
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Migration-mitigated crossover of organic redox anions across a proton-exchange membrane†
Penghui
Ding
a,
Mikhail
Vagin
 *ab,
Mohammad Javad
Jafari
d,
Aleksandar Y.
Mehandzhiyski
a,
Viktor
Gueskine
ac,
Tobias
Abrahamsson
a,
Igor
Zozoulenko
ac,
Thomas
Ederth
d and
Reverant
Crispin
abc
*ab,
Mohammad Javad
Jafari
d,
Aleksandar Y.
Mehandzhiyski
a,
Viktor
Gueskine
ac,
Tobias
Abrahamsson
a,
Igor
Zozoulenko
ac,
Thomas
Ederth
d and
Reverant
Crispin
abc
aLaboratory of Organic Electronics, Department of Science and Technology, Linköping University, 60174 Norrköping, Sweden. E-mail: mikhail.vagin@liu.se; Tel: +46 702753087
bWallenberg Initiative Materials Science for Sustainability, Department of Science and Technology, Linköping University, Norrköping 60174, Sweden
cWallenberg Wood Science Center, Linköping University, 60174 Norrköping, Sweden
dDepartment of Physics, Chemistry and Biology, Linköping University, 58183 Linköping, Sweden
First published on 9th September 2024
Abstract
The two-electron oxygen reduction reaction (ORR), powered by affordable renewable energy, presents a more promising and sustainable approach to hydrogen peroxide production than traditional methods. In this study, we introduce a membrane electrolyzer for ORR-to-H2O2 generation. The conducting polymer poly(3,4-ethylenedioxythiophene):poly(styrene sulfonate) (PEDOT:PSS) acts as the cathode that aids the oxygen reduction reaction through a two-electron pathway to produce H2O2. At the anode, we employed the oxidation of a model organic molecule, 4,5-dihydroxy-1,3-benzenedisulfonic acid disodium salt monohydrate (tiron). This catalyst-free anode process, as an alternative to the sluggish water oxidation reaction commonly used in classical electrolyzers, reduces voltage loss to release protons, cross the membrane, and feed the ORR at the cathode. Our study investigated the often-neglected issue of organic crossover during electrolyzer operation and its significant impact on transport behavior. This research paves the way for the development of crossover-free flow cells, extending the realm of electrochemical devices based on the electrolyte fed and the membrane.
1 Introduction
Hydrogen peroxide (H2O2) is a powerful oxidant considering its relatively high oxidation potential (e.g. for the half reaction H2O2 + 2H+ + 2e− → 2H2O, the equilibrium potential (Eeq) is +1.763 V vs. reversible hydrogen electrode (RHE)). It is utilized in wood pulp bleaching processes to increase the brightness of paper, which accounts for more than 40% of worldwide production. The combination of the liquid form of oxidants with water as the only product of oxidation makes H2O2 an increasingly attractive reagent in a wide range of oxidative transformations in the synthesis of fine chemicals.1–4 Moreover, H2O2 is considered an advantageous discharge product in the emerging field of high energy-density rechargeable metal–air batteries.5–7The anthraquinone oxidation process, the industrialized route for H2O2 production, suffers from many disadvantages due to the use of hydrogen gas for the unsafe transportation of highly concentrated H2O2. Electrification of the chemical industry is one of the biggest steps towards a carbon neutral economy, which uses electricity from renewable sources to synthesize desired products compared to the processes driven by fossil fuels.8 This technological route enables the production of desired chemicals in a sustainable, decentralized and on-site/-demand way. The electrochemical synthesis of H2O2 combines O2 from air, water and green electricity through a two-electron oxygen reduction reaction (O2-to-H2O2).
The recent surge in studies on H2O2 electrosynthesis is mainly focused on the design of cheap and advanced catalysts aiming to achieve high activity and selectivity for O2-to-H2O2 conversion. Metal alloys, oxidized carbons, and macrocyclic complexes are representative catalysts for ORR-to-H2O2 generation.9
Conducting polymers (CPs), such as PEDOT:PSS, have been recently identified as another group of materials for ORR-to-H2O2 generation.10–12 Their molecular defect-free structures can be synthetically designed to tune electronic transport, and primary and secondary dopants define ionic transport. The modulation of either electrical or ionic charge within a CP phase results in the appearance of compensational ionic or electrical transport, respectively, which defines the CP as a mixed ionic–electronic conductor.13,14 These properties make CPs a versatile self-suspended electrode15 for the elucidation of reaction mechanisms.16–19
In parallel to the catalyst development, the optimization of the electrolyzer's design is needed for the commercialization of such sustainable technology.20,21 Here, little attention is paid to the oxidation at anodes, the auxiliary half-reaction for the main productive ORR-to-H2O2 process. The oxygen evolution reaction (OER), the most used anode oxidation process in H2O2 electrolyzers, has a significant voltage loss and needs expensive catalysts based on platinum group metals.22 The use of the organic oxidation process with the possible generation of value-added products instead of the OER could reduce the costs of technology.23–25
The forest and paper industries are the major sources of biopolymers. The lignin-rich waste product called black liquor with a global production of ca. 170 million tonnes per year is a side-product of paper making. The de-polymerization of lignin started to attract attention due to the possibility of obtaining aromatic alcohols26,27 including benzenediols such as catechols.28Ortho- and para-benzenediols are the molecules of high energy density because they can accommodate and release two electrons per aromatic ring via two proton-coupled electron transfers: benzenediol ↔ benzenedioloxidized + 2e− + 2H+. The proceeding of this reaction on the electrode in acidic media, which enables the carbon-neutral use of wood-derived organics, has few advantages in comparison with the OER, which are as follows: (1) it is fast enough on graphite enabling catalyst-free electrodes in contrast to the OER, which relies on the noble catalysts; (2) it proceeds at potentials lower than the onset of the OER (e.g. for tiron: Eeq = +0.908 V vs. RHE, for OER: Eeq = +1.23 V vs. RHE), which reduces the voltage losses; and (3) it is reversible, which opens the possibility for the redox regeneration of anode reagents.
Importantly, the strategy to use a specific reagent to drive the anode oxidation implies the asymmetry in its concentrations applied across the membrane of the electrolyzer. The cross-contamination of the cathode compartment with the anode reagent is unfavorable for H2O2 production. This specifies the membrane selectivity: to maintain the transport of ions involved in charge compensation between the reactions at the cathode and the anode and to prevent the contamination of cathode compartment by anode reagents.
In this work, we chose tiron as an anode oxidation reagent because of its low cost and high solubility in water. The oxidation of tiron on graphite was used as an auxiliary process to compensate the ORR-to-H2O2 half-reaction occurring on the cathode. The goal of this study is to investigate the transport of tiron across the proton-exchange membrane (Nafion 115) in the absence and presence of membrane electrolysis (Scheme 1). Crossover is a collective term describing all unwanted transport of species through the membrane-based electrochemical devices. Most membrane studies focus the diffusional transport under zero current conditions only. However, crossover has another contributing factor: migration. For the charged molecules during membrane cell operation, the former could not be ignored. We found a remarkably different transport behavior of the dianion tiron between diffusion and operation cells, demonstrating the importance of migration in the design of membrane electrolyzers fed with electrolytes.
