
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A guide to lignin valorization in biorefineries: traditional, recent, and forthcoming approaches to convert raw lignocellulose into valuable materials and chemicals
Filippo
Brienza
ab,
David
Cannella
c,
Diego
Montesdeoca
a,
Iwona
Cybulska
*b and
Damien P.
Debecker
 *a
*a
aInstitute of Condensed Matter and Nanoscience (IMCN), UCLouvain, Place Louis Pasteur 1, 1348 Louvain-La-Neuve, Belgium. E-mail: damien.debecker@uclouvain.be
bApplied Microbiology Division, Earth and Life Institute (ELI), UCLouvain, Croix du Sud 2, 1348 Louvain-La-Neuve, Belgium. E-mail: iwona.cybulska@uclouvain.be
cPhotoBioCatalysis Unit (CPBL) and Biomass Transformation Lab (BTL), École Interfacultaire de Bioingénieurs (EIB), Université Libre de Bruxelles, Avenue Franklin D. Roosevelt 50, 1050 Bruxelles, Belgium
First published on 9th November 2023
Abstract
Lignin is the most abundant source of renewable aromatics on Earth, yet its enormous potential remains underexploited in current biorefinery and pulping processes. The extensive degree of condensation of the lignin fractions produced via the most widely adopted biomass pretreatments (i.e. “technical lignin”) poses a prominent limitation to their subsequent conversion toward valuable products. In this work, a broad range of methods for biomass pretreatment are reviewed, illustrating the impact of each strategy on the properties of the isolated lignin and carbohydrate fractions. The main pathways for the valorization of the obtained lignin streams (i.e. toward polymeric materials or chemicals) are critically discussed, and the relationship existing between (i) native lignin structure, (ii) pretreatment conditions, and (iii) lignin processability is rationalized. A key aspect for producing lignin streams amenable to further upgrading is the prevention of condensation reactions between lignin fragments during biomass fractionation. In this respect, a class of so-called “lignin-first” pretreatments, targeting the prompt stabilization of reactive lignin intermediates to minimize lignin condensation, has recently gained momentum. Herein, lignin-first approaches are reviewed, discussing in detail the fate of lignin, cellulose, and hemicellulose for each strategy. The potential of lignin-first biorefineries to realize a more complete valorization of lignocellulose and the current limitations of each method are highlighted. Overall, this work provides a comprehensive overview of the technologies that are available or currently emerging for lignin isolation and subsequent valorization.
Sustainability spotlightLignin is the most abundant source of renewable aromatics on Earth; yet its enormous potential remains largely underexploited in current biorefinery and pulping processes. Most large-scale industrial processes that use plant polysaccharides from lignocellulose used to burn lignin to generate power. With the advent of the integrated biorefinery concept, more lignin is being produced and therefore efforts are underway to transform it to value-added products: materials, fuels, and chemicals. This tutorial review provides a comprehensive overview of the technologies that are available or emerging for lignocellulose pretreatment, for lignin isolation and valorization, and for lignin depolymerization and valorization of the monomers. Research on lignocellulose and lignin valorization align with UN SDG number 9, 12, and 15. |
1. Introduction
Driven by the growing environmental concerns related to massive exploitation of fossil resources and the consequent dramatic increase of CO2 emissions (from about 6 billion tons in 1950 to over 36 billion tons in 2021),1 the development of biorefineries for the conversion of biomass into energy, chemicals, and materials, is playing a pivotal role in the transition toward a more circular, sustainable economy.2–4 From this point of view, lignocellulosic biomass, which comprises byproducts from the forestry and the wood industries, as well as agricultural residues and dedicated crops, represents the largest fraction of available terrestrial biomass and a promising candidate for the replacement of fossil resources.5,6Lignocellulose is made of three main components: cellulose and hemicellulose, which are carbohydrate polymers, and lignin, which is an aromatic polymer.7 On the one hand, the inherent heterogeneity of lignocellulose implies a large potential of this raw material for the manufacturing of a broad range of renewable, high-value products. On the other hand, it considerably complicates it processability.8,9 For this reason, lignocellulose biorefineries commonly implement an initial pretreatment step, with the goal of separating the different components of lignocellulosic biomass prior to further upgrading.10
Traditionally, pretreatments have been almost exclusively focused on the valorization of the carbohydrate fraction of lignocellulose, while lignin was regarded as an obstacle, hindering (hemi)cellulose valorization (e.g. in the pulping industry, in second-generation bioethanol production, …).9 Thus, the principal objective of many fractionation processes is the removal of lignin from biomass to yield a processable carbohydrate fraction, regardless of the properties of the isolated lignin stream.11 As a result, the valorization of the lignin fractions obtained from conventional pretreatments (often referred to as “technical lignins”) has been found to be particularly challenging.12
In spite of the difficulties related to its valorization, lignin represents the largest source of renewable aromatics available on Earth and, therefore, an alluring platform for the sustainable production of aromatic chemicals and polymers.13 With this perspective, research on lignin valorization flourished in the last few years, and an increasing number of studies have been conducted, providing a better understanding of lignin chemistry and leading to the development of a wide range of strategies for lignin isolation and upgrading.14 Recent notable reviews in this field include works by Rinaldi et al.,15 Galkin and Samec,11 Sun et al.,16 Schutyser et al.,14 Questell-Santiago et al.,17 Luo et al.,18 Abu-Omar et al.,.19 and Zhang et al.20 In this context, the conversion of lignin to chemicals and materials constitutes the focus of the present review. This work aims at providing a comprehensive overview of the strategies existing for the isolation and subsequent valorization of lignin in the landscape of lignocellulose biorefining. Rather than an exhaustive analysis of all the work being reported in the literature this field, the review is presented as a didactical guide, that we hope will be useful for newcomers and experts alike. We propose to cover the approaches that can be implemented to convert raw lignocellulose into valuable materials and chemicals, via a complete overview along the entire value chain and considering diverse methodologies; from the raw biomass to the valuable products.
After a brief introduction on the main compositional and structural properties of lignocellulose and its constituents (Section 2), the most largely adopted methods for biomass pretreatment are reviewed (Section 3), discussing in detail the impact of each strategy on the disassembly of lignocellulose and on the chemistry of lignin and (hemi)cellulose. We also discuss the pros and cons of the different technologies with respect to the subsequent processability of the isolated fractions. Thereafter, the main pathways existing for the valorization of the so-formed lignin streams (i.e. toward polymers or chemicals) are examined (Section 4), providing insight into the potential applications of lignin-derived products. The successful conversion of lignin toward chemicals and materials is strongly dependent on (i) native lignin properties, (ii) pretreatment methods and conditions, and (iii) lignin processing methods and conditions. The interrelation between these aspects is unraveled and critically discussed in Section 5. A key aspect for enhancing the processability of the lignin fractions isolated from biomass pretreatment is the suppression of recondensation reactions occurring between lignin fragments.17 Centered on this objective, a new class of lignocellulose pretreatments, termed “lignin-first” biorefineries has recently gained momentum.19 These lignin-first methods are reviewed in Section 6, wherein the advantages and disadvantages of each method and the current challenges for the upscaling of these strategies are illustrated.
2. Lignocellulosic biomass
Lignocellulose is generally recognized as the most abundant source of terrestrial organic matter on Earth.21 It is the main component of plant biomass and it is principally found in the secondary cell wall of plant cells, the thick and robust layer that is formed toward the inside of the primary cell wall, once plant cells ceased to grow.22 The main constituents of lignocellulose are cellulose, hemicellulose, and lignin, which are present in quantities that vary depending on the biomass source (plant species, tissue, environmental conditions, …).23 In addition, minor constituents such as proteins, lipids, tannins, pectin, non-structural polysaccharides, waxes, and inorganic materials may also be present in lignocellulose.23–25Table 1 summarizes typical ranges for the composition of lignocellulose from hardwoods (angiosperms), softwoods (gymnosperms) and grasses.| Compositiona (wt%) | |||
|---|---|---|---|
| Cellulose | Hemicellulose | Lignin | |
| a The compositional analysis of lignocellulose is commonly carried out as described in by Sluiter et al.28 Lignin is quantified based on the so-called Klason lignin analysis. This method is generally recognized as reliable for woody biomass, but is known to be subject to errors when applied to other biomass types (grasses, bark, leaves, …). For a more exhaustive discussion on the subject, the reader is invited to examine the works by Bunzel et al.,29,30 and the recent review by Abu-Omar et al.19 | |||
| Hardwoods | 45–55 | 25–40 | 18–25 |
| Softwoods | 45–50 | 25–35 | 25–35 |
| Grasses | 25–40 | 25–50 | 10–30 |
From a structural point of view, lignocellulose possesses a complex tridimensional arrangement in which cellulose, hemicellulose, and lignin are deeply interconnected.31 Cellulose strands are bundled together to form microfibrils, which are surrounded by hemicellulose and lignin.31,32 In turn, microfibrils are arranged into larger clusters, termed macrofibrils.33 Within these domains, cellulose is linked to hemicellulose and lignin principally by hydrogen bonding.34 On the other hand, hemicellulose and lignin are coupled via covalent bonds (ether and ester linkages).34 The structure of lignocellulose is schematically outlined in Fig. 1.
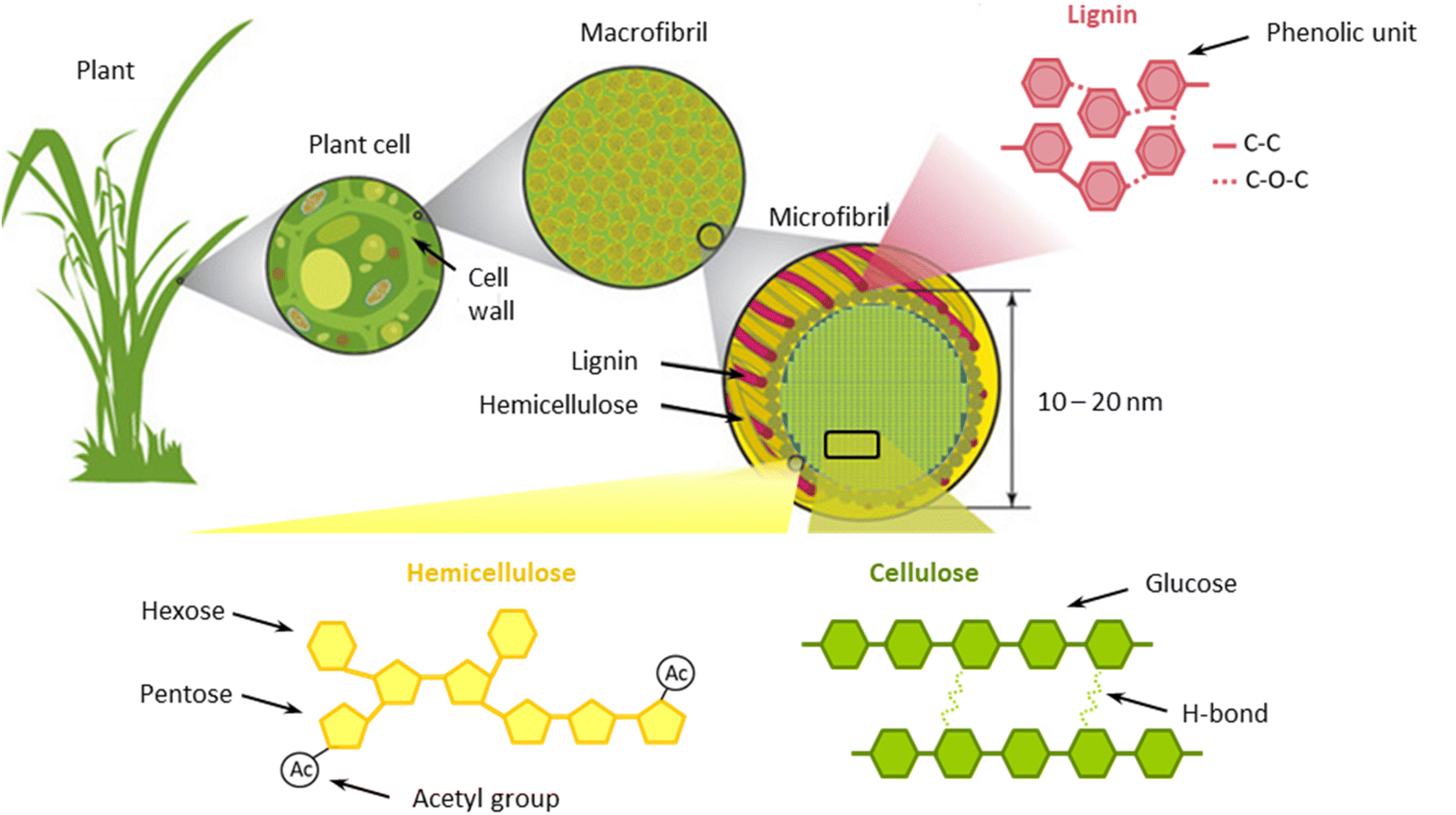 | ||
| Fig. 1 Schematic structure of lignocellulose components. Modified from Potters et al.33 | ||
2.1. Cellulose
Cellulose is a linear syndiotactic polymer of glucose units, linked together by β-1,4 glycosidic bonds. The degree of polymerization may vary between few hundreds to tens of thousands, depending on plant species, tissue type, growth stage of the plant, and other environmental factors.35,36 The glucose monomers in cellulose possess three free hydroxyl groups, which form both intramolecular and intermolecular hydrogen bonds, as schematized in Fig. 2. By virtue of this extensive hydrogen bonding, the polymer chains are strongly linked to one another and stack together to form microfibrils (Fig. 2), consisting of crystalline domains surrounded by paracrystalline or disordered domains.37 The tight packing of cellulose chains within crystalline regions hinders both its solubility in most common solvents, as well as its enzymatic digestibility. Conversely, disordered domains are less densely packed, thus more accessible to solvents and hydrolytic enzymes.38,39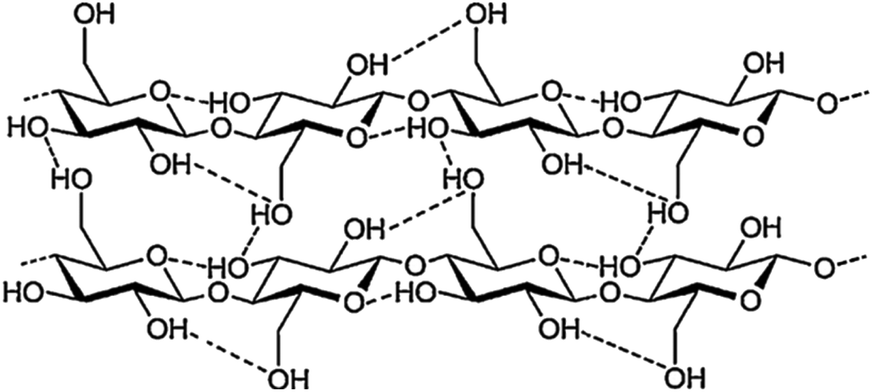 | ||
| Fig. 2 Molecular structure of cellulose. Adapted from Liao et al.39 | ||
2.2. Hemicellulose
Hemicellulose comprises diverse linear and branched short polysaccharides that differ broadly in terms of composition based on plant species, tissue type, growth stage of the plant, and environmental factors.40 Hemicellulose polymers consist of a backbone containing β-1,4-linked pentoses (mainly β-D-xylose) or hexoses (mainly β-D-mannose and β-D-glucose), branched with single or longer glycosyl residues (Fig. 3).40,41 In addition, uronic acids such as α-D-glucuronic and α-D-galacturonic acids can be found in hemicellulose chains. Typically, xylans are the most abundant hemicellulose components in grasses and hardwoods, while mannans are the major hemicellulose components of softwoods (Fig. 3).23 The degree of polymerization of hemicellulose is lower compared to that of cellulose, and is usually in the range of 100–200 units.42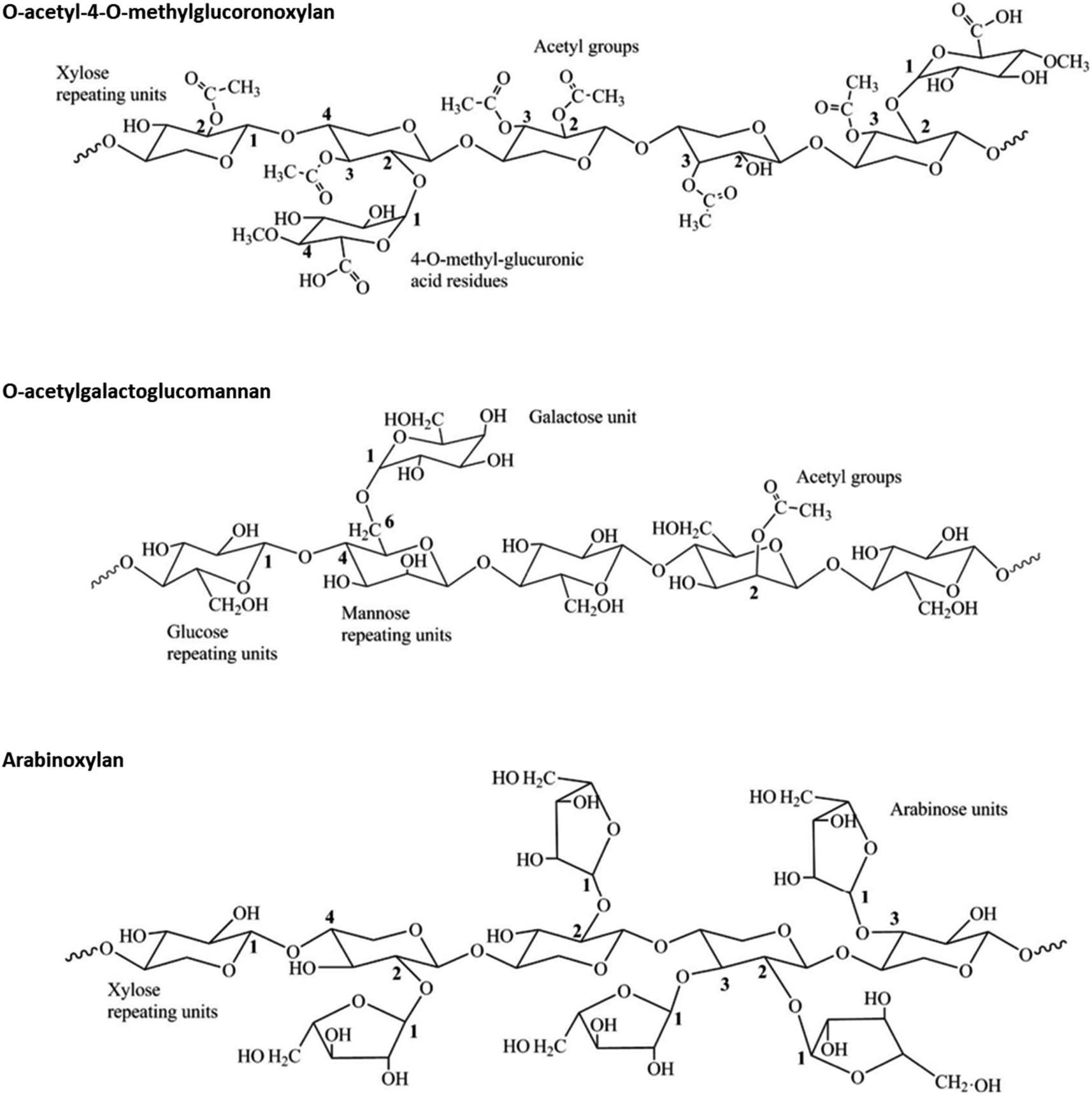 | ||
| Fig. 3 Examples of hemicellulose structures typically found in hardwoods (O-acetyl-4-O-methylglucoronoxylan), softwoods (O-acetylgalactoglucomannan) and herbaceous crops (arabinoxylan). Adapted from Farhat et al.43 | ||
Hemicellulose plays the role of a binding agent in the plant cell wall, linking cellulose bundles, lignin, cell wall proteins, pectin, and non-structural polysaccharides. Due to the high extent of branching, hemicellulose possesses an amorphous structure and is more easily subjected to depolymerization by the action of solvents or enzymes, compared to cellulose. Furthermore, hydroxyl groups in hemicellulose are acetylated to some extent and, during biomass pretreatment, the acetic acid formed upon cleavage of acetyl groups can further enhance hemicellulose decomposition.23
2.3. Lignin
Lignin is the most abundant non-carbohydrate component in the plant cell wall where it provides plant cells with rigidity, hydrophobicity, and resistance to microbial attack.7 Owing to these properties, it is regarded as the principal contributor to lignocellulose recalcitrance.9 It is an amorphous polymer of oxygenated p-propylphenol units and possesses a networked three-dimensional structure. The three main building blocks of lignin are p-coumaryl alcohol which forms p-hydroxyphenyl units (H-units), coniferyl alcohol which forms guaiacyl units (G-units), and sinapyl alcohol which forms syringyl units (S-units). These three types of units differ from one another based on the number of methoxy groups in the ortho-positions of aromatic rings: zero for H-units, one for G-units, and two for S-units (Fig. 4).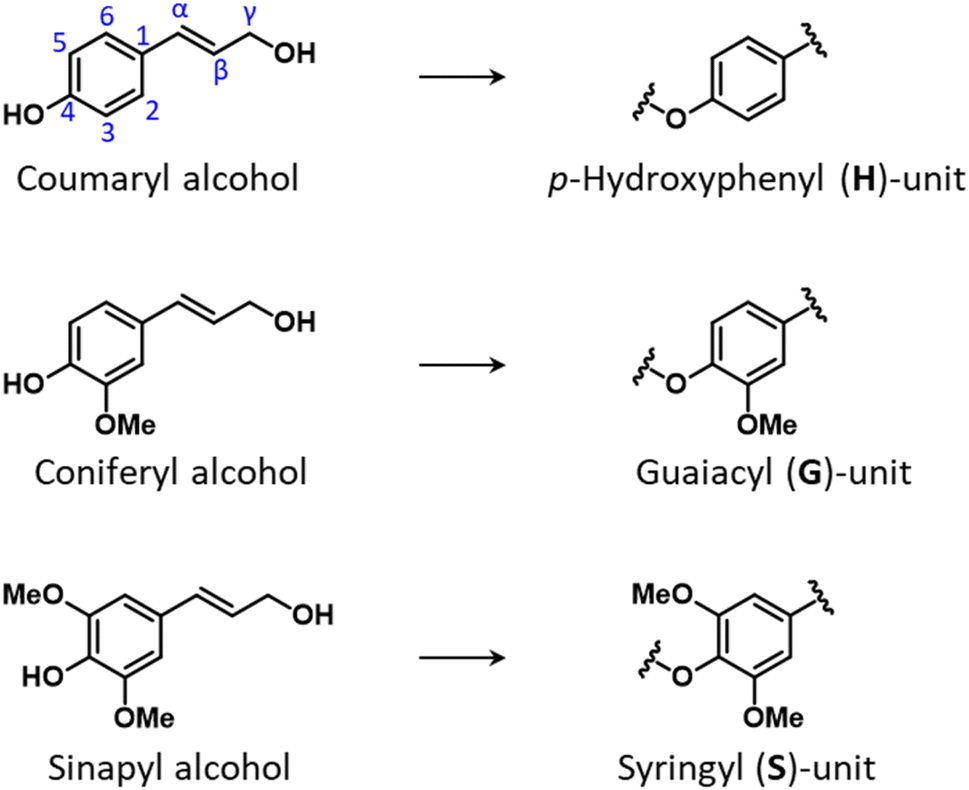 | ||
| Fig. 4 Chemical structures of monolignols, and of the derived lignin units. | ||
Following their synthesis in the cytosol via the phenylpropanoid pathway, monolignols are directed to the cell wall, where radical polymerization takes place, initiated by laccase and peroxidase enzymes, and the lignin polymer is formed.15 An alternative view to the long-established model of random radical coupling of monolignols was proposed which postulates that so-called dirigent proteins catalyze a stereo-oriented biosynthesis of inter-unit lignin linkages.44 The relative amount of H-, G- and S-units in the polymeric network varies substantially for different plant species and plant parts.25 In general, softwood lignin is composed exclusively of G-units, whereas hardwood lignin contains both G- and S-units, and lignin from herbaceous biomass may comprise all three types of monolignols. In addition, other compounds may be incorporated in lignin structures, such as hydroxycinnamates (p-coumarate, p-ferulate), p-hydroxybenzoate, tricin and other minor components.45–49 Lignin composition is also largely dependent on the specific plant part of origin and on environmental factors to which the plant has been exposed to during growth. For instance, compression wood, which is typically formed in the lower side of branches and stems of coniferous trees (softwoods) in response to mechanical stress, possesses a higher lignin content than normal softwood and a lignin structure that comprises significant amounts of H-units.50 Another example is lignin from bark, which often displays a much higher degree of heterogeneity as compared to lignin in wood, comprising S-, G-, and H- units, as well as variable amounts of hydroxycinnamates, hydroxystilbenes, flavonoids, and other constituents.51–53
As illustrated in Fig. 5, various types of inter-unit linkages can be formed during the radical coupling of monolignols, including ether bonds, such as β-O-4′ (in β-aryl ether and dibenzodioxicin motifs), α-O-4′ (in phenylcoumaran and dibenzodioxicin motifs), 4-O-5′ (in diaryl ether motifs), γ-O-α′ (in resinol motifs), and α-O-α′ (in spirodienone motifs), as well as C–C bonds, such as β-5′ (in phenylcoumaran motifs), β-1′ (in 1,2-diarylpropane and spirodienone motifs), β-β′ (in resinol motifs), and 5-5′ (in biphenyl and dibenzodioxicin motifs). The content of inter-unit linkages also varies for different feedstocks.15 A correlation has been proposed between lignin composition in terms of H-/G-/S-units and the content of C–C inter-unit bonds. Indeed, methoxy-substituted ortho-positions on aromatic rings of lignin units cannot participate in the formation of β-5′ and 5-5′ bonds during lignin polymerization, so the relative content of C–C bonds would be lower for lignin polymers richer in S-units.14 However, besides the composition of lignin units, other factors, such as the transport of monolignols from the cytoplasm to the cell wall, have been suggested to affect the content of C–O and C–C inter-unit linkages in native lignin.54 Feedstocks possessing lignin polymers with a low content of C–C bonds and a high amount of labile β-O-4′ linkages are the ideal target of processes that aim at depolymerizing lignin.14
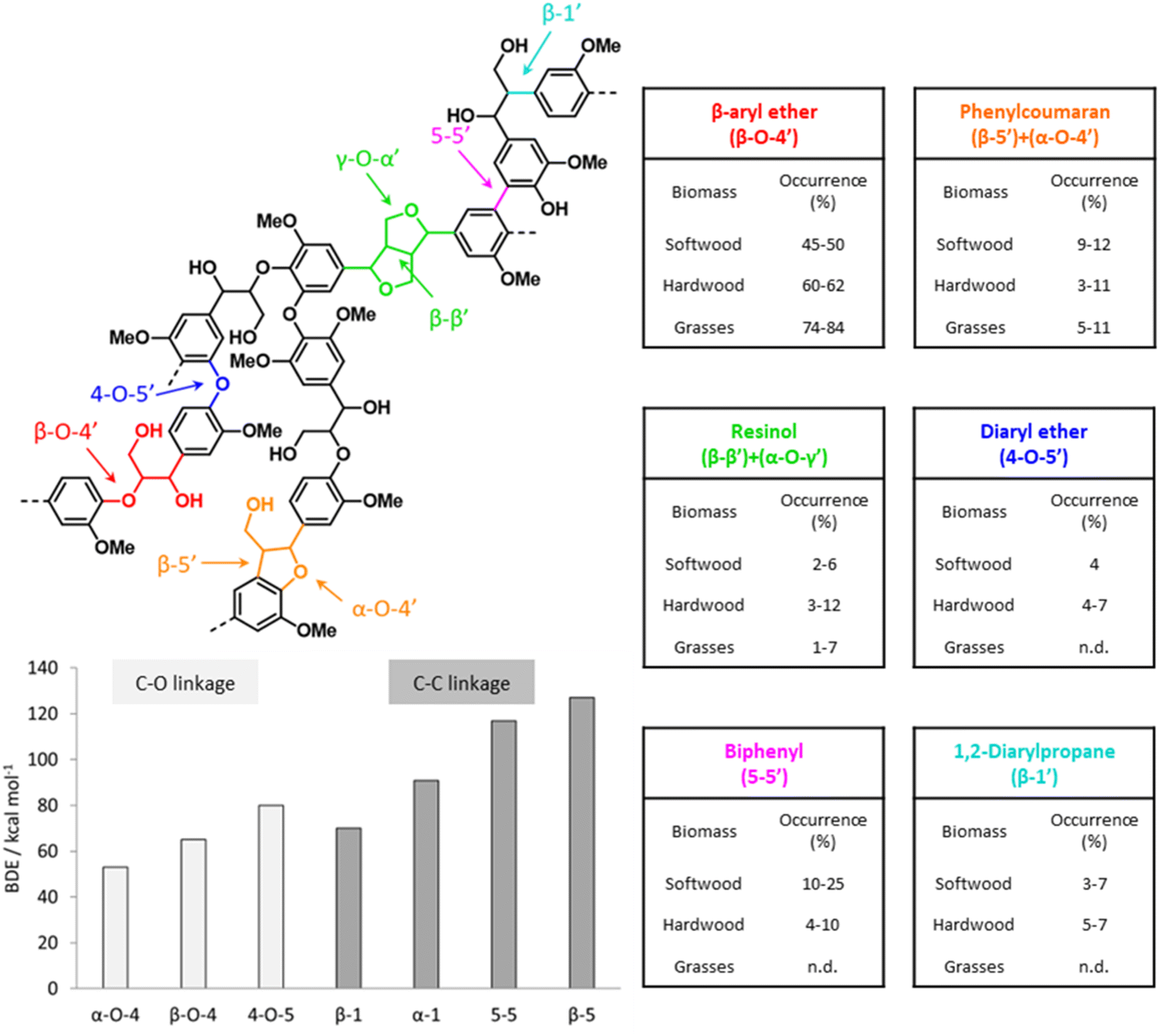 | ||
| Fig. 5 Main lignin inter-unit linkage motifs with relative occurrence in different types of lignocellulose feedstocks and their bond dissociation energy (BDE). Based on Rinaldi et al.,15 Wang and Wang,55 and Parthasarathi et al.56 | ||
Within the lignocellulose matrix, lignin is also linked to hemicellulose via ether and ester bonds to form hybrid structural motifs known as lignin–carbohydrate complexes (LCCs).57–60 The main types of linkages that are typically found in LCCs include benzyl ether, benzyl ester, ferulate ester, phenyl glycosidic, and diferulate ester (Fig. 6). Benzyl ether and phenyl glycosidic linkages connect glycosyl or mannosyl units of carbohydrates to phenolic or hydroxyl groups in lignin, whereas benzyl ester bonds link sugar uronic acids in hemicellulose to hydroxyl groups in lignin structures.58 The abundance of the different LCC motifs in lignocellulose varies depending on the feedstock: hardwood LCCs are characterized by a high content of phenyl glycosidic linkages,61,62 whereas softwood LCCs feature a high amount of benzyl ether structures,61,63 and herbaceous LCCs possess high contents of (di)ferulate and phenyl glycosidic bonds.64 As a general trend, herbaceous biomass possesses higher amounts of LCCs compared to woody biomass.58,65 Even though the influence of LCCs on lignocellulose pretreatment has not been fully elucidated yet, it has been shown that a high presence of LCCs may hamper biomass delignification during pretreatment and the enzymatic hydrolysis of the isolated (hemi)cellulose.57,58
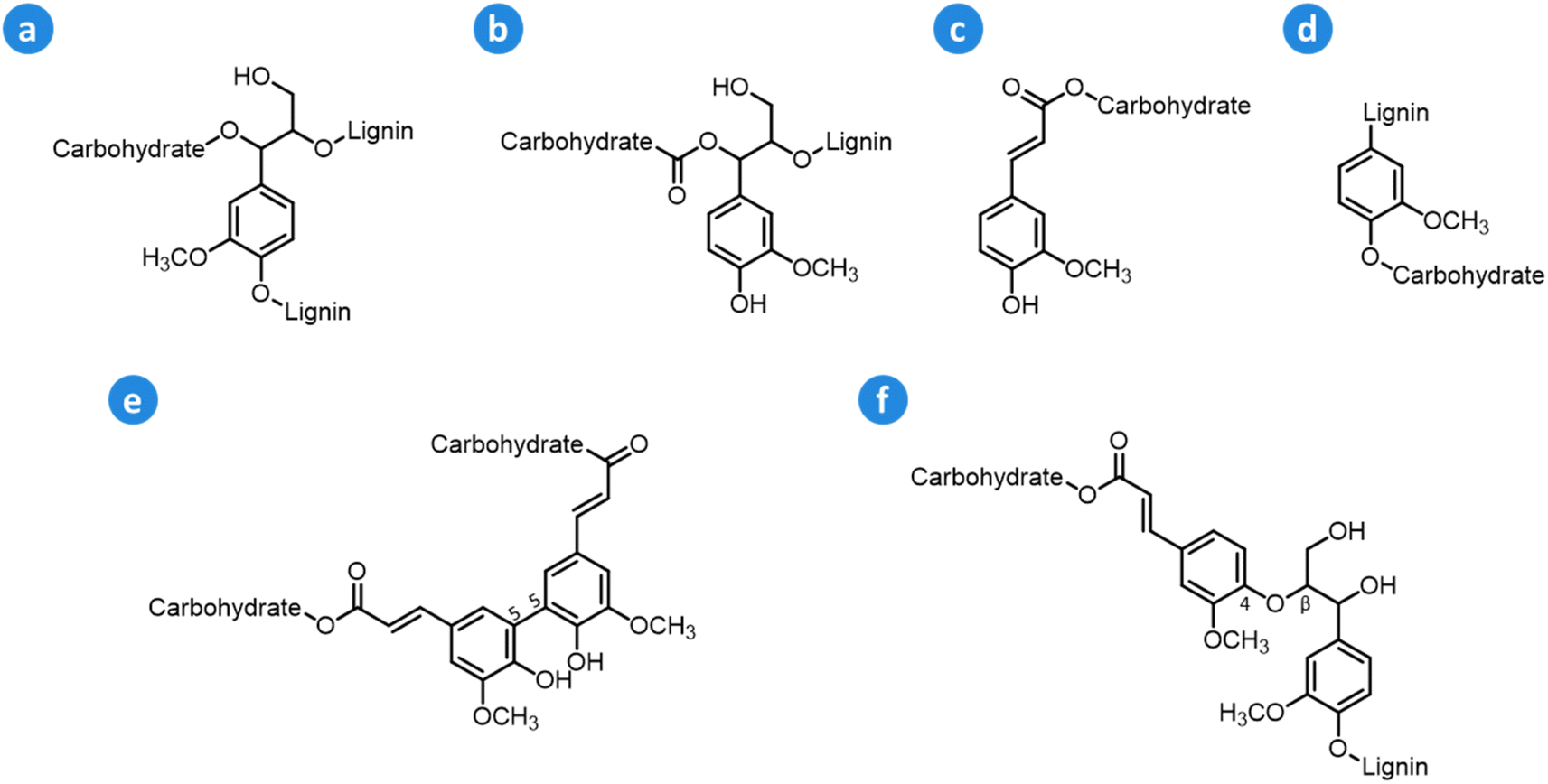 | ||
| Fig. 6 Main types of LCC linkages: (a) benzyl ether, (b) benzyl ester, (c) ferulate ester, (d) phenyl glycosidic, (e) diferulate ester (5-5′), and (f) diferulate ester (β-O-4′). Based on Zhao et al.57 and Tarasov et al.58 | ||
Due to the strong bonds existing between lignin and hemicellulose, which complicate the isolation of pure native lignin, the molecular weight (MW) of lignin polymers is difficult to assess. However, close-to-native lignin fractions have been reported to possess MWs in the range: 2500–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1.14
000 g mol−1.14
3. Lignocellulose pretreatment as a source of lignin
One of the major obstacles to lignocellulose valorization in a biorefinery is its recalcitrance toward enzymatic hydrolysis. Several factors affect lignocellulose recalcitrance, including both physical and chemical factors. Cellulose crystallinity correlates negatively with enzymatic hydrolysis, due to the tight packing of cellulose chains in crystalline regions, and a consequently reduced enzymatic accessibility.9 The biomass particle size exerts as well an impact on cellulose hydrolysis potential; smaller biomass particles have an enhanced enzymatic digestibility.66 Similarly, substrates with a higher porosity and with larger pore sizes possess a better processability, in view of a larger surface area of cellulose available for enzymatic attack.67 On the contrary, the presence of hemicellulose and lignin hinders the enzymatic digestion of cellulose, since they both constitute a physical barrier that limits enzymatic accessibility, and they can also interfere with enzyme recognition or irreversibly adsorb enzymes.9Thus, producing energy, chemicals and materials from renewable lignocellulose requires the development of effective pretreatment strategies that could reduce its inherent recalcitrance toward further processing. In general, such biomass pretreatments are aimed at the disruption of lignocellulose structure by the partial removal of lignin and hemicellulose and by a partial amorphization of cellulose crystalline domains.68,69
Lignocellulose pretreatments have long been employed in the pulping industry to convert woody biomass into pulp for papermaking. In this case, the pretreatment typically aims at removing lignin and hemicellulose from the biomass to yield a pulp with a high cellulose purity. The amorphization and the depolymerization of cellulose are undesirable, as they negatively affect the mechanical resistance of the pulps. In the context of “integrated biorefining”, in view of the various applications envisaged for lignocellulosic biomass, several pretreatment approaches have been developed, including physical, physicochemical, chemical, and biological methods.39,68–70 A common feature of many of them is the isolation of a solid carbohydrate fraction, amenable to further processing (saccharification or papermaking), and a lignin fraction, possibly comprising hemicellulose as well. The following sections explore the main aspects of the pretreatment strategies that are most widely adopted in biorefineries and in the pulping industry, with a focus on the properties of the isolated lignin fractions.
