
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Na0.5Bi0.5TiO3 perovskite anode for lithium-ion batteries†
Sridivya
Chintha
a,
Shahan
Atif
 a,
Anshuman
Chaupatnaik
a,
Alexander
Golubnichiy
b,
Artem M.
Abakumov
b and
Prabeer
Barpanda
*acd
a,
Anshuman
Chaupatnaik
a,
Alexander
Golubnichiy
b,
Artem M.
Abakumov
b and
Prabeer
Barpanda
*acd
aFaraday Materials Laboratory (FaMaL), Materials Research Center, Indian Institute of Science, Bangalore 560012, India. E-mail: prabeer@iisc.ac.in
bCenter for Energy Science and Technology, Skolkovo Institute of Science and Technology, Bolshoi blv. 30-1, 121205 Moscow, Russia
cHelmholtz Institute Ulm (HIU), Electrochemical Energy Storage, Ulm 89081, Germany
dInstitute of Nanotechnology, Karlsruhe Institute of Technology (KIT), Karlsruhe, 76021, Germany
First published on 30th September 2024
Abstract
Lithium-ion battery technology, currently the most popular form of mobile energy storage, primarily uses graphite as the anode. However, the graphite anode, owing to its low working voltage at high current density, is susceptible to lithium plating and related safety risks. In this direction, perovskite oxides like CaSnO3, more recently PbTiO3, have been explored as alternate anode materials due to their higher operational voltage. Extending this family of perovskites, we introduce a widely used lead-free piezoelectric ceramic Na0.5Bi0.5TiO3 (NBT) as a potential anode for lithium-ion batteries. NBT has an average voltage of 0.7 V and a high capacity of 220 mA h g−1. Ex situ diffraction and spectroscopy tools were used to understand the charge storage mechanism. The oxide undergoes an irreversible conversion reaction in the first discharge, followed by reversible (de)alloying of Bi with Li in the subsequent cycles. This material is airstable, with a capacity retention of 82% up to 50 cycles at a high current of 100 mA g−1 without any optimization. Furthermore, limiting the voltage window increases the cycle life to 200 cycles. Perovskite-type Na0.5Bi0.5TiO3 is proposed as a new Bi-based conversion alloying anode for lithium-ion batteries.
1. Introduction
The growing demand for Li-ion batteries (LIBs) with better performance for applications like portable electronics, electric vehicles, and grid storage is constantly driving us to explore newer chemistries.1 Battery design involves exploration and optimization of a wide variety of (in)organic materials for various components of batteries. One such candidate is the anode. To date, graphite and spinel Li4Ti5O12 (LTO) are the most successful anode intercalation hosts for LIBs.2,3 LTO is a high-rate anode, but offers a low energy density in a full cell due to enhanced working voltage (1.55 V vs. Li/Li+), whereas, despite high capacity of 372 mA h g−1, graphite poses serious safety concerns at high current rates as its working potential is low and close to Li plating/stripping (0.2 V vs. Li/Li+). This inspires the search for alternate anode materials for LIBs with a combination of high-power density and operational safety with a decent energy density.In this context, alternate anode materials that alloy or undergo conversion reactions with Li are attractive. Alloying elements (such as Si, Ge, Pb, Bi, Sn, etc.)4 have high theoretical capacities and working potential above graphite, but suffer from significant volume changes leading to cracking and failure.5 In contrast, conversion compounds without alloying elements, like transition metal oxides (TMOs like CoO, NiO, FeO, etc.),6 suffer very large voltage hysteresis due to the formation of insulating phases like Li2O, which impede their kinetics and reversibility.7 Here, conversion–alloying materials (CAMs), typically amorphous/crystalline compounds having an alloying element, combine both processes in a single compound.8,9 CAMs generate a “plum-pudding” phase dispersion during their first discharge having the following synergistic advantage. The converted “pudding” phases like Li2O formed during discharge buffer the large volume expansions during the alloying step, while the metal/alloy “plum” phases enhance the electronic conductivity in addition to storing charge. It is worth mentioning here that early advances on CAM anodes were triggered in the 1990s by the commercial success of amorphous tin composite oxide (ATCO) CAM by Fujifilm Celltec Co. Ltd9 following Miyasaka's work.10
Diversification of widely known functional ABX3-type perovskites such as CaSnO3,11 PbMO3 (M = Ti/Zr),12 CH3NH3PbX3 (X = I, Cl, Br)13 and APbO3 (A = Ba/Sr)14 as CAM based anodes in lithium-ion batteries has paved the path to a research direction employing the “perovskite frameworks” in energy storage applications.15 In this context, we repurpose yet another widely used lead-free piezoelectric ceramic Na0.5Bi0.5TiO3 (NBT) perovskite oxide as an anode material for LIBs. Like other CAMs, NBT delivers a high irreversible capacity of 700 mA h g−1 during the first discharge from a conversion reaction at ∼0.6 V, followed by an alloying reaction at lower potentials. Low current densities like 5 mA g−1 give a reversible first charge capacity as high as 380 mA h g−1, albeit with faster capacity fade. In contrast, at high current densities, the cycle life performance is improved with relatively lower capacities of about 150–220 mA h g−1. Furthermore, a detailed study of the charge storage mechanism using multiple electroanalytical and material characterization techniques shows that the active redox center is Bi. This work opens up the possibility of exploring numerous other lead-free Bi-based perovskite framework compounds as anodes in rechargeable (post) lithium-ion batteries.