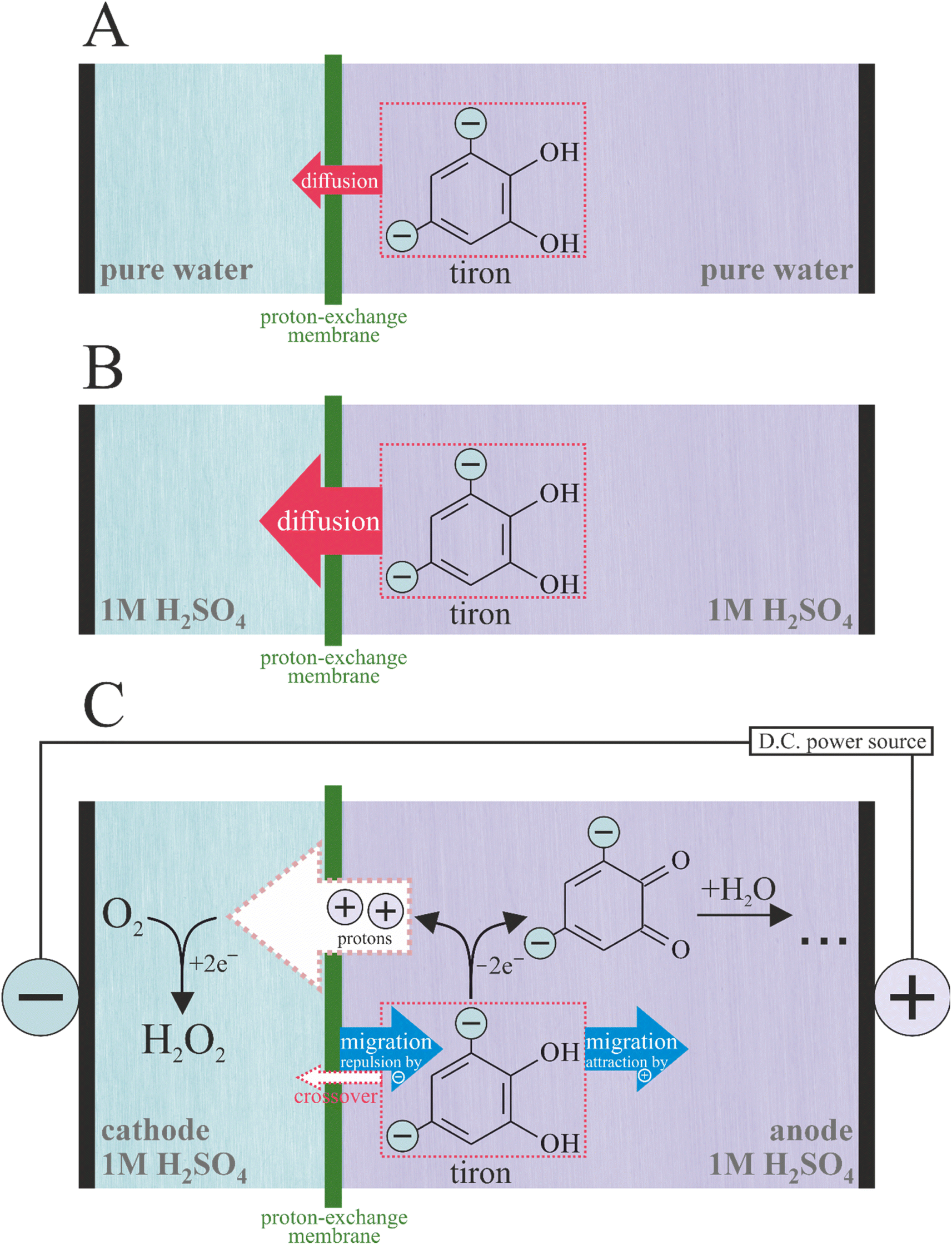 | ||
| Scheme 1 Processes occurring in the electrolyzer under zero current ((A) pure water; (B) 1 M H2SO4) and non-zero current (C) conditions. | ||
2 Experimental section
2.1 Reagents and chemicals
Dimethyl sulfoxide (99.9%), 4,5-dihydroxy-1,3-benzenedisulfonic acid disodium salt monohydrate (tiron, 97%), and Nafion 115 membrane were provided by Sigma-Aldrich. PEDOT:PSS (Clevios PH 1000) was purchased from Heraeus. High-purity oxygen (O2) was supplied by Linde Gas AB. All reagents and chemicals were used directly without any purification processes. Deionized (DI) water was used in all experiments.2.2 Electrolyzers
The cathode of the electrolyzer was fabricated by spray-coating 25 mL of PEDOT:PSS containing 2% DMSO onto a piece of carbon paper (5 × 5 cm, AvCarb MGL 190, Fuel Cell Store Inc.) followed by drying at 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C overnight. The anode was unmodified carbon paper (5 × 5 cm, AvCarb MGL 190, Fuel Cell Store Inc.). The electrolyzer (Scheme 2) was assembled using a C-Flow LAB 5 × 5 plastic electrochemical cell (C-Tech Innovation Ltd). The graphite felt (AvCarb G200, 6.5 mm thickness, FuelCellStore (TX, USA)) used as the diffusion layer of the electrolyzer was hydrophilized by immersion in concentrated H2SO4 for about 5 seconds followed by washing with an excess of water. A solid graphite plate (6 mm thickness) was used as an endplate. Constant current electrolysis at 2 mA (current density of 0.08 mA cm−2) was carried out at room temperature using a BioLogic SP-200 potentiostat. The Nafion 115 membrane is pre-treated according to the established procedures.29 The geometrical area of Nafion membrane (6.2 × 6.2 cm) was defined by a square-shaped stencil provided by C-Tech Innovation, which is slightly larger than the electrode area (5 × 5 cm). The PEDOT:PSS-modified carbon paper was placed on a membrane (the modified side facing the membrane). The cathode compartment was supplied with 45 mL of liquid (water or 1 M H2SO4) by a peristaltic pump with constant oxygen flow bubbled through the liquid in the tank. The anode compartment was supplied with 45 mL of 0.05 M tiron solution in water or in 1 M H2SO4 by a peristaltic pump. The flow rate is 7.8 mL per minute for both cathode and anode compartments.
