 12 h of chronopotentiometry measurements at 10 mA cm−2, NCMO/NF maintains its excellent catalytic performance (Fig. 6c).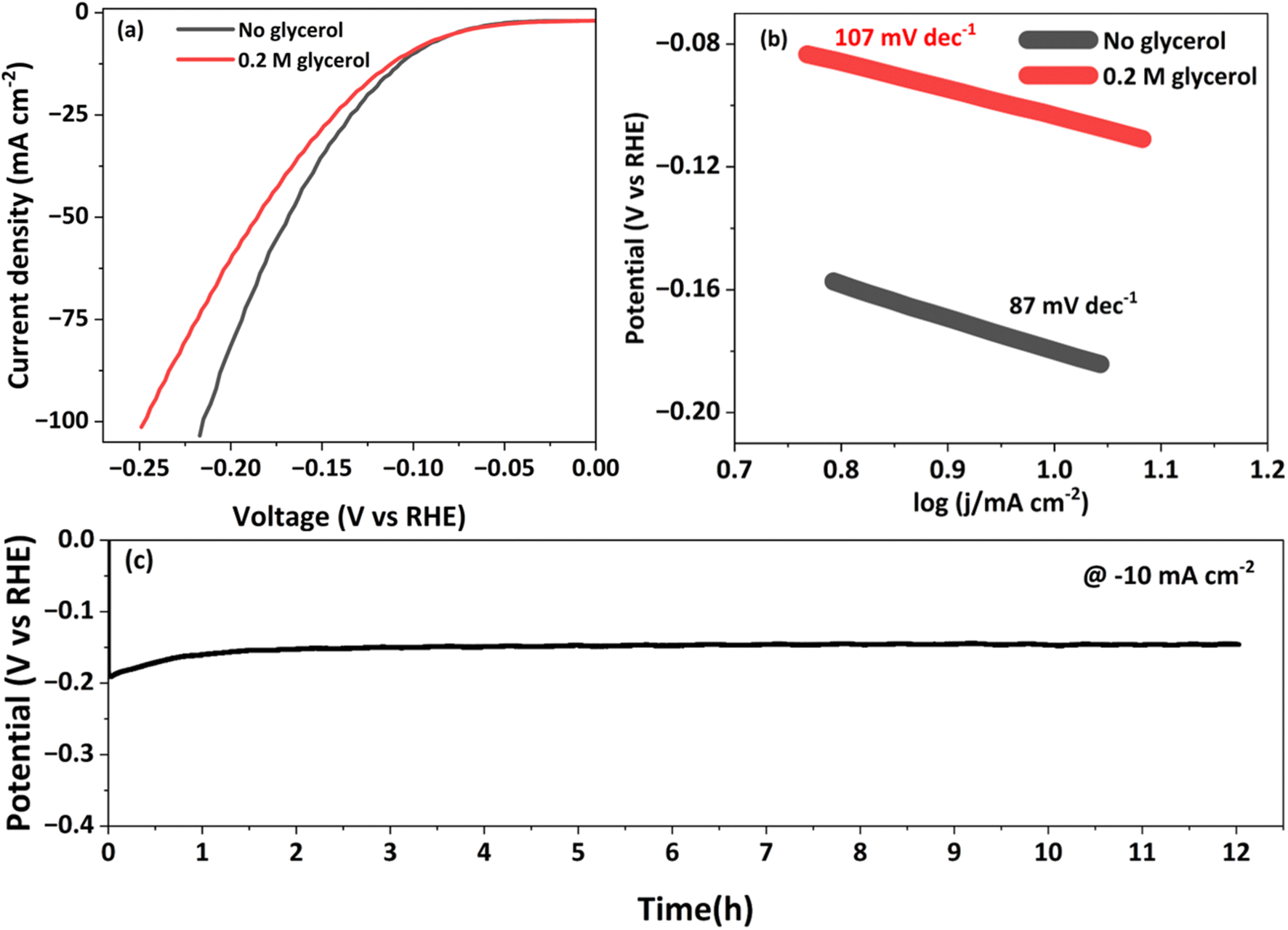 | ||
| Fig. 6 (a) Cathodic electrocatalytic performances of NCMO/NF for the HER measured through the LSV plots in 1.0 M KOH, and with 0.2 M glycerol. Scan rate employed is 5 mV s−1. (b) Tafel plots calculated with NCMO/NF in the presence and absence of glycerol. (c) Twelve-hour sustainability test conducted with NCMO/NF for cathodic HERs at a current density of 10 mA cm−2. | ||
Bifunctional behavior of the NiCoMn oxide for GORs and HER
An industry-friendly, membrane-free, two-electrode electrolyzer setup was initially utilized based on the electrochemical performance of NCMO/NF for GORs and HERs in a three-electrode system. A digital photograph of the setup is shown in Fig. 7a and b for membrane-free and with membrane setup, respectively. NCMO/NF was employed as both the anode and the cathode (full cell denoted as NCMO|NF‖NCMO|NF in the electrolyzer setup with 1 M KOH solution containing 0.2 M glycerol as the electrolyte). Simultaneous hydrogen and formate productions were observed at room temperature. In the two-electrode setup, 10 mA cm−2 (100 mA cm−2) current density was observed at a cell potential of 1.38 V (1.64 V) (Fig. 7c). In the electrolyzer setup, 10 mA cm−2 (100 mA cm−2) was achieved at a cell potential of 1.41 V (1.94 V) in the presence of glycerol; under these conditions, HER and GOR performances are observed concurrently. Conventional water electrolysis