2. Experimental section
2.1 Synthesis
Na0.5Bi0.5TiO3 was synthesized by a conventional solid-state (dry) synthesis route. The Na2CO3 (99.5%, Merck), Bi2O3 (99%, SDFCL), and anatase TiO2 (98.5%, Merck) precursors were intimately mixed using an agate mortar pestle in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:4. The precursor mixture was pelletized using uniaxial press. These pellets were calcined (in air) at 850 °C for 5 h, followed by sintering at 1100 °C for 2 h (ramp rate = 5 °C min−1) in a muffle furnace to get the final product.
:1:4. The precursor mixture was pelletized using uniaxial press. These pellets were calcined (in air) at 850 °C for 5 h, followed by sintering at 1100 °C for 2 h (ramp rate = 5 °C min−1) in a muffle furnace to get the final product.
2.2 Material characterization
The powder X-ray diffraction (PXRD) pattern of Na0.5Bi0.5TiO3 was obtained using a PANalytical X'pert pro diffractometer (Cu Kα source with λ1 = 1.5405 Å and λ2 = 1.5443 Å operated at 40 kV/30 mA) in the 2θ range of 5–80°. Rietveld refinement was performed using Fullprof software,16 and the crystal structure was visualized using the VESTA program.17 The particle morphology was observed using an Ultra 55 Field emission scanning electron microscope (FESEM, W-source) operated at 0.1–30 kV. X-ray photoelectron spectroscopy (XPS) analysis of the pristine, charged, and discharged samples was conducted using a Thermo-Scientific K-Alpha X-ray Photoelectron Spectrometer with monochromatic Al Kα radiation (operated at 12 kV/6 mA). The XPS spectra were calibrated with the carbon signal as the reference at 284.6 eV binding energy after the Shirley background subtraction.For transmission electron microscopy (TEM) analysis, samples were prepared by crushing the powders in a mortar in dimethyl carbonate (DMC) under an argon atmosphere. A few drops of the suspension were deposited on the lacey carbon-supported copper TEM grids. To avoid contact with the atmosphere, a Gatan vacuum holder was used to transfer the samples from the glove box to a TEM column. High-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) images, high-resolution transmission electron microscopy (HRTEM) images, selected area electron diffraction (SAED) patterns, and energy-dispersive X-ray maps in the STEM mode (EDX-STEM) were acquired with a Thermo Fischer Titan Themis Z transmission electron microscope operated at 200 kV and equipped with a Super-X system for EDX analysis. For the electron energy loss spectroscopy (EELS) experiment, the microscope was operated at 200 kV in a TEM mode using a monochromatic filtered beam. The energy resolution, defined as the full width at half-maximum height of the zero-loss peak, was 0.21 eV. Measurements were conducted in Dual EELS mode using a 2.5 mm aperture and 0.05 eV per channel dispersion.
2.3 Electrochemical study
A mixture of active material (perovskite NBT), Super P carbon black (Alfa Aesar, 99%), and Sodium Carboxy Methyl Cellulose (Na-CMC) salt (Sigma) taken in a 70:20:10 ratio was hand mixed in distilled water to form a consistent slurry. Before dissolving in the binder, NBT and Super P carbon black were ball-milled for 4 h at 400 rpm to ensure thorough mixing of both components. The slurry was cast on a battery-grade Cu foil using a doctor blade and was dried in a vacuum oven at 80 °C overnight to remove water. These composite anode electrodes, with a typical mass loading of 2–4 mg cm−2, were used to study the electrochemical performance in the LIB in a coin-type half-cell configuration. CR2032-type half cells were assembled inside an Ar-filled glovebox (MBraun LabStar GmbH, O2 and H2O levels <0.5 ppm) to avoid any air/moisture contact. Li metal foil was used as the counter/reference electrode, and glass fiber composite (GFC sheet-Whatman) was used as the separator. A commercial electrolyte of 1 M LiPF6 dissolved in 1:1:1 v/v% of ethylene carbonate/dimethyl carbonate/diethyl carbonate (EC: DMC: DEC) (Sigma) mixture was used. Galvanostatic charge–discharge cycling was performed using a Neware BTS-4000 battery tester (Shenzhen, China) at ambient temperature (25 °C).
