
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fuel-range liquid hydrocarbon products from catalytic deoxygenation of mixtures of fatty acids obtained from the hydrolysis of rapeseed oil†
Morenike A.
Peters
,
Jude A.
Onwudili
 * and
Jiawei
Wang
* and
Jiawei
Wang
Energy and Bioproducts Research Institute, College of Engineering and Physical Sciences, Aston University, Aston Triangle, Birmingham B4 7ET, UK. E-mail: j.onwudili@aston.ac.uk
First published on 13th September 2024
Abstract
The combined hydrolysis–deoxygenation method reported here demonstrates the efficiency of hydrogen-free catalytic conversion of lipid-derived multi-fatty acids into renewable drop-in hydrocarbon biofuels. Using a 5 wt% Pt on carbon (5 wt% Pt/C) catalyst for a detailed deoxygenation study, experiments were conducted in a batch reactor under nitrogen atmosphere at various temperatures (350–400 °C), reaction times (0–3 h) and catalyst/feedstock mass ratios (0–0.2). The Pt/C catalyst showed remarkable selectivity towards the decarboxylation mechanism, as evidenced by substantial CO2 formation. The Pt/C-catalysis optimal conditions were: 400 °C, 2 h reaction time and a catalyst/feedstock mass ratio of 0.2, resulting in a yield of 3.76 wt% gasoline, 14.7 wt% kerosene and 53.7 wt% diesel range hydrocarbons. Under the set of optimal conditions, five other catalysts with different supports were tested and the results showed that 5 wt% Pt/MgSiO3 and 5 wt% Pt/Al2O3 catalysts enabled complete conversion of fatty acids (total acid number = 0 mg KOH g−1). In contrast, 5 wt% Pt/SiO2, 5 wt% Pd/MgSiO3 and bimetallic 10 wt% Ni–Cu/Al2O3 performed poorly, indicating the effects of both the active metal and metal–support interaction on the conversion of fatty acids. In all cases, the oil products were dominated by heptadecane, originating from decarboxylation and in situ hydrogenation of the dominant oleic acid (74.4 wt%) and other C18 fatty acids present in the hydrolysed feedstock. Visually, only the Pt/C catalyst produced light-coloured liquids with direct-use fuel appeal, possibly due to its mildly acidic nature and comparably much larger surface area of 650 m2 g−1.
1 Introduction
The decarbonisation of the chemical and energy sectors is essential for mitigating climate change due to their significant greenhouse gas emissions.1,2 A promising strategy involves producing and utilising green hydrocarbons from biomass, potentially aiding in the achievement of net zero emissions by 2050.3,4The complexity of producing these hydrocarbons depends on the type of biomass feedstock,5 such as sugars,6 fats and oils,7 and lignocellulosic biomass.8 Key pathways for producing liquid hydrocarbons from biomass include biomass gasification Fischer–Tropsch (FT),9 hydroprocessed renewable jet (HRJ) or hydrotreated esters and fatty acids (HEFA),10 direct sugar to hydrocarbon (DSHC),11 alcohol to jet (ATJ),12 and catalytic hydrothermolysis (CH).13 Current research is focused on renewable aviation fuels, though these pathways can also produce hydrocarbons for other uses.14
Most routes, aside from biomass gasification FT, require substantial hydrogen gas input, which contributes to high production costs.15,16 In addition, steam methane reforming (SMR), currently the cheapest hydrogen source, has a high carbon footprint, emitting approximately 5.5 kg of CO2 per 1 kg of hydrogen, stoichiometrically.17 While green hydrogen from electrolysis is seen as a future fuel, its production costs remain high at up to £8 per kg compared to around £1.50 per kg for SMR.18,19 Thus, greening the hydrogen supply chain for sustainable hydrocarbon production from biomass will likely incur higher costs in the near term.20
Lipids, with high hydrogen-to-carbon (H/C) ratios, are promising feedstock materials for hydrocarbon production. However, their relatively high oxygen-to-carbon (O/C) ratios require further oxygen removal (deoxygenation) to obtain hydrocarbon molecules. Converting the triglycerides in vegetable oils, algal oils and animal fats offers a sustainable, efficient route to green hydrocarbons. This process may involve direct hydrogenolysis/hydrogenation or a combination of hydrolysis, deoxygenation, and cracking steps, allowing flexibility to co-produce valuable chemicals like glycerol.21 Recent studies focused on optimising each step, including hydrolysis,22,23 decarboxylation,24,25 and cracking.26,27 One-pot systems for direct conversion of triglycerides to alkanes have also been developed.28,29
Hydrolysis, which breaks down triglycerides with water into fatty acids and glycerol, has long been used in the oleochemical industry, with processes like Eisenlohr,30 Colgate-Emery,31 and Twitchell.32 Recent advancements use hot-pressurised water at above 200 °C to achieve similar hydrolysis results without additional external acid or alkaline catalysts. Hydrothermal hydrolysis at 300 °C, with an oil-to-water mass ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, achieved nearly 100% conversion of rapeseed oil into fatty acids, with over 98% theoretical yields in 1 hour.33–35 The main oxygen atoms in fatty acids exist in the carboxylic acid groups (–COOH) and can be removed via specific deoxygenation reactions. Decarboxylation is a thermo-catalytic method targeted by many researchers as the main deoxygenation mechanism that removes oxygen from fatty acids as CO2, while preserving most of the carbon chain in hydrocarbon molecules.18,36,37 The decarboxylation reaction for saturated fatty acids can be represented by eqn (1).
:2, achieved nearly 100% conversion of rapeseed oil into fatty acids, with over 98% theoretical yields in 1 hour.33–35 The main oxygen atoms in fatty acids exist in the carboxylic acid groups (–COOH) and can be removed via specific deoxygenation reactions. Decarboxylation is a thermo-catalytic method targeted by many researchers as the main deoxygenation mechanism that removes oxygen from fatty acids as CO2, while preserving most of the carbon chain in hydrocarbon molecules.18,36,37 The decarboxylation reaction for saturated fatty acids can be represented by eqn (1).
| CxHyCOOH → CxHy+1 + CO2 | (1) |
Eqn (1) is therefore a simple deoxygenation reaction that results in higher quality fuels due to enhanced H/C ratios, while improving chemical stability and energy density.38,39 Yang et al.16 investigated the decarboxylation of stearic acid using Pt/C, Pt/MWCNT, and Ru/C catalysts, achieving conversions of 54.9%, 54.8%, and 47.5%, respectively. The highest heptadecane selectivity (97%) was obtained with Pt/MWNTs at 330 °C. Similarly, Yang et al.40 used several supported Pt catalysts for oleic acid decarboxylation at 320 °C with 20 bar hydrogen and a 1:1 oleic acid/catalyst ratio, achieving 81% heptadecane selectivity after 2 hours when Pt was supported on microporous ZIF-67 beads. Edeh et al.37 examined the hydrothermal deoxygenation of hexadecanoic acid (palmitic acid) using a Pd/C catalyst, achieving 71.2% conversion at 290 °C after 1 hour, with a pentadecane selectivity of 54.6%. Fu et al.36 also studied hydrothermal deoxygenation of palmitic acid using Pd/C and Pt/C catalysts, reporting high pentadecane selectivities without external hydrogen input. The Pt/C catalyst showed higher selectivity for pentadecane.36
Most reported decarboxylation studies have focused on the catalytic conversion of individual fatty acids to evaluate their effectiveness.16,36,40 Direct use of lipid hydrolysis products containing various fatty acids for combined deoxygenation into hydrocarbons is rare with only a few reports in the literature.41,42 Hence, there is a need to further explore this line of research to generate more comprehensive data for process development. Many studies use hydrogen gas atmospheres at high pressures to target alkanes as the major products16 and/or maintain catalyst performance.40 For instance, Jeništová et al.43 achieved 99% conversion during the deoxygenation of stearic acid using a Ni–Al2O3 catalyst at 60 bar hydrogen pressure for 6 hours. However, recently advancements involving the use of reduced hydrogen pressure (e.g., 20 bar) while maintaining high conversion rates have been reported.44,45 Madsen et al.46 obtained 100% conversion of oleic acid in the presence of Pt/MgSiO3 and 20 bar hydrogen, but they still required a reaction time of 5 hours. Additionally, some researchers have emphasised the need to optimise catalyst performance under varying hydrogen pressures to reduce costs and enhance sustainability.47,48
The literature49,50 showed that Pt-based catalysis can promote the in situ generation of reactive hydrogen species for the direct hydrogenation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds, leading to predominantly saturated hydrocarbon products. Therefore, avoiding the use of expensive external hydrogen can improve the sustainability and efficiency of converting lipids to hydrocarbon fuels, leading to lower production costs.51,52 This present study is an innovative attempt to simultaneously deoxygenate the hydrolysis product of rapeseed oil (HRSO), consisting of various C16–C22 fatty acids,35 without using expensive external hydrogen gas. Using 5 wt% platinum supported on carbon (5 wt% Pt/C), various reaction conditions (temperature, time, and catalyst loading) were investigated to optimise HRSO conversion, yielding high selectivity for fuel-range liquid hydrocarbons. Five other heterogeneous catalysts with different supports were also tested to compare their catalytic activities with 5 wt% Pt/C as part of a screening exercise in preparation for future optimisation studies.53,54 High yields of fuel-range liquid hydrocarbons from two-step hydrolysis–deoxygenation of lipids without external hydrogen can promote low-cost renewable hydrocarbon fuel production from various sustainable lipid-based feedstock materials. This process, free from possible interferent side-reactions of the glycerol moiety in triglycerides, offers simplicity in design and controlled production of gasoline, diesel, or kerosene for the sustainable liquid fuel market.
C bonds, leading to predominantly saturated hydrocarbon products. Therefore, avoiding the use of expensive external hydrogen can improve the sustainability and efficiency of converting lipids to hydrocarbon fuels, leading to lower production costs.51,52 This present study is an innovative attempt to simultaneously deoxygenate the hydrolysis product of rapeseed oil (HRSO), consisting of various C16–C22 fatty acids,35 without using expensive external hydrogen gas. Using 5 wt% platinum supported on carbon (5 wt% Pt/C), various reaction conditions (temperature, time, and catalyst loading) were investigated to optimise HRSO conversion, yielding high selectivity for fuel-range liquid hydrocarbons. Five other heterogeneous catalysts with different supports were also tested to compare their catalytic activities with 5 wt% Pt/C as part of a screening exercise in preparation for future optimisation studies.53,54 High yields of fuel-range liquid hydrocarbons from two-step hydrolysis–deoxygenation of lipids without external hydrogen can promote low-cost renewable hydrocarbon fuel production from various sustainable lipid-based feedstock materials. This process, free from possible interferent side-reactions of the glycerol moiety in triglycerides, offers simplicity in design and controlled production of gasoline, diesel, or kerosene for the sustainable liquid fuel market.
2 Materials and methods
2.1 Materials
The present study is based on the use of total fatty acids produced in-house from the hydrolysis of commercial rapeseed oil (RSO). RSO has recently been the most widely used vegetable oil for biodiesel production in the EU and UK, which, according to 2019 figures, accounted for 37% of biodiesel feedstock, followed by palm oil (30%) and used cooking oil (18.5%).55 As one of the most consumed vegetable oils in the UK, RSO also contributes to the waste cooking oil feedstock used for the UK's 340 million-litre yearly biodiesel production.56 The RSO was purchased from a local grocery and directly hydrolysed under hydrothermal conditions without any further pre-treatment.35 Analytical grade dichloromethane (99.8%) solvent, hydrochloric acid (0.1 M), absolute ethanol (99.8%), sodium hydroxide (pellets), and phenolphthalein indicator (1 wt% in ethanol) were purchased from Fisher Scientific, Leicester (UK), for acid value determination of the hydrolysed samples. After the hydrolysis experiment, two phases were obtained: a semi-solid phase composed mainly of fatty acids and a slightly viscous aqueous phase containing mostly glycerol. Furthermore, water washing of the semi-solid phase was carried out to remove any entrained glycerol.35 The obtained HRSO product (semi-solid) was transferred into a 500 ml bottle and kept in a desiccator overnight before further use.While Pt/C and Pd/C catalysts have been extensively researched for deoxygenation reactions,57–59 there are engineering problems associated with the use of powered carbon supports in terms of their industrial applicability in fixed bed reactors. These problems include high pressure drops in long fixed beds due to powders becoming compacted under high pressure operations, or when used in fluidised bed reactors, the powders may be blown out by high gas velocities. Recent developments indicate that pelletised carbon can be used as catalyst supports; however, there is also the challenge of regenerating coke-poisoned carbon supported catalysts by oxidation or combustion, which leads to burn-off of the support and the recovery of metals for new catalyst synthesis. The regeneration of carbon supports from coke deactivation via selective oxidation60 still needs to be optimised. Hence, using supports that are easily pelletised and regenerated by oxidation would be ideal for large-scale industrial operations.