in a two-electrode setup in the same NCMO/NF‖NCMO/NF cell, but without the addition of glycerol, was also conducted under the same conditions; however, a significantly higher potential of 1.63 V (1.93 V) was necessary to obtain the identical current density of 10 mA cm−2 (100 mA cm−2). Such a low cell voltage is associated with NCMO/NF as biomass-component-assisted water splitting is an attractive feature of most reported systems.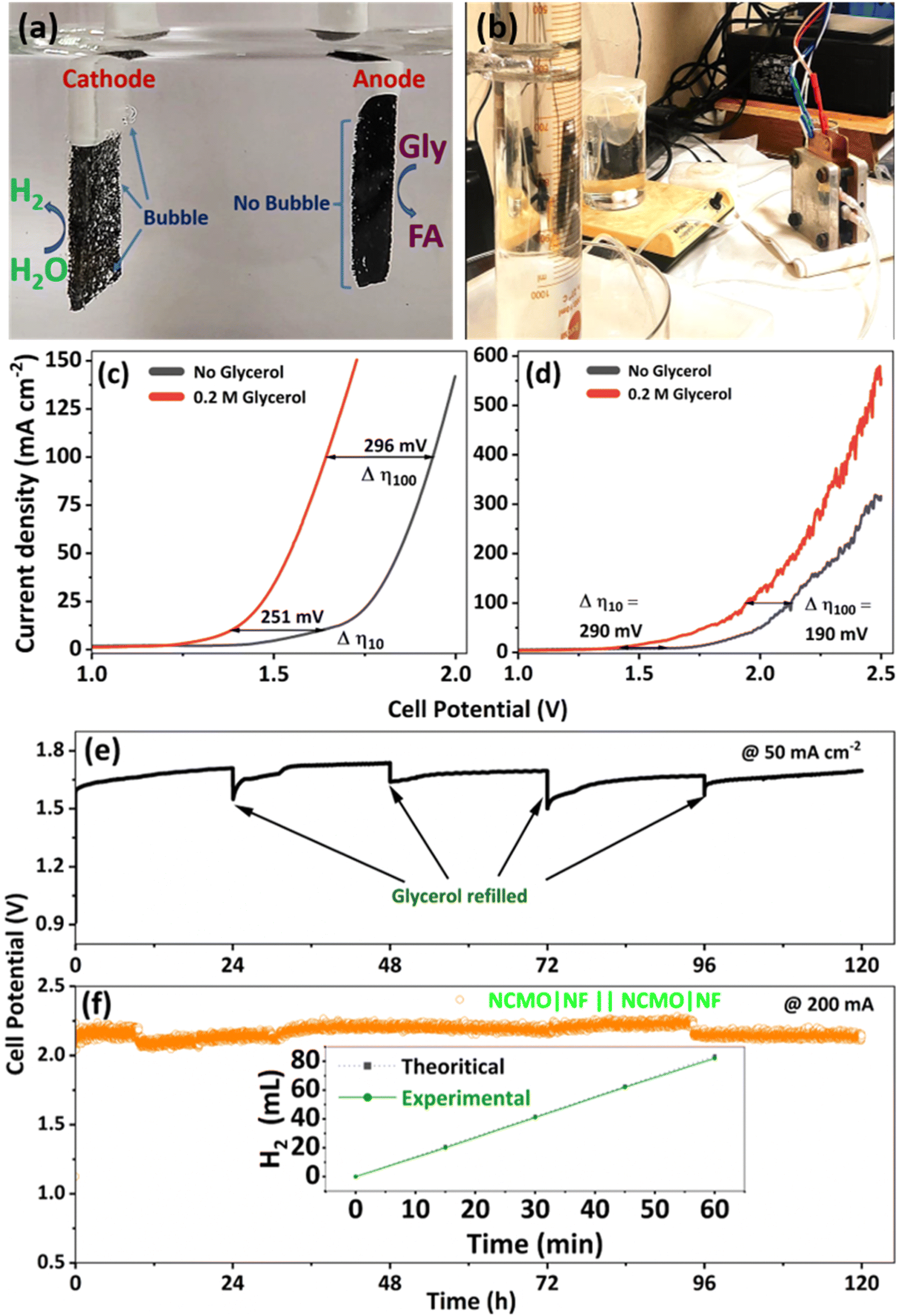 | ||
| Fig. 7 Electrochemical performances of NCMO/NF‖NCMO/NF. (a) Digital photograph showing concurrent electrolytic hydrogen and formate production from an alkaline glycerol solution, and displaying small bubbles on and near the cathode catalyst, whereas no bubbles are seen on the anode owing to liquid product formation. (b) Digital photograph of the closed electrolyzer setup with inverted burette for H2 collection. LSV curves for the NCMO/NF‖NCMO/NF cell in 1.0 M KOH solution with and without alkaline 0.2 M glycerol solution, (c) in the membrane-free system, and (d) membrane-added electrolyzer setup. Scan rate is 5 mV s−1. Stability test for the bifunctional NCMO|NF‖NCMO|NF setup with (e) membrane-free system and (f) membrane-added electrolyzer setup carried out at constant currents of 50 and 200 mA, respectively. | ||
Contact angle measurements were carried out on NF and NCMO/NF; the results are shown in Fig. S20a and b,† respectively. These measurements indicate a hydrophobic nature of NF with a contact angle of 120.8°, while NCMO/NF shows zero