3.1. Physical pretreatment methods
Physical pretreatments include mechanical comminution, extrusion, and irradiation. In contrast to chemical and physicochemical pretreatments, physical pretreatments do not target the removal of lignin and hemicellulose from the produced pulp, thus they do not realize an actual fractionation of biomass. Instead, physical pretreatments aim at the partial disruption of the lignocellulose structure to enhance its subsequent processability.In biorefineries, the main goal of mechanical pretreatments is the reduction of the particle size and crystallinity of lignocellulosic biomass, to enhance the enzymatic accessibility of cellulose, thus more intense milling can be applied compared to what is done for pulping. Previous works reported a substantial increase of (hemi)cellulose digestibility (yields of monosaccharides up to 80–90%) upon the application of ball milling to herbaceous and woody biomass.73–75 The most critical shortcomings related to the adoption of ball milling for lignocellulose pretreatment are the high energy demand of this method, and the large equipment cost.69,76,77 Moreover, even though cellulose is made more accessible through milling, the residual presence of lignin may still hinder further processing.39,75
On the other hand, the use of milling as a pretreatment is of paramount importance for research efforts aimed at investigating lignocellulose structural properties. For instance, mechanical pretreatment at room temperature coupled with lignin extraction in an organic solvent constitute the basis for the isolation of native-like lignin (termed “milled wood lignin”).78,79
Extrusion pretreatment is often coupled with other chemical pretreatments.82–84 For instance, an extrusion in hot water, or in the presence of an acid or basic catalyst can result in a partial removal of amorphous components (in acidic environment: hemicellulose hydrolysis; in alkaline environment: lignin solubilization), and consequently in a higher enzymatic digestibility of the cellulosic pulp.80,84
Low-energy microwave radiation excites molecular vibrational and rotational modes, thereby providing thermal energy which, ultimately contributes to the disruption of lignocellulose structure.87,91–93 Microwave pretreatment induces a rapid heating,69 and is often used to assist other pretreatment strategies.39,94–97 However, the treatment of heterogeneous substrates may result in a non-uniform temperature profile in the biomass.98 This can lead to the creation of hot spots, where lignocellulose degradation may occur, together with the formation of enzyme inhibitors. The high energy consumption also represents a limitation to the industrial application of microwave irradiation.68,91,98
Ultrasound irradiation creates pressure differences in the reaction medium and cavitation phenomena, resulting in shear forces that break down the lignocellulose structure and enhance mass transfer and mixing processes.87,99 Additionally, the cavitation of microbubbles generates radicals that contribute to the cleavage of intermolecular bonds between lignin, hemicellulose, and cellulose.87,99 Ultrasonication is often used in combination with chemical or physicochemical pretreatments to improve the extraction of desired lignocellulose components in the reaction medium.100–104 Similarly to other irradiation pretreatments, ultrasonication is energy intensive and difficult to implement at large scale.69
3.2. Physicochemical pretreatment methods
In physicochemical pretreatments, the lignocellulose structure is decomposed either by the hydrolytic action of water or by pressure shocks (“explosions”).Generally, the severity of LHW pretreatments is described by means of a severity factor log(R0) (eqn (1)).39,108–110
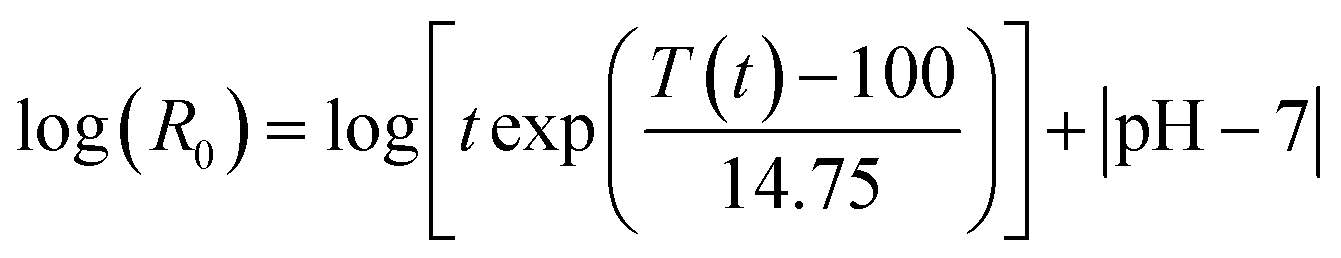 | (1) |
Although LHW pretreatment solubilizes only a limited amount of lignin, it causes considerable modifications of the lignin chemical structure. Most prominently, β-O-4′ ether linkages undergo acid-catalyzed cleavage, as illustrated in Fig. 7.39 The first step of the reaction pathway involves the cleavage of the hydroxyl group in the α-position with the formation of an intermediate benzylic carbenium ion (1).114–117 The latter can then transform into two enol–ether structures (2 and 3 in pathways A and B, respectively), prone to undergo hydrolysis to yield C3-ketone-substituted phenolics (4, known as Hibbert's ketones) or C2-aldehyde-substituted phenolics (5).115–117 Hibbert's ketones, together with C2-aldehyde-substituted phenolics and carbenium ions (pathway C) participate in repolymerization reactions, ultimately leading to the formation of condensed recalcitrant lignin structures.39,118 Furthermore, lignin can undergo melting at high temperature, followed by redeposition on the surface of cellulose fibers in the form of lignin droplets upon cooling, potentially reducing the accessibility of cellulose.119
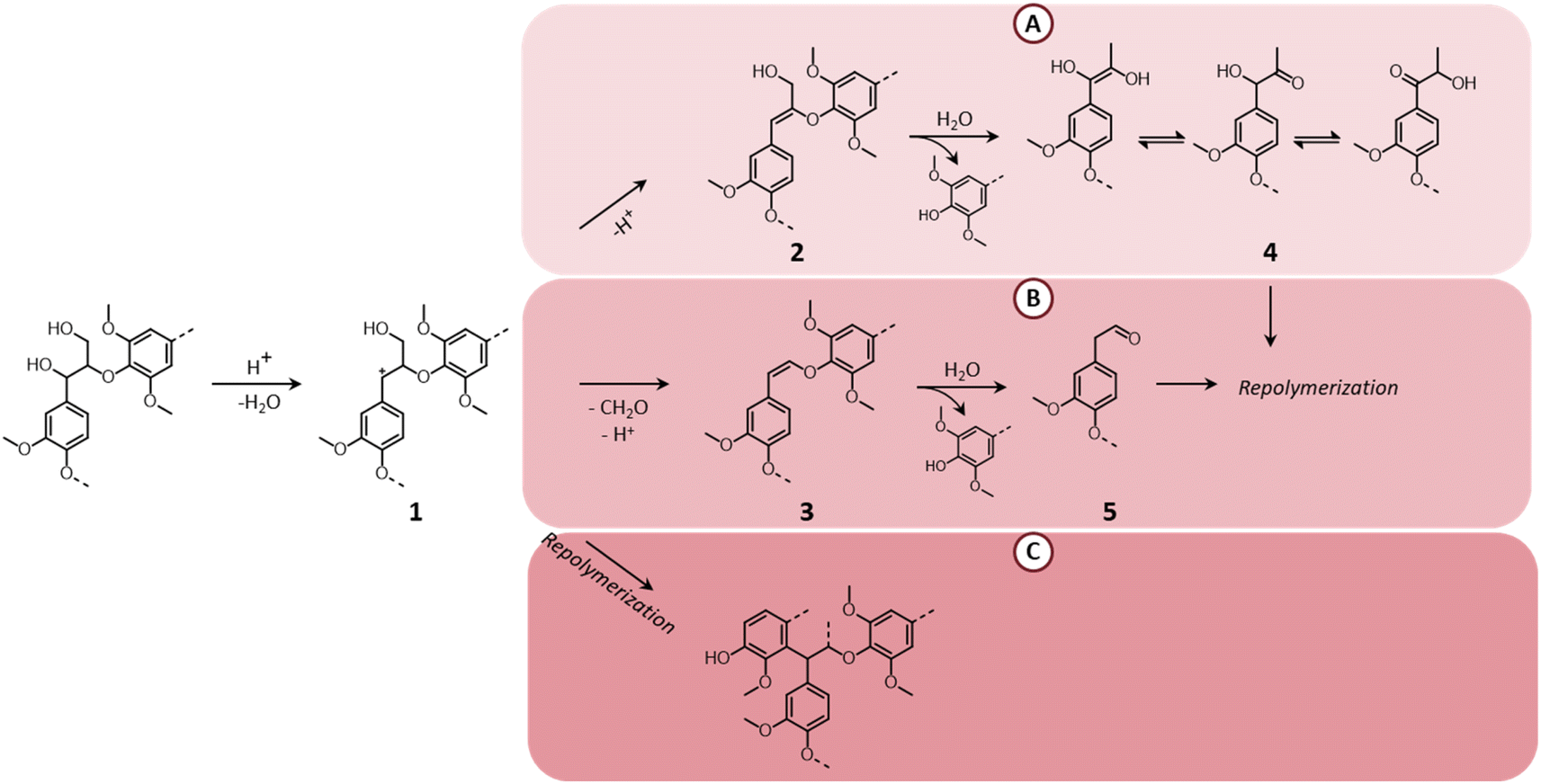 | ||
| Fig. 7 Lignin chemistry in acidic conditions. Pathway A: formation of Hibbert's ketones; pathway B: formation of C2-aldehyde substituted phenolics; pathway C: repolymerization. Based on Liao et al.39 | ||
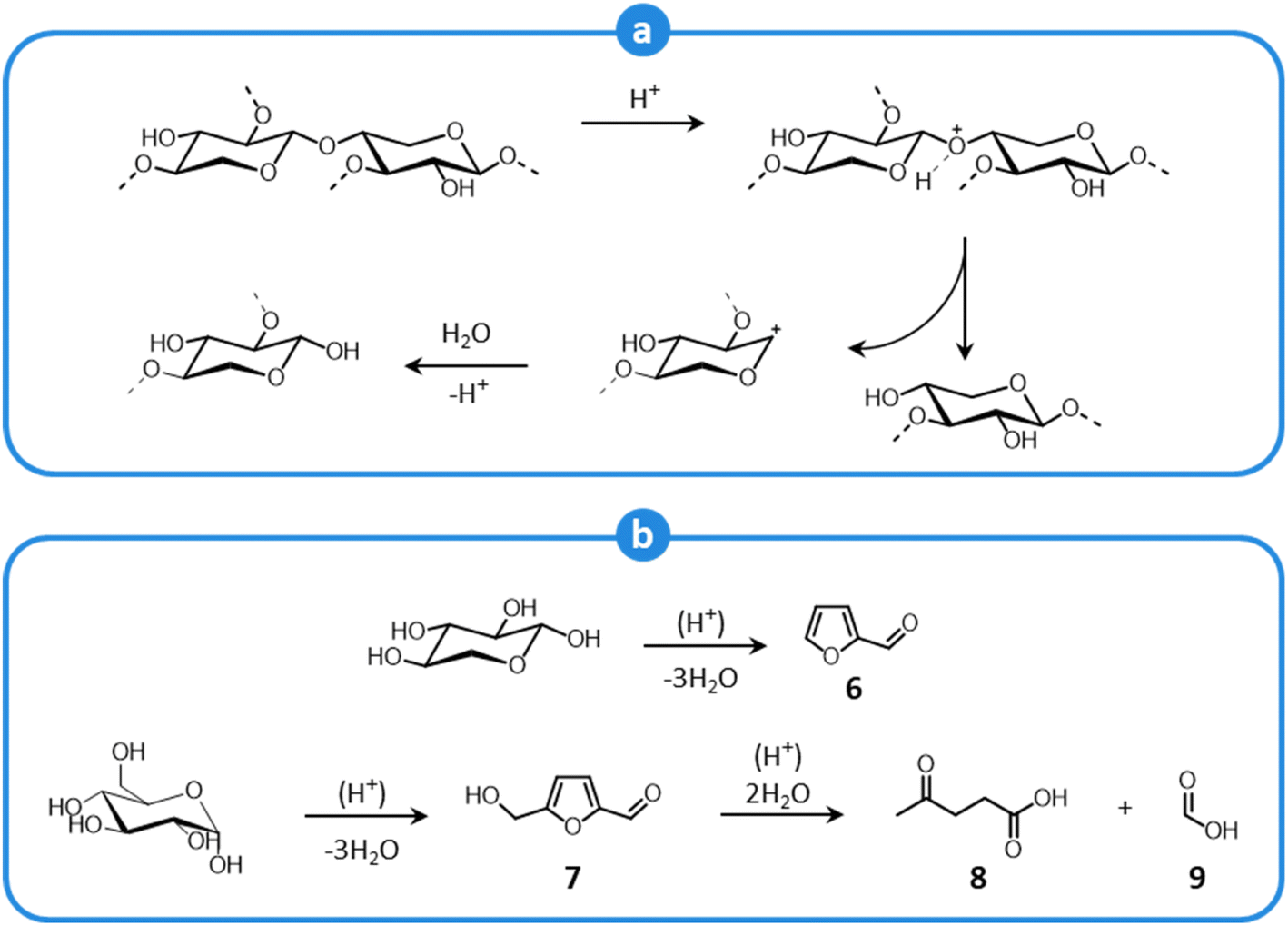
Carbohydrate polymers are also affected by the acidic environment of LHW pretreatment, particularly hemicellulose, which is more exposed to the action of solvents. Glycosidic bonds undergo hydrolysis to yield oligo- and monosaccharides,108,109,120 according to the mechanism shown in Fig. 8a. The released sugar fragments are subject to further acid-catalyzed degradation reactions (Fig. 8b), resulting in the formation of short aldehydes (furfural (6), 5-hydroxymethylfurfural (7)) and organic acids (levulinic (8), formic acid (9)).108,109,120 These products inhibit the subsequent fermentation of the pulp (post-enzymatic saccharification).39,108 Additionally, products from carbohydrate degradation may generate so-called pseudolignin (also termed humins),121,122 which deposits on the pulp surface, limiting the accessibility of cellulose fibers.108,123
During SE pretreatment, the linkages between cellulose and hemicellulose, and between hemicellulose and lignin are broken down by the action of temperature and pressure, resulting in a partial solubilization of hemicellulose and (to a minor extent) lignin.124,126,132 The extracted lignin possesses features analogous to the lignin obtained from LHW pretreatment, since it undergoes similar acid-catalyzed depolymerization and repolymerization reactions.130 Thus, depending on the treatment severity, lignin can be subject to moderate or extensive chemical modifications.126,133
The hydrolysis of hemicellulose and the degradation of the released sugars to form aldehydes and organic acids occur, which can cause inhibition of fermentative microorganisms during the downstream processing of the isolated pulp.82,127,133 Despite this, the low energy requirements, the absence of the use of expensive chemicals (with the associated recycling/disposal cost), and the effective deconstruction of lignocellulose structure make SE pretreatment an attractive method for biorefineries.124,125,134,135
During AFEX, ammonolytic and hydrolytic cleavage of LCCs occurs, with the partial solubilization of hemicellulose and lignin, which, upon the release of the applied pressure, are redistributed in the pretreated solids.14,39 Even though AFEX does not delignify biomass, it facilitates downstream lignin extraction, for example with an organic or alkaline solution.14,139 In contrast to the lignin obtained from LHW or SE processes, the structure of AFEX lignin is usually composed of oligomeric fragments, with well-preserved β-O-4′ bonds, thus amenable to further upgrading.14
Another advantage of AFEX lies in the minor degradation of hemicellulose and lignin, with a negligible formation of fermentation inhibitory compounds (e.g. furans, organic acids, phenols).69,82 On the other hand, the need to recycle ammonia increases the operating cost. Also, the method is unsuitable for the pretreatment of biomasses possessing high lignin contents (e.g. softwood or hardwood).39,140 Nevertheless, when applied to herbaceous plants with low lignin contents, AFEX showed better results compared to other pretreatments, yielding a pulp with enhanced digestibility.69,140,141 Thus, it is regarded as a promising method for overcoming the high enzyme cost of many current biorefinery processes.142
Compared to SE and AFEX pretreatments, the principal advantages offered by CO2 explosion are the lower energy requirements, and the non-flammability, non-toxicity and low cost of CO2.69,144,148 Despite this, the high cost of the equipment necessary to withstand the elevated pressure constitutes an obstacle for industrial implementation.148
3.3. Chemical pretreatment methods
In chemical pretreatments lignocellulosic biomass is processed in the presence of an exogenous chemical agent. These treatments include common pulping methods such as kraft, sulfite, and soda pulping, as well as acid, oxidative, organosolv, ionic liquid and deep-eutectic solvent pretreatments.Lignocellulosic biomass is treated in an aqueous solution of sodium hydroxide (NaOH) and sodium sulfide (Na2S) termed white liquor (pH ∼14), at a temperature comprised between 150 and 170 °C.149 Under such conditions, lignin and part of the hemicellulose are solubilized, leaving behind a cellulosic pulp. Delignification takes place by the action of the OH− and HS− ions, present in the white liquor, that cause the cleavage of LCCs and the depolymerization of lignin into fragments, which are soluble in the alkaline medium. Ester bonds between hemicellulose and lignin are cleaved by saponification reactions.39,69 On the other hand, lignin depolymerization involves mainly the cleavage of β-O-4′ inter-unit linkages, with a mechanism that follows different pathways for phenolic units (possessing free phenolic OH-groups) and non-phenolic lignin units (possessing etherified phenolic OH-groups).149,150,152–155
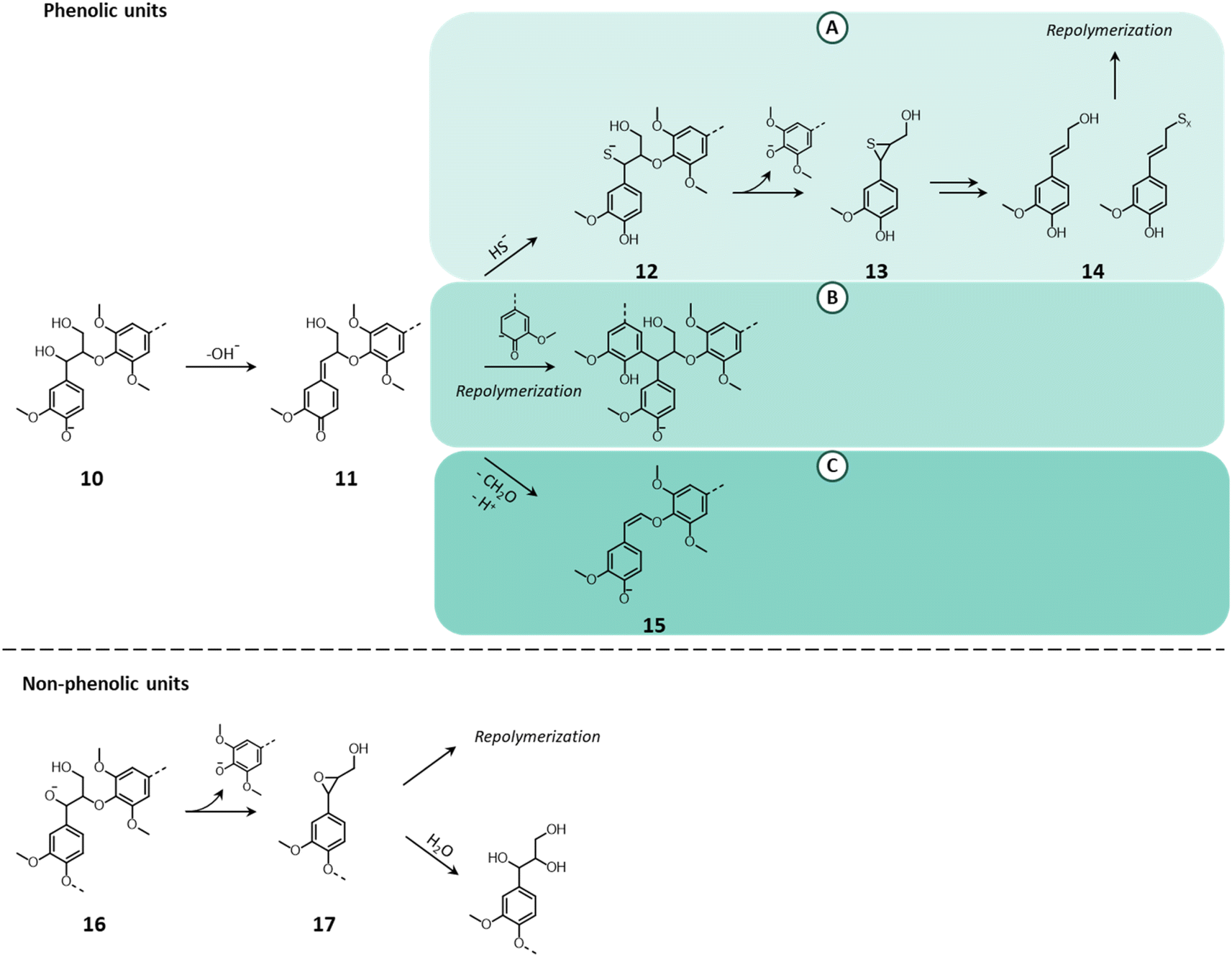
As schematized in Fig. 9, under the harsh alkaline conditions applied during kraft pulping, a phenolic unit (10) that possess a suitable leaving group on the α-position (e.g. –OH, –OR) is transformed into a quinone methide (11).149,150,153 In the presence of HS−, the quinone methide undergoes nucleophilic attack to restore the aromaticity (pathway A), forming a benzyl mercaptide (12). The ionized form of the thiol attacks the β-carbon breaking the β-O-4′ bond and leading to the formation of an episulfide (13).149,150,153 The latter may undergo a variety of reactions, ultimately leading to the formation of compounds with unsaturated (thio)/(hydroxy)alkenyl side chains (14) that are prone to undergo repolymerization.15,149,153 Alternatively, the quinone methide can also react with a lignin nucleophile (phenolate), leading to the recondensation of lignin (pathway B).150,154,155 Hence, for kraft pulping, the addition of HS− to quinone methides is in competition with lignin condensation. Indeed, the presence of HS− contributes to prevent excessive lignin repolymerization, thereby ensuring the solubility of lignin fragments and the successful biomass delignification.149,154 Finally, the quinone methide can restore the aromaticity via retro-aldol reaction and the loss of the γ–CH2–OH in the form of formaldehyde to yield an alkali-stable enol ether (15) (pathway C).153,154 However, this reaction pathway is more likely to occur in the absence of a strong nucleophile (as is HS−).15 For non-phenolic units (16), the cleavage of β-O-4′ bonds is due to the partial dissociation of the –OH groups at the α- or γ-positions (predominantly at the α-position, which possesses a higher acidity).149,150,153 The ionization of these groups causes a nucleophilic attack on the β-carbon and the cleavage of the β-O-4′ bond to generate an epoxide (17), which quickly opens by reaction with water or with another lignin unit.149,150,153
 | ||
| Fig. 9 Lignin chemistry in alkaline conditions. Pathway A: nucleophilic attack by HS−; pathway B: repolymerization; pathway C: loss of formaldehyde and enol ether formation. Based on Lachenal149 and Rinaldi et al.15 | ||
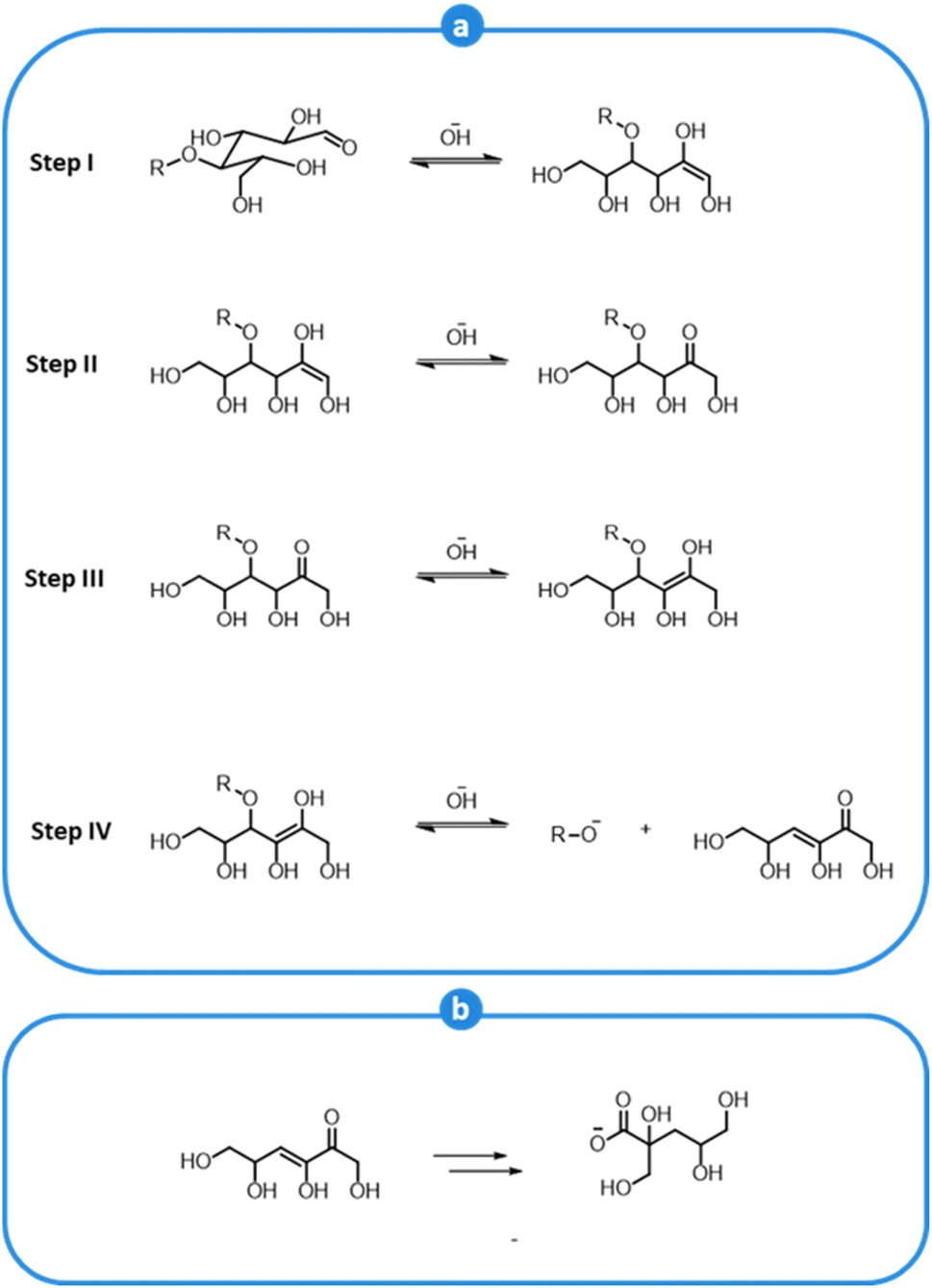
Cellulose and, most prominently, hemicellulose are also affected by the alkaline environment of kraft pulping. Carbohydrate polymers undergo extensive depolymerization and a substantial amount of hemicellulose is solubilized during pulping. Carbohydrate degradation occurs via a so-called peeling reaction, which causes the removal of sugar units one by one from the reducing ends of the polymer chains (cellulose and hemicelluloses), as shown in Fig. 10a. The peeling mechanisms is initiated by the reaction of OH− with the H-atom at the α-carbon of an aldehyde group, which is slightly acidic (Step I).149,150 This causes an alkaline-induced isomerization of an aldose to a ketose (Step II). The latter is then transformed into an enediol (Step III), which can go through β-alkoxy elimination, leading to the cleavage of the glycosidic bond (Step IV).149,150 The cleaved end-unit ultimately rearranges into a carboxylic acid and is solubilized, causing the concomitant undesired neutralization of the alkaline pulping medium (Fig. 10b).149 The peeling process goes on until competing reactions that convert the reducing end groups into stable carboxylic acid groups become preponderant.149,156
 | ||
| Fig. 10 Peeling reaction of carbohydrates in alkaline conditions (a), and fate of the eliminated units (b). Based on Lachenal149 and Fearon et al.156 | ||
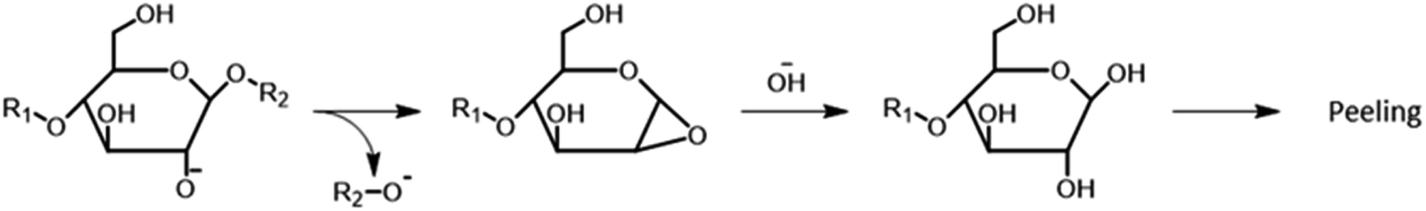
Alkaline hydrolysis of polysaccharides also contributes to polysaccharides decomposition. Under kraft pulping conditions, the hydroxyl groups of carbohydrate molecules may partially dissociate to give alkoxide anions, which can perform an intramolecular nucleophilic attack leading to the cleavage of glycosidic bonds and to the formation of oxirane structures (Fig. 11).149,156,157 Ultimately, the hydrolysis of such oxiranes gives new reducing end groups, which can undergo peeling reaction (this process is termed secondary peeling).149
 | ||
| Fig. 11 Alkaline hydrolysis of carbohydrates in alkaline conditions. Based on Lachenal149 and Gierer.157 | ||
As a result of the considerable lignin and carbohydrate degradation occurring during kraft pulping, the spent process liquor, termed black liquor, contains partially condensed lignin structures, heavily contaminated with carbohydrate and sulfur derivatives.11,15 Black liquor is commonly combusted to recover thermal energy and to fuel the pulping process.149,150 Alternatively, lignin can be precipitated from the liquor by acidification.11,14 However, the low content of residual β-O-4′ linkages, the high degree of condensation and the presence of contaminants (e.g. 1–3% of sulfur)11 make kraft lignin particularly recalcitrant toward further upgrading.11,14,15 Despite the excellent quality of kraft pulps and the near-complete recovery of chemicals and energy in modern kraft mills,158 the limited possibilities for the valorization of the lignin and hemicellulose fractions of biomass represent critical shortcomings for an effective valorization of lignocellulose during the kraft process. Recently, research efforts were spent with the goal of better elucidating the structural motifs in kraft lignin.159,160 Arguably, such work will serve as a means for the development of improved valorization strategies for kraft lignin.
Sulfite pulping relies on the treatment of lignocellulosic biomass in an aqueous solution of salts of sulfurous acid, at a temperature in a range between 130 and 180 °C.161 The salts employed in the process can be formed by different cations, such as calcium, magnesium, sodium and ammonium, and the choice of the cation ultimately affects the pH of the pulping medium (typically comprised between 2 and 12).11,150,161 In turn, the pH of the medium determines the active sulfur species (H2SO3, HSO3−, or SO32−).161
During sulfite pulping biomass delignification is achieved by two mechanisms: sulfonation and hydrolysis. Sulfonation introduces polar sulfonate groups in lignin structures, thereby increasing lignin solubility in the aqueous medium, while hydrolysis breaks LCCs and intermolecular ether linkages between lignin units (i.e. β-O-4′ and α-O-4′ bonds).150,161,163
Lignin sulfonation follows different pathways under acidic and neutral/alkaline conditions. In an acidic medium, benzylic carbenium ions are formed according to a mechanism illustrated in Fig. 7, which implies a nucleophilic attack by bisulfite ions (Fig. 12a), ultimately yielding α-sulfonic acid structures (18).150,161,163,164 On the other hand, under neutral or basic conditions, sulfonation involves only phenolic units, which generate quinone methides (Fig. 9). In the presence of sulfite ions, quinone methides (11) are subjected to nucleophilic attack (Fig. 12b), and α-sulfonic acids are formed (19).150,161,163,164 Moreover, other inter-unit linkages can undergo sulfonation, and β- and γ-sulfonic groups are introduced as well in lignin units, both under acid and alkaline conditions.150,161,163,165–167
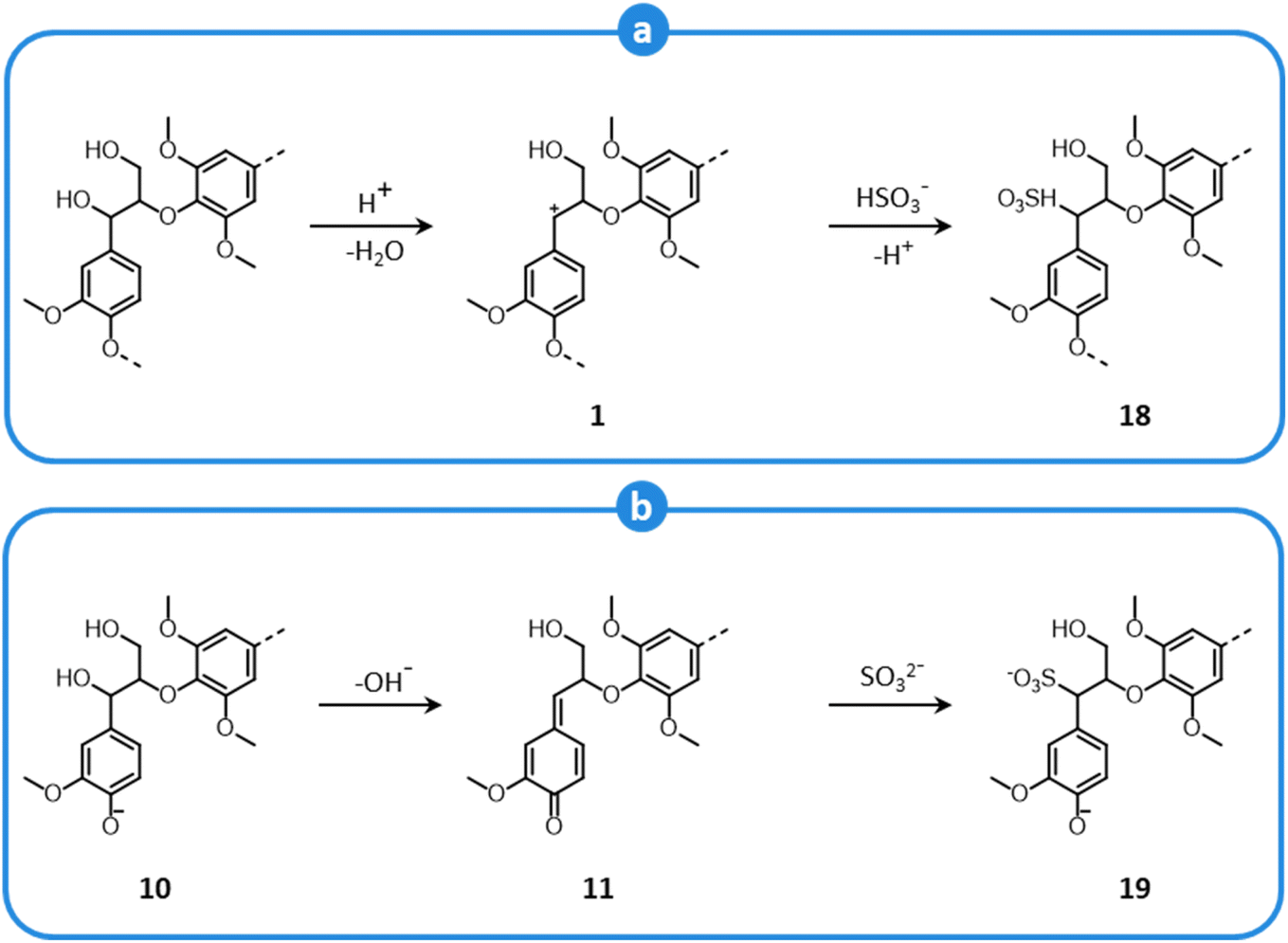 | ||
| Fig. 12 Sulfonation of β-O-4′ lignin motifs during sulfite pulping at acid conditions (a), and neutral/basic conditions (b). Based on Sjöström,150 and Evtuguin.161 | ||
The cleavage of lignin inter-unit bonds and the recondensation of lignin fragments principally follow the previously discussed acid-catalyzed or a base-catalyzed chemistry depending on the pH of the pulping medium (Fig. 7 and 9, respectively).161,163 Additionally, in neutral/alkaline sulfite pulping the cleavage of β-O-4′ linkages through the displacement of the β-substituent by a sulfite ion can also occur, in a mechanism known as “sulfitolysis” (Fig. 13), ultimately leading to the formation of lignin units with β-sulfonated, unsaturated side chains, prone to undergo repolymerization (20).150,161,163,164
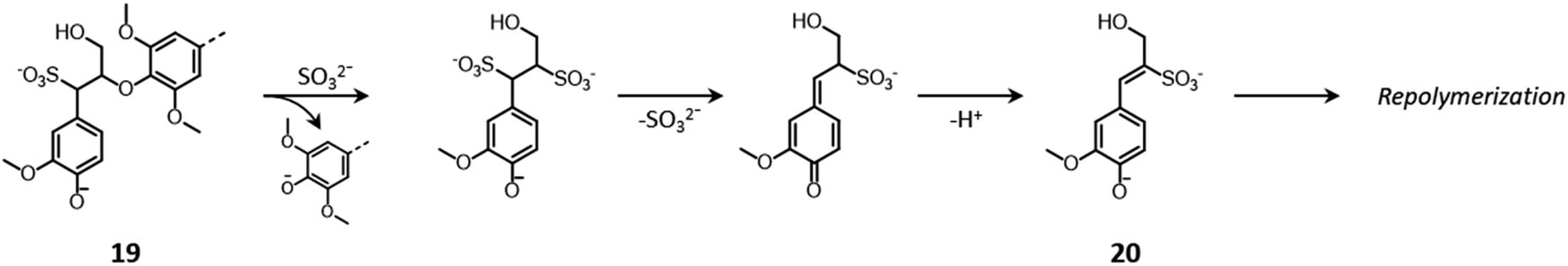 | ||
| Fig. 13 Sulfitolysis of β-O-4′ linkages during neutral/alkaline sulfite pulping. Based on Sjöström,150 and Evtuguin.161 | ||
Hemicellulose and cellulose are impacted as well by sulfite pulping and undergo acid-catalyzed hydrolysis and degradation (Fig. 8), or alkaline hydrolysis and peeling reactions (Fig. 10 and 11) in acid and basic medium, respectively.150,161
Overall, sulfite pulping has become gradually less popular as a pulping method due to the low strength of the produced pulps.150,162 Other shortcomings include the extensive degradation of hemicellulose and the low quality of the produced lignin streams (lignosulfonates), which are heavily condensed and contaminated by sulfur (3–8%),11 carbohydrates, and other impurities (e.g. minerals, extractives, …).11,149 Importantly, while the high degree of condensation and the presence of impurities hinder the upgrading of lignosulfonates toward high-value aromatics, the unique properties of these lignin products (chiefly the molecular weight and the degree of sulfonation) make them effective materials for the manufacturing of composites, dispersants, or flocculants.168 Currently, lignosulfonates account for nearly 90% of the total market of commercial lignin,169 and represent a prominent example of lignin valorization at the industrial scale.
In some configurations, soda pulping implements the use of anthraquinone that simultaneously stabilizes polysaccharides against peeling reactions and catalyzes the cleavage of β-O-4′ bonds in lignin.150,174,175 Anthraquinone (21) acts as a redox shuttle between carbohydrates and lignin (Fig. 14). On the one hand, it oxidizes the reducing ends of polysaccharides (23) toward alkali-stable aldonic acid groups (24), with the concomitant formation of anthrahydroquinone (22).150,174 On the other hand, the latter performs a nucleophilic attack on quinone methides (11), ultimately causing the reductive cleavage of β-O-4′ bonds and the regeneration of anthraquinone.150,174
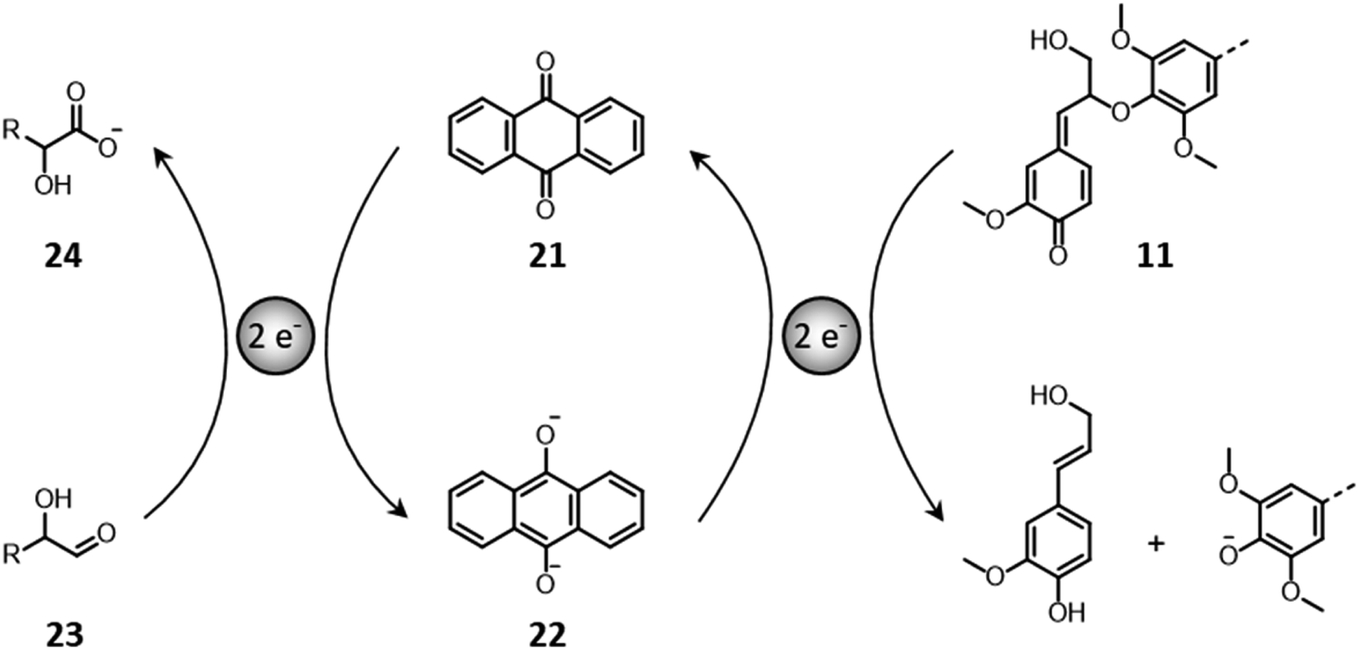 | ||
| Fig. 14 Anthraquinone–anthrahydroquinone reactions with carbohydrates and lignin in alkaline conditions. Based on Sjöström.150 | ||
Despite the improvement deriving from the introduction of anthraquinone, soda pulps possess lower mechanical resistance compared to kraft pulps.150 A major advantage of soda pulping is the production of sulfur-free lignin, which can be recovered by precipitation.11,171 This makes soda lignin more attractive as a raw material, compared to kraft lignin or lignosulfonates. Nonetheless, the high degree of condensation still represents an obstacle for the upgrading of soda lignin.11
Concentrated acid pretreatment targets the complete conversion of polysaccharides in the biomass to fermentable sugars, circumventing the need of an enzymatic hydrolysis of the pulp.176 However, extensive washing, detoxification and neutralization of the solution are required before fermentation.176 Moreover, concentrated acids are toxic, highly corrosive compounds, which are difficult to recycle when in solution.39,69 Thus, the stringent safety/equipment requirements and the large amount of neutralization waste deriving from the use of concentrated acids represent important drawbacks of concentrated acid pretreatments.39,69,176 In general, the use of dilute acid solutions is a more suitable option for the pretreatment of lignocellulose at industrial scale, as the enzymatic digestibility of the biomass can be greatly improved, and the process is less costly and more environmentally friendly.69,135,177
One drawback of acid pretreatment is that the released sugars may undergo acid-catalyzed degradation reactions, leading to the formation of furans (e.g. furfural, 5-hydroxymethylfurfural), organic acids (e.g. formic acid, levulinic acid) and condensation products (pseudolignin), which inhibit the subsequent downstream processing of the cellulosic pulp.39,69,176 Nonetheless, furans and levulinic acid can also represent attractive platforms for the manufacturing of polymers, chemicals, and fuels.6,178–181 Therefore, severe acid pretreatment configurations are sometimes adopted with the goal of converting (hemi)cellulose toward these intermediates.39
During acid pretreatment, lignin is partially solubilized and undergoes depolymerization and repolymerization reactions, following the pathways discussed for LHW pretreatment. The residual amount of inter-unit ether linkages in the isolated lignin decreases with the severity of the pretreatment.14 At higher severity, lignin fragments exhibit a greater tendency to undergo recondensation, and a larger redeposition of lignin on cellulose fibers occurs.14 This represents a crucial shortcoming of acid pretreatment, as it negatively affects both cellulose and lignin valorization downstream. Overall, the selection of optimal process conditions for the acid pretreatment of lignocellulosic biomass is dependent on the purpose of the pretreatment and on the targeted products.