°C overnight. The anode was unmodified carbon paper (5 × 5 cm, AvCarb MGL 190, Fuel Cell Store Inc.). The electrolyzer (Scheme 2) was assembled using a C-Flow LAB 5 × 5 plastic electrochemical cell (C-Tech Innovation Ltd). The graphite felt (AvCarb G200, 6.5 mm thickness, FuelCellStore (TX, USA)) used as the diffusion layer of the electrolyzer was hydrophilized by immersion in concentrated H2SO4 for about 5 seconds followed by washing with an excess of water. A solid graphite plate (6 mm thickness) was used as an endplate. Constant current electrolysis at 2 mA (current density of 0.08 mA cm−2) was carried out at room temperature using a BioLogic SP-200 potentiostat. The Nafion 115 membrane is pre-treated according to the established procedures.29 The geometrical area of Nafion membrane (6.2 × 6.2 cm) was defined by a square-shaped stencil provided by C-Tech Innovation, which is slightly larger than the electrode area (5 × 5 cm). The PEDOT:PSS-modified carbon paper was placed on a membrane (the modified side facing the membrane). The cathode compartment was supplied with 45 mL of liquid (water or 1 M H2SO4) by a peristaltic pump with constant oxygen flow bubbled through the liquid in the tank. The anode compartment was supplied with 45 mL of 0.05 M tiron solution in water or in 1 M H2SO4 by a peristaltic pump. The flow rate is 7.8 mL per minute for both cathode and anode compartments.
 | ||
| Scheme 2 Membrane electrolyzer based on the proton-exchange membrane. | ||
2.3 H2O2 detection
First, 10 μL of aliquot from the cathode compartment was collected every hour. Then 290 μL of a stirred freshly prepared solution of HRP (0.75 ng mL−1) and TMB (30 mg mL−1) in a 0.1 M phosphate-citrate buffer solution of pH 6 was added to the collected analytes. Then the absorbance of the mixture was measured (at 653 nm) using a plate reader (BioTek Synergy H1 Hybrid Multi-Mode Reader). Finally, the concentration of H2O2 was determined from the calibration line with known H2O2 concentrations (0 μM, 10 μM, 20 μM, 30 μM, and 40 μM).2.4 Crossover experiments
Tiron crossover experiments in the absence and presence of current were conducted using an electrolyzer, as shown above.30 The anode compartment was supplied with 45 mL of 0.05 M tiron either in pure water or in 1 M H2SO4 using a peristaltic pump. The cathode compartment was supplied with 45 mL of pure solvent (pure water or 1 M H2SO4) using a peristaltic pump. An aliquot was collected from the cathode compartment for further analysis every one hour. The UV-vis absorbance spectra were recorded using a spectrometer (Lambda 900, PerkinElmer). The actual tiron concentration was calculated from the calibration curve obtained for the specific media (pure water or 1 M H2SO4).2.5 Attenuated total reflection-Fourier-transform infrared (ATR-FTIR) measurements
For ex situ measurements, a Nafion 115 film was clamped onto an ATR prism. Measurements were carried out using a PIKE MIRacle ATR accessory in a Bruker Vertex 70 spectrometer, with a DLaTGS detector. IR spectra were recorded at 4 cm−1 resolution. Baseline correction and vector normalization were applied to all spectra. To observe the changes, all spectra were subtracted from the pristine spectrum. The in situ measurements were carried out using a Nafion 115 film clamped using a plastic lid comprising cathode and anode compartments (Fig. 3A), enabling ATR-FTIR data acquisition combined with chronopotentiometry on the applied constant current. The distance from the anode compartment to the detection zone of ATR-FTIR prism was 1 mm, while the similar distance to cathode was ca. 1 cm. Two flags of graphite paper were utilized as electrodes. A tiron solution in 1 M H2SO4 was used in the anode compartment. The solution of alizarine red S in 1 M H2SO4 was used in the cathode compartment to enable the proton-coupled electron donor process similar to the ORR in the electrolyzer. The cell was under open circuit conditions for 15 minutes and then a constant current (0.5 mA) was applied, while the FTIR spectra acquisition in the spectral range of 3700–600 cm−1 was carried out continuously.2.6 1H-nuclear magnetic resonance (1H-NMR) spectroscopy
The samples of electrolyte from the anode compartment (200 μl) were diluted with 900 μl of D2O. 1H-NMR measurements were carried out for the dry samples obtained after solvent removal by rotary evaporation at 500 MHz using an Inova Varian Oxford AS500 spectrometer. The analysis was performed in CDCl3 with chemical shifts referenced against the CDCl3 residual peak at 7.26 ppm or DMF at 8.02 ppm.31,32 The data were processed using MestReNova 12.04-22023 (Mestrlab Research).2.7 Modelling
To model the flux of species across the membrane and the device using COMSOL Multiphysics® v. 6.2., the 1D model based on the generalized Poisson–Nernst–Planck equations was created (ESI Note 1†). The model consists of two porous electrode domains (anode and cathode) and a membrane domain placed in-between the two electrodes, as depicted in Fig. 4A. The anode and cathode consist of solid (carbon felt) and liquid (electrolyte) phases with volume fractions of 0.1 and 0.9, respectively. In our model, the electrolyte in the cathode contains icathode = H+, HSO−4, SO42−, O2 and the electrolyte in the anode contains ianode = H+, HSO4−, SO42−, tiron2− species. The tertiary current distribution, Nernst–Plank–Poisson interface was used for simulating the transport of species.3 Results and discussion
