
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning 2D perovskite–graphene layered composite for photocatalysis†
Haozhe
Zhang
a,
Yanjie
Wang
ab,
Wentian
Niu
a,
Tatchamapan
Yoskamtorn
a,
Mingyu
Luo
a,
Robert
Tayler
c,
Sarah
Day
d and
Shik Chi
Edman Tsang
 *a
*a
aWolfson Catalysis Centre, Department of Chemistry, University of Oxford, Oxford, OX1 3QR, UK. E-mail: edman.tsang@chem.ox.ac.uk
bCAS Key Laboratory of Nanosystem and Hierarchical Fabrication, National Centre for Nanoscience and Technology, Beijing 100190, China
cClarendon Laboratory, Department of Physics, University of Oxford, China
dDiamond Light Source Ltd, Harwell Science and Innovation Campus, Didcot OX11 0DE, UK
First published on 27th June 2024
Abstract
The augmentation of photocatalytic activity in layered perovskite oxides via the integration of graphene-like materials presents a promising pathway for the optimization of solar energy conversion. The electron-rich nature of graphene, coupled with its high electron conductivity, functions as an effective photosensitizer, thereby enhancing visible light harvesting. In this investigation, we have, for the first time, assembled ultrathin exfoliated Dion–Jacobson perovskite layers with reduced graphene oxide (rGO) layers, resulting in a high surface area layered nanocomposite, achieved through a tailored electrostatic approach. To further refine the electron properties of the layered perovskite–reduced graphene oxide composites, we have explored the use of various lanthanides as A-site cations in the Dion–Jacobson perovskites, including LaNb2O7 (LNO), PrNb2O7 (PNO), and NdNb2O7 (NNO). The synthesized composites demonstrate exceptional performance in photocatalytic H2 production, with rGO/NNO exhibiting the highest activity, achieving a hydrogen evolution rate (HER) of 835 μmol g−1 under light illumination, attributable to optimal interfacial effects. Our experimental and theoretical analyses indicate that hydrogen production is predominantly influenced by the A-site cation charge density at the materials' interface, as dictated by the charge transfer dynamics. This research potentially contributes to the comprehension and enhancement of photocatalytic processes for applications in solar energy conversion.
1. Introduction
The photocatalytic water splitting reaction offers a significant avenue for the direct conversion of solar energy into hydrogen, a clean and efficient energy medium. Since Fujishima and Honda's groundbreaking work in 1972,1 semiconductor materials have been extensively studied as photocatalysts for water splitting reactions, encompassing both the overall process and specific half-reactions. Among these, layered perovskite oxides have emerged as a promising class of materials.2 The layered perovskite oxides exhibit distinct structures, including the Aurivillius phase (Bi2O2)(An−1BnO3n+1), Ruddlesden–Popper phase An+1BnO3n+1 or A′2An−1BnO3n+1 and Dion–Jacobson (DJ) phase A′An−1BnO3n+1,3–5 where A′ and A represent alkali or rare-earth metals, B denotes transition metals such as Ta5+, Nb5+, or Ti5+, and n is the number of BO6 octahedra arranged perpendicular to the layers. To enhance the photocatalytic performance of layered perovskite oxides for hydrogen production, various strategies have been employed to modify their structure and properties. Notably, Wakayama et al. synthesized Rb2NdNb2O6N by nitriding RbNdNb2O7 with Rb2CO3, effectively narrowing the bandgap and enhancing light absorption.6 Similarly, Ida et al. successfully exfoliated CsCaTa3O10−xNy into CaTa3O10−xNy nanosheets, resulting in a substantial increase in the hydrogen evolution rate (HER) under full strong arc irradiation with 10 mg of catalysts.7 The exfoliation of layered perovskites into nanosheets has been shown to significantly enhance photocatalytic performance. This improvement is attributed to the increased surface area, which provides more active sites for the water splitting reaction.8,9 Additionally, the reduced size of the catalysts enables shorter diffusion distances for charge carriers, facilitating their participation in the water splitting process.7The aggregation of perovskite nanosheets and the swift recombination of charge carriers continue to present substantial obstacles in the field of perovskite nanosheet photocatalysts. Zhou et al. proposed a solution to the aggregation issue by introducing a core–shell structure, in which perovskite nanosheets were dispersed on SiO2 spheres. However, this method resulted in a slight decrease in activity, a consequence of the light-scattering properties of SiO2 spheres that obstructed optimal light penetration.10 In light of this, the creation of nanocomposites that incorporate perovskite oxide nanosheets and photosensitizers is gaining importance. The objective of these nanocomposites is to augment light-harvesting capability and nanosheet stability. Carbon-based materials, renowned for their exceptional structural and electronic properties, are emerging as promising candidates.11,12 For example, multi-wall carbon nanotubes, capable of sustaining high current density (∼109 A cm−2),13 can be utilized to synthesize nanocomposites with perovskite. Shu et al. successfully anchored CsPbBr3 quantum dots on multi-wall carbon nanotubes, leading to a threefold enhancement in the photocatalytic performance of CO2 reduction.14 Graphitic carbon nitride (g-C3N4), characterized by a band gap of 2.7 eV with delocalized electrons during illumination, is another key material for forming heterostructures with perovskite.15 Jiang et al. constructed a 2D-2D g-C3N4/KCa2Nb3O10 heterojunction, which demonstrated superior photocatalytic activity in the degradation of tetracycline hydrochloride.16 Amongst conjugated carbon materials, graphene is regarded as an excellent supporting matrix for hybridization with perovskite due to its electron-rich characteristics, which facilitate high electrical conductivity (15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 m2 V−1 s−1 at room temperature) and high surface area (2600 m2 g−1).17 Li et al. synthesized a nanohybrid by integrating Ca2Nb3O10 perovskite nanosheets with graphene, which exhibited a significant enhancement in photocatalytic activity. This improvement was attributed to the charge transfer from graphene and efficient visible light absorption.12 Xian et al. also reported that anchoring CaTiO3 on graphene could enhance the photocatalytic performance towards methyl orange degradation.18 However, the advantageous effects of such layered perovskite–graphene composites at the materials interface in photocatalysis have not been thoroughly examined. Moreover, it is worth noting that perovskite oxide and graphene nanohybrids are typically fabricated through a hydrothermal step, which eliminates the need for high temperatures and extended reaction time. Nevertheless, the negative zeta potential in both perovskite nanosheets and graphene in water may not facilitate their higher dispersion easily.19,20