Therefore, in this present work, four Pt-based catalysts on various powered supports (carbon, alumina, silica and magnesium silicate) have been tested, along with powdered Pd/MgSiO3 and powdered Ni–Cu/Al2O3 for comparison. Alumina, silica and magnesium silicate were selected as supports as they can be used to make pelletised catalysts for potential large-scale industrial operations. In addition, alumina and magnesium silicate supported catalysts can be more easily regenerated by oxidation (carbon burn-off) compared to carbon supports. All the catalysts were supplied by Catal International Limited, Sheffield (United Kingdom), characterised and used. Some properties of the catalysts used for these tests in this present work are shown in Table 1.
| Catalyst | d ave | Surface area (m2 g−1) | Pore volume (cc g−1) | Pore diameter (nm) |
|---|---|---|---|---|
| a d ave = average particle size. | ||||
| 5 wt% Pt/C | 4.8 nm | 650 | 0.26 | 3.84 |
| 5 wt% Pt/silica | 7.9 nm | 210 | 0.97 | 16.09 |
| 5 wt% Pt/Al2O3 | 5.4 nm | 160 | 0.39 | 6.72 |
| 5 wt% Pt/MgSiO3 | 12.1 nm | 116 | 0.18 | 19.26 |
| 5 wt% Pd/MgSiO3 | 45.3 nm | 102 | 0.16 | 19.36 |
| 10 wt% Ni–Cu/Al2O3 | 17.8–18.1 nm | 110 | 0.27 | 6.69 |
The highest surface area of 650 m2 g−1 was observed for 5 wt% Pt/C, and the surface areas of the other catalysts were similar, with 5 wt% Pd/MgSiO3 having the lowest surface area of 102 m2 g−1. The silica-based catalysts had the highest pore volumes, while the MgSiO3-based catalysts had the highest pore diameters.
Fig. 1 shows the powder XRD patterns of the seven catalysts used in this research. The Al2O3 peaks identified at 43.1°, 45.9°, and 67.1° in Ni–Cu/Al2O3 match those reported by Suryawanshi et al.54 (JCPDS file No. 10-0425). The Ni–Cu phase was observed at 48° and 76°.61,62 The broad XRD peaks of SiO2 were observed in the range 20–30° in the Pt/Silica catalyst.46 The XRD diffraction peaks at 40°, 46°, and 68° identified in Pt/Al2O3, Pt/C, and Pt/SiO2 correspond to the (111), (200), and (220) characteristic planes of a typical face-centred structure of Pt.63 The peak observed for graphitic carbon on the XRD for Pt/C was assigned to the (002) plane at 26°.64 The peak at 40° for Pd/MgSiO3 was assigned to Pd,65 while the MgSiO3 peaks were identified in Pt/MgSiO3 and Pd/MgSiO3.66,67 The Ni–Cu/Al2O3 catalyst used in this study contained 10 wt% of each metal.
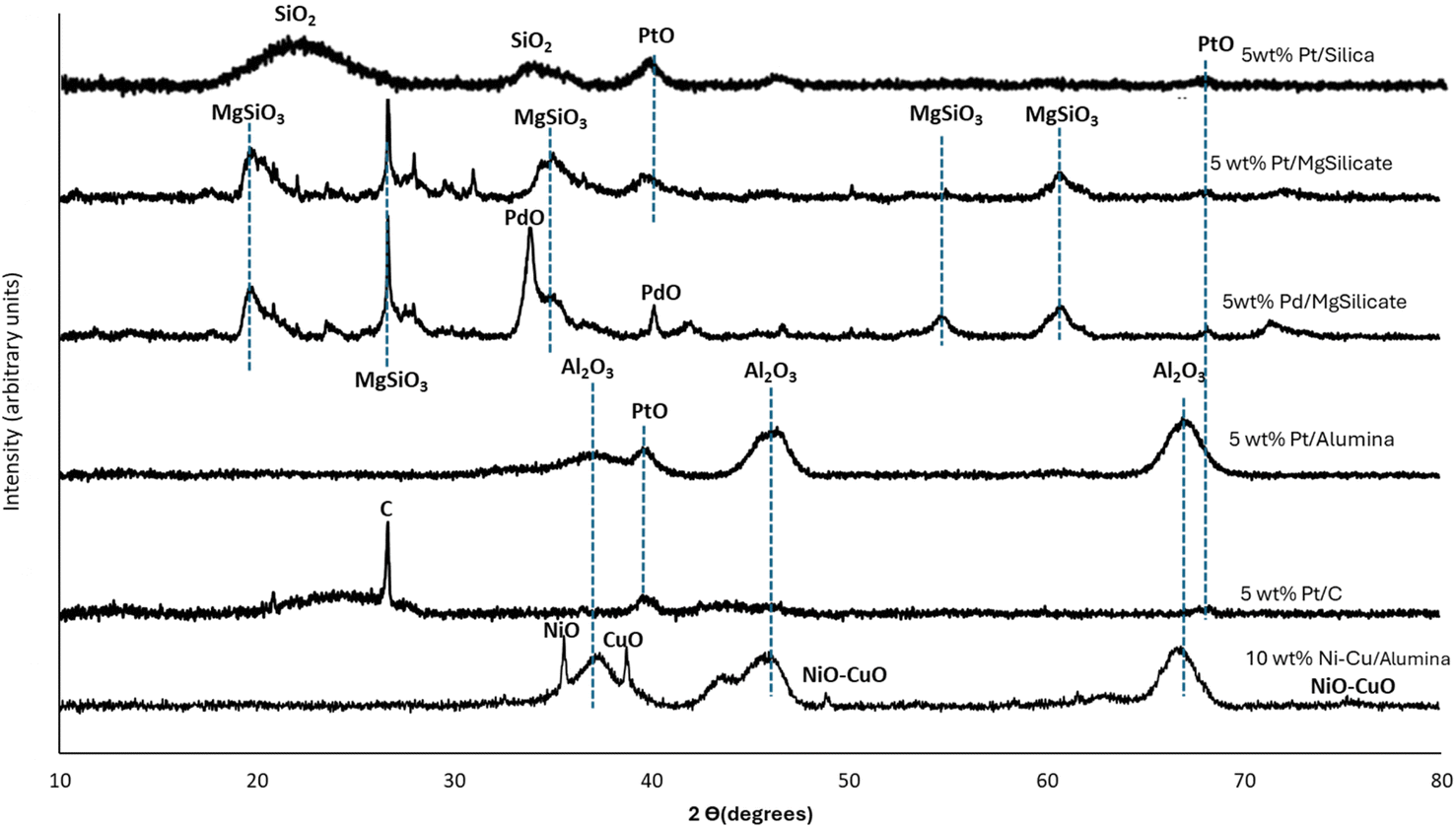 | ||
| Fig. 1 XRD patterns of the fresh catalysts used in this work. | ||
The elemental compositions of the HRSO are shown in Table 2 (based on the ASTM D5291 Method). Table 2 also shows the higher heating value (HHV) of the HRSO based on Dulong's formula68 according to eqn (2) as well as the acid numbers.69 The high acid numbers (TANs) were a good indication that the hydrolysis of the RSO occurred successfully under non-catalytic hydrothermal conditions as reported previously.35
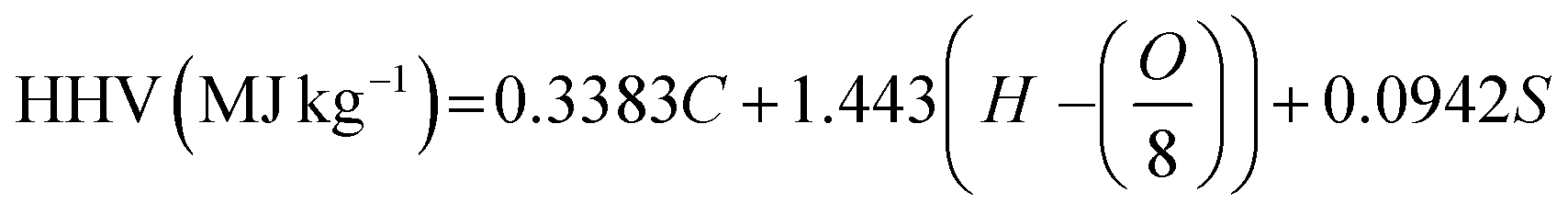 | (2) |
| Sample | C (wt%) | H (wt%) | N (wt%) | S (wt%) | O (wt%) | HHV (MJ kg−1) | TAN (mg KOH g−1) | Ash (wt%) |
|---|---|---|---|---|---|---|---|---|
| a nd = not detected. | ||||||||
| RSO | 77.00 ± 1.02 | 11.0 ± 0.15 | 0.13 ± 0.01 | nd | 10.9 ± 1.42 | 41.00 ± 0.65 | 1.40 ± 0.02 | nd |
| HRSO | 74.89 ± 1.49 | 11.9 ± 0.81 | 0.15 ± 0.02 | nd | 13.1 ± 0.83 | 40.16 ± 1.52 | 173.44 ± 0.81 | nd |
The compositions of the fatty acids in the HRSO are presented in Table 3.35 Clearly, oleic acid was identified as the main fatty acid (74.4 wt%) in HRSO, which agrees with literature data on the fatty acid profiles of RSO.69
| Systematic name | Common name | Composition (wt%) |
|---|---|---|
| 9-Hexadecenoic acid | Palmitoleic acid | 0.36 |
| Hexadecanoic acid | Palmitic acid | 6.89 |
| 9-Octadecenoic acid | Oleic acid | 74.4 |
| Octadecanoic acid | Stearic acid | 6.46 |
| 9,11-Octadecadienoic acid | Linoleic acid | 5.11 |
| 9,12,15-Octadecatrienoic acid | α-Linolenic acid | 1.92 |
| Eicosanoic acid | Arachidic acid | 2.55 |
| 13-Docosenoic acid (Z) | Erucic acid | 0.86 |
| Docosanoic acid | Behenic acid | 0.59 |
2.2 Experimental methods for catalytic deoxygenation
 | ||
| Fig. 2 Schematic of the experimental procedure for the deoxygenation of HRSO. | ||
Thereafter, the reactor was opened to sample any liquid products and solid residues. About 80% of the liquid product was first recovered without any solvent. Subsequently, dichloromethane (DCM) was used to wash the reactor to recover the remaining liquid product and solid residues. The solid residues were separated via vacuum filtration using a KNF LABOPORT vacuum filtration system, dried and weighed. Furthermore, the DCM solvent was removed by evaporation at 40 °C. The oil product obtained was weighed and stored separately from the one recovered without DCM. The total mass of the two oil product fractions was noted for mass balance.
The yields of each reaction product were calculated based on eqn (3) and (4).
 | (3) |
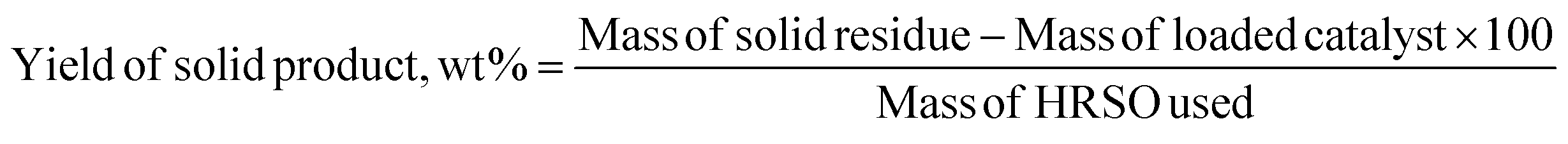 | (4) |
2.3 Analysis of reaction products
Each of the reaction products obtained was characterised in detail using a suite of analytical techniques, wet chemical methods and physical methods as described in sections 2.3.1–2.3.4.Table 3 shows that oleic acid was the dominant fatty acid in the rapeseed oil, accounting for 74.4 wt%.35 Therefore, the fatty acid contents of the oil products obtained after the catalytic deoxygenation tests were calculated based on the molecular weight of oleic acid according to eqn (5).
 | (5) |
The acid contents of the deoxygenated or partially deoxygenated oil products were calculated using eqn (6):
 | (6) |
The volume percent of each gas produced was calculated from their peak area and was used to determine the masses of gases produced according to the ideal gas eqn (7), followed by gas yield calculation using eqn (8).
 | (7) |
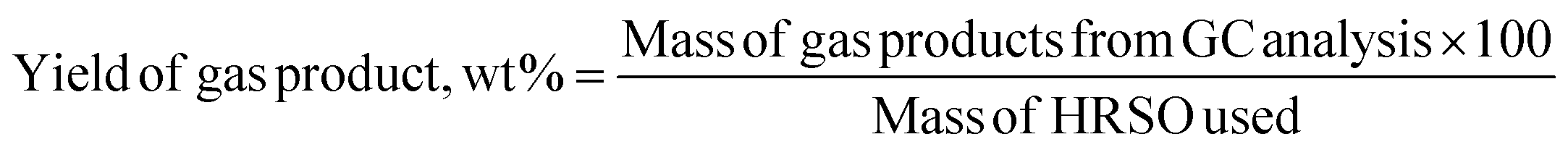 | (8) |
:20. The oven programme was as follows: started at 40 °C and held for 5 min, ramped at 8 °C min−1 to 185 °C, and then ramped at 14 °C min−1 to 280 °C, and held for 2 min, resulting in a total analysis time of 30.66 min. Due to the large numbers of compounds present in the oil products, the internal standard method was used for the quantification of the compounds.71–73 Diphenyl ether was used as the internal standard.
