
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unveiling the reactivity of CO2 with carbanions: a theoretical analysis of the carboxylation step†
Catia
Nicoletti
 *,
Manuel
Orlandi
,
Luca
Dell'Amico
and
Andrea
Sartorel
*
*,
Manuel
Orlandi
,
Luca
Dell'Amico
and
Andrea
Sartorel
*
Department of Chemical Sciences, University of Padova, via Marzolo 1, 35131 Padova, Italy. E-mail: Catia.nicoletti@phd.unipd.it; Andrea.sartorel@unipd.it
First published on 26th September 2024
Abstract
The synthetic insertion of carbon dioxide into organic scaffolds typically requires the reaction of CO2 with a carbanion (carboxylation step), with the latter being generated through chemical, electrochemical, or photochemical routes. Still, little is known about the energetic and structural requirements of this step. In this work, we unveil the reactivity of CO2 with a selected set of 28 carbanions through DFT calculations and provide linear free-energy relationships that correlate the ΔG0 and the ΔG‡ of the carboxylation step. These reveal a Leffler–Hammond parameter α = 0.26 ± 0.02 and an intrinsic barrier ΔG‡0 = 12.7 ± 0.3 kcal mol−1 (ωb97XD/aug-cc-pvtz//ωb97XD/def2tzvp level of theory), indicative of smooth reactivity of carbanions with CO2. This reactivity is further associated with the basicity of the carbanions (expressed as the pKaH of the conjugate acid), in a linear Brønsted plot between calculated ΔG‡ and experimental pKaH (slope β = 0.40 ± 0.04 kcal mol−1). According to the Mayr–Patz equation, calculations allow the extrapolation of electrophilicity values for CO2 in the range from −15.3 to −18.7, in good agreement with a single reported experimental value of −16.3. Concerning the structural changes occurring in the transition state, the major energy penalty comes from the distortion of CO2. These findings can be useful in designing novel reactivity targeting carbon dioxide fixation.
Introduction
Carbon dioxide is a by-product of many anthropogenic activities, and its current increase in the atmosphere is causing severe climate changes.1 On the other hand, CO2 is a cheap, non-toxic and renewable single carbon atom building block that can be exploited for fuel production or for organic synthesis. Strategies to capture and reconvert CO2 into fuels or chemicals are thus important targets for achieving carbon neutrality,2 in line with the United Nations sustainable development goals.3 Current carbon dioxide capturing technologies exploit amine solutions, with the formation of carbamate salts;4,5 CO2 is then released by an energy demanding thermal step involving heating of the solution up to 100–125 °C. Electrochemical methods are considered as possible alternatives to avoid the need for an external high temperature energy source; they can operate through electrochemically induced displacement reactions (such as in the case of electrochemically modulated amine regeneration) or through CO2 binding to electrochemically reduced nucleophilic capturing agents, such as quinones.4,6Concerning carbon dioxide reconversion, some strategies target CO2 reduction to C1 or C2+ products.7–9 For instance, a cobalt phthalocyanine molecular catalyst was exploited to mediate CO2 to CO formation in a zero-gap membrane flow reactor with selectivity >95% at an impressive current density of 150 mA cm−2.10 Examples of reduction of CO2 to C2 products include the use of a 3D dendritic copper–cuprous oxide composite for acetic acid and ethanol production11 and the use of single site metal catalysts. In this field, the use of DFT calculations for a thermodynamic and kinetic analysis of the relevant steps is considered a powerful tool for catalyst design.8,12
Finally, carbon dioxide is a single carbon atom building block to be exploited in organic synthesis.13–18 In this regard, processes involving CO2 are already industrialised for the preparation of linear and cyclic carbonates (from alkynes or epoxides), for urea synthesis,19 and in the Kolbe–Schmitt reaction20,21 for the preparation of salicylic acid and 3-hydroxy-2-naphthoic acid (from sodium phenolate and 2-naphtholate, respectively).
CO2 is however a thermodynamic sink of the carbon cycle and its conversion requires highly reactive chemicals, transition metal catalysis22,23 and/or severe reaction conditions.13,24 An appealing strategy is the formation of new C–C bonds through the polar reaction of CO2 with nucleophilic carbanions generated through chemical,25,26 electrochemical,27,28 or photochemical routes.29–31 This strategy enabled the preparation of products of pharmaceutical interest – although obtained as racemic mixtures – such as amino acids,32,33 the anti-epilepsy and absence seizure drug ethosuximide precursor (synthesised via electrochemical hydrocarboxylation in flow),34 and benzylic carboxylic acids such as fenoprofen, flurbiprofen, ibuprofen and naproxen.35
However, while earlier reports rationalised the conversion of CO2 to formate using metal and organic hydrides,36 its reactivity with carbanion nucleophiles is still poorly understood, due to very limited kinetic data available.37 In a seminal work by Ofial and Mayr, a rate constant for the carboxylation step was reported only for the indenide anion (bimolecular k = 2.09 ± 0.07 105 M−1 s−1, in dimethylsulfoxide at 20 °C).37 Therefore, the rationalisation of the thermodynamic, kinetic and structural requirements that govern the reactivity of carbanions with CO2 is an important target, from the perspective of designing novel carboxylation processes. In particular, the prediction of free energy barriers and reaction rates on the basis of physical parameters of carbanions would be a powerful tool for an a priori evaluation of the envisaged reactivity.