Lignin oxidation follows a radical reaction mechanism, involving a multitude of possible depolymerization and repolymerization pathways.184–187 In contrast to the previously discussed acid- and base-catalyzed lignin reactions, in which lignin depolymerization only proceeds via the cleavage of inter-unit ether bonds, in this case the rupture of C–C linkages can also take place.184–187 C–C bonds in the side chains of lignin units can be broken, with the formation of phenolic aldehydes (25), acids (26) or quinones (27) (Fig. 15).184–187 Additionally, aromatic rings can be cleaved, with the consequent formation of aliphatic carboxylic acids (28).184,185 Condensation reactions between lignin radicals can yield biphenyl structures (29).184,185
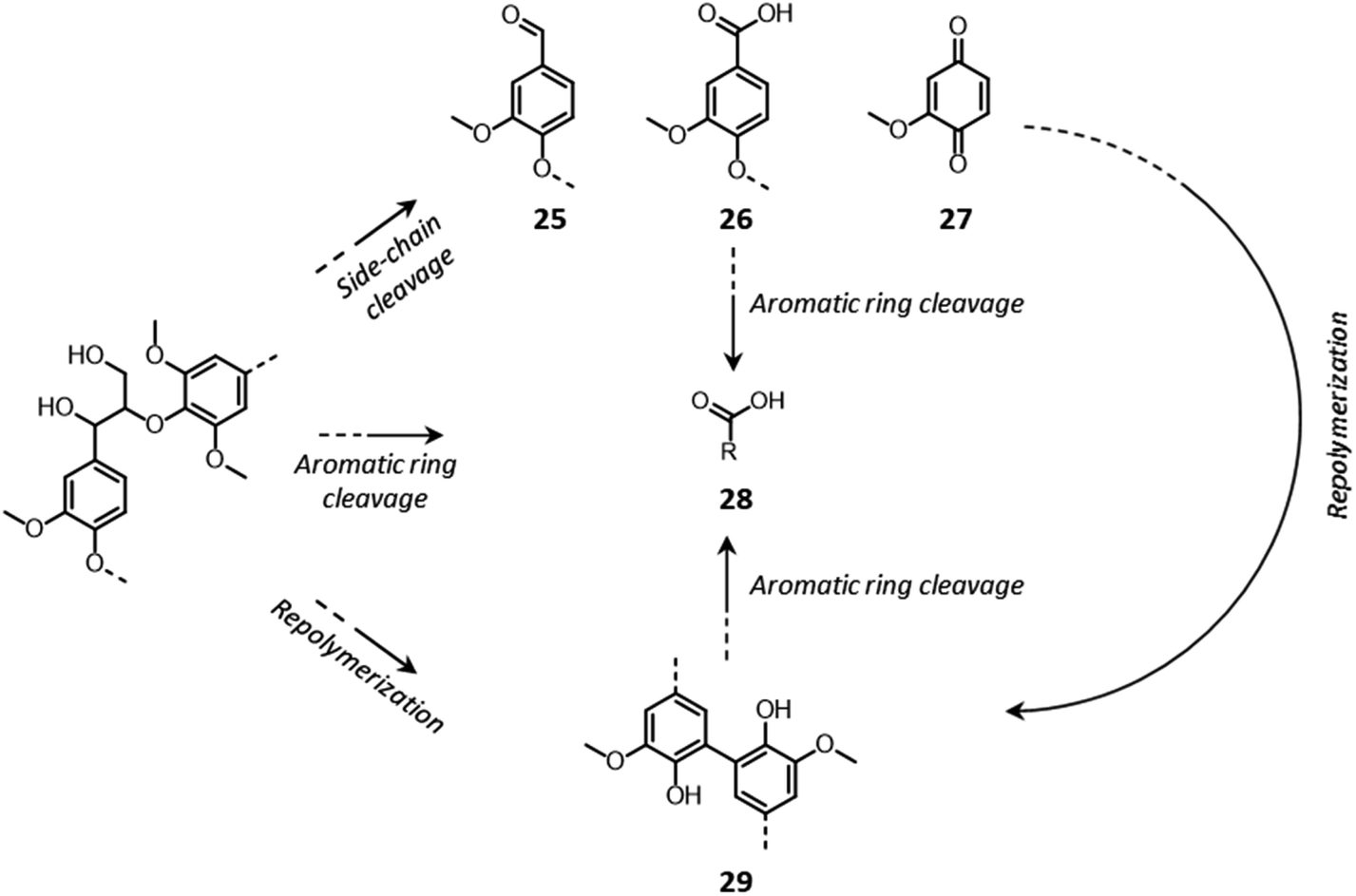 | ||
| Fig. 15 Lignin chemistry under oxidative conditions. Based on Schutyser et al.14 | ||
Along with lignin, hemicellulose and cellulose undergo substantial degradation under the non-selective action of the free radical species formed during the process. As a result, various organic acid byproducts are formed (e.g. succinic acid, glycolic acid, formic acid, acetic acid, …), which act as inhibitors of the subsequent enzymatic and fermentative processing, thereby limiting the effectiveness of the oxidative pretreatment.39,68,176
In view of this, a mild oxidative pretreatment is usually preferred over harsh conditions, since partial delignification can be achieved with low degradation of lignin and carbohydrate products, leading to an enhancement of the pulp processability via enzymatic or chemical methods.188,189 Moreover, the oxidized lignin obtained from a mild pretreatment can be further upgraded toward aromatic chemicals, thereby constituting an additional valuable product stream.190–193 In spite of this, the high cost of the most commonly adopted oxidizing agents currently represents an obstacle to the scale-up of oxidative pretreatments.68,82
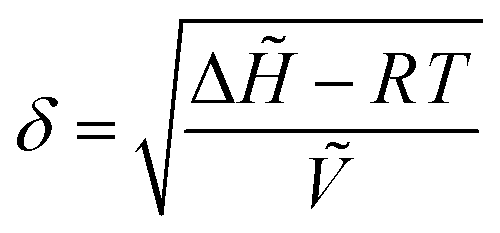 | (2) |
![[H with combining tilde]](https://www.rsc.org/images/entities/i_char_0048_0303.gif) is the molar heat of vaporization of the solvent (J mol−1), R is the ideal gas constant (J mol−1 K−1), T is the temperature (K), and Ṽ is the molar volume of the solvent (m3 mol−1). For optimal solvency, the Hildebrand solubility parameter of the solvent should match that of the solute, which, in the case of lignin, is about 23.5 MPa1/2.196,197,199 Depending on the solvent used, and on the use of an aqueous or a non-aqueous solution, only lignin or both lignin and hemicellulose are solubilized. Concomitantly, cellulose swelling occurs due to the action of the organic solvent (and water, when present), which disrupts the hydrogen bonds in the amorphous regions of cellulose, leading to an increased enzymatic accessibility.39
is the molar heat of vaporization of the solvent (J mol−1), R is the ideal gas constant (J mol−1 K−1), T is the temperature (K), and Ṽ is the molar volume of the solvent (m3 mol−1). For optimal solvency, the Hildebrand solubility parameter of the solvent should match that of the solute, which, in the case of lignin, is about 23.5 MPa1/2.196,197,199 Depending on the solvent used, and on the use of an aqueous or a non-aqueous solution, only lignin or both lignin and hemicellulose are solubilized. Concomitantly, cellulose swelling occurs due to the action of the organic solvent (and water, when present), which disrupts the hydrogen bonds in the amorphous regions of cellulose, leading to an increased enzymatic accessibility.39
The treatment temperature and time are generally comprised in the ranges of 100–200 °C and 30–150 min, respectively, and they are related to the solvent adopted.196 Moreover, acid co-catalysts (e.g. H2SO4, H3PO4, oxalic acid, …) or basic co-catalysts (e.g. NaOH, …) are often employed within organosolv processes to increase the rate of delignification or hemicellulose dissolution, and reduce the treatment duration or the operating temperature.194–197 The use of an acid co-catalyst can also enhance the depolymerization of cellulose. However, the formation of inhibitors of fermentative microorganisms (e.g. furfural, 5-hydroxymethylfurfural, organic acids, phenols, …) is favored under acid-catalyzed conditions, implying that a tradeoff should be found between lignocellulose deconstruction and the formation of degradation products.82,195,197
During organosolv pretreatment, LCCs are ruptured by the action of the organic solvent, contributing to the solubilization of large lignin fragments. Such lignin oligomers are further broken down by cleavage of β-O-4′ inter-unit bonds, following acid- or base-catalyzed reactions (Fig. 7 and 9).209 Repolymerization reactions between lignin moieties occur as well, and both the extent of the cleavage of ether linkages, and that of lignin recondensation increase with the process severity.196 In the presence of aliphatic alcohols solvents, lignin etherification may take place via nucleophilic attack of solvent molecules at the α-carbon of β-aryl ether motifs leading to the formation of α-etherified lignin structures (Fig. 16).17,199,210–214 The latter were shown to possess a considerably lower tendency to undergo condensation reactions and β-O-4′ bond cleavage as compared to benzylic carbenium ions or parent β-aryl ether motifs with α-hydroxyl groups.118,199,211,215 Noteworthy, lignin etherification is a reversible reaction and native-like lignin structures may be obtained by subjecting etherified lignin to mildly acidic conditions. Lignin stabilization by aliphatic alcohols was reported to be an effective strategy for protecting lignin against repolymerization during pretreatment,17,213,214,216 ultimately opening the way to the successful downstream conversion of lignin to chemicals.118,211,213,215–218
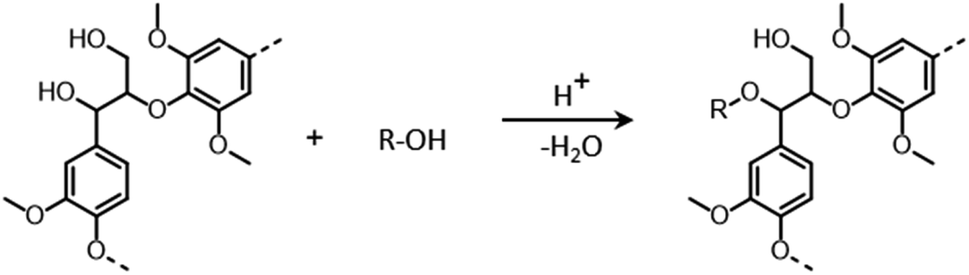 | ||
| Fig. 16 Lignin stabilization by etherification during organosolv pretreatment in the presence of aliphatic alcohols. | ||
A great advantage offered by organosolv pretreatment compared to other fractionation methods is the possibility of producing three separate product streams: a cellulose-enriched pulp, an aqueous fraction enriched with hemicellulose derivatives (e.g. sugars, alkylated sugars, furans, organic acids, …) and an organic fraction enriched with lignin derivatives (polyphenols), as illustrated in Fig. 17.39,194–198 This feature enables the valorization of all lignocellulose constituents, provided that no excessive degradation takes place during the pretreatment.
 | ||
| Fig. 17 Schematic outline of the organosolv process for the solvolytic extraction of lignin (and hemicellulose) derivatives from lignocellulosic biomass. Note: the isolation of the product streams enclosed in colored boxes on the right-hand side of the figure can be achieved only after a series of downstream operations. These may include a filtration step to isolate the solid pulp from the liquid extract, followed by precipitation of lignin derivatives from the liquid fraction, before further filtration to separate insoluble lignin derivatives from soluble hemicellulose derivatives, and concentration/purification of hemicellulose and lignin streams. | ||
On the other hand, the high cost of organic solvents imposes a downstream recovery step. Moreover, when organosolv fractionation is used as a pretreatment for further enzymatic/fermentative conversion of the pulps, organic solvents should be completely removed from the pulp by extensive washing, otherwise they would cause inhibition of the subsequent saccharification and fermentation steps.39,195,197 Clearly, this leads to the need of additional unit operations, and, therefore, additional costs. From this point of view, the selection of the solvent has a crucial impact on the economics of the organosolv process. The use of low-boiling solvents, such as methanol or ethanol, results in high operating pressures, and strict requirements for the reactor design, but allows a facile solvent recovery by distillation.194–198 Conversely, the adoption of high-boiling solvents, such as ethylene glycol or glycerol, entails low operating pressure and less stringent reactor requirements, but also more energy-intensive solvent recovery by distillation.198 In addition, for the solvent selection, the possible formation of azeotropes should also be taken into account (e.g. ethanol–water, n-butanol–water, tetrahydrofuran–water…), as it could complicate the recovery of the solvent by distillation.198 Other aspects that are to be taken into account for the choice of a suitable organic solvent include its flammability, toxicity and sustainability.196Table 2 summarizes important features of some solvents commonly adopted in organosolv processes. Overall, ethanol is among the preferred solvents for organosolv pretreatment, since it possesses a high ability to solubilize lignin (δ = 26.5 MPa1/2), a low toxicity, and can be produced from fermentation of the hydrolyzed pulp, creating an integrated biorefinery process.219 Another alluring option is the adoption of biphasic solvent mixtures (e.g. n-butanol and water), which have been reported to facilitate the downstream products separation.198,212,220–222
| Organic solvent | Boiling point (°C) | Flash point (°C) | δ (MPa1/2) | Green solvent ratingb | |||
|---|---|---|---|---|---|---|---|
| Safety scorec | Health scored | Environment scoree | Rankingf | ||||
| a Hildebrand solubility parameter. b From 1 (greenest) to 10 (least green). c The safety score is determined based on the flash point temperature of the solvent.223 d The health score reflects the occupational hazard. It is established based on the hazard statements in the Globally Harmonized System of Classification and Labelling of Chemicals.223 e The environment score takes into account the toxicity toward aquatic life, bioaccumulation, the tendency to form volatile organic compounds, and the carbon footprint of the synthesis, recycling, and disposal of the solvent.223 f The overall ranking is established based on the criteria: (i) a solvent is classified as hazardous (H) if one of the scores is greater than or equal to 8, or if two of the scores are greater than or equal to 7; (ii) a solvent is classified as problematic (P) if one of the scores is equal to 7, or if two of the scores are comprised between 3 and 7; (iii) in all other cases the solvent is classified as recommended (R).223 g Methyl isobutyl ketone. | |||||||
| Methanol | 65 | 12 | 29.6 | 4 | 7 | 5 | P |
| Ethanol | 78 | 13 | 26.5 | 4 | 3 | 3 | R |
| Propanol | 97 | 24 | 24.6 | 4 | 3 | 3 | R |
| n-Butanol | 118 | 35 | 23.2 | 3 | 4 | 3 | R |
| Ethylene glycol | 197 | 111 | 32.9 | 1 | 2 | 5 | R |
| Acetone | 56 | −17 | 20.0 | 5 | 3 | 5 | P |
| MIBKg | 117 | 18 | 17.0 | 4 | 2 | 3 | R |
| Acetic acid | 118 | 44 | 21.4 | 3 | 7 | 3 | P |
Organosolv pretreatment has attracted a lot of attention in the recent years and, currently, different processes have been scaled-up to the pilot or industrial scale (e.g. organocell process, lignol process, CIMV process).39,127,195,197 Nevertheless, the effective conversion of the lignin fraction into valuable products still represents a challenge. Organosolv lignin possesses a higher purity compared to lignin streams obtained from other fractionation methods (i.e. less contamination by carbohydrate residues or chemical additives), and a more native-like structure (i.e. lower degree of condensation).11,15 These features pave the way for the upgrading of the lignin isolated from organosolv pretreatment toward aromatic chemicals. The future success of industrial scale organosolv pretreatments depends on the development of processes that allow upgrading each isolated fraction into marketable end-products.195 Ideally operated at low pressure and with inexpensive, recoverable, green solvents, these processes must combine an effective lignin (and hemicellulose) extraction and the preservation of a digestible cellulose fraction.207,224,225
The use of ILs for the pretreatment of lignocellulosic biomass has become a popular strategy, since these solvents possess excellent performance with respect to the solubilization of biomass components at low severity conditions, with operating temperatures typically in the range of 90–130 °C, and durations of 1–24 hours.82,176 More specifically, ILs can dissolve cellulose, hemicellulose and lignin, and their selectivity with respect to the dissolution of different biomass components can be tuned by modifying the cation and the anion.226–228
Cellulose solubility depends strongly on the nature of the anion, since the cellulose dissolving power of ILs is related to the capacity of their anions to form hydrogen bonds with hydroxyl groups in the cellulose structure.82,226,228 In addition to the anion, the choice of the cation affects cellulose solubility, which decreases with the lengthening of alkyl chains in the cation.229,230 The use of protic cations dramatically reduces cellulose solubility, due to increased interactions between cations and the anions that limit the hydrogen bonding capacity of the latter.230
The solubility of hemicellulose and lignin is also predominantly dependent on the type of anion used, involved in the cleavage of LCCs and inter-unit bonds (glycosidic bonds in hemicellulose and ether bonds in lignin).226,228 During IL pretreatment, the solubilized lignin fragments undergo depolymerization and recondensation reactions, following pathways analogous to those previously discussed for LHW and acid pretreatment, with the initial formation of benzylic carbenium ions that react with the anions of the IL to generate enol ether structures, ultimately leading to the production of Hibbert's ketones (dominant pathway in the presence of strongly coordinating anions) or C2-aldehyde-substituted phenolics (dominant pathway in the presence of weakly coordinating anions), which can then undergo further recondensation.231,232
Two main strategies have been developed based on the use of ionic solvents for lignocellulose pretreatment: (i) a dissolution approach aimed at dissolving the totality of lignocellulose in the ionic liquid for the subsequent recovery of the isolated fractions, and (ii) an ionosolv approach targeting the solubilization of lignin (and hemicellulose in some cases) to leave behind a cellulosic pulp.226 In the dissolution approach, after pretreatment, the different biomass fractions are recovered with a two-stage process. Firstly, (hemi)cellulose is precipitated by addition of an antisolvent, usually an organic solvent or a mixture of an organic solvent and water. Subsequently, lignin can be precipitated in acidified water.233–235 Conversely, in the ionosolv approach, lignin is directly precipitated after pretreatment by addition of water, while hemicellulose derivatives can be removed from the IL by solvent extraction.236–238
The properties of the recovered fractions are greatly modified after IL pretreatment. In the dissolution approach, the regenerated cellulose possesses a considerably lower crystallinity compared to the initial biopolymer, which results in a fast enzymatic conversion in the subsequent saccharification step.228,233,235 On the contrary, in the ionosolv approach cellulose crystallinity remains mostly unaltered after pretreatment.226 Hemicellulose is usually partially hydrolyzed by ILs, and is recovered in the form of oligo- and monosaccharides.226,237 Degradation products such as furans are also formed due to the occurrence of dehydration reactions. The lignin fraction recovered after IL pretreatment possesses a lower content of inter-unit ether bonds and is partly condensed.239,240 Additionally, condensation reactions between the lignin and the IL may occur, at an extent that increases with the operating temperature.239,240 Overall, the IL lignin fractions possess similar features to organosolv lignin streams and are regarded as a promising substrate for further upgrading toward aromatic chemicals.171,241
Despite the advantages offered by ILs in terms of the selective solubilization and deconstruction of biomass, few bottlenecks exist for the development of IL-based biorefineries. First, ILs are expensive solvents, hence their effective recovery is imperative for the economic viability of the pretreatment.176,226,228,237 While different strategies have been explored for the recovery and recycling of ILs, including liquid–liquid separation and distillation, the presence of residual biomass components (e.g. sugars, minerals, extractives, …) in the recovered ILs reduces their performance upon reuse.226 Moreover, most ILs cause inactivation of saccharification enzymes and are toxic to fermentative microorganisms. Thus, a complete removal of ILs from the recovered pulp is required before further conversion, resulting in the need of extensive washing, and additional processing costs.82,176,228
Similarly to ILs, DESs are extremely versatile and their properties can be tuned by changing the hydrogen bond acceptor or donor.242 In view of their ability of selectively dissolving biomass components, DESs have been recently explored as solvents for the pretreatment of lignocellulose.243–247 The disruption of the lignocellulose structure follows a mechanism that is analogous to that discussed for IL pretreatment (see above). Cellulose can be dissolved in DESs thanks to the formation of extensive hydrogen bonding between the hydrogen bond acceptor of the DES and the hydroxyl groups in cellulose structure.243,244,248 Additionally, LCCs can be cleaved during pretreatment with DESs, facilitating the solubilization of hemicellulose and lignin. Partial hydrolysis and degradation of hemicellulose may occur, depending on the properties of the DES.243,248 Lignin undergoes depolymerization (mainly cleavage of inter-unit ether bonds) and repolymerization pathways similar to those described for the use of ILs.246,249 Ultimately, the residual content of β-O-4′ linkages and the degree of condensation of the isolated lignin depend on the properties of the solvent and the operating temperature.249–252
DESs offers several advantages for the pretreatment of lignocellulosic biomass. First, in contrast to ILs, the components of DESs are inexpensive and readily available chemicals. For instance, one of the most adopted hydrogen bond acceptors, choline chloride, is a common additive used in animal feeds, and urea, a frequently adopted hydrogen bond donor, is used in fertilizers.243,248 Second, DESs possess a much lower inhibitory effect toward cellulolytic enzymes and fermentative microorganisms, implying that the complete purification of the pulp from the solvent is not necessary after DES pretreatment.247,248 One drawback of DESs is their high viscosity, which entails high pumping costs.243 In addition, DESs recycling is challenging and the lack of effective methods represents a prominent limitation to the technoeconomic viability of DESs for biorefinery applications.247,248,253,254 Nevertheless, these green solvents are considered promising candidates for the pretreatment of lignocellulose and for the subsequent valorization of the isolated lignin.246,247,255,256
3.4. Biological pretreatment methods
Biological pretreatments rely on the delignification of lignocellulosic biomass performed by microorganisms or enzymes. These methods are attractive since they have a low energy demand (they are carried out at ambient conditions), do not require expensive chemicals, are environmentally friendly, and do not lead to the formation of inhibitors of the subsequent saccharification and fermentation steps.69,257–260Microbial decomposition of lignin can be carried out both using bacteria or fungi (principally, white-, brown-, and soft-rot fungi).257–260 In general, white rot fungi are preferred, since they are particularly active in lignin degradation and they do not cause excessive decomposition of (hemi)cellulose.257–260 During pretreatment, white rot fungi secrete a range of enzymes, such as laccases, lignin peroxidases, and manganese peroxidases, which contribute to depolymerize lignin by an oxidative action.257,259 Laccases attack phenolic lignin units to form phenoxy radicals and, concomitantly, reduce oxygen to water (Fig. 18a). Due to their low redox potential, laccases cannot oxidize non-phenolic lignin units.257,258 However, in the presence of a suitable low-MW mediator, such as syringaldehyde, acetosyringone, and other lignin-derived monophenolics, the oxidation of non-phenolic units can occur as well.257 Lignin peroxidases possess a high redox potential and can directly attack both phenolic and non-phenolic units, generating cation radicals that can react further to cause the breakdown of C–C bonds, leading to side-chain cleavage or ring-opening reactions.257,258 In this case, the final electron acceptor is hydrogen peroxide (Fig. 18b). Manganese peroxidases oxidize Mn2+ to Mn3+, which forms a complex with a chelator, such as oxalate or malonate, and diffuses away from the enzyme to attack phenolic lignin units, forming phenoxy radicals (Fig. 18c).257,258 In the presence of unsaturated lipids or thiols, their oxidation by manganese peroxidases can produce peroxyl radicals, which act as mediators in the oxidation of non-phenolic lignin units.257 Along with these enzymes, others contribute to lignin oxidation, such as versatile peroxidases, dye-decolorizing peroxidases, and various accessory enzymes, including oxidases that produce hydrogen peroxide, and dehydrogenases that reduce lignin depolymerization derivatives, preventing their subsequent recondensation.257,258
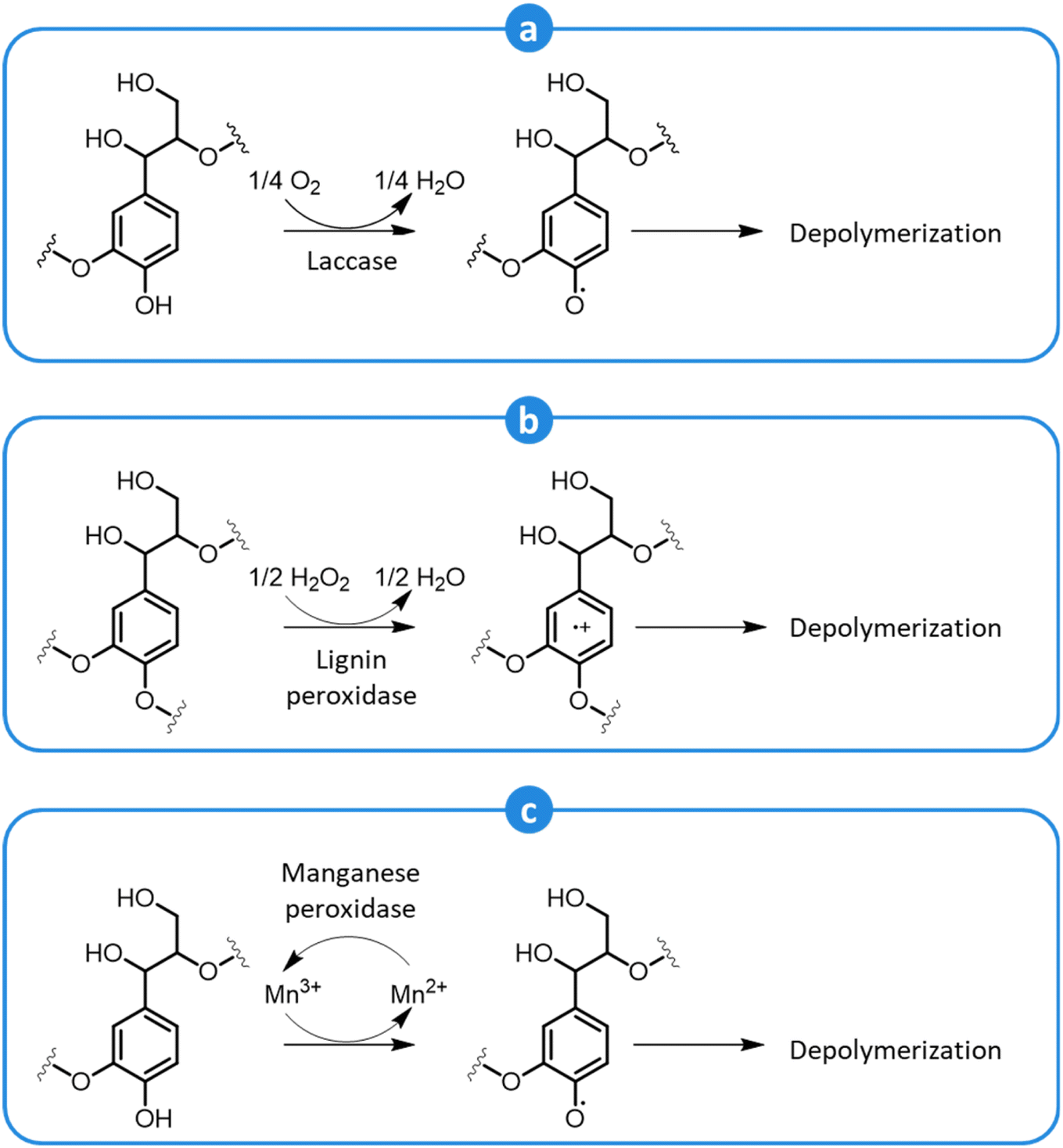 | ||
| Fig. 18 Oxidative depolymerization of lignin by the action of laccase (a), lignin peroxidase (b), and manganese peroxidase (c). Adapted from Vasco-Correa et al.257 | ||
Biological pretreatments lead to variable degrees of delignification, depending on the structural complexity of the feedstocks, on the bacterial or fungal strains employed, and process conditions, such as the operating temperature, the pH, the moisture content, the substrate particle size, the aeration conditions, and the incubation time.258–260 Typically, the lignin recovered from biological pretreatments possesses a lower MW compared to native lignin, a decreased content of inter-unit ether linkages, and is partially condensed.257,261 Noteworthy, the so-formed lignin fractions were recently reported to act as suitable electron donors for the redox cellulolytic enzymes such as lytic polysaccharide monooxygenases, paving the way for the exploration of synergies between oxidase enzymes for the simultaneous decomposition of lignin and polysaccharides.262,263
In spite of the inherent advantage of offering a low-severity strategy for lignocellulose pretreatment, biological methods suffer important shortcomings. The sterilization of biomass that is frequently required before microbial pretreatment to prevent outcompetition of lignolytic microbes by the indigenous microbes that are naturally present in the biomass, is an energy-intensive operation.257,258 Moreover, the long incubation time needed for microbial growth (from few days to several weeks) is incompatible with industrial needs.257–260 In addition, most microorganisms cannot use lignin as a carbon source and consume part of the (hemi)cellulose for their growth, leading to an undesirable loss of polysaccharides.257–259 Such limitations can be overcome by the adoption of lignolytic enzymes for the pretreatment.257 However, this approach requires a separate unit for the production of enzymes. Furthermore, lignolytic enzymes are typically produced in low amounts by lignin-degrading microbes and, even though they are extracellular, their recovery from the fermentation broth is challenging.257 Overall, these drawbacks currently hamper the implementation of biological pretreatments at the industrial scale.
3.5. Comment
From the overview of pretreatments methods presented above, it appears that no simple method currently stands out as a “one-size-fits-all” solution, able to fully embrace the complexity of the task. Nevertheless, important progress is currently being made in the development of novel approaches to biomass processing. In this perspective, lignin depolymerization will probably play a major role, giving access to (industrially) useable lignin streams, but complementary methods will be needed for certain biomasses or for certain applications. In this context, the research community is rightfully focused on developing the knowledge that will allow scaling up the most effective pretreatment methods.4. Valorization of the isolated lignin fractions
Lignocellulose pretreatments produce large amounts of lignin as a byproduct. While pulp and paper facilities generate about 50–70 million tons of lignin per year, lignin production from lignocellulose biorefineries is expected to reach nearly 225 million tons per year by 2030, in view of to their rapid diffusion.264 Thus, the development of processes for the effective valorization of lignin is essential for improving the economic viability of second generation biorefineries.Although in pulping processes the lignin fraction is often employed as an energy source, alternative conversion routes for lignin streams isolated from biomass pretreatment have been developed, thanks to which a range of lignin-derived products can be manufactured (Fig. 19), including materials (e.g. thermosets, thermoplastics, elastomers, …), fine chemicals (e.g. vanillin, eugenol, …), and bulk chemicals (e.g. benzene, toluene, xylene, phenol, …).265–268 The choice of the most suitable valorization routes for lignin streams isolated from biomass pretreatment is primarily dependent on the features of lignin fractions (e.g. MW, degree of condensation, presence of contaminants, content of β-O-4′ linkages, …). Lignin streams possessing a high purity are suitable for high-value materials applications, such as the manufacture of carbon fibers or resins (Fig. 20).265,267,269 In addition, the residual presence of a high content of labile inter-unit ether linkages in the isolated lignin is convenient for its further conversion toward chemicals (Fig. 20).16,265,267,270 On the other hand, highly condensed, contaminated lignin streams typically require extensive purification and harsher treatment conditions for their upgrading to chemicals and materials, which is why low-value applications, such as the production of syngas and power, are often preferred in this case (Fig. 20).264,265,267
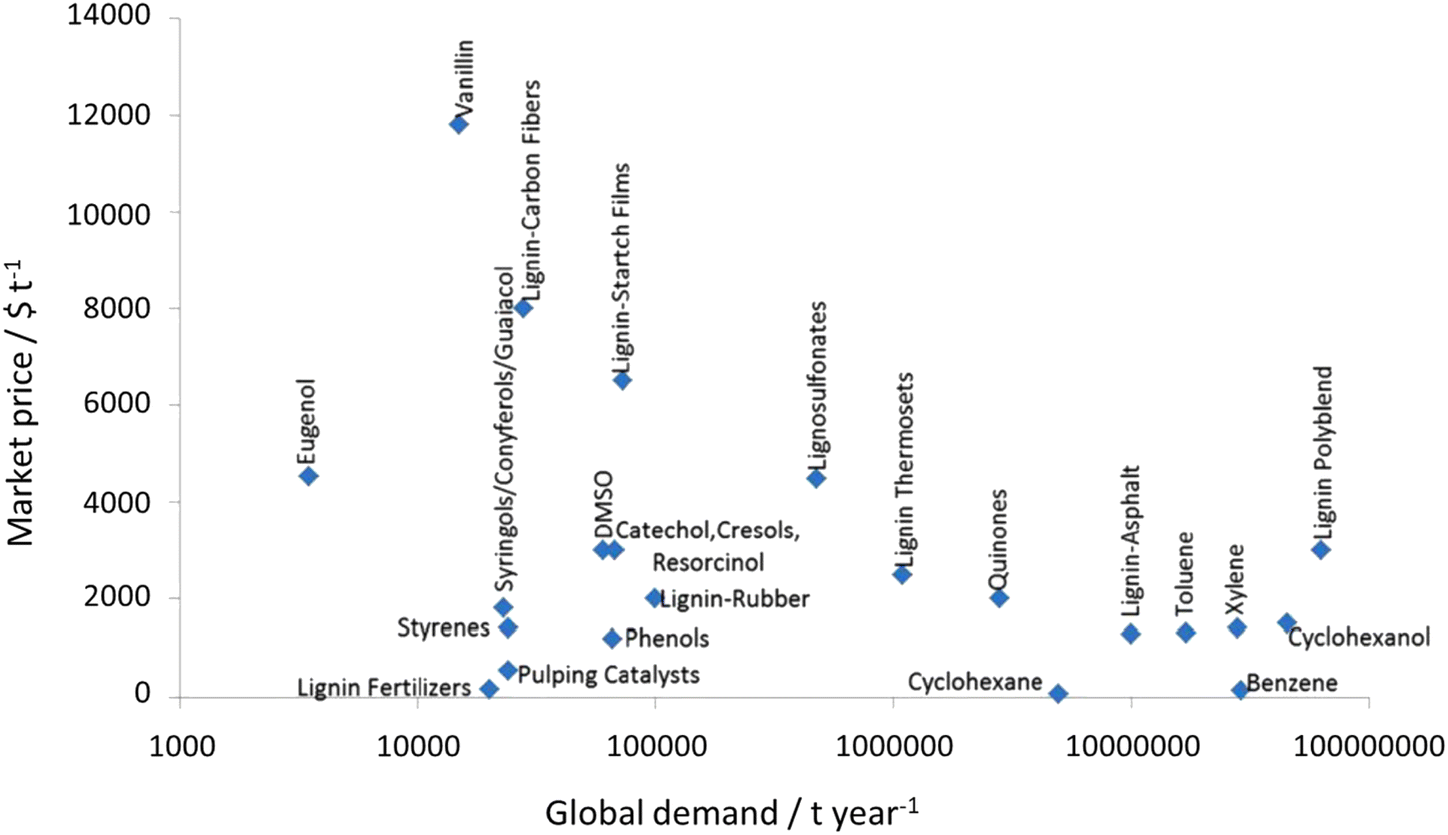 | ||
| Fig. 19 Market price and global demand of some lignin products (price estimations are from 2006). Adapted from Gillet et al.265 | ||
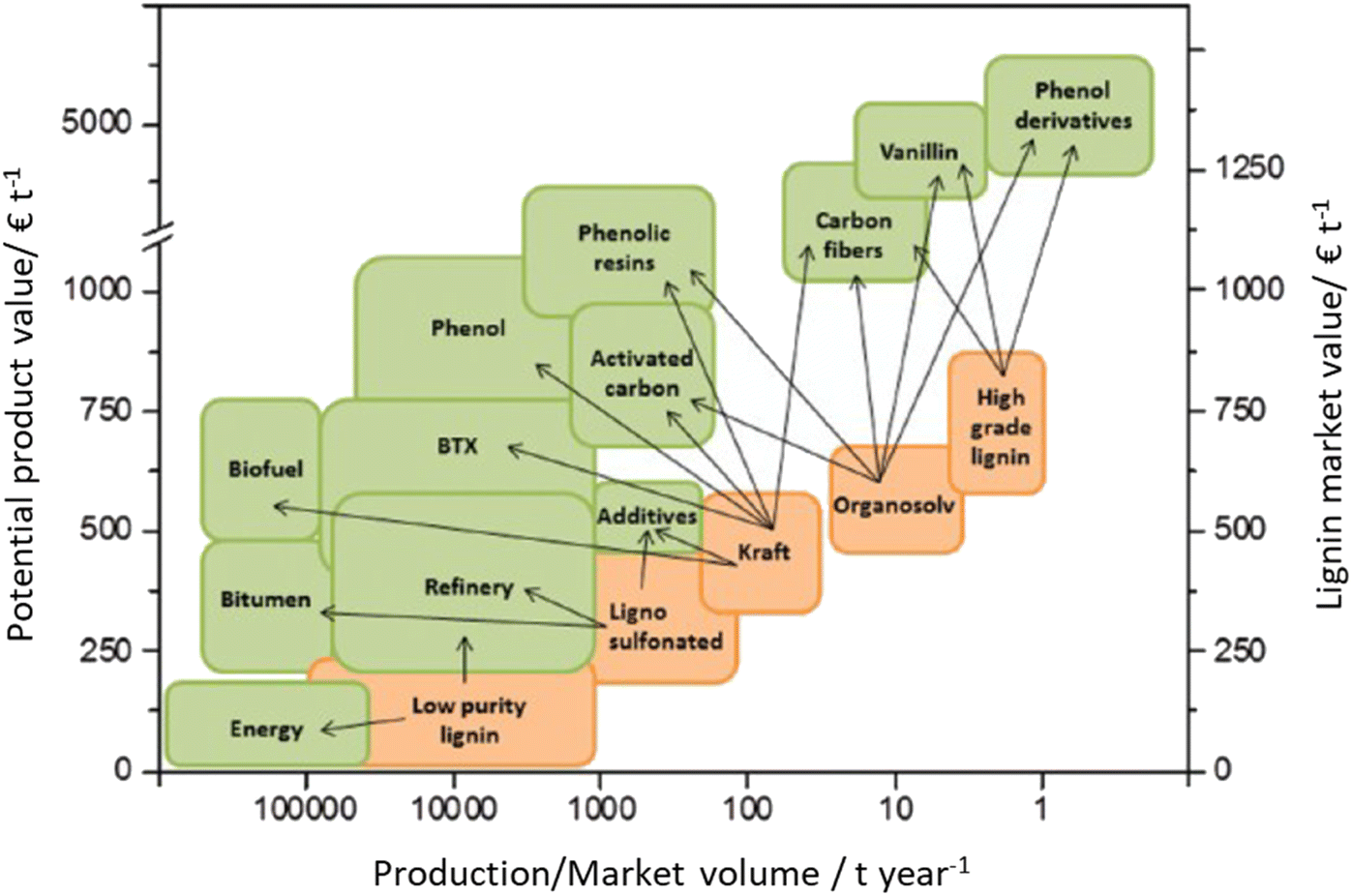 | ||
| Fig. 20 Market value of isolated lignin streams (orange) and potential market value for lignin-based products (green) with respect to their production/market volume (price estimations are from 2011). Adapted from Behling et al.272 Note: high-grade lignin indicates lignin fractions that were subjected to extensive purification after isolation using techniques such as liquid–liquid extraction, vacuum distillation, liquid chromatography, crystallization, etc.273 | ||
The most alluring options for lignin valorization include the manufacturing of polymers and chemicals, as they entail a high economic potential, in terms of market price and size (Fig. 19). The production of polymers may represent a direct valorization route, in which the lignin streams isolated from lignocellulose pretreatment can be employed in the manufacturing process as they are, or after minor purification/chemical modification. Conversely, the production of chemicals involves an extensive depolymerization of lignin into monomeric products before commercialization of lignin derivatives or further upgrading toward selected chemicals.264,266,271 An overview of the strategies adopted for the utilization of isolated lignin streams in polymer manufacturing and for their depolymerization into low-MW aromatics is presented in the following sections.
