
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCarboxylation reactions for the sustainable manufacture of chemicals and monomers
Laura
Faba
 and
Salvador
Ordóñez
*
and
Salvador
Ordóñez
*
Catalysis, Reactors and Control Research Group (CRC), Dept. of Chemical and Environmental Engineering, University of Oviedo, Oviedo 33006, Spain. E-mail: sordonez@uniovi.es
First published on 30th September 2024
Abstract
Carboxylation stands out as one of the most versatile and viable routes for carbon dioxide fixation, a crucial chemical transformation essential for advancing capture technologies and fostering a sustainable industry. The carboxylic acids and derivatives produced through this process hold considerable interest for various sectors, including pharmaceuticals and polymers. Presently, most of these chemicals are derived from non-renewable resources, underscoring the imperative need to develop sustainable pathways for their synthesis. The inherent stability of the CO2 molecule, owing to its high oxidation state and linear configuration, poses significant challenges for activation. Diverse approaches, including photochemical, electrochemical, enzymatic, and thermochemical carboxylation have been explored. While noteworthy results have been achieved with these methods, substantial efforts are still required to facilitate their scalability. This review provides a comprehensive overview of each of these routes, elucidating their respective strengths and weaknesses. Emphasis is placed on thermochemical routes, given their proximity to potential industrial-scale application.
Sustainability spotlightThe current efforts for CO2 capture and sequestration require the development of new chemical processes devoted to using this carbon dioxide as a raw material. Carboxylation reactions are of key interest in industrial chemistry, requiring the use of CO2 and a reactant and allowing the incorporation of these carbon atoms into chemical products. In this article, we review the current situation of thermo-, photo-, and electrochemical processes for upgrading CO2via carboxylation reactions. This article is related to the following UN sustainable development goals: affordable and clean energy (SDG 7), industry, innovation, and infrastructure (SDG 9), and climate action (SDG 13). |
Introduction
Carbon dioxide (CO2) is a non-toxic, non-combustible and non-flammable gas. It is a crucial component of Earth's atmosphere, playing a pivotal role in regulating the planet's temperature and sustaining life. However, the rapid and constant increase in atmospheric CO2 concentration, primarily due to human activities (combustion of fossil fuels, deforestation, industrial processes, and agriculture), has raised serious environmental concerns, including climate change, ocean acidification, and impacts on biodiversity. Thus, the decline in CO2 concentration in the atmosphere may be considered one of the most significant challenges of the early decades of the 21st century.Several alternatives have been proposed for its capture (absorption, adsorption, membranes, etc.).1,2 Although they are technically viable solutions, the economic viability of these technologies is dependent on the development of efficient routes for the direct (i.e., not chemically altered) or indirect (i.e., transformed) use of this CO2.3 Chemically, this compound provides an environmentally friendly and readily available source of carbon. Its use as a C1 building block in organic synthesis (emulating natural photosynthesis) is the most attractive alternative for a sustainable industry, combining climate change mitigation with a reduction in the need for fossil fuels as raw materials in various industrial applications.4
CO2 is a linear and highly oxidized molecule, two properties that hinder its reactivity and restrict its utility as a regent in industrial chemical processes. However, CO2 participates in well-stablished and relevant processes, such as the synthesis of urea and its derivatives,5,6 the salicylic acid production;7,8 and, in recent years, the production of organic carbonates.9
Despite its low reactivity, the oxygen atoms in CO2 show weak Lewis basicity, while the carbon atom is electrophilic. Therefore, the reactions involving CO2 are dominated by nucleophilic attachment at the carbon atom and reactions with electron-donating reagents.10 In other words, CO2 is necessarily reduced in any chemical carbon fixation reaction. Organic chemistry offers different reactions that fulfil these conditions, see Scheme 1, and the optimization of these reactions could expand the range of possibilities for CO2 utilization.
 | ||
| Scheme 1 Summary of the most promising chemical routes for CO2 valorisation, highlighting carboxylation, which is the primary objective of this review. Adapted from (ref. 11). | ||
Almost all these alternatives are still at the lab scale, requiring strong efforts to optimize them and make them attractive for scale-up. Because of the versatility and high value of the chemicals that could be obtained, carboxylation, the topic of this review, is identified as one of the most promising routes for CO2 chemical valorisation.
This work reviews the current state of scientific research on carboxylation using CO2, including electrochemical, photochemical, enzymatic, and catalytic approaches. General aspects of the mechanisms, reaction conditions, and the current advances and drawbacks of each alternative are discussed, highlighting the specific processes that have greater potential for near future scale-up to industrial applications. Catalytic carboxylation is examined in greater detail since, as will be discussed in the subsequent sections, it is the only pathway currently providing sufficient feasibility conditions to be considered as a potential sustainable route for the industrial production of chemical products.