000 m2 V−1 s−1 at room temperature) and high surface area (2600 m2 g−1).17 Li et al. synthesized a nanohybrid by integrating Ca2Nb3O10 perovskite nanosheets with graphene, which exhibited a significant enhancement in photocatalytic activity. This improvement was attributed to the charge transfer from graphene and efficient visible light absorption.12 Xian et al. also reported that anchoring CaTiO3 on graphene could enhance the photocatalytic performance towards methyl orange degradation.18 However, the advantageous effects of such layered perovskite–graphene composites at the materials interface in photocatalysis have not been thoroughly examined. Moreover, it is worth noting that perovskite oxide and graphene nanohybrids are typically fabricated through a hydrothermal step, which eliminates the need for high temperatures and extended reaction time. Nevertheless, the negative zeta potential in both perovskite nanosheets and graphene in water may not facilitate their higher dispersion easily.19,20
In this study, we initially introduce a straightforward synthetic approach to anchor perovskite nanosheets onto reduced graphene oxide (rGO), followed by an attempt to elucidate the advantageous interfacial effects during photocatalysis. We employ poly diallyldimethylammonium chloride (PDDA), a UV light-sensitive mediator, on the rGO surface, which induces a shift in the zeta potential from −34.1 mV (anionic) of rGO to 41.5 mV (cationic) at approximately pH 7. We discovered that the nanohybrids can be assembled effortlessly at room temperature within a brief reaction time of 10 minutes. This method represents an efficient means of synthesizing perovskite nanosheets and graphene nanocomposites under ambient conditions, thereby addressing a significant gap in existing methodologies. Furthermore, through meticulous characterization using X-ray diffraction (XRD) via Simple Rietveld refinement, X-ray photoelectron spectroscopy (XPS), and X-ray absorption spectroscopy (XAS) experiments with a synchrotron light source, as well as density functional theory (DFT) calculations, we observe a significant effect on bandgap reduction via lattice contraction and octahedra tilting of the layered perovskites. This effect is achieved by increasing the charge density of lanthanide as A-site, even in the absence of rGO. Intriguingly, the accumulated charge and the structural distortion at the rGO/perovskite interface could miraculously amplify the further distortion in octahedra. This accounts for the more subtle effect on bandgap reduction via the A-site cations. Therefore, our combination of experimental and theoretical studies allows for a comprehensive understanding of charge dynamics at the interface of these layered composites. The findings of this study are particularly significant for advancing the field of layered perovskite oxide and graphene-analogous materials composites in catalysis.21–23
2. Results and discussion
Scheme 1 presents a schematic representation of the procedure for creating an rGO/perovskite nanocomposite. To ensure the anchoring of Dion–Jacobson layered perovskites on the rGO with a high surface area, we employed a two-step exfoliation approach involving protonation and intercalation. We selected the perovskite precursors RbLnNb2O7 (Ln = La, Pr, and Nd), as illustrated by their XRD patterns in Fig. S1a.† We also explored the use of other lanthanide elements such as Ce, Pm, Sm, and Eu in the A-site. However, their tolerance factor values fell outside the range of 0.925 to 0.955, preventing the achievement of the desired layered arrangement. RbLaNb2O7 exhibits orthorhombic symmetry with the Imma space group (ICSD = 75456; a = 5.4941 Å, b = 21.9901 Å, c = 5.4925 Å). Similarly, RbPrNb2O7 (ICSD = 238794; a = 5.4533 Å, b = 5.4503 Å, c = 21.9932 Å) and RbNdNb2O7 (ICSD = 258765; a = 5.4422 Å, b = 5.4297 Å, c = 21.9632 Å) both display orthorhombic symmetry with the I2cm space group.
 | ||
| Scheme 1 The overall synthetic approach towards nanohybrids of rGO and perovskite nanosheet. | ||
As shown in Fig. S2,† protonation of RbLnNb2O7 results in HLnNb2O7, with the diffraction peak (001) plane shifting from 2θ = ∼8.0° to a higher angle 2θ = ∼8.4°. This shift is due to the decrease in interlayer distance caused by the smaller radius of H+ compared to Rb+. The XRD patterns also exhibit significant changes after protonation, reflecting the tetragonal symmetry of the protonated perovskites with the P4/mmm space group. Detailed Rietveld refinement information is provided in Fig. S1b–d and Tables S1–S3.† We obtained perovskite nanosheets through sequential exfoliation along (00l) planes using TBAOH solution. The XRD pattern of the resulting perovskite nanosheet is presented in Fig. S3a–c.† The nanosheet sample exhibits a very weak (001) diffraction peak and a significant shift to a lower angle due to the presence of TBA+ ions in the interlayers to compensate for the negatively charged nanosheets. Furthermore, (002) peaks at 16.84° for HLaNb2O7 (Fig. S3a†), at 16.89° for HPrNb2O7 (Fig. S3b†), and at 16.87° for HNdNb2O7 (Fig. S3c†) disappear after exfoliation. This absence of (00l) (l ≥ 2) peaks indicates the loss of the periodic layered structure of protonated perovskites following the exfoliation with TBAOH solution. We gained further insights into the microstructure and morphologies of NNO (exfoliated NdNb2O7) through high-resolution TEM, HAADF-STEM, and SEM imaging in ESI.† Comparative analysis with the crystallised layered perovskite RbNdNb2O7 TEM image (Fig. S4†) reveals that NNO (Fig. S5†) manifests as a nearly transparent sheet with an ultrathin lamellar nanosheet morphology. This observation is corroborated by the SEM image in Fig. S6,† highlighting the sheet-like nanostructure of NNO. Fig. S7† presents the HAADF-STEM images of the NNO monolayer, affirming that the nanosheets consist of a single-crystal structure.