To investigate the processes occurring during the membrane electrolysis, we constructed an electrolyzer (Scheme 2) based on the proton-exchange membrane (PEM) sandwiched between two electrodes. The compression applied between two electrodes assures minimization of ohmic losses during electrolysis. The electrodes were integrated in two compartments separated by PEM and fed independently by two flows of aqueous media. The application of the electricity from the external source on the positive and negative electrodes of electrolyzer results in oxidation and reduction, respectively. The functionality of PEM includes (1) maintenance of the proton transport to achieve electroneutrality on the electrolyzer and (2) prevention of the cross-contamination of two flows of aqueous media. To estimate the contributions of the different tiron transport processes across the PEM, such as diffusion and migration, we investigated the time dependencies of cross-transported tiron concentration under the conditions of zero and non-zero current (the absence and presence of electrolysis, respectively).3.1 Zero current conditions
We utilized the electrolyzer cell to estimate the rate of the tiron transport in the absence of electrolysis (Scheme 1A and B), i.e. without applying any current. Both cathode and anode compartments of electrolyzers were fed with identical solvents, either pure water or 1 M H2SO4. Additionally, the anode compartment contains 0.05 M of tiron solution to create the concentration gradient across PEM as a driving force for the tiron diffusion. The appearance of tiron at the cathode compartment of the electrolyzer after crossing over the membrane was quantified by ultraviolet-visible (UV-vis) spectrophotometry (Fig. 1A, B and ESI Note 2†). Tiron has a specific optical absorption band at 208 nm. The difference in tiron absorption spectra obtained in pure water and 1 M H2SO4 implies the different interactions with the media.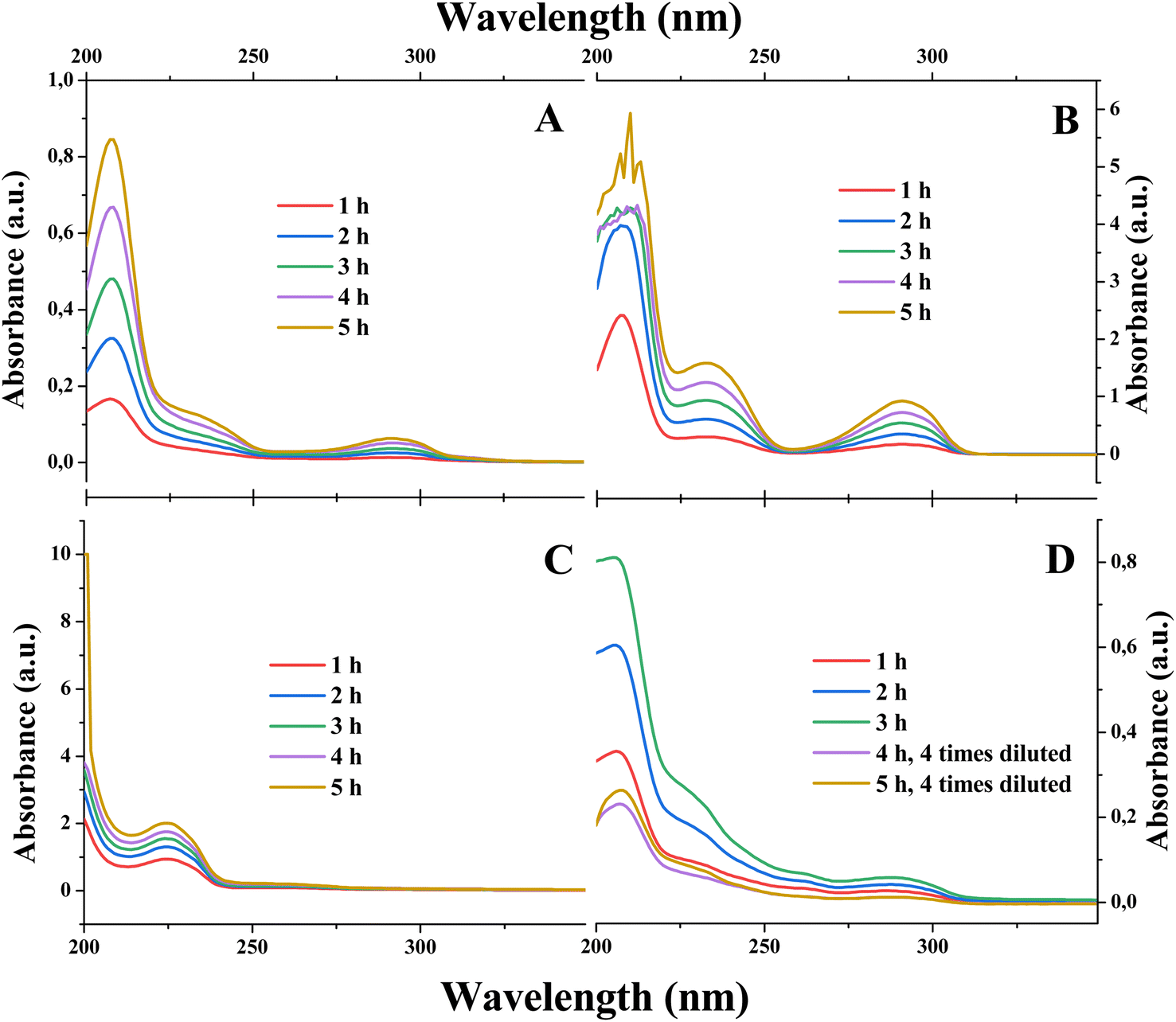 | ||
| Fig. 1 The effect of electrolysis and electrolytes on tiron transport across the PEM. The UV-vis spectra of cathode electrolyte samples obtained after (A) with water (in both anode and cathode compartments) and absence of electrolysis (Scheme 1A); (B) with 1 M H2SO4 (in both anode and cathode compartments) and absence of electrolysis (Scheme 1B); (C) with water (in both anode and cathode compartments) and active electrolysis (0.08 mA cm−2); and (D) with 1 M H2SO4 (in both anode and cathode compartments) and active electrolysis (0.08 mA cm−2; Scheme 1C). | ||
We investigated the rates of tiron transport in the absence of electrolysis (Fig. 1A and B). Assuming the constant flux of transported tiron assured by constant gradient of concentration across PEM, the permeability of tiron was estimated as follows:33
 | (1) |
The appearance of dissociated sulfonic groups as a result of the hydration of Nafion in the absence of electrolysis was observed by ex situ ATR-FTIR study of the PEMs (ESI Note 3†).
3.2 Non-zero current conditions
We performed similar experiments on the identical electrolyzer under the electrolysis conditions (Scheme 1C). The proton transport across the PEM in the parallel direction to tiron transport was established by the proton-coupled donor process, namely tiron oxidation at the anode (Eeq = +0.908 V vs. RHE):| Tiron → tironoxidized + 2e− + 2H+ | (2) |
| O2 + 2H+ + 2e− → H2O2 | (3) |
| Tiron + O2 → tironoxidized + H2O2 |
The H2O2 was detected in cathode liquids under both water and acid conditions (Fig. 2A). The linear increase in the H2O2 concentration observed in both cases implies the pseudo-steady-state conditions of electrolysis with time. The faradaic efficiency (FE) of the H2O2 electrosynthesis process was calculated as follows:
 485.3 A mol s−1), CH2O2 is the concentration of H2O2 (mol L−1), I is the current (A) and t is the time of electrolysis (s). The electrolysis in pure water showed very low faradaic efficiency (below 20%, Fig. 2B) of the H2O2 electrosynthesis. Persuasively, the addition of electrolyte, 1 M H2SO4, led to the visible mitigation of the electrical losses on H2O2 electrosynthesis represented as ca. 20% cell voltage decrease (Fig. S3A†) as well as higher rate capabilities and efficiency improvement (up to ca. 50% in Fig. 2B).
485.3 A mol s−1), CH2O2 is the concentration of H2O2 (mol L−1), I is the current (A) and t is the time of electrolysis (s). The electrolysis in pure water showed very low faradaic efficiency (below 20%, Fig. 2B) of the H2O2 electrosynthesis. Persuasively, the addition of electrolyte, 1 M H2SO4, led to the visible mitigation of the electrical losses on H2O2 electrosynthesis represented as ca. 20% cell voltage decrease (Fig. S3A†) as well as higher rate capabilities and efficiency improvement (up to ca. 50% in Fig. 2B).
 | ||
| Fig. 2 H2O2 electrosynthesis at the cathode. The time dependencies of H2O2 concentration (A) and faradaic efficiency (B) of H2O2 electrosynthesis in pure water and 1 M H2SO4. | ||
In the electrolyzer fed by ionically non-conducting media, pure water (with only a low concentration of tiron as the electrolyte), faradaic reactions (2) and (3) can only proceed at the triple points of physical contact of electronic (porous electrode), ionic (PEM), and reagent/product (solution) transporting phases, which are located at a very thin close-to-2D zone defined by the direct contact between the porous electrode and the PEM. However, when the liquid phase is a good electrolyte itself (tiron in 1 M H2SO4), the whole electrode surface is made available for the reaction by additional ionic transport to a thicker 3D region, which is visualized by the higher capacitive currents recorded at short times after the beginning of electrolysis (Fig. S3B†). The transition from 2D to 3D caused by electrolyte addition leads, at the same applied current, to a notable decrease in current density delivered to the reaction zones. As electrosynthesized H2O2 is a product of the ORR under pure kinetic control (H2O2 should not appear under thermodynamic control), a lower current density supresses the contributions from side reactions and results in a higher faradaic efficiency.