3 Results and discussion
3.1 Overall product yields and mass balances
Table 4 presents a comprehensive list of experiments carried out in this present study along with the yields of reaction products and the overall mass balances during the conversion of HRSO. In all cases, the reactions produced solids, liquids and gases, while in a few cases, wax was produced instead of liquid. The results from Table 4 and detailed analysis of the reaction products are presented in different sections to discuss the influence of reaction temperature, catalyst/HRSO mass ratios and reaction times in relation to the use of a 5 wt% Pt/C catalyst. Thereafter, the results from the effects of the three other Pt-based catalysts, the Pd/MgSiO3 and the Ni–Cu/Al2O3 catalyst are subsequently discussed.| Experiment | Temperature (°C) | Reaction time (h) | Catalyst type | Catalyst amount (g) | Solid (wt%) | Liquid (wt%) | Wax (wt%) | Gas (wt%) | Balance (wt%) |
|---|---|---|---|---|---|---|---|---|---|
| a “0 h” is the reaction time when the reactor was removed from the heater and cooled on reaching the set reaction temperature. | |||||||||
| 1 | 350 | 1 | — | 0.00 | 0.60 | 95.91 | — | 0.83 | 97.34 |
| 2 | 380 | 1 | — | 0.00 | 0.60 | 91.81 | — | 2.30 | 94.70 |
| 3 | 400 | 1 | — | 0.00 | 2.60 | 86.14 | — | 4.49 | 93.23 |
| 4 | 350 | 1 | 5 wt% Pt/C | 2.00 | 7.21 | — | 84.28 | 4.70 | 96.19 |
| 5 | 400 | 1 | 5 wt% Pt/C | 2.00 | 5.37 | 77.81 | 7.08 | 90.26 | |
| 6 | 400 | 1 | 5 wt% Pt/C | 0.50 | 2.30 | — | 85.40 | 4.50 | 92.20 |
| 7 | 400 | 1 | 5 wt% Pt/C | 1.00 | 4.89 | — | 84.13 | 4.99 | 94.02 |
| 8a | 400 | 0 | 5 wt% Pt/C | 2.00 | 18.38 | — | 76.79 | 1.46 | 96.64 |
| 9 | 400 | 2 | 5 wt% Pt/C | 2.00 | 8.35 | 73.76 | — | 7.94 | 90.05 |
| 10 | 400 | 3 | 5 wt% Pt/C | 2.00 | 6.73 | 64.66 | — | 8.30 | 79.69 |
| 11 | 400 | 2 | 5 wt% Pt/MgSiO3 | 2.00 | 3.78 | 84.18 | 6.97 | 94.93 | |
| 12 | 400 | 2 | 5 wt% Pt/SiO2 | 2.00 | 4.98 | 82.57 | 6.18 | 93.73 | |
| 13 | 400 | 2 | 5 wt% Pt/Al2O3 | 2.00 | 6.08 | 81.36 | 6.78 | 94.22 | |
| 14 | 400 | 2 | 5 wt% Pd/MgSiO3 | 2.00 | 12.00 | 79.28 | 4.78 | 96.01 | |
| 15 | 400 | 2 | 10 wt% Ni–Cu/Al2O3 | 2.00 | 4.39 | 87.03 | 6.98 | 98.40 | |
3.2 Influence of reaction temperature on the deoxygenation of HRSO
This section presents information on the effect of varying reaction temperature on the deoxygenation of fatty acids in hydrolysis products. The reactions were carried out for 1 h from 350 to 400 °C, with and without a catalyst. 2 g of 5 wt% Pt/C was used in the reactions involving a catalyst such as Pt/C which has been well reported in the literature as effective for the decarboxylation of fatty acid reactions to give high yields of hydrocarbons.36,67 These are presented as experiments 1–5 in Table 4.Compared to the corresponding non-catalytic test, the formation of wax at 350 °C with Pt/C suggested the formation of saturated long-chain hydrocarbons, which appeared as wax at room temperature.75 Although liquid/wax products obtained in the absence of the catalyst looked similar in terms of their colours, the oil from the reaction at 350 °C was very viscous, and this viscosity reduced with the increase in temperature. Overall, all the oil/wax products obtained were relatively darker in colour compared to the feed (ESI Fig. SI1†).
With the 5 wt% Pt/C catalysed reaction at 350 °C, it was impossible to separate the catalyst from white solid organic particles, and a hence higher (34%) solid yield was recorded during this test compared to the catalytic test at 400 °C. Indeed, a significant amount of DCM-insoluble white solid particles was formed during the catalytic experiment at 350 °C, which melted after heating on a hot plate to 50 °C, showing that they were organic in nature and therefore would be solid hydrocarbons (wax). On the other hand, the solid fractions obtained from the non-catalysed reactions at 350 °C, 380 °C, and 400 °C were essentially charred materials. However, the subsequent detailed characterisation of reaction products focused mainly on the gas and liquid/wax products, as the yield of char was small and of no direct benefit to the present study.
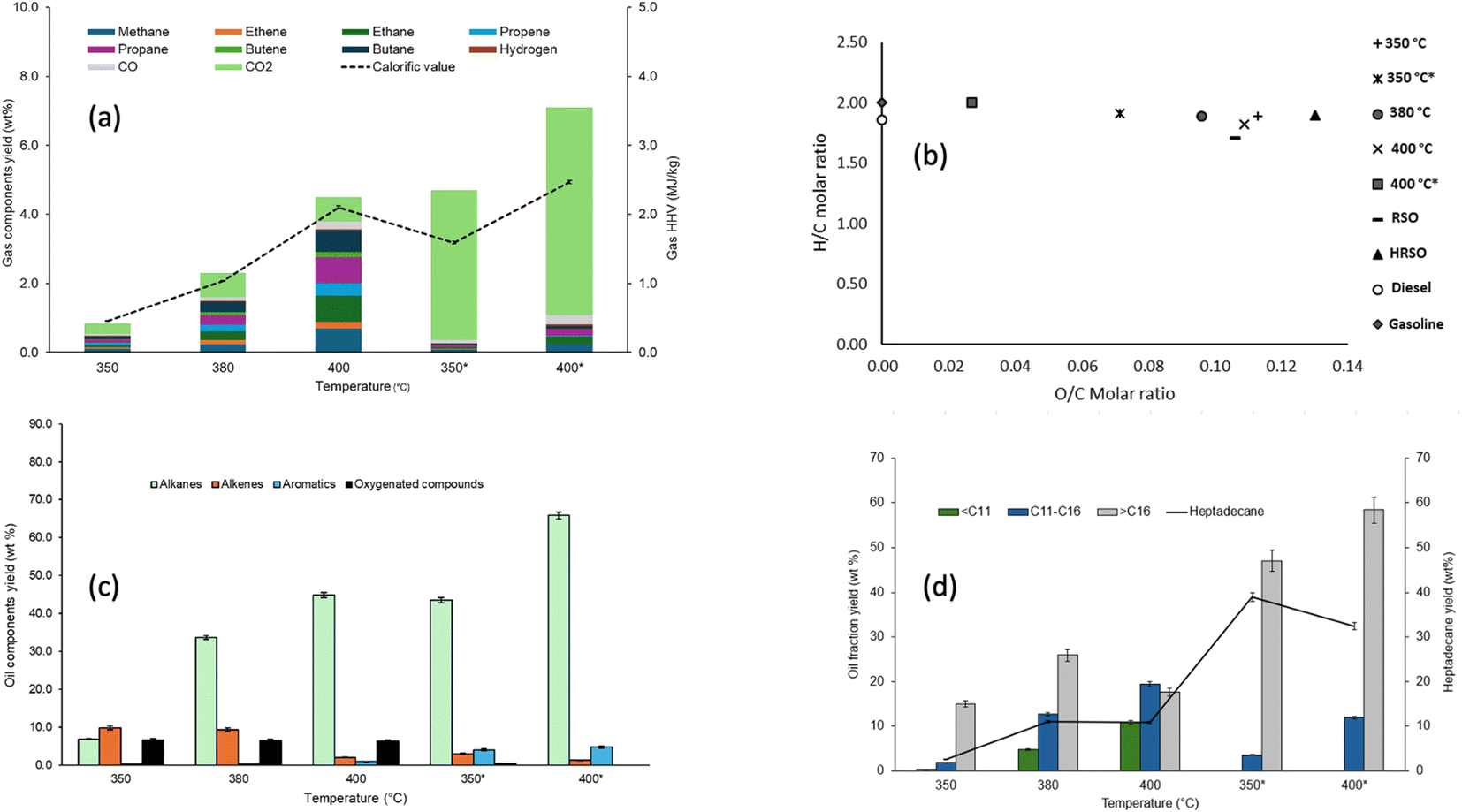 | ||
| Fig. 3 Influence of reaction temperature on (a) the compositions and calorific values of gas products, (b) the van Krevelen H/C and O/C molar ratios of the oil products, (c) the yields of groups of compounds in the light oils and (d) the yields of hydrocarbons and heptadecane in the light oils with and without 2 g of 5 wt% Pt/C catalyst. | ||
The selectivity of the catalyst towards decarboxylation and hence CO2 production is clearly shown in Fig. 3a between 350 °C and 400 °C. This may be the reason why decarboxylation reactions are commonly carried out between 350 °C and 400 °C in the literature53,76 and Pt/C is well-known to promote the decarboxylation of fatty acids. For example, Fu et al.57 reported over 90% selectivity towards hydrocarbons from the decarboxylation of C12, C16, and C18 saturated fatty acids. There is also an increase in the yield of hydrocarbon gases with an increase in temperature, with or without a catalyst. However, the yields of gas products indicated that gas formation in the absence of a catalyst was a non-selective process, with apparent increases in the yields of different gas components. Hence, the effect of temperature alone was insufficient to make gas formation selective towards the targeted decarboxylation reaction. Instead, the cracking of C–C bonds in fatty acids or their decarboxylation products is an endothermic process, which can be promoted at elevated temperatures.