contact angle with the superhydrophilic nature of the catalyst (ESI Video S1†). Contact angle measurements show promising adaptability of NCMO in the aqueous medium, facilitating remarkable electrocatalytic performance in the two-electrode setup. It is to be underscored that the two-electrode setup (in an open beaker) does not employ any membrane for selective anion/proton diffusion and gas separation (Fig. 7a); it should also be noted that hydrogen is the only gaseous product observed without any oxygen or carbon-containing products (CO and CO2). In addition, sustainable electrocatalytic performance was demonstrated for 120 h without any activity deterioration, and the results are shown in Fig. 7e. Throughout the reaction, the amount of evolved H2 was measured, and it showed a good agreement with the theoretical estimation; this indicates a very high electron transfer efficiency from H2O to H2 during the overall electrolysis process. In the electrolyzer setup, the amount of hydrogen was measured by employing an inverted measuring cylinder, and the hydrogen yield observed was 100 mL h−1 (25 mL hydrogen in 15 min), as shown in Fig. 7f, inset. The average hydrogen FE was calculated to be 98%, while the selectivity for formate is 90% at 1.4 V with no OER observed. In conclusion, the established membrane-free electrolytic approach with NCMO/NF as both anode and cathode electrocatalysts in an alkaline glycerol solution is a low-cost and highly efficient strategy for the simultaneous electrochemical hydrogen and formate production.