In this work, we sought to unveil the reactivity of CO2 with carbanions by exploiting DFT calculations. We thus considered a selected set of 28 organic carbanions (Ri−) and calculated both the standard free energy (ΔG0) and the activation free energy (ΔG‡) of the carboxylation step (eqn (1) and Scheme 1). Products and transition states will be denoted as Ri-COO− and Ri-TS, respectively.
| Ri− + CO2 ⇌ Ri-COO− ΔG0, ΔG‡ | (1) |
 | ||
| Scheme 1 Schematic representation of the carboxylation step of a carbanion Ri− to Ri-COO−, and representation of the transition state Ri-TS. Linear free-energy relationship and distortion–interaction analysis explored in this work. | ||
Computational methods
Calculations were performed with Gaussian16 software.41 Most carbanions included in this study possess few rotatable bonds. In the cases of anions with possible multiple conformations that may impact the target C-site reactivity (i.e. anions R23−, R24−, R25−, and R272−), an initial conformational search was performed with a semiempirical pm3 method42–44 including a solvation model for acetonitrile to identify the most stable conformers that were then optimized with DFT methods (see ESI, Fig. S1–S4†). In the manuscript and in the correlations presented, we report ΔG‡ and ΔG0 data for the most stable conformer of these anions. We have corroborated this assumption by performing calculations and analysing the ΔG‡ and ΔG0 data also for the second most stable conformer of R23−, R24− and R25−. From the results reported in Table S1 in the ESI,† the variations of the calculated activation and standard free energy of carboxylation resulting from changes in the nature of the initial conformer are in a range of 0.16–0.65 and 0.73–2.14 kcal mol−1, respectively, minimally impacting the correlations presented in this work.Structures were optimized with DFT methods including a continuum solvation model for acetonitrile (or dimethylsulfoxide) using the integral equation formalism variant (IEFPCM).45–47 Analysis of the frequencies was performed to assess the nature of the stationary points; in particular, for the transition states, the presence of a single imaginary frequency was confirmed, and the reliability of the transition states was corroborated by intrinsic reaction coordinate (IRC) analysis. For all calculations, a pruned (99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 590) integration grid was used. For all the species, a correction of +1.90 kcal mol−1 was applied to the computed free energy values to convert the standard state from 1 atmosphere to a 1 M solution.37,48 When specified, corrections for entropy were applied according to the Truhlar model,49 using a frequency cut-off value of 100.0 wavenumber.
590) integration grid was used. For all the species, a correction of +1.90 kcal mol−1 was applied to the computed free energy values to convert the standard state from 1 atmosphere to a 1 M solution.37,48 When specified, corrections for entropy were applied according to the Truhlar model,49 using a frequency cut-off value of 100.0 wavenumber.
The following levels of theory were employed:
ωb97XD/def2tzvp//ωb97XD/def2tzvp: geometries optimised using the ωb97XD50 hybrid functional taking advantage of Grimme's dispersion model with Ahlrichs' triple-zeta basis set def2tzvp;51,52
ωb97XD/aug-cc-pvtz//ωb97XD/def2tzvp: geometries optimized at the ωb97XD/def2tzvp level, followed by a single point energy calculation performed using the same functional and the aug-cc-pvtz basis set53,54 including diffuse functions;
b2plypd/aug-cc-pvtz//ωb97XD/def2tzvp: geometries optimized at the ωb97XD/def2tzvp level, followed by a single point energy calculation performed using the double hybrid b2plypd functional,55 with the aug-cc-pvtz basis set;
b3lyp/6–311g(d,p)//b3lyp/6–311g(d,p): geometries optimised using the b3lyp functional (without dispersion correction) with a Pople56 6–311g(d,p) basis set (not including diffuse functions).
Results and discussion
Evaluation of the level of theory