Fig. 3a shows that the calorific values of the gas products increased with increasing reaction temperatures for the non-catalysed reaction because of random cracking, which enhanced the yields of hydrocarbon gases. Hence, a calorific value of 15 MJ kg−1 was obtained with the non-catalysed reaction at 400 °C. The reactions with Pt/C at 350 °C and 400 °C produced mostly CO2, which increased with temperature due to the targeted decarboxylation mechanism. Carbon dioxide was the dominant gas product observed in these reactions. This provided evidence that decarboxylation was the main deoxygenation mechanism for the conversion of fatty acids to hydrocarbons as reported in several publications,57–59 While the existence of CO2 in the product gas could be confounded by other reactions that produce and consume CO2, such as methanation, water–gas shift reaction and Boudouard reaction (CO disproportionation), the compositions of the oil products (see Section 3.1.3) can provide valuable support for the predominance of decarboxylation as the main source of CO2. However, Fig. 3a also shows that a higher temperature of 400 °C promoted cracking in the presence of the Pt/C, albeit to a more controlled extent compared to the corresponding non-catalysed reaction. Hence, the calorific value of the gas produced increased 5 times to 5 MJ kg−1 compared to 1.07 MJ kg−1 at 350 °C.
| Temperature (°C) | Catalyst | TAN (mg KOH g−1) | C (wt%) | H (wt%) | N (wt%) | O (wt%) | HHV (MJ kg−1) |
|---|---|---|---|---|---|---|---|
| 350 | — | 75.16 | 76.32 ± 0.34 | 12.06 ± 0.23 | 0.11 ± 0.02 | 11.5 ± 0.21 | 41.15 |
| 380 | — | 45.81 | 77.69 ± 0.70 | 12.26 ± 0.02 | 0.11 ± 0.01 | 9.94 ± 0.10 | 42.18 |
| 400 | — | 37.1 | 77.04 ± 0.84 | 11.72 ± 0.09 | 0.09 ± 0.01 | 11.2 ± 0.13 | 40.95 |
| 350 | Pt/C | 39.32 | 79.58 ± 0.64 | 12.74 ± 0.70 | 0.10 ± 0.01 | 7.59 ± 0.16 | 43.94 |
| 400 | Pt/C | 7.49 | 83.00 ± 0.10 | 13.9 ± 0.31 | 0.11 ± 0.01 | 2.99 ± 0.11 | 47.60 |
The oxygen contents in the oil products obtained from the non-catalysed experiments exhibited a different trend to the TAN numbers. Essentially, the oxygen contents remained fairly stable as temperature increased, even though the values decreased consistently. As the GC/MS analysis of the oils would reveal, this was due to the transformation of the fatty acids into other oxygenated compounds like alcohols, ketones and esters (ESI Table SI1†). In contrast, the oxygen contents of the oil products derived from the catalysed reactions showed similar trends to the respective TAN values, such that oxygen contents decreased from 7.59 wt% in the oil at 350 °C to 2.99 wt% in the oil at 400 °C.
Overall, there was a gradual increase in the carbon contents of all the oil products obtained in the absence of a catalyst, with contents of around 76–78 wt%. However, in the presence of Pt/C a dramatic increase in carbon contents was observed. For instance, at 350 °C, the carbon content was nearly 80 wt% in the presence of Pt/C and this increased to 83 wt% at 400 °C. Considering the carbon contents in the oils obtained at 400 °C alone, the use of the catalyst led to a nearly 10% increase in carbon contents compared to the non-catalysed test. Hence, the observed increase in carbon contents in the presence of Pt/C corresponded to both an increase in the calorific value and a decrease in both oxygen content and TAN values, indicating increased formation of hydrocarbons in the oils.
The elemental composition data shown in Table 5 were used to plot the Van Krevelen diagram of the oil/wax products shown in Fig. 3b. The reactions carried out in the absence of the catalysts had the highest O/C ratios and the H/C ratios increased with increasing reaction time. For the tests with Pt/C catalysts, the O/C ratios decreased while their H/C ratios increased with increasing temperature, so that the reaction at 400 °C resulted in the least oxygen content. The major findings from these tests at varying temperature were that an increase in reaction severity in terms of temperature favoured the conversion of fatty acids to hydrocarbons. This is observed with the decrease in fatty acid yields and the increase in hydrocarbon yields as temperature increased. This result also supports the expectation that increasing reaction temperature will cause an increase in the reaction rate due to the increase in kinetic energy, which causes increased collision of molecules and faster conversions in a given time.
The GC/MS results of the light oil obtained in the absence of 5 wt% Pt/C showed that the trend in the conversions of HRSO was as follows: 350 °C (24.84 wt%) < 380 °C (54.19 wt%) < 400 °C (62.9 wt%). In the presence of the catalyst, conversion to light oil was still low at 350 °C (60.68%) but increased dramatically to 92.5% at 400 °C after 1 h of reaction in each case. The results in Fig. 3c show that there was an increase in the yield of alkanes with increasing temperature with and without a catalyst, but higher yields of alkanes were observed in the presence of 5 wt% Pt/C, showing the selective effect of the catalyst towards decarboxylation as temperature increased. The GC/MS results showed that the light oil products obtained in the presence of a catalyst contained higher proportions of alkanes. Indeed, no oxygenated compounds were detected at 400 °C. However, at 350 °C in the presence of Pt/C, about 0.5 wt% oxygenates remained in the light oil product.
In the presence of 5 wt% Pt/C, there was also an increase in the yield of aromatic hydrocarbons as shown in Fig. 3c, with the test at 400 °C giving the highest aromatic yield of 4.83 wt%. As reported in the literature, aromatics can be produced from the combined process of cyclisation and aromatisation of alkanes, with the subsequent release of hydrogen gas.76 However, the low yields of hydrogen gas in the presence of Pt/C indicated that it was being used for the hydrogenation of unsaturated aliphatic hydrocarbons, so that alkanes were the dominant compounds in the oils.76,77 Platinum-based catalysts are known to be suitable for the hydrogenation of alkenes but rarely effective in hydrogenating aromatic rings. The presence of aromatics may be important for fuel properties, but the amounts produced in these tests were lower than those found in conventional gasoline and kerosene fuels.78
While the carbon numbers of different fuel fractions (gasoline, kerosene and diesel) overlap significantly,79,80 the compounds contained in the oil products from this present work have been presented in three different groups, including compounds with <C11, compounds with C11–C16, and compounds with >C16. With the HRSO feedstock being dominated by oleic acid (74.4 wt%), the formation of shorter chain alkanes (ESI Table SI1†) could be from one of two possible pathways: (1) cracking of long-chain alkanes after the decarboxylation of fatty acids or (2) cracking of the fatty acid carbon chains followed by decarboxylation.36,57
The results in Fig. 3d mirrored those in Fig. 3c, showing that the increase in reaction severity (temperature) and the use of catalysts led to increased decarboxylation and subsequently produced higher alkane yields. In particular, the yield of heptadecane was a good indication of the decarboxylation selectivity of the reactions. Fig. 3d shows the yields of hydrocarbons in the light oil products based on the carbon chain length over the temperature range of 350–400 °C. The light oil products were dominated by hydrocarbons with >C16 chains, which reflected the compositions of fatty acids in HRSO. Table 3 shows that RSO contained fatty acids within the C16 to C22 range.35 Being dominated by oleic acid and other >C18 fatty acids (≈88 wt% in total), decarboxylation produced >C16 hydrocarbons, of which heptadecane was the main product. The formation of alkanes rather than alkenes has been explained by the hydrogenation of CC bonds by in situ generated hydrogens,53 as shown in eqn (9).
| C17H33COOH + H2 → C17H36 + CO2 | (9) |
As the reaction temperature increased, a wider range of production distribution was observed as shown in Fig. 3d, with the increased formation of <C11 and C11–C16 hydrocarbons due to increased thermal cracking. Therefore, the formation of hydrocarbons with chain lengths of less than C15 indicated that some C–C bond cracking occurred in addition to the main decarboxylation mechanism. For example, hardly any compounds within the <C11 range were found at 350 °C with or without the catalyst. However, the formation of total hydrocarbons with <C16 became significant at 380 °C but the light oil was still dominated by >C16 hydrocarbons in the non-catalysed tests. Indeed, the absence of the catalyst promoted random C–C bond cracking so that at 400 °C, the yields of >C16 hydrocarbons decreased by 31% compared to their yields at 380 °C. Simultaneously, the yields of both the <C11 and >C11–C16 ranges of compounds increased by 132% and 53%, respectively, when the reaction temperature increased from 380 °C to 400 °C in the absence of the catalyst.
With 5 wt% Pt/C, no hydrocarbons within the <C11 range were found. Instead, there was over a three-fold increase in the yields of C11–C16 hydrocarbons when the reaction temperature was increased from 350 °C to 400 °C. Correspondingly, the yields of the >C16 hydrocarbon fraction increased by 24.1% over the same temperature range. Hence, the Pt/C catalyst was both effective in the decarboxylation of fatty acids and highly selective towards the range of hydrocarbons produced. The results in Fig. 3d therefore confirmed that the Pt/C catalyst prevented random C–C bond breaking compared to the non-catalysed test and that increased temperature was the main source of activation energy for C–C bond breaking. Hence, based on the promising results obtained with the use of the 5 wt% Pt/C catalyst, further studies of the effects of the catalyst/feed ratio and reaction times on the deoxygenation of the mixtures of fatty acids in HRSO were performed.
3.3 Effect of 5 wt% Pt/C catalyst/HRSO mass ratios on the conversion of fatty acids
From the work conducted on the effect of temperature, the reaction conditions of 400 °C and 1 h with 2 g of 5 wt% Pt/C (a catalyst/feed mass ratio of 0.2) seemed to produce the best results in terms of oil yield and hydrocarbon contents. Therefore, experiments were carried out to test the effect of catalyst loading using identical conditions to investigate the possibility of achieving desirable results with lower catalyst loadings. Catalyst/feed mass ratios of 0.0, 0.05, 0.1 and the baseline 0.2 were explored based on experiments 3 and 5–7 in Table 4.Table 4 shows the yields of the different phases of products obtained, with mass balance closures over 90 wt%. For a basis of comparison, an experiment was carried out in the absence of any catalyst, which produced a liquid with similar yields to the yields of oil/wax from the experiments with Pt/C (Table 4). Liquids and waxes from these reactions were the dominant products and followed the order of catalyst/feed ratios 0 > 0.05 > 0.1 > 0.2. Solid product yields increased with increasing catalyst/feed ratios and a similar trend was observed for the gas products.
Table 4 indicates that increased catalyst loading led to increased cracking to lower molecular weight compounds with higher volatility, thus leading to losses and lower mass balance closures. The formation of wax products with catalyst/feed ratios of 0.05 and 0.1 implied the formation of long-chain saturated hydrocarbons, while the liquid products formed with catalyst/feed ratios of 0 and 0.2 suggested the formation of shorter chain hydrocarbons due to random cracking of C–C bonds (ESI Table SI2†). Hence, the selectivity of the Pt/Catalyst was lacking during the reactions carried out with catalyst/feed ratios of 0 and 0.2, so that decarboxylation was not the main mechanism for HRSO conversion.
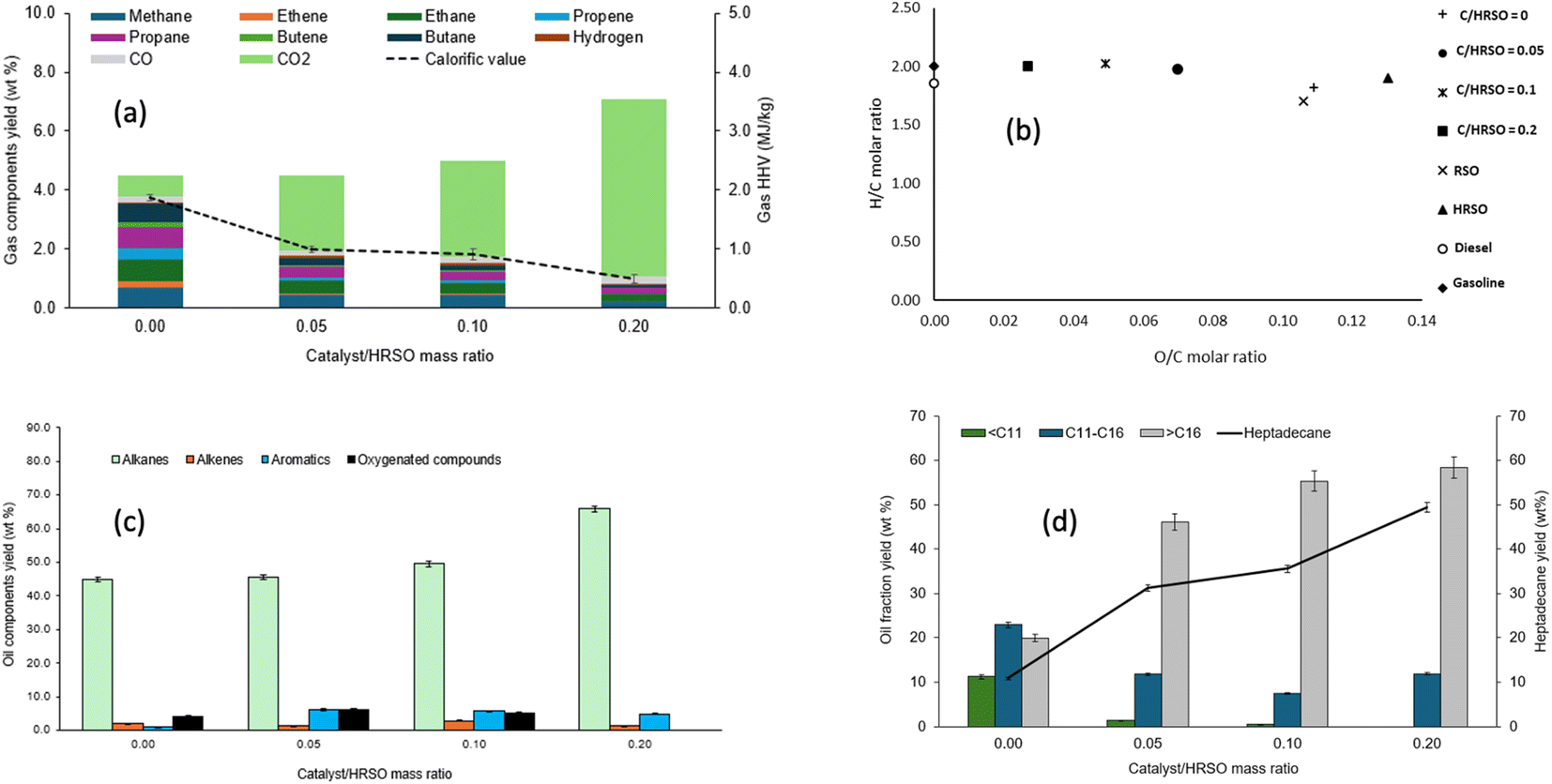 | ||
| Fig. 4 Influence of 5 wt% Pt/C catalyst/HRSO mass ratios on (a) the compositions and calorific values of gas products, (b) the van Krevelen H/C and O/C molar ratios of the oil products, (c) the yields of groups of compounds in the light oils and (d) the yields of hydrocarbons and heptadecane in the light oil. | ||
The test without the catalyst gave the highest combined yield of 3.6 wt% for CH4, C2H4, and C2H6, which implied that thermal cracking occurred. Fig. 4a therefore shows that the dominance of CO2 with increasing catalyst led to a decrease in the calorific value of the gas products.