An activity comparison of some of the top electrocatalysts reported for organic-assisted oxidation reaction has been made, and the summary is provided in Table 1, along with the present result.19,47–57 Although few bifunctional catalysts are reported, the cell potential is the lowest for the presently reported system. Most of the reported reactions involve the oxidation of methanol or ethanol to formate or acetate. However, the challenge with glycerol oxidation involves C–C cleavage, along with selective formate production, which is reported in this work. Nonetheless, the Co–Ni alloy reported for glucose oxidation52 is interesting, as the challenge with glucose oxidation is higher than glycerol; in this case, sustainability has also been demonstrated at 10 mA cm−2. The present catalyst system was demonstrated for 120 h at a high current density of 50 mA cm−2, which has not been demonstrated to the best of our knowledge.
| Cathodic catalyst | Anodic catalyst | Electrolyte (+1 M KOH) | Main product at anode | Cell voltage at 10 mA cm−2 (V) | Ref. |
|---|---|---|---|---|---|
| Ni3S2/NF | Ni3S2/NF | 0.01 M 5-hydroxy-methyl furfural | 2,5-FDCA | 1.46 | 19 |
| NC/Ni–Mo–N/NF | NC/Ni–Mo–N/NF | 0.1 M glycerol | Formate | 1.38 | 47 |
| Co(OH)2@HOS/CP | Co(OH)2@HOS/CP | 3 M methanol | Formate | 1.49 | 48 |
| Ni(OH)2/NF | Ni(OH)2/NF | 0.5 M methanol | Formate | 1.52 | 49 |
| CoS2 NA/Ti | CoS2 NA/Ti | 0.3 M urea | N2, CO2 | 1.59 | 50 |
| Ni2P-UNMs/NF | Ni2P-UNMs/NF | 0.125 M benzylamine | Benzonitrile | 1.41 | 51 |
| Co–Ni alloy | Co–Ni alloy | 0.1 M glucose | Gluconolactone gluconic acid | 1.39 | 52 |
| Co–S–P/CC | Co–S–P/CC | 1.0 M ethanol | Acetic acid | 1.63 | 53 |
| Ni0.33Co0.67(OH)2/NF | Ni0.33Co0.67(OH)2/NF | 0.5 M methanol | Formate | 1.50 | 54 |
| Co3S4-NSs/Ni–F | Co3S4-NSs/Ni–F | 0.5 M ethanol | Acetate | 1.48 | 55 |
| Ni2P | F-β-FeOOH | 0.33 M ethanol | Acetic acid | 1.46 | 56 |
| NiS@Ni3S2/NiMoO4 | NiS@Ni3S2/NiMoO4 | 0.5 M urea | N2, CO2 | 1.40 | 57 |
| NCMO/NF | NCMO/NF | 0.1 M glycerol | Formate | 1.36 | Present work |
Table 2 compares the sustainable performance reported for top-performing electrocatalysts for selective glycerol oxidation to formate at a relatively high current density and elongated time span. Other reported catalysts were demonstrated with 0.1 M glycerol, while our catalyst works well with 0.2 M glycerol, and sustainability was demonstrated for 120 h. Moreover, no decrease in current density underscores that the sustainability is likely to continue for a long reaction period (Fig. 7f). Indeed, one of the previously reported NiVRu-LDH catalyst exhibits a very high current density of 1 A cm−2 at 2 V; it may be noted that the catalyst contains Ru.61
| Catalyst | Electrolyte | Two electrode operating conditions | Ref. | |
|---|---|---|---|---|
| Current density (mA cm−2) or voltage (V vs. RHE) | Time | |||
| NiCoMn/NF | 1 M KOH + 0.2 M gly | 50 mA cm−2 | 120 h | Present work |
| NiCo2O4/NF | 1 M KOH + 0.1 M gly | 1.75 V vs. RHE | >200 h | 58 |
| NiCo hydroxide | 1 M KOH + 0.1 M gly | 100 mA cm−2 | 110 h | 59 |
| HEA-CoNiCuMnMo | 1 M KOH + 0.1 M gly | 50 mA cm−2 | 12 days | 60 |
| NiVRu LDH | 1 M KOH + 0.1 M gly | 1000 mA cm−2 at 2 V | 120 h | 61 |
| NiCo2O4/NF | 1 M NaOH + 0.1 M gly | 1.4 V vs. RHE | 120 h | 7 |
However, there is more scope to increase the performance and sustainability of the catalysts, and more work is required in this direction.