The construction of rGO/perovskite nanosheets is accomplished via an assembly approach, which is driven by a specifically tailored electrostatic method. Fig. 1a depicts the negatively charged surfaces of both the perovskite nanosheets and graphene oxide. Their zeta potential values are measured at −41.9 and −34.1 mV, respectively, at a pH of approximately 7. This electrostatic repulsion presents a challenge to the assembly process. To counteract this, graphene oxide is modified with PDDA to introduce positive charges, resulting in a zeta potential value of 41.5 mV at a pH of approximately 7, due to the presence of quaternary ammonium cations. It is important to highlight that perovskite nanosheets have a tendency to restack in acidic environments due to the presence of protons, necessitating the assembly process to occur at a pH of 7 or higher. Fig. 1b demonstrates the variation in zeta potential of perovskite nanosheets, rGO/PDDA, and graphene oxide within the pH range of 7 to 11. The zeta potential values of perovskite nanosheets and rGO/PDDA remain negative and positive, respectively, within this range, suggesting that the assembly process can be carried out in both neutral and basic environments. Subsequently, the resulting composites are exposed to UV light to photochemically convert the easily manipulated PDDA into NH4+ and other soluble products. FTIR measurements were utilized to monitor this process. As depicted in Fig. 1c, IR did not detect any signal from perovskite nanosheets. In contrast, rGO/PDDA/perovskite nanosheet displayed strong signals in the range of 1200 to 1300 cm−1, which are assigned to C–N stretching. However, after UV irradiation, these peaks vanish (due to the removal of PDDA by UV illumination) in the IR spectra of rGO/perovskite nanosheets in solution. Fig. 1d presents a comparison between the self-assembly approach and physical mixing. As can be observed, NNO suspensions are well dispersed in water due to their reduced thickness. When NNO suspension is mixed with rGO/PDDA, suspension is no longer observed after 10 minutes, as NNO is anchored on the rGO surface, rapidly forming a precipitate. Similar observations are made when exfoliated PrNb2O7 (PNO) or LaNb2O7 (LNO) suspension is mixed with rGO/PDDA. However, when NNO is physically mixed with unmodified graphene oxide, no precipitate is observed, indicating that perovskite nanosheets cannot be directly incorporated with graphene at room temperature without prior modification.
 | ||
| Fig. 1 (a) Zeta potential of GO, rGO/PDDA and exfoliated perovskite nanosheet dispersed in deionized water at pH 7; (b) zeta potential diagram of GO, rGO/PDDA and exfoliated perovskite nanosheet as a function of pH in aqueous solution; (c) FT-IR spectra of exfoliated perovskite nanosheet, rGO/perovskite nanosheet and rGO/PDDA/perovskite nanosheet; (d) a photograph showing graphene, exfoliated perovskite nanosheet and nanohybrid suspension. | ||
The synthesis of rGO/NNO nanohybrids is visually depicted in Fig. 2a, as evidenced by HAADF-STEM imaging. The image clearly delineates an interface between rGO and NNO. A selected region of the composite displays distinct crystal lattice fringes attributable to NNO. However, due to its ultrathin structure, the rGO region does not exhibit a clear crystal lattice fringe. Notably, the interplanar spacing of approximately 0.388 nm (indicated by the yellow arrow) corresponds to the (100) crystal plane of HNdNb2O7. This finding strongly implies that the perovskite nanosheets are anchored onto the rGO surface via c-axis stacking. Fig. 2d illustrates that PNO is also anchored on the rGO surface, revealing a coherent and clear interface between PNO and rGO regions. The rGO region exhibits a typical wrinkled 2D nanosheet morphology. Within the PNO region, a distinct fringe pattern is observed with an interplanar spacing of approximately 0.387 nm (indicated by yellow arrows), corresponding to the (100) crystal plane of HPrNb2O7. Moreover, a line scan profile in Fig. 2e, applied in the white arrow range in Fig. 2d, enables the visualization of element distribution. In the rGO region, carbon signals predominate, with minimal oxygen signals. Conversely, within the PNO region, there is a significantly high oxygen signals, accompanied by the emergence of Pr element signals. Although carbon signals decrease, they do not completely vanish due to the carbon film coating on the copper grid. Fig. 2b presents the SEM image of rGO/NNO, which displays a sheet-like structure. Its corresponding energy dispersive X-ray analysis (EDX) in Fig. 2c confirms the presence of carbon, oxygen, neodymium, and niobium elements. Additionally, the EDX elemental mapping visualizes a homogeneous distribution of carbon, oxygen, neodymium, and niobium in the rGO/NNO nanohybrid.
 | ||
| Fig. 2 (a) HAADF-STEM images of rGO/NNO nanohybrid; (b) SEM images of rGO/NNO and the corresponding (c) EDX mapping of C, O, Nb and Nd elements; (d) HAADF-STEM images of rGO/PNO nanohybrid; (e) corresponding line profile in the white arrow range with C, O and Pr element signals. | ||
The XPS survey scan spectra in Fig. 3a reveal that the binding energy peaks of these nanohybrids correspond to Nb 3d, Nb 3p, O 1s and lanthanide (La 3d, Pr 3d and Nd 3d), indicating the stability of perovskite nanosheets against decomposition on the rGO. The XRD patterns in Fig. S8† confirm the formation of rGO/LnNO nanohybrids without other impurities. The peaks marked by a star at approximately 26° represent the presence of rGO. Furthermore, EXAFS analysis has been employed to investigate the structural characteristics of the nanohybrids. Fig. 3b displays the Fourier-transformed EXAFS spectra of Nb K-edge for rGO/LNO and rGO/NNO, respectively. The intensity of peaks at approximately 1.5 Å and 3.2 Å are attributed to the Nb–O and Nb–Nb scattering, respectively. In Table S4,† the average Nb–O coordination numbers in rGO/LNO, rGO/PNO, and rGO/NNO are determined to be 5.97, 5.98, and 5.77, respectively within experimental errors. These values suggest the preservation of an octahedral coordination environment, albeit in a slightly distorted manner.