4.1. From lignin to polymeric materials
The use of lignin as a substitute for fossil-derived building blocks in the manufacture of polymers is a convenient way for reducing the environmental impact of these products, while improving material properties such as flame-retardant ability, UV- and thermo-oxidative resistance, and antimicrobial resistance.269 In this context, the most widespread applications include the manufacturing of thermoplastic blends, phenol-formaldehyde resins, epoxy resins, polyurethanes, and carbon fibers.269,271,274–277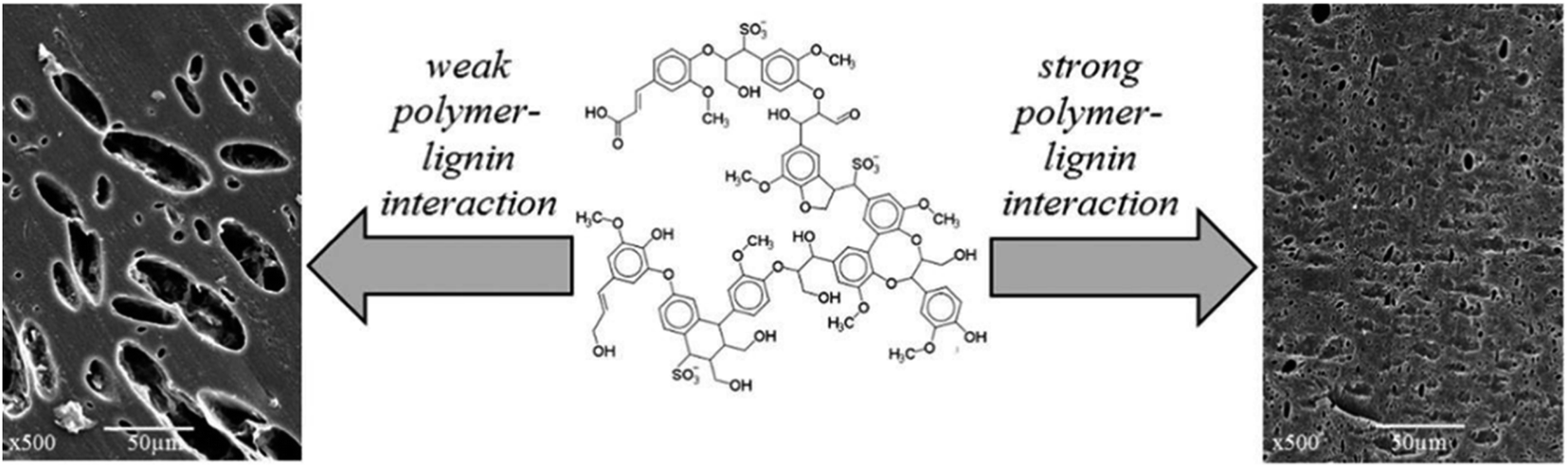 | ||
| Fig. 21 Possible morphologies of lignin–polymer blends: heterogeneous blend with large dispersed particles (left) and homogeneous blend with no observable particles. Adapted from Kun et al.275 | ||
Lignin blends with polyolefins, such as polyethylene and polypropylene, have been studied with the goal of exploiting the radical scavenging ability of lignin to enhance the stability of polyolefins against UV radiation and thermal oxidation.269,275 While a low lignin content in the blend can effectively stabilize polyolefins, the introduction of larger amounts of lignin results in a higher tensile modulus of the blends, due to the inherent stiffness of lignin molecules, but also in a lower strength and deformability, determined by the poor interactions between the components in the blend.280 An important point to consider with respect to the manufacturing of blends with lignin is the compatibility of the different components in the blends. The presence of a large amount of hydroxyl groups in lignin structures imparts lignin with a polar character, which results in weak interactions with apolar polymers, such as polyolefins, and, typically, in the formation of heterogeneous blends with large dispersed particles.279,281,282 A better compatibility can be expected for the blending of lignin with polymers containing aromatic rings (e.g. polystyrene, polyethylene terephthalate, …), by virtue of π-stacking interactions.283 However, also in this case, a complete miscibility between lignin and aromatic polymers is seldomly achieved and the produced blends usually exhibit poor deformability.275,282,283 A similar scenario is observed also for blends between lignin and more polar polymers, such as polyvinyl chloride, polyvinyl alcohol or polylactic acid, despite their ability to form hydrogen bonds.275,282
To improve the homogeneity (and the mechanical properties) of lignin–polymer blends different approaches have been developed. The introduction of a plasticizer in the blend is often adopted as a method to reduce lignin–lignin interactions, favoring the dispersion of lignin in the blend.274,275,280 Additionally, chemical modification of the lignin (e.g. via esterification) can be used to reduce its polarity and increase its compatibility with apolar polymers.275,280,281,284,285 Lignin depolymerization and/or fractionation can also be used to generate lignin fractions possessing a narrowed molecular-weight distribution and specific structural features that enhance compatibility.279,286
Another crucial challenge in the manufacture of thermoplastic blends with lignin is the high glass transition temperature of lignin streams.274,280 While lignin exhibits a thermoplastic behavior at low temperature, at high temperature crosslinking reactions are initiated and lignin acts as a thermoset.269,274 This transformation is observed already at temperatures close to the glass transition temperature of lignin, which makes thermal blending particularly complicated, as it leads to blends that possess an inherent brittleness.269 However, the use of a plasticizer, or chemical modification/depolymerization of lignin are strategies that can be adopted also for lowering the glass transition temperature of lignin, thereby facilitating the blending process.269 Overall, the manufacturing of thermoplastic polymer blends is regarded as a rather simple and inexpensive way of producing lignin-based materials for high added-value applications.269,287
Traditionally, PF resins are prepared by reacting phenol with formaldehyde. More specifically, formaldehyde reacts with the aromatic ring in the ortho- or para-position to yield hydroxymethyl groups, which subsequently condense to form methylene or ether bridges.289 Upon curing of the resin, further crosslinking between residual free hydroxymethyl groups polymer chains occurs, and a rigid thermoset is obtained.289
Since the phenol employed for the fabrication of PF resins, is typically derived from fossil resources via the cumene hydroperoxide process, the use of lignin as an alternative to phenol represents an attractive opportunity to simultaneously decrease the price and increase the sustainability of PF resins.288,290–292 Nevertheless, the poor solubility of lignin in common organic solvents complicates its incorporation in the polymers.288,293 In addition, the reactivity of the isolated lignin fractions with formaldehyde is lower than that of phenol, due to the presence of methoxy groups in the ortho-positions of lignin units, and also due to the higher steric hindrance of large lignin molecules.269,271,288,294 For these reasons, only part of the phenol can be replaced by lignin (usually less than 30%), and a higher reaction temperature as well as longer reaction times are often required to manufacture resins with satisfactory mechanical properties.288,290,295 Moreover, the low reactivity of lignin with formaldehyde can constitute an issue since it may lead to the presence of residual formaldehyde after curing, and to the subsequent release of this toxic chemical.292,296
Several strategies have been developed to overcome these limitations and improve the reactivity of lignin fractions. Chemical modification of the raw lignin streams via hydroxymethylation, demethylation, or phenolation can be adopted to increase lignin reactivity with formaldehyde.284,288,290,291,293,294,297–299 In addition, the partial depolymerization of lignin into low-MW aromatics is another way to increase the number of active sites available for crosslinking reactions.288,290,300 Thanks to these modifications of the lignin structure, a higher degree of phenol replacement by lignin in PF resins is achieved (>50%) and the adhesive properties of the lignin-based resins are improved.290,291,294,300
Currently, about 80–90% of commercial epoxy resins are prepared by reacting bisphenol-A (BPA) with epichlorohydrin.301
During the last three decades, the use of BPA for the fabrication of epoxy resins has raised increasing concerns, since BPA is a renowned endocrine disruptor, and leaching of this chemical from polymeric materials had been reported.303 From this point of view, the adoption of lignin as a replacement for BPA in epoxy resins constitutes an attractive option for eliminating the risks related to the use of such harmful chemical, while concomitantly reducing the environmental impact of these polymeric products.304–306 However, few downsides exist with respect to the use of isolated lignin fractions as a replacement for BPA. The low purity and the high content of minerals constitute a limitation to the use of kraft lignin, lignosulfonates, and soda lignin, imposing an initial purification step.269,301,302,307 Moreover, the low solubility of lignin in both aqueous and organic media constitutes another limitation to the employment of lignin for the production of epoxy resins.293,301,305 In addition, lignin possesses a lower reactivity compared to BPA, due to the typically high MW and the lower content of hydroxyl groups.301,304 For these reasons, chemical modification (e.g. via hydroxyalkylation, demethylation, phenolation)304,308,309 and partial depolymerization of isolated lignin streams are often adopted to increase lignin solubility and the content of reactive functional groups in lignin molecules.269,284,293,301,310,311 Overall, lignin can be employed to replace over 50% of the BPA, to yield epoxy resins with electrical, mechanical, and thermal properties comparable to lignin-free polymers.269,301,307,310–312 Most promisingly, the use of high-purity, low-MW lignin streams has been recently reported to allow for the full replacement of BPA in the production of bio-based epoxy resins.307,313–317
Driven by the increasing awareness of the impact of fossil-based materials on climate change, as well as by an escalation of the prices of fossil-based polyols, the use of lignin as a sustainable, inexpensive alternative has attracted conspicuous attention.271,302,318,319 Isolated lignin fractions can be employed for the synthesis of PUs either directly or after chemical modification/fractionation.321–323 The latter is aimed at increasing lignin solubility and reactivity with cyanate groups.318,319,324,325 In particular, oxypropylation is commonly employed to extend the distance between hydroxyl groups in the lignin matrix and reduce steric hindrance effects.284,319,324,326 Lignin depolymerization is also adopted prior to polymer synthesis to increase the number of reactive groups in lignin molecules.318,319,327 When unmodified lignin is employed for PUs synthesis, it can replace only a limited fraction of the polyol (typically below 30%) without compromising the polymer performance.271,318,328 On the contrary, the prior chemical modification of the lignin makes it possible to achieve a higher degree of incorporation of the biopolymer in PUs (>50%).302,318,319,329 In general, the introduction of lignin in PUs results in polymers with increased rigidity (larger tensile modulus) and a higher thermal stability.302,318,319 In addition, the use of lignin in PU foams advantageously reduces the polymer flammability, which is one of the major drawbacks of these polymeric materials.319
On the other hand, the high cost of carbon fibers considerably restricts the range of applications of these materials.271,330,331,334,335 It is estimated that over 50% of the cost of carbon fibers is related to the production of their precursor, which is typically polyacrylonitrile, a fossil-based polymer made by condensation of acrylonitrile, methyl methacrylate and itaconic acid.271,334 The adoption of lignin as an alternative precursor for carbon fibers is a promising solution to simultaneously reduce the cost and improve the sustainability of final products.336–339
The preparation of carbon fibers follows three main steps: a spinning of the precursor, a thermal stabilization step, and a carbonization step.330–332,336 When lignin is adopted as a precursor, the properties of the isolated lignin fraction, such as the presence of contaminants and the degree of condensation have a substantial impact on the requirements for the manufacturing process as well as on the features of the final products.271,330–332 The lignin feedstock has to be purified to remove minerals and sulfur, as well as to reduce the heterogeneity of the precursor prior to spinning.271,337,338,340 Hardwood lignin possesses a less crosslinked structure and a lower glass transition temperature compared to softwood lignin, which makes the latter less suitable for spinning.341 To enhance the spinnability of softwood lignin, the use of a plasticizer or chemical modification (e.g. acetylation) are usually adopted.269,271,335,338 After spinning, the lignin fibers undergo a thermal stabilization during which crosslinking occurs and lignin glass transition temperature increases, as lignin gains a thermosetting behavior.330,331 Thermal stabilization is performed at temperatures comprised between 200 and 300 °C, and a slow heating rate is typically applied to enhance the fibers stability.269,330,331,336 In this step, the oxygen content of the lignin initially increases due to oxidation reactions, then decreases due to dehydration, condensation, and crosslinking reactions.331 Subsequently, the stabilized lignin fibers are carbonized at high temperature (≥1000 °C) and under inert atmosphere to eliminate all elements other than carbon.330,331 During carbonization a considerable weight loss occurs, due to the release of volatile components, which can also result in microstructural defects. A fast heating rate produces more stable and less brittle fibers.331,337
Lignin-based carbon fibers with tensile strength up to 1.07 GPa and modulus up to 82.7 GPa have been reported, which, however, do not reach the mechanical performance of synthetic carbon fibers (tensile strength ∼7 GPa, and modulus ∼900 GPa).271,331 This is mainly due to a less ordered structure of the lignin-based fibers, ultimately ascribable to the nonuniform structure of the lignin precursor.271,342 Thus, overcoming lignin heterogeneity still represents a critical challenge for the manufacturing of lignin-based carbon fibers with higher performance.337,338
4.2. Depolymerization of isolated lignin
The conversion of isolated lignin streams into chemicals encompasses an extensive depolymerization of lignin to produce monomeric intermediates that can subsequently be funneled toward target end-products. The goal of lignin depolymerization methods is to maximize the yield of phenolic monomers, which are obtained by the disruption of inter-unit linkages in lignin polymers. However, not all these linkages can be easily disrupted without causing extensive defunctionalization of lignin derivatives. As previously illustrated, ether bonds in lignin structures possess a lower dissociation energy and are more easily cleavable compared to C–C bonds.11,15,55 For this reason, ether linkages are the primary target of lignin depolymerization methods. The maximum yield of monomers that can be achieved during lignin depolymerization is directly dependent on the amount of β-O-4′ bonds in the isolated lignin.11,15,16 Mathematically, assuming an infinite linear structure for the lignin polymer, the maximum theoretical yield of monomers corresponds to the square of the fraction of cleavable linkages (β-O-4′ bonds) contained in the lignin polymer.343 Several strategies have been developed for the depolymerization of isolated lignin streams, which can be divided into five main groups, based on the exploited cleavage mechanism: reductive, oxidative, acid- or base-catalyzed, solvolytic, and thermal depolymerization. The following sections provide an overview of these methods.Hydroprocessing is further divided into three subcategories, according to the applied process conditions: mild, harsh, and bifunctional hydroprocessing (Fig. 22).152,351 Mild hydroprocessing is performed at relatively low temperatures (≤300 °C), in the presence of a solvent (either water, an organic solvent, or a combination of the two). Methoxy groups in lignin units are preserved, and the obtained pool of monomeric products comprises mainly p-alkylated methoxyphenols, which are obtained with a high selectivity and a yield that is generally below 20% (Fig. 22).152,351 Harsh hydroprocessing is conducted at higher temperatures (≥320 °C) and, most often, in the absence of a solvent. Extensive demethoxylation of lignin derivatives occurs, and a much broader distribution of monomeric products is obtained, comprising phenols, alkylated phenols, along with various amounts of mono- and polycyclic deoxygenated aromatics, alkanes, catechols, and methoxyphenols.349–351 The yield of monomers is usually below 20%.14,152,351 In bifunctional hydroprocessing an acid catalyst is introduced in the system, in such a way that hydrolysis and dehydration reactions occur, along with hydrogenolysis and hydrogenation reactions.16,349–351 The operating temperature is usually below 320 °C.152,351 Under these conditions, extensive HDO of lignin derivatives is achieved, with the selective formation of cycloalkanes.351 Monomer yields of up to 50% toward C6–C18 cycloalkanes (including both monomers and dimers) have been reported for these processes (Fig. 22).14,152,351
 | ||
| Fig. 22 Overview of phenolic monomers produced upon mild, harsh, and bifunctional hydroprocessing, and liquid-phase reforming of isolated lignin. Based on Schutyser et al.14 | ||
As opposed to hydroprocessing, liquid-phase reforming is performed under inert atmosphere, and in the presence of a hydrogen donor.344,347–351 The operating temperature range spans from 150 °C to 400 °C.349,351 Hydrogen donating solvents commonly employed in liquid-phase reforming include isopropanol, tetralin, formic acid, methanol, and ethanol.344,347–351 The advantages of this method are a lower operating pressure (i.e. less stringent reactor requirements), and thus reduced safety concerns.152 Notably, the utilization of hydrogen gas is usually still required for the subsequent reduction of the oxidized hydrogen donor.152 A large pool of monomeric products is obtained from liquid-phase reforming, comprising alkyl-substituted phenols and methoxyphenols (possibly with oxygenated side-chains), catechols, deoxygenated aromatics, and cycloalkanes (Fig. 22).16,349–351 Monomer yields up to 30% are frequently reported for liquid-phase reforming processes.14,351
An additional subcategory of reductive depolymerization processes includes recently developed two-step methods for the breakdown of β-O-4′ linkages relying on an initial oxidation of lignin by conversion of the hydroxyl group at the α-position into a ketone, followed by a reductive cleavage of the ether bond.191,352–355 The oxidation step reduces the dissociation energy of β-O-4′ bonds, facilitating their subsequent rupture.191,352–355 One of the key advantages of these methods is that they allow achieving lignin depolymerization at ambient or near ambient conditions (without external pressurization, and at temperatures below 100 °C),191,192,352–355 thereby reducing the energy and equipment requirements. Lignin oxidation can be performed using 2,2,6,6-tetramethylpiperidinyloxy (TEMPO),352,355–357 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ),353,356,358–360 electrocatalytic oxidation,354,361 or a mild oxidative biomass pretreatment.191,192 The reductive cleavage of oxidized lignin can then be conducted in the presence of different reducing agents, such as zinc,353 photo-reductive iridium complexes,354,355 or nucleophilic thiols.191,192 A redox neutral process for the cleavage of oxidized lignin in the presence of formic acid and sodium formate has also been proposed.352 The monomeric products produced with these depolymerization methods mainly include methoxyphenols possessing oxidized (hydroxy)alkyl side chains, typically obtained with a high selectivity, and a yield that can vary from ∼2% to over 60%, depending on the features of the isolated lignin fraction, and on the oxidation/depolymerization method applied (Fig. 23).191,192,352–355
 | ||
| Fig. 23 Overview of phenolic monomers produced upon two-step reductive depolymerization of isolated lignin. Based on Schutyser et al.14 and Sun et al.16 | ||
Other methods rely on the etherification of the hydroxyl group at the α-position of β-O-4′ motifs, followed by the catalytic hydrogenolysis of β-O-4′ bonds.362,363 The initial etherification reduces the dissociation energy of β-O-4′ linkages, thereby favoring their subsequent cleavage.362,363 The use of an acidic environment in the presence of methanol was shown to promote the nucleophilic substitution of the α-hydroxyl groups in lignin β-aryl ether motifs by the alcohol molecules.364 The so-formed α-methoxylated intermediates were reported to possess enhanced susceptibility toward catalytic hydrogenolysis, ultimately leading to higher yields of monophenolics possessing (hydroxy)alkyl side chains (Fig. 23).362,363
Alternatively, non-metal-based systems for the reductive depolymerization of lignin streams have been developed. For instance, silanes (e.g. Et3SiH, polymethylhydrosiloxane, and tetramethyldisiloxane)365–367 in combination with Lewis acids (e.g. B(C6F5)3)365 or bases (KOt-Bu)367 were reported to act as effective reducing agents for the cleavage of inter-unit ether linkages in lignin structures, affording silylated monoaromatics, which could be subsequently purified and hydrolyzed to form phenolic products with yields up to about 25%.366 The use of sodium or potassium dithionite as a reducing agent was also reported to enhance the depolymerization of pre-isolated lignin fractions.368
Oxidative depolymerization of lignin is most commonly performed in aqueous alkaline media (e.g. NaOH, KOH), but acids (e.g. acetic acid, H2SO4), and organic solvents (e.g. methanol) can also be applied.349,350,369,370 Additionally, a large number of homogeneous and heterogeneous catalysts have been investigated to enhance the yield and selectivity of oxidative depolymerization methods.185,272,349,350,369,370 The frequent selection of alkaline media for oxidative methods is related to the rapid formation of phenolate ions at high pH, which facilitates the subsequent oxidation to generate phenoxy radicals.272,370 The latter is considered a pivotal step for the initiation of C–C bonds rupture.14,152 In addition, a high pH slows down the degradation of the produced aromatic aldehydes.14
When the target products are phenolic monomers, the operating temperature is usually between 120 and 190 °C, and the oxidant is either oxygen or air (with a partial pressure of oxygen comprised between 2 and 14 bar).272,369,370 The obtained monomeric products include aromatic aldehydes (vanillin, syringaldehyde, 4-hydroxybenzaldehyde), and their acetyl-substituted (acetovanillone, acetosyringone, 4-hydroxyacetophenone), and acid (vanillic acid, syringic acid, 4-hydroxybenzoic acid) analogs (Fig. 24).272,370,371 The most renowned example of oxidative depolymerization of lignin toward monophenolics is the Borregaard process, which produces vanillin from sulfite liquor, and currently contributes to about 15% of the global supply of vanillin.379,380
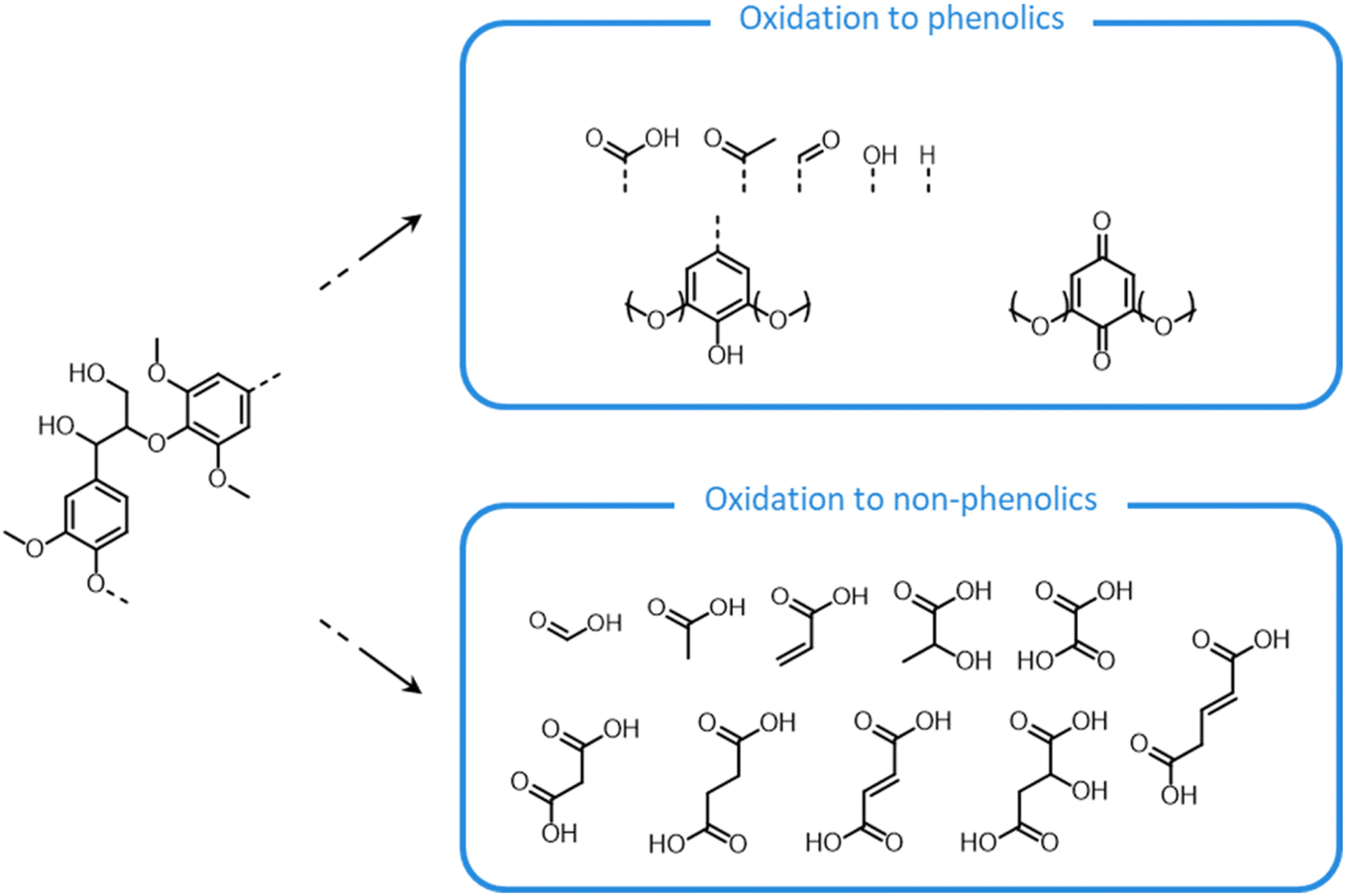 | ||
| Fig. 24 Overview of phenolic monomers produced upon oxidative depolymerization of isolated lignin. Based on Schutyser et al.14 Note: bonds other than β-O-4′ linkages are cleaved during oxidative depolymerization, including both C–O and C–C bonds. | ||
While the final product distribution depends on the properties of the lignin fraction and on the conditions of the depolymerization process, these compounds are prone to undergo repolymerization reactions.14,371 As a result, the yields of monophenolics for oxidative depolymerization processes are typically below 10%.14,152
An alternative strategy for the oxidative depolymerization of lignin relies on targeting the further conversion of the phenolic compounds initially obtained from lignin oxidation toward carboxylic acids, via the cleavage of aromatic rings.371,381,382 This can be achieved by the adoption of prolonged reaction times, higher temperatures, or by the use of stronger oxidants (e.g. H2O2).369,370,381,382 The process can be performed in neutral, acidic, or alkaline media, and the operating temperature is commonly in the range of 60–225 °C.272,371 Under such conditions, the main products are organic acids such as, formic, acetic, succinic, oxalic, and malonic acid, with yields that vary between ∼10% and ∼60%, depending on the properties of the isolated lignin stream and on the applied process conditions (Fig. 24).14,152
Acid-catalyzed lignin depolymerization is usually performed at temperatures in the range of 250–400 °C, and various types of acids can be employed in the process, including Lewis acids, Brønsted acids, zeolites, and ionic liquids.350,383,384 The chemistry of acid-catalyzed cleavage of β-O-4′ linkages follows the pathways illustrated in Fig. 7, and the final monomeric products principally include methoxyphenols possessing side chains with alkyl, alkenyl, carbonyl, and carboxyl functionalities (Fig. 25).344,350,384 Increasing the process temperature causes a gradual shift of the product selectivity toward catechols and alkylcatechols.152,344,384 In general, the monomer yields obtained for acid-catalyzed depolymerization do not exceed 20%.14,152
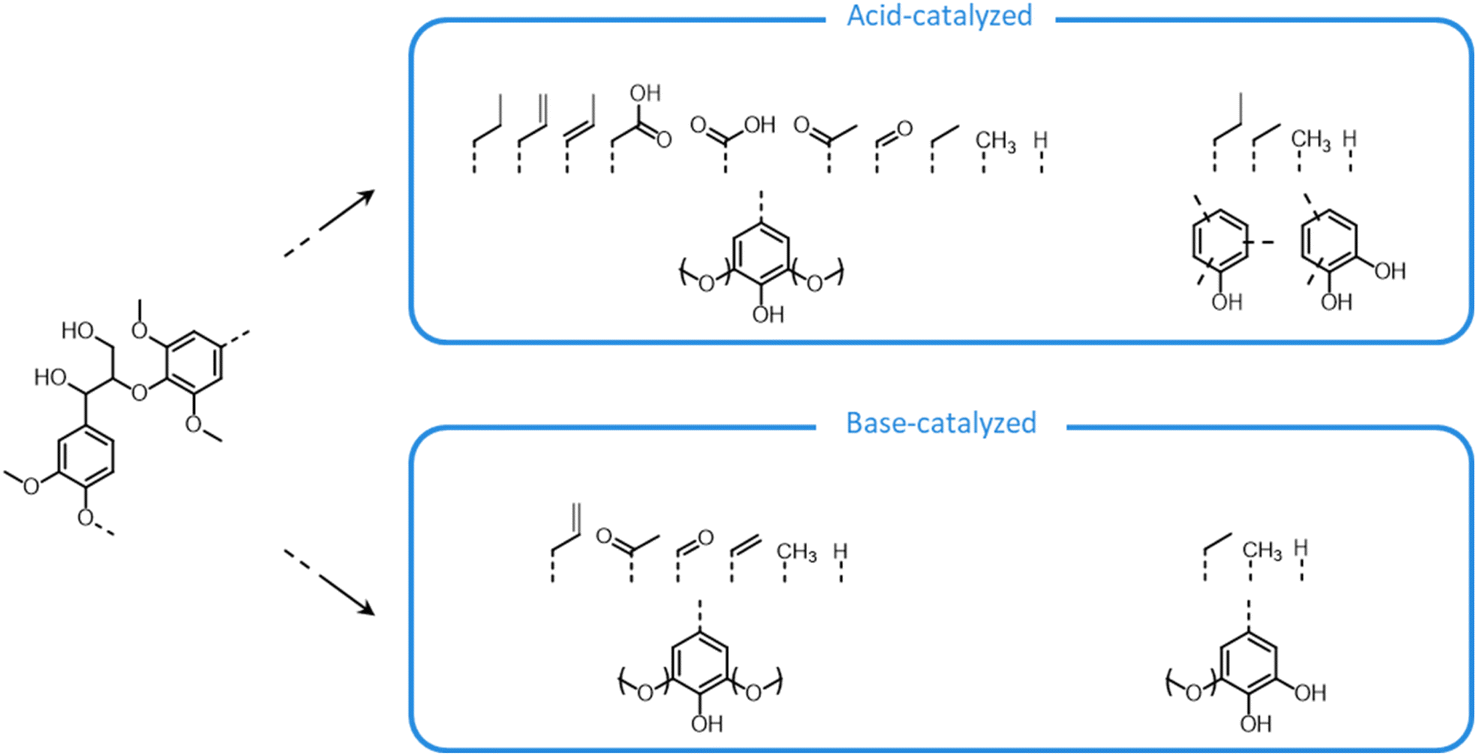 | ||
| Fig. 25 Overview of phenolic monomers produced upon acid- and base-catalyzed depolymerization of isolated lignin. Based on Schutyser et al.14 | ||
Base-catalyzed lignin depolymerization is commonly performed at temperatures comprised between 240 and 330 °C, and in the presence of soluble or insoluble base catalysts.350,383–385 Under alkaline conditions, the cleavage of β-O-4′ linkages follows the pathways illustrated in Fig. 9, and monophenolic products with functionalities analogous to those described for acid-catalyzed lignin depolymerization are obtained (Fig. 25).350,384,385 Most frequently, the monomer yields achieved with base-catalyzed depolymerization are below 10%.14,152
An important limitation is the fact that the high temperature and the presence of acid and base catalysts do not only enhance the cleavage of inter-unit ether bonds, but also promote the repolymerization of the so-formed lignin moieties, which tend to recondense yielding insoluble products.344,347,350,383–385 In order to partially overcome this issue, organic solvents can be adopted in the place of pure water to increase the solubility of lignin products, and, ultimately, improve the monomer yield.14,152 Moreover, the introduction of redox catalysts in the reaction system is another way to quench the reactive lignin fragments and to improve the yield of low-MW products.16,349 In addition, in the case of acid-catalyzed depolymerization, the yield of monophenolics may be enhanced through the stabilization of C2-aldehyde-substituted phenolics, which otherwise possess a high tendency to undergo repolymerization.118,152 Suitable routes for trapping these reactive intermediates include hydrogenation, decarbonylation, or reaction with glycols to form stable acetal structures (Fig. 26).16,118 Recently, acid-catalyzed depolymerization was reported to be effective for the depolymerization of pre-oxidized lignin, leading to a monomer yield of up to about 30%.386
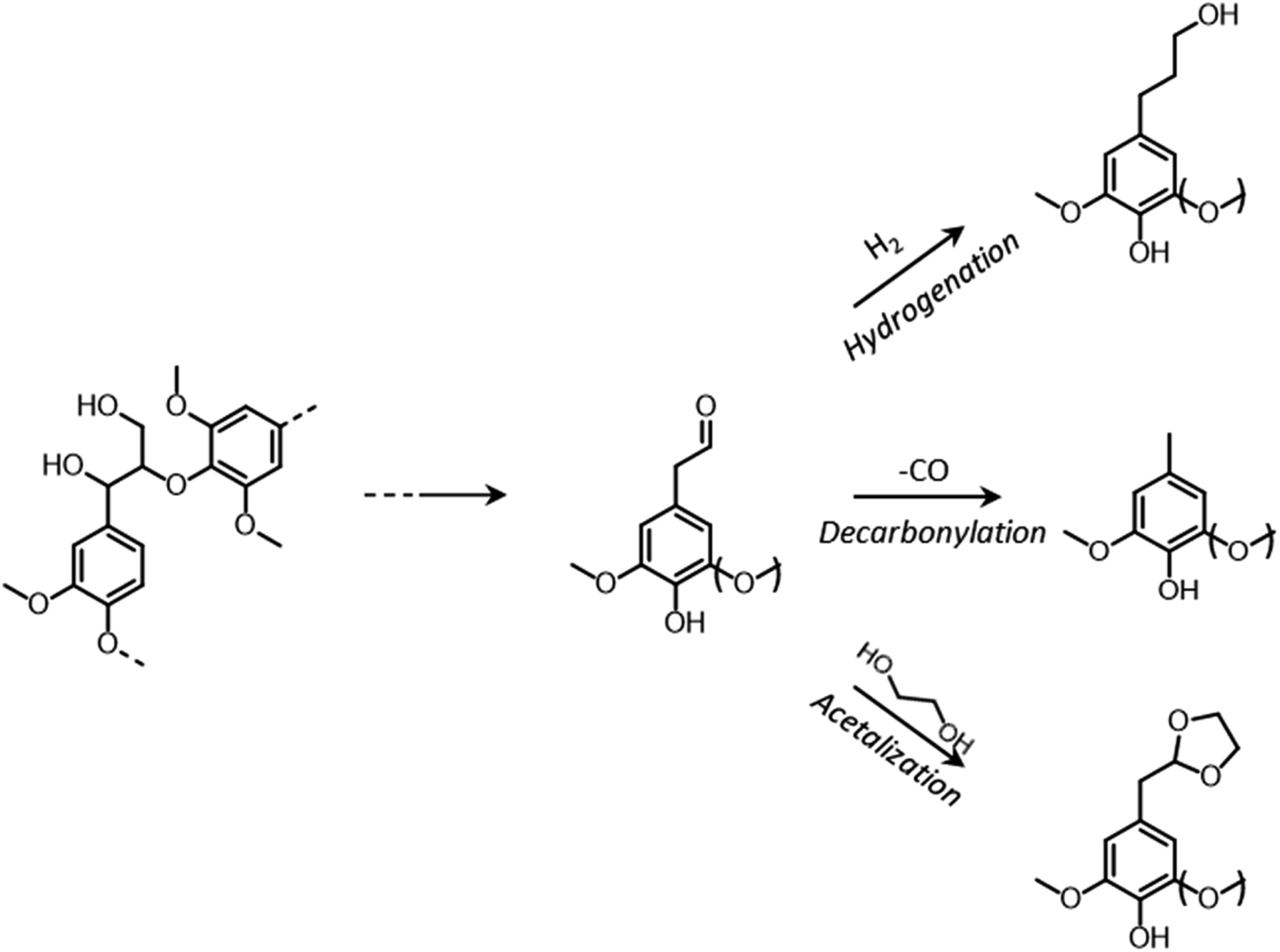 | ||
| Fig. 26 Possible routes for the stabilization of C2-aldehyde-substituted phenolics during acid-catalyzed depolymerization of isolated lignin. | ||
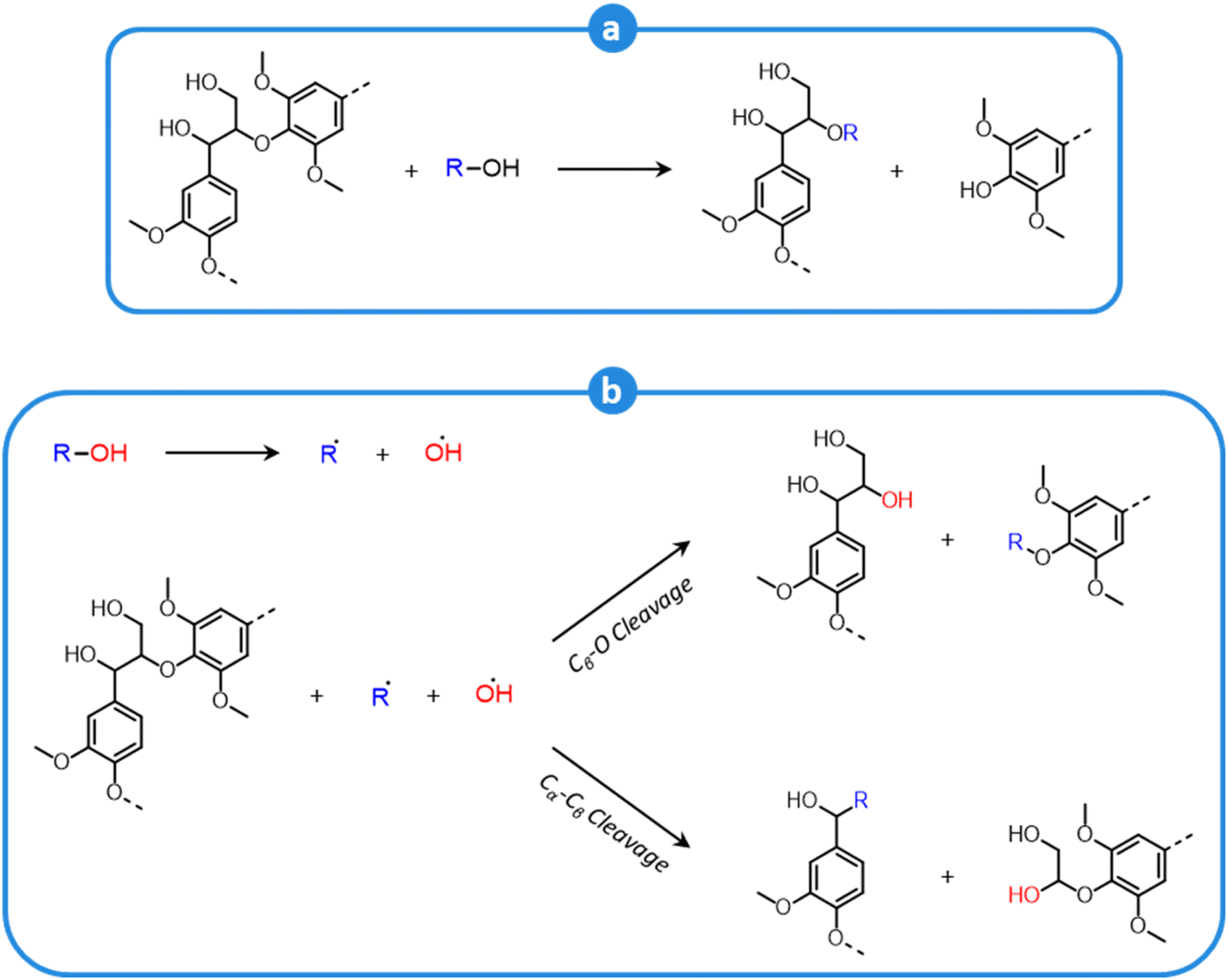 | ||
| Fig. 27 Possible pathways for the non-radical- (a) and radical-mediated (b) cleavage of lignin inter-unit linkages during the solvolytic depolymerization of lignin in the presence of alcohols. Based on Jensen et al.387 | ||
Overall, a broad spectrum of monomeric products is obtained from solvolytic depolymerization of lignin, mainly comprising unsubstituted methoxyphenols or methoxyphenols possessing saturated, unsaturated, or oxygenated side chains (Fig. 28).383,384,387 Notably, processing at higher temperatures or in the presence of a larger fraction of water in the solvent results in a higher selectivity toward unsubstituted/alkylated methoxyphenols and catechols.14 A limitation of solvolytic processes lies in the lack of stabilization routes for the reactive intermediates produced upon the disruption of inter-unit ether linkages in lignin, which ultimately restrain the yield of monomeric products.348 Thus, the yields of phenolic monomers generally do not exceed 10%.152,270
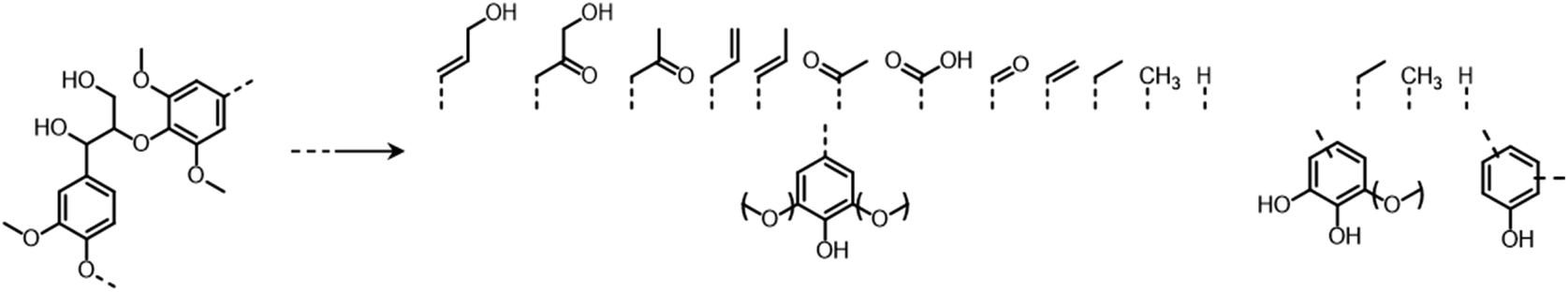 | ||
| Fig. 28 Overview of phenolic monomers produced upon solvolytic depolymerization of isolated lignin. Based on Schutyser et al.14 | ||
During lignin pyrolysis, the treatment time and the operating temperature have a critical impact in steering the selectivity toward liquid, solid, or gaseous products. Slow pyrolysis, with a heating rate well below 100 °C h−1 and a duration greater than 30 minutes, favors the formation of gaseous products and biochar.345,391 On the other hand, fast pyrolysis, with higher heating rates and durations in the order of seconds, promotes the formation of liquid bio-oil.345,391 Within fast pyrolysis, a temperature comprised between 400 and 600 °C is commonly required to maximize the yield of bio-oil and low-MW phenolic products.345,390,392,393 In view of this, fast pyrolysis represents the main thermal approach for the depolymerization of lignin into monomers.
Several chemical reactions, typically involving the formation of free radical species,350,394 take place over the broad temperature range of pyrolytic depolymerization processes.395,396 Below 400 °C, the cleavage of labile inter-unit ether bonds occurs, while C–C bonds and methoxy substituents on aromatic rings are stable.350,383,390 As the temperature increases above 400 °C, C–C linkages are broken, and methoxy, hydroxyl and methyl substituents on aromatic rings are cleaved.350,383,390 At the same time, the higher temperature boosts lignin recondensation and the formation of biochar and polycyclic aromatic hydrocarbons.383 Finally, at temperatures greater than 550 °C, aromatic rings are broken down and an increasingly larger amount of gaseous products is obtained.350,383,390
Due to the complex network of reactions occurring during fast pyrolysis, a broad pool of monomeric products is generally obtained, comprising unsubstituted and substituted (methoxy)phenols and catechols, possessing unsaturated, saturated, or oxygenated side chains (Fig. 29).350,383,392,397 Overall, as a result of the extensive recondensation of the unstable lignin moieties formed during the depolymerization process, the monomer yield from fast pyrolysis of lignin does not exceed 10%.14,152
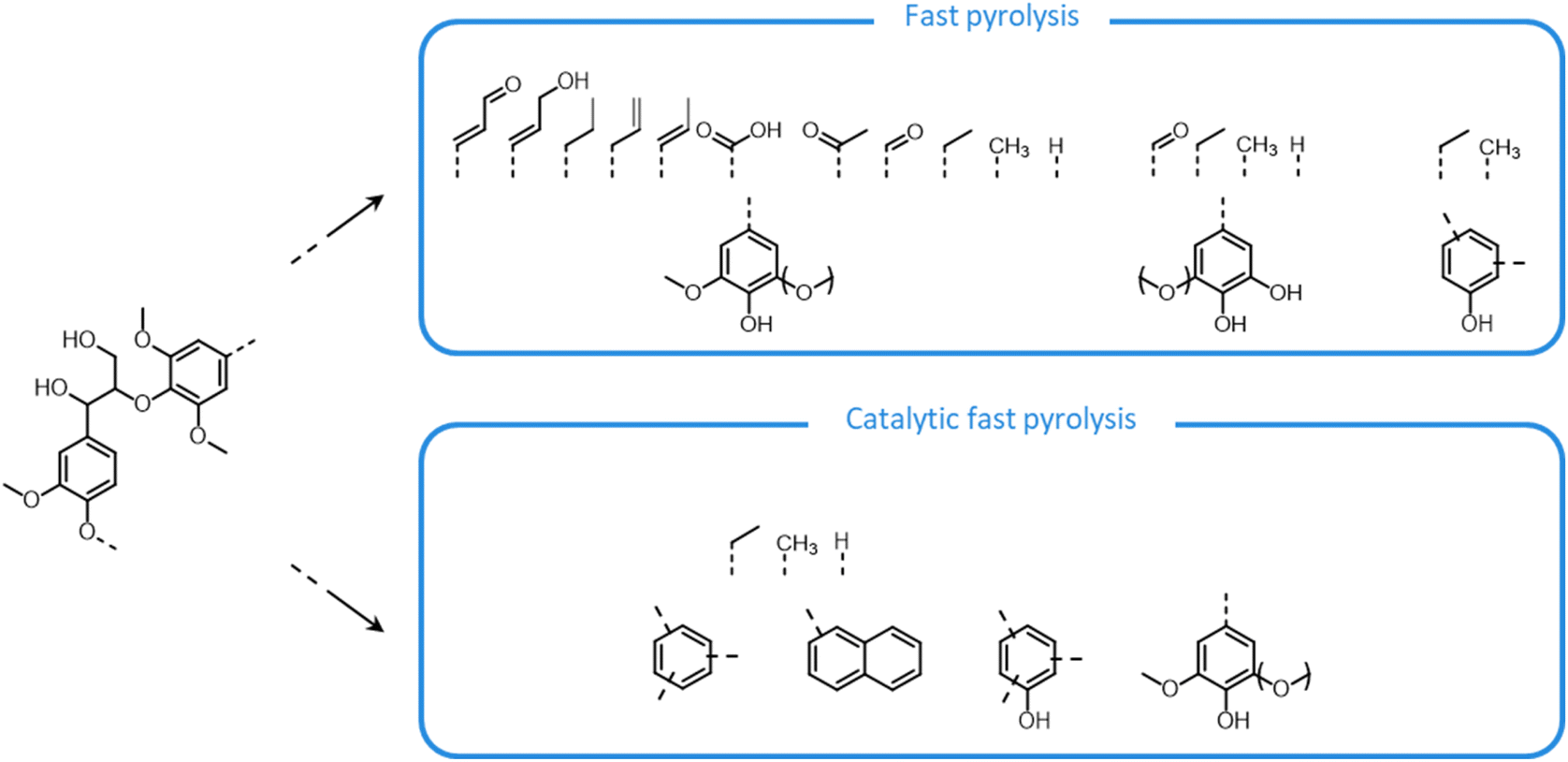 | ||
| Fig. 29 Overview of phenolic monomers produced upon fast pyrolysis and catalytic fast pyrolysis of isolated lignin. Based on Schutyser et al.14 Note: bonds other than β-O-4′ linkages are cleaved during pyrolysis, including both C–O and C–C bonds. | ||
Fast pyrolysis can also be performed in the presence of a catalyst, which improves the stability of the produced monomeric species, reducing excessive repolymerization and biochar formation, and increasing the selectivity toward target products.270,350,383 Most frequently, acid zeolites (e.g. H-ZSM-5) are employed, and tuning the acidity and the pore size of the zeolites can drastically enhance the selectivity toward deoxygenated aromatics (e.g. benzene, toluene, xylene) (Fig. 29).270,350,383,398 Namely, a higher density of acid sites boosts deoxygenation reactions, while a small pore size may limit the access of methoxyphenols to the active catalytic sites.152,350,397 Along with acid zeolites, other catalysts such as mesoporous silica, metal oxides, and supported metals have been explored within catalytic fast pyrolysis.350,383,397,399 A common issue of these processes is the considerable catalyst deactivation that occurs due to the formation of char on the catalyst surface (coking), which imposes frequent catalyst regeneration.350,383 In general, despite a higher selectivity, monomer yields from catalytic fast pyrolysis remain lower than 20%.152
4.3. Upgrading lignin monomers to chemicals
The phenolic monomers obtained after lignin depolymerization typically possess structures resembling native lignin units, with aromatic rings featuring methoxy and hydroxyl groups, as well as side chains comprising alkyl, alkenyl, carbonyl, carboxyl, or hydroxyl functionalities. Although such mixtures of monophenolics are substantially less complex than the native lignin polymer, further purification and/or chemical modification is required before they can be commercialized.120,267,268 The series of transformations to which these monomers are subjected prior to being marketed are termed “upgrading”. Two distinct approaches exist for upgrading phenolic monomers: (i) functionalization strategies, which aim at introducing new chemical groups in lignin moieties to yield high-value fine chemicals and materials,400,401 and (ii) defunctionalization strategies, which target the removal of functional groups from lignin derivatives to yield bulk chemicals and fuels (Fig. 30).16,267,268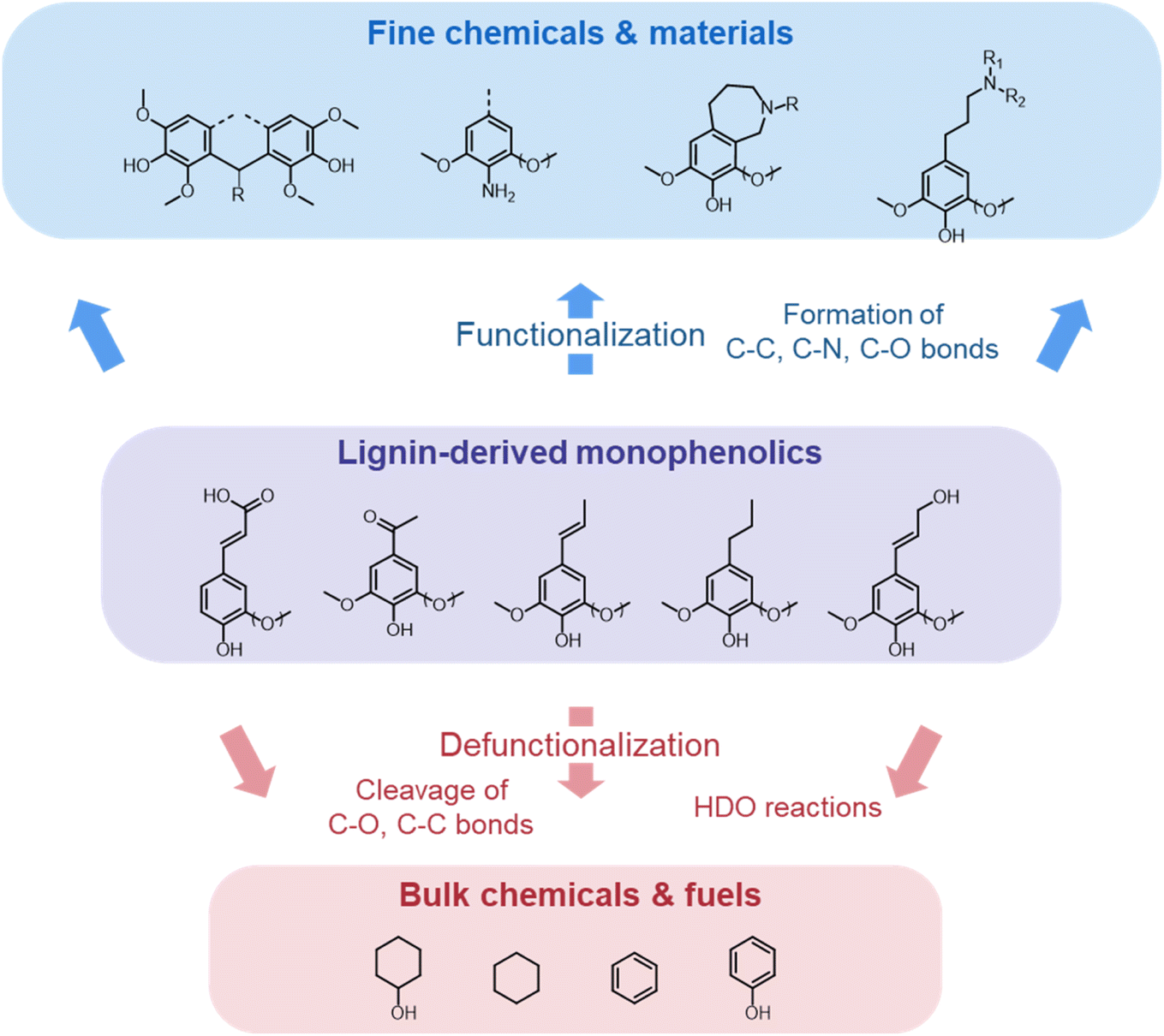 | ||
| Fig. 30 Strategies for the conversion of lignin-derived monophenolics into fine and bulk chemicals. | ||
Functionalization strategies are emerging approaches that rely on the conversion of aromatic or aliphatic hydroxyl groups in lignin monomers toward amines, or on the formation of new C–C or C–O bonds exploiting aromatic and side chain reactive functionalities.268,402–404 Various value-added polymer building blocks can be obtained in this way, as precursors for epoxy resins, polyurethanes, and polyesters.267,268,405 One example is the dimerization of lignin monomers to form bisphenols that can be employed as renewable alternatives to BPA. The acid-catalyzed hydroxyalkylation of stoichiometric amounts of monophenolics and formaldehyde is a suitable route for the synthesis of lignin-derived bisphenols (Fig. 31a).313,316,406,407 In this context, the selective preservation of natural lignin functionalities in the building blocks (e.g. methoxy groups, side chains, …) may be exploited to improve the features of the bisphenols, as reported for bisguaiacols or bissyringols, which possess a substantially reduced endocrine disrupting activity compared to BPA, while their use in the synthesis of polyesters and epoxy resins gives materials possessing properties similar to their synthetic counterparts.313,316,408 Another example of functionalization pathway is the amination of phenolic hydroxyl groups of lignin monomers to yield anilines (Figure 31b),409,410 which are employed as building blocks for the synthesis of polyurethanes, as well as in the fabrication of dyes.411
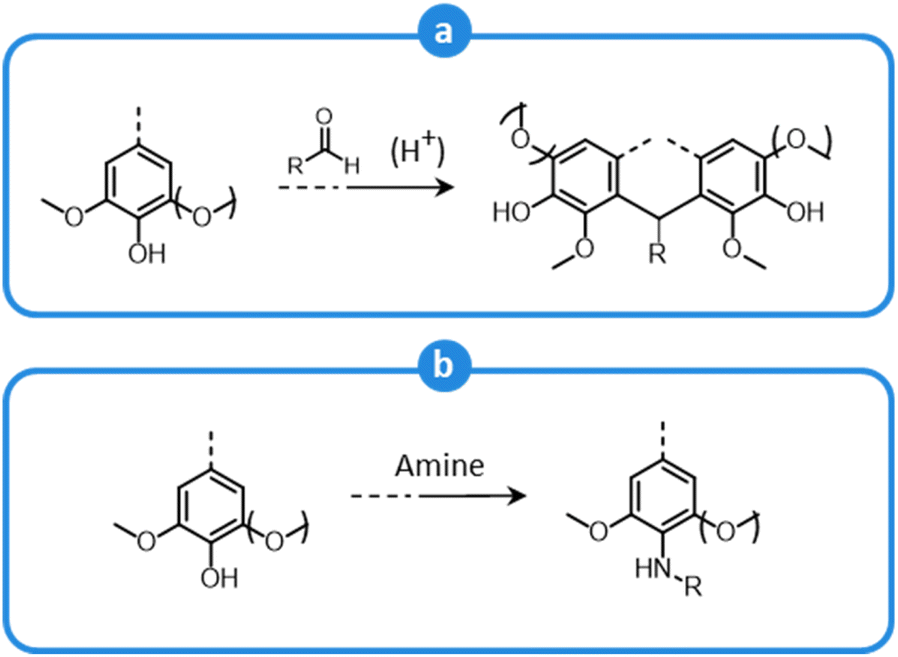 | ||
| Fig. 31 Formation of bisphenols (a) and aniline derivatives (b) from lignin monomers. The structure of the R-group in aniline derivatives is dependent on the synthesis conditions. | ||
Alternatively, the chemical functionalization of phenolic monomers can be directed toward the synthesis of high-value bioactive compounds, such as benzazepines, benzoxepines, carbazoles, and many others.412–415 For instance, the amination of terminal hydroxyl groups in the alkyl side chains of methoxyphenols can be used to form aminoalkyl-substituted monomers, which, upon cyclization, yield benzazepine-like structures (Fig. 32a).412,414 Recently, valuable tertiary amines with proven antioxidant properties have been obtained from series of lignin monomers, using an atom-efficient Cu-catalyzed “hydrogen borrowing” strategy (Fig. 32b).416,417 Although the market volume of these bioactive compounds and pharmaceuticals is smaller compared to that of bulk chemicals or commodity polymers, the value of such compounds is considerably higher.413,414 Such approach may be a promising way to significantly improve the profitability of existing and future biorefineries.