General aspects of carboxylation
Carboxylation is a chemical reaction that involves the incorporation of a carboxyl group (–COOH) into an organic compound, typically in the form of a CO2 molecule, as represented in eqn (1):| R−H + CO2 → R−COOH | (1) |
The carboxylic group consists of a carbonyl group (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and a hydroxyl group (OH) connected to the same carbon atom. This unique structure imparts several key properties, being one of the most versatile functional groups. The carboxylic group is highly polar, and the oxygen atoms can form hydrogen bonds, making carboxylic acids and related compounds excellent solvents for other polar molecules. When dissolved in water, they release H+ ions, making them acids. Based on these properties, carboxylic acids are fundamental in various industrial sectors, including chemistry, biochemistry, pharmacology, and plastics.
O) and a hydroxyl group (OH) connected to the same carbon atom. This unique structure imparts several key properties, being one of the most versatile functional groups. The carboxylic group is highly polar, and the oxygen atoms can form hydrogen bonds, making carboxylic acids and related compounds excellent solvents for other polar molecules. When dissolved in water, they release H+ ions, making them acids. Based on these properties, carboxylic acids are fundamental in various industrial sectors, including chemistry, biochemistry, pharmacology, and plastics.
The history of carboxylation can be traced back to the late 19th century when scientists began to explore the reaction of carbon dioxide (CO2) with various organic compounds. The Kolbe–Schmitt reaction, discovered by Hermann Kolbe and Rudolf Schmitt in 1860, marked an early milestone in carboxylation.12 Although interest in this reaction has remained constant, the current environmental pressure has led to an increasing interest in this reaction, not only from a chemical viewpoint but also from biotechnological and industrial viewpoints.
When CO2 participates in a reaction, the thermodynamic barrier (ΔH ≈ −400 kJ mol−1) that must be overcome requires homogeneous or heterogeneous catalysts, energetic reaction partners, or energy inputs into the process. The different alternatives to overcome this energy barrier have led to approaches to carboxylation through electrochemical, photochemical, enzymatic, and catalytic pathways.
Electrochemical carboxylation
Electrochemical fixation of CO2 to generate carboxylic acids is a feasible method in which the redox reaction replaces the utilization of traditional reducing agents. It is performed at atmospheric pressure and room temperature, decreasing the reaction costs, and suppressing side reactions, i.e., achieving high atom economy, and being environmentally friendly. However, from an industrial viewpoint, the use of electrocarboxylation is not very promising, due to the difficulty of scaling up the results obtained in batch configurations or microreactors, which are usually performed on a milligram scale.Electrocarboxylation can occur via two general pathways, as shown in Scheme 2. If the reduction potential of an organic substrate is more positive than that of CO2, electrochemical reduction of the organic compound predominantly occurs, yielding anionic species. The subsequent nucleophilic attack on carbon dioxide yields the carboxylic acid. In contrast, if the reduction potential of the organic molecule is more negative than that of CO2, one-electron reduction of carbon dioxide predominantly takes place, generating the radical anion of CO2 that reacts with the organic substrate. This second route typically occurs with alkenes.
 | ||
| Scheme 2 Two different pathways in electrochemical fixation of carbon dioxide with C–C bond formation. | ||
Electrochemical carboxylation has received attention since the last decades of the 20th century, with findings indicating that it takes place efficiently when a sacrificial anode such as magnesium or aluminium is used as an anode.13 After several studies with different organic molecules, such as alkenes, alkynes, enynes, aromatic and aliphatic halides, benzyl carbonates, aldehydes, and ketones, this configuration remains the optimal one.14–17 The oxidation of the anode metal, resulting in its dissolution proceeds at the anode during electrolysis and it prevents any species from oxidizing at the anode. Furthermore, the formation of a metal salt of the carboxylate ion with the metal ion generated from the anode is a crucial factor for obtaining carboxylic acids in good yields. However, contamination due to these metals is a serious drawback from the viewpoint of green chemistry.