2.4 Ex situ analysis
For post-mortem analysis, Swagelok-type cells were assembled and were cycled at a low current density of 10 mA g−1. After cycling to different states of (dis)charge, these Swageloks containing NBT electrodes were immediately transferred to the glove box to avoid any reaction with the atmosphere. They were disassembled, and the recuperated electrodes were washed with dimethyl carbonate (DMC, anhydrous, ≥99%), and dried for a day inside the glovebox. Sealed electrodes were used to carry out the ex situ diffraction (XRD), spectroscopy (XPS) and microscopy (TEM) studies.3. Results and discussion
3.1 Synthesis
A stoichiometric mixture of Na2CO3, Bi2O3 and TiO2 was subjected to two step heat treatment (calcination at 850 °C for 5 h followed by sintering at 1100 °C for 2 h) to prepare the target perovskite Na0.5Bi0.5TiO3 (NBT). The phase purity of as-synthesized NBT powders was confirmed by the Rietveld analysis of its powder XRD pattern (Fig. 1a). NBT assumes a trigonal perovskite structure (having R3c symmetry) with disordered Bi and Na occupying the A-site and tilted TiO6 octahedra at the B-site (Fig. 1a, inset). Additionally, the perovskite structure could be directly visualized (Fig. 1b) from the HAADF-STEM images from 1 to 5 micron sized irregular particles (Fig. 1c and d). The [2![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 1]-zone axis projection is equivalent to the 〈100〉 projection of the perovskite basic structure (Fig. 1b). Atomic columns with higher intensity correspond to A-positions of the perovskite structure due to the high atomic number of Bi. Fainter dots stand for the mixed Ti/O columns. Importantly, one can note that the intensity associated with the Na/Bi columns at the A-sites is not uniform and locally can be very low, being comparable to that of the Ti/O column. It indicates that the distribution of Na and Bi is not totally random, and there are Na-enriched and Na-depleted columns. The a, b and c lattice parameters calculated from Rietveld refinement are 5.484 Å, 5.484 Å and 13.503 Å, respectively (Table S1†), which agree with previous reports.18 Additionally, the SAED patterns along the main directions of the perovskite sub-cell could also be indexed in a hexagonal unit cell with a ≈ 5.5 Å, c ≈ 13.5 Å (Fig. 1e–g). Systematic reflection conditions hkl: −h + k + l = 3n and 00l: l = 6n are compatible with the R3c space group. Phase-pure NBT perovskite can be easily prepared at a large scale and is air stable. Next, we investigated the Li storage performance of NBT in a lithium half-cell.