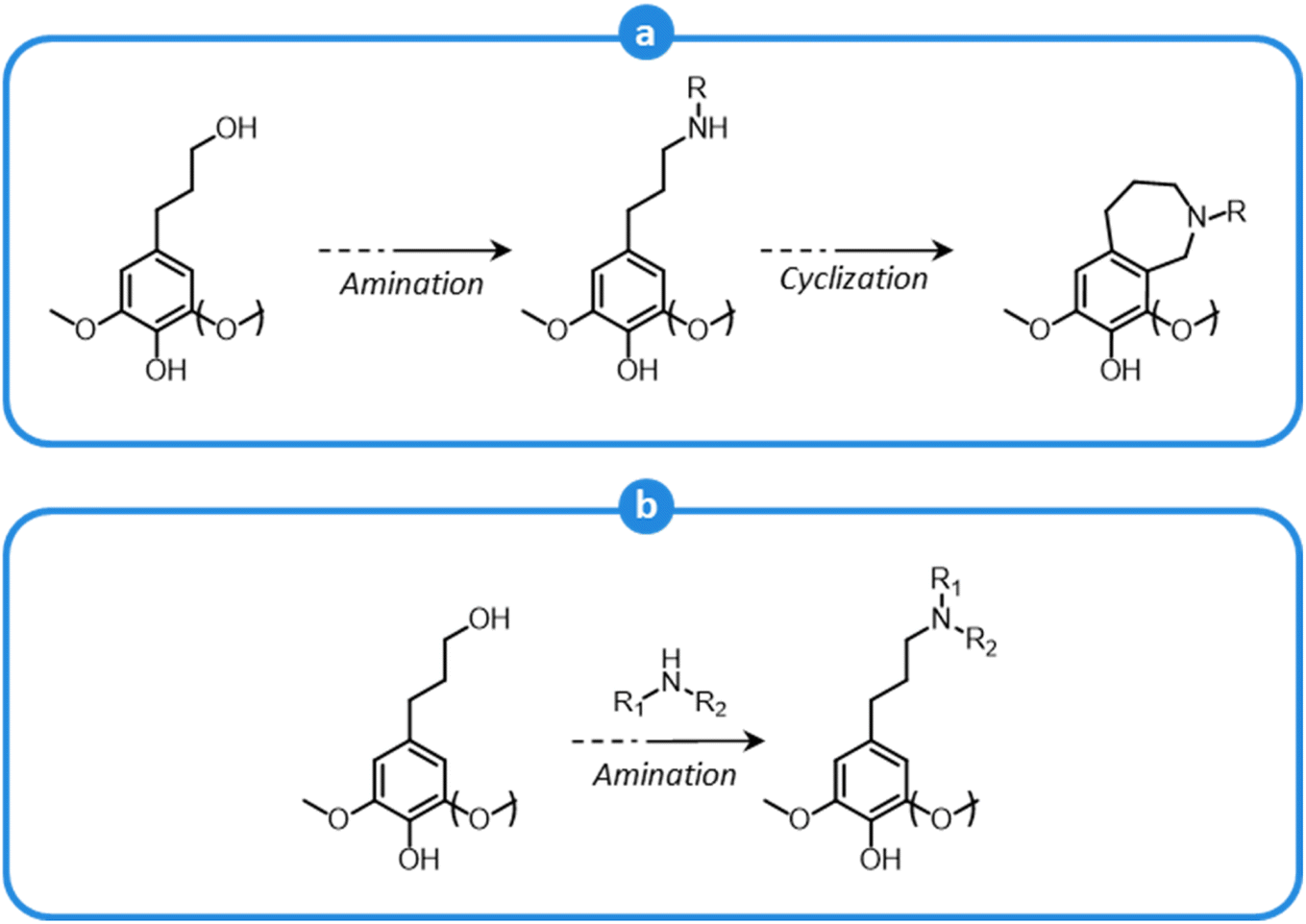 | ||
| Fig. 32 Formation of benzazepines (a) via amination and subsequent cyclization of lignin monomers and (b) formation of tertiary amines from lignin monomers, as examples of upgrading toward effective antioxidants. | ||
However, a crucial limitation of such functionalization strategies lies in the need of isolating and purifying single components from the complex mixtures of monomers usually obtained upon lignin depolymerization. In particular, for processes characterized by a low product selectivity, the separation step(s) may be technically challenging, energy-intensive, and, ultimately, not economically viable. A defunctionalization approach may be preferable for funneling the heterogeneous mixture of monomers into a handful of drop-in platform chemicals, which can serve for a variety of applications (e.g. fuels, polymers manufacturing, solvents, …), thereby reducing the cost associated with downstream product isolation.267,268 From this point of view, both chemocatalytic and biological pathways have been developed for the defunctionalization of lignin monomers.267,405,418–421
Among the possible routes for chemocatalytic upgrading of phenolic monomers, their defunctionalization through HDO reactions is particularly attractive for decreasing the complexity of the mixture of monomeric products, as well as the oxygen content of the produced molecules.267,405,422,423 Depending on the applied process conditions, HDO of phenolic monomers can yield alkanes, aromatic hydrocarbons, phenols, or cyclohexanols (Fig. 33).419,420,422,423 Lignin-derived alkanes (e.g. alkyl cyclohexane) can be produced from substituted methoxyphenols by the combined action of a redox catalyst (noble- or base metal) and an acid catalyst (homogeneous or heterogeneous).405,422,423 The obtained mixture of alkanes may be directly exploited as fuel additives in midrange fuels.16,152 Alternatively, aromatic hydrocarbons (e.g. alkylbenzene) can be produced when reaction conditions are tuned in such a way to prevent the hydrogenation of aromatic rings.405,422 To achieve this goal, gas-phase reactions at high temperature, with a low hydrogen pressure, and in the presence of redox catalysts are often employed.419,422,424 The preservation of phenolic hydroxyl groups can be achieved when the HDO conditions are adjusted to target the selective demethoxylation of lignin-derived monophenolics, yielding alkylphenols.422,423 Further dealkylation of alkylbenzenes and alkylphenols yields benzene/phenol and short olefins (e.g. ethylene, propylene), which can be exploited as building blocks for the production of chemicals, fuels, or polymer precursors.405,420 Finally, alkylated cyclohexanols can be obtained from lignin monomers via demethoxylation and aromatic ring hydrogenation.405,422 Notably, alkylated cyclohexanols can be oxidized into the corresponding cyclohexanones, which may be further transformed into (alkylated) caprolactone, caprolactam, or adipic acid.267,268,405,425 The latter are widely adopted polyester building blocks. In this context, as mentioned above, the presence of alkyl side chains may impart new properties to the polymeric materials, possibly resulting in an added value.311,423,426
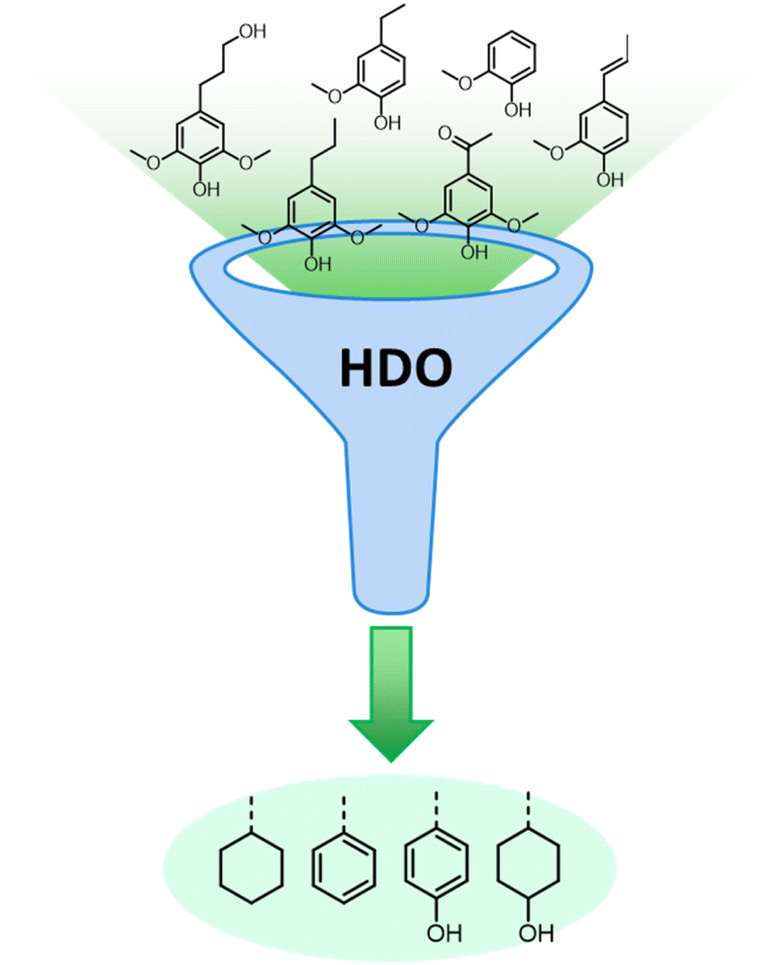 | ||
| Fig. 33 Schematic representation of the chemocatalytic upgrading of lignin monomers via HDO reactions to yield substituted alkanes, aromatics, phenols and cyclohexanols. | ||
As an alternative to chemocatalytic funneling, biological approaches have been investigated for the conversion of lignin monomers toward valuable platform chemicals.418,427,428 These strategies rely on the exploitation of metabolic pathways that defunctionalize aromatic compounds to yield few central intermediates, which then undergo ring-opening and ultimately enter central carbon metabolism.427–431 Microbial strains (e.g. Pseudomonas putida, Rhodococcus opacus, …) have been genetically engineered to convert lignin-derived monomers toward value-added chemicals, such as vanillin, muconic acid, triglycerides, and others (Fig. 34).418,430 These chemicals can then be directly marketed or transformed into polymer precursors or fuels. Although promising, biological pathways require further research efforts to improve the concentration of final products in fermentation broths and the resistance of microbes and enzymes against toxic aromatic chemicals.428,430
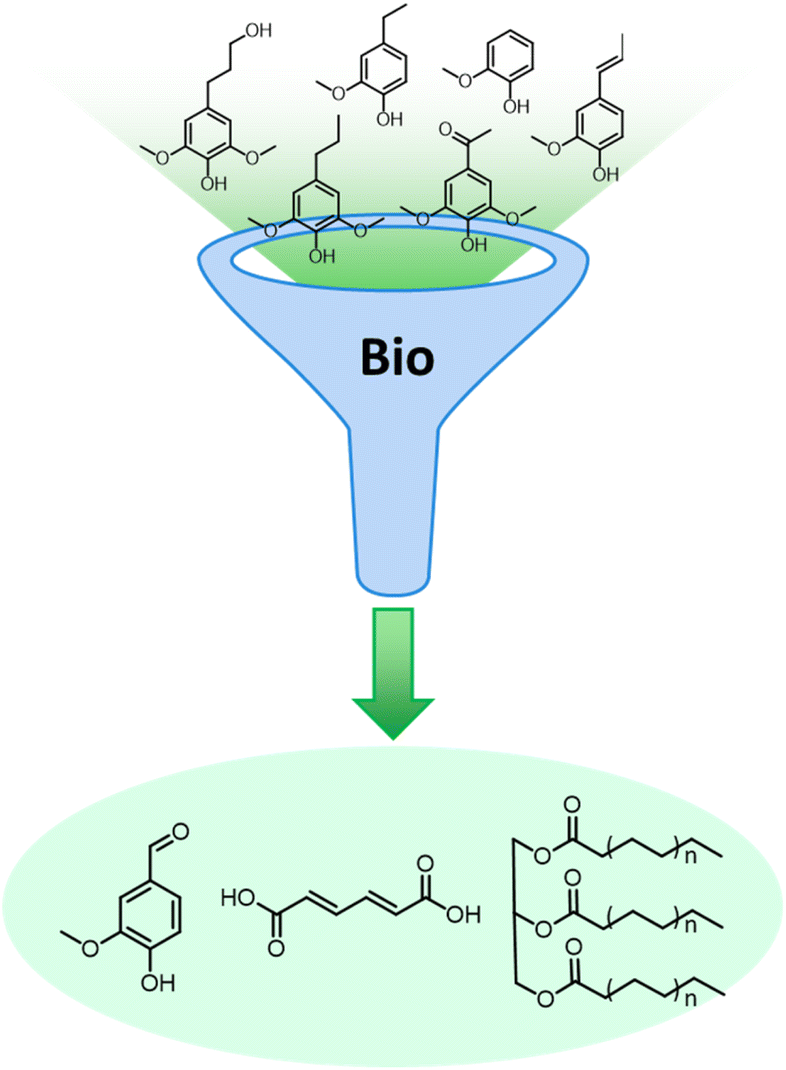 | ||
| Fig. 34 Schematic representation of the biological upgrading of lignin monomers toward platform chemicals. | ||
5. Critical considerations about isolated lignin valorization
The sections above illustrate a plethora of routes that have been explored for the valorization of lignin streams isolated from biomass after pretreatment. While the direct utilization of lignin fractions for the manufacturing of polymeric materials represents a relatively facile and alluring route for generating value from lignin, it has been shown that the implementation of lignin within the polymeric frameworks is typically hindered by the low reactivity and solubility of isolated lignin streams. The manufacturing of polymer blends possessing large contents of lignin (>∼30%) often requires a prior chemical modification or a partial depolymerization of the isolated lignin in order to attain mechanical properties comparable to those of synthetic materials.An alternative strategy is to subject lignin to a depolymerization process, and to selectively upgrade the low-MW lignin derivatives toward target high-value products. This approach entails the potential of developing fully lignin-based chemicals, pharmaceuticals, and materials, exploiting the natural functionalities of lignin units. However, the low susceptibility of isolated lignin fractions toward depolymerization constitutes a significant limitation to the yield and selectivity of low-MW products, and, eventually, to the yield of (de)functionalized products after upgrading. In this respect, it is important to recognize that lignin isolation, depolymerization, and upgrading are consecutive processes, and each one of them affects the following steps (Fig. 35). Namely, biomass fractionation induces chemical modifications in the isolated lignin streams which may reduce the efficacy of lignin depolymerization (e.g. reduced content of β-O-4′ linkages, increased degree of condensation, presence of contaminants), resulting in a lower yield toward phenolic monomers and, ultimately, undermining the production of final end-products from lignin upgrading.
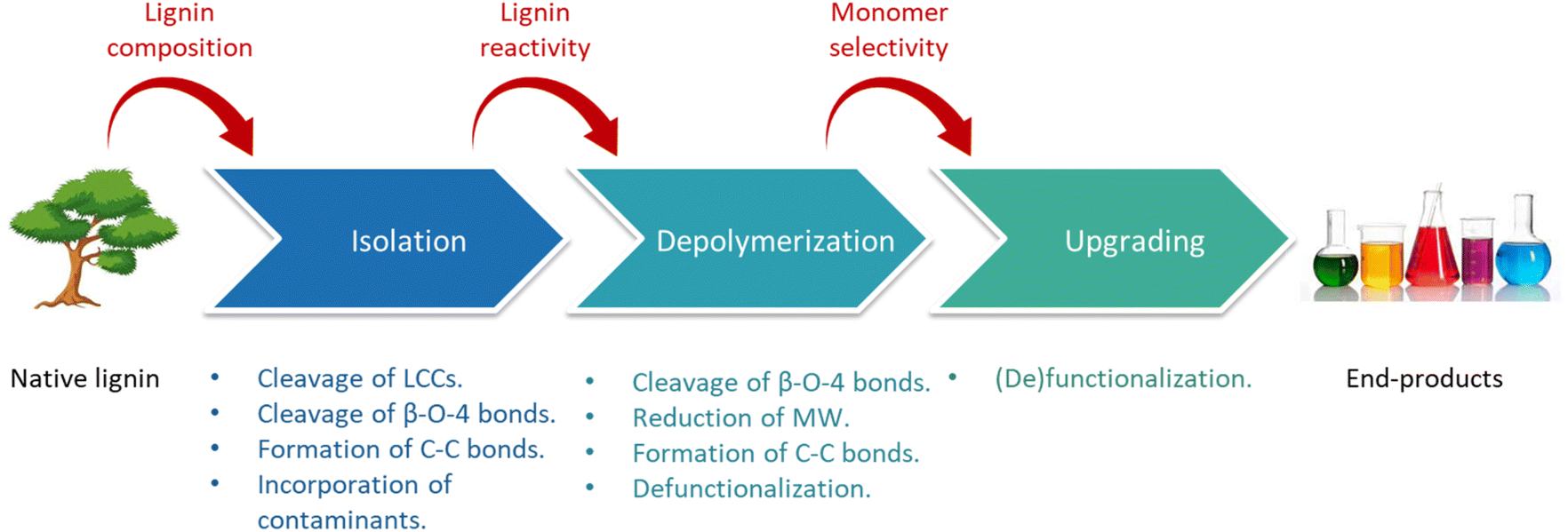 | ||
| Fig. 35 Outline of the sequence of operations involved in the valorization of lignin to chemicals: lignin isolation, depolymerization and upgrading. The main structural modifications occurring in the lignin polymer and its derivatives during the different operations are reported below each block. The properties of lignin streams affecting subsequent steps are reported above the red arrows. | ||
Overall, the yield of valuable lignin-derived products that can be obtained from biomass processing is influenced by the biomass source from which the lignin is extracted, by the fractionation method applied for lignin isolation, by the depolymerization method adopted, and by the effectiveness of the (de)functionalization strategy employed.
A crucial parameter correlated with lignin reactivity toward depolymerization is the content of labile β-O-4′ bonds in the lignin structure. The lower the content of β-O-4′ linkages, the less susceptible the isolated lignin is toward depolymerization.11,15 In addition, the incorporation of impurities deriving from additives (e.g. sulfur), carbohydrates (e.g. pseudolignin), or minerals during biomass pretreatment may also increase lignin recalcitrance toward further depolymerization and upgrading.11,12,15 Indeed, such impurities may cause deactivation of the catalysts that are typically employed in these processing steps.11,12 From this point of view, the drawback of many of the methods adopted for biomass fractionation within biorefining and the pulping industry is that they are predominantly focused on the valorization of the carbohydrate fraction, while little attention is given to preventing excessive lignin degradation.12,15
The formation of valuable lignin products may be enhanced by following two leading principles during biomass pretreatment. The first one relies on minimizing lignin degradation reactions by preserving β-O-4′ linkages and preventing the incorporation of contaminants. This objective may be realized by adopting mild fractionation methods, with a low process severity and, ideally, not employing chemical agents that can irreversibly bind lignin derivatives hindering their subsequent processing.14 Notably, a compromise should be established between biomass delignification, and the isolation of well-preserved lignin. The second principle is based on minimizing lignin recondensation reactions and in particular the formation of new C–C bonds. One way to achieve this goal is to adopt flow-through reactor configurations instead of a conventional batch configurations for biomass pretreatment, so that the reactive intermediates formed during fractionation are promptly removed from the heating zone, thereby reducing their tendency to undergo repolymerization.14,17
Importantly, these two principles can also be applied upstream of biomass fractionation, when biomass feedstocks are selected or designed on purpose for their subsequent conversion to chemicals and materials. Genetic modification of plants may be exploited to create lignin structures that are less networked, with larger contents of labile β-O-4′ linkages, and possessing a higher ratio of S-units, which are less prone to undergo recondensation due to the presence of methoxy groups in the ortho-positions.432–436 In addition, labile ester bonds may be introduced in the lignin backbone to facilitate its fragmentation. A well-known example is the incorporation of ferulate linkages to create a so-called zip-lignin,436–438 which can be more easily extracted and depolymerized during biomass pretreatment.438 However, these alterations can typically be realized only up to a certain extent, since they also exert an impact on other key biological functions of the lignin polymer, such as imparting structural resistance to plant cells, ultimately hampering plant growth, and reducing biomass yields.432,434–436,439 The improvement of the yield and selectivity of lignin derivatives from biomass processing also encompasses the design of the depolymerization step. In this context, the suppression of recondensation reactions that occur at the high temperatures typically applied for lignin depolymerization processes is a crucial objective.
Finally, developing strategies for upgrading lignin-derived monomers toward marketable fine or bulk chemicals and materials also entails expanding the market of lignin-derived products, finding applications which benefit of the unique functionalities of lignin derivatives that are not found in their fossil-derived counterparts.152 The latter is certainly a decisive aspect for creating added-value from lignin and for drawing an increased attention on this subject from industrial players in the field of biomass processing.
Recognizing the interdependence of native lignin properties, lignin isolation conditions, depolymerization conditions, upgrading methods, and the yield and selectivity of end-products, naturally implies that a process aimed at valorizing lignin should take all these aspects into account since its initial design. In other words, biomass fractionation should be performed in such a way to produce a lignin stream that is suitable for the subsequent depolymerization. Similarly, the depolymerization process should be tuned to maximize the yield and selectivity toward target monomers in view of further upgrading.
Nevertheless, the conversion of lignin to products should not be carried out at the expense of the valorization of cellulose and hemicellulose. To achieve the fullest utilization of biomass potential, a holistic approach is required that considers the interplay of biomass components and their derivatives in each stage of the process. In this regard, biomass pretreatment represents a pivotal aspect of biomass valorization, being the first step of the processing sequence in which all biomass components are present and potentially subject to alteration. Driven by the need of alternative pretreatment approaches that could facilitate the upgrading of all biomass fractions, new biorefinery schemes termed “lignin-first” biorefineries have been recently developed, which aim at extracting lignin from biomass while preventing the occurrence of repolymerization reactions.17,19 The next section discusses in detail these innovative methods.
6. Lignin-first biorefineries
Throughout the last few decades, a wide variety of biomass pretreatment methods have been developed with the main goal of delignifying lignocellulose to enhance the processability of the residual carbohydrate fraction toward materials (e.g. paper), or chemicals and fuels (e.g. bioethanol). More recently, the perspective of exploiting also the lignin fraction as a source of renewable aromatics directed an increased attention toward the effect of biomass pretreatment on the properties of the isolated lignin streams. In this context, the adoption of organosolv fractionation has been shown to effectively extract lignin from lignocellulose with minor chemical modifications compared to many other pretreatment methods (e.g. kraft, sulfite, hydrothermal, …), while producing a processable carbohydrate pulp.14,15,39 Yet, the acidic conditions frequently applied within organosolv treatments cause a partial depletion of native ether bonds in lignin structures, leading to the formation of reactive intermediates that tend to recondense creating C–C linkages, and, ultimately, boosting lignin recalcitrance toward further depolymerization.12,15,39With the goal to produce isolated lignin streams possessing an improved reactivity, a new class of so-called “lignin-first” processes has been developed. Lignin-first approaches are biomass fractionation methods that aim at (i) extracting a relatively pure lignin stream from lignocellulosic biomass in high yields, while concomitantly (ii) suppressing recondensation reactions between the lignin fragments liberated in the medium, ultimately yielding a lignin fraction (typically in the form of a lignin oil) that is either depolymerized or prone to undergo subsequent depolymerization, purification by fractionation, or upgrading. These methods typically rely on catalysis or protection-group chemistry to achieve the active stabilization of lignin derivatives.19,440,441 The lignin-first concept was clearly defined in a recent review by Abu-Omar et al. as “an active stabilization approach that liberates lignin from the plant cell wall and prevents condensation reactions through either catalysis or protection-group chemistry”.19 Importantly, the stabilization strategies that characterize lignin-first processes are often suitable also for suppressing undesirable condensation of the released carbohydrate fragments toward humins.18,441–443 Hence, lignin-first methods are integral approaches, designed for the valorization of both lignin and (hemi)cellulose, thus aiming at a more complete utilization of lignocellulose potential.19,441
Lignin-first processes commonly involve a solvolytic extraction of lignin, analogous to that occurring during organosolv processes, followed by a stabilization of the reactive lignin (and carbohydrate) intermediates and, finally, by the depolymerization of the isolated lignin fraction (when this is not achieved concomitantly with lignin stabilization).19,441 The passivation of reactive lignin (and carbohydrate) moieties may be realized through physical protection, as in the case of flow-through reactors, via chemical functionalization (e.g. hydrogenation, acetalization, …), or by a combination of the two.17,18,216,441,442 The lignin oil can also undergo purification through fractionation (i.e. successive solvent extractions).417
Different strategies have been reported within the scope of lignin-first pretreatment. The following sections provide an overview of the most widely acknowledged methods: reductive catalytic fractionation (RCF), aldehyde-assisted fractionation (AAF), and diol-assisted fractionation (DAF). We also present an emerging method based on the use of sodium dithionite. It should be mentioned that alternative methods are currently being developed and could arguably lead to further progress in the field in the years to come (e.g. shape-selective catalysis).444–446
6.1. Reductive catalytic fractionation
Reductive catalytic fractionation, also termed Early-Stage Catalytic Conversion of Lignin (ECCL) or Catalytic Upstream Biorefining (CUB), relies on catalytic hydrogenation for stabilizing the reactive intermediates released during lignocellulose solvolysis.447–449 This method was originally exploited in the 1940s as a means to gain insight in the structural composition of the lignin from different biomass sources.450–453 However, the potential of the RCF as a biomass pretreatment for the development of integrated biorefineries was recognized only in the last decade, leading to a renewed interest in this approach.448,454–457RCF is performed in an organic solvent (typically an alcohol), possibly in combination with water or an acid catalyst, in the presence of a reducing agent (most often H2) and a heterogeneous redox catalyst (e.g. Ru/C, Pd/C, Raney® Ni, …), at a temperature in the range between 180 and 250 °C.458,459 Similarly to organosolv pretreatment, during the RCF, LCCs are ruptured by the action of the organic solvent, and large lignin fragments are released in the medium, which undergo further solvolysis and hydrogenolysis of labile inter-unit ether linkages to generate smaller lignin moieties (Fig. 36).460,461 The latter are characterized by reactive alkenyl functionalities in the side chains of lignin units, which make them prone to undergo repolymerization.460 Notably, the formation of (hydroxy)alkenyl-substituted lignin derivatives implies the occurrence of a reductive cleavage of ether linkages, in which hydrogen gas or a hydrogen donor act as reducing agents.460 When water is employed as a co-solvent, or when an acid co-catalyst is used, acidolysis of ether bonds occurs along with solvolysis, and lignin fragments possessing reactive carbonyl functionalities, prone to recondensation, are produced as well (i.e. Hibbert's ketones and C2-aldehyde-substituted phenolics).17,462 However, by virtue of the presence of the redox catalyst and H2 (or a H-donor), the catalytic hydrogenation of reactive moieties is favored over repolymerization, and stable (hydroxy)alkyl-substituted phenolic compounds are ultimately formed (e.g. 4-propanol, 4-propyl, 4-ethyl guaiacol/syringol) (Fig. 36).458,460 Since the stabilization of lignin derivatives occurs after the cleavage of inter-unit bonds, RCF integrates lignin isolation and depolymerization in a single operation, producing a low-MW lignin stream, rich in phenolic monomers.458
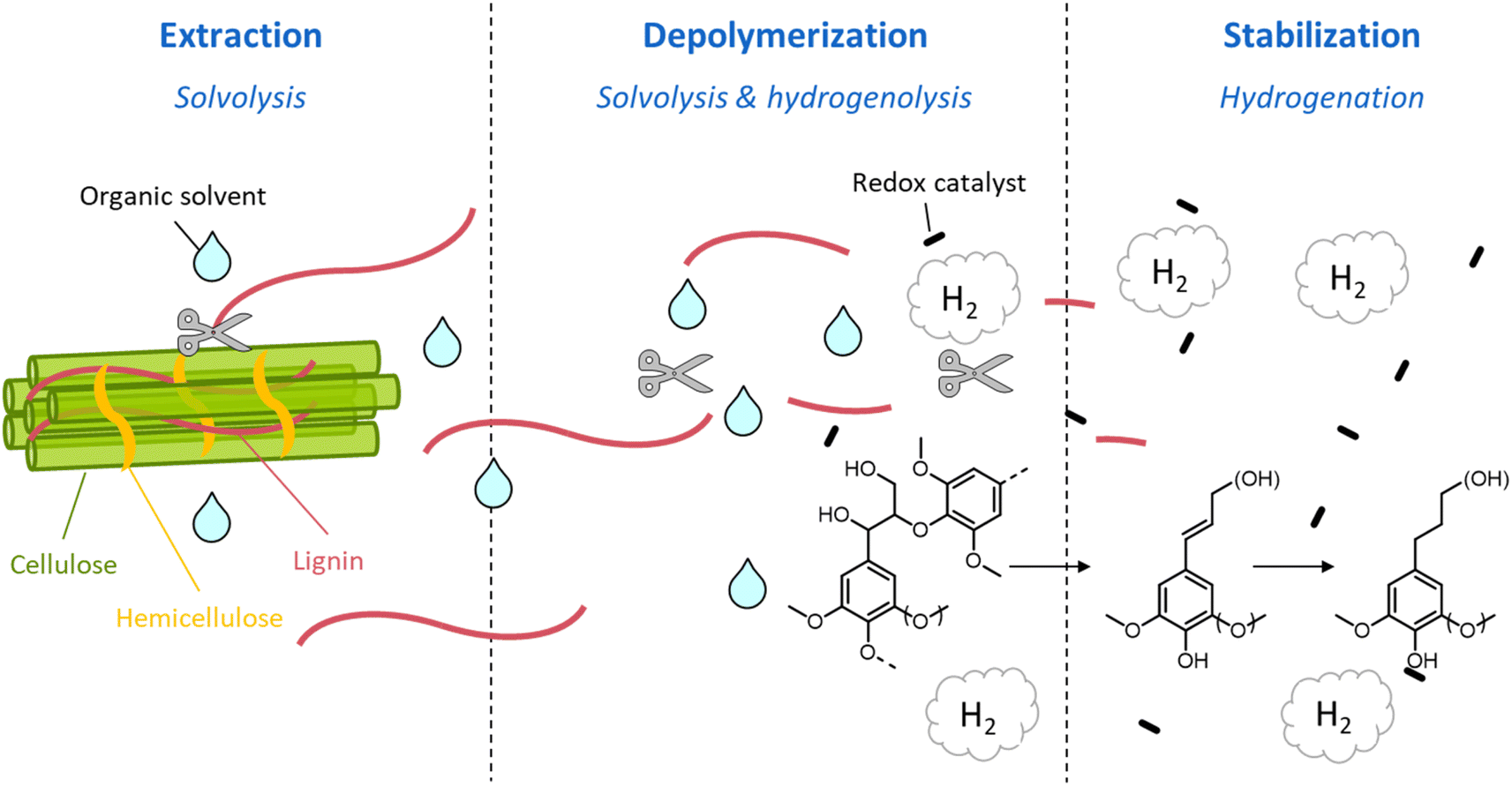 | ||
| Fig. 36 Schematic representation of lignin extraction, depolymerization, and stabilization during the RCF. Based on Van den Bosch et al.460 | ||
Thus, as illustrated in Fig. 36, the RCF can essentially be divided into three main steps: lignin extraction, depolymerization, and stabilization. While the extraction of lignin is only determined by solvolysis (and acidolysis),460 and the stabilization of reactive intermediates is exclusively governed by redox catalysis,460,463,464 the depolymerization step involves an interplay of the two phenomena.460,461,465,466 In particular, the lignin oligomers released during solvolysis (and acidolysis) may reach the catalyst surface to undergo further catalytic hydrogenolysis of residual ether bonds.461,465,466 The contribution of hydrogenolysis to lignin depolymerization is more evident when the RCF is performed under low severity conditions, at which solvolysis (and acidolysis) are less effective.466
Although RCF shares some similarities with organosolv pretreatment concerning the solvolytic disassembly of lignin, a key difference exists in the criterion to be adopted for solvent selection. While for organosolv processes the best solvents are typically weakly polar solvents, exhibiting the highest lignin solubility, for the RCF more polar solvents (e.g. methanol, ethylene glycol) are preferred in view of their improved ability to depolymerize lignin.467 Moreover, the type of solvent also contributes to affect the features of the phenolics products obtained from the RCF, such as their molecular weight distribution,467 and the residual presence of alkenyl or hydroxyl functionalities in the side chains of lignin moieties.468 Other factors affecting the properties of the RCF lignin streams are the type of metal catalyst employed and the hydrogen pressure (when H2 is used). For instance, the use of Ru/C or Rh/C has been shown to afford a high selectivity toward alkyl-substituted phenolics,469,470 whereas Pd/C preferentially yields hydroxyalkyl-substituted phenolics,469,470 and Raney® Ni leads to the partial hydrogenation of aromatic rings.448,468 Additionally, the adoption of a low hydrogen pressure favors the hydrogenolysis of terminal hydroxyl groups in the side chains of lignin units, ultimately leading to alkyl-substituted phenolics, while at higher hydrogen pressure terminal hydroxyl groups are preferentially preserved.470 The process severity is another element that influences the features of the isolated lignin fraction, with lignin extraction and defunctionalization of lignin derivatives both being favored at higher severity conditions.462,471,472
A schematic outline of the RCF process summarizing the features of each product fraction is displayed in Fig. 37. Notably, high yields of delignification (up to ∼90%) and near-theoretical yields of monophenolics (based on the content of β-O-4′ linkages) have been reported for the RCF of hardwood biomass (e.g. birch,447,454,460 poplar,448,455,466 eucalyptus,470,473,474 …). The treatment of softwood biomass (e.g. pine,315,446,475 spruce,447,476,477 …) resulted in lower yields of lignin extraction (≤∼60%)447,475 and in a higher molecular weight of the isolated lignin streams, due to a lower fraction of cleavable β-O-4′ linkages and to a typically more networked structure of softwood lignin.447,477 The RCF of herbaceous feedstocks (e.g. miscanthus,465,478 corn stover,472,479 wheat straw,478,480,481 …) has been explored as well, and relatively high yields of delignification have been reported (up to ∼90%),478 along with a yield of monophenolics that varies in the range of ∼10 –∼60% of the initial lignin content.465,478,482
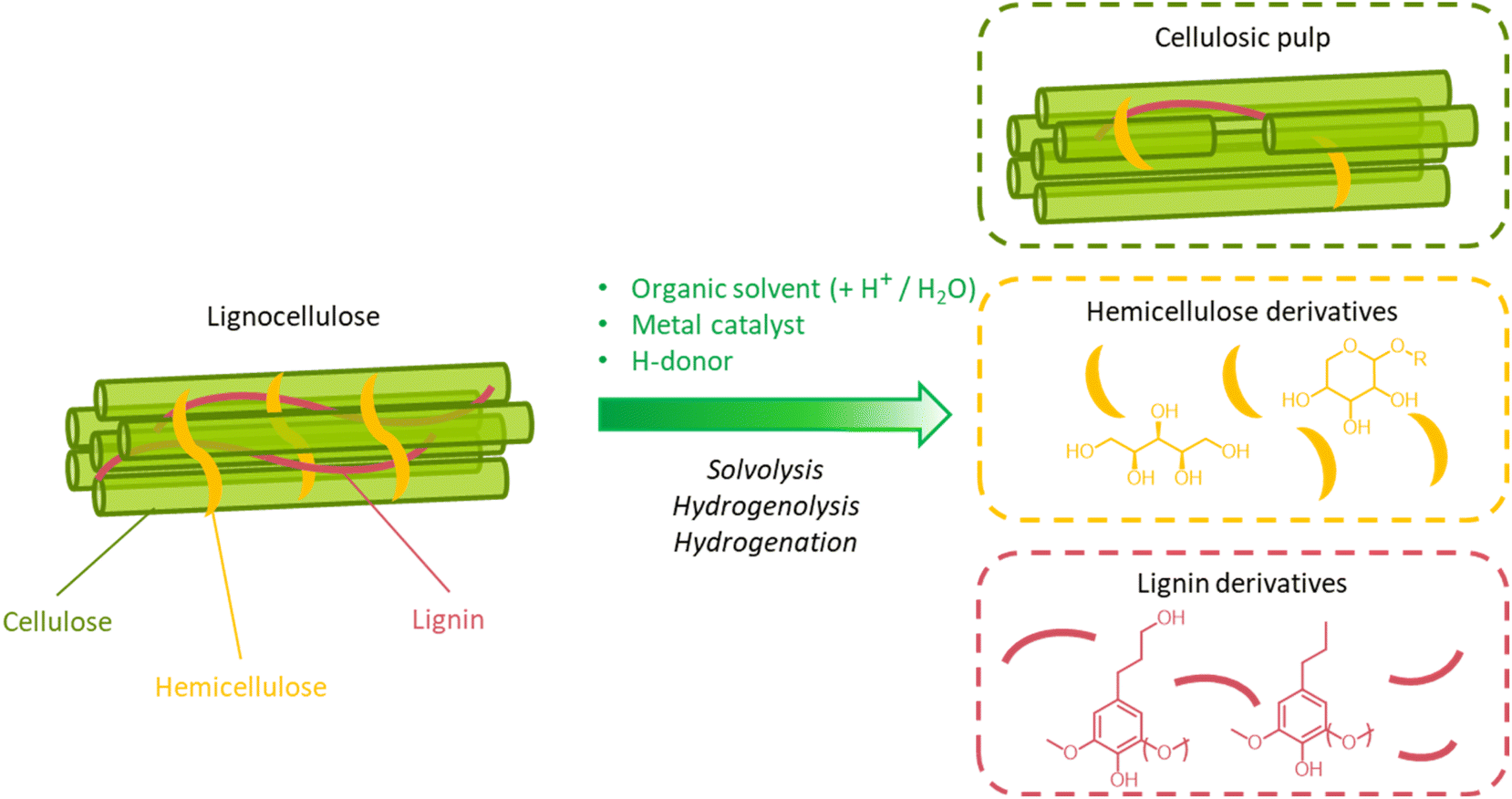 | ||
| Fig. 37 Schematic outline of the RCF process for the extraction, depolymerization, and stabilization of lignin (and hemicellulose) derivatives from lignocellulosic biomass via solvolysis, solvolysis/hydrogenolysis, and hydrogenation, respectively. Note: when RCF is performed in the absence of water or without an acid co-catalyst, hemicellulose is mainly retained in the solid pulp. | ||
Next to lignin, the fate of hemicellulose is also greatly dependent on RCF conditions. Similarly to organosolv pretreatment, RCF can extract hemicellulose from lignocellulosic biomass when water or an acid co-catalyst are added to the system.462,467,470,472,477 Under such conditions, hemicellulose is hydrolyzed and the so-formed sugars are typically alkylated by the action of the alcoholic solvent,462,472,477 or hydrogenated,462,470 to yield alkyl glycosides or polyols, respectively. Importantly, the latter are more stable toward dehydration and condensation reactions compared to monosaccharides, which reduces the formation of pseudolignin.17
Cellulose is not hydrolyzed in RCF processes, and is mostly retained in the form of solid pulp. Nonetheless, cellulose morphology is partially altered during the treatment, as swelling occurs due to the disruption of lignocellulose structure, with a consequent increase of cellulose surface area.39,483 The size of the isolated cellulose fibers varies depending on the solvent adopted for the RCF treatment.467 In contrast to organosolv fractionation, the redeposition of condensed pseudolignin on the pulp surface is largely prevented within RCF, thanks to the active stabilization of lignin and hemicellulose derivatives formed in the process.483 As a result, the cellulosic pulps produced by RCF commonly possess an excellent enzymatic digestibility (up to over 90% of glucan converted to glucose).472,477,483
A few challenges exist that need to be addressed to facilitate upscaling. In this respect, the design of continuous RCF process configurations may help reducing processing costs.458,459,484 Furthermore, the use of hydrogen gas as a reducing agent constitutes a liability of RCF, imposing strict safety, environmental, and equipment requirements.485 The solvent selection is another crucial parameter impacting the operation performance and processing costs.485 Moreover, the development of ad hoc catalysts that are able to steer the process selectivity toward the desired products, and to preserve catalytic activity over time is important for ensuring operational stability.484,486,487 Current research efforts are directed toward addressing these potential limitations. The adoption of semi-continuous flow-through reactors, in which the biomass and the heterogeneous catalyst are loaded in two separate compartments to facilitate the post-fractionation catalyst recovery and re-use, has been explored for RCF processes.466,488,204,489–492 In addition, various works explored the development of H2-free RCF configurations in which the solvent or hemicellulose itself act as alternative reducing agents, reporting moderately high yields of lignin and carbohydrate products,448,468,477,481,491,493–495 and a positive impact on the overall biorefinery economy.485 Recently, the influence of solvent selection and on the RCF performance was inspected, and the choice of high-boiling solvents was reported to minimize capital costs.485 The use of membrane separation instead of the conventional distillation for solvent recuperation was indicated as a potentially convenient approach to further mitigate energy consumption.268,485,496 Alternatively, for the use of low-boiling solvents, the recycling of a portion of the fractionation liquor was reported as an effective strategy to reduce the heat duty for distillation.497 Various supported redox catalysts were explored for RCF at lab scale, as alternatives to the widely adopted commercial ones.486 Transition metal carbide catalysts were synthesized which exhibit improved stability of the metal active phase and a higher activity toward lignin hydrogenolysis.498,499 Transition metal sulfides were reported to excellently adsorb oxygen atoms on sulfur vacancies, boosting ether bonds cleavage in lignin species.479,500 Solid catalysts with adjustable acidity (e.g. with supports based on zeolites, niobium phosphate, silica, alumina, …) were shown to substantially enhance C–O reductive cleavage in lignin species.486,501–503 Notably, while most studies focused on the development of redox catalysts that could favor the hydrogenolysis of lignin toward monophenolics, the catalyst properties may also be tuned to achieve the stabilization of macromolecular intermediates released during lignin depolymerization, such as lignin dimers, trimers or tetramers.446 This approach is particularly intriguing as it entails the idea of using the redox catalyst for selectively controlling the fate of lignin species during RCF.