The performance characteristics of the ORR-to-H2O2 electrolyzer are comparable with the values reported for the systems based on the oxidation of organics as an auxiliary process (Table S2†). In contrast to them, the oxidation of tiron yields the products with the confined quinone aromatics, which implies the possibility of regeneration.36
To visualize the proton-coupled donor process of electrolyzer, namely tiron oxidation, we performed an ex situ1H-NMR study of aliquot samples of tiron-in-water-fed anode compartment collected at different elapsed times of electrolysis (ESI Note 4†). The concentrations of tiron (Fig. S5A†) and the products of its oxidation (Fig. S5B†) accompanied by 1,4 Michael addition reactions (Scheme 1S†) showed the evolution with the elapsed time of electrolysis because of the actual anode process. The decrease in tiron concentration is still small enough to assume its minor change due to electrolysis, which implies the validity of the use of eqn (1):
To study the transport phenomena in the bulk of PEM induced by electrolysis, we carried out in situ ATR-FTIR measurements on the PEM-based cell (Fig. 3A). The set of spectra averaged for 30 seconds were recorded for 2 hours. First, to equilibrate the system and to track any possible changes due to hydration, the spectra were recorded for 15 minutes without the application of current. Second, a constant current of 5 mA was applied. The set of time-resolved spectra were recorded with 200 seconds time interval (Fig. S6A†). The set of difference spectra (Fig. 3B) were obtained by the subtraction of each spectrum from the pristine membrane spectrum. The analysis of acquired data was performed in three spectral regions (Fig. 3B), (1) O–H stretching region (3800–2700 cm−1), (2) H–O–H bending region (2000–1500 cm−1) and (3) Nafion fingerprint region (1400–900 cm−1).
 | ||
| Fig. 3 Electrolysis-induced changes in the ATR-FTIR spectra of the bulk of Nafion 115: (A) scheme of the in situ ATR-FTIR cell; (B) time-resolved ATR-FTIR difference spectra of the Nafion membrane under electrolysis conditions (for the O–H stretching band: (a) 3370 cm−1, (b) 3235 cm−1; for O–H stretching band: (c) shifts from 1730 cm−1 to 1714 cm−1, (d) 1640 cm−1; region for Nafion fingerprint region: C–F bond symmetric vibration (νs(C–F)) shifts from 1201 cm−1 to 1211 cm−1; C–F bond asymmetric vibration (νa(C–F)) shifts from 1145 cm−1 to 1149 cm−1; S–O bond symmetric vibration (νs(S–O)) shifts from 1060 cm− to 1056 cm−1; C–O–C group vibration bands (ν(C–O–C)) shifts from 1000 cm−1 to 950 cm−1); (C) loading plot in the O–H stretching region (PC1: 98.0%, PC2: 1.6%; (a) 3380 cm−1, (b) 3245 cm−1, (c) 3043 cm−1, (d) 3750 cm−1, (e) 3678 cm−1 and (f) 3518 cm−1); (D) loading plot in the H–O–H bending region (PC1: 70.2%, PC2: 28.8%; bands a and b correspond to 1635 cm−1 and 1750 cm−1); (E) loading plot in the Nafion fingerprint region (PC1: 94.0%, PC2: 5.5%; bands (a) 1191 cm−1, (b) 1128 cm−1, (c) 1053 cm−1 and (d) 968 cm−1); (F–H) score plots (the dependencies of the first principal components on the time of electrolysis) for the O–H stretching region, H–O–H bending region and Nafion fingerprint region, respectively. | ||
The launching of electrolysis leads to an increase in the amount of unbound water inside the membrane. This is concluded from the shift of the centre of the broad O–H stretching band to lower wavenumbers visible on time-resolved ATR-FTIR spectra (from 3470 to 3395 cm−1, Fig. S6B†) and the increase in the intensity of the broad band at the difference spectra (two main centres at 3370 and 3235 cm−1, Fig. 3B). Coherently, the difference spectra acquired in the H–O–H bending region show an increase in the intensity of the broad overlapped peak with two centres (bending of protonated water, shifts from 1730 cm−1 to 1714 cm−1, bending of unprotonated water at 1640 cm−1, Fig. 3B) with the proceeding of electrolysis. The hydration of the membrane due to the electrolysis was visible in the Nafion fingerprint region by the change in the surroundings of polymer chains (Fig. 3B). In particular, the bands corresponding to the C–F bond vibrations showed the changes in intensity and positions (symmetric vibration: from 1201 cm−1 to 1211 cm−1; asymmetric vibration: from 1145 cm−1 to 1149 cm−1).37 The different spectra showed shift of the symmetric S–O bond vibration to a lower wavenumber (from 1060 cm−1 to 1056 cm−1; Fig. 3B), which is assigned to the change in the equilibrium between bound and unbound cations by the increase in the amount of shielding water.38,39 The membrane hydration caused the influence on the C–O–C group vibration bands (from 1000 cm−1 to 950 cm−1, Fig. 3B), which can be related to the dilution of the C–O–C group in the hydrophilic domain37 and the change in the polarization of the sulfonate groups.40
Principal component analysis (PCA) was applied to investigate the ATR-FTIR spectra in all the three spectral regions to understand the changes in membrane imposed by electrolysis. Such data processing for both the OH stretching region (loading plot: 3C) and the Nafion fingerprint region (loading plot: 3E) showed the domination of hydration process over proton transport, which is illustrated by much higher PC1 contributions of variation assigned to water transport (OH stretching region: PC1 and PC2: 98% and 1.6% of the variation, respectively; Nafion fingerprint region: PC1 and PC2: 94.5% and 5.5% of the variation, respectively). On the contrary, the proton transport in PCA showed higher visibility in the H–O–H bending region (PC1 and PC2: 70.2% and 28.8% of the variation, respectively). In this spectral region, the loading spectrum for PC1 (Fig. 3D) has a peak at 1635 cm−1, which corresponds to the H–O–H bending of water molecules transported within the membrane film by electrolysis. PC2 in this spectral region (loading spectrum Fig. 3D) showed a peak at 1750 cm−1, which is related to the H–O–H bending of protonated water appearing under the applied potential.
The dependences of PC1 and PC2 (score plot Fig. 3F–H) on time assigned to the transports water and protons, respectively, showed similar trends in all three spectral regions. As it is illustrated by PC1 dynamics, water molecules begin to enter inside the membrane immediately after the current is applied and continue to do so linearly with time until the end of the measurement. On the contrary, the rate of water–proton interactions, represented by the PC2 dynamics, shows a non-linear increase featured with the saturation state.