1]-zone axis projection is equivalent to the 〈100〉 projection of the perovskite basic structure (Fig. 1b). Atomic columns with higher intensity correspond to A-positions of the perovskite structure due to the high atomic number of Bi. Fainter dots stand for the mixed Ti/O columns. Importantly, one can note that the intensity associated with the Na/Bi columns at the A-sites is not uniform and locally can be very low, being comparable to that of the Ti/O column. It indicates that the distribution of Na and Bi is not totally random, and there are Na-enriched and Na-depleted columns. The a, b and c lattice parameters calculated from Rietveld refinement are 5.484 Å, 5.484 Å and 13.503 Å, respectively (Table S1†), which agree with previous reports.18 Additionally, the SAED patterns along the main directions of the perovskite sub-cell could also be indexed in a hexagonal unit cell with a ≈ 5.5 Å, c ≈ 13.5 Å (Fig. 1e–g). Systematic reflection conditions hkl: −h + k + l = 3n and 00l: l = 6n are compatible with the R3c space group. Phase-pure NBT perovskite can be easily prepared at a large scale and is air stable. Next, we investigated the Li storage performance of NBT in a lithium half-cell.
 | ||
| Fig. 1 Synthesis of Na0.5Bi0.5TiO3 (NBT). (a) Rietveld refinement of the NBT phase (inset shows the crystal structure, (Na, Bi, Ti and O atoms are shown as dark yellow, black, white and red spheres, respectively)). (b) [21] HAADF-STEM image showing the perovskite structure of NBT with atomic resolution indicating Na-rich and Na-lean zones. (c) Low-magnification TEM and (d) SEM images of NBT powders. Bar size is equal to 2 and 200 nm for figures (b) and (c). (e–g) Selected area diffraction patterns (SADPs) of Na0.5Bi0.5TiO3 along the primary directions of the perovskite sub-cell. | ||
3.2 Electrochemical performance
The working electrode was prepared by casting a handmixed slurry of NBT, carbon and NaCMC binder in the ratio 70:20:10 on a copper foil. The galvanostatic voltage profiles in a voltage window of 0.01–3.0 V at 10 mA g−1 indicate that almost 5 moles of Li were consumed during the first discharge, corresponding to 628 mA h g−1 capacity (Fig. 2a). The first discharge curve follows a large voltage plateau around 0.6 V, implying a biphasic conversion process as explained later. Subsequent cycles indicated a voltage hysteresis of 0.3 V with reversible (de)insertion of nearly 2.5 Li (hinting at a reversible LixBi alloying reaction with x < 3). Fig. 2b shows the voltage profiles after first discharge at a higher current of 100 mA g−1. The charge capacity is 250 mA h g−1, with higher capacity fading during the first 3–4 cycles, similar to other Bi-based CAMs.19 A very low first cycle coulombic efficiency of 43.4% could be attributed to the irreversible SEI formation reaction in the first cycle. This value rises to 95.3% in the second cycle, reaching 98.7% in the subsequent cycles, suggesting a stable SEI formation (Fig. 2c). The rate performance of NBT (Fig. 2d) shows that this anode can deliver capacities of 185, 158, 138, 120, and 102 mA h g−1 at current densities of 0.02, 0.05, 0.1, 0.2, and 0.4 A g−1, respectively, which could be restored upon progressively decreasing