In spite of the current challenges, the RCF is considered a promising method for future lignin-first biorefineries, as highlighted in recent techno-economic and environmental assessments of this technology.485,497,504,505 Presently, the RCF technology is being scaled-up to multi-kg scale at the BioCon Pilot facilities (KULeuven, Belgium), with the prospect of bringing the process to the multi-ton scale by 2030.506,507
6.2. Aldehyde-assisted fractionation
Aldehyde-assisted fractionation entails the use of aldehydes (e.g. formaldehyde, acetaldehyde, propionaldehyde, …) within the organosolv treatment of biomass under acidic conditions, to suppress the repolymerization of the solubilized lignin and hemicellulose intermediates by formation of relatively stable acetal structures.508–515 AAF is carried out in acidified aqueous mixtures of aprotic organic solvents (e.g. dioxane), at temperatures in the range of 80–120 °C.508,509,511 Aprotic organic solvents are the most suitable, since hydroxyl groups in protic solvents would react with aldehydes causing their depletion.17 Under these conditions, lignin is extracted from lignocellulose by solvolysis and acidolysis of LCCs, similarly to an acid organosolv pretreatment, and large lignin fragments are released in solution (Fig. 38). However, in this approach, the cleavage of β-O-4′ linkages is prevented. The aldehyde molecules react with the α- and γ-hydroxyl groups in the side chains of lignin moieties to yield 1,3-dioxane structures within β-O-4′ linkages, trapping the hydroxyl groups in the form of a stable cyclic acetal, thereby avoiding the generation of reactive benzylic carbenium ions, and the occurrence of lignin repolymerization reactions.508–512,516,517 The formation of acetals is a reversible reaction that releases H2O, and is favored in water-deficient environments.508,509,512,517 | ||
| Fig. 38 Schematic representation of lignin extraction and stabilization during AAF. | ||
Moreover, when formaldehyde is employed, it contributes to lignin stabilization also by blocking reactive electron-rich meta-positions on aromatic rings of lignin units to form hydroxymethyl groups.508,509,511 While this can enhance repolymerization in the presence of small amounts of formaldehyde (e.g. only generated in situ by cleavage of lignin side chains), an excess of formaldehyde completely depletes free meta-positions, effectively suppressing the formation of methylene bridges, and lignin recondensation.511
The features of the different product streams obtained via AAF are summarized in Fig. 39. Lignin is extracted in high yields (up to ∼80% delignification for hardwood biomass),508 and the isolated lignin fractions possess a high content of reactive inter-unit ether linkages, prone to be cleaved during a subsequent depolymerization step.508–512 The latter is commonly realized by subjecting the lignin stream to reductive depolymerization at conditions analogous to those applied for the RCF,508–510,512,514,518 as illustrated in Fig. 40a. In this way, near-theoretical yields of monophenolics (up to 40–50% for hardwood biomass) are obtained.508–510,512 One limitation to this approach is that, when formaldehyde is utilized as a protecting agent, the hydroxymethylation of aromatic rings leads to additional (hydroxy)methyl groups in lignin units after reductive depolymerization, decreasing product selectivity.508 The use of longer aldehydes (e.g. acetaldehyde, propionaldehyde), which do not result in electrophilic substitution on aromatic rings, provides a solution to this issue, and allows achieving a high selectivity (over 90% for 4-propanolsyringol/guaiacol for the treatment of hardwood biomass) with analogous near-theoretical monomer yields.510
 | ||
| Fig. 39 Schematic outline of the AAF process for the extraction and stabilization of lignin (and hemicellulose) derivatives from lignocellulosic biomass via solvolysis/acidolysis and acetalization, respectively. Note: the organic solvent should be an aprotic solvent to avoid reaction of the solvent's hydroxyl groups with the aldehyde.17 Moreover, the water fraction should be limited (<20% w/w) to achieve effective acetalization of lignin and carbohydrate derivates.17,18 | ||
 | ||
| Fig. 40 Cleavage of β-O-4′ acetals structures by tandem hydrogenolysis and hydrogenation (a), and by consecutive oxidation and depolymerization (b). | ||
An alternative strategy for the depolymerization of AAF lignin is the adoption of a two-step oxidation and depolymerization of acetal-stabilized β-O-4′ structures (Fig. 40b). The oxidation is realized by treating the isolated lignin in a wet organic solvent solution (with 5% H2O), in the presence of DDQ and HNO3, under O2 pressure.519 Under such conditions, the acetal is cleaved, and the α-hydroxyl group is oxidized into a ketone.519 The oxidized lignin subsequently undergoes depolymerization in an aqueous solution of formic acid/sodium formate, to give diketones with high yield (30–35%) and selectivity (over 90% for syringyl/guaiacyl propane dione) when treating hardwood biomass.519
Along with lignin, hemicellulose is also extensively hydrolyzed due to the acidic conditions applied within AAF. The released monosaccharides promptly react with the aldehydes to form acetal structures that are stable against dehydration and subsequent recondensation, thus limiting the formation of pseudolignin during the fractionation process.508,509,513 The acetal functionalities can be subsequently removed by dilute acid hydrolysis to give monosaccharides.508,509,513,520 Alternatively, the acetal-functionalized sugars can be directly converted to furfural by hydrothermal treatment (at a temperature of ∼160 °C), in the presence of a Brønsted acid catalyst (e.g. sulfuric acid).513 Importantly, acetal-functionalized sugars possess a high volatility (e.g. diformyl-xylose exhibits a boiling temperature of 250 °C)508 compared to the parent sugars (e.g. xylose exhibits a boiling temperature of 415 °C), which makes it possible to purify these compounds by distillation before further upgrading, thereby circumventing the need of expensive chromatography separations.17,513
The extraction of lignin and hemicellulose leaves behind a highly pure cellulosic pulp, possessing a good enzymatic digestibility.508,509 The latter can be further improved (up to over 90% of glucan to glucose conversion) by subjecting the pulp to dilute acid hydrolysis prior to enzymatic processing to remove the aldehyde molecules grafted on the pulp surface.508,509 Another possibility is to perform an aldehyde-assisted acid depolymerization of cellulose in an organic aprotic solvent (e.g. γ-valerolactone) to produce glucose acetals (with yields of up to 70% and concentrations of ∼5% w/w).513 In this case, a higher temperature (∼200 °C) is required compared to that needed for hemicellulose hydrolysis. The acetal protection groups can subsequently be removed by treatment in aqueous conditions, and the aldehyde can be recovered.513
Overall, AAF offers the advantage of allowing to optimize lignin extraction and depolymerization as two separate steps. Thus, mild fractionation conditions can be applied to minimize degradation of biomass components, while harsher conditions can be used for the subsequent lignin depolymerization. On the other hand, an important limitation of this methodology lies in the toxicity of the organic solvents (e.g. dioxane) and of the aldehydes that are employed.216,509,511 Moreover, the recovery of aldehyde derivatives upon deprotection of the products is not straightforward. From this point of view, the aldehydes incorporated in lignin acetals are transformed into alkanes during reductive depolymerization (e.g. formaldehyde is converted into methane), implying the need to reconvert alkanes into aldehydes in a separate process, or to feed fresh aldehydes for every run.508 The fate of aldehyde derivatives during the oxidative deprotection of acetal-stabilized lignin has been less investigated, but it has been postulated that the cleavage of the acetal groups would lead to the release of aldehyde molecules in the medium.519 Thus, the protecting agent may be recovered thereafter, provided that aldehyde molecules are not irreversibly incorporated in lignin structures (e.g. in the form of hydroxymethyl groups in aromatic rings) under the applied conditions. Despite these challenges, a preliminary economic assessment of formaldehyde-assisted fractionation highlighted the potential profitability of such technology.513 Notworthy, the AAF process is currently being upscaled by the Swiss company Bloom Biorenewables, with the aim of exploring commercial opportunities for the obtained product streams.512
6.3. Diol-assisted fractionation
Diol-assisted fractionation (DAF) relies on the adoption of diols within the acid-catalyzed organosolv pretreatment of lignocellulose to stabilize lignin and hemicellulose derivatives against recondensation through the formation of acetals.118,521–525 While diol-assisted acidolysis has been initially explored as an approach to depolymerize isolated lignin fractions,118,523,525 it has been recently shown that it can be effectively integrated within biomass fractionation to develop a lignin-first process.521,522,524,526DAF is typically performed in organic aprotic solvents (e.g. 1,4-dioxane, dimethylcarbonate, ethylene carbonate),118,521,524 in the presence of an acid catalyst (e.g. triflic acid, H2SO4),118,521,524,525 and a diol (e.g. ethylene glycol, 1,3-propanediol, …),118,521,524,525 at mild temperature (∼140 °C).521,524 Under such conditions, LCCs are cleaved by the combined action of the organic solvent and the acid catalyst and lignin is extracted in the medium (Fig. 41).521,524 Lignin fragments are further cleaved, most prominently by acidolysis of inter-unit β-O-4′ linkages, which encompasses the formation of benzylic carbenium ions, and, ultimately, the formation of Hibbert's ketones and C2-aldehyde-substituted phenolics.115–118,521–523,527–530 Once formed, these species are prone to participate in repolymerization reactions. In particular, C2-aldehyde-substituted phenolics are extremely reactive, which is why they are only found in trace amounts in typical acid-catalyzed organosolv processes.118,523 On the other hand, during the DAF, these species react with diols to form cyclic acetals, thus their recondensation is avoided (Fig. 41).118,521–523 Similar acetal structures can be formed as well by reaction of Hibbert's ketones with diols.521
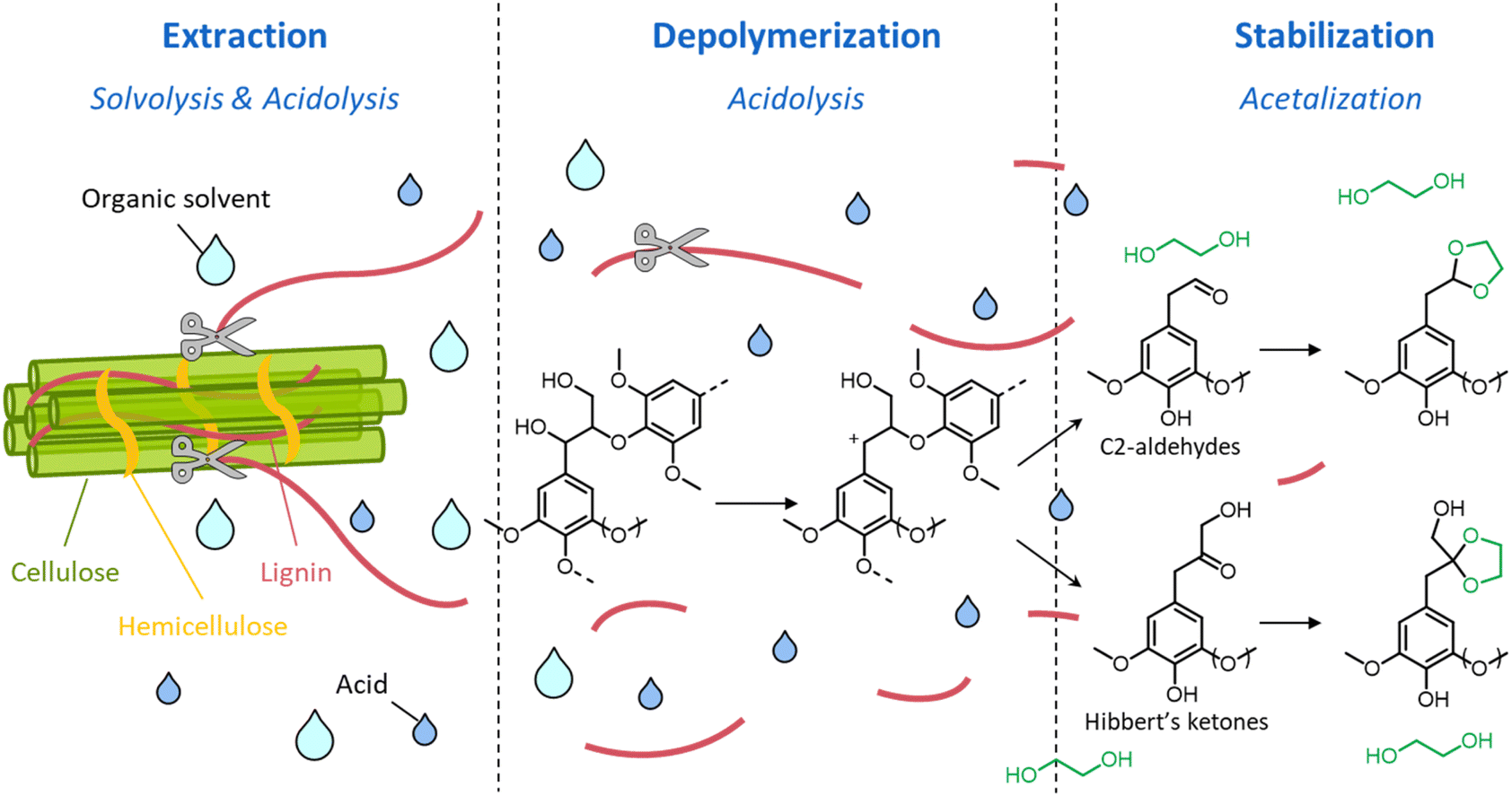 | ||
| Fig. 41 Schematic representation of lignin extraction, depolymerization, and stabilization during the DAF. Note: in this example, ethylene glycol is shown as a diol, but other diols can be adopted as well. | ||
By virtue of the stabilization of reactive lignin derivatives, the yield of monophenolics obtained from DAF is considerably larger compared to that achieved for an acid organosolv pretreatment, albeit lower than theoretical monomer yields.17,118,521,522 From this point of view, even though benzylic carbenium ions can also be stabilized by etherification with the diol (Fig. 42),217,521 the repolymerization of reactive lignin derivatives is not completely suppressed during DAF, posing a limitation to the yield of monophenolics.217,521,522 Notably, the extent of formation of C2-aldehydes and Hibbert's ketones is largely dependent on reaction conditions; hence, it is difficult to determine individual theoretical monomer yields for C2- and C3-acetals, respectively. Overall, for softwood biomass, DAF enables to effectively extract lignin from lignocellulosic biomass (up to about 80%) and to simultaneously depolymerize it, resulting in moderate yields of phenolic monomers (up to about 9%).
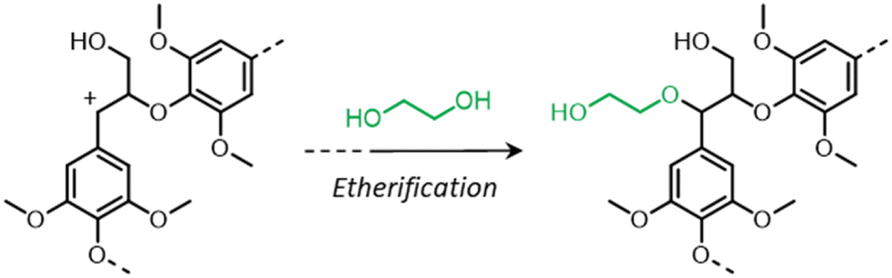 | ||
| Fig. 42 Stabilization of benzylic carbenium ions by etherification with the diol during the DAF. Note: in this example, ethylene glycol is shown as a diol, but other diols can be adopted as well. | ||
In addition to lignin, hemicellulose is also extracted during DAF. The acidic environment contributes to realize an extensive depolymerization of the released sugar fragments, yielding monosaccharides, which are hydroxyalkylated by further reaction with the diol.521,524 Notably, the modified sugars are more stable against dehydration and recondensation reactions.17 This reduces the formation of pseudolignin, thereby improving the quality and the processability of the recovered pulp. Hydroxyalkylated sugars can subsequently be converted back to their parent sugar forms by dilute acid hydrolysis in aqueous medium.521,524
The cellulosic pulp obtained from DAF possesses a high cellulose purity, and superior digestibility (up to ∼90% of glucan recovered as glucose upon enzymatic hydrolysis).521,524 Incorporation of the diol in the pulp may occur, due to glycosylation reactions, and the diol can be subsequently removed by dilute acid hydrolysis of the pulp.521 Importantly, the presence of the hydroxyalkylated sugars within the cellulose backbone does not have adverse effects on enzymatic hydrolysis.521
The features of the different product streams obtained via DAF are summarized in Fig. 43. In general, this process offers the advantage of affording moderately high yields of stable lignin and carbohydrate derivatives without requiring the use of a heterogeneous metal catalyst, thereby circumventing the issues related to catalyst deactivation and recovery-regeneration. However, the incorporation of diols in the products implies the need to recover this reagent downstream, or to feed fresh diols for every run, which result in additional costs. In spite of this, the DAF represents a relatively facile lignin-first process that is compatible with current industrial organosolv pretreatments.17 This aspect potentially simplifies further scale-up of the DAF.
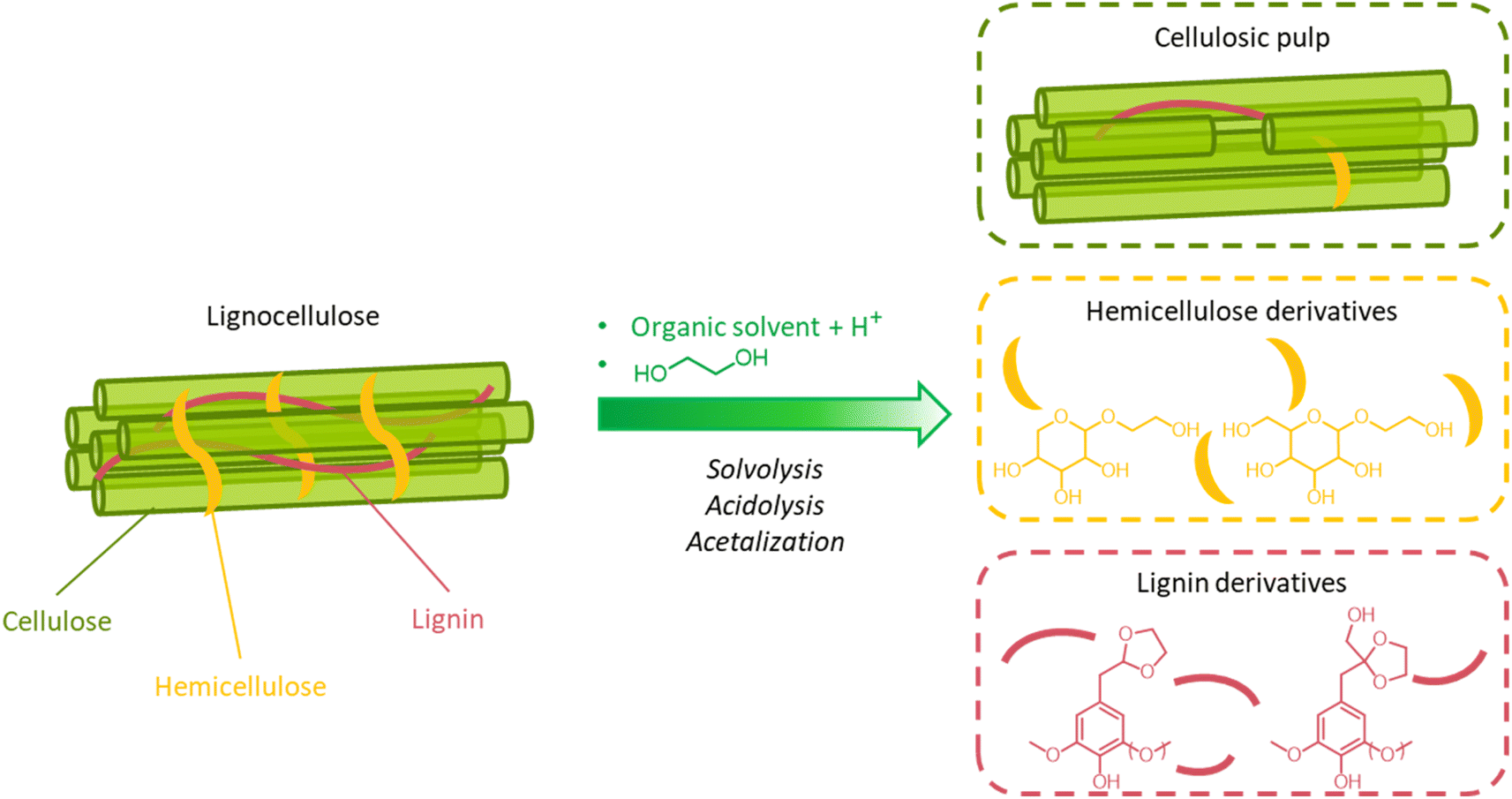 | ||
| Fig. 43 Schematic outline of the DAF process for the extraction, depolymerization and stabilization of lignin (and hemicellulose) derivatives from lignocellulosic biomass via solvolysis/acidolysis, acidolysis, and acetalization, respectively. Note: in this example, ethylene glycol is shown as a diol, but, in general, other diols can be adopted as well. The organic solvent should be an aprotic solvent, to prevent possible condensation reactions with the diol. | ||
6.4. Dithionite-assisted organosolv fractionation
Dithionite-assisted organosolv fractionation (DAOF) is based on the use of sodium dithionite (Na2S2O4) as a reducing agent within the organosolv pretreatment of lignocellulosic biomass to enhance the reductive depolymerization of lignin and to suppress the occurrence of lignin recondensation.531–533 Typically, DAOF is carried out in mixtures of n-butanol and water, at temperatures in a range of 150 and 250 °C.531–533 Lignin is extracted from lignocellulose by solvolysis of LCCs, similarly to an organosolv pretreatment, and large lignin fragments are released in solution. The latter are further broken down by the action of dithionite, which promotes the reductive cleavage of β-O-4′ linkages in lignin structures, leading to the formation of lignin moieties characterized by the presence of alkenyl functionalities in the side chains of phenolic units (Fig. 44). Such functionalities may ultimately be stabilized against recondensation by the reducing effect of dithionite. Along the formation of alkenyl and alkyl-substituted phenolics, lignin units possessing oxidized side chains are observed as well during DAOF and their presence has been explained by the occurrence of acid-catalyzed cleavage reactions of β-O-4′ linkages under DAOF conditions. | ||
| Fig. 44 Schematic representation of lignin extraction, depolymerization, and stabilization during DAOF. Based on Brienza et al.531 | ||
Thanks to the action of dithionite, the yield of phenolic monomers achieved during DAOF is superior to that obtained during typical organosolv pretreatments, but remains below the theoretical monomer yield, due to an incomplete prevention of lignin recondensation. Overall, DAOF has been shown to be partially effective for the treatment of hardwood and herbaceous biomass, resulting in excellent yields of delignification (up to about 80%)532 and moderately high yields of phenolic monomers (up to about 20% for hardwood biomass).532
Next to the lignin fraction, hemicellulose is also extensively extracted from the biomass. However, only a minor fraction of hemicellulose is converted toward sugars, glycols, or other non-condensed carbohydrate derivatives, whereas a considerable part of it is recovered as humic products in the lignin stream.531 Conversely, cellulose is almost completely preserved in the form of a highly pure cellulosic pulp possessing an excellent digestibility (up to ∼90% of glucan recovered as glucose upon enzymatic hydrolysis).531
Importantly, the use of various organic solvents may be envisaged for DAOF.533 The adoption of the n-butanol/water solvent system has been illustrated to be effective for enhancing lignin extraction and depolymerization (which would be hampered in the pure solvents)532 and to facilitate downstream product separation. The immiscibility of n-butanol and water at ambient conditions allows achieving the effective isolation of three distinct product fractions after DAOF: a solid fraction containing cellulose, an organic liquid fraction containing extracted lignin and humin derivatives (monomers and oligomers), and an aqueous fraction containing non-condensed carbohydrate derivatives (sugars, glycols, and organic acids).531
The outcomes of DAOF are schematically summarized in Fig. 45. Overall, DAOF is a less mature technology compared to other lignin-first pretreatments such as the RCF, AAF, or DAF. As such, some limitations currently exist for the adoption of DAOF. For instance, dithionite derivatives have been reported to be incorporated in the product streams,531,532 thereby complicating their subsequent upgrading, and making the potential recuperation of the reducing agent more challenging. In addition, the valorization of hemicellulose is hampered by the extensive condensation of carbohydrate derivatives and the consequent formation of humins.531 Nevertheless, a recent techno-economic assessment of the DAOF of birch wood has shown that this method could be economically feasible, especially in wood-abundant regions.532 Most promisingly, the flexibility of this method with respect to the treatment of various lignocellulosic feedstocks makes it an attractive candidate for the lignin-first pretreatment of highly heterogeneous waste streams (e.g. agricultural residues, waste wood).
 | ||
| Fig. 45 Schematic outline of the DAOF process for the extraction, depolymerization, and stabilization of lignin derivatives from lignocellulosic biomass via reductive cleavage, and hydrogenation, respectively. | ||
7. Conclusions and outlook
Traditionally, biomass pretreatment approaches applied in the pulping industry and in biorefineries have been principally focused on the valorization of the carbohydrate fraction of biomass, while much less attention has been dedicated to the conversion of the lignin fraction. (Hemi)cellulose is typically employed for papermaking, or for the production of biofuels (e.g. bioethanol). On the other hand, lignin streams isolated from biomass pretreatment are mainly exploited for energy production (e.g. via combustion), but can actually be employed also for other higher-value applications, such as the manufacturing of polymeric materials (e.g. via blending or copolymerization with synthetic polymers), or the production of bulk and fine chemicals (e.g. via lignin depolymerization and upgrading). From this point of view, the choice of the most suitable pathway for lignin valorization is related to the properties of the isolated lignin fractions.More recently, the perspective of utilizing lignin as a renewable source of aromatic chemicals and polymer precursors stimulated a renewed interest in the development of strategies for the depolymerization and upgrading of isolated lignin streams. Thus, a variety of methods have been developed for the depolymerization of pre-isolated lignin fractions toward phenolic monomers, and for the upgrading of the latter into marketable products through functionalization or defunctionalization pathways. However, lignin conversion has often been found to be tedious due to the high degree of condensation of lignin structures or to the presence of impurities (deriving from other biomass components or from additives employed within the pretreatment), which makes “technical lignin” recalcitrant toward further depolymerization, limiting the yield and the selectivity for target monomeric products. Clearly, achieving an effective transformation of lignin into products cannot prescind from taking into consideration the impact of the structural features of the isolated lignin fractions on the subsequent processing steps.
Studies on biomass fractionation highlighted that the reactivity of isolated lignin streams depends on the properties of the biomass feedstock employed (e.g. lignin content, β-O-4′ linkages content, G-/S-/H-units composition, …) and on the conditions applied within the pretreatment (e.g. process severity, use of chemical additives, …). The interplay between these factors ultimately determines the extent of cleavage of native labile inter-unit ether linkages in lignin matrices, and that of undesirable repolymerization reactions occurring during the fractionation. On the one hand, the genetic modification of plants can serve as a powerful means for creating less networked lignin structures, possessing an elevated content of cleavable inter-unit ether linkages. On the other hand, the careful design of biomass pretreatment methods is of paramount importance for taking full advantage of the potential reactivity of native lignin toward depolymerization. In this respect, suppressing the formation of stable C–C bonds during lignin isolation (and depolymerization) is a key aspect with respect to the production of lignin streams amenable toward further processing. While the adoption of low-severity fractionation methods may appear as a suitable solution, in that it allows to better preserve ether linkages in lignin structures, a tradeoff typically exists between the preservation of native β-O-4′ bonds and lignin extraction. Thus, low severity pretreatment methods are often coupled with low yields of isolated lignin derivatives.
A better response to the need of extracting non-condensed lignin streams from lignocellulosic biomass requires a thoughtful, on-purpose design of the fractionation process. In the last decade, novel so-called lignin-first strategies have emerged that rely on physical protection strategies (e.g. flow-through reactors), chemical protection strategies (e.g. catalytic hydrogenation, reduction, acetalization), or a combination thereof, to achieve a passivation of reactive lignin derivatives, while concomitantly allowing to effectively extract lignin from biomass, leaving behind a carbohydrate fraction amenable to further upgrading. Moreover, when hemicellulose is extracted along with lignin, carbohydrate fragments in solution are stabilized as well against recondensation. As a result, lignin-first approaches allow for a more complete valorization of lignocellulose compared to traditional pretreatments. In particular, lignin depolymerization can be achieved either concomitantly with its extraction, when inter-unit ether linkages are cleaved and the so-formed lignin fragments are stabilized (as in the RCF DAF, or DAOF), or in a subsequent step, when the protection strategy targets the stabilization of inter-unit ether linkages (as in the AAF). For both scenarios, the yield and selectivity toward monomeric products are substantially higher compared to those obtained upon the depolymerization of lignin streams isolated from traditional pretreatments. In addition, native lignin functionalities are better preserved within lignin-first processes, which allows to expand the range of potential products.
Currently, the upscaling of lignin-first strategies remains challenging for different reasons. For instance, the use of heterogeneous catalysts, which need to be recovered and regenerated, within some lignin-first approaches (e.g. RCF) represents a considerable complication compared to a typical organosolv pretreatment, and requires ad hoc solutions. One way could be envisaging facile and easily scalable strategies for catalyst recuperation (e.g. the use of flow-through reactors). Next to that, the development of new lignin-first approaches that do not require heterogeneous catalysis is also highly desirable for expanding the range of processing opportunities (e.g. DAF or DAOF). Moreover, suitable replacements for the toxic or explosive chemicals (e.g. aldehydes, H2, …) adopted within lignin-first processes should be envisaged to reduce the environmental and safety risks associated to their industrial implementation. Finally, the profitability of lignin-first pretreatments should be thoroughly investigated. From this perspective, the choice of the final market applications for lignin and carbohydrate derivatives has a crucial impact on the economic viability of lignin-first biorefineries, which can generate a wide spectrum of functionalized platform molecules. Hence, the exploration of new market sectors for the produced biomass derivatives represents a key strategy for maximizing the revenues from lignin-first processes.
Future research efforts should therefore be oriented toward improving the compatibility of lignin-first strategies with existing industrial pretreatments to facilitate their implementation within biorefinery schemes. In parallel, further studies should explore the use of lignin and carbohydrate derivatives for the manufacturing of high-value chemicals, pharmaceuticals, or polymers, to highlight what applications would benefit the most from the unique properties of such biomass-derived intermediates, and, ultimately, to pave the way for new, remunerative application routes.
In spite of the existing challenges, lignin valorization has come a long way throughout the last few decades. The initial (mis)conception that “one can make anything from lignin except money”, which used to be widespread within the pulp and paper sector, is much less popular today, as lignin-first processes are steps away from entering the industry. The long-desired waste-to-resource transition of lignin is gradually becoming a reality and the promising advancements made in the last few years project an exciting future for lignin valorization.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
F. B. thanks the FSR (Fonds spéciaux de recherche) from UCLouvain and the Innoviris research grant BRIDGE-RE4BRU for funding his PhD fellowship. D. P. D. thanks the Francqui Foundation for his Francqui Research Professor chair.References
- International Energy Agency (IEA), Global Energy Review: CO2 Emissions in 2021, Paris, 2022 Search PubMed.
- E. de Jong, A. Higson, P. Walsh and M. Wellisch, IEA Bioenergy – Task 42 Biorefinery, Bio Based Chemicals Value Added Products from Biorefineries, 2012 Search PubMed.
- F. Cherubini, Energy Convers. Manage., 2010, 51, 1412–1421 CrossRef CAS.
- F. Cherubini, G. Jungmeier, M. Wellisch, T. Willke, I. Skiadas, R. Van Ree and E. de Jong, Biofuels, Bioprod. Biorefin., 2009, 3, 534–546 CrossRef CAS.
- N. Dahmen, I. Lewandowski, S. Zibek and A. Weidtmann, GCB Bioenergy, 2019, 11, 107–117 CrossRef.
- R. Gérardy, D. P. Debecker, J. Estager, P. Luis and J.-C. M. Monbaliu, Chem. Rev., 2020, 120, 7219–7347 CrossRef PubMed.
- M. D. Smith, in ACS Symposium Series, ed. M. D. Smith, American Chemical Society, Washington, DC, 2019, vol. 1338, pp. 1–15 Search PubMed.
- M. S. Singhvi and D. V. Gokhale, Appl. Microbiol. Biotechnol., 2019, 103, 9305–9320 CrossRef CAS PubMed.
- A. Zoghlami and G. Paës, Front. Chem., 2019, 7, 874 CrossRef CAS PubMed.
- M. S. Singhvi, S. Chaudhari and D. V. Gokhale, RSC Adv., 2014, 4, 8271 RSC.
- M. V. Galkin and J. S. M. Samec, ChemSusChem, 2016, 9, 1544–1558 CrossRef CAS PubMed.
- G. Calvaruso, M. T. Clough, M. D. K. Rechulski and R. Rinaldi, ChemCatChem, 2017, 9, 2691–2700 CrossRef CAS.
- Q. Meng, J. Yan, R. Wu, H. Liu, Y. Sun, N. Wu, J. Xiang, L. Zheng, J. Zhang and B. Han, Nat. Commun., 2021, 12, 4534 CrossRef CAS PubMed.
- W. Schutyser, T. Renders, S. Van den Bosch, S.-F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. A. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 8164–8215 CrossRef CAS PubMed.
- Z. Sun, B. Fridrich, A. de Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef CAS PubMed.
- Y. M. Questell-Santiago, M. V. Galkin, K. Barta and J. S. Luterbacher, Nat. Rev. Chem, 2020, 4, 311–330 CrossRef CAS PubMed.
- X. Luo, Y. Li, N. K. Gupta, B. Sels, J. Ralph and L. Shuai, Angew. Chem., 2020, 132, 11800–11812 CrossRef.
- M. M. Abu-Omar, K. Barta, G. T. Beckham, J. S. Luterbacher, J. Ralph, R. Rinaldi, Y. Román-Leshkov, J. S. M. Samec, B. F. Sels and F. Wang, Energy Environ. Sci., 2021, 262–292 RSC.
- C. Zhang, X. Shen, Y. Jin, J. Cheng, C. Cai and F. Wang, Chem. Rev., 2023, 123, 4510–4601 CrossRef CAS PubMed.
- A. Yousuf, D. Pirozzi and F. Sannino, in Lignocellulosic Biomass to Liquid Biofuels, Elsevier, 2020, pp. 1–15 Search PubMed.
- P. E. Marriott, L. D. Gómez and S. J. McQueen-Mason, New Phytol., 2016, 209, 1366–1381 CrossRef CAS PubMed.
- N. Sorek, T. H. Yeats, H. Szemenyei, H. Youngs and C. R. Somerville, BioScience, 2014, 64, 192–201 CrossRef.
- W. B. Betts, R. K. Dart, A. S. Ball and S. L. Pedlar, in Biodegradation, ed. W. B. Betts, Springer London, London, 1991, pp. 139–155 Search PubMed.
- H. Chen, in Biotechnology of Lignocellulose, Springer Netherlands, Dordrecht, 2014, pp. 25–71 Search PubMed.
- J. Cai, Y. He, X. Yu, S. W. Banks, Y. Yang, X. Zhang, Y. Yu, R. Liu and A. V. Bridgwater, Renewable Sustainable Energy Rev., 2017, 76, 309–322 CrossRef CAS.
- V. Menon and M. Rao, Prog. Energy Combust. Sci., 2012, 38, 522–550 CrossRef CAS.
- A. Sluiter, B. Hames, R. Ruiz, C. Scarlata, J. Sluiter, D. Templeton and D. Crocker, Determination of Structural Carbohydrates and Lignin in Biomass, National Renewable Energy Laboratory, Golden, CO, USA, 2012 Search PubMed.
- M. Bunzel and J. Ralph, J. Agric. Food Chem., 2006, 54, 8352–8361 CrossRef CAS PubMed.
- M. Bunzel, A. Schüßler and G. Tchetseubu Saha, J. Agric. Food Chem., 2011, 59, 12506–12513 CrossRef CAS PubMed.
- N. Sorek, T. H. Yeats, H. Szemenyei, H. Youngs and C. R. Somerville, BioScience, 2014, 64, 192–201 CrossRef.
- P. E. Marriott, L. D. Gómez and S. J. McQueen-Mason, New Phytol., 2016, 209, 1366–1381 CrossRef CAS PubMed.
- G. Potters, D. Van Goethem and F. Schutte, Nat. Educ., 2010, 3, 14 Search PubMed.
- C. Yang and X. Lü, in Advances in 2nd Generation of Bioethanol Production, Elsevier, 2021, pp. 71–85 Search PubMed.
- R. W. Lenz, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 2401 CrossRef.
- C. Somerville, Annu. Rev. Cell Dev. Biol., 2006, 22, 53–78 CrossRef CAS PubMed.
- K. Kulasinski, S. Keten, S. V. Churakov, D. Derome and J. Carmeliet, Cellulose, 2014, 21, 1103–1116 CrossRef CAS.
- N. Lavoine, I. Desloges, A. Dufresne and J. Bras, Carbohydr. Polym., 2012, 90, 735–764 CrossRef CAS PubMed.
- Y. Liao, B. O. de Beeck, K. Thielemans, T. Ennaert, J. Snelders, M. Dusselier, C. M. Courtin and B. F. Sels, Mol. Catal., 2020, 487, 110883 CrossRef CAS.
- M. Pauly, S. Gille, L. Liu, N. Mansoori, A. de Souza, A. Schultink and G. Xiong, Planta, 2013, 238, 627–642 CrossRef CAS PubMed.
- H. V. Scheller and P. Ulvskov, Annu. Rev. Plant Biol., 2010, 61, 263–289 CrossRef CAS PubMed.
- T. Rodrigues Mota, D. Matias de Oliveira, R. Marchiosi, O. Ferrarese-Filho and W. Dantas dos Santos, AIMS Bioeng., 2018, 5, 63–77 Search PubMed.
- W. Farhat, R. A. Venditti, M. Hubbe, M. Taha, F. Becquart and A. Ayoub, ChemSusChem, 2017, 10, 305–323 CrossRef CAS PubMed.
- L. B. Davin and N. G. Lewis, Plant Physiol., 2000, 123, 453–462 CrossRef CAS PubMed.
- J. Ralph, Phytochem. Rev., 2010, 9, 65–83 CrossRef CAS.
- F. Lu, J. Ralph, K. Morreel, E. Messens and W. Boerjan, Org. Biomol. Chem., 2004, 2, 2888 RSC.
- W. Lan, F. Lu, M. Regner, Y. Zhu, J. Rencoret, S. A. Ralph, U. I. Zakai, K. Morreel, W. Boerjan and J. Ralph, Plant Physiol., 2015, 167, 1284–1295 CrossRef CAS PubMed.
- R. Vanholme, K. Morreel, C. Darrah, P. Oyarce, J. H. Grabber, J. Ralph and W. Boerjan, New Phytol., 2012, 196, 978–1000 CrossRef CAS PubMed.
- J. C. del Río, J. Rencoret, A. Gutiérrez, H. Kim and J. Ralph, in Advances in Botanical Research, Elsevier, 2022, p. S0065229622000131 Search PubMed.
- T. E. Timell, Wood Sci. Technol., 1982, 16, 83–122 CrossRef CAS.
- D. M. Neiva, J. Rencoret, G. Marques, A. Gutiérrez, J. Gominho, H. Pereira and J. C. Del Río, ChemSusChem, 2020, 13, 4537–4547 CrossRef CAS PubMed.
- J. Dou, H. Kim, Y. Li, D. Padmakshan, F. Yue, J. Ralph and T. Vuorinen, J. Agric. Food Chem., 2018, 66, 7294–7300 CrossRef CAS PubMed.
- J. Rencoret, D. Neiva, G. Marques, A. Gutiérrez, H. Kim, J. Gominho, H. Pereira, J. Ralph and J. C. Del Río, Plant Physiol., 2019, 180, 1310–1321 CrossRef CAS PubMed.
- E. M. Anderson, M. L. Stone, R. Katahira, M. Reed, W. Muchero, K. J. Ramirez, G. T. Beckham and Y. Román-Leshkov, Nat. Commun., 2019, 10, 2033 CrossRef PubMed.
- M. Wang and F. Wang, Adv. Mater., 2019, 31, 1901866 CrossRef PubMed.
- R. Parthasarathi, R. A. Romero, A. Redondo and S. Gnanakaran, J. Phys. Chem. Lett., 2011, 2, 2660–2666 CrossRef CAS.
- Y. Zhao, U. Shakeel, M. Saif Ur Rehman, H. Li, X. Xu and J. Xu, J. Cleaner Prod., 2020, 253, 120076 CrossRef CAS.
- D. Tarasov, M. Leitch and P. Fatehi, Biotechnol. Biofuels, 2018, 11, 269 CrossRef PubMed.
- N. Giummarella, Y. Pu, A. J. Ragauskas and M. Lawoko, Green Chem., 2019, 21, 1573–1595 RSC.
- D. M. D. Carvalho, M. H. Lahtinen, M. Lawoko and K. S. Mikkonen, ACS Sustainable Chem. Eng., 2020, 8, 11795–11804 CrossRef.