Electrocatalyst characterizations post GOR and HER
To gain a better understanding of the catalytic activity after the synchronized GOR to formate and H2 evolution for 120 h, post-GOR characterization was performed by employing LSV, chronopotentiometry, XRD, FESEM, and XPS. It is evident from the LSV that a faster reaction kinetics was observed, with reference to the fresh catalyst before the 120 h stability test, as shown in Fig. 8a. The faster rate of the reaction indicates that more active sites are exposed under the reaction conditions, possibly due to changes within the catalyst. This shows the long-term acceptability of NCMO for the GOR. However, to verify the integrity of the catalyst, a potentiostatic chronopotentiometry experiment was conducted for 1 h (after 120 h of stability test); the result is shown in Fig. 8b. It shows a steady voltage input under operating conditions with minimum deflection. Surprisingly, morphology retention was observed in the FESEM image shown in Fig. 8c–e. After 120 h of the reaction, the nanoneedle structure was retained with minor changes. In the case of the anode (Fig. 8d), where the oxidation reaction takes place, multiple oxidation states work in synergy to promote the active sites (NiOOH) and form a connected oxide network, which is not only active for the oxidation reaction but also shifts overpotential towards a lower voltage, as implied by Corby et al.62 In the case of the cathode (Fig. 8e), a significant densification of nanoneedles was observed to the existing cactus-like morphology. In contrast, densification occurs to a lesser extent on the anode material. A careful analysis of FESEM results reveals that the density of nanoneedles present on the surface of the fresh catalyst decreases on the anode and further decreases on the cathode material. This suggests that surface modification could be occurring under reaction conditions. To further confirm the structural integrity of the material on a molecular scale, we performed TEM. The microscopic images appeared with clear lattice distribution of lattice fringes corresponding to the prepared oxides (Fig. S21a–d†). This demonstrates the overall integrity and sustainability of the NCMO material for glycerol oxidation in an electrolyzer setup, with a significant increase in activity at the cost of surface modification. | ||
| Fig. 8 Electrochemical performances of NCMO/NF‖NCMO/NF after 120 h of concurrent GORs and HERs. (a) LSV recorded for the fresh catalyst and after 120 h of the GOR. (b) Chronopotentiometry results recorded after 120 h under the same condition of electrolyzers for 120 h. The FESEM image of NCMO (c) fresh catalyst and after 120 h of (d) the GOR anode and (e) HER cathode. While densification of cactus occurs on the cathode, densification of surface with nanoneedles occurs on the NF on the anode. | ||
Conclusions
Using the NCMO/NF electrocatalyst as both the anode and the cathode, a method for the simultaneous and highly efficient electrolytic generation of H2 and value-added formate with high selectivity was demonstrated in the alkaline electrolysis of glycerol. At a current density of 100 mA cm−2 (1.36 V), the trimetallic catalyst demonstrates very decent electrocatalytic performance for GORs. Interestingly, the two-electrode configuration requires a low cell voltage of 1.64 V for 100 mA cm−2 for H2 production in an alkaline glycerol solution, which is 260 mV lower than that of conventional water electrolysis under the same conditions with NCMO/NF at both the anode and the cathode. Additionally, by carefully manipulating trimetallic combinations, the ultrahigh selective electro-oxidation conversion of glycerol into value-added formate with high selectivity and yield has been achieved. The formate production selectivity by glycerol oxidation on the anode is 90%, and the faradaic efficiency for the generation of H2 on the cathode is 98% for chronopotentiometry carried out at 200 mA. By further optimizing the electrolysis conditions, it is possible to achieve exclusive formate production, which would avoid the product separation step; indeed, this is important because of the high energy involved in product separation. High and sustainable catalytic activity (for 5 days or 120 h) was also demonstrated. More importantly, it has been established that the production of formate results from a sequential reaction during protracted electrolysis and that both the secondary and primary glycerol carbons contribute to the formate generation. The present work is complimentary to a recent work based on different morphologies of Pd employed as the electrocatalyst and operating at a significantly lower potential, but produces a mixture of acids.63 This work offers a non-noble metal oxide electrocatalyst, NCMO/NF, for a practical, energy-efficient, and potentially commercially viable system for generating H2 and formate. Moreover, the catalyst is stable enough to operate at a higher current density. Higher carbon chains containing biomolecules could also be employed to obtain value-added products in the green electrochemical process. In the industry, formic acid is produced from fossil fuel–based resources; however, the present method employed a waste to generate formate. Moreover, hydrogen produced using the current process is carbon-negative green hydrogen. Due to the excellent electrocatalytic performance of the NCMO/NF electrocatalyst, it could be evaluated to oxidize more robust biomass components such as cellulose, where prior base modification is needed.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Arindam Saha acknowledges CSIR for the senior research fellowship. All the authors acknowledge the financial support from CSIR for a NCP project (HCP-44) through the H2T program.References
- G. Supran, S. Rahmstorf and N. Oreskes, Science, 2023, 379, eabk0063 CrossRef CAS PubMed
.