The set of broad overlapped peaks visible at the difference spectra in the H–O–H bending region and collected at different times of electrolysis were subjected to the spectral deconvolution analysis (Fig. S7†). Four individual band components were identified. The band at 1835 ± 5 cm−1 can be related to the asymmetric vibration of (H2O)nH+ (where n = 3 or 5).41 In coherence with the visibility of difference spectra, the bands at 1738 ± 10 and 1635 ± 5 cm−1 correspond to the HOH bending of (H2O)nH+ and the H–O–H bending of water molecules, respectively.37,38,42 Interestingly, the band at 1708 ± 3 cm−1 is assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond stretching vibration, which is present only in quinone molecules,43–45 namely the tiron oxidation product. The presence of tiron-associated bands was evidenced in PCA of the spectra collected in the Nafion fingerprint region (Fig. 3E). While PC1 is assigned to water transport, the PC2 loading spectrum demonstrates the presence of four bands (at 1191 cm−1, 1128 cm−1, 1053 cm−1 and 968 cm−1) with a broad negative band between 1300 and 1200 cm−1 (Fig. 3E). By comparing the PC2 loading spectrum with the spectra of tiron in powder form and in solution (Fig. S8†), it is possible to observe the similarity in positions of few peaks. Therefore, the peaks in the PC2 loading spectrum (Fig. 3E) can be assigned to tiron within the membrane film. The broad negative band in the PC2 loading spectrum can be assigned to the C–C ring and C–O vibration region of pristine tiron. The decrease in the intensity of this broad peak can represent the consumption of tiron by oxidation under electrolysis conditions, which is coherent with the increase in the quinoidal peak of the CO double bond at 1708 cm−1. The appearance of tiron-associated peaks and their growth with the beginning and proceeding of electrolysis illustrate the tiron transport, which defines its crossover through the PEM.
O double bond stretching vibration, which is present only in quinone molecules,43–45 namely the tiron oxidation product. The presence of tiron-associated bands was evidenced in PCA of the spectra collected in the Nafion fingerprint region (Fig. 3E). While PC1 is assigned to water transport, the PC2 loading spectrum demonstrates the presence of four bands (at 1191 cm−1, 1128 cm−1, 1053 cm−1 and 968 cm−1) with a broad negative band between 1300 and 1200 cm−1 (Fig. 3E). By comparing the PC2 loading spectrum with the spectra of tiron in powder form and in solution (Fig. S8†), it is possible to observe the similarity in positions of few peaks. Therefore, the peaks in the PC2 loading spectrum (Fig. 3E) can be assigned to tiron within the membrane film. The broad negative band in the PC2 loading spectrum can be assigned to the C–C ring and C–O vibration region of pristine tiron. The decrease in the intensity of this broad peak can represent the consumption of tiron by oxidation under electrolysis conditions, which is coherent with the increase in the quinoidal peak of the CO double bond at 1708 cm−1. The appearance of tiron-associated peaks and their growth with the beginning and proceeding of electrolysis illustrate the tiron transport, which defines its crossover through the PEM.
The quantification of tiron transported across PEM under the electrolysis conditions on an electrolyzer fed with pure water was not possible. This is due to the conversion of transported tiron to unknown products under the conditions of ORR (Fig. 1C). We failed to identify the structure of a new tiron product transported thought the PEM by electrolysis in pure water using 1H-NMR (data not shown). On the contrary, the presence of electrolyte, 1 M H2SO4, in the electrolyzer maintained transported tiron intact (Fig. 1D), which allowed its quantification. The absence of additional products except transported tiron illustrates the stability of electrocatalyst under the conditions of ORR. Surprisingly, the permeability of tiron estimated under the conditions of parallel proton transport driven by electrolysis at 0.08 mA cm−2 was 2.60 × 10−8 cm2 min−1, which is ca. 15 times smaller than that in the absence of electrolysis (Fig. 1D). In other words, the application of constant current enabling cation (proton) transport across the PEM supresses the transport of di-anions (tiron) in a parallel direction. Importantly, E. J. Latchem et al.46 observed the coherent effect on the PEM electrolyzer, namely redox flow battery during the charging process, under identical conditions to our measurements. The anode process was single-electron oxidation of ferrocyanide (K4[Fe(CN)6]) liberating one potassium ion and ferricyanide (K3[Fe(CN)6]). The cathode process was a di-electron reduction of di-anion anthraquinone (namely, 2,6-dihydroxyanthraquinone) accommodating two potassium ions. The transport rate of cathode reduction reagent across the PEM increased upon launching the electrolysis. These data enable us to complete the puzzle of reagent transport across the PEM: under identical operational conditions the transport of the anode reagent decreased, while the transport of the cathode reagent increased. These contrasting behaviours of anode- and cathode-charged reagents are only due to the coulombic forces maintained by the electrical field appearing in the electrolyzer and distributed between the electrodes and the membrane. Anions are repulsed by the anode and attracted by the cathode. Such effect of the electrical field on the movement of ions known as migration is present in the bulk of the electrolyte. In contrast, the electrical field is shielded at the electrode surface by the electric double layer formed. As soon as the compartments of the electrolyzer are filled with a supporting electrolyte (1 M H2SO4), the bulks of the porous anode and cathode are shielded by the electrical double layers. Therefore, we would locate the zones under the influence of the electrical field on the narrow 2D interface formed by the direct electrolyte-free contact between PEM and the electrodes.
3.3 Modeling
We performed the computational study of the tiron concentrations in the 1D model of electrolyzers under zero and non-zero current conditions (Fig. 4A) using the value of tiron diffusion coefficient estimated from the experiment. All initial concentrations of the species are set according to the experimental concentrations: tiron concentrations are 50 mM and 0 mM in the anode and cathode electrolytes, respectively, HSO4− concentration is 1 M, oxygen concentration is 50 mM in the cathode electrolyte, and H+ concentrations are 1.1 M and 1 M in the anode and cathode electrolytes, respectively. The excess of H+ concentration was due to the charge compensation of tiron dianion. We used the constant concentration boundary condition for the background electrolyte at both boundaries (x = 0 and x = L) and the concentrations are set to the same values given above. This is consistent with the experiments where the concentrations are constant in the tanks and therefore at the inlets of both electrodes. No flux boundary condition for tiron was used at the same boundaries (x = 0 and x = L). The electrolyte (φl) and electrode (φs) potentials are set to 0 for case 1 and for case 2 as follows:| φl = φs = −0.5 V, x = 0 |
| φl = φs = 0.5 V, x = L |
The tiron concentrations were computed after the achievement of steady-state regime in the middle of the electrode domain (x = 2.5 × 10−3 m), where the mass transport is driven by diffusion only. Although the membrane has a substantial amount of immobile negative charges, the electrostatic repulsion in not enough to block the transport of tiron dianion across the membrane, which illustrates the loss of ion selectivity in electrolyte solutions. This is due to the screening of immobile charges by excess of electrolyte ions. After one hour, the tiron concentration increases linearly with time for both simulation and experiment (Fig. 4B) illustrating the achievement of steady-state diffusion across the membrane. The distribution of tiron concentrations computed for different elapsed times (Fig. 4C and D) showed the slow increase due to the diffusional mass transport. Importantly, the application of potential (1 V) imposing non-zero current conditions led to the suppression of tiron (Fig. 4E and F). This can be explained with the strong attraction of tiron dianion. Under such conditions, tiron migrates to the electrode surface at x = L, where the potential is applied (Fig. 4E), while its concentration at the cathode electrolyte is negligible.