The availability of an experimental ΔG‡ value of 10.0 kcal mol−1 for the carboxylation of the indenide anion R2− from a bimolecular rate constant of 2.09 ± 0.07 105 M−1 s−1 (in dimethylsulfoxide at 20 °C)37 allowed the evaluation and the selection of the suitable level of theory among the four levels discussed in the “Computational methods” section. The four levels of theory (continuum model for dimethylsulfoxide solvent) provided calculated ΔG‡ for the carboxylation of R2− in the range 11.9–15.2 kcal mol−1, in good agreement with the experimental value (Table S2 in the ESI†). The best match was found at the ωb97XD/aug-cc-pvtz//ωb97XD/def2tzvp level (calculated ΔG‡ = 11.9 kcal mol−1), which was chosen as the level for the calculations discussed in this work. Corrections for entropy according to the Truhlar model49 lead to an increase of +0.7 kcal mol−1 in the calculated activation free energies and thus were not further applied.Calculations of ΔG0 and ΔG‡ for the carboxylation step
The carbanions Ri− were selected from our previous investigation,57 and the corresponding carboxylated species Ri-COO− are represented in Fig. 1. Since several reports exploit acetonitrile as the solvent for carboxylation reactions, this was chosen as the medium for the calculations presented in this work. Fig. 2A reports the plot of ΔG‡vs. ΔG0 values for the carboxylation step (ωb97XD/aug-cc-pvtz//ωb97XD/def2tzvp level of theory, including a continuum model for acetonitrile solvent; data including the calculations at the other levels are reported in Tables S3–S7 and Fig. S5 in the ESI†). The geometries of the transition states R4-TS and R17-TS are also shown in Fig. 2A as representative cases.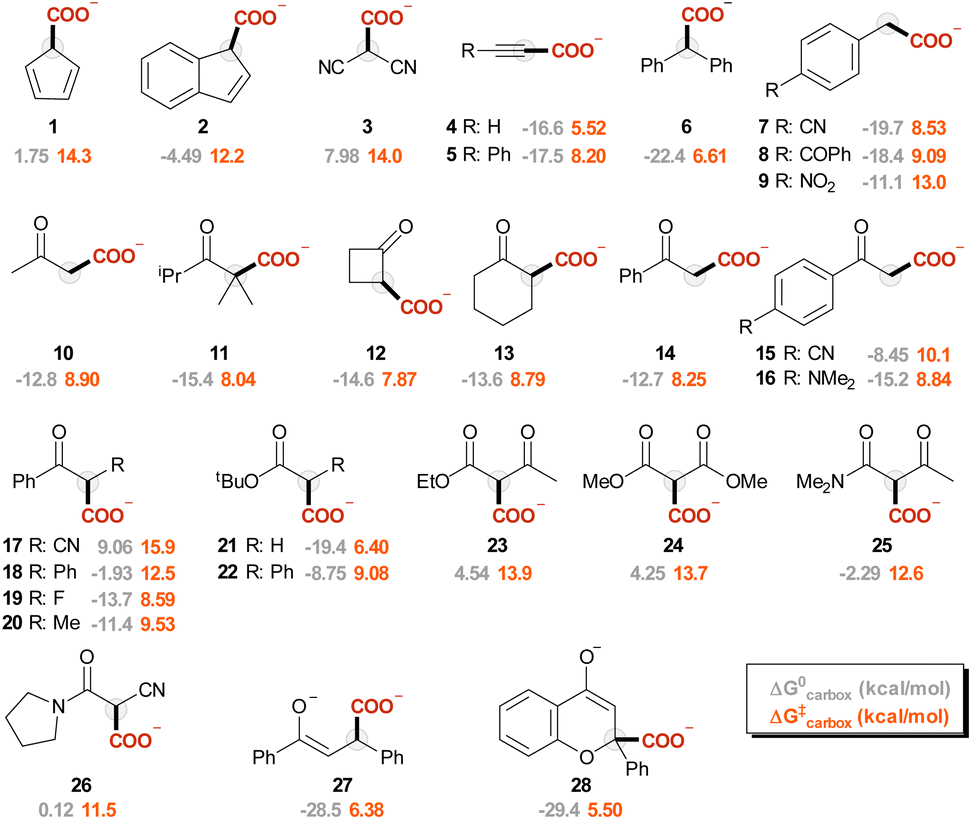 | ||
| Fig. 1 Representation of Ri-COO− considered in this work (i is indicated below each structure), together with ΔG0 and ΔG‡ values for the carboxylation step at the ωb97XD/aug-cc-pvtz//ωb97XD/def2tzvp level of theory. | ||
 | ||
| Fig. 2 (A) Plot of ΔG‡vs. ΔG0 values for the carboxylation of carbanions Ri− (ωb97XD/aug-cc-pvtz//ωb97XD/def2tzvp level of theory, including a continuum model for acetonitrile solvent). The shaded ranges represent the 95% confidence interval of the correlations. Geometries of R4-TS and R17-TS (ωb97xd/def2tzvp level) are shown as representative cases of transition states with low and high ΔG‡. (B) Plot of ΔG‡ (ωb97XD/aug-cc-pvtz//ωb97XD/def2tzvp) vs. experimental pKaH of carbanions Ri−. | ||
Clearly, considering that charged species are involved in eqn (1), solvation impacts the ΔG0 and ΔG‡ values. While this was addressed by the inclusion of an implicit solvation model, any possible deviation from an ideal explicit solvation could be neglected assuming that such an effect would be similar through the anion series, thus preserving the sought-after linear correlations. Also, the impact of cations possibly present under real reaction conditions could impact the structure and the reactivity of carbanions; this is particularly relevant for the case of enolate like anions in the presence of small alkali cations.58
The data plot of ΔG‡vs. ΔG0 values shows a good linear trend according to eqn (2) (R2 = 0.88).
| ΔG‡ = α × ΔG0 + ΔG‡0 | (2) |
| α = 0.26 ± 0.02; ΔG‡0 = 12.7 ± 0.3 kcal mol−1 |
The slope α of 0.26 ± 0.02 corresponds to the Leffler–Hammond parameter α = δΔG‡/δΔG0, describing the fraction of the change in the Gibbs energy of the reaction reflected in the change of the Gibbs energy of activation.39 It is worth noting that α has long been exploited as an indication of the position of the transition state along the reaction coordinate (with α close to 0 indicating an early transition state and α close to 1 indicating a late transition state), until Bordwell's observation of an α value of 1.31 for the deprotonation of substituted 1-nitro-1-phenylethanes (nitroalkane anomaly)59 evidenced that the former interpretation was wrong and that α cannot be a measure for the position of the transition state.