In the last decade, sacrificial anode-free electrocarboxylation was also demonstrated as a promising strategy.18,19 For example, the carboxylation of benzylic halides using a quasi-divided cell (an undivided cell equipped with a much smaller counter electrode than the working electrode) with a Pt plate cathode and a Pt wire anode achieved yields of up to 78% at 0 °C in a THF medium, see Scheme 3.
 | ||
| Scheme 3 Sacrificial anode-free electrocarboxylation of benzylic halides using a quasi-divided cell.18 | ||
Considering these relevant results, this sacrificial anode-free reaction has been also tested using a microreactor, a configuration that has several advantages, such as high-speed mixing, a large surface-to-volume ratio, and precise temperature control. Once the electrode distance was optimized (20 μ), a 90% yield was achieved working with a 0.6 mL min−1 flow rate.20 Other studies have extended this no sacrificial anode configuration to the C–H carboxylation of arenes, using graphite felt electrodes, achieving yields of up to 87%.21
Among all the molecules tested, the electrochemical fixation of CO2 is of great importance for synthesizing nonsteroidal anti-inflammatory drugs (NSAIDs), such as naproxen, ibuprofen, and their precursors and derivatives, see Scheme 4.
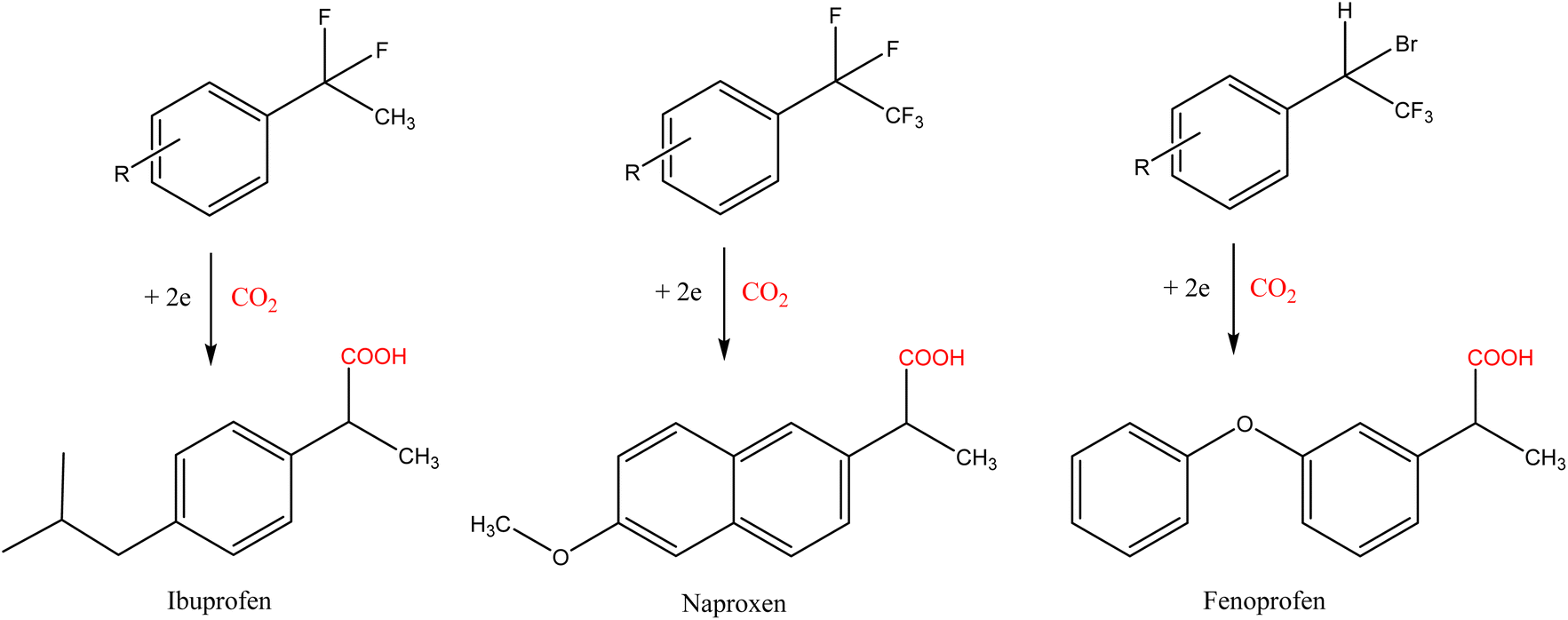 | ||
| Scheme 4 Potential synthesis of NSAIDs from organic halides.16 | ||
According to the latest data available, ibuprofen was prescribed more than 16.5 million times in 2020, while total prescriptions for naproxen exceeded 8.1 million.22 The classical synthesis of ibuprofen (developed in the 1960s) involves six steps, including the use of toxic aluminium chloride.23 This original method has been improved to the current three-step procedure (Hoechst process), which is based on cumene (coal and oil-derived compounds) and has several drawbacks from an environmental viewpoint.24 The traditional synthesis of naproxen (1976) was based on the traditional Friedel–Crafts alkylation, resulting in naproxen after a four-step process using homogeneous catalysts (strong acid, NaOH, chlorides, and iodides).25 In the last few decades, the development of simpler, more efficient, stereospecific, and sustainable routes for synthesizing these two drugs has been a hot topic in the pharmaceutical sector.26 In both cases, electrochemical carboxylation offers promising results, being a friendly alternative that does not require stoichiometric metals or external reducing agents. As relevant results, Yang and co-workers achieved yields of up to 87% for ibuprofen by electrochemical carboxylation of benzyl trimethylammonium bromide using commercially available carbon cloth and platinum on carbon (Pt/C) as the cathode and anode, respectively, and bubbling CO2 in a DMF solution (room temperature, −4.5 V).27 The electrocarboxylation route for synthesizing naproxen has not been proposed since the asymmetric electrochemical carboxylation (required in this case because of the optical activity of naproxen) is one of the issues that remains to be investigated and resolved.19 However, preliminary studies with aromatic ketones28,29 and aryl epoxides30 suggest that naproxen synthesis will be possible shortly.