| Catalyst/HRSO ratio | Density (kg m−3) | TAN (mg KOH g−1) | C (wt%) | H (wt%) | N (wt%) | O (wt%) | HHV (MJ kg−1) |
|---|---|---|---|---|---|---|---|
| 0.00 | 923 | 37.10 | 77.04 ± 0.84 | 11.72 ± 0.09 | 0.09 ± 0.01 | 11.2 ± 0.13 | 40.95 |
| 0.05 | 910 | 30.65 | 79.31 ± 0.03 | 13.14 ± 0.03 | 0.15 ± 0.06 | 7.39 ± 0.40 | 44.46 |
| 0.10 | 907 | 24.98 | 80.90 ± 0.89 | 13.69 ± 0.11 | 0.12 ± 0.00 | 5.30 ± 0.02 | 46.17 |
| 0.20 | 906 | 7.49 | 83.00 ± 0.10 | 13.90 ± 0.31 | 0.11 ± 0.01 | 2.99 ± 0.11 | 47.60 |
The chain lengths of the components of the light components of the oil/wax product obtained from different catalyst to feed ratios are compared in Fig. 4d. The percentage yields of heptadecane at catalyst/feed ratios of 0.0, 0.05, 0.1 and 0.2 were found to be 10.9 wt%, 31.3 wt%, 35.6 wt%, and 49.4 wt%, respectively. This indicated that in addition to the increasing conversion with catalyst loading, the catalyst also favoured selectivity to heptadecane from the decarboxylation of the dominant C18 fatty acids in HRSO.
The reaction carried out in the absence of the catalyst also showed a wide range of product distribution, which implied that thermal cracking occurred possibly due to the high reaction temperature used. Cracking can produce hydrogen, which can enhance the decarboxylation reaction.53 However, cracking can also lead to the generation of shorter chain hydrocarbons, the consumption of energetic C–H bonds, and the coking of the catalyst surface.59
Nonetheless, the increase in the catalyst/HRSO mass ratio produced longer chain hydrocarbons, which indicated that decarboxylation reactions were favoured over cracking reactions. Overall, with a higher catalyst/feed mass ratio, higher yields of hydrocarbons were achieved. Yang et al.40 reported 98.7% oleic acid conversion over a Pt/zeolite catalyst and 81% heptadecane selectivity in the presence of a 0.01 catalyst metal/feed ratio and 20 bar hydrogen at 320 °C after 2 h of reaction. In this present work, the reaction at 400 °C and 1 h achieved 92.5% conversion of multiple fatty acids in HRSO, with a heptadecane selectivity of 84%, without adding external hydrogen. Hence, the present study has provided a potentially more cost-effective pathway to produce a wide distribution of fuel-range hydrocarbons (ESI Table SI2†) from fatty acids.
3.4 Effect of reaction time on the conversion of fatty acids in HRSO with the 5 wt% Pt/C catalyst
To determine the reaction time to maximise the degree of decarboxylation, experiments were carried out in the time range of 0 h to 3 h. These are experiments 5 and 8–10 in Table 4. The reaction time here referred to the time the reaction was kept running once the reaction temperature was reached. Hence, the time of reaction “0 h” simply means that the reaction was stopped once the set reaction temperature was reached. These reactions were carried out at 400 °C, which gave the highest conversion reported in Section 3.2.The reaction at 0 h gave the highest yields of solid residues (18.38%), and this was partly due to the presence of embedded unreacted feedstock with the catalyst. As explained earlier in Section 3.1, some heavy oil molecules formed white solids, which were insoluble in the dichloromethane solvent. At 0 h, the product obtained was a semi-solid but as the severity of the reaction increased in terms of reaction time, the products became liquids with much lighter colours.
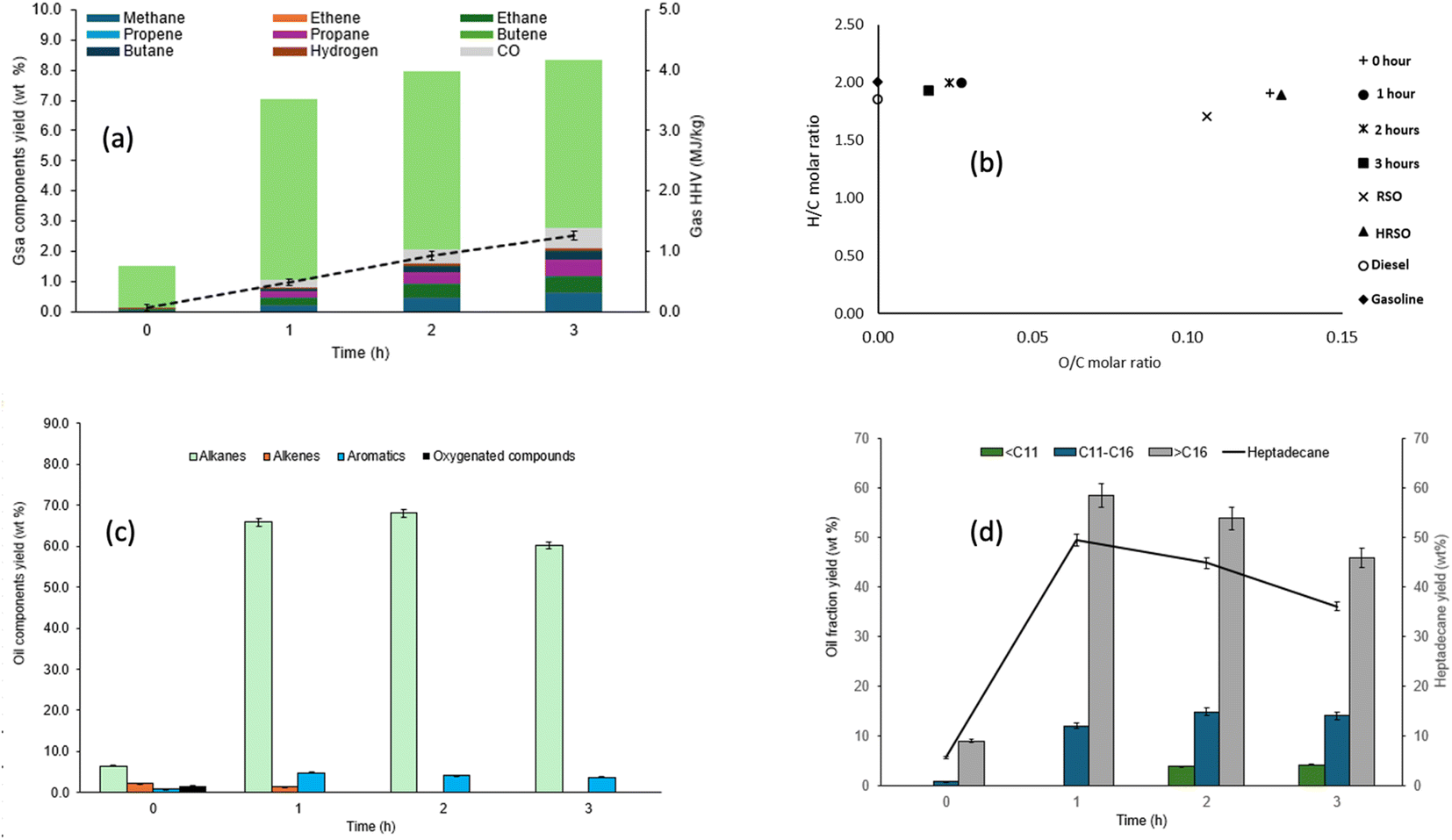 | ||
| Fig. 5 Influence of reaction time on (a) the compositions and calorific values of gas products, (b) the van Krevelen H/C and O/C molar ratios of the oil products, (c) the yields of groups of compounds in the light oils and (d) the yields of hydrocarbons and heptadecane in the light oil obtained from the conversion of HRSO with 2 g of 5 wt% Pt/C catalyst at 400 °C. | ||
From a reaction time of 1 h, the yield of CO2 stayed fairly constant, while the yields of hydrogen and other hydrocarbon gases became more significant, suggesting that increased cracking reactions occurred with longer reaction times. The calorific values of the gas products reflected their compositions. The increase in the yields of hydrogen and the hydrocarbon gases under more severe reaction conditions led to a higher calorific value of the gas products with a seventeen-fold increase between 0 h and 2 h.
| Time (h) | Density (kg m−3) | TAN (mg KOH g−1) | C (wt%) | H (wt%) | N (wt%) | O (wt%) | HHV (MJ kg−1) |
|---|---|---|---|---|---|---|---|
| 0 | 923 | 85.43 | 75.08 ± 1.63 | 12.00 ± 0.24 | 0.27 ± 0.00 | 12.66 ± 0.04 | 40.43 |
| 1 | 906 | 7.49 | 83.00 ± 0.1 | 13.90 ± 0.31 | 0.11 ± 0.01 | 2.99 ± 0.11 | 47.60 |
| 2 | 883 | 2.08 | 83.31 ± 3.20 | 13.93 ± 0.51 | 0.20 ± 0.00 | 2.56 ± 0.08 | 47.82 |
| 3 | 879 | 1.15 | 84.2 ± 3.83 | 13.60 ± 0.63 | 0.14 ± 0.01 | 1.86 ± 0.16 | 46.80 |
Fig. 5b shows the effect of reaction times on the O/C and H/C ratios of the oil/wax products obtained. The results show that increasing the reaction time to 1 h and above had a significant impact on the O/C ratios, causing the values to approach those of hydrocarbon fuels (gasoline and diesel).
Fig. 5c shows the results of the GC/MS analysis of liquid oil products based on the group of compounds obtained from the decarboxylation reactions carried out at different reaction times. Oxygenated compounds only appeared at 0 h reaction time. In addition to the increase in conversion with the increased reaction time, the yield of alkanes, which was only 6.48 wt% at 0 h, reached 68.12 wt% after 2 h.
The yield of aromatics remained at approximately 4–5 wt% at reaction times 1–3 h. This implied there was little to no effect on the aromatisation of the compounds as the reaction time increased. Little amounts of alkenes were present at 0 h (2.28 wt%) and 1 h (1.32 wt%) but none was detected at longer reaction times, which showed that increasing the reaction time minimised the production of olefins, possibly via catalytic hydrogenation with the in situ hydrogen gas.53,77 As the reaction time increased from 2 to 3 h at 400 °C, there was very little change in the conversion as well as the profile of compounds produced, and more gas was produced, reducing the yield of oil. Therefore, choosing a reaction time of 2 h at 400 °C could be more beneficial as the energy consumed in extending the reaction time would only translate to the minor differences observed.