- C. W. Callahan and J. S. Mankin, Science, 2023, 380, 1064 CrossRef CAS PubMed
- IEA – International Energy Agency, https://www.iea.org.
- P. C. K. Vesborg and T. F. Jaramillo, RSC Adv., 2012, 2, 7933 RSC
- H. Nishiyama, T. Yamada, M. Nakabayashi, Y. Maehara, M. Yamaguchi, Y. Kuromiya, Y. Nagatsuma, H. Tokudome, S. Akiyama, T. Watanabe, R. Narushima, S. Okunaka, N. Shibata, T. Takata, T. Hisatomi and K. Domen, Nature, 2021, 598, 304 CrossRef CAS PubMed
- K. N. Salgaonkar, H. Bajpai, N. B. Mhamane, N. Nalajala, I. Chauhan, K. Thakkar, K. Joshi and C. S. Gopinath, J. Mater. Chem. A, 2023, 11, 15168 RSC
- G. Wu, X. Dong, J. Mao, G. Li, C. Zhu, S. Li, A. Chen, G. Feng, Y. Song, W. Chen and W. Wei, Chem. Eng. J., 2023, 468, 143640 CrossRef CAS
- X. Han, H. Sheng, C. Yu, T. W. Walker, G. W. Huber, J. Qiu and S. Jin, ACS Catal., 2020, 10, 6741 CrossRef CAS
- D. Liu, J.-C. Liu, W. Cai, J. Ma, H. B. Yang, H. Xiao, J. Li, Y. Xiong, Y. Huang and B. Liu, Nat. Commun., 2019, 10, 1779 CrossRef PubMed
- L. J. Enman, M. S. Burke, A. S. Batchellor and S. W. Boettcher, ACS Catal., 2016, 6, 2416 CrossRef CAS
- D. A. Kuznetsov, B. Han, Y. Yu, R. R. Rao, J. Hwang, Y. Román-Leshkov and Y. Shao-Horn, Joule, 2018, 2, 225 CrossRef CAS
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977 CrossRef CAS PubMed
- H. Bajpai, I. Chauhan, K. N. Salgaonkar, N. B. Mhamane and C. S. Gopinath, RSC Sustainability, 2023, 1, 481 RSC
- R. Venkatkarthick, J. Niu, A. Srikhaow, C. Sriprachuabwong, S. Vasudevan, A. Tuantranont and J. Qin, ACS Appl. Energy Mater., 2021, 4, 6520 CrossRef CAS
- G. P. Kharabe, R. Illathvalappil, S. Barik, F. Kanheerampockil, P. S. Walko, S. K. Bhat, R. N. Devi and S. Kurungot, Sustainable Energy Fuels, 2023, 7, 2428 RSC
- J. Dong, N. Lv, X. Kang, X. Liu, H. Li, T. Li, Z. Guo and J. Luo, Mater. Res. Bull., 2024, 173, 112681 CrossRef CAS
- Y. H. Fang and Z. P. Liu, J. Am. Chem. Soc., 2010, 132, 18214 CrossRef CAS PubMed
- B. You, X. Liu, N. Jiang and Y. Sun, J. Am. Chem. Soc., 2016, 138, 13639 CrossRef CAS PubMed
- B. You, N. Jiang, X. Liu and Y. Sun, Angew. Chem., Int. Ed., 2016, 55, 9913 CrossRef CAS PubMed
- W. Liu, Y. Cui, X. Du, Z. Zhang, Z. Chao and Y. Deng, Energy Environ. Sci., 2016, 9, 467 RSC
- S. A. Patil, A. C. Khot, K. D. Kadam, H. T. Bui, H. Im and N. K. Shrestha, Inorg. Chem. Front., 2023, 10, 7204 RSC
- N. K. Shrestha, S. A. Patil, A. S. Salunke, A. I. Inamdar and H. Im, Dalton Trans., 2023, 52, 10933 RSC
- S. A. Patil, S. Cho, Y. Jo, N. K. Shrestha, H. Kim and H. Im, Chem. Eng. J., 2021, 426, 130773 CrossRef CAS
- A. A. A. Raman, H. W. Tan and A. Buthiyappan, Front. Chem., 2019, 7, 774 CrossRef PubMed