Despite these promising results, the scale-up of electrocarboxylation faces several challenges related to low CO2 solubility under working conditions, mass transfer issues, the high cost of electrodes (more relevant in the sacrificial anode configuration), and, and more relevant nowadays, the involved energy costs. At this point, the wider extent of renewable energy opens a new window for these kinds of processes.
Photochemical carboxylation
Photocatalytic carboxylation relies on the photochemical reduction of CO2, often using sunlight or artificial light sources. The photocatalyst, typically a semiconductor material, absorbs photons and generates electron–hole pairs, which can then be used to drive the carboxylation. Photo-carboxylation is a very attractive approach because of its mild conditions, high activity, facile operation, and scalability. It can also be integrated into energy storage systems since using solar energy to convert CO2 into energy-dense molecules, such as formate or other organic fuels, contributes to the development of renewable energy storage technologies. Thus, relevant contributions in this field have been reported in the last two decades, which have been summarized and analysed in comprehensive reviews.31–34Photochemical carboxylation can occur via two mechanisms: (1) the insertion of CO2 as an electrophile or, (2) the single-electron reduction of CO2, generating a radical-anion species that acts as a nucleophile.35 In the latter case, the reduction potential of CO2 is quite high (E0 = −2.21 V),36 which triggers the process of CO2 assimilation under mild conditions. Today, photocatalytic carboxylation has made significant advances in the selective functionalization of benzylic bonds (C(sp3)-H) as well as C(sp2)-H, and C(sp)-H bonds.
One of the most interesting studies was presented in 2015 by Murakami and co-workers, dealing with the photocatalytic carboxylation of alkylphenyl ketones to produce acylphenylacetic acids using an LED lamp (365 nm, 2 h).37 This reaction occurs at room temperature in DMSO solvent, and high yields (61–95%) are obtained, even when working with CO2 atmospheric pressure, see Scheme 5. The reaction is so effective that there is no need for any catalyst or sacrificial reagent. Even if LED light is substituted with solar irradiation, a relevant yield of 72% is achieved after 7 h. The most interesting application of this reaction is the production of 2,3-benzodiazepines, an extended intermediate in some biologically active pharmaceutical molecules, including anxiolytics.38 The extensive market of this drug (valued at USD 2.96 billion in 2021 (ref. 39)) justifies the high relevance of substituting the current petrol-derived synthesis process with a sustainable alternative.
 | ||
| Scheme 5 Photoinduced carboxylation of o-alkylphenyl ketones with CO2 (ref. 37). | ||
Based on these studies, this strategy has been extended to the photoinduced carboxylation of benzylic and aliphatic C–H bonds with CO2 using aromatic ketones and nickel complex catalysts under UV light,40 an interesting and selective route to obtain mono-carboxylated products even in those compounds that have more than one benzylic C–H bond. However, using benzene solvent is a relevant drawback of these studies.