- M. Balakshin, E. Capanema, H. Gracz, H. Chang and H. Jameel, Planta, 2011, 233, 1097–1110 CrossRef CAS.
- B.-C. Zhao, B.-Y. Chen, S. Yang, T.-Q. Yuan, A. Charlton and R.-C. Sun, ACS Sustainable Chem. Eng., 2017, 5, 1113–1122 CrossRef CAS.
- N. Giummarella, L. Zhang, G. Henriksson and M. Lawoko, RSC Adv., 2016, 6, 42120–42131 RSC.
- L. Yao, C. Chen, X. Zheng, Z. Peng, H. Yang and Y. Xie, BioResources, 2016, 11, 6692–6707 CAS.
- D. M. de Oliveira, A. Finger-Teixeira, T. Rodrigues Mota, V. H. Salvador, F. C. Moreira-Vilar, H. B. Correa Molinari, R. A. Craig Mitchell, R. Marchiosi, O. Ferrarese-Filho and W. Dantas dos Santos, Plant Biotechnol. J., 2015, 13, 1224–1232 CrossRef CAS PubMed.
- A. Barakat, C. Mayer-Laigle, A. Solhy, R. A. D. Arancon, H. de Vries and R. Luque, RSC Adv., 2014, 4, 48109–48127 RSC.
- Z.-H. Liu, L. Qin, B.-Z. Li and Y.-J. Yuan, ACS Sustainable Chem. Eng., 2015, 3, 140–146 CrossRef CAS.
- P. Bajpai, in Pretreatment of Lignocellulosic Biomass for Biofuel Production, Springer Singapore, Singapore, 2016, pp. 17–70 Search PubMed.
- J. Baruah, B. K. Nath, R. Sharma, S. Kumar, R. C. Deka, D. C. Baruah and E. Kalita, Front Energy Res, 2018, 6, 141 CrossRef.
- M. Galbe and O. Wallberg, Biotechnol. Biofuels, 2019, 12, 294 CrossRef PubMed.
- B. Lönnberg, Papermaking Science and Technology. 5: Mechanical Pulping, ed. B. Lönnberg, Paperi ja Puu Oy, Helsinki, 2nd edn, totally updated version, 2009 Search PubMed.
- J. Blechschmidt and S. Heinemann, in Handbook of Pulp, ed. H. Sixta, Wiley-VCH Verlag GmbH, Weinheim, Germany, 2006, pp. 1079–1111 Search PubMed.
- M. H. Sipponen, S. Laakso and S. Baumberger, Ind. Crops Prod., 2014, 61, 130–136 CrossRef CAS.
- Z. Wang, X. Zhu and P. J. Deuss, Ind. Crops Prod., 2021, 167, 113493 CrossRef CAS.
- Y. Wu, S. Ge, C. Xia, C. Mei, K.-H. Kim, L. Cai, L. M. Smith, J. Lee and S. Q. Shi, Renewable Sustainable Energy Rev., 2021, 136, 110442 CrossRef CAS.
- A. T. W. M. Hendriks and G. Zeeman, Bioresour. Technol., 2009, 100, 10–18 CrossRef CAS PubMed.
- A. S. da Silva, R. S. Sobral Teixeira, R. de Oliveira, V. Santana, R. R. O. de Barros, M. Antonieta and E. P. da Silva Bo, in Sustainable Degradation of Lignocellulosic Biomass – Techniques, Applications and Commercialization, ed. A. Chandel, InTech, 2013 Search PubMed.
- K. M. Holtman, H. Chang, H. Jameel and J. F. Kadla, J. Wood Chem. Technol., 2006, 26, 21–34 CrossRef CAS.
- Y. W. Sitotaw, N. G. Habtu, A. Y. Gebreyohannes, S. P. Nunes and T. Van Gerven, Biomass Convers. Biorefin., 2021 DOI:10.1007/s13399-021-01800-7.
- A. Duque, P. Manzanares and M. Ballesteros, Renewable Energy, 2017, 114, 1427–1441 CrossRef CAS.
- A. Gallos, G. Paës, F. Allais and J. Beaugrand, RSC Adv., 2017, 7, 34638–34654 RSC.
- L. Capolupo and V. Faraco, Appl. Microbiol. Biotechnol., 2016, 100, 9451–9467 CrossRef CAS PubMed.
- E. Gatt, L. Rigal and V. Vandenbossche, Ind. Crops Prod., 2018, 122, 329–339 CrossRef CAS.
- K. S. Guiao, A. Gupta, C. Tzoganakis and T. H. Mekonnen, J. Cleaner Prod., 2022, 355, 131840 CrossRef CAS.
- M. R. Al-Masri and M. Zarkawi, Radiat. Phys. Chem., 1994, 44, 661–663 CrossRef CAS.
- M. Kumakura and I. Kaetsu, Biotechnol. Bioeng., 1978, 20, 1309–1315 CrossRef CAS.
- Y.-L. Loow, T. Y. Wu, G. H. Yang, J. Md. Jahim, W. H. Teoh and A. W. Mohammad, Cellulose, 2016, 23, 2761–2789 CrossRef CAS.
- Y. Liu, L. Guo, L. Wang, W. Zhan and H. Zhou, Bioresour. Technol., 2017, 232, 270–277 CrossRef CAS.
- X. Fei, W. Jia, J. Wang, T. Chen and Y. Ling, Int. J. Biol. Macromol., 2020, 145, 733–739 CrossRef CAS PubMed.
- X. Wu, L. Chen, W. He, H. Qi, Y. Zhang, Y. Zhou, X. Su, M. Deng and K. Wang, Ind. Crops Prod., 2020, 150, 112228 CrossRef CAS.
- A. Aguilar-Reynosa, A. Romaní, R. Ma. Rodríguez-Jasso, C. N. Aguilar, G. Garrote and H. A. Ruiz, Energy Convers. Manage., 2017, 136, 50–65 CrossRef CAS.
- H. Li, Y. Qu, Y. Yang, S. Chang and J. Xu, Bioresour. Technol., 2016, 199, 34–41 CrossRef CAS PubMed.
- S. Ethaib, R. Omar, S. M. Kamal and D. A. Biak, J. Eng. Sci. Technol., 2015, 10, 97–109 Search PubMed.
- V. Sorn, K.-L. Chang, P. Phitsuwan, K. Ratanakhanokchai and C.-D. Dong, Bioresour. Technol., 2019, 293, 121929 CrossRef CAS PubMed.
- K. Kohli, S. Katuwal, A. Biswas and B. K. Sharma, Bioresour. Technol., 2020, 303, 122897 CrossRef CAS PubMed.
- A. Isci, G. M. Erdem, S. Bagder Elmaci, O. Sakiyan, A. Lamp and M. Kaltschmitt, Cellulose, 2020, 27, 8949–8962 CrossRef CAS.
- S. Shangdiar, Y.-C. Lin, V. K. Ponnusamy and T.-Y. Wu, Bioresour. Technol., 2022, 361, 127724 CrossRef CAS PubMed.
- A. T. Hoang, S. Nižetić, H. C. Ong, M. Mofijur, S. F. Ahmed, B. Ashok, V. T. V. Bui and M. Q. Chau, Chemosphere, 2021, 281, 130878 CrossRef CAS PubMed.
- V. Z. Ong and T. Y. Wu, Renewable Sustainable Energy Rev., 2020, 132, 109924 CrossRef CAS.
- G. Yang and J. Wang, Bioresour. Technol., 2019, 275, 10–18 CrossRef CAS PubMed.
- V. Sharma, P. Nargotra and B. K. Bajaj, Bioresour. Technol., 2019, 285, 121319 CrossRef CAS PubMed.
- W. Zhang, J. Liu, Y. Wang, J. Sun, P. Huang and K. Chang, Biomass Convers. Biorefin., 2021, 11, 1749–1757 CrossRef CAS.
- X. Yu, X. Bao, C. Zhou, L. Zhang, A. E.-G. A. Yagoub, H. Yang and H. Ma, Ultrason. Sonochem., 2018, 41, 410–418 CrossRef CAS PubMed.
- Q. Ji, X. Yu, A. E. A. Yagoub, L. Chen, A. T. Mustapha and C. Zhou, Renewable Energy, 2021, 172, 304–316 CrossRef CAS.
- N. S. Mosier, in Aqueous Pretreatment of Plant Biomass for Biological and Chemical Conversion to Fuels and Chemicals, ed. C. E. Wyman, John Wiley & Sons, Ltd, Chichester, UK, 2013, pp. 129–143 Search PubMed.
- X. Zhuang, W. Wang, Q. Yu, W. Qi, Q. Wang, X. Tan, G. Zhou and Z. Yuan, Bioresour. Technol., 2016, 199, 68–75 CrossRef CAS PubMed.
- L. Yan, R. Ma, L. Li and J. Fu, Chem. Eng. Technol., 2016, 39, 1759–1770 CrossRef CAS.
- I. Cybulska, G. Brudecki and H. Lei, in Green Biomass Pretreatment for Biofuels Production, ed. T. Gu, Springer, Netherlands, Dordrecht, 2013, pp. 87–106 Search PubMed.
- L.-P. Xiao, G.-Y. Song and R.-C. Sun, in Hydrothermal Processing in Biorefineries, ed. H. A. Ruiz, M. Hedegaard Thomsen and H. L. Trajano, Springer International Publishing, Cham, 2017, pp. 45–94 Search PubMed.
- M. Pedersen and A. S. Meyer, New Biotechnol., 2010, 27, 739–750 CrossRef CAS PubMed.
- J. Larsen, M. Østergaard Petersen, L. Thirup, H. Wen Li and F. Krogh Iversen, Chem. Eng. Technol., 2008, 31, 765–772 CrossRef CAS.
- D. Cannella and H. Jørgensen, Biotechnol. Bioeng., 2014, 111, 59–68 CrossRef CAS PubMed.
- K. Birikh, A. Michine, M. Heikkilä and P. Ihalainen, in Biorefineries – Selected Processes, ed. K. Biernat, IntechOpen, 2022 Search PubMed.
- K. Lundquist, R. Lundgren, J. Danielsen, A. T. Haaland and S. Svensson, Acta Chem. Scand., 1972, 26, 2005–2023 CrossRef CAS.
- M. R. Sturgeon, S. Kim, K. Lawrence, R. S. Paton, S. C. Chmely, M. Nimlos, T. D. Foust and G. T. Beckham, ACS Sustainable Chem. Eng., 2014, 2, 472–485 CrossRef CAS.
- T. Yokoyama and Y. Matsumoto, Holzforschung, 2008, 62, 164–168 CrossRef CAS.
- H. Ito, T. Imai, K. Lundquist, T. Yokoyama and Y. Matsumoto, J. Wood Chem. Technol., 2011, 31, 172–182 CrossRef CAS.
- P. J. Deuss, M. Scott, F. Tran, N. J. Westwood, J. G. de Vries and K. Barta, J. Am. Chem. Soc., 2015, 137, 7456–7467 CrossRef CAS PubMed.
- B. S. Donohoe, S. R. Decker, M. P. Tucker, M. E. Himmel and T. B. Vinzant, Biotechnol. Bioeng., 2008, 101, 913–925 CrossRef CAS.
- I. K. M. Yu, H. Chen, F. Abeln, H. Auta, J. Fan, V. L. Budarin, J. H. Clark, S. Parsons, C. J. Chuck, S. Zhang, G. Luo and D. C. W. Tsang, Crit. Rev. Environ. Sci. Technol., 2021, 51, 1479–1532 CrossRef CAS.
- M. Yu. Balakshin, E. A. Capanema, I. Sulaeva, P. Schlee, Z. Huang, M. Feng, M. Borghei, O. J. Rojas, A. Potthast and T. Rosenau, ChemSusChem, 2021, 14, 1016–1036 CrossRef CAS PubMed.
- B. Cheng, X. Wang, Q. Lin, X. Zhang, L. Meng, R.-C. Sun, F. Xin and J. Ren, J. Agric. Food Chem., 2018, 66, 11981–11989 CrossRef CAS PubMed.
- C. Fang, J. E. Schmidt, I. Cybulska, G. P. Brudecki, C. G. Frankær and M. H. Thomsen, BioMed Res. Int., 2015, 2015, 1–13 Search PubMed.
- A. T. Hoang, X. P. Nguyen, X. Q. Duong, Ü. Ağbulut, C. Len, P. Q. P. Nguyen, M. Kchaou and W.-H. Chen, Bioresour. Technol., 2023, 385, 129398 CrossRef CAS PubMed.
- Y. Yu, J. Wu, X. Ren, A. Lau, H. Rezaei, M. Takada, X. Bi and S. Sokhansanj, Renewable Sustainable Energy Rev., 2022, 154, 111871 CrossRef CAS.
- T. Auxenfans, D. Crônier, B. Chabbert and G. Paës, Biotechnol. Biofuels, 2017, 10, 36 CrossRef PubMed.
- I. Cybulska, T. Chaturvedi and M. H. Thomsen, in Biorefinery, ed. J.-R. Bastidas-Oyanedel and J. E. Schmidt, Springer International Publishing, Cham, 2019, pp. 153–165 Search PubMed.
- A. Duque, P. Manzanares, I. Ballesteros and M. Ballesteros, in Biomass Fractionation Technologies for a Lignocellulosic Feedstock Based Biorefinery, Elsevier, 2016, pp. 349–368 Search PubMed.
- D. Sulzenbacher, D. Atzmüller, F. Hawe, M. Richter, A. Cristobal-Sarramian and A. Zwirzitz, Biomass Convers. Biorefin., 2023, 13, 1035–1046 CrossRef CAS.
- T. R. Sarker, F. Pattnaik, S. Nanda, A. K. Dalai, V. Meda and S. Naik, Chemosphere, 2021, 284, 131372 CrossRef CAS PubMed.
- D. Kim, Molecules, 2018, 23, 309 CrossRef PubMed.
- F. Rodríguez, A. Sanchez and C. Parra, ACS Sustainable Chem. Eng., 2017, 5, 5234–5240 CrossRef.
- C. Martín, G. Wu, Z. Wang, S. Stagge and L. J. Jönsson, Bioresour. Technol., 2018, 262, 242–250 CrossRef.
- N. Jacquet, G. Maniet, C. Vanderghem, F. Delvigne and A. Richel, Ind. Eng. Chem. Res., 2015, 54, 2593–2598 CrossRef CAS.
- N. R. Baral and A. Shah, Bioresour. Technol., 2017, 232, 331–343 CrossRef CAS PubMed.
- C. Zhao, Q. Shao and S. P. S. Chundawat, Bioresour. Technol., 2020, 298, 122446 CrossRef CAS.
- V. Balan, B. Bals, S. P. S. Chundawat, D. Marshall and B. E. Dale, in Biofuels, ed. J. R. Mielenz, Humana Press, Totowa, NJ, 2009, vol. 581, pp. 61–77 Search PubMed.
- S. P. S. Chundawat, B. S. Donohoe, L. da Costa Sousa, T. Elder, U. P. Agarwal, F. Lu, J. Ralph, M. E. Himmel, V. Balan and B. E. Dale, Energy Environ. Sci., 2011, 4, 973 RSC.
- J. R. Meyer, S. B. Waghmode, J. He, Y. Gao, D. Hoole, L. Da Costa Sousa, V. Balan and M. B. Foston, Biomass Bioenergy, 2018, 119, 446–455 CrossRef CAS.
- Y. Sun and J. Cheng, Bioresour. Technol., 2002, 83, 1–11 CrossRef CAS PubMed.
- B. Kamm, S. Leiß, P. Schönicke and M. Bierbaum, ChemSusChem, 2017, 10, 48–52 CrossRef CAS.
- M. Jin, L. da Costa Sousa, C. Schwartz, Y. He, C. Sarks, C. Gunawan, V. Balan and B. E. Dale, Green Chem., 2016, 18, 957–966 RSC.
- T. Gu, in Green Biomass Pretreatment for Biofuels Production, ed. T. Gu, Springer Netherlands, Dordrecht, 2013, pp. 107–125 Search PubMed.
- H. Li, H. Wu, Z. Yu, H. Zhang and S. Yang, ChemSusChem, 2020, 13, 3565–3582 CrossRef CAS PubMed.
- A. R. C. Morais and R. M. Lukasik, in Hydrothermal Processing in Biorefineries, ed. H. A. Ruiz, M. Hedegaard Thomsen and H. L. Trajano, Springer International Publishing, Cham, 2017, pp. 353–376 Search PubMed.
- M. Zhao, Q. Xu, G. Li, Q. Zhang, D. Zhou, J. Yin and H. Zhan, J. Energy Chem., 2019, 31, 39–45 CrossRef.
- A. Navarro, C. Montiel, J. Gracia-Fadrique, A. Tecante and E. Bárzana, Biomass Convers. Biorefin., 2023, 13, 5691–5699 CrossRef CAS.
- K. H. Kim and J. Hong, Bioresour. Technol., 2001, 77, 139–144 CrossRef CAS PubMed.
- D. Lachenal, in Lignocellulosic Fibers and Wood Handbook, ed. N. Belgacem and A. Pizzi, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2016, pp. 207–223 Search PubMed.
- E. Sjöström, in Wood Chemistry, Elsevier, 1993, pp. 114–164 Search PubMed.
- H. Sixta, A. Potthast and A. W. Krotschek, in Handbook of Pulp, ed. H. Sixta, Wiley-VCH Verlag GmbH, Weinheim, Germany, 2006, pp. 109–229 Search PubMed.
- S. Van den Bosch, S.-F. Koelewijn, T. Renders, G. Van den Bossche, T. Vangeel, W. Schutyser and B. F. Sels, Top. Curr. Chem., 2018, 376, 36 CrossRef CAS PubMed.
- F. S. Chakar and A. J. Ragauskas, Ind. Crops Prod., 2004, 20, 131–141 CrossRef CAS.
- J. Gierer, Wood Sci. Technol., 1985, 19, 289–312 CrossRef CAS.
- Pulping Chemistry and Technology, ed. M. Ek, G. Gellerstedt and G. Henriksson, De Gruyter, 2009, pp. 91–120 Search PubMed.
- O. Fearon, V. Nykänen, S. Kuitunen, K. Ruuttunen, R. Alén, V. Alopaeus and T. Vuorinen, AIChE J., 2020, 66(8), e16252 CrossRef CAS.
- J. Gierer, Wood Sci. Technol., 1980, 14, 241–266 CrossRef CAS.
- P. Bajpai, in Biermann's Handbook of Pulp and Paper, Elsevier, 2018, pp. 425–451 Search PubMed.
- C. S. Lancefield, H. L. J. Wienk, R. Boelens, B. M. Weckhuysen and P. C. A. Bruijnincx, Chem. Sci., 2018, 9, 6348–6360 RSC.
- S. Constant, H. L. J. Wienk, A. E. Frissen, P. de Peinder, R. Boelens, D. S. van Es, R. J. H. Grisel, B. M. Weckhuysen, W. J. J. Huijgen, R. J. A. Gosselink and P. C. A. Bruijnincx, Green Chem., 2016, 18, 2651–2665 RSC.
- D. V. Evtuguin, in Lignocellulosic Fibers and Wood Handbook, ed. N. Belgacem and A. Pizzi, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2016, pp. 225–244 Search PubMed.
- Handbook of Pulp, ed. H. Sixta, Wiley-VCH Verlag GmbH, Weinheim, Germany, 2006, pp. 366–509 Search PubMed.
- G. Gellerstedt, Sven. Papperstidn., 1976, 79, 537 CAS.
- G. Gellerstedt, J. Gierer, O. Buchardt and T. Norin, Acta Chem. Scand., 1968, 22, 2510–2518 CrossRef CAS.
- G. Gellerstedt, J. Gierer, B. Reinhammar and P. H. Nielsen, Acta Chem. Scand., 1968, 22, 2029–2031 CrossRef CAS.
- G. Gellerstedt, J. Gierer, B. Lüning, S. Liaaen-Jensen, A. Lamvik, E. Sunde and N. A. Sørensen, Acta Chem. Scand., 1970, 24, 1645–1654 CrossRef CAS.
- G. Gellerstedt, Acta Chem. Scand., 1972, 26, 701 CrossRef CAS.
- T. Aro and P. Fatehi, ChemSusChem, 2017, 10, 1861–1877 CrossRef CAS PubMed.
- P. Fatehi and Y. Ni, in ACS Symposium Series, ed. J. Zhu, X. Zhang and X. Pan, American Chemical Society, Washington, DC, 2011, vol. 1067, pp. 409–441 Search PubMed.
- H. Sixta, in Handbook of Pulp, ed. H. Sixta, Wiley-VCH Verlag GmbH, Weinheim, Germany, 2006, pp. 2–19 Search PubMed.
- A. Vishtal and A. Kraslawski, BioResources, 2011, 6, 3547–3568 Search PubMed.
- F. G. Calvo-Flores, J. A. Dobado, J. Isac-García and F. J. Martín-MartíNez, in Lignin and Lignans as Renewable Raw Materials, John Wiley & Sons, Ltd, Chichester, UK, 2015, pp. 113–144 Search PubMed.
- G.-H. Kim and B.-H. Um, Int. J. Biol. Macromol., 2020, 158, 443–451 CrossRef CAS PubMed.
- P. Bajpai, in Biermann's Handbook of Pulp and Paper, Elsevier, 2018, pp. 295–351 Search PubMed.
- S. Anderson, D. Dimmel and P. Izsak, J. Wood Chem. Technol., 2003, 23, 141–159 CrossRef.
- G. Brodeur, E. Yau, K. Badal, J. Collier, K. B. Ramachandran and S. Ramakrishnan, Enzyme Res., 2011, 2011, 1–17 CrossRef.
- J. Zhang and J. Bao, in Handbook of Biorefinery Research and Technology, ed. J. M. Park, Springer Netherlands, Dordrecht, 2018, pp. 1–14 Search PubMed.
- R. Bielski and G. Grynkiewicz, Green Chem., 2021, 23, 7458–7487 RSC.
- X. Tong, Y. Ma and Y. Li, Appl. Catal., A, 2010, 385, 1–13 CrossRef CAS.
- K. Yan, C. Jarvis, J. Gu and Y. Yan, Renewable Sustainable Energy Rev., 2015, 51, 986–997 CrossRef CAS.
- F. D. Pileidis and M.-M. Titirici, ChemSusChem, 2016, 9, 562–582 CrossRef CAS PubMed.
- H. U. Suess, Pulp Bleaching Today, De Gruyter, Berlin, New York, 2010 Search PubMed.
- H. Sixta, H.-U. Sss, A. Potthast, M. Schwanninger and A. W. Krotscheck, in Handbook of Pulp, ed. H. Sixta, Wiley-VCH Verlag GmbH, Weinheim, Germany, 2006, pp. 609–708 Search PubMed.
- J. Gierer, Wood Sci. Technol., 1986, 20, 1–33 CrossRef CAS.
- R. Ma, Y. Xu and X. Zhang, ChemSusChem, 2015, 8, 24–51 CrossRef CAS PubMed.
- V. E. Tarabanko, Yu. V. Hendogina, D. V. Petuhov and E. P. Pervishina, React. Kinet. Catal. Lett., 2000, 69, 361–368 CrossRef CAS.
- V. E. Tarabanko and D. V. Petukhov, Chem. Sustainable Dev., 2003, 11, 655–667 Search PubMed.
- Y. Han, Y. Bai, J. Zhang, D. Liu and X. Zhao, Bioresour. Bioprocess., 2020, 7, 24 CrossRef.
- Z. Li, C. H. Chen, T. Liu, V. Mathrubootham, E. L. Hegg and D. B. Hodge, Biotechnol. Bioeng., 2013, 110, 1078–1086 CrossRef CAS PubMed.
- Z. Li, N. Bansal, A. Azarpira, A. Bhalla, C. H. Chen, J. Ralph, E. L. Hegg and D. B. Hodge, Biotechnol. Biofuels, 2015, 8, 123 CrossRef PubMed.
- G. E. Klinger, Y. Zhou, J. A. Foote, A. M. Wester, Y. Cui, M. Alherech, S. S. Stahl, J. E. Jackson and E. L. Hegg, ChemSusChem, 2020, 13, 4394–4399 CrossRef CAS PubMed.
- Z. Fang, M. G. Flynn, J. E. Jackson and E. L. Hegg, Green Chem., 2021, 23, 412–421 RSC.
- H. Luo, E. P. Weeda, M. Alherech, C. W. Anson, S. D. Karlen, Y. Cui, C. E. Foster and S. S. Stahl, J. Am. Chem. Soc., 2021, 143, 15462–15470 CrossRef CAS PubMed.
- X. Zhao, K. Cheng and D. Liu, Appl. Microbiol. Biotechnol., 2009, 82, 815–827 CrossRef CAS PubMed.
- M. N. Borand and F. Karaosmanoğlu, J. Renewable Sustainable Energy, 2018, 10, 033104 CrossRef.
- D. Wei Kit Chin, S. Lim, Y. L. Pang and M. K. Lam, Biofuels, Bioprod. Biorefin., 2020, 14, 808–829 CrossRef CAS.
- Z. Zhang, M. D. Harrison, D. W. Rackemann, W. O. S. Doherty and I. M. O'Hara, Green Chem., 2016, 18, 360–381 RSC.
- P. P. Thoresen, L. Matsakas, U. Rova and P. Christakopoulos, Bioresour. Technol., 2020, 306, 123189 CrossRef CAS.
- D. S. Zijlstra, C. W. Lahive, C. A. Analbers, M. B. Figueirêdo, Z. Wang, C. S. Lancefield and P. J. Deuss, ACS Sustainable Chem. Eng., 2020, 8, 5119–5131 CrossRef CAS.
- I. Salapa, C. Katsimpouras, E. Topakas and D. Sidiras, Biomass Bioenergy, 2017, 100, 10–16 CrossRef CAS.
- T. A. Pham, D. S. Ngo and K. A. To, Sugar Tech, 2022, 24, 779–787 CrossRef CAS.
- M. Hu, J. Chen, Y. Yu and Y. Liu, Molecules, 2022, 27, 6359 CrossRef CAS PubMed.
- A. T. Smit, M. Verges, P. Schulze, A. van Zomeren and H. Lorenz, ACS Sustainable Chem. Eng., 2022, 10, 10503–10513 CrossRef CAS.
- J. Tao, O. Hosseinaei, L. Delbeck, P. Kim, D. P. Harper, J. J. Bozell, T. G. Rials and N. Labbé, RSC Adv., 2016, 6, 79228–79235 RSC.
- C. M. Cai, T. Zhang, R. Kumar and C. E. Wyman, Green Chem., 2013, 15, 3140 RSC.
- E. Viola, F. Zimbardi, M. Morgana, N. Cerone, V. Valerio and A. Romanelli, Processes, 2021, 9, 2051 CrossRef CAS.
- D. M. Alonso, S. H. Hakim, S. Zhou, W. Won, O. Hosseinaei, J. Tao, V. Garcia-Negron, A. H. Motagamwala, M. A. Mellmer, K. Huang, C. J. Houtman, N. Labbé, D. P. Harper, C. T. Maravelias, T. Runge and J. A. Dumesic, Sci. Adv., 2017, 3, e1603301 CrossRef PubMed.
- M. Granatier, H. Q. Lê, E. C. González and H. Sixta, RSC Sustainability, 2023, 1, 97–106 RSC.
- T. J. McDonough, Tappi J., 1993, 76, 186–193 CAS.
- C. Lai, M. Tu, C. Xia, Z. Shi, S. Sun, Q. Yong and S. Yu, Energy Fuels, 2017, 31, 12317–12326 CrossRef CAS.
- C. S. Lancefield, I. Panovic, P. J. Deuss, K. Barta and N. J. Westwood, Green Chem., 2017, 19, 202–214 RSC.
- D. S. Zijlstra, J. de Korte, E. P. C. de Vries, L. Hameleers, E. Wilbers, E. Jurak and P. J. Deuss, Front. Chem., 2021, 9, 655983 CrossRef CAS PubMed.
- D. S. Zijlstra, A. De Santi, B. Oldenburger, J. De Vries, K. Barta and P. J. Deuss, J. Visualized Exp., 2019, 58575 Search PubMed.
- K. J. Yong and T. Y. Wu, Bioresour. Technol., 2023, 384, 129238 CrossRef CAS PubMed.
- C. W. Lahive, P. J. Deuss, C. S. Lancefield, Z. Sun, D. B. Cordes, C. M. Young, F. Tran, A. M. Z. Slawin, J. G. De Vries, P. C. J. Kamer, N. J. Westwood and K. Barta, J. Am. Chem. Soc., 2016, 138, 8900–8911 CrossRef CAS PubMed.
- W. Lan and J. S. Luterbacher, Chimia, 2019, 73, 591–598 CrossRef CAS PubMed.
- C. Dong, X. Meng, S.-Y. Leu, L. Xu, Z. Wu, G. Cravotto and Z. Fang, Ind. Crops Prod., 2022, 185, 115130 CrossRef CAS.
- Z. Zhang, C. W. Lahive, D. S. Zijlstra, Z. Wang and P. J. Deuss, ACS Sustainable Chem. Eng., 2019, 7, 12105–12116 CAS.
- Z. Zhou, F. Lei, P. Li and J. Jiang, Biotechnol. Bioeng., 2018, 115, 2683–2702 CrossRef CAS PubMed.
- H. Teramura, K. Sasaki, T. Oshima, H. Kawaguchi, C. Ogino, T. Sazuka and A. Kondo, Bioresour. Technol., 2018, 252, 157–164 CrossRef CAS PubMed.
- H. Teramura, K. Sasaki, T. Oshima, F. Matsuda, M. Okamoto, T. Shirai, H. Kawaguchi, C. Ogino, K. Hirano, T. Sazuka, H. Kitano, J. Kikuchi and A. Kondo, Biotechnol. Biofuels, 2016, 9, 27 CrossRef PubMed.
- Q. Schmetz, H. Teramura, K. Morita, T. Oshima, A. Richel, C. Ogino and A. Kondo, ACS Sustainable Chem. Eng., 2019, 7, 11069–11079 CrossRef CAS.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC.
- D. Wei Kit Chin, S. Lim, Y. L. Pang and M. K. Lam, Biofuels, Bioprod. Biorefin., 2020, 14, 808–829 CrossRef CAS.
- R. Nitzsche, M. Budzinski and A. Gröngröft, Bioresour. Technol., 2016, 200, 928–939 CrossRef CAS PubMed.
- A. Brandt, J. Gräsvik, J. P. Hallett and T. Welton, Green Chem., 2013, 15, 550 RSC.
- J. L. Espinoza-Acosta, P. I. Torres-Chávez, E. Carvajal-Millán, B. Ramírez-Wong, L. A. Bello-Pérez and B. Montaño-Leyva, BioResources, 2014, 9, 3660–3687 CrossRef.
- C. G. Yoo, Y. Pu and A. J. Ragauskas, Curr. Opin. Green Sustainable Chem., 2017, 5, 5–11 CrossRef.
- H. Zhao, G. A. Baker, Z. Song, O. Olubajo, T. Crittle and D. Peters, Green Chem., 2008, 10, 696 RSC.
- B. Lu, A. Xu and J. Wang, Green Chem., 2014, 16, 1326–1335 RSC.
- B. J. Cox, S. Jia, Z. C. Zhang and J. G. Ekerdt, Polym. Degrad. Stab., 2011, 96, 426–431 CrossRef CAS.
- J. Dai, A. F. Patti and K. Saito, in Encyclopedia of Ionic Liquids, ed. S. Zhang, Springer Singapore, Singapore, 2020, pp. 1–12 Search PubMed.
- I. Kilpeläinen, H. Xie, A. King, M. Granstrom, S. Heikkinen and D. S. Argyropoulos, J. Agric. Food Chem., 2007, 55, 9142–9148 CrossRef PubMed.
- W. Li, N. Sun, B. Stoner, X. Jiang, X. Lu and R. D. Rogers, Green Chem., 2011, 13, 2038 RSC.
- K. Ikeguchi, K. Yoshida and H. Nonaka, BioResources, 2020, 15, 1587–1599 CAS.
- S. H. Lee, T. V. Doherty, R. J. Linhardt and J. S. Dordick, Biotechnol. Bioeng., 2009, 102, 1368–1376 CrossRef CAS PubMed.
- A. Brandt-Talbot, F. J. V. Gschwend, P. S. Fennell, T. M. Lammens, B. Tan, J. Weale and J. P. Hallett, Green Chem., 2017, 19, 3078–3102 RSC.
- E. Achinivu, Int. J. Mol. Sci., 2018, 19, 428 CrossRef PubMed.
- A. V. Belesov, A. V. Ladesov, I. I. Pikovskoi, A. V. Faleva and D. S. Kosyakov, Molecules, 2020, 25, 2479 CrossRef CAS PubMed.
- A. Brandt, L. Chen, B. E. van Dongen, T. Welton and J. P. Hallett, Green Chem., 2015, 17, 5019–5034 RSC.
- J.-Y. Kim, E.-J. Shin, I.-Y. Eom, K. Won, Y. H. Kim, D. Choi, I.-G. Choi and J. W. Choi, Bioresour. Technol., 2011, 102, 9020–9025 CrossRef CAS PubMed.
- E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060–11082 CrossRef CAS PubMed.
- A. Satlewal, R. Agrawal, S. Bhagia, J. Sangoro and A. J. Ragauskas, Biotechnol. Adv., 2018, 36, 2032–2050 CrossRef CAS PubMed.
- Y. Chen and T. Mu, Green Energy Environ., 2019, 4, 95–115 CrossRef.
- P. Bajpai, Deep Eutectic Solvents for Pretreatment of Lignocellulosic Biomass, Springer Singapore, Singapore, 2021 Search PubMed.
- Z. Chen, A. Ragauskas and C. Wan, Ind. Crops Prod., 2020, 147, 112241 CrossRef CAS.
- M. del Mar Contreras-Gámez, Á. Galán-Martín, N. Seixas, A. M. da Costa Lopes, A. Silvestre and E. Castro, Bioresour. Technol., 2023, 369, 128396 CrossRef.
- Á. Lobato-Rodríguez, B. Gullón, A. Romaní, P. Ferreira-Santos, G. Garrote and P. G. Del-Río, Bioresour. Technol., 2023, 388, 129744 CrossRef PubMed.
- P. Li, Z. Zhang, X. Zhang, K. Li, Y. Jin and W. Wu, RSC Adv., 2023, 13, 3241–3254 RSC.
- S. Wang, H. Li, L.-P. Xiao and G. Song, Front Energy Res, 2020, 8, 48 CrossRef.
- Y. Liu, N. Deak, Z. Wang, H. Yu, L. Hameleers, E. Jurak, P. J. Deuss and K. Barta, Nat. Commun., 2021, 12, 5424 CrossRef CAS.
- J. Cheng, C. Huang, Y. Zhan, S. Han, J. Wang, X. Meng, C. G. Yoo, G. Fang and A. J. Ragauskas, Chem. Eng. J., 2022, 443, 136395 CrossRef CAS.
- J. Peng, H. Xu, W. Wang, Y. Kong, Z. Su and B. Li, Ind. Crops Prod., 2021, 172, 114036 CrossRef CAS.
- G. Zang, A. Shah and C. Wan, Biofuels, Bioprod. Biorefin., 2020, 14, 326–343 CrossRef CAS.
- J. Cheng, C. Huang, Y. Zhan, X. Liu, J. Wang, X. Meng, C. G. Yoo, G. Fang and A. J. Ragauskas, Green Chem., 2023, 25, 1571–1581 RSC.
- Z. Guo, Q. Zhang, T. You, X. Zhang, F. Xu and Y. Wu, Green Chem., 2019, 21, 3099–3108 RSC.
- J. Vasco-Correa, X. Ge and Y. Li, in Biomass Fractionation Technologies for a Lignocellulosic Feedstock Based Biorefinery, Elsevier, 2016, pp. 561–585 Search PubMed.
- R. Sindhu, P. Binod and A. Pandey, Bioresour. Technol., 2016, 199, 76–82 CrossRef CAS PubMed.
- H. K. Sharma, C. Xu and W. Qin, Waste Biomass Valorization, 2019, 10, 235–251 CrossRef CAS.
- S. B. Ummalyma, R. D. Supriya, R. Sindhu, P. Binod, R. B. Nair, A. Pandey and E. Gnansounou, in Second and Third Generation of Feedstocks, Elsevier, 2019, pp. 197–212 Search PubMed.
- A. Tolbert, H. Akinosho, R. Khunsupat, A. K. Naskar and A. J. Ragauskas, Biofuels, Bioprod. Biorefin., 2014, 8, 836–856 CrossRef CAS.
- B. Westereng, D. Cannella, J. Wittrup Agger, H. Jørgensen, M. Larsen Andersen, V. G. H. Eijsink and C. Felby, Sci. Rep., 2015, 5, 18561 CrossRef CAS PubMed.
- L. Brenelli, F. M. Squina, C. Felby and D. Cannella, Biotechnol. Biofuels, 2018, 11, 10 CrossRef PubMed.
- D. S. Bajwa, G. Pourhashem, A. H. Ullah and S. G. Bajwa, Ind. Crops Prod., 2019, 139, 111526 CrossRef CAS.
- S. Gillet, M. Aguedo, L. Petitjean, A. R. C. Morais, A. M. da Costa Lopes, R. M. Łukasik and P. T. Anastas, Green Chem., 2017, 19, 4200–4233 RSC.
- J. E. Holladay, J. F. White, J. J. Bozell and D. Johnson, Top Value-Added Chemicals from Biomass. Volume II—Results of Screening for Potential Candidates from Biorefinery Lignin, Pacific Northwest National Laboratory (PNNL), Richland, WA (United States), 2007 Search PubMed.
- S. S. Wong, R. Shu, J. Zhang, H. Liu and N. Yan, Chem. Soc. Rev., 2020, 49, 5510–5560 RSC.
- Z. Sun, J. Cheng, D. Wang, T. Yuan, G. Song and K. Barta, ChemSusChem, 2020, 13, 5199–5212 CrossRef CAS PubMed.
- M. N. Collins, M. Nechifor, F. Tanasă, M. Zănoagă, A. McLoughlin, M. A. Stróżyk, M. Culebras and C.-A. Teacă, Int. J. Biol. Macromol., 2019, 131, 828–849 CrossRef CAS PubMed.
- P. Azadi, O. R. Inderwildi, R. Farnood and D. A. King, Renewable Sustainable Energy Rev., 2013, 21, 506–523 CrossRef CAS.
- T. Li and S. Takkellapati, Biofuels, Bioprod. Biorefin., 2018, 12, 756–787 CrossRef CAS PubMed.
- R. Behling, S. Valange and G. Chatel, Green Chem., 2016, 18, 1839–1854 RSC.
- A. Vigneault, D. K. Johnson and E. Chornet, Can. J. Chem. Eng., 2008, 85, 906–916 CrossRef.
- W. G. Glasser, Front. Chem., 2019, 7, 565 CrossRef CAS PubMed.
- D. Kun and B. Pukánszky, Eur. Polym. J., 2017, 93, 618–641 CrossRef CAS.
- N. Sallem-Idrissi, M. Sclavons, D. P. Debecker and J. Devaux, J. Appl. Polym. Sci., 2016, 133(6), 42963 CrossRef.
- N. Sallem-Idrissi, C. Vanderghem, T. Pacary, A. Richel, D. P. Debecker, J. Devaux and M. Sclavons, Polym. Degrad. Stab., 2016, 130, 30–37 CrossRef CAS.
- J. Parameswaranpillai, S. Thomas and Y. Grohens, in Characterization of Polymer Blends, ed. S. Thomas, Y. Grohens and P. Jyotishkumar, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2014, pp. 1–6 Search PubMed.
- C. Pouteau, S. Baumberger, B. Cathala and P. Dole, C. R. Biol., 2004, 327, 935–943 CrossRef CAS PubMed.
- C. Wang, S. S. Kelley and R. A. Venditti, ChemSusChem, 2016, 9, 770–783 CrossRef CAS PubMed.
- S. Yang, T.-Q. Yuan, Q. Shi and R.-C. Sun, in Encyclopedia of Sustainability Science and Technology, ed. R. A. Meyers, Springer New York, New York, NY, 2018, pp. 1–22 Search PubMed.
- J. F. Kadla and S. Kubo, Composites, Part A, 2004, 35, 395–400 CrossRef.
- V. Romhányi, D. Kun and B. Pukánszky, ACS Sustainable Chem. Eng., 2018, 6, 14323–14331 CrossRef.
- S. Laurichesse and L. Avérous, Prog. Polym. Sci., 2014, 39, 1266–1290 CrossRef CAS.
- A. Orebom, D. Di Francesco, P. Shakari, J. S. M. Samec and C. Pierrou, Molecules, 2021, 26, 3219 CrossRef CAS PubMed.
- T. Pang, G. Wang, H. Sun, W. Sui and C. Si, Ind. Crops Prod., 2021, 165, 113442 CrossRef CAS.
- A. Grossman and W. Vermerris, Curr. Opin. Biotechnol., 2019, 56, 112–120 CrossRef CAS PubMed.
- C. Xu and F. Ferdosian, in Conversion of Lignin into Bio-Based Chemicals and Materials, Springer Berlin Heidelberg, Berlin, Heidelberg, 2017, pp. 91–109 Search PubMed.
- S. Lin-Gibson and J. S. Riffle, in Synthetic Methods in Step-Growth Polymers, ed. M. E. Rogers and T. E. Long, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2003, pp. 375–430 Search PubMed.
- Z. Gao, X. Lang, S. Chen and C. Zhao, Energy Fuels, 2021, 35, 18385–18395 CrossRef CAS.
- Z. Peng, X. Jiang, C. Si, A. Joao Cárdenas-Oscanoa and C. Huang, ChemSusChem, 2023, 16, e202300174 CrossRef CAS PubMed.
- C. Huang, Z. Peng, J. Li, X. Li, X. Jiang and Y. Dong, Ind. Crops Prod., 2022, 187, 115388 CrossRef CAS.
- S. Zhao and M. M. Abu-Omar, ACS Sustainable Chem. Eng., 2021, 9, 1477–1493 CrossRef CAS.
- D. Kai, M. J. Tan, P. L. Chee, Y. K. Chua, Y. L. Yap and X. J. Loh, Green Chem., 2016, 18, 1175–1200 RSC.
- M. Kienberger, in Economics of Bioresources, ed. Y. Krozer and M. Narodoslawsky, Springer International Publishing, Cham, 2019, pp. 183–193 Search PubMed.
- S. Yang, Y. Zhang, T.-Q. Yuan and R.-C. Sun, J. Appl. Polym. Sci., 2015, 132(36), 42493 CrossRef.
- S. Yang, J.-L. Wen, T.-Q. Yuan and R.-C. Sun, RSC Adv., 2014, 4, 57996–58004 RSC.
- H. Younesi-Kordkheili and A. Pizzi, Polymers, 2021, 13, 3502 CrossRef CAS PubMed.
- B. Venkatesagowda and R. F. H. Dekker, Biomass Convers. Biorefin., 2020, 10, 203–225 CrossRef CAS.
- P. Solt, B. Rößiger, J. Konnerth and H. van Herwijnen, Polymers, 2018, 10, 1162 CrossRef PubMed.
- C. Xu and F. Ferdosian, in Conversion of Lignin into Bio-Based Chemicals and Materials, Springer Berlin Heidelberg, Berlin, Heidelberg, 2017, pp. 111–131 Search PubMed.
- J. H. Lora and W. G. Glasser, J. Polym. Environ., 2002, 10, 39–48 CrossRef CAS.
- A. V. Krishnan, P. Stathis, S. F. Permuth, L. Tokes and D. Feldman, Endocrinology, 1993, 132, 2279–2286 CrossRef CAS PubMed.
- C. Pappa, E. Feghali, K. Vanbroekhoven and K. S. Triantafyllidis, Curr. Opin. Green Sustainable Chem., 2022, 38, 100687 CrossRef CAS.
- X. Lu and X. Gu, Int. J. Biol. Macromol., 2023, 229, 778–790 CrossRef CAS PubMed.