The intercept of the fitting in Fig. 2 ΔG‡0 = 12.7 ± 0.3 kcal mol−1 provides the intrinsic barrier, i.e. the activation free energy for an isoergonic reaction (with ΔG0 = 0).‡ Importantly, quadratic fitting of the data in Fig. 2 according to the Marcus model for group transfer reactions (eqn (3)) leads to consistent values of ΔG‡0 of 14.2 ± 0.4 kcal mol−1, see ESI Fig. S6.† According to the Marcus model for group transfer in bimolecular reactions, the ΔG‡0 is associated with the reorganization energy λ (λ = 4 × ΔG‡0), with λ being estimated to be 50.8 ± 1.2 kcal mol−1.
| ΔG‡ = ΔG‡0 + 0.5 × ΔG0 + {(ΔG0)2/(16 × ΔG‡0)} | (3) |
Comparable α and ΔG‡0 values were found with the other levels of theory employed (Fig. S5 in the ESI†). Importantly, α and ΔG‡0 parameters determined from the fittings are in line with those reported for the decarboxylation reaction of organic carboxylates in water (i.e. the microscopically reverse reaction of eqn (1));60 the value of α reported in this case was 0.76/0.78 (experimental/computed; note that the sum of α for the forward and for the backward step must be 1.00,39 while the intrinsic barrier was 15.5/16.8 kcal mol−1 (experimental/computed).60 The α and ΔG‡0 parameters extrapolated from the data in Fig. 2A indicate smooth reactivity of carbanions with CO2,61 characterised by low energy barriers and by a slight dependence on ΔG0; these identify CO2 as a powerful electrophile, comparable to carbonyl compounds37,62 and support the recent observation of the exchange of the COO group from carboxylate salts, occurring under an atmosphere of CO2 at room temperature and in the absence of catalysts.63,64 As a comparison, in nucleophilic substitutions α values between 0.48 and 0.73 were found for the reaction of several types of nucleophiles with benzhydrylium ions,39 while the intrinsic barriers ΔG‡0 reach up to 45 kcal mol−1 for carbanion nucleophiles.65,66 An intrinsic barrier of 14.1 kcal mol−1 was reported for the addition of enolate ions to the carbonyl group of benzaldehyde.67 Lower intrinsic barriers have been observed for proton and hydride transfer (1–10 kcal mol−1).68–70
Four further points are worth mentioning.
(i) We previously reported a linear correlation between variations in ΔG0 and the change of the basicity of the carbanion Ri− expressed as the pKaH of the conjugate acid (eqn (4));57 combination of eqn (2) and (4) allows the direct correlation of variations in ΔG‡ with pKaH, in a Brønsted type plot (eqn (5)). Indeed, a good linear correlation of calculated ΔG‡ with experimental pKaH71 is observed in Fig. 2B (R2 = 0.83), providing a powerful prediction tool of ΔG‡.
| δ(ΔG0) = −1.30 kcal mol−1 × δ(pKaH) | (4) |
| ΔG‡ = −β × pKaH + const | (5) |
(ii) The calculated ΔG‡ values can be exploited to contextualise the carboxylation step on a more general nucleophilic/electrophilic scale, exploiting the Mayr–Patz equation (eqn (6)),72
| logk (20 °C) = sN(N + E) | (6) |
k (20 °C) from ΔG‡ through the Eyring equation and exploiting the reported SN and N parameters for carbanions R2−, R3−, R17−, R23− and R24−,73 it is possible to extrapolate electrophilicity values for CO2 in the range from −15.3 to −18.7, while a value of −16.3 was reported according to a single experimental kinetic data with the indenide anion (R2−)37 (Table S8 in the ESI†). Many reports have discussed the theoretical prediction of nucleophilicity/electrophilicity.48,74–76
(iii) The variations of ΔG0 and ΔG‡ values for the carboxylation step represented in Fig. 2 are mainly related to enthalpic contributions, while the TΔS0 and TΔS‡ terms are found to be almost constant in the carbanion series (Tables S4–S7 and Fig. S7 in the ESI†), with average values of −10.6 ± 0.8 and −9.95 ± 0.8 kcal mol−1, respectively (the error is given as the standard deviation).
(iv) In the reactivity investigated, the formation of a reactant complex between the anion and carbon dioxide could in principle occur,63,77 in particular for highly basic carbanions. Based on our calculations and IRC analysis, we do not have evidence for the formation of such a reactant complex; the calculated energy barriers refer then to the isolated reactants.
Distortion interaction analysis
We rationalised the molecular factors that impact the energetic barrier of the carboxylation step by employing the distortion/interaction model (also known as the activation/strain model) for bimolecular reactions, developed independently by the Bickelhaupt and Houk groups.40 Accordingly, the potential energy ΔE along the reaction coordinate is given by the sum of the energy due to the distortion of the reactants (ΔEdistortion > 0) and the energy given by the interaction of the two reactants (ΔEinteraction < 0), eqn (7).| ΔE = ΔEdistortion + ΔEinteraction | (7) |
Fig. 3A reports the ΔE versus the C(Ri−)⋯C(CO2) distance as the reaction coordinate for the carboxylation step of R3−, R4−, R17−, R21− and R282− as representative cases, spanning the entire range of ΔG‡ explored. As can be visually perceived, the profiles show a switch from a late to an early transition state along the series R17− ∼ R3− → R4−→ R21− → R282−, consistent with the progressive decrease of activation energy. When the ΔE is dissected into the ΔEdistortion and ΔEinteraction contributions (Fig. 3B and Table S9 in the ESI†), the profiles suggest that the carboxylation step is both distortion- and interaction-controlled,78,79 with an evident contribution from the interaction of the carbanions with CO2.