As to the single-electron reduction strategy, the synthesis of α-amino acids from different amines, which is highly relevant for the food sector, is one of the most interesting applications. The reduction potential of most transition metal photocatalysts is insufficient in this case; so these reactions require an organic photosensitizer para-terphenyl, which in addition to being an electron donor acts as a free radical precursor.41 Preliminary analyses in batch configurations achieved yields up to 92% in 10 min, whereas continuous bubbling of CO2 (Scheme 6) provided only a 30% yield after 2 h of irradiation, requiring the development of more active sensitizers or catalysts.
 | ||
| Scheme 6 Photocatalytic α-carboxylation of amines with CO2 in continuous flow41 (SET = single-electron transfer). | ||
Modifications of this strategy define the hydrogen atom transfer (HAT), which enables the production of bioactive molecules, such as fenoprofen, naproxen, and flurbiprofen, from benzylic compounds, but in moderate yields.42 For example, 53% of fenoprofen, or 38% of naproxen are obtained using high-power blue LED light for 24 h, in the presence of a photosensitizer, and complex catalysts, when the reaction is carried out in DMS at 0 °C. This strategy has been deeply studied for many different chemicals, resulting in the identification of a huge number of catalysts (based on Ni, Ti, and Zn, among others). Despite the chemical interest and extensive mechanistic studies proposed in the literature, discussed in detail in (ref. 32) these routes are not specifically defined for the synthesis of any valuable chemical, and the complexity, high price, and potential toxicity of the chemicals involved hinder their scalability.
By combining SET and HAT steps, the photoinduced carboxylation concept has also been proposed for the carboxylation of alkyl halides at remote C(sp3)-H sites, as well as for activated and common C(sp3)-X (X = H, N) bonds, C(sp2)-X (X = H, N, (pseudo)halide) bonds and, to a lesser extent, C(sp)-H bonds. The C(sp2)-X group includes the hydrocarboxylation of alkenes, imines, and aromatic halocarbons. In most cases, using stoichiometric amounts of organometallic reductants is indispensable to provide active metal hydride species, including Zn, Al, Ni, Ir, Pd, Ru, and Rh complexes and photocatalysts. These reactions have been extensively studied in excellent reviews published on this topic.31–34 Despite the high academic relevance of these studies, the industrial interest in these reactions is quite low.
The carboxylation of alkynes (C(sp)-H) using CO2 is a promising method to prepare α,β-unsaturated acids. Acrylic acid is the most relevant example of this kind of acid, and there is no doubt about the industrial interest in developing a sustainable route for its synthesis. This route has been studied since the 1980s43 and the recent studies offer interesting results using visible light with the aid of iridium/cobalt dual catalysis, thus avoiding the utilization of metallic reagents.44
To sum up, while photocatalytic carboxylation holds significant promise, the efficiency of the process using UV light must be improved. Relevant results are achieved with C(sp3)-X (X = N, H) bonds and styrene, whereas olefins without activation have not showed the same success. The selectivity of carboxylation products and the stability of the photocatalysts are among the key areas of concern. Avoiding the utilization of noble-metal photosensitizers and organic solvents is in favour of these systems to meet the need for green chemistry. There are promising results for its application to many different substrates, but currently, photocatalytic carboxylation is of more academic than industrial interest.
A possible scale-up of these routes presents several challenges, primarily due to the complexity and sensitivity of photochemical reactions. Requirements in terms of light penetration, distribution and stability are difficult to fulfil on a large scale, requiring complex reactor designs (geometry, materials, and cooling systems). Photochemical reactions are highly sensitive to variations in light intensity, wavelength, and reaction conditions, parameters that are more difficult to control at an industrial scale, challenging the quantum efficiency. The energy and material costs are also very relevant, especially when working with UV light.
Enzymatic carboxylation
Enzymatic carbon dioxide fixation is one of the most important metabolic reactions as it allows the capture of inorganic carbon from the atmosphere. Natural enzymatic reactions also include carboxylation, and the knowledge of these mechanisms has inspired many studies to extend these procedures to biotechnology contexts.45–47 Biocatalytic carboxylation is applied to overcome the most relevant obstacles of using CO2 in a chemical reaction: its highest oxidation state and the difficulty in activating it, the CO2 low concentration in nature that does not allow direct use of atmospheric air, and its reactivity with water, which hinders the reactions in aqueous solutions because of the formation of bicarbonates. The high efficiency of enzymes and their substrate affinity enable operations under low CO2 concentrations and the broad range of carboxylases allows different binding and activation modes of CO2 (including as a bicarbonate) and different attack modes of the carbon nucleophile, yielding highly selective reactions.48Irrespective of the carboxylase used, almost all the enzymatic carboxylation reactions follow the same general five-step mechanism,49 shown in Scheme 7: (1) generation of an enol/enolate to create a nucleophile, (2) binding and stabilization of the enol/enolate; (3) accommodation and activation of CO2