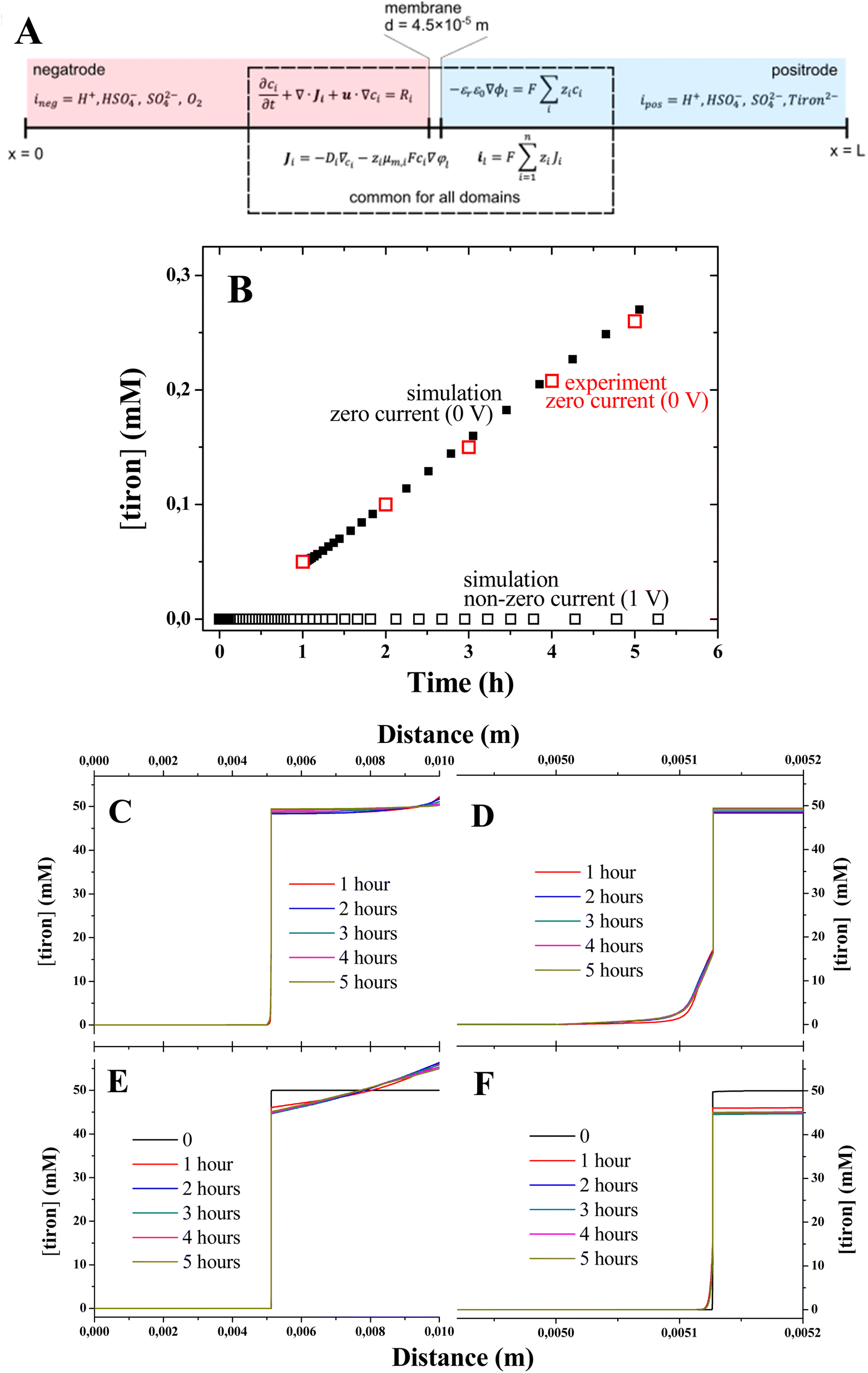 | ||
| Fig. 4 (A) Schematic representation of the 1D model used for the calculations; (B) comparison of experimental and computed time dependencies of tiron concentration for the conditions of zero current (0 V) and non-zero current (1 V); the evolution of computed concentration profiles for tiron across the electrolyzer with time under zero and non-zero current conditions (C and E), respectively; the concentration is computed in the middle of the negative electrode domain (x = 2.5 × 10−3 m); (D and F) computed concentration profiles at smaller distances at zero and non-zero current conditions, respectively. | ||
4 Conclusions
In this study, we fabricated a membrane-based H2O2 electrosynthesis device consisting of an ORR cathode and a model catalyst-free anode driving the oxidation of organics. The quinone dianion crossover through the membrane was measured by UV-vis spectroscopy and modelled to simulate the mass transport without and with electricity. The results indicated that the dianion crossover was greatly suppressed under electrolysis conditions due to migration effects, which indicated that simple and routine diffusional tests could not account for the mass transport in electrochemical devices in operation. This study might be helpful for the crossover-free design of other membrane-based electrochemical systems fed by electrolytes.Data availability
The data for this article are partly included in the manuscript (e.g.Fig. 2 and 3, S3, S5 and S6 are linked with original data in submitted manuscript) and partly in the ESI.† The rest of the data are available upon request from the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank VINNOVA (Digital Cellulose Center, 308634, 308635), the Knut and Alice Wallenberg foundation (KAW 2019.0604, KAW 2021.0195, Wallenberg Wood Science Center (WWSC), Wallenberg Initiative Materials Science for Sustainability (WISE), Wallenberg Launchpad (WALP), KAW Project Grant 2018 ‘Hydrogen Peroxide Fuel and Energy Technology for the Future’), the Swedish Energy Agency (52023-1), Vetenskaprådet (2016-05990, 2019-05577) for financial funding. This project is financed through the Swedish Electricity Storage and Balancing Centre (SESBC). The Centre is funded by the Swedish Energy Agency together with five academic and twenty-six non-academic partners. In particular, the authors acknowledge RedoxMe AB for their contribution and support to the project. The computations were conducted on resources provided by the Swedish National Infrastructure for Computing (SNIC) at NSC and HPC2N.References
- Z. Li, H. S. Park, J. X. Qiao, K. S. Yeung and J. Q. Yu, J. Am. Chem. Soc., 2022, 144, 18109–18116 CrossRef CAS PubMed.
- R. J. Lewis, K. Ueura, X. Liu, Y. Fukuta, T. E. Davies, D. J. Morgan, L. W. Chen, J. Z. Qi, J. Singleton, J. K. Edwards, S. J. Freakley, C. J. Kiely, Y. Yamamoto and G. J. Hutchings, Science, 2022, 376, 615–620 CrossRef CAS PubMed.
- J. Kim, J. H. Kim, C. Oh, H. Yun, E. C. Lee, H. S. Oh, J. H. Park and Y. J. Hwang, Nat. Commun., 2023, 14, 4704 CrossRef CAS.
- L. Fan, Y. L. Zhao, L. Chen, J. Y. Chen, J. M. Chen, H. Z. Yang, Y. K. Xiao, T. Y. Zhang, J. Y. Chen and L. Wang, Nat. Catal., 2023, 6, 585–595 CrossRef CAS.
- K. S. Exner, Energy Adv., 2023, 2, 522–529 RSC.
- W. Sun, F. Wang, B. Zhang, M. Y. Zhang, V. Kupers, X. Ji, C. Theile, P. Bieker, K. Xu, C. S. Wang and M. Winter, Science, 2021, 371, 46–51 CrossRef CAS.