Fig. 5d shows that up to 1 h reaction time only hydrocarbons within the range of C11–C16 > C16 were present in the light oils obtained. Between 0 h and 1 h, the total yield of C11–C16 compounds increased 10 times, while that of >C16 increased nearly seven times due to increased decarboxylation. Shorter chain hydrocarbons in the carbon range < C11 were formed only after 2 h of reaction and their yields increased by 10.11% when the reaction time was increased from 2 h to 3 h. In general, the formation of the shorter chain compounds led to a reduction in the yields of the longer chain compounds within the >C16 range of hydrocarbons. After 2 h of reaction, the middle range compounds exhibit a marginal increase to 14.7 wt% from nearly 12 wt% after 1 h reaction time. Further increase in the reaction time to 3 h led to a considerable reduction in the total yields of hydrocarbons, due mainly to the reduction of the >C16 compounds. The highest yield of the fraction occurred after 1 h (58.4 wt%), reduced to 53.7 wt% after 2 h and further reduced to 45.8 wt% after 3 h, indicating the consistent conversion of these longer chain compounds to the highly volatile >C11 fraction and gas products (Table 7). A trend similar to the yields of the >C16 compounds was observed with heptadecane, with its highest yields of 49.4 wt% achieved after 1 h reaction but consistently reduced with increasing reaction times (ESI Table SI3†).
Wu et al.81 reported 75 wt% heptadecane yield from stearic acid using Ni/C as the catalyst at 330 °C after 5 h of reaction. Using stearic acid (saturated C18 fatty acid), the authors could produce heptadecane via pure decarboxylation, whereas with oleic acid (the main fatty acid in HRSO), obtaining heptadecane showed the occurrence of in situ hydrogenation along with the decarboxylation.53,76 In addition, the authors used a combination of lower temperature (300 °C) and long reaction time (5 h) to achieve their result. In contrast, a higher reaction temperature of 400 °C used in this present work, while still giving a comparable total hydrocarbon yield of 72.1 wt% after 2 h, led to the formation of useful three potential replacement fuel fractions – gasoline, kerosene and diesel – through a combination of decarboxylation, in situ hydrogenation and cracking.
3.5 Influence of different catalysts on the conversion of HRSO
The literature has consistently shown that Pt-based catalysts have been effective for the decarboxylation of fatty acids.53,57 So far in this present study, results have proved the effectiveness of the 5 wt% Pt/C catalyst in the deoxygenation (mainly via decarboxylation) of fatty acids to produce hydrocarbon-rich oils. However, it is often challenging to recover metal catalysts from carbon supports by simple methods like recalcination due to support destruction by carbon burn-off. Therefore, further investigation was carried out using 5 wt% Pt metal supported on silica, magnesium silicate and alumina as the catalyst for the conversion of HRSO. In addition, the Ni–Cu/Al2O3 catalyst was tested as a potentially cheaper catalyst than the Pd- and Pt-based ones; however, it gave a very low catalytic activity towards fatty acid conversion. The reactions were carried out at 400 °C for 2 h, being the best conditions obtained from the test with the 5 wt% Pt/C (compared to 1 h and 3 h reactions at the same temperature), the results of which have been included for ease of comparison. These are experiments 5 and 11–15 in Table 4.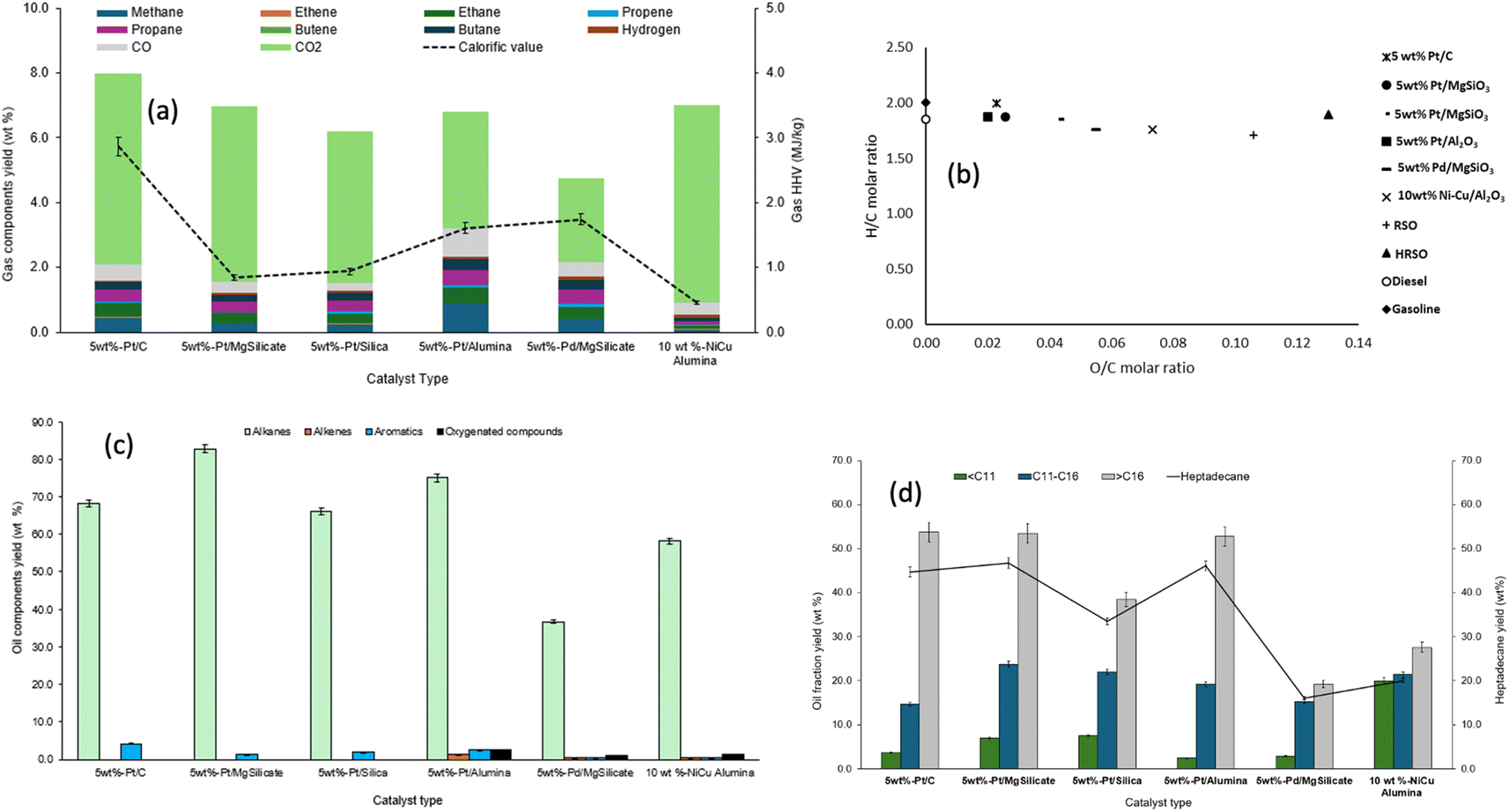 | ||
| Fig. 6 Influence of different catalysts on (a) the compositions and calorific values of gas products, (b) the van Krevelen H/C and O/C molar ratios of the oil products, (c) the yields of groups of compounds in the light oils and (d) the yields of hydrocarbons and heptadecane in the light oil obtained from the conversion of HRSO at 400 °C and 2 h reaction time. | ||
| Catalyst | Density (kg m−3) | TAN (mg KOH g−1) | C (wt%) | H (wt%) | N (wt%) | O (wt%) | HHV (MJ kg−1) |
|---|---|---|---|---|---|---|---|
| 5 wt% Pt/C | 883 | 2.08 | 83.31 ± 3.20 | 13.93 ± 0.51 | 0.20 ± 0.00 | 2.56 ± 0.08 | 47.82 |
| 5 wt% Pt/MgSiO3 | 797 | 0.00 | 83.76 ± 0.40 | 13.13 ± 0.02 | 0.22 ± 0.03 | 2.89 ± 0.14 | 46.76 |
| 5 wt% Pt/Silica | 899 | 17.66 | 82.28 ± 2.75 | 12.76 ± 0.30 | 0.20 ± 0.03 | 4.76 ± 0.21 | 45.39 |
| 5 wt% Pt/Al2O3 | 880 | 0.00 | 84.33 ± 2.13 | 13.22 ± 0.16 | 0.20 ± 0.03 | 2.25 ± 0.19 | 47.20 |
| 5 wt% Pd/MgSiO3 | 966 | 51.63 | 81.76 ± 0.85 | 12.04 ± 0.10 | 0.20 ± 0.02 | 6.00 ± 0.12 | 43.95 |
| 10 wt% Ni–Cu/Al2O3 | 942 | 49.54 | 80.09 ± 2.54 | 11.82 ± 0.40 | 0.24 ± 0.02 | 7.85 ± 0.15 | 42.73 |
However, Table 8 shows that the extent of deoxygenation varied among the catalysts, with some of the oil products having little to no fatty acid contents left while some still had acid values or TANs of over 50 mg KOH g−1. For instance, catalysts such as Pt/C, Pt/MgSiO3, and Pt/SiO2, which gave low TAN values and low oxygen contents, had superior deoxygenation capacities over catalysts such as Pd/MgSiO3, and Ni–Cu/Al2O3. The densities and calorific contents (HHVs) of the oils were similar to those of conventional fuels, ranging from 797 kg m−3 to 966 kg m−3 and 46–50 MJ kg−1 respectively.
A van Krevelen diagram was generated using the H/C and O/C molar ratios based on the elemental analysis data. This diagram is shown in Fig. 6b, where the data for the oil products have been compared to those of common fuels (gasoline, kerosene and diesel). Fig. 6b shows that the supported Ni–Cu and Pd catalysts gave the highest O/C ratios, which correlated with their relatively high oxygen contents reported in Table 8. The oils produced using Pt/C, Pt/SiO2, and Pt/Al2O3 had the greatest potential of comparison with conventional fuels, as they had the highest H/C ratios and the lowest O/C ratios.
While 5 wt% Pt/MgSiO3 gave the highest yield of oil products, Pt/C produced oil with the lightest colour, which may be an indication of a comparably much larger surface area of the latter (650 m2 g−1) compared to the former (116 m2 g−1). In contrast, all the other catalysts produced dark coloured liquids (ESI Fig. SI4†), which could cause issues with further processing and use of the products. The dark-coloured liquids produced may be due to the low surface areas as well as the acidity/alkalinity of the catalysts, which could promote hotspots for the formation of high molecular weight carbon compounds or coke that eventually dissolved in the oils.13
Fig. 6d shows the range of hydrocarbons obtained according to their carbon chain lengths (also see ESI Table SI4†). The supported Pt catalysts produced the largest proportion of fractions with >C16, with 5 wt% Pt/C being the most prominent. Pt/C, Pt/MgSiO3 and Pt/Al2O3 produced similar yields of >C16 alkanes of about 53 wt%. For these three Pt-based catalysts, heptadecane with yields of around 47 wt accounted for over 86% of their >C16 fractions, which indicated that these catalysts favoured direct decarboxylation36 and CC bond saturation53,57 over random cracking of C–C bonds. However, Pt/MgSiO3 and Pt/SiO2 produced the highest yields of kerosene range compounds with around 23 wt% each; this may indicate that basic supports favoured this fuel range. In contrast, in the presence of the Ni–Cu catalyst, nearly equal yields of the three fractions were produced. This could be due to random reactions due to little or no catalytic selectivity, with the results being similar to those obtained from the non-catalysed reaction in Fig. 3d (see Section 3.1). Therefore, it could be argued that the Ni–Cu catalyst had little or no catalytic effect on the reactions leading to the products obtained.
For the Pt-based catalysts, both the metal particle size and the support material seemed to have influenced their selectivity towards the yields and compositions of liquid hydrocarbons. While the Pt metal particle sizes ranged from 4.8 nm (Pt/C) to 12.1 nm (Pt/MgSiO3), the latter gave overall better yields of extensively deoxygenated liquid products, showing that metal–support interaction was more favourable than particle size in this case. However, Pd/MgSiO3 (45.3 nm) with the largest particle size gave the lowest yield of deoxygenated fuel, when compared to all the Pt-based catalysts and even the poorly catalytic Ni–Cu/Al2O3 (≈18 nm) (Fig. 6c and d). In general, therefore, the results from the use of these different catalysts indicated the effects of the combination of active metal, particle size and metal–support interaction on the conversion of fatty acids to hydrocarbons. In a comprehensive review paper, Mäki-Arvela and Murzin82 reported that the particle size of the active metal catalyst was a function of many factors including the type of precursor salts and catalyst preparation procedures (e.g., pH, temperature, ageing time, precipitation agent and type of support) as well as the post-synthesis methods of washing, calcination and reduction. In addition, metal particle sizes <10 nm have been reported to be most effective for catalysis.83 These ideas and findings from this present work will be explored during future optimisation studies.