 | ||
| Fig. 3 (a) Overall XPS spectra of rGO/LNO, rGO/PNO and rGO/NNO; (b) Fourier transformed EXAFS spectra for Nb K-edge of rGO/LNO and rGO/NNO; (c) photocatalytic hydrogen production rate over layered perovskite RbLnNb2O7, perovskite nanosheets and rGO/perovskite nanosheets; (d) H2 evolution reaction in 3 hours. | ||
The photocatalytic efficacy of the fabricated samples was evaluated by tracking hydrogen evolution in a methanol solution under light exposure. Fig. 3c illustrates the catalytic activity of the crystalline layered perovskite RbLnNb2O7, which exhibits negligible catalytic activity. Prior research conducted by Wakayama et al. underscored the difficulties encountered when attempting to enhance the activity of Rb2NdNb2O6N by loading it with Pt as a co-catalyst (0.5 wt%), which only yielded a modest hydrogen evolution rate (HER) of 0.4 μmol g−1 under a 300 W xenon lamp (λ > 400 nm).6 The primary factor contributing to this limited activity is the inherently small surface area of these bulk crystalline materials, which suggests a dearth of reaction sites and consequently results in low photocatalytic activity. Following exfoliation, perovskite nanosheets exhibit a substantial improvement in photocatalytic activity for hydrogen production, primarily due to the increased surface area. Intriguingly, the incorporation of rGO into the nanosheets significantly enhances the photocatalytic activity. Notably, all nanohybrids containing reduced graphene oxide demonstrate excellent stability over a 3 hour testing period, with no significant changes in HER (Fig. 3d). Control photocatalytic activity tests were conducted in the absence of light or catalyst materials to verify any potential hydrogen production reaction, and no hydrogen was detected without light or catalyst samples. Additionally, the photocatalytic activity of layered perovskite RbLnNb2O7 (Ln = La, Pr, and Nd) was evaluated in the presence of TBAOH and PDDA for comparison. The results indicated that neither TBAOH nor PDDA could enhance the catalytic performance of RbLnNb2O7. Besides, NNO was mechanically mixed with GO. The HER activity did not alter within experimental errors (in Fig. S16†), which indicates GO and NNO are not contacted with each other too well, and the GO would also have blocked or scattered the incident light. This result are in line with previous reports, in which perovskite nanosheets were loaded on rGO and SiO2.10,12 The increase in activity following the incorporation of rGO is not directly proportional to their surface areas, but is critically dependent on the specific Ln in the A site. The rGO/PNO (310 μmol g−1) exhibits slightly higher activity than rGO/LNO (273 μmol g−1), but rGO/NNO demonstrates the highest photocatalytic activity (835 μmol g−1).
In general, perovskite oxides, with their unique advantages such as flexible chemical compositions, diverse crystal structures, and adjustable physicochemical properties, have been demonstrated to offer a remarkable platform for tuning catalytic performance. In most cases, the catalytic properties are intrinsically dictated by the nature of B-site cations or B–O bonding.21,22 Our calculations, conducted using the non-SOC DFT-D3+U method, suggest that the total density of states (DOS) and partial density of states (pDOS) for the valence band (VB) and conduction band (CB) of perovskite nanosheets near the Fermi level are primarily contributed by the 2p orbitals of oxygen atoms and 4d orbitals of niobium atoms as B site, respectively (Fig. S9a–c†).23 Interestingly, it is observed that lanthanide cations, despite being non-interacting A sites with no significant electronic influence, can also substantially affect catalytic activity. As depicted in Fig. 4a, meticulous Rietveld refinements of the HLaNb2O7, HPrNb2O7, and HNdNb2O7 powders, devoid of reduced graphene, reveal a progressive lattice contraction and octahedral tilting with the average O–Nb–O bond angle decreasing from 180°. This observation aligns with numerous experimental and theoretical studies on Ln-containing perovskites, where the increasing charge density (+3 oxidation state/cation radius) of Ln3+ at the A site can distort the Nb–O octahedra for charge polarization via electrostatic means, rather than involving their contracted f-electrons in the band structure.24–26 Both lattice contraction and octahedral tilting seem to decrease the Nb–O–Nb bond angles, thereby narrowing the bandgap to facilitate charge separation in photocatalysis, as reflected by the measured bandgaps shown in Fig. 4b. It is plausible that the reducing of the Nb–O–Nb bond angle due to structural distortions can stabilise CB minimum which is the t2g antibonding orbitals shown in Fig. S9d,† resulting in a narrower bandgap for excitation (Fig. S9e† and references therein). However, the extent of structural change due to the A site prior to exfoliation and without rGO inclusion is relatively minimal. Interestingly, as seen in Fig. 4a, the perovskite–graphene, exhibits the greatest reduction in Nb–O bond overlap, suggesting that the introduction of the graphene interface can amplify the tilting effects. It is shown that perovskite can be tilted through both tensile and compressive interfacial strain.27 When nanocomposites cooling down to room temperature after UV-vis treatment in Scheme 1, rGO would distort the perovskite nanosheets because it would expand in different directions.28 Moreover, this interfacial strain depends on the thermal expansion coefficient difference between rGO (−70 × 10−6 K−1) and perovskite (7–9 × 10−6 K−1). As the A site cation radius declines, the increasing thermal expansion coefficient of perovskite would further amplify the interfacial strain effect.29,30 This is aligned with our EXAFS in Fig. 3b, where rGO/NNO experiences greater distortion on Nb–Nb which indicates a reduced Nb–O–Nb bond angle with a longer and a shorter Nb–Nb distances.31 And this would keep narrowing the bandgaps. It is also hypothesized that the rGO, acting as an electron photosensitizer, may contribute a higher charge density to the Nb–O octahedra at the interface, thereby inducing lattice distortion and altering the O–Nb–O bond angle via electrostatic means to reduce the bandgaps in the composites. Such precise control in composite layers offers an additional and effective strategy to modulate the electron-accepting capability of perovskite by the A site cations.