- S. Siahrostami, ACS Energy Lett., 2022, 7, 2717–2724 CrossRef CAS.
- National Academies of Sciences, and Medicine, New Directions for Chemical Engineering, The National Academies Press, Washington DC, 2022, DOI:10.17226/26342.
- S. C. Perry, D. Pangotra, L. Vieira, L. I. Csepei, V. Sieber, L. Wang, C. P. de Leon and F. C. Walsh, Nat. Rev. Chem, 2019, 3, 442–458 CrossRef CAS.
- E. Mitraka, M. Gryszel, M. Vagin, M. J. Jafar, A. Singh, M. Warczak, M. Mitrakas, M. Berggren, T. Ederth, I. Zozoulenko, X. Crispin and E. D. Głowacki, Adv. Sustain. Syst., 2019, 3, 1800110 CrossRef.
- Y. L. Wang, G. I. N. Waterhouse, L. Shang and T. R. Zhang, Adv. Energy Mater., 2021, 11, 2003323 CrossRef CAS.
- Z. X. Wu, P. H. Ding, V. Gueskine, R. Boyd, E. D. Glowacki, M. Oden, X. Crispin, M. Berggren, E. M. Bjork and M. Vagin, Energy Environ. Mater., 2023, e12551 Search PubMed.
- B. D. Paulsen, K. Tybrandt, E. Stavrinidou and J. Rivnay, Nat. Mater., 2020, 19, 13–26 CrossRef CAS.
- J. Rivnay, S. Inal, B. A. Collins, M. Sessolo, E. Stavrinidou, X. Strakosas, C. Tassone, D. M. Delongchamp and G. G. Malliaras, Nat. Commun., 2016, 7, 11287 CrossRef PubMed.
- J. K. Lee, G. Anderson, A. W. Tricker, F. Babbe, A. Madan, D. A. Cullen, J. D. Arregui-Mena, N. Danilovic, R. Mukundan, A. Z. Weber and X. Peng, Nat. Commun., 2023, 14, 4592 CrossRef PubMed.
- A. Agee, T. M. Gill, G. Pace, R. Segalman and A. Furst, J. Electrochem. Soc., 2023, 170, 016509 CrossRef.
- K. Wijeratne, U. Ail, R. Brooke, M. Vagin, X. J. Liu, M. Fahlman and X. Crispin, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 11899–11904 CrossRef PubMed.
- M. Rudolph and E. L. Ratcliff, Nat. Commun., 2017, 8, 1048 CrossRef PubMed.
- L. Lal and M. C. Toroker, Curr. Opin. Electrochem., 2023, 40, 101342 CrossRef.
- S. C. Perry, C. P. de Leon and F. C. Walsh, J. Electrochem. Soc., 2020, 167, 155525 CrossRef.
- D. Pletcher, Industrial Electrochemistry, Springer, Dordrecht, 1984 Search PubMed.
- A. Vass, A. Kormanyos, Z. Koszo, B. Endrodi and C. Janaky, ACS Catal., 2022, 12, 1037–1051 CrossRef CAS.
- L. Q. Li, C. Tang, Y. Zheng, B. Q. Xia, X. L. Zhou, H. L. Xu and S. Z. Qiao, Adv. Energy Mater., 2020, 10, 2000789 CrossRef CAS.
- S. K. Geng, Y. Zheng, S. Q. Li, H. Su, X. Zhao, J. Hu, H. B. Shu, M. Jaroniec, P. Chen, Q. H. Liu and S. Z. Qiao, Nat. Energy, 2021, 6, 904–912 CrossRef CAS.
- M. S. Dhawan, G. D. Yadav and S. C. Barton, Sustainable Energy Fuels, 2021, 5, 2972–2984 RSC.
- P. Azadi, O. R. Inderwildi, R. Farnood and D. A. King, Renewable Sustainable Energy Rev., 2013, 21, 506–523 CrossRef CAS.
- L. P. Christopher, B. Yao and Y. Ji, Energy Environ. Mater., 2014, 2, 12 Search PubMed.
- M. J. Hidajat, A. Riaz, J. Park, R. Insyani, D. Verma and J. Kim, Chem. Eng. J., 2017, 317, 9–19 CrossRef.
- C. Spiegel, Designing and Building Fuel Cells, McGraw Hill Professional, 2007 Search PubMed.
- B. Hu, M. W. Hu, J. Luo and T. L. Liu, Adv. Energy Mater., 2022, 12, 2102577 CrossRef.
- H. E. Gottlieb, V. Kotlyar and A. Nudelman, J. Org. Chem., 1997, 62, 7512–7515 CrossRef.
- F. Malz and H. Jancke, ACS Catal., 2005, 38, 813–823 Search PubMed.
- Z. J. Li and Y. C. Lu, Nat. Energy, 2021, 6, 517–528 CrossRef.
- Y. S. Xia, H. Y. Cao, F. Xu, Y. X. Chen, Y. Xia, D. Z. Zhang, L. H. Dai, K. Qu, C. Lian, K. Huang, W. H. Xing, W. Q. Jin and Z. Xu, Nat Sustainability, 2022, 5, 1080–1091 CrossRef.
- FC The Chemours Company FC, LLC, Product Bulletin P-12: Nafion N115, N117, N1110 (Ion Exchange Materials Extrusion Cast Membranes), 2016 Search PubMed.
- H. Ji, Z. Zhao, C. Zhang and X. Li, Chem. Sci., 2024, 15, 13185–13190 RSC.
- D. T. Hallinan and Y. A. Elabd, J. Phys. Chem. B, 2009, 113, 4257–4266 Search PubMed.
- K. Kunimatsu, B. Bae, K. Miyatake, H. Uchida and M. Watanabe, J. Phys. Chem. B, 2011, 115, 4315–4321 CrossRef CAS.
- S. R. Lowry and K. A. Mauritz, J. Am. Chem. Soc., 1980, 102, 4665–4667 Search PubMed.
- K. M. Cable, K. A. Mauritz and R. B. Moore, J. Polym. Sci., Part B: Polym. Phys., 1995, 33, 1065–1072 CrossRef CAS.
- H. B. Yu and Q. Cui, J. Chem. Phys., 2007, 127, 234504 CrossRef PubMed.
- T. Shimoaka, C. Wakai, T. Sakabe, S. Yamazaki and T. Hasegawa, Phys. Chem. Chem. Phys., 2015, 17, 8843–8849 RSC.
- F. N. Ajjan, M. J. Jafari, T. Rebis, T. Ederth and O. Inganäs, J. Mater. Chem. A, 2015, 3, 12927–12937 Search PubMed.
- H. Makita and G. Hastings, Biochim. Biophys. Acta, Bioenerg., 2020, 1861, 148173 CrossRef CAS.
- G. Socrates, Infrared and Raman Characteristic Group Frequencies: Tables and Charts, Wiley, 3rd edn, 2004 Search PubMed.
- E. J. Latchem, T. Kress, P. A. A. Klusener, R. V. Kumar and A. C. Forse, J. Phys. Chem. Lett., 2024, 15, 1515–1520 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4se00682h |
| This journal is © The Royal Society of Chemistry 2024 |