- S. C. Chang, Y. Ho and M. J. Weaver, J. Am. Chem. Soc., 1991, 113, 9506 CrossRef CAS
- D. Samul, K. Leja and W. Grajek, Ann. Microbiol., 2014, 64, 891 CrossRef CAS PubMed
- M. R. Monteiro, C. L. Kugelmeier, R. S. Pinheiro, M. O. Batalha and A. da Silva César, Renewable Sustainable Energy Rev., 2018, 88, 109 CrossRef CAS
- H. Wang, L. Thia, N. Li, X. Ge, Z. Liu and X. Wang, ACS Catal., 2015, 5, 3174 CrossRef CAS
- S. H. Shi, Y. Liang and N. Jiao, Chem. Rev., 2021, 121, 485 CrossRef CAS PubMed
- Y. Li, X. Wei, L. Chen, J. Shi and M. He, Nat. Commun., 2019, 10, 5335 CrossRef PubMed
- D. A. Bulushev, J. R. H. Ross and C. Sus, Chem, 2018, 11, 821 CAS
- J. E. N. Brainer, D. C. S. Sales, E. B. M. Medeiros, N. M. Lima Filho and C. A. M. Abreu, Braz. J. Chem. Eng., 2014, 31, 913 CrossRef
- J. Zhang, S. Guo, B. Xiao, Z. Lin, L. Yan, D. Du and S. Shen, Chem. Eng. J., 2021, 416, 129127 CrossRef CAS
- R. Ranjan, J. Tekawadia, R. Jain, N. B. Mhamane, T. Raja and C. S. Gopinath, Chem. Eng. J., 2023, 471, 144459 CrossRef CAS
- B. Tudu, N. Nalajala, K. Prabhakar Reddy, P. Saikia and C. S. Gopinath, ACS Sustain. Chem. Eng., 2021, 9, 13915 CrossRef CAS
- D. J. Miller, M. C. Biesinger and N. S. McIntyre, Surf. Interface Anal., 2002, 33, 299 CrossRef CAS
- E. S. Ilton, J. E. Post, P. J. Heaney, F. T. Ling and S. N. Kerisit, Appl. Surf. Sci., 2016, 366, 475 CrossRef CAS
- K. M. Shaju, G. V. Subba Rao and B. V. R. Chowdari, Electrochim. Acta, 2002, 48, 145 CrossRef CAS
- D. Zhu, C. Guo, J. Liu, L. Wang, Y. Du and S.-Z. Qiao, Chem. Commun., 2017, 53, 10906 RSC
- Z. Zhang, L. Xin, J. Qi, Z. Wang and W. Li, Green Chem., 2012, 14, 2150 RSC
- Y. Xu, T. Liu, K. Shi, H. Yu, K. Deng, Z. Wang, X. Li, L. Wang and H. Wang, Chem. Commun., 2023, 59, 1817 RSC
- H. Yu, W. Wang, Q. Mao, K. Deng, Z. Wang, Y. Xu, X. Li, H. Wang and L. Wang, Appl. Catal., B, 2023, 330, 122617 CrossRef CAS
- Y. Ye, G. Zhou, K. Li and Y. Tong, Catalysts, 2023, 13, 1064 CrossRef CAS
- X. Li, M. Xin, S. Guo, T. Cai, D. Du, W. Xing, L. Zhao, W. Guo, Q. Xue and Z. Yan, Electrochim. Acta, 2017, 253, 302 CrossRef CAS
- S. S. Kanungo, A. K. Mishra, N. B. Mhamane, U. K. Marelli, D. Kumar and C. S. Gopinath, Inorg. Chem., 2022, 61, 19577 CrossRef CAS PubMed
- Y. Xu, M. Liu, S. Wang, K. Ren, M. Wang, Z. Wang, X. Li, L. Wang and H. Wang, Appl. Catal., B, 2021, 298, 120493 CrossRef CAS
- K. Xiang, D. Wu, X. Deng, M. Li, S. Chen, P. Hao, X. Guo, J.-L. Luo and X.-Z. Fu, Adv. Funct. Mater., 2020, 30, 1909610 CrossRef CAS
- J. Hao, J. Liu, D. Wu, M. Chen, Y. Liang, Q. Wang, L. Wang, X.-Z. Fu and J.-L. Luo, Appl. Catal., B, 2021, 281, 119510 CrossRef CAS