- P. Ortiz, R. Vendamme and W. Eevers, Molecules, 2020, 25, 1158 CrossRef CAS PubMed.
- S. Nikafshar, J. Wang, K. Dunne, P. Sangthonganotai and M. Nejad, ChemSusChem, 2021, 14, 1184–1195 CrossRef CAS PubMed.
- X. Zhen, H. Li, Z. Xu, Q. Wang, J. Xu, S. Zhu, Z. Wang and Z. Yuan, Ind. Crops Prod., 2021, 173, 114135 CrossRef CAS.
- X. Jiang, J. Liu, X. Du, Z. Hu, H. Chang and H. Jameel, ACS Sustainable Chem. Eng., 2018, 6, 5504–5512 CrossRef CAS.
- E. Feghali, D. J. Van De Pas, A. J. Parrott and K. M. Torr, ACS Macro Lett., 2020, 9, 1155–1160 CrossRef CAS PubMed.
- E. Feghali, D. J. van de Pas and K. M. Torr, Biomacromolecules, 2020, 21, 1548–1559 CrossRef CAS PubMed.
- C. Gioia, M. Colonna, A. Tagami, L. Medina, O. Sevastyanova, L. A. Berglund and M. Lawoko, Biomacromolecules, 2020, 21, 1920–1928 CrossRef CAS PubMed.
- S.-F. Koelewijn, C. Cooreman, T. Renders, C. Andecochea Saiz, S. Van den Bosch, W. Schutyser, W. De Leger, M. Smet, P. Van Puyvelde, H. Witters, B. Van der Bruggen and B. F. Sels, Green Chem., 2018, 20, 1050–1058 RSC.
- Y. Jiang, D. Ding, S. Zhao, H. Zhu, H. I. Kenttämaa and M. M. Abu-Omar, Green Chem., 2018, 20, 1131–1138 RSC.
- K. Van Aelst, E. Van Sinay, T. Vangeel, Y. Zhang, T. Renders, S. Van den Bosch, J. Van Aelst and B. F. Sels, Chem. Commun., 2021, 57, 5642–5645 RSC.
- S.-F. Koelewijn, S. Van den Bosch, T. Renders, W. Schutyser, B. Lagrain, M. Smet, J. Thomas, W. Dehaen, P. Van Puyvelde, H. Witters and B. F. Sels, Green Chem., 2017, 19, 2561–2570 RSC.
- D. J. van de Pas and K. M. Torr, Biomacromolecules, 2017, 18, 2640–2648 CrossRef CAS PubMed.
- C. Xu and F. Ferdosian, in Conversion of Lignin into Bio-Based Chemicals and Materials, Springer Berlin Heidelberg, Berlin, Heidelberg, 2017, pp. 133–156 Search PubMed.
- X. Ma, J. Chen, J. Zhu and N. Yan, Macromol. Rapid Commun., 2021, 42, 2000492 CrossRef CAS PubMed.
- J. O. Akindoyo, M. D. H. Beg, S. Ghazali, M. R. Islam, N. Jeyaratnam and A. R. Yuvaraj, RSC Adv., 2016, 6, 114453–114482 RSC.
- T. Wu, X. Li, X. Ma, J. Ye, L. Shen and W. Tan, Mater. Res. Express, 2022, 9, 105302 CrossRef.
- M. Alinejad, C. Henry, S. Nikafshar, A. Gondaliya, S. Bagheri, N. Chen, S. Singh, D. Hodge and M. Nejad, Polymers, 2019, 11, 1202 CrossRef PubMed.
- Y. Chen, S. Fu and H. Zhang, Colloids Surf., A, 2020, 585, 124164 CrossRef CAS.
- Y.-Y. Wang, C. Cai and A. Ragauskas, Tappi J., 2017, 16, 203–207 CAS.
- Y.-Y. Wang, C. E. Wyman, C. M. Cai and A. J. Ragauskas, ACS Appl. Polym. Mater., 2019, 1, 1672–1679 CrossRef CAS.
- X. Zhang, Y. Kim, I. Elsayed, M. Taylor, T. L. Eberhardt, E. B. Hassan and R. Shmulsky, Ind. Crops Prod., 2019, 141, 111797 CrossRef CAS.
- Y. Yang, H. Cao, R. Liu, Y. Wang, M. Zhu, C. Su, X. Lv, J. Zhao, P. Qin and D. Cai, Ind. Crops Prod., 2023, 193, 116213 CrossRef CAS.
- A. Gondaliya and M. Nejad, Molecules, 2021, 26, 2302 CrossRef CAS PubMed.
- H. Luo, X. Hua, W. Liu, Y. Xu, F. Cao, S. Nikafshar and Z. Fang, ACS Appl. Polym. Mater., 2023, 5, 6061–6068 CrossRef CAS.
- C. Xu and F. Ferdosian, in Conversion of Lignin into Bio-Based Chemicals and Materials, Springer Berlin Heidelberg, Berlin, Heidelberg, 2017, pp. 55–79 Search PubMed.
- S. Chatterjee and T. Saito, ChemSusChem, 2015, 8, 3941–3958 CrossRef CAS PubMed.
- P. Bajpai, in Carbon Fibre from Lignin, Springer Singapore, Singapore, 2017, pp. 17–23 Search PubMed.
- P. Bajpai, in Carbon Fibre from Lignin, Springer Singapore, Singapore, 2017, pp. 25–28 Search PubMed.
- H. Mainka, O. Täger, E. Körner, L. Hilfert, S. Busse, F. T. Edelmann and A. S. Herrmann, J. Mater. Res. Technol., 2015, 4, 283–296 CrossRef CAS.
- P. Bajpai, in Carbon Fibre from Lignin, Springer Singapore, Singapore, 2017, pp. 43–61 Search PubMed.
- S. Wang, J. Bai, M. T. Innocent, Q. Wang, H. Xiang, J. Tang and M. Zhu, Green Energy Environ., 2022, 7, 578–605 CrossRef CAS.
- W. Qu, J. Yang, X. Sun, X. Bai, H. Jin and M. Zhang, Int. J. Biol. Macromol., 2021, 189, 768–784 CrossRef CAS PubMed.
- S.-C. Sun, Y. Xu, J.-L. Wen, T.-Q. Yuan and R.-C. Sun, Green Chem., 2022, 24, 5709–5738 RSC.
- F. Hermansson, M. Janssen and M. Svanström, J. Cleaner Prod., 2019, 223, 946–956 CrossRef CAS.
- R. Zhang, Q. Du, L. Wang, Z. Zheng, L. Guo, X. Zhang, X. Yang and H. Yu, Green Chem., 2019, 21, 4981–4987 RSC.
- Q. Li, C. Hu, M. Li, P. Truong, J. Li, H.-S. Lin, M. T. Naik, S. Xiang, B. E. Jackson, W. Kuo, W. Wu, Y. Pu, A. J. Ragauskas and J. S. Yuan, Green Chem., 2021, 23, 3725–3739 RSC.
- M. Föllmer, S. Jestin, W. Neri, V. S. Vo, A. Derré, C. Mercader and P. Poulin, Adv. Sustainable Syst., 2019, 3, 1900082 CrossRef.
- N. Yan, C. Zhao, P. J. Dyson, C. Wang, L. Liu and Y. Kou, ChemSusChem, 2008, 1, 626–629 CrossRef CAS PubMed.
- C. Xu, R. A. D. Arancon, J. Labidi and R. Luque, Chem. Soc. Rev., 2014, 43, 7485–7500 RSC.
- L. Cao, I. K. M. Yu, Y. Liu, X. Ruan, D. C. W. Tsang, A. J. Hunt, Y. S. Ok, H. Song and S. Zhang, Bioresour. Technol., 2018, 269, 465–475 CrossRef CAS PubMed.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- Y. Cao, S. S. Chen, S. Zhang, Y. S. Ok, B. M. Matsagar, K. C.-W. Wu and D. C. W. Tsang, Bioresour. Technol., 2019, 291, 121878 CrossRef CAS PubMed.
- X. Liu, F. P. Bouxin, J. Fan, V. L. Budarin, C. Hu and J. H. Clark, ChemSusChem, 2020, 13, 4296–4317 CrossRef CAS PubMed.
- D. Bourbiaux, J. Pu, F. Rataboul, L. Djakovitch, C. Geantet and D. Laurenti, Catal. Today, 2021, 373, 24–37 CrossRef CAS.
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed.
- Y. Song, in Chemical Catalysts for Biomass Upgrading, ed. M. Crocker and E. Santillan-Jimenez, Wiley, 1st edn, 2020, pp. 395–437 Search PubMed.
- A. Rahimi, A. Ulbrich, J. J. Coon and S. S. Stahl, Nature, 2014, 515, 249–252 CrossRef CAS PubMed.
- C. S. Lancefield, O. S. Ojo, F. Tran and N. J. Westwood, Angew. Chem., Int. Ed., 2015, 54, 258–262 CrossRef CAS PubMed.
- I. Bosque, G. Magallanes, M. Rigoulet, M. D. Kärkäs and C. R. J. Stephenson, ACS Cent. Sci., 2017, 3, 621–628 CrossRef CAS PubMed.
- J. D. Nguyen, B. S. Matsuura and C. R. J. Stephenson, J. Am. Chem. Soc., 2014, 136, 1218–1221 CrossRef CAS PubMed.
- T. Vangeel, W. Schutyser, T. Renders and B. F. Sels, Top. Curr. Chem., 2018, 376, 30 CrossRef PubMed.
- M. Wang, J. Lu, X. Zhang, L. Li, H. Li, N. Luo and F. Wang, ACS Catal., 2016, 6, 6086–6090 CrossRef CAS.
- H. Guo, D. M. Miles-Barrett, A. R. Neal, T. Zhang, C. Li and N. J. Westwood, Chem. Sci., 2018, 9, 702–711 RSC.
- C. Zhang, H. Li, J. Lu, X. Zhang, K. E. MacArthur, M. Heggen and F. Wang, ACS Catal., 2017, 7, 3419–3429 CrossRef CAS.
- M. Wang, X. Zhang, H. Li, J. Lu, M. Liu and F. Wang, ACS Catal., 2018, 8, 1614–1620 CrossRef CAS.
- C. Yang, S. Maldonado and C. R. J. Stephenson, ACS Catal., 2021, 11, 10104–10114 CrossRef CAS.
- J. Chen, F. Lu, X. Si, X. Nie, J. Chen, R. Lu and J. Xu, ChemSusChem, 2016, 9, 3353–3360 CrossRef CAS PubMed.
- G. Zhu, X. Qiu, Y. Zhao, Y. Qian, Y. Pang and X. Ouyang, Bioresour. Technol., 2016, 218, 718–722 CrossRef CAS PubMed.
- H. Li and G. Song, ACS Catal., 2019, 9, 4054–4064 CrossRef CAS.
- E. Feghali and T. Cantat, Chem. Commun., 2014, 50, 862–865 RSC.
- E. Feghali, G. Carrot, P. Thuéry, C. Genre and T. Cantat, Energy Environ. Sci., 2015, 8, 2734–2743 RSC.
- A. Fedorov, A. A. Toutov, N. A. Swisher and R. H. Grubbs, Chem. Sci., 2013, 4, 1640 RSC.
- A. Orebom, C. Dahlstrand, J. Löfstedt and J. Samec, in Depolymerisation of Lignin, 2015, at, https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015199608 Search PubMed.
- R. Ma, M. Guo and X. Zhang, Catal. Today, 2018, 302, 50–60 CrossRef CAS.
- S. Kumaravel, P. Thiruvengetam, K. Karthick, S. S. Sankar, A. Karmakar and S. Kundu, Biotechnol. Prog., 2021, 37, 13 CrossRef PubMed.
- O. Y. Abdelaziz, I. Clemmensen, S. Meier, C. A. E. Costa, A. E. Rodrigues, C. P. Hulteberg and A. Riisager, ChemSusChem, 2022, 15(20), e202201232 CrossRef CAS PubMed.
- C. Yang, H. Chen, T. Peng, B. Liang, Y. Zhang and W. Zhao, Chin. J. Catal., 2021, 42, 1831–1842 CrossRef CAS.
- D. Gao, D. Ouyang and X. Zhao, Green Chem., 2022, 24, 8585–8605 RSC.
- M. G. A. da Cruz, R. Gueret, J. Chen, J. Piątek, B. Beele, M. H. Sipponen, M. Frauscher, S. Budnyk, B. V. M. Rodrigues and A. Slabon, ChemSusChem, 2022, 15(15), e202200718 CrossRef CAS PubMed.
- Z. Xiang, W. Han, J. Deng, W. Zhu, Y. Zhang and H. Wang, ChemSusChem, 2020, 13, 4199–4213 CrossRef CAS PubMed.
- J. Xu, M. Li, J. Qiu, X.-F. Zhang and J. Yao, Int. J. Biol. Macromol., 2021, 180, 403–410 CrossRef CAS PubMed.
- H. Wang, G. J. Giardino, R. Chen, C. Yang, J. Niu and D. Wang, ACS Cent. Sci., 2023, 9, 48–55 CrossRef CAS PubMed.
- M. Alherech, S. Omolabake, C. M. Holland, G. E. Klinger, E. L. Hegg and S. S. Stahl, ACS Cent. Sci., 2021, 7, 1831–1837 CrossRef CAS PubMed.
- M. Fache, B. Boutevin and S. Caillol, ACS Sustainable Chem. Eng., 2016, 4, 35–46 CrossRef CAS.
- P. C. Rodrigues Pinto, E. A. Borges da Silva and A. E. Rodrigues, in Biomass Conversion, ed. C. Baskar, S. Baskar and R. S. Dhillon, Springer Berlin Heidelberg, Berlin, Heidelberg, 2012, pp. 381–420 Search PubMed.
- U. Junghans, J. J. Bernhardt, R. Wollnik, D. Triebert, G. Unkelbach and D. Pufky-Heinrich, Molecules, 2020, 25, 2717 CrossRef CAS PubMed.
- A. C. Kokossis, M. Tsakalova and K. Pyrgakis, Comput. Chem. Eng., 2015, 81, 40–56 CrossRef CAS.
- C. Chio, M. Sain and W. Qin, Renewable Sustainable Energy Rev., 2019, 107, 232–249 CrossRef CAS.
- H. Wang, M. Tucker and Y. Ji, J. Appl. Chem., 2013, 2013, 1–9 CrossRef.
- B. Rößiger, G. Unkelbach and D. Pufky-Heinrich, in Lignin – Trends and Applications, ed. M. Poletto, InTech, 2018 Search PubMed.
- M. Rafiee, M. Alherech, S. D. Karlen and S. S. Stahl, J. Am. Chem. Soc., 2019, 141, 15266–15276 CrossRef CAS PubMed.
- A. Jensen, J. B. Nielsen, A. D. Jensen and C. Felby, in Energy and Environment Series, ed. G. T. Beckham, Royal Society of Chemistry, Cambridge, 2018, pp. 74–107 Search PubMed.
- M. Puig-Arnavat, T. P. Thomsen, G. Ravenni, L. R. Clausen, Z. Sárossy and J. Ahrenfeldt, in Biorefinery, ed. J.-R. Bastidas-Oyanedel and J. E. Schmidt, Springer International Publishing, Cham, 2019, pp. 79–110 Search PubMed.
- E. T. Liakakou, B. J. Vreugdenhil, N. Cerone, F. Zimbardi, F. Pinto, R. André, P. Marques, R. Mata and F. Girio, Fuel, 2019, 251, 580–592 CrossRef CAS.
- M. P. Pandey and C. S. Kim, Chem. Eng. Technol., 2011, 34, 29–41 CrossRef CAS.
- H. Tan, C. T. Lee, P. Y. Ong, K. Y. Wong, C. P. C. Bong, C. Li and Y. Gao, IOP Conf. Ser.: Mater. Sci. Eng., 2021, 1051, 012075 CAS.
- A. Singh-Morgan, A. Puente-Urbina and J. A. van Bokhoven, ChemSusChem, 2022, 15(14), e202200343 CrossRef CAS PubMed.
- Z. Luo, K. Lu, Y. Yang, S. Li and G. Li, RSC Adv., 2019, 9, 31960–31968 RSC.
- M. Kosa, H. Ben, H. Theliander and A. J. Ragauskas, Green Chem., 2011, 13, 3196 RSC.
- A. J. Yanez, P. Natarajan, W. Li, R. Mabon and L. J. Broadbelt, Energy Fuels, 2018, 32, 1822–1830 CrossRef CAS.
- D. K. Ojha, D. Viju and R. Vinu, Bioresour. Technol., 2017, 241, 142–151 CrossRef CAS PubMed.
- L. Fan, Y. Zhang, S. Liu, N. Zhou, P. Chen, Y. Cheng, M. Addy, Q. Lu, M. M. Omar, Y. Liu, Y. Wang, L. Dai, E. Anderson, P. Peng, H. Lei and R. Ruan, Bioresour. Technol., 2017, 241, 1118–1126 CrossRef CAS PubMed.
- Z. Ma, A. Ghosh, N. Asthana and J. van Bokhoven, ChemCatChem, 2017, 9, 954–961 CrossRef CAS.
- X. Lu and X. Gu, Biotechnol. Biofuels, 2022, 15, 106 CrossRef CAS PubMed.
- A. M. Da Costa Lopes, A. J. D. Silvestre and J. A. P. Coutinho, Curr. Opin. Green Sustainable Chem., 2023, 43, 100850 CrossRef CAS.
- H. Li, A. Bunrit, N. Li and F. Wang, Chem. Soc. Rev., 2020, 49, 3748–3763 RSC.
- N. Giummarella, C. Gioia and M. Lawoko, Green Chem., 2018, 20, 2651–2662 RSC.
- B. Zheng, H. Wu, J. Song, W. Wu, X. Mei, K. Zhang, C. Xu, J. Xu, M. He and B. Han, Green Chem., 2021, 23, 8441–8447 RSC.
- X. Wu, M. De bruyn and K. Barta, ChemSusChem, 2022, 15(18), e202200914 CrossRef CAS PubMed.
- C. S. Lancefield, B. M. Weckhuysen and P. C. A. Bruijnincx, in Energy and Environment Series, ed. G. T. Beckham, Royal Society of Chemistry, Cambridge, 2018, pp. 159–198 Search PubMed.
- H. A. Meylemans, T. J. Groshens and B. G. Harvey, ChemSusChem, 2012, 5, 206–210 CrossRef CAS PubMed.
- P. Ferrini, S.-F. Koelewijn, J. Van Aelst, N. Nuttens and B. F. Sels, ChemSusChem, 2017, 10, 2249–2257 CrossRef CAS PubMed.
- E. D. Hernandez, A. W. Bassett, J. M. Sadler, J. J. La Scala and J. F. Stanzione, ACS Sustainable Chem. Eng., 2016, 4, 4328–4339 CrossRef CAS.
- Z. Sun, G. Bottari, A. Afanasenko, M. C. A. Stuart, P. J. Deuss, B. Fridrich and K. Barta, Nat. Catal., 2018, 1, 82–92 CrossRef CAS.
- Z. Chen, H. Zeng, S. A. Girard, F. Wang, N. Chen and C. Li, Angew. Chem., Int. Ed., 2015, 54, 14487–14491 CrossRef CAS PubMed.
- M. Anjalin, N. Kanagathara and A. R. Baby Suganthi, Mater. Today: Proc., 2020, 33, 4751–4755 Search PubMed.
- S. Elangovan, A. Afanasenko, J. Haupenthal, Z. Sun, Y. Liu, A. K. H. Hirsch and K. Barta, ACS Cent. Sci., 2019, 5, 1707–1716 CrossRef CAS PubMed.
- J. Park, M. A. Kelly, J. X. Kang, S. S. Seemakurti, J. L. Ramirez, M. C. Hatzell, C. Sievers and A. S. Bommarius, Green Chem., 2021, 23, 7488–7498 RSC.
- A. Afanasenko and K. Barta, iScience, 2021, 24, 102211 CrossRef CAS PubMed.
- M. Vinardell and M. Mitjans, Int. J. Mol. Sci., 2017, 18, 1219 CrossRef PubMed.
- D. Ruijten, T. Narmon, H. De Weer, R. van der Zweep, C. Poleunis, D. P. Debecker, B. U. W. Maes and B. F. Sels, ChemSusChem, 2022, 15(19), e202200868 CrossRef CAS PubMed.
- D. Ruijten, T. Narmon, K. Van Aelst, H. De Weer, R. van der Zweep, T. Hendrickx, C. Poleunis, L. Li, K. M. Van Geem, D. P. Debecker and B. F. Sels, ACS Sustainable Chem. Eng., 2023, 11, 4776–4788 CrossRef CAS.
- L. D. Eltis and R. Singh, in Energy and Environment Series, ed. G. T. Beckham, Royal Society of Chemistry, Cambridge, 2018, pp. 290–313 Search PubMed.
- A. K. Singh, S. Jang, J. Y. Kim, S. Sharma, K. C. Basavaraju, M.-G. Kim, K.-R. Kim, J. S. Lee, H. H. Lee and D.-P. Kim, ACS Catal., 2015, 5, 6964–6972 CrossRef CAS.
- Z. Zhang, D. S. Zijlstra, C. W. Lahive and P. J. Deuss, Green Chem., 2020, 22, 3791–3801 RSC.
- G. Jeantelot, S. P. Følkner, J. I. S. Manegold, M. G. Ingebrigtsen, V. R. Jensen and E. Le Roux, ACS Omega, 2022, 7, 31561–31566 CrossRef CAS PubMed.
- M. Saidi, F. Samimi, D. Karimipourfard, T. Nimmanwudipong, B. C. Gates and M. R. Rahimpour, Energy Environ. Sci., 2014, 7, 103–129 RSC.
- A. Kumar, M. Jindal, S. Maharana and B. Thallada, Energy Fuels, 2021, 35, 16965–16994 CrossRef CAS.
- S. Li, B. Liu, J. Truong, Z. Luo, P. C. Ford and M. M. Abu-Omar, Green Chem., 2020, 22, 7406–7416 RSC.
- W. Schutyser, S. Van den Bosch, J. Dijkmans, S. Turner, M. Meledina, G. Van Tendeloo, D. P. Debecker and B. F. Sels, ChemSusChem, 2015, 8, 1805–1818 CrossRef CAS PubMed.
- H. Takeshima, K. Satoh and M. Kamigaito, Macromolecules, 2017, 50, 4206–4216 CrossRef CAS.
- Z. Xu, P. Lei, R. Zhai, Z. Wen and M. Jin, Biotechnol. Biofuels, 2019, 12, 32 CrossRef PubMed.
- X. Li and Y. Zheng, Biotechnol. Prog., 2020, 36(1), e2922 CrossRef CAS PubMed.
- S. C. Seaton and E. L. Neidle, in Energy and Environment Series, ed. G. T. Beckham, Royal Society of Chemistry, Cambridge, 2018, pp. 252–289 Search PubMed.
- G. T. Beckham, C. W. Johnson, E. M. Karp, D. Salvachúa and D. R. Vardon, Curr. Opin. Biotechnol., 2016, 42, 40–53 CrossRef CAS PubMed.
- E. Erickson, A. Bleem, E. Kuatsjah, A. Z. Werner, J. L. DuBois, J. E. McGeehan, L. D. Eltis and G. T. Beckham, Nat. Catal., 2022, 5, 86–98 CrossRef CAS.
- A. Eudes, Y. Liang, P. Mitra and D. Loqué, Curr. Opin. Biotechnol., 2014, 26, 189–198 CrossRef CAS PubMed.
- A. S. Pazhany and R. J. Henry, Ind. Eng. Chem. Res., 2019, 58, 16190–16203 CrossRef CAS.
- M. Baucher, C. Halpin, M. Petit-Conil and W. Boerjan, Crit. Rev. Biochem. Mol. Biol., 2003, 38, 305–350 CrossRef CAS PubMed.
- B. De Meester, R. Vanholme, T. Mota and W. Boerjan, Plant Commun., 2022, 3, 100465 CrossRef PubMed.
- J. Ralph, C. Lapierre and W. Boerjan, Curr. Opin. Biotechnol., 2019, 56, 240–249 CrossRef CAS PubMed.
- C. G. Wilkerson, S. D. Mansfield, F. Lu, S. Withers, J.-Y. Park, S. D. Karlen, E. Gonzales-Vigil, D. Padmakshan, F. Unda, J. Rencoret and J. Ralph, Science, 2014, 344, 90–93 CrossRef CAS PubMed.
- S. Zhou, T. Runge, S. D. Karlen, J. Ralph, E. Gonzales-Vigil and S. D. Mansfield, ChemSusChem, 2017, 10, 3565–3573 CrossRef CAS PubMed.
- L. de Vries, S. Guevara-Rozo, M. Cho, L.-Y. Liu, S. Renneckar and S. D. Mansfield, Biotechnol. Biofuels, 2021, 14, 167 CrossRef CAS PubMed.
- M. Galkin, Curr. Opin. Green Sustainable Chem., 2021, 28, 100438 CrossRef CAS.
- T. Renders, S. Van den Bosch, S.-F. Koelewijn, W. Schutyser and B. F. Sels, Energy Environ. Sci., 2017, 10, 1551–1557 RSC.
- P. Sudarsanam, D. Ruijten, Y. Liao, T. Renders, S.-F. Koelewijn and B. F. Sels, Trends Chem., 2020, 2, 898–913 CrossRef CAS.
- S. Liu, Y. Zhu, Y. Liao, H. Wang, Q. Liu, L. Ma and C. Wang, Appl. Energy Combust. Sci., 2022, 100062 Search PubMed.
- E. Subbotina, A. Velty, J. S. M. Samec and A. Corma, ChemSusChem, 2020, 13, 4528–4536 CrossRef CAS PubMed.
- A. Kramarenko, D. Etit, G. Laudadio and F. N. D'Angelo, ChemSusChem, 2021, 14, 3838–3849 CrossRef CAS PubMed.
- S. Qiu, X. Guo, Y. Huang, Y. Fang and T. Tan, ChemSusChem, 2019, 12, 944–954 CrossRef CAS PubMed.
- S. Van den Bosch, W. Schutyser, R. Vanholme, T. Driessen, S.-F. Koelewijn, T. Renders, B. De Meester, W. J. J. Huijgen, W. Dehaen, C. M. Courtin, B. Lagrain, W. Boerjan and B. F. Sels, Energy Environ. Sci., 2015, 8, 1748–1763 RSC.
- P. Ferrini and R. Rinaldi, Angew. Chem., Int. Ed., 2014, 53, 8634–8639 CrossRef CAS PubMed.
- R. Rinaldi, in Energy and Environment Series, ed. G. T. Beckham, Royal Society of Chemistry, Cambridge, 2018, pp. 108–127 Search PubMed.
- H. P. Godard, J. L. McCarthy and H. Hibbert, J. Am. Chem. Soc., 1940, 62, 988 CrossRef CAS.
- J. M. Pepper and H. Hibbert, J. Am. Chem. Soc., 1948, 70, 67–71 CrossRef CAS PubMed.
- C. P. Brewer, L. M. Cooke and H. Hibbert, J. Am. Chem. Soc., 1948, 70, 57–59 CrossRef CAS PubMed.
- H. P. Godard, J. L. McCarthy and H. Hibbert, J. Am. Chem. Soc., 1941, 63, 3061–3066 CrossRef CAS.
- Q. Song, F. Wang, J. Cai, Y. Wang, J. Zhang, W. Yu and J. Xu, Energy Environ. Sci., 2013, 6, 994 RSC.
- T. Parsell, S. Yohe, J. Degenstein, T. Jarrell, I. Klein, E. Gencer, B. Hewetson, M. Hurt, J. I. Kim, H. Choudhari, B. Saha, R. Meilan, N. Mosier, F. Ribeiro, W. N. Delgass, C. Chapple, H. I. Kenttämaa, R. Agrawal and M. M. Abu-Omar, Green Chem., 2015, 17, 1492–1499 RSC.
- C. Li, M. Zheng, A. Wang and T. Zhang, Energy Environ. Sci., 2012, 5, 6383–6390 RSC.
- K. M. Torr, D. J. Van De Pas, E. Cazeils and I. D. Suckling, Bioresour. Technol., 2011, 102, 7608–7611 CrossRef CAS PubMed.
- T. Renders, G. Van den Bossche, T. Vangeel, K. Van Aelst and B. Sels, Curr. Opin. Biotechnol., 2019, 56, 193–201 CrossRef CAS PubMed.
- W. Arts, D. Ruijten, K. Van Aelst, L. Trullemans and B. Sels, in Advances in Inorganic Chemistry, Elsevier, 2021, vol. 77, pp. 241–297 Search PubMed.
- S. Van den Bosch, T. Renders, S. Kennis, S.-F. Koelewijn, G. Van den Bossche, T. Vangeel, A. Deneyer, D. Depuydt, C. M. Courtin, J. M. Thevelein, W. Schutyser and B. F. Sels, Green Chem., 2017, 19, 3313–3326 RSC.
- M. V. Galkin and J. S. M. Samec, ChemSusChem, 2014, 7, 2154–2158 CrossRef CAS PubMed.
- T. Renders, W. Schutyser, S. Van den Bosch, S.-F. Koelewijn, T. Vangeel, C. M. Courtin and B. F. Sels, ACS Catal., 2016, 6, 2055–2066 CrossRef CAS.
- S. Qiu, M. Wang, Y. Fang and T. Tan, Sustainable Energy Fuels, 2020, 4, 5588–5594 RSC.
- L. Chen, A. P. van Muyden, X. Cui, Z. Fei, N. Yan, G. Laurenczy and P. J. Dyson, JACS Au, 2021, 1, 729–733 CrossRef CAS PubMed.
- H. Luo, I. M. Klein, Y. Jiang, H. Zhu, B. Liu, H. I. Kenttämaa and M. M. Abu-Omar, ACS Sustainable Chem. Eng., 2016, 4, 2316–2322 CrossRef CAS.
- E. M. Anderson, M. L. Stone, R. Katahira, M. Reed, G. T. Beckham and Y. Román-Leshkov, Joule, 2017, 1, 613–622 CrossRef CAS.
- W. Schutyser, S. Van den Bosch, T. Renders, T. De Boe, S.-F. Koelewijn, A. Dewaele, T. Ennaert, O. Verkinderen, B. Goderis, C. M. Courtin and B. F. Sels, Green Chem., 2015, 17, 5035–5045 RSC.
- P. Ferrini, C. Chesi, N. Parkin and R. Rinaldi, Faraday Discuss., 2017, 202, 403–413 RSC.
- S. Van den Bosch, W. Schutyser, S.-F. Koelewijn, T. Renders, C. M. Courtin and B. F. Sels, Chem. Commun., 2015, 51, 13158–13161 RSC.
- T. Renders, E. Cooreman, S. Van den Bosch, W. Schutyser, S.-F. Koelewijn, T. Vangeel, A. Deneyer, G. Van den Bossche, C. M. Courtin and B. F. Sels, Green Chem., 2018, 20, 4607–4619 RSC.
- R. Rinaldi, R. Woodward, P. Ferrini and H. Rivera, J. Braz. Chem. Soc., 2019, 30(3), 479–491 CAS.
- E. M. Anderson, R. Katahira, M. Reed, M. G. Resch, E. M. Karp, G. T. Beckham and Y. Román-Leshkov, ACS Sustainable Chem. Eng., 2016, 4, 6940–6950 CrossRef CAS.
- X. Chen, K. Zhang, L.-P. Xiao, R.-C. Sun and G. Song, Biotechnol. Biofuels, 2020, 13, 2 CrossRef CAS PubMed.
- X. Liu, H. Li, L.-P. Xiao, R.-C. Sun and G. Song, Green Chem., 2019, 21, 1498–1504 RSC.
- K. Van Aelst, E. Van Sinay, T. Vangeel, E. Cooreman, G. Van den Bossche, T. Renders, J. Van Aelst, S. Van den Bosch and B. F. Sels, Chem. Sci., 2020, 11, 11498–11508 RSC.
- Z. Cao, M. Dierks, M. T. Clough, I. B. Daltro de Castro and R. Rinaldi, Joule, 2018, 2, 1118–1133 CrossRef CAS PubMed.
- M. V. Galkin, A. T. Smit, E. Subbotina, K. A. Artemenko, J. Bergquist, W. J. J. Huijgen and J. S. M. Samec, ChemSusChem, 2016, 9, 3280–3287 CrossRef CAS PubMed.
- O. E. Ebikade, N. Samulewicz, S. Xuan, J. D. Sheehan, C. Wu and D. G. Vlachos, Green Chem., 2020, 22, 7435–7447 RSC.
- S. Li, W. Li, Q. Zhang, R. Shu, H. Wang, H. Xin and L. Ma, RSC Adv., 2018, 8, 1361–1370 RSC.
- F. Brienza, K. Van Aelst, F. Devred, D. Magnin, B. F. Sels, P. A. Gerin, I. Cybulska and D. P. Debecker, ACS Sustainable Chem. Eng., 2022, 10, 11130–11142 CrossRef CAS.
- F. Brienza, K. Van Aelst, F. Devred, D. Magnin, B. F. Sels, P. Gerin, I. Cybulska and D. P. Debecker, ChemSusChem, 2023, 16, e202300103 CrossRef CAS PubMed.
- J. H. Jang, A. R. C. Morais, M. Browning, D. G. Brandner, J. K. Kenny, L. M. Stanley, R. M. Happs, A. S. Kovvali, J. I. Cutler, Y. Román-Leshkov, J. R. Bielenberg and G. T. Beckham, Green Chem., 2023, 25, 3660–3670 RSC.
- P. Ferrini, C. A. Rezende and R. Rinaldi, ChemSusChem, 2016, 9, 3171–3180 CrossRef CAS PubMed.
- E. Cooreman, T. Vangeel, K. Van Aelst, J. Van Aelst, J. Lauwaert, J. W. Thybaut, S. Van den Bosch and B. F. Sels, Ind. Eng. Chem. Res., 2020, 59, 17035–17045 CrossRef CAS.
- A. W. Bartling, M. L. Stone, R. J. Hanes, A. Bhatt, Y. Zhang, M. J. Biddy, R. Davis, J. S. Kruger, N. E. Thornburg, J. S. Luterbacher, R. Rinaldi, J. S. M. Samec, B. F. Sels, Y. Román-Leshkov and G. T. Beckham, Energy Environ. Sci., 2021, 14, 4147–4168 RSC.
- H. Zhang, S. Fu, X. Du and Y. Deng, ChemSusChem, 2021, 14, 2268–2294 CrossRef CAS PubMed.
- G. Van den Bossche, T. Vangeel, K. Van Aelst, W. Arts, L. Trullemans, K. Navare, S. Van den Bosch, K. Van Acker and B. F. Sels, in ACS Symposium Series, ed. C. G. Yoo and A. Ragauskas, American Chemical Society, Washington, DC, 2021, vol. 1377, pp. 37–60 Search PubMed.
- D. S. Zijlstra, C. A. Analbers, J. de Korte, E. Wilbers and P. J. Deuss, Polymers, 2019, 11, 1913 CrossRef CAS PubMed.
- D. G. Brandner, J. S. Kruger, N. E. Thornburg, G. G. Facas, J. K. Kenny, R. J. Dreiling, A. R. C. Morais, T. Renders, N. S. Cleveland, R. M. Happs, R. Katahira, T. B. Vinzant, D. G. Wilcox, Y. Román-Leshkov and G. T. Beckham, Green Chem., 2021, 23, 5437–5441 RSC.
- I. Kumaniaev, E. Subbotina, J. Sävmarker, M. Larhed, M. V. Galkin and J. S. M. Samec, Green Chem., 2017, 19, 5767–5771 RSC.
- A. Adler, I. Kumaniaev, A. Karacic, K. R. Baddigam, R. J. Hanes, E. Subbotina, A. W. Bartling, A. J. Huertas-Alonso, A. Moreno, H. Håkansson, A. P. Mathew, G. T. Beckham and J. S. M. Samec, Joule, 2022, 6, 1845–1858 CrossRef CAS.
- F. Brandi, B. Pandalone and M. Al-Naji, RSC Sustainability, 2023, 1, 459–469 RSC.
- J. K. Kenny, D. Brandner, S. R. Neefe, W. E. Michener, Y. Roman-Leshkov, G. T. Beckham and W. Medlin, React. Chem. Eng., 2022, 7, 2527–2533 RSC.
- T. Ren, S. You, Z. Zhang, Y. Wang, W. Qi, R. Su and Z. He, Green Chem., 2021, 23, 1648–1657 RSC.
- X. Ouyang, X. Huang, J. Zhu, M. D. Boot and E. J. M. Hensen, ACS Sustainable Chem. Eng., 2019, 7, 13764–13773 CrossRef CAS.
- Z. Sultan, I. Graça, Y. Li, S. Lima, L. G. Peeva, D. Kim, M. A. Ebrahim, R. Rinaldi and A. G. Livingston, ChemSusChem, 2019, 12, 1203–1212 CrossRef CAS PubMed.
- W. Arts, K. Van Aelst, E. Cooreman, J. Van Aelst, S. Van den Bosch and B. F. Sels, Energy Environ. Sci., 2023, 16, 2518–2539 RSC.
- X. Yang, M. Feng, J.-S. Choi, H. M. Meyer and B. Yang, Fuel, 2019, 244, 528–535 CrossRef CAS.
- Y.-Y. Wang, L.-L. Ling and H. Jiang, Green Chem., 2016, 18, 4032–4041 RSC.
- N. Li, L. Wei, R. bibi, L. Chen, J. Liu, L. Zhang, Y. Zheng and J. Zhou, Fuel, 2016, 185, 532–540 CrossRef CAS.
- J. Hu, M. Zhao, B. Jiang, S. Wu and P. Lu, Energy Fuels, 2020, 34, 9754–9762 CrossRef CAS.
- L. Jiang, H. Guo, C. Li, P. Zhou and Z. Zhang, Chem. Sci., 2019, 10, 4458–4468 RSC.
- Q. Xia, Z. Chen, Y. Shao, X. Gong, H. Wang, X. Liu, S. F. Parker, X. Han, S. Yang and Y. Wang, Nat. Commun., 2016, 7, 11162 CrossRef PubMed.
- Y. Liao, S.-F. Koelewijn, G. Van den Bossche, J. Van Aelst, S. Van den Bosch, T. Renders, K. Navare, T. Nicolaï, K. Van Aelst, M. Maesen, H. Matsushima, J. M. Thevelein, K. Van Acker, B. Lagrain, D. Verboekend and B. F. Sels, Science, 2020, 367, 1385–1390 CrossRef CAS PubMed.
- M. Tschulkow, T. Compernolle, S. Van den Bosch, J. Van Aelst, I. Storms, M. Van Dael, G. Van den Bossche, B. Sels and S. Van Passel, J. Cleaner Prod., 2020, 266, 122022 CrossRef CAS.
- Towards a Sustainable Value Chain of Lignin-Based Bio-Aromatics in Flanders [White Paper], Catalisti, 2022 Search PubMed.
- E. Cooreman, T. Nicolaï, W. Arts, K. V. Aelst, T. Vangeel, S. V. den Bosch, J. V. Aelst, B. Lagrain, K. Thiele, J. Thevelein and B. F. Sels, ACS Sustainable Chem. Eng., 2023, 11, 5440–5450 CrossRef CAS.
- L. Shuai, M. T. Amiri, Y. M. Questell-Santiago, F. Héroguel, Y. Li, H. Kim, R. Meilan, C. Chapple, J. Ralph and J. S. Luterbacher, Science, 2016, 354, 329–333 CrossRef CAS PubMed.
- M. Talebi Amiri, G. R. Dick, Y. M. Questell-Santiago and J. S. Luterbacher, Nat. Protoc., 2019, 14, 921–954 CrossRef CAS PubMed.
- W. Lan, M. T. Amiri, C. M. Hunston and J. S. Luterbacher, Angew. Chem., Int. Ed., 2018, 57, 1356–1360 CrossRef CAS PubMed.
- L. Shuai and B. Saha, Green Chem., 2017, 19, 3752–3758 RSC.
- J. Behaghel de Bueren, F. Héroguel, C. Wegmann, G. R. Dick, R. Buser and J. S. Luterbacher, ACS Sustainable Chem. Eng., 2020, 8, 16737–16745 CrossRef CAS.
- Y. M. Questell-Santiago, R. Zambrano-Varela, M. Talebi Amiri and J. S. Luterbacher, Nat. Chem., 2018, 10, 1222–1228 CrossRef CAS PubMed.
- R. N. Nishide, J. H. Truong and M. M. Abu-Omar, ACS Omega, 2021, 6, 8142–8150 CrossRef CAS PubMed.
- R. Vendamme, J. Behaghel de Bueren, J. Gracia-Vitoria, F. Isnard, M. M. Mulunda, P. Ortiz, M. Wadekar, K. Vanbroekhoven, C. Wegmann, R. Buser, F. Héroguel, J. S. Luterbacher and W. Eevers, Biomacromolecules, 2020, 21, 4135–4148 CrossRef CAS PubMed.
- S. Bertella and J. S. Luterbacher, Green Chem., 2021, 23, 3459–3467 RSC.
- G. R. Dick, A. O. Komarova and J. S. Luterbacher, Green Chem., 2022, 24, 1285–1293 RSC.
- W. Lan, Y. P. Du, S. Sun, J. Behaghel de Bueren, F. Héroguel and J. S. Luterbacher, Green Chem., 2021, 23, 320–327 RSC.
- W. Lan, J. B. de Bueren and J. S. Luterbacher, Angew. Chem., Int. Ed., 2019, 58, 2649–2654 CrossRef CAS PubMed.
- Y. M. Questell-Santiago, J. H. Yeap, M. Talebi Amiri, B. P. Le Monnier and J. S. Luterbacher, ACS Sustainable Chem. Eng., 2020, 8, 1709–1714 CrossRef CAS.
- A. De Santi, M. V. Galkin, C. W. Lahive, P. J. Deuss and K. Barta, ChemSusChem, 2020, 13, 4468–4477 CrossRef CAS PubMed.
- A. De Santi, S. Monti, G. Barcaro, Z. Zhang, K. Barta and P. J. Deuss, ACS Sustainable Chem. Eng., 2021, 9, 2388–2399 CrossRef CAS PubMed.
- P. J. Deuss, C. S. Lancefield, A. Narani, J. G. de Vries, N. J. Westwood and K. Barta, Green Chem., 2017, 19, 2774–2782 RSC.
- Z. Zhang, I. M. O'Hara, D. W. Rackemann and W. O. S. Doherty, Green Chem., 2013, 15, 255–264 RSC.
- Z. Zhang, C. Lahive, J. Winkelman, K. Barta and P. J. Deuss, Green Chem., 2022, 24, 3193–3207 RSC.
- O. Yu, C. G. Yoo, C. S. Kim and K. H. Kim, ACS Omega, 2019, 4, 16103–16110 CrossRef CAS PubMed.
- T. Yokoyama and Y. Matsumoto, J. Wood Chem. Technol., 2010, 30, 269–282 CrossRef CAS.
- T. Imai, T. Yokoyama and Y. Matsumoto, J. Wood Sci., 2011, 57, 219–225 CrossRef CAS.
- T. Yokoyama, J. Wood Chem. Technol., 2015, 35, 27–42 CrossRef.
- Q. Ye and T. Yokoyama, J. Wood Sci., 2020, 66, 80 CrossRef CAS.
- F. Brienza, K. Van Aelst, K. Thielemans, B. F. Sels, D. P. Debecker and I. Cybulska, Green Chem., 2021, 23, 3268–3276 RSC.
- F. Brienza, K. Van Aelst, F. Devred, D. Magnin, M. Tschulkow, P. Nimmegeers, S. Van Passel, B. F. Sels, P. Gerin, D. P. Debecker and I. Cybulska, Chem. Eng. J., 2022, 450, 138179 CrossRef CAS.
- I. Cybulska, F. Brienza and D. P. Debecker, Process for Producing Lignin Components from Lignocellulosic Biomass, 2021, at, https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021058483 Search PubMed.
| This journal is © The Royal Society of Chemistry 2024 |