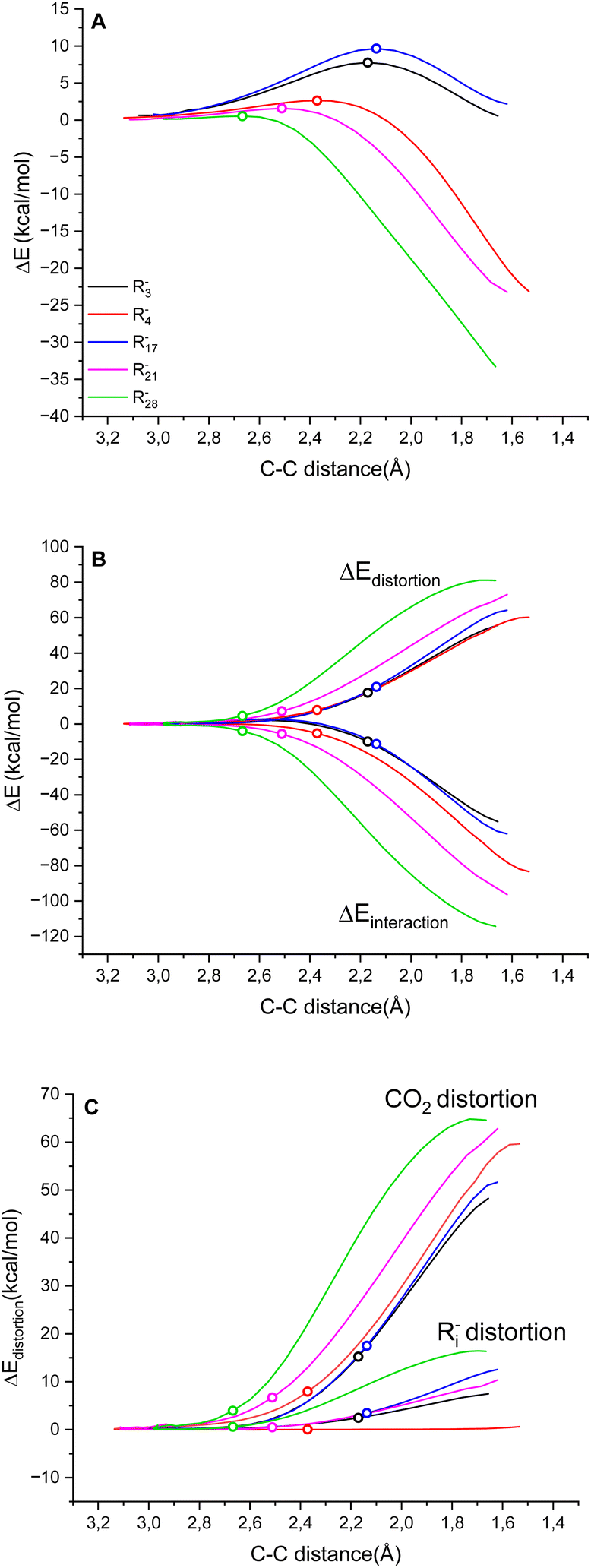 | ||
| Fig. 3 (A) Plot of ΔE vs. the C(Ri−)⋯C(CO2) distance as the reaction coordinate for R3−, R4−, R17−, R21− and R282− as representative cases. (B) Dissection of ΔE into ΔEdistortion and ΔEinteraction. (C) Dissection of ΔEdistortion into ΔE‡distortion (Ri−) and ΔE‡distortion (CO2). In all graphs, the empty dots indicate the transition states. | ||
According to energy decomposition analysis, with some software the ΔEinteraction could be dissected into separate contributions from electrostatic interaction, Pauli repulsion, orbital interaction and dispersion interaction.79 In the present case, the previous correlation between the activity and basicity of carbanions (Fig. 2B) identifies pKaH as the physical parameter impacting the interaction. This is also supported by a smooth correlation between nucleophilicity and proton affinity among nucleophiles belonging to the same class.75
Concerning the distortion, the ΔEdistortion can be decomposed into the contributions of the carbanion Ri− and CO2 reactants (eqn (8)); the ΔEdistortion for each reactant along the reaction coordinate is given by the single point energy difference between the geometry of the reactant at each point and the geometry of the isolated reactant.40,78,79
| ΔEdistortion = ΔEdistortion (Ri−) + ΔEdistortion (CO2) | (8) |
Splitting the ΔEdistortion along the reaction coordinate according to eqn (8) shows that the major energy penalty arises from CO2 for the five cases investigated, Fig. 3C. Consistently, by expanding the analysis by evaluating the ΔEdistortion (Ri−) and ΔEdistortion (CO2) contributions with the transition state geometries of all Ri− species, it is observed that the ΔEdistortion of CO2 exceeds the ΔEdistortion of carbanions by ca. 5 times (Fig. S8 in the ESI;† note that for rigid “sp” carbanions such as R4− and R5− the ΔEdistortion (Ri−) is almost negligible).
From a structural point of view, the distortion of carbon dioxide in the transition states is obviously described by the bending of CO2 and by the contraction of the O–C–O angle, while the carbanion nucleophile approaches and the C(Ri−)⋯C(CO2) distance decreases. Calculations show indeed a linear trend in the carbanion series between the ΔG‡ of carboxylation and the O–C–O angle (see Fig. S9 in the ESI†), predicting that one kcal mol−1 of penalty in the ΔG‡ is associated with a decrease of 1.41 ± 0.09° of the O–C–O angle in the transition state.