the current. Also, capacity fading observed at 100 mA g−1 was less rapid than that at lower 10 mA g−1, possibly due to undesirable side reactions. Additionally, higher capacity retention at 100 mA g−1 could be further improved by narrowing the voltage window to solely involve the Bi (de)alloying activity, i.e., 0.2–1.2 V (Fig. S1†). This agrees well with previous studies on CAMs where cycling these materials in a wider potential window is detrimental to stability and cycle life, likely owing to parasitic conversion reactions or SEI/electrolyte oxidation over the oxidized metallic alloying element.20
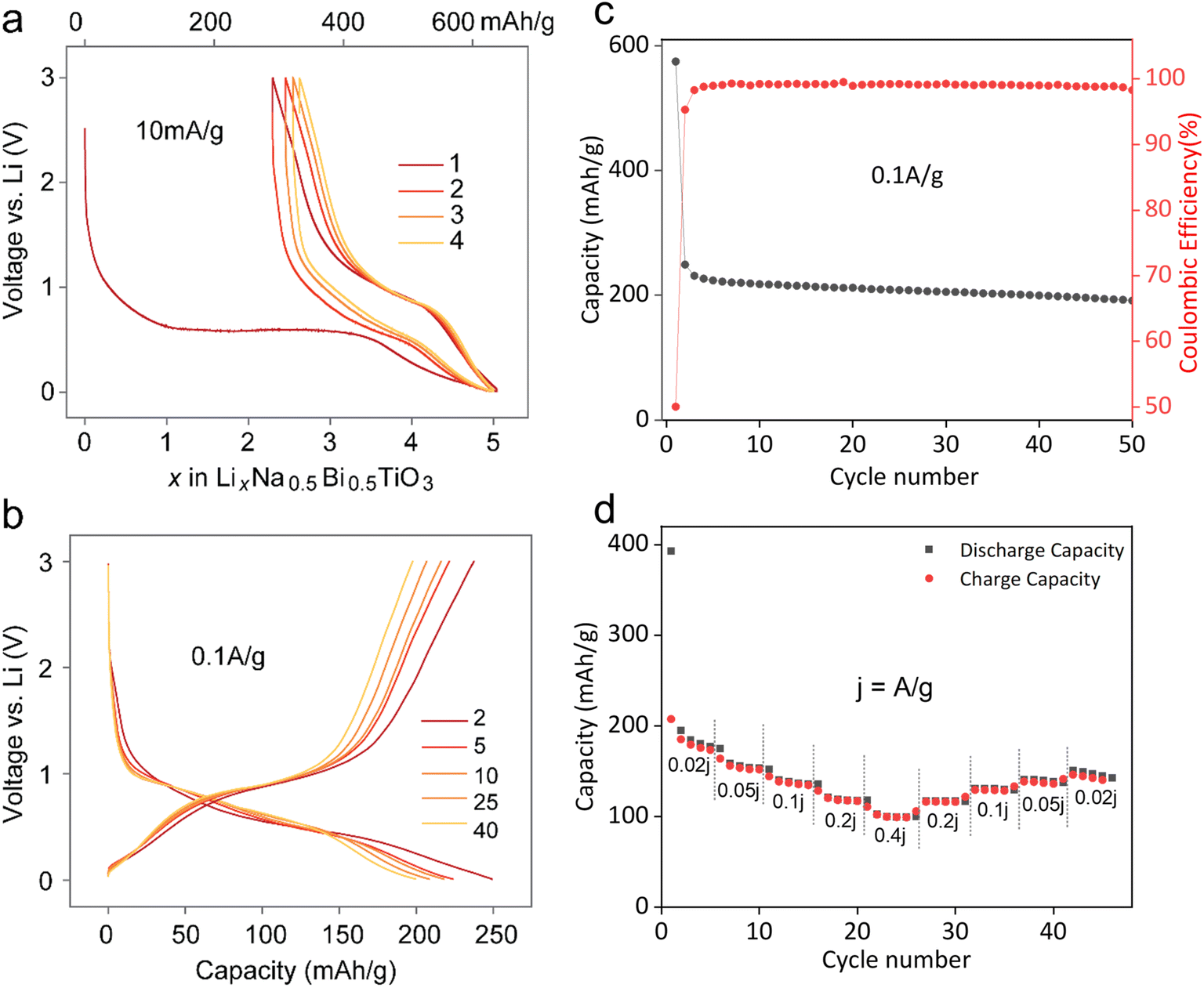 | ||
| Fig. 2 Electrochemical performance of lithium insertion in the NBT perovskite. Galvanostatic voltage profiles at (a) slow 10 mA g−1 and (b) fast 0.1 A g−1 current densities, (c) cycle life, and (d) rate performance of Li-NBT half cells. | ||
3.3 Lithium insertion mechanism
The preliminary investigation of the lithium insertion mechanism in NBT was based on cyclic voltammograms (CVs) and potentiostatic intermittent titration (PITT) profiles. The PITT curves in Fig. 3(a and b) show a biphasic region in the first discharge and solid solution behavior in consecutive charge and the second cycle, suggesting an irreversible conversion reaction during the first discharge. Fig. 3(c and d) show a direct comparison of cyclic voltammograms scanned at 0.01 V s−1 and dQ/dV plots extracted from charge–discharge curves at a low current density of 10 mA g−1, indicating that the same processes are occurring in both cases with a negligible difference. It is well-known from previous reports that the reduction of Bi to LiBi and Li3Bi occurs at around 0.77 and 0.68 V, respectively.21,22 However, only one oxidation peak exists at ∼0.9 V due to the overlapping of two expected peaks. The exact origin of the peak with reducing intensity in Fig. 3c at 0.55 V is unknown and is possibly linked to reversible SEI formation reactions which significantly decreases in the first few cycles.23 With respect to the above results, the conversion–alloying mechanism may be expressed as:| Irreversible conversion: Na0.5Bi0.5TiO3 + 1.5Li+ + 1.5e− → 0.25Na2O + TiO2 + 0.75Li2O + 0.5Bi… | (1) |
| Reversible alloying: Bi + Li+ + e− ↔ LiBi…, | (2) |
| LiBi + 2 Li+ + 2e− ↔ Li3Bi… | (3) |
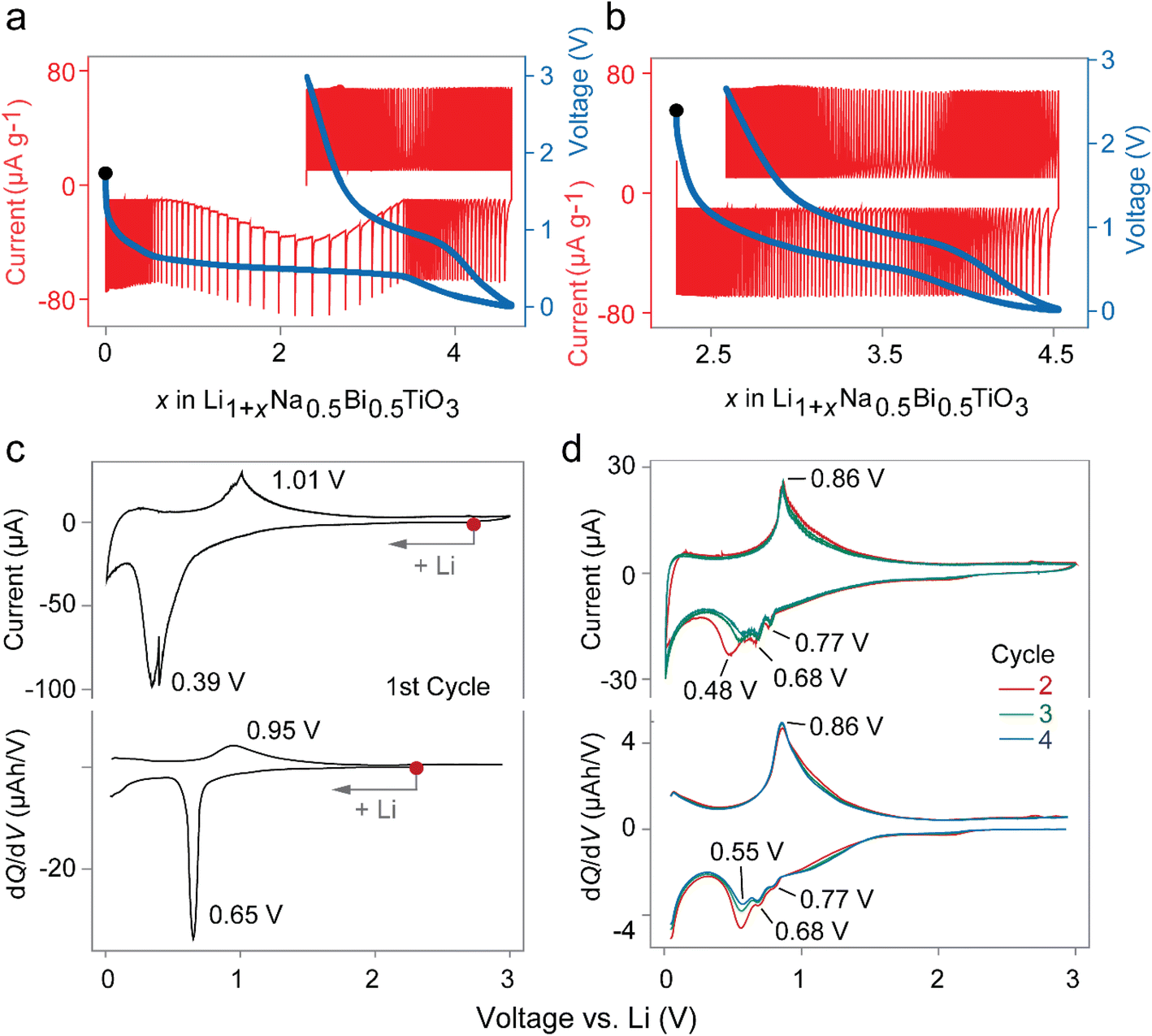 | ||
| Fig. 3 Comparison of Li insertion in perovskite NBT in the first and subsequent cycles. PITT curves for the (a) first and (b) second cycle. CV and dQ/dV curves, at 0.01 V s−1 and 10 mA g−1 respectively, of the (c) first cycle, and (d) second, third and fourth cycles. The voltage window was fixed at 0.01–3 V (vs. Li) for these Li-NBT half-cells. | ||
In order to further understand the exact (de)lithiation mechanism of NBT, a comprehensive set of ex situ analyses involving XRD, TEM, and XPS was performed on sealed NBT samples recovered at various states of first (dis)charge cycle in the 0.01–3.0 V range.