- S. Wei, X. Wang, J. Wang, X. Sun, L. Cui, W. Yang, Y. Zheng and J. Liu, Electrochim. Acta, 2017, 246, 776 CrossRef CAS
- Y. Ding, B.-Q. Miao, S.-N. Li, Y.-C. Jiang, Y.-Y. Liu, H.-C. Yao and Y. Chen, Appl. Catal., B, 2020, 268, 118393 CrossRef CAS
- C. Lin, P. Zhang, S. Wang, Q. Zhou, B. Na, H. Li, J. Tian, Y. Zhang, C. Deng, L. Meng, J. Wu, C. Liu, J. Hu and L. Zhang, J. Alloys Compd., 2020, 823, 153784 CrossRef CAS
- S. Sheng, K. Ye, L. Sha, K. Zhu, Y. Gao, J. Yan, G. Wang and D. Cao, Inorg. Chem. Front., 2020, 7, 4498 RSC
- M. Li, X. Deng, K. Xiang, Y. Liang, B. Zhao, J. Hao, J. L. Luo and X. Z. Fu, ChemSusChem, 2020, 13, 914 CrossRef CAS PubMed
- Y. Ding, Q. Xue, Q. L. Hong, F.-M. Li, Y. C. Jiang, S. N. Li and Y. Chen, ACS Appl. Mater. Interfaces, 2021, 13, 4026 CrossRef CAS PubMed
- G. F. Chen, Y. Luo, L. X. Ding and H. Wang, ACS Catal., 2018, 8, 526 CrossRef CAS
- L. Sha, T. Liu, K. Ye, K. Zhu, J. Yan, J. Yin, G. Wang and D. Cao, J. Mater. Chem. A, 2020, 8, 18055 RSC
- W. Luo, H. Tian, Q. Li, G. Meng, Z. Chang, C. Chen, R. Shen, X. Yu, L. Zhu, F. Kong, X. Cui and J. Shi, Adv. Funct. Mater., 2024, 34, 2306995 CrossRef CAS
- Z. He, J. Hwang, Z. Gong, M. Zhou, N. Zhang, X. Kang, J. W. Han and Y. Chen, Nat. Commun., 2022, 13, 3777 CrossRef CAS PubMed
- L. Fan, Y. Ji, G. Wang, J. Chen, K. Chen, X. Liu and Z. Wen, J. Am. Chem. Soc., 2022, 144, 7224 CrossRef CAS PubMed
- Q. Qian, X. He, Z. Li, Y. Chen, Y. Feng, M. Cheng, H. Zhang, W. Wang, C. Xiao, G. Zhang and Y. Xie, Adv. Mater., 2023, 35, 2300935 CrossRef CAS PubMed
- S. Corby, R. R. Rao, L. Steier and J. R. Durrant, Nat. Rev. Mater., 2021, 6, 1136 CrossRef CAS
- I. Chauhan, H. Bajpai, B. Ray, S. K. Kolekar, S. Datar, K. K. Patra and C. S. Gopinath, ACS Appl. Mater. Interfaces, 2024, 16, 26130–26141 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: ESI contains FESEM images (S1), wide-scan XPS (S2), XPS spectra of Ni 2p on a Cu foam (S3), FESEM and HRTEM images (S4), EDS elemental mappings (S5), glycerol optimization (S6), comparison of potential vs. current density (S7), LSV results (S8), Tafel slope (S9), EIS spectra (S10), double-layer capacitance and ECSA (S11), ECSA-normalized GOR LSV (S12), product selectivity (S13), chronoamperometry (S14), reactant and product distribution (S15), addition of glycerol effect (S16), 13C NMR (S17), LSV result (S18), ECSA-normalized LSV HER (S19), contact angle measurement (S20), TEM image after 120 h stability test (S21), and video for contact angle measurement (Video 1). See DOI: https://doi.org/10.1039/d4se00434e |
| This journal is © The Royal Society of Chemistry 2024 |