Fig. 4 shows the ΔG‡, the O–C–O angle and the C⋯C distance in a single graph, demonstrating that the higher the free energy of activation, the higher the required distortion of CO2 in a “late” transition state, with a small O–C–O angle and a short C(Ri−)⋯C(CO2) distance.
 | ||
| Fig. 4 Plot of the ΔG‡ for the carboxylation of the carbanions versus the O–C–O angle and the C(Ri−)⋯C(CO2) distance in the transition states. | ||
Conclusions and perspectives
In conclusion, we have reported energy and structural correlations for the carboxylation step of carbanions with CO2. These allow linking the basicity of the carbanion (pKaH), the standard and activation free energies of the carboxylation step (ΔG0 and ΔG‡) and the distortion of CO2 in the transition state (O–C–O angle and C(Ri−)⋯C(CO2) distance), deriving reactivity descriptors such as the Leffler–Hammond coefficient, the intrinsic barrier and the electrophilicity of CO2.These tools are instrumental for the design of experimental chemical, electrochemical or photochemical fixation of CO2 into organic compounds. The a priori evaluation of the reaction barrier (and consequently the rate constant) between a carbanion and CO2 should be pivotal in driving the envisaged carboxylation reactivity with respect to undesired competitive pathways. In particular, these may include irreversible protonation of the carbanion or its reactivity with other electrophiles (as documented in the case of electrochemical carboxylation of α,β-unsaturated ketones57). The regioselectivity of the carboxylation step could also be a challenge and should be considered when involving extended organic scaffolds, as in the case of electrochemical carboxylation of dienes80 and naphthalene derivatives.81 Thus, we hope to provide a guide for future studies on carbon dioxide reconversion.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
All authors conceptualised the research and contributed to the discussion of the results. C. N., M. O. and A. S. performed the calculations. C. N. and A. S. co-wrote the manuscript. L. D. A. and A. S. were responsible for funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are indebted to Prof. Herbert Mayr (Department Chemie Ludwig-Maximilians-Universität München) for very constructive and useful discussions. Financial support from the Italian Ministero dell’Università e della Ricerca (Grants “PROMETEO” 2022KPK8WM and 2020927WY3) and from the European Union – Next Generation EU – Bando PRIN 2022 – M4.C2.1.1 (Grants “PHOTOCORE” P2022ZSPWF and P2022WLAY7) is greatly acknowledged.Notes and references
- (a) D. I. A. McKay, A. Staal, J. F. Abrams, R. Winkelmann, B. Sakschewski, S. Loriani, I. Fetzer, S. E. Cornell, J. Rockström and T. M. Lenton, Science, 2022, 377, eabn7950 CrossRef CAS; (b) S. I. Zandalinas, F. B. Fritschi and R. Mittler, Trends Plant Sci., 2021, 26, 588–599 CrossRef CAS PubMed.
- (a) IEA International Energy Agency, Carbon Capture, Utilisation and Storage, https://www.iea.org/energy-system/carbon-capture-utilisation-and-storage; (b) K. Kohse-Höinghaus, Chem. Rev., 2023, 123, 5139–5219 CrossRef; (c) P. Friedlingstein and M. O. Sullivan, et al. , Earth Syst. Sci. Data, 2023, 15, 5301–5369 CrossRef; (d) L. Chen, G. Msigwa, M. Yang, A. I. Osman, S. Fawzy, D. W. Rooney and P.-S. Yap, Environ. Chem. Lett., 2022, 20, 2277–2310 CrossRef CAS PubMed.
- United Nations 17, Sustainable Development Goals, https://www.un.org/sustainabledevelopment/.
- (a) T. L. Biel-Nielsen, T. A. Hatton, S. N. B. Villadsen, J. S. Jakobsen, J. L. Bonde, A. M. Spormann and P. L. Fosbøl, ChemSusChem, 2023, e202202345 CrossRef CAS; (b) R. J. Detz, C. J. Ferchaud, A. J. Kalkman, J. Kemper, C. Sanchez-Martinez, M. Saric and M. V. Shinde, Sustainable Energy Fuels, 2023, 7, 5445–5472 RSC.
- K. M. Diederichsen, R. Sharifian, J. S. Kang, Y. Liu, S. Kim, B. M. Gallant, D. Vermaas and T. A. Hatton, Nat. Rev. Methods Primers, 2022, 2, 68 CrossRef CAS.
- A. Hasanzadeh, S. Ghazanfari, M. Janbazvatan, H. Rashidpour, A. Chitsaz and M. Khalilian, J. CO2 Util., 2023, 67, 102306 CrossRef CAS.
- J. Albero, Y. Peng and H. García, ACS Catal., 2020, 10, 5734–5749 CrossRef CAS.
- B. Chang, H. Pang, F. Raziq, S. Wang, K.-W. Huang, J. Ye and H. Zhang, Energy Environ. Sci., 2023, 16, 4714–4758 RSC.
- S. Fang, M. Rahaman, J. Bharti, E. Reisner, M. Robert, G. A. Ozin and Y. H. Hu, Nat. Rev. Methods Primers, 2023, 3, 61 CrossRef CAS.
- S. Ren, D. Joulié, D. Salvatore, K. Torbensen, M. Wang, M. Robert and C. P. Berlinguette, Science, 2019, 365, 367–369 CrossRef CAS PubMed.
- Q. Zhu, X. Sun, D. Yang, J. Ma, X. Kang, L. Zheng, J. Zhang, Z. Wu and B. Han, Nat. Commun., 2019, 10, 3851 CrossRef PubMed.
- M. Han, X. Fu, A. Cao, C. Guo, W. Chu and J. Xiao, Adv. Theory Simul., 2019, 2, 1800200 CrossRef.
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef.
- Q. W. Song, R. Ma, P. Liu, K. Zhang and L. N. He, Green Chem., 2023, 25, 6538–6560 RSC.
- S. Pimparkar, A. K. Dalvi, A. Koodan, S. Maiti, S. A. Al-Thabaiti, M. Mokhtar, A. Dutta, Y. R. Lee and D. Maiti, Green Chem., 2021, 23, 9283–9317 RSC.