4 Conclusions
This novel work is based on the initial pre-treatment of rapeseed oil to its hydrolysed form (HRSO) containing mixtures of fatty acids. This study has demonstrated the catalytic deoxygenation of the inherent mixtures of fatty acids in lipid feedstock, such as rapeseed oil, to produce fuel-range liquid hydrocarbons. More importantly, commercially relevant high yields of hydrocarbons have been achieved without the addition of external hydrogen gas. There was undeniable evidence of in situ hydrogenation of alkenes to alkanes via catalytically promoted hydrogen generation. The results indicated that increasing the reaction temperature from 350 °C to 400 °C significantly enhanced the yield and selectivity of hydrocarbons, with a notable increase in the production of C11–C16 hydrocarbons and a reduction in the formation of <C11 hydrocarbons. During effective catalysis, the yields of carbon dioxide in the product gas correlated positively with both the conversion of HRSO and the yields of hydrocarbons. While the existence of CO2 in the product gas could be confounded by other reactions that produce and consume CO2, such as methanation, water–gas shift reaction and Boudouard reaction (CO disproportionation), there was sufficient evidence that it was formed directly from decarboxylation. For instance, the oil products were dominated by heptadecane, which resulted from the decarboxylation of the predominant oleic acid and other C18 fatty acids in the HRSO feedstock. This could indicate that decarboxylation was the main deoxygenation mechanism for the conversion of fatty acids to hydrocarbons as reported in several publications. The 5 wt% Pt/C catalyst exhibited the highest selectivity towards decarboxylation, which correlated with a decrease in oxygen content and total acid number values in the resulting oils. Additionally, increasing the reaction time, catalyst loading, and reaction temperature resulted in higher fatty acid conversion and improved selectivity for hydrocarbons, especially alkanes. Heptadecane was a key hydrocarbon product having been formed from the deoxygenation of the dominant oleic acid and other C18 fatty acids. This study also highlighted that the optimal reaction time at 400 °C is 2 hours, which balanced the high conversion rates with the yields of gasoline, kerosene and diesel fuel fractions. These findings suggested that a combination of appropriate catalysts and pre-treatment of lipid feedstock (e.g., to avoid the interferent side reactions of glycerol) can enhance the selectivity of liquid fuel-range hydrocarbons. This could provide a promising pathway for converting various types of lipid feedstock materials into valuable hydrocarbon fuels under moderate reaction conditions, presenting a potentially efficient method for green hydrocarbon-based biofuel production. While this present work has shown good promise, further work on the development and application of cheaper catalysts and testing for catalyst stability are needed to enhance the commercial viability of the process.Data availability
All relevant data generated from this study have been included in the main article and ESI.† Additional data will be provided on request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the Energy & Bioproducts Research Institute (EBRI) and Aston University, UK, for all support received, especially for the PhD student (M. Peters).References
- S. Luh, S. Budinis, T. J. Schmidt and A. Hawkes, Decarbonisation of the industrial sector by means of fuel switching, electrification and CCS, Computer Aided Chemical Engineering, ed. A. Friedl, J. J. Klemeš, S. Radl, P. S. Varbanov and T. Wallek, Elsevier, 2018, vol. 43, pp. 1311–1316 Search PubMed.
- C. Chung, J. Kim, B. K. Sovacool, S. Griffiths, M. Bazilian and M. Yang, Decarbonising the chemical industry: A systematic review of sociotechnical systems, technological innovations, and policy options, Energy Res. Soc. Sci., 2023, 96, 102955 CrossRef.
- I. Awudu and J. Zhang, Uncertainties and sustainability concepts in biofuel supply chains: A review, Renewable Sustainable Energy Rev., 2019, 88, 401–412 Search PubMed.
- N. Pour, M. E. Webber and C. W. King, Scenarios for linking water conservation, energy consumption, and food production, Environ. Sci. Technol., 2021, 55(5), 3029–3037 Search PubMed.
- W. Deng, Y. Feng, J. Fu, H. Guo, Y. Guo, B. Han, Z. Jiang, L. Kong, C. Li, H. Liu, P. T. T. Nguyen, P. Ren, F. Wang, S. Wang, Y. Wang, Y. Wang, S. S. Wong, K. Yan, N. Yan, X. Yang, Y. Zhang, Z. Zhang, X. Zeng and H. Zhou, Catalytic conversion of lignocellulosic biomass into chemicals and fuels, Green Energy Environ., 2023, 8, 10–114 CrossRef CAS.
- K. Przybysz, E. Małachowska, D. Martyniak, P. Boruszewski, H. Kalinowska and P. Przybysz, Production of sugar feedstocks for fermentation processes from selected fast-growing grasses, Energies, 2019, 12(16), 3129 CrossRef CAS.
- M. Mittelbach, Fuels from oils and fats: Recent developments and perspectives, Eur. J. Lipid Sci. Technol., 2015, 11, 1832–1846 CrossRef.
- L. Guangyi, R. Wang, J. Pang, A. Wang, N. Li and T. Zhang, Production of renewable hydrocarbon biofuels with lignocellulose and its derivatives over heterogeneous catalysts, Chem. Rev., 2024, 124, 2889–2954 CrossRef PubMed.
- A. A. Adesina, Hydrocarbon synthesis via Fischer-Tropsch reaction: travails and triumphs, Appl. Catal., A, 1999, 138, 345–367 CrossRef.
- S. Doliente, A. Narayan, F. Tapia, N. J. Samsatli, Y. Zhao and S. Samsatli, Bio-aviation fuel: A comprehensive review and analysis of the supply chain components, Front. energy res., 2020, 8, 110 CrossRef.
- J. G. Speight, Catalytic Cracking, Heavy and Extra-heavy Oil Upgrading Technologies, 2013, pp. 39–67 Search PubMed.
- J. Pechstein, U. Neuling, J. Gebauer and M. Kaltschmitt, Alcohol-to-Jet (AtJ), Biokerosene, 2017, 543–574 Search PubMed.
- S. Eswaran, S. Subramaniam, S. Geleynse, K. Brandt, M. Wolcott and X. Zhang, Techno-economic analysis of catalytic hydrothermolysis pathway for jet fuel production, Renewable Sustainable Energy Rev., 2021, 151, 111516 CrossRef CAS.
- W.-J. Liu and H.-Q. Yu, Thermochemical conversion of lignocellulosic biomass into mass-producible fuels: Emerging technology progress and environmental sustainability evaluation, ACS Environ. Au, 2021, 2(2), 98–114 CrossRef PubMed.
- D. R. Vardon, B. K. Sharma, H. Jaramillo, D. W. Kim, J. Choe and P. N. Ciesielski, Hydrothermal catalytic processing of saturated and unsaturated fatty acids to hydrocarbons with glycerol for in situ hydrogen production, Green Chem., 2014, 16(3), 1507–1520 RSC.
- Y. Yang, Q. Wang, X. Zhang, L. Wang and G. Li, Hydrotreating of C18 fatty acids to hydrocarbons on sulphided NiW/SiO2–Al2O3, Fuel Process. Technol., 2013, 116, 165–174 CrossRef CAS.
- A. Christensen, S. Searle, N. Pavlenko, The cost of supporting alternative jet fuels in the European Union, International Council on Clean Transportation, 2019, Available at: https://theicct.org/publication/the-cost-of-supporting-alternative-jet-fuels-in-the-european-union/ Search PubMed.
- J. Ogden, A. M. Jaffe, D. Scheitrum, Z. McDonald and M. Miller, Natural gas as a bridge to hydrogen transportation fuel: Insights from the literature, Energy Policy, 2018, 115, 317–329 CrossRef CAS.
- J. L. L. C. C. Janssen, M. Weeda, R. J. Detz and B. van der Zwaan, Country-specific cost projections for renewable hydrogen production through off-grid electricity systems, Appl. Energy, 2022, 309, 118398 CrossRef.
- S. J. Davis, N. S. Lewis, M. Shaner, S. Aggarwal, D. Arent, I. L. Azevedo, S. M. Benson, T. Bradley, J. Brouwer, Y. M. Chiang, C. T. M. Clack, A. Cohen, S. Doig, J. Edmonds, P. Fennell, C. B. Field, B. Hannegan, B. M. Hodge, M. I. Hoffert, E. Ingersoll, P. Jaramillo, K. S. Lackner, K. J. Mach, M. Mastrandrea, J. Ogden, P. F. Peterson, D. L. Sanchez, D. Sperling, J. Stagner, J. E. Trancik, C. J. Yang and K. Caldeira, Net-zero emissions energy systems, Science, 2018, 29, 360 Search PubMed.
- V. K. Mishra and R. Goswami, A review of production, properties and advantages of biodiesel, Biofuels, 2017, 9(2), 273–289 CrossRef.
- S. He, T. Kramer, S. S. Dian, A. Heeres and E. Heeres, Catalytic conversion of glycerol and co-feeds (fatty acids, alcohols, and alkanes) to bio-based aromatics: remarkable and unprecedented synergetic effects on catalyst performance, Green Chem., 2022, 24(2), 941–949 RSC.
- J. Sun and H. Liu, Selective hydrogenolysis of biomass-derived xylitol to ethylene glycol and propylene glycol on Ni/C and basic oxide-promoted Ni/C catalysts, Catal. Today, 2014, 234, 75–82 CrossRef CAS.
- P. Lahijani, M. Mohammadi, A. R. Mohamed, F. Ismail, K. T. Lee and G. Amini, Upgrading biomass-derived pyrolysis bio-oil to bio-jet fuel through catalytic cracking and hydrodeoxygenation: A review of recent progress, Energy Convers. Manage., 2022, 268, 115956 CrossRef CAS.
- Y. Shi, E. Xing, K. Wu, J. Wang, M. Yang and Y. Wu, Recent progress on upgrading of bio-oil to hydrocarbons over metal/zeolite bifunctional catalysts, Catal. Sci. Technol., 2017, 7(12), 2385–2415 RSC.
- S. Bao, G. Liu, X. Zhang, L. Wang and Z. Mi, New Method of catalytic cracking of hydrocarbon fuels using a highly dispersed nano-HZSM-5 Catalyst, Ind. Eng. Chem. Res., 2010, 49(8), 3972–3975 CrossRef CAS.
- X. Qu, Y. Li, S. Li, J. Wang, H. Xu and Z. Li, Thermal cracking, aquathermolysis, and their upgrading effects of Mackay River oil sand, J. Pet. Sci. Eng., 2021, 201, 108473 CrossRef CAS.
- W. T. Hartati, R. R. Mukti, I. Kartika, B. Dea and S. D. Sumbogo, et al., Highly selective hierarchical ZSM-5 from kaolin for catalytic cracking of Calophyllum inophyllum oil to biofuel, J. Energy Inst., 2020, 93(6), 2238–2246 CrossRef CAS.
- M. M. Naeem, E. G. Al-Sakkari, D. C. Boffito, M. A. Gadalla and F. H. Ashour, One-pot conversion of highly acidic waste cooking oil into biodiesel over a novel bio-based bi-functional catalyst, Fuel, 2021, 283, 118914 CrossRef CAS.
- B. Casali, E. Brenna, F. Parmeggiani, D. Tessaro and F. Tentori, Enzymatic methods for the manipulation and valorization of soapstock from vegetable oil refining processes, Sustainable Chem., 2021, 2(1), 74–91 CrossRef CAS.
- H. L. Barnebey and A. C. Brown, Continuous fat splitting plants using the colgate-emery process, J. Am. Oil Chem. Soc., 1948, 25(3), 95–99 CrossRef CAS.
- K. Fukuzumi and Y. Koyama, Fat splitting by the twitchell process at low temperature, J. Am. Oil Chem. Soc., 1957, 34(10), 500–503 CrossRef CAS.
- Y. Sugami, E. Minami and S. Saka, Renewable diesel production from rapeseed oil with hydrothermal hydrogenation and subsequent decarboxylation, Fuel, 2016, 166, 376–381 CrossRef CAS.
- S. K. Bhatia, S. S. Jagtap, A. A. Bedekar, R. K. Bhatia, A. K. Patel and D. Pant, et al., Recent developments in pretreatment technologies on lignocellulosic biomass: Effect of key parameters, technological improvements, and challenges, Bioresour. Technol., 2020, 300, 122724 CrossRef CAS PubMed.