Fig. 4 shows the ex situ XRD patterns taken at different points in the first cycle, with the first discharge and subsequent charge cycle represented by 1D and 1C, respectively. It further confirms the structural breakdown of NBT via a conversion reaction, as indicated by amorphization of the powder XRD pattern of pristine NBT (black) into an almost featureless XRD pattern (blue) in the first discharge 0.6 V plateau. While the presence of LiBi or Li3Bi could not be pinpointed from XRD patterns (green) recorded after full discharge to 0.01 V (1D), the charged electrode (purple) confirmed dealloyed metallic Bi as expected from earlier CV, HAADF-STEM and XPS results and previous reports.24
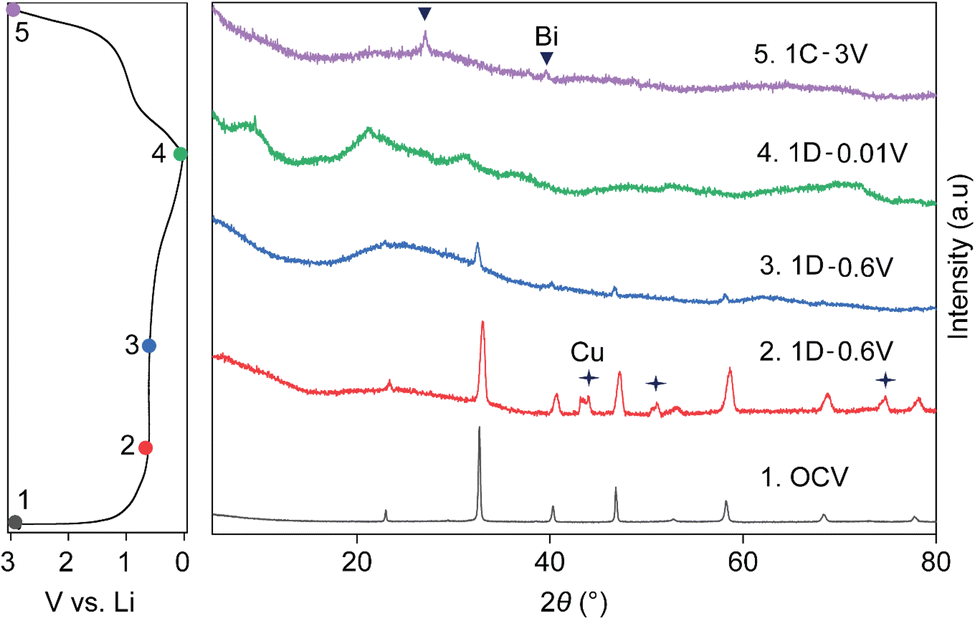 | ||
| Fig. 4 Ex situ XRD analysis of the perovskite NBT anode after lithium (de)insertion, taken at the following points: (1) OCV (black); (2) first discharge to 0.6 V at the start of conversion (red); (3) first discharge to 0.6 V at the end of the conversion step (blue); (4) full first discharge to 0.01 V (green); (5) full charge to 3.0 V (lavender). | ||
HAADF-STEM images indeed show that the surface of the crystals is covered with numerous nanoparticles with the size of 1–10 nm (Fig. 5b and d). The size of nanoparticles is larger for the sample charged up to 3 V (labelled as 1C). Atomic resolution HAADF-STEM imaging of these nanoparticles reveals their crystalline structure (Fig. S2†), which was more precisely identified as metallic Bi by Fourier transformation. Moreover, EDX-STEM elemental maps of the 1C charged sample (as indicated in Fig. 5e and f) clearly show that these Bi-metal nanoparticles decorate the surface and contain no oxygen, while titanium and oxygen are present together (possibly as TiO2) in the bulk of the former Na0.5Bi0.5TiO3 crystal. Furthermore, the charged sample 1C also contains conductive carbon and another electron beam-sensitive material (Fig. S2†). In Fig. S2,† the following three components are depicted: spherical-like particles of conductive carbon in the upper part of the image (the magnified particle is shown in Fig. S2b†); parent Na0.5Bi0.5TiO3 crystal as a dark particle in the lower right part of the image (the magnified surface with numerous nanoparticles is shown in Fig. S2c†); foamy beam sensitive material in direct contact with the parent Na0.5Bi0.5TiO3 crystal in the central lower part of the image in Fig. S2a.† To reveal the nature of the latter component, EELS spectra were recorded (Fig. 5g). According to the reference spectra,25 this edge corresponds to the lithium K-edge, where lithium is present in the form of Li2O. Thus, ex situ TEM analysis of the charged sample could unambiguously certify the presence of the large dealloyed Bi particles dispersed in a matrix of Li2O and TiO2 as “plum-pudding”.