- S. Bierbaumer, M. Nattermann, L. Schulz, R. Zschoche, T. J. Erb, C. K. Winkler, M. Tinzl and S. M. Glueck, Chem. Rev., 2023, 123, 5702–5754 CrossRef CAS PubMed.
- M. M. F. Hasan, L. M. Rossi, D. P. Debecker, K. C. Leonard, Z. Li, B. C. E. Makhubela, C. Zhao and A. Kleij, ACS Sustainable Chem. Eng., 2021, 9, 12427–12430 CrossRef CAS.
- P. K. Sahoo, Y. Zhang and S. Das, ACS Catal., 2021, 11, 3414–3442 CrossRef CAS.
- J. Ding, R. Ye, Y. Fu, Y. He, Y. Wu, Y. Zhang, Q. Zhong, H. H. Kung and M. Fan, Nat. Commun., 2023, 14, 4586 CrossRef CAS PubMed.
- H. Kolbe, Adv. Cycloaddit., 1860, 113, 125–127 Search PubMed.
- R. Schmitt, J. Prakt. Chem., 1885, 2, 397–410 CrossRef.
- A. Tortajada, F. Juliá-Hernández, M. Börjesson, T. Moragas and R. Martin, Angew. Chem., Int. Ed., 2018, 57, 15948–15982 CrossRef CAS.
- J. Davies, J. R. Lyonnet, B. Carvalho, B. Sahoo, C. S. Day, F. Juliá-Hernández, Y. Duan, N. Álvaro Velasco-Rubio, M. Obst, P. O. Norrby, K. H. Hopmann and R. Martin, J. Am. Chem. Soc., 2024, 146, 1753–1759 CrossRef CAS.
- C. Qiao, W. Shi, A. Brandolese, J. Benet-Buchholz, E. C. Escudero-Adán and A. W. Kleij, Angew. Chem., Int. Ed., 2022, 61, e202205053 CrossRef CAS.
- Y. Yan, J. Hao, F. Xie, F. Han, L. Jing and P. Han, J. Org. Chem., 2023, 88, 14640–14648 CrossRef CAS.
- S. Zheng, T. Zhang and H. Maekawa, J. Org. Chem., 2022, 87, 7342–7349 CrossRef CAS PubMed.
- J. Wang, Z. F. Wei, Y. X. Luo, C. H. Lu and R. J. Song, SynOpen, 2024, 8, 116–124 CrossRef CAS.
- H. Senboku, Chem. Rec., 2021, 21, 2354–2374 CrossRef CAS PubMed.
- P. Franceschi, E. Rossin, G. Goti, A. Scopano, A. Vega-Penaloza, M. Natali, D. Singh, A. Sartorel and L. Dell'Amico, J. Org. Chem., 2023, 88, 6454–6464 CrossRef CAS.
- M. Schmalzbauer, T. D. Svejstrup, F. Fricke, P. Brandt, M. J. Johansson, G. Bergonzini and B. König, Chem, 2020, 6, 2658–2672 CAS.
- Z. Zhang, J. H. Ye, T. Ju, L. L. Liao, H. Huang, Y. Y. Gui, W. J. Zhou and D. G. Yu, ACS Catal., 2020, 10, 10871–10885 CrossRef CAS.
- H. Seo, M. H. Katcher and T. F. Jamison, Nat. Chem., 2017, 9, 453–456 CrossRef CAS PubMed.
- Y. Naito, Y. Nakamura, N. Shida, H. Senboku, K. Tanaka and M. Atobe, J. Org. Chem., 2021, 86, 15953–15960 CrossRef CAS.
- A. Sheta, A. Alkayal, M. Mashaly, S. Said, S. Elmorsy, A. V Malkov and B. R. Buckley, Angew. Chem., Int. Ed., 2021, 60, 21832–21837 CrossRef CAS.
- Q. Y. Meng, T. E. Schirmer, A. L. Berger, K. Donabauer and B. König, J. Am. Chem. Soc., 2019, 141, 11393–11397 CrossRef CAS.
- K. M. Waldie, A. L. Ostericher, M. H. Reineke, A. F. Sasayama and C. P. Kubiak, ACS Catal., 2018, 8, 1313–1324 CrossRef CAS.
- Z. Li, R. J. Mayer, A. R. Ofial and H. Mayr, J. Am. Chem. Soc., 2020, 142, 8383–8402 CrossRef CAS.
- F. G. Bordwell and D. L. Hughes, J. Org. Chem., 1980, 45, 3314–3320 CrossRef CAS.
- H. Mayr and A. R. Ofial, Acc. Chem. Res., 2016, 49, 952–965 CrossRef CAS PubMed.
- F. M. Bickelhaupt and K. N. Houk, Angew. Chem., Int. Ed., 2017, 56, 10070–10086 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- J. J. P. Stewart, J. Comput. Chem., 1989, 10, 209–220 CrossRef CAS.
- J. J. P. Stewart, J. Comput. Chem., 1989, 10, 221–264 CrossRef CAS.
- E. Anders, R. Koch and P. Freunscht, J. Comput. Chem., 1993, 14, 1301–1312 CrossRef CAS.
- S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef.
- S. Miertuš and J. Tomasi, Chem. Phys., 1982, 65, 239–245 CrossRef.
- J. L. Pascual-Ahuir, E. Silla and I. Tuñón, J. Comput. Chem., 1994, 15, 1127–1138 CrossRef CAS.
- J. N. Harvey, F. Himo, F. Maseras and L. Perrin, ACS Catal., 2019, 9, 6803–6813 CrossRef CAS.