 | ||
| Fig. 4 (a) Average unit cell volume, O–Nb–O bond angle and Nb–O bond length of HLaNb2O7, HPrNb2O7, HNdNb2O7 and average Nb–O bond length of rGO/LNO, rGO/PNO, rGO/NNO against Ln3+ charge density at A site; (b) band alignments of LNO, PNO, NNO with rGO derived from UV-vis and XPS measurements. | ||
Density Functional Theory (DFT) calculations depicted in Fig. 5a, d, g reveal a depletion of charge density on rGO and a corresponding accumulation of charge density on the interface oxygens of the layered perovskite at the interface. Notably, the charge density of rGO/NNO in Fig. 5g is significantly larger than that of rGO/PNO in Fig. 5d and rGO/LNO in Fig. 5a. This suggests that the interfacial oxygen atoms of NbO6 octahedra, particularly on the NNO with stronger electronegativity, can more readily retain excited electrons from rGO. Utilizing the Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional within the generalized gradient approximation (GGA), the calculated Bader charge on the interfacial oxygen represents the electron accepting ability of the perovskite nanosheets.32 The Bader analysis for the periodic Ln series, shown in Fig. S10,† indicates that the degree of electrons transferred from rGO to the perovskites progressively increases along with the Ln cations at the interface, yielding 0.56 |e| for rGO/GdNO, 0.54 |e| for rGO/NNO, 0.43 |e| for rGO/PNO, 0.50 |e| for rGO/CeNO, and 0.42 |e| for rGO/LNO, respectively. From our DFT calculations, we did not see any influence on band states from Ln 4f electrons of lanthanides. This agrees with the general consensus that the 4f electrons are more contracted to the nuclei of Ln3+ making no direct electronic involvement to the H2 evolution. Fig. 5h and i show that the binding energy of O 1 s and Nb 3d of rGO/NNO XPS exhibit a clear negative shift of 0.29 and 0.31 eV (electron richer), respectively. It is noteworthy that rGO/NNO also displays a much larger binding energy shift than rGO/PNO (Fig. 5e and f) and rGO/LNO (Fig. 5b and d). Additionally, the C 1s (Fig. S11†) of the graphitic Cg-O interface of rGO/NNO (286.3 eV) exhibits a more pronounced positive shift compared to rGO/LNO (286.0 eV) and rGO/PNO (286.1 eV), due to more h+ (holes) residing in the region. This is attributed to the transfer of some excited electrons through the Cg-O interface between NNO and rGO.12 To mitigate the influence of peak shift by charging effect, all the XPS peaks are referenced to the adventitious C 1s spectra which is distinguishable from C 1s of rGO in Fig. S11.† In addition, 3d spectra of lanthanides shown in Fig. S11 and S15† including La 3d, Pr 3d and Nd 3d display no shift in their binding energy after the incorporation with rGO giving the same values of 834.5 eV, 933.7 eV and 982.9 eV, respectively.
 | ||
| Fig. 5 Differential charge distribution of modelled (a) rGO/LNO; (d) rGO/PNO; (g) rGO/NNO. The yellow and blue regions represent net electron accumulation and depletion, respectively. (La: blue; Pr: orange; Nd: purple; Nb: green; O: pink; C: black); corresponding shift in O 1s XPS spectra for (b) rGO/LNO; (e) rGO/PNO; (h) rGO/NNO; corresponding shift Nb 3d XPS spectra for (c) rGO/LNO; (f) rGO/PNO; (i) rGO/NNO with reference to adventitious C 1s (no shift). | ||
Fig. 6a illustrates the dynamics of charge carriers in these nanohybrids during light activation. The rapid decay of fluorescence intensities, as evaluated by Time-Resolved Photoluminescence (TRPL) spectroscopy, indicates a swift interfacial electron transfer from rGO to NNO in rGO/NNO. This leads to a much quicker charge carrier decay compared to rGO/LNO and rGO/PNO, respectively. As the electron accepting ability of perovskite nanosheets increases (higher Bader charge of interfacial oxygen), the rGO can supply excited mobile electrons for charge recombination and their exciton lifetime becomes shortened. NNO with the highest electron accepting ability by structural distortion could obtain more excited electrons from rGO, which makes it become a visible-light-driven photocatalyst, further boosting its photocatalytic activity.33 To assess the photocatalytic activity in visible light range, we have conducted the HER measurements of rGO/NNO under different wavelengths without any sacrificial reagent (Fig. S14†). It clearly demonstrates that rGO/NNO still gives H2 evolution activity after 550 nm even with a very low light intensity (15.7 mW). This observation aligns with our expectation that more mobile electrons can be readily transferred from rGO to NNO in the nanohybrids.12,34 The charge carrier lifetime was determined from biexponential function fitting to the TRPL,12,35 and the average lifetime was calculated by  , where A1 and A2 are the corresponding amplitude of the decay time. Steady-state PL spectra in Fig. S12† confirm the Burstein–Moss effect in rGO/NNO, resulting in a blue shift of the emission peak due to a significant increase in charge carrier concentration,14 which will lead to greater distortion in Nb–O and bandgap narrowing by the Nd sites, as previously stated. The activity order also appears to show a strong correlation with the inverse lifetime of excitons and Bader charge on oxygen atoms (Fig. 6b).