 | ||
Fig. 5
Ex situ TEM and XPS analyses of NBT following lithium (de)insertion. SAED patterns and HAADF-STEM images of first discharged (a and b) and charged (c and d) samples. Bar size is equal to 5 nm−1 and 5 nm for the SAED patterns and HAADF-STEM images, respectively. (d) Fourier transformation of nanoparticles in the charged sample indexed to the R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m structure of metallic Bi. (e and f) Color coded mixed EDS map of the charged sample, clearly showing the presence of bismuth metal and TiO2 matrix that can be picturized as the plum-pudding model. (g) EELS spectrum of the foamy beam sensitive material (Fig. S2†) in the 1C sample near the Li K-edge region. (h) Bi 4f XPS spectra of pristine NBT (bottom) after first discharge (middle) and charge (top). m structure of metallic Bi. (e and f) Color coded mixed EDS map of the charged sample, clearly showing the presence of bismuth metal and TiO2 matrix that can be picturized as the plum-pudding model. (g) EELS spectrum of the foamy beam sensitive material (Fig. S2†) in the 1C sample near the Li K-edge region. (h) Bi 4f XPS spectra of pristine NBT (bottom) after first discharge (middle) and charge (top). | ||
Similar results were obtained from the XPS study as shown in Fig. 5h. The binding energies of Bi 4f doublet peaks in the pristine NBT are centered at 159.7 eV and 164.9 eV. The discharged electrode gives corresponding values of 158.8 eV and 164 eV indicating that Bi is reduced to lower oxidation states due to alloying with Li. Further reduction of binding energies is observed upon complete charge. However, the charged electrode exhibits two doublets corresponding to Bi in metallic and oxidized states with binding energies of 156.9 eV, 162 eV, 159 eV, and 164.4 eV, respectively. Furthermore, Fig. S3a† shows the XPS peaks of C 1s with distinct peaks at binding energies of 284.6 eV, 286.8 eV, and 289.5 eV due to the presence of C–C, C–O, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds. Due to changes in C–O bonds, the 286 eV peak slightly varies for discharged and charged states.24 The XPS peaks of O 1s (Fig. S3b†) at binding energies of about 531 eV and 532 eV in discharge and charge correspond to OC and O–C bonds in the discharged and charged states.26
O bonds. Due to changes in C–O bonds, the 286 eV peak slightly varies for discharged and charged states.24 The XPS peaks of O 1s (Fig. S3b†) at binding energies of about 531 eV and 532 eV in discharge and charge correspond to OC and O–C bonds in the discharged and charged states.26
4. Conclusions
The current work advances on the pathway towards utilizing perovskite frameworks containing an alloying element as a conversion–alloy-type anode material for electrochemical energy storage. More precisely, a lead-free widely used piezoelectric Na0.5Bi0.5TiO3 (NBT) perovskite oxide was successfully prepared using a solid-state route and was assessed as an anode for Li-ion batteries for the first time. This anode material gives a capacity of 200 mA h g−1 and 120 mA h g−1 after 50 cycles at a high current of 50 and 500 mA g−1, respectively. A high working potential of 0.7 V with respect to lithium makes NBT a safer alternative to graphite. Subsequently, its electrochemical redox mechanism was probed using ex situ analytical tools. Similar to most conversion–alloying materials, lithium is reversibly stored in the perovskite NBT host in the form of LixBi alloys dispersed in a Li2O and TiO2 matrix, which is visually identical to a “plum-pudding” phase dispersion. This matrix is formed after an initial irreversible conversion reaction. When cycled in a narrow voltage window, the material exhibits better stability with a capacity retention of 81% even after 200 cycles, despite micron-sized Bi particles. The NBT perovskite can be harnessed as a safer high-rate anode material for Li-ion batteries with further optimization in the form of coating, particle size reduction and electrolytes. Overall, this work paves way for the exploration of numerous other Bi-based perovskites as anode materials for rechargeable batteries.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
S. C.: data curation, formal analysis, investigation, methodology, writing – original draft; S. A.: data curation, investigation, writing – original draft; A. C.: conceptualization, formal analysis, investigation, writing – review & editing; A. G.: TEM work, formal analysis, writing – original draft; A. M. A.: formal analysis, supervision, writing – review & editing; P. B.: conceptualization, funding acquisition, formal analysis, project administration, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the Department of Science and Technology (Govt of India) under the Core Research Grant (CRG/2022/000963). S. C. and S. A. thank Prime Minister Research Fellowships from the Ministry of Human Resource Development (MHRD, Government of India). A. C. acknowledges the Raman Charpak Fellowship-2019 (RCF-IN-00125) by Centre Franco-Indien pour la Promotion de la Recherche Avancee (CEFIPRA). A. G. and A. M. A. are grateful to the Russian Science Foundation for financial support (grant 23-73-30003). A. G. and A. M. A. are also grateful to the Advanced Imaging Core Facility (AICF) at Skoltech for providing access to electron microscopy equipment. P. B. is grateful to Alexander von Humboldt Foundation (Bonn, Germany) for a 2022 Humboldt fellowship for experienced researchers.References
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef PubMed.
- T. Ohzuku, A. Ueda and N. Yamamoto, J. Electrochem. Soc., 1995, 142, 1431–1435 CrossRef.
- T. Ohzuku, Y. Iwakoshi and K. Sawai, J. Electrochem. Soc., 1993, 140, 2490–2498 CrossRef.
- A. N. Dey, J. Electrochem. Soc., 1971, 118, 1547 CrossRef.
- J. O. Besenhard, J. Yang and M. Winter, J. Power Sources, 1997, 68, 87–90 CrossRef.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J. M. Tarascon, Nature, 2000, 407, 496–499 CrossRef PubMed.
- J. Cabana, L. Monconduit, D. Larcher and M. R. Palacín, Adv. Mater., 2010, 22, E170–E192 CrossRef PubMed.
- D. Bresser, S. Passerini and B. Scrosati, Energy Environ. Sci., 2016, 9, 3348–3367 RSC.
- R. A. Huggins, Solid State Ionics, 1998, 113–115, 57–67 CrossRef.
- Y. Idota, T. Kubota, A. Matsufuji, Y. Maekawa and T. Miyasaka, Science, 1997, 276, 1395–1397 CrossRef.
- N. Sharma, K. M. Shaju, G. V. Subba Rao and B. V. R. Chowdari, Electrochem. Commun., 2002, 4, 947–952 CrossRef.
- A. Chaupatnaik and P. Barpanda, Electrochem. Commun., 2021, 127, 107038 CrossRef.
- H. R. Xia, W. T. Sun and L. M. Peng, Chem. Commun., 2015, 51, 13787–13790 RSC.
- S. Atif, A. Chaupatnaik, A. Rao, A. Padhy, S. Chintha, P. Nukala, M. Fichtner and P. Barpanda, Electrochim. Acta, 2024, 502, 144838 CrossRef.
- A. S. Bhalla, R. Guo and R. Roy, Mater. Res. Innovat., 2000, 4, 3–26 CrossRef.
- J. Rodriguez-Carjaval, Phys. B Condens. Matter, 1993, 192, 55–69 CrossRef.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef.
- G. O. Jones and P. A. Thomas, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 168–178 CrossRef PubMed.
- C. F. Sun, J. Hu, P. Wang, X. Y. Cheng, S. B. Lee and Y. Wang, Nano Lett., 2016, 16, 5875–5882 CrossRef PubMed.
- S. D. Beattie, T. Hatchard, A. Bonakdarpour, K. C. Hewitt and J. R. Dahn, J. Electrochem. Soc., 2003, 150, A701 CrossRef.
- O. Crosnier, T. Brousse, X. Devaux, P. Fragnaud and D. M. Schleich, J. Power Sources, 2001, 94, 169–174 CrossRef.
- W. Xianming, T. Nishina and I. Uchida, J. Power Sources, 2002, 104, 90–96 CrossRef.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J. M. Tarascon, Ionics, 2000, 6, 321–330 CrossRef.
- K. K. Bharathi, B. Moorthy, H. K. Dara, L. Durai and D. K. Kim, J. Mater. Sci., 2019, 54, 13236–13246 CrossRef.
- R. Qiao, Y.-D. Chuang, S. Yan and W. Yang, PLoS One, 2012, 7, e49182 CrossRef PubMed.
- T. Sun, Z. Zhang, J. Xiao, C. Chen, F. Xiao, S. Wang and Y. Liu, Sci. Rep., 2013, 3, 2527 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4se00935e |
| This journal is © The Royal Society of Chemistry 2024 |