- R. F. Ribeiro, A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2011, 115, 14556–14562 CrossRef CAS PubMed.
- J. Da Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- R. A. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- D. E. Woon and T. H. Dunning, J. Chem. Phys., 1993, 98, 1358–1371 CrossRef CAS.
- T. Schwabe and S. Grimme, Phys. Chem. Chem. Phys., 2007, 9, 3397–3406 RSC.
- J. A. Pople, P. M. W. Gill and B. G. Johnson, Chem. Phys. Lett., 1992, 199, 557–560 CrossRef CAS.
- P. Franceschi, C. Nicoletti, R. Bonetto, M. Bonchio, M. Natali, L. Dell'Amico and A. Sartorel, Front. Chem., 2021, 9, 783993 CrossRef CAS.
- O. Larrañaga, A. De Cõzar, F. M. Bickelhaupt, R. Zangi and F. P. Cossío, Chem.–Eur. J., 2013, 19, 13761–13773 CrossRef.
- F. Bordwell, W. J. J. Boyle, J. A. Hautala and K. C. Yee, J. Am. Chem. Soc., 1969, 91, 4002–4003 CrossRef CAS.
- S. Zhou, Y. Wang and J. Gao, JACS Au, 2021, 1, 233–244 CrossRef CAS PubMed.
- Y. Fu, X. Suo, Z. Yang, S. Dai and D. E. Jiang, J. Phys. Chem. B, 2022, 126, 6979–6984 CrossRef CAS PubMed.
- F. M. Bickelhaupt and I. Fernández, Chem. Sci., 2024, 15, 3980–3987 RSC.
- D. Kong, P. J. Moon, E. K. J. Lui, O. Bsharat and R. J. Lundgren, Science, 2020, 369, 557–561 CrossRef CAS.
- G. Destro, K. Horkka, O. Loreau, D. A. Buisson, L. Kingston, A. Del Vecchio, M. Schou, C. S. Elmore, F. Taran, T. Cantat and D. Audisio, Angew. Chem., Int. Ed., 2020, 59, 13490–13495 CrossRef CAS PubMed.
- H. Mayr, M. Breugst and A. R. Ofial, Angew. Chem., Int. Ed., 2011, 50, 6470–6505 CrossRef CAS.
- S. Hoz, H. Basch, J. L. Wolk, T. Hoz and E. Rozental, J. Am. Chem. Soc., 1999, 121, 7724–7725 CrossRef CAS.
- J. P. Richard, T. L. Amyes and M. M. Toteva, Acc. Chem. Res., 2001, 34, 981–988 CrossRef CAS PubMed.
- A. Kresge, Acc. Chem. Res., 1975, 8, 354–360 CrossRef CAS.
- C. R. Reinhardt, T. C. Jaglinski, A. M. Kastenschmidt, E. H. Song, A. K. Gross, A. J. Krause, J. M. Gollmar, K. J. Meise, Z. S. Stenerson, T. J. Weibel, A. Dison, M. R. Finnegan, D. S. Griesi, M. D. Heltne, T. G. Hughes, C. D. Hunt, K. A. Jansen, A. H. Xiong, S. Hati and S. Bhattacharyya, J. Mol. Model., 2016, 22, 199 CrossRef.
- C. F. Bernasconi and P. J. Wenzel, J. Am. Chem. Soc., 2001, 123, 7146–7153 CrossRef CAS.
- Bordwell pKa table, collected by prof. Hans Reich (UW-Madison) https://organicchemistrydata.org/hansreich/resources/pka/last update April 2024.
- H. Mayr and M. Patz, Angew. Chem., Int. Ed., 1994, 33, 938–957 CrossRef.
- H. Mayr and A. R. Ofial, Mayr's Database of Reactivity Parameters, https://www.cup.lmu.de/oc/mayr/reaktionsdatenbank2/.
- C. Wang, Y. Fu, Q. X. Guo and L. Liu, Chem.–Eur. J., 2010, 16, 2586–2598 CrossRef CAS PubMed.
- M. Orlandi, M. Escudero-Casao and G. Licini, J. Org. Chem., 2021, 86, 3555–3564 CrossRef CAS PubMed.
- M. Vahl and J. Proppe, Phys. Chem. Chem. Phys., 2023, 25, 2717–2728 RSC.
- M. A. Zambri and R. Kluger, J. Am. Chem. Soc., 2024, 146, 1403–1409 CrossRef CAS.
- I. Fernández and F. M. Bickelhaupt, Chem. Soc. Rev., 2014, 43, 4953–4967 RSC.
- P. Vermeeren, S. C. C. van der Lubbe, C. Fonseca Guerra, F. M. Bickelhaupt and T. A. Hamlin, Nat. Protoc., 2020, 15, 649–667 CrossRef CAS PubMed.
- A. M. Sheta, M. A. Mashaly, S. B. Said, S. S. Elmorsy, A. V Malkov and B. R. Buckley, Chem. Sci., 2020, 11, 9109–9114 RSC.
- V. K. Rawat, H. Hayashi, H. Katsuyama, S. R. Mangaonkar and T. Mita, Org. Lett., 2023, 25, 4231–4235 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of calculations and additional tables and figures. See DOI: https://doi.org/10.1039/d4se01065e |
| ‡ The existence of a linear correlation between ΔG‡ and ΔG0 is indicative of a constant ΔG0‡ throughout the reaction series or of a ΔG‡0 varying proportionally to ΔG0. |
| This journal is © The Royal Society of Chemistry 2024 |