, where A1 and A2 are the corresponding amplitude of the decay time. Steady-state PL spectra in Fig. S12† confirm the Burstein–Moss effect in rGO/NNO, resulting in a blue shift of the emission peak due to a significant increase in charge carrier concentration,14 which will lead to greater distortion in Nb–O and bandgap narrowing by the Nd sites, as previously stated. The activity order also appears to show a strong correlation with the inverse lifetime of excitons and Bader charge on oxygen atoms (Fig. 6b).
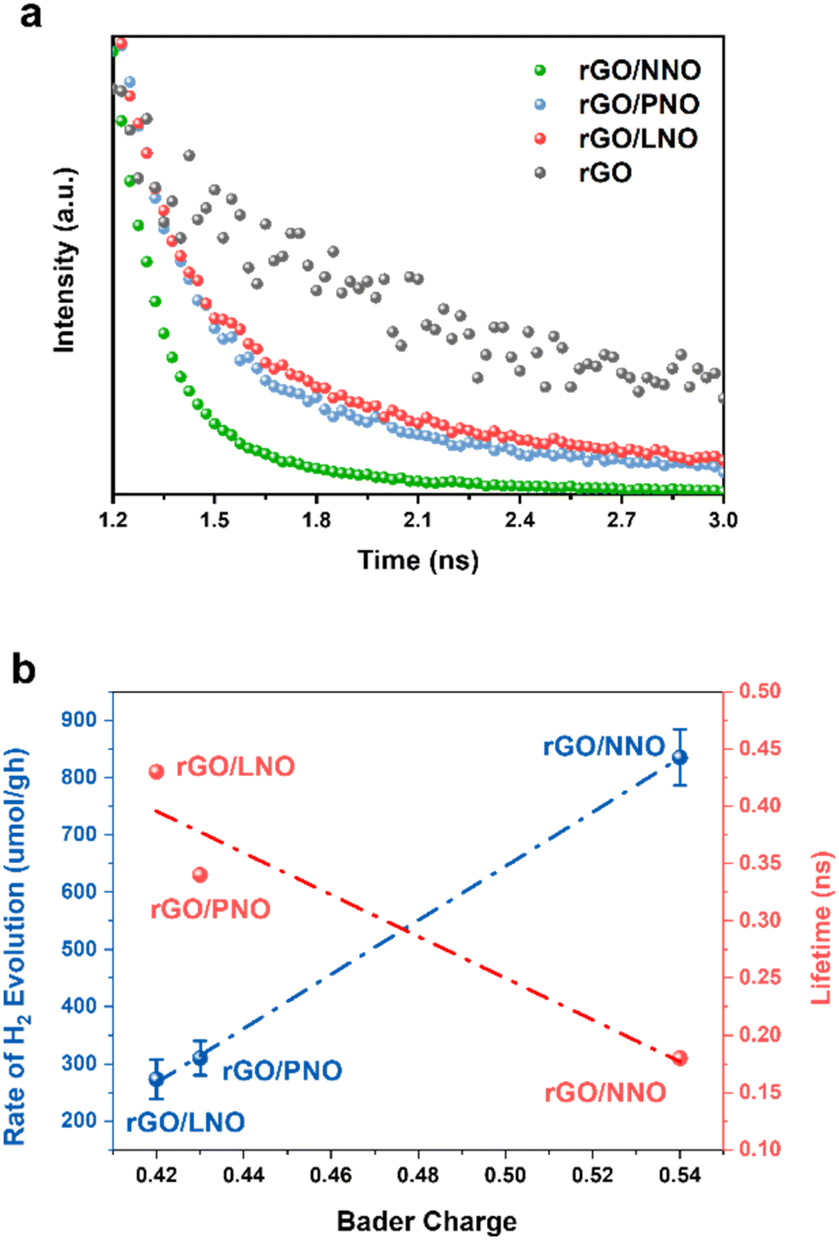 | ||
| Fig. 6 (a) TRPL of rGO compared with substantial shorter lifetimes for nanohybrids; (b) inversed correlation of HER activity with average exciton lifetime of nanohybrids plotted against calculated Bader charge of interface oxygen atoms. | ||
The above characterizations clearly highlight the importance of optimal interfacial interactions between perovskite nanosheets and rGO to facilitate electron transfer from rGO to perovskite nanosheets. This enhances the screening effect and reduces the binding energy of O 1s and Nb 3d. As a result, rGO plays a crucial role as a photosensitizer, providing excited electrons to perovskite nanosheets and transforming UV-light-driven perovskite into visible light-driven photocatalysts.12,36,37 Therefore, this difference in charge dynamics at the materials interface can amplify the A site effect in the distortion of perovskite, providing a new means to tune photocatalysis.
3. Conclusions
In conclusion, we have successfully presented a large surface area of Dion–Jacobson perovskite to rGO through exfoliation and an electrostatic-driven assembly approach. These nanohybrids demonstrated enhanced photocatalytic activity for hydrogen evolution due to the beneficial interfacial electron transfer. By modifying the A site charge density, the lattice contraction and octahedral tilting at the materials interface could dramatically change the band structure of perovskite nanosheets. NNO, with its reduced bandgap and lower level of conduction band minimum due to distortion, could capture more excited electrons from rGO for charge separation required in photocatalysis. This synergistic effect of A-site charge density, combined with interfacial photosensitizer material, can greatly tune the electron accepting ability of layered perovskite. This study reveals the design rules for fine-tuning targeted band structures.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Haozhe Zhang: methodology, synthesis, refinement, photocatalytic activity test, writing, editing. Yanjie Wang and Mingyu Luo: DFT calculation. Wentian Niu: XAS analysis. Tatchamapan Yoskamtorn, Robert Taylor and Sarah Day: resources. Edman Tsang: supervision, resources, methodology, writing, editing and reviewing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Chinese Scholarship Council of People's Republic of China for the scholarship to Haozhe Zhang. The authors acknowledge the technical supports from the Research Complex at Harwell, the BRICS STI Framework Program (52261145703) and the European Union's Horizon 2020 Research under Grant agreement no. 823717 – ESTEEM3. We also thank Dr Yiyang Li and Baron Ho in Tsang group for their helps in TEM and TRPL experiments.References
- A. Fujishima and K. Honda, Electrochemical photolysis of water at a semiconductor electrode, Nature, 1972, 238(5358), 37–38 CrossRef CAS PubMed.
- T. Oshima, T. Ichibha, K. Oqmhula, K. Hibino, H. Mogi, S. Yamashita, K. Fujii, Y. Miseki, K. Hongo and D. Lu, Two-Dimensional Perovskite Oxynitride K2LaTa2O6N with an H+/K+ Exchangeability in Aqueous Solution Forming a Stable Photocatalyst for Visible-Light H2 Evolution, Angew. Chem., Int. Ed., 2020, 59(24), 9736–9743 CrossRef CAS PubMed.
- Y. Hu, L. Mao, X. Guan, K. A. Tucker, H. Xie, X. Wu and J. Shi, Layered perovskite oxides and their derivative nanosheets adopting different modification strategies towards better photocatalytic performance of water splitting, Renewable Sustainable Energy Rev., 2020, 119, 109527 CrossRef CAS.
- C. Hanmandlu, A. Singh, K. M. Boopathi, C.-S. Lai and C.-W. Chu, Layered perovskite materials: key solutions for highly efficient and stable perovskite solar cells, Rep. Prog. Phys., 2020, 83(8), 086502 CrossRef CAS PubMed.
- G. Zhang, G. Liu, L. Wang and J. T. Irvine, Inorganic perovskite photocatalysts for solar energy utilization, Chem. Soc. Rev., 2016, 45(21), 5951–5984 RSC.
- H. Wakayama, K. Hibino, K. Fujii, T. Oshima, K. Yanagisawa, Y. Kobayashi, K. Kimoto, M. Yashima and K. Maeda, Synthesis of a layered niobium oxynitride, Rb2NdNb2O6N·H2O, showing visible-light photocatalytic activity for H2 evolution, Inorg. Chem., 2019, 58(9), 6161–6166 CrossRef CAS PubMed.
- S. Ida, Y. Okamoto, M. Matsuka, H. Hagiwara and T. Ishihara, Preparation of tantalum-based oxynitride nanosheets by exfoliation of a layered oxynitride, CsCa2Ta3O10–xNy, and their photocatalytic activity, J. Am. Chem. Soc., 2012, 134(38), 15773–15782 CrossRef CAS PubMed.
- M. S. Khan, Z. Diao, M. Osada and S. Shen, Nitrogen doped ultrathin calcium/sodium niobate perovskite nanosheets for photocatalytic water oxidation, Sol. Energy Mater. Sol. Cells, 2020, 205, 110283 CrossRef CAS.
- M. Hojamberdiev, M. F. Bekheet, E. Zahedi, H. Wagata, Y. Kamei, K. Yubuta, A. Gurlo, N. Matsushita, K. Domen and K. Teshima, New Dion–Jacobson phase three-layer perovskite CsBa2Ta3O10 and its conversion to nitrided Ba2Ta3O10 nanosheets via a nitridation–protonation–intercalation–exfoliation route for water splitting, Cryst. Growth Des., 2016, 16(4), 2302–2308 CrossRef CAS.
- H. Zhou, E. M. Sabio, T. K. Townsend, T. Fan, D. Zhang and F. E. Osterloh, Assembly of Core–Shell Structures for Photocatalytic Hydrogen Evolution from Aqueous Methanol, Chem. Mater., 2010, 22(11), 3362–3368 CrossRef CAS.
- S. Acharya, S. Martha, P. C. Sahoo and K. Parida, Glimpses of the modification of perovskite with graphene-analogous materials in photocatalytic applications, Inorg. Chem. Front., 2015, 2(9), 807–823 RSC.
- D. Li, H. Zhao, L. Li, B. Mao, M. Chen, H. Shen, W. Shi, D. Jiang and Y. Lei, Graphene-sensitized perovskite oxide monolayer nanosheets for efficient photocatalytic reaction, Adv. Funct. Mater., 2018, 28(52), 1806284 CrossRef.
- V. Gupta and T. A. Saleh, Syntheses of carbon nanotube-metal oxides composites; adsorption and photo-degradation, Carbon Nanotubes: Res. Appl., 2011, 17, 295–312 Search PubMed.
- M. Shu, Z. Zhang, Z. Dong and J. Xu, CsPbBr3 perovskite quantum dots anchored on multiwalled carbon nanotube for efficient CO2 photoreduction, Carbon, 2021, 182, 454–462 CrossRef CAS.
- X. Xu, G. Liu, C. Randorn and J. T. Irvine, g-C3N4 coated SrTiO3 as an efficient photocatalyst for H2 production in aqueous solution under visible light irradiation, Int. J. Hydrogen Energy, 2011, 36(21), 13501–13507 CrossRef CAS.
- D. Jiang, T. Wang, Q. Xu, D. Li, S. Meng and M. Chen, Perovskite oxide ultrathin nanosheets/g-C3N4 2D-2D heterojunction photocatalysts with significantly enhanced photocatalytic activity towards the photodegradation of tetracycline, Appl. Catal., B, 2017, 201, 617–628 CrossRef CAS.
- M. D. Stoller, S. Park, Y. Zhu, J. An and R. S. Ruoff, Graphene-based ultracapacitors, Nano Lett., 2008, 8(10), 3498–3502 CrossRef CAS PubMed.
- T. Xian, H. Yang and Y. Huo, Enhanced photocatalytic activity of CaTiO3-graphene nanocomposites for dye degradation, Phys. Scr., 2014, 89(11), 115801 CrossRef.
- Y.-S. Han, I. Park and J.-H. Choy, Exfoliation of layered perovskite, KCa2Nb3O10, into colloidal nanosheets by a novel chemical process, J. Mater. Chem., 2001, 11(4), 1277–1282 RSC.
- A. H. Mevold, W.-W. Hsu, A. Hardiansyah, L.-Y. Huang, M.-C. Yang, T.-Y. Liu, T.-Y. Chan, K.-S. Wang, Y.-A. Su and R.-J. Jeng, Fabrication of gold nanoparticles/graphene-PDDA nanohybrids for bio-detection by SERS nanotechnology, Nanoscale Res. Lett., 2015, 10, 1–7 CrossRef CAS PubMed.
- L. Lu, S. Ni, G. Liu and X. Xu, Structural dependence of photocatalytic hydrogen production over La/Cr co-doped perovskite compound ATiO3 (A = Ca, Sr and Ba), Int. J. Hydrogen Energy, 2017, 42(37), 23539–23547, DOI:10.1016/j.ijhydene.2017.01.064.
- A. Kudo, H. Kato and S. Nakagawa, Water splitting into H2 and O2 on new Sr2M2O7 (M = Nb and Ta) photocatalysts with layered perovskite structures: factors affecting the photocatalytic activity, J. Phys. Chem. B, 2000, 27(3), 571–575 CrossRef.
- S. Di Tommaso, F. Giannici, A. Mossuto Marculescu, A. Martorana, C. Adamo and F. Labat, Toward tailorable surfaces: a combined theoretical and experimental study of lanthanum niobate layered perovskites, J. Chem. Phys., 2014, 141, 024704 CrossRef PubMed.
- R. Prasanna, A. Gold-Parker, T. Leijtens, B. Conings, A. Babayigit, H. G. Boyen, M. F. Toney and M. D. McGehee, Band Gap Tuning via Lattice Contraction and Octahedral Tilting in Perovskite Materials for Photovoltaics, J. Am. Chem. Soc., 2017, 139(32), 11117–11124 CrossRef CAS PubMed.
- A. G. Bispo-Jr, A. J. de Morais, C. M. Calado, I. O. Mazali and F. A. Sigoli, Lanthanide-doped luminescent perovskites: a review of synthesis, properties, and applications, J. Lumin., 2022, 252, 119406 CrossRef CAS.
- T. Hasegawa, T. Ueda, Y. Asakura and S. Yin, Cerium (III) Niobate Layered Perovskites: Abnormal Optical Absorption Modulations by Tuning of B-Site Composition and Perovskite Layer Charge Control, Inorg. Chem., 2022, 61(50), 20636–20646 CrossRef CAS PubMed.
- H. S. Kim and N. G. Park, Importance of tailoring lattice strain in halide perovskite crystals, NPG Asia Mater., 2020, 12(1), 78 CrossRef CAS.
- W. Pan, J. Xiao, J. Zhu, C. Yu, G. Zhang, Z. Ni, K. Watanabe, T. Taniguchi, Y. Shi and X. Wang, Biaxial compressive strain engineering in graphene/boron nitride heterostructures, Sci. Rep., 2012, 2(1), 893 CrossRef PubMed.
- S. Y. Ju, W. I. Lee and H. S. Kim, Enhanced Phase Stability of Compressive Strain-Induced Perovskite Crystals, ACS Appl. Mater. Interfaces, 2022, 14(35), 39996–40004 CrossRef CAS PubMed.
- A. Løken, S. Ricote and S. Wachowski, Thermal and chemical expansion in proton ceramic electrolytes and compatible electrodes, Crystals, 2018, 8(9), 365 CrossRef.
- Z. Weng, L. Liu, Y. Hu, Y. Wei, P. Da, Z. Wu, Z. Mu, P. Xi and C. H. Yan, Significance of Engineering the MnO6 Octahedral Units to Promote the Oxygen Reduction Reaction of Perovskite Oxides, Adv. Mater., 2023, 25, 2311102 Search PubMed.
- T. Jaouen, P. Aebi, S. Tricot, G. Delhaye, B. Lépine, D. Sébilleau, G. Jézéquel and P. Schieffer, Induced work function changes at Mg-doped MgO/Ag (001) interfaces: Combined Auger electron diffraction and density functional study, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90(12), 125433 CrossRef.
- P. Zhou, H. Chen, Y. Chao, Q. Zhang, W. Zhang, F. Lv, L. Gu, Q. Zhao, N. Wang, J. Wang and S. Guo, Single-atom Pt-I3 sites on all-inorganic Cs2SnI6 perovskite for efficient photocatalytic hydrogen production, Nat. Commun., 2021, 12(1), 4412 CrossRef CAS PubMed.
- J. Tang, L. Brzozowski, D. A. Barkhouse, X. Wang, R. Debnath, R. Wolowiec, E. Palmiano, L. Levina, A. G. Pattantyus-Abraham, D. Jamakosmanovic and E. H. Sargent, Quantum dot photovoltaics in the extreme quantum confinement regime: the surface-chemical origins of exceptional air-and light-stability, ACS Nano, 2010, 4(2), 869–878 CrossRef CAS PubMed.
- Y. Yamamoto, T. Fukushima, Y. Suna, N. Ishii, A. Saeki, S. Seki, S. Tagawa, M. Taniguchi, T. Kawai and T. Aida, Photoconductive coaxial nanotubes of molecularly connected electron donor and acceptor layers, Science, 2006, 314(5806), 1761–1764 CrossRef CAS PubMed.
- Y. Zhang, N. Zhang, Z. R. Tang and Y. J. Xu, Graphene transforms wide band gap ZnS to a visible light photocatalyst. The new role of graphene as a macromolecular photosensitizer, ACS Nano, 2012, 6(11), 9777–9789 CrossRef CAS PubMed.
- G. Xie, K. Zhang, B. Guo, Q. Liu, L. Fang and J. R. Gong, Graphene-based materials for hydrogen generation from light-driven water splitting, Adv. Mater., 2013, 25(28), 3820–3839 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4se00630e |
| This journal is © The Royal Society of Chemistry 2024 |