- M. A. Peters, C. T. Alves, J. Wang and J. A. Onwudili, Subcritical water hydrolysis of fresh and waste cooking oils to fatty acids followed by esterification to fatty acid methyl esters: detailed characterization of feedstocks and products, ACS Omega, 2022, 7(50), 46870–46883 CrossRef CAS PubMed.
- J. Fu, X. Lu and P. E. Savage, Catalytic hydrothermal deoxygenation of palmitic acid, Energy Environ. Sci., 2010, 3(3), 311 RSC.
- I. Edeh, T. Overton and S. Bowra, Renewable diesel production by hydrothermal decarboxylation of fatty acids over platinum on carbon catalyst, Biofuels, 2019, 1–8 Search PubMed.
- A. R. K. Gollakota, N. Kishore and S. Gu, A review on hydrothermal liquefaction of biomass, Renewable Sustainable Energy Rev., 2018, 81, 1378–1392 CrossRef.
- Y. Zheng, F. Wang, X. Yang, Y. Huang, C. Liu and Z. Zheng, Study on aromatics production via the catalytic pyrolysis vapor upgrading of biomass using metal-loaded modified H-ZSM-5, J. Anal. Appl. Pyrolysis, 2017, 126, 169–179 CrossRef CAS.
- L. Yang, K. L. Tate, J. B. Jasinski and M. A. Carreon, Decarboxylation of oleic ccid to heptadecane over Pt-Supported on zeolite 5A beads, ACS Catal., 2015, 5(11), 6497–6502 CrossRef CAS.
- J. Wang, X. Yao, Y. Li, J. Zhang, C. Zhao and T. J. Strathmann, Catalytic hydrothermal deoxygenation of stearic acid with Ru/C: effects of alcohol- and carboxylic acid-based hydrogen donors, ACS Omega, 2023, 8(22), 19969–19975 CrossRef CAS PubMed.
- Z. He and X. Wang, Hydrocarbon production from carboxylic acids via catalytic deoxygenation: Required catalytic properties, ACS Symp. Ser., 2013, 301–329 CrossRef CAS.
- K. Jeništová, I. Hachemi, P. Mäki-Arvela, N. Kumar, M. Peurla and L. Čapek, Hydrodeoxygenation of stearic acid and tall oil fatty acids over Ni-alumina catalysts: Influence of reaction parameters and kinetic modelling, Chem. Eng. J., 2017, 316, 401–409 CrossRef.
- K. Hengst, M. Arend, R. Pfützenreuter and W. F. Hoelderich, Deoxygenation and cracking of free fatty acids over acidic catalysts by single step conversion for the production of diesel fuel and fuel blends, Appl. Catal., B, 2015, 174–175, 383–394, DOI:10.1016/j.apcatb.2015.03.009.
- R. Bartosz, M.-A. Päivi, A. Tokarev, A. R. Leino, K. Eränen and Y. Dmitry, Influence of hydrogen in catalytic deoxygenation of fatty acids and their derivatives over Pd/C, Ind. Eng. Chem. Res., 2012, 51(26), 8922–8927 CrossRef.
- A. T. Madsen, A. El Hadi, C. H. Christensen, R. Fehrmann and A. Riisager, Hydrodeoxygenation of waste fat for diesel production: Study on model feed with Pt/alumina catalyst, Fuel, 2011, 90(11), 3433–3438 CrossRef CAS.
- S. Janampelli and S. Darbha, Selective and reusable Pt-WOx/Al2O3 catalyst for deoxygenation of fatty acids and their esters to diesel-range hydrocarbons, Catal. Today, 2018, 309, 219–226 CrossRef CAS.
- Y. Yan, H. Zhang, Q. Liao, Y. Liang and J. Yan, Roadmap to hybrid offshore system with hydrogen and power co-generation, Energy Convers. Manage., 2021, 247, 114690 CrossRef CAS.
- E. O. Lee, M. Y. Choo, V. L. Hwei, H. T.-Y. Yun, K. C. Chin and C. J. Joon, Catalytic deoxygenation of triolein to green fuel over mesoporous TiO2 aided by in situ hydrogen production, Int. J. Hydrogen Energy, 2020, 45(20), 11605–11614 CrossRef.
- X. Gao and S. Kawi, Catalytic hydrogen production, Heterogeneous catalysis for sustainable energy, 2022, pp. 1–32, DOI:10.1002/9783527815906.ch1.
- H. I. Mahdi, A. Bazargan, G. McKay, N. I. Azelee and L. Meili, Catalytic deoxygenation of palm oil and its residue in green diesel production: A current technological review, Chem. Eng. Res. Des., 2021, 174, 158–187, DOI:10.1016/j.cherd.2021.07.009.
- M. Tabandeh, C. K. Cheng, G. Centi, P. L. Show, W.-H. Chen and T. C. Ling, et al., Recent advancement in deoxygenation of fatty acids via homogeneous catalysis for biofuel production, Mol. Catal., 2022, 523, 111207, DOI:10.1016/j.mcat.2020.111207.
- M. Z. de Hossain, M. B. I. Chowdhury, A. K. Jhawar, W. Z. Xu and P. A. Charpentier, Continuous low-pressure decarboxylation of fatty acids to fuel-range hydrocarbons with in situ hydrogen production, Fuel, 2018, 212, 470–478 CrossRef.
- Y. R. Suryawanshi, M. Chakraborty, S. Jauhari, S. Mukhopadhyay, K. T. Shenoy and D. Sen, Selective hydrogenation of Dibenzo-18-crown-6 ether over highly active monodisperse Ru/γ-Al2O3 nanocatalyst, Bull. Chem. React. Eng. Catal., 2015, 10, 23–29 CrossRef.
- Used Cooking Oil (UCO) as biofuel feedstock in the EU, https://cedelft.eu/wp-content/uploads/sites/2/2021/04/CE_Delft__200247_UCO_as_biofuel_feedstock_in_EU_FINAL-v5.pdf Search PubMed.
- Renewable fuel statistics 2022: Second provisional report, https://www.gov.uk/government/statistics/renewable-fuel-statistics-2022-second-provisional-report/renewable-fuel-statistics-2022-second-provisional-report Search PubMed.
- J. Fu, X. Lu and P. E. Savage, Hydrothermal decarboxylation and hydrogenation of fatty acids over Pt/C, ChemSusChem, 2011, 4(4), 481–486 CrossRef CAS PubMed.
- S. Lestari, I. Simakova, A. Tokarev, P. Mäki-Arvela, K. Eränen and D. Y. Murzin, Synthesis of biodiesel via deoxygenation of stearic acid over supported Pd/C catalyst, Catal. Lett., 2008, 122(3–4), 247–251 CrossRef CAS.
- M. Snåre, I. Kubičková, P. Mäki-Arvela, K. Eränen and M. DYu, Heterogeneous Catalytic Deoxygenation of Stearic Acid for Production of Biodiesel, Ind. Eng. Chem. Res., 2006, 45(16), 5708–5715 CrossRef.
- T. Yeh, S. Linic and P. E. Savage, Deactivation of Pt Catalysts during Hydrothermal Decarboxylation of Butyric Acid, ACS Sustain. Chem. Eng., 2014, 2(10), 2399–2406 CrossRef CAS.
- M. G. Jeong, K. Zhuo, S. Cherevko and C. H. Chung, Formation of nanoporous nickel oxides for supercapacitors prepared by electrodeposition with hydrogen evolution reaction and electrochemical dealloying, Korean J. Chem. Eng., 2012, 29(12), 1802–1805 CrossRef CAS.
- J. He, L. Zu, X. Liu, L. Zhang and B. Duan, Porous NiCu alloy cathode with oriented pore structure for hydrogen evolution reaction by freeze casting, J. Porous Mater., 2019, 26(5), 1533–1539 CrossRef CAS.
- A. Singh and K. Miyabayashi, Novel continuous flow synthesis of Pt NPs with narrow size distribution for Pt@carbon catalysts, RSC Adv., 2020, 10(1), 362–366 RSC.
- R. Pandya, R. Mane and C. V. Rode, Cascade dehydrative amination of glycerol to oxazoline, Catal. Sci. Technol., 2018, 8(11), 2954–2965 RSC.
- Y. Dong, Q. Chen, X. Cheng, H. Li, J. Chen, X. Zhang, Q. Kuang and Z. Xie, Optimization of gold-palladium core-shell nanowires towards H2O2 reduction by adjusting shell thickness, Nanoscale Adv., 2019, 2(2), 785–791 RSC.
- I. Bouaoun, H. Hammi, A. Aît-Mokhtar, A. El and M. Adel, Effect of calcination temperature of magnesium silicate on the properties of magnesium phosphate cement, J. Aust. Ceram. Soc., 2017, 53(2), 351–359 CrossRef CAS.
- B. S. Chen, Y. Y. Zeng, L. Liu, L. Chen, P. Duan and R. Luque, et al., Advances in catalytic decarboxylation of bioderived fatty acids to diesel-range alkanes, Renewable Sustainable Energy Rev., 2022, 158, 112178 CrossRef CAS.
- D. Castello, B. Rolli, A. Kruse and L. Fiori, Supercritical water gasification of biomass in a ceramic reactor: long-time batch experiments, Energies, 2017, 10(11), 1734 CrossRef.
- A. Sagan, A. Blicharz-Kania, M. Szmigielski, D. Andrejko, P. Sobczak, K. Zawiślak and A. Starek, Assessment of the Properties of Rapeseed Oil Enriched with Oils Characterized by High Content of α-linolenic Acid, Sustainability, 2019, 11(20), 5638 CrossRef CAS.
- Official methods and recommended practices of the AOCS, 2024, https://search.worldcat.org/title/Official-methods-and-recommended-practices-of-the-AOCS/oclc/286476642 Search PubMed.
- K. Lissitsyna, S. Huertas, R. Morales, L. C. Quintero and L. M. Polo, Determination of trace levels of fatty acid methyl esters in aviation fuel by GC × GC–FID and comparison with the reference GC–MS method, Chromatographia, 2012, 75(21–22), 1319–1325 CrossRef CAS.
- X. Xu, J. Fillos, K. Ramalingam and A. Rosenthal, Quantitative analysis of methanol in wastewater by GC-MS with direct injection or headspace SPME sample introduction, Anal. Methods, 2012, 4(11), 3688 RSC.
- T. Capoun and J. Krykorkova, Internal standards for quantitative analysis of chemical warfare agents by the GC/MS Method: nerve agents, J. Anal. Methods Chem., 2020, 8857210 CAS.
- Standard Test Method for Specific Gravity of Oils and Liquid Fats, 2017, https://www.astm.org/d5355-95r01.html Search PubMed.
- B. Oliver-Tomas, M. Renz and A. Corma, High quality biowaxes from fatty acids and fatty esters: catalyst and reaction mechanism for accompanying reactions, Ind. Eng. Chem. Res., 2017, 56(45), 12870–12877 CrossRef CAS.
- S. Wang and G. Song, A pathway to bio-based aromatics, Nat Sustainability, 2023, 1–2 Search PubMed.
- J. Feng, W. Xiong, H. Ding and B. He, Hydrogenolysis of glycerol over Pt/C catalyst in combination with alkali metal hydroxides, Open Chem., 2016, 14, 279–286 CAS.
- C. Saggese, A. V. Singh, X. Xue, C. Chu, M. R. Kholghy and T. Zhang, et al., The distillation curve and sooting propensity of a typical jet fuel, Fuel, 2019, 235, 350–362 CrossRef CAS.
- Y. Kar, D. Ş. Göksu and Y. Yalman, Characterization of light diesel fraction obtained from upgraded heavy oil, Egypt. J. Pet., 2018, 27(4), 1301–1304 CrossRef.
- A. Demirbas and H. S. Bamufleh, Optimization of crude oil refining products to valuable fuel blends, Pet. Sci. Technol., 2017, 35(4), 406–412 CrossRef CAS.
- J. Wu, J. Shi, J. Fu, J. A. Leidl, Z. Hou and X. Lu, Catalytic decarboxylation of fatty acids to aviation fuels over nickel supported on activated carbon, Sci. Rep., 2016, 6, 27820 CrossRef CAS PubMed.
- P. Mäki-Arvela and D. Y. Murzin, Effect of catalyst synthesis parameters on the metal particle size, Appl. Catal., A, 2013, 451, 251–281 CrossRef.
- J. D. Aiken and R. G. Finke, A review of modern transition-metal nanoclusters: their synthesis, characterization, and applications in catalysis, J. Mol. Catal. A: Chem., 1999, 145, 1–44 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4se00864b |
| This journal is © The Royal Society of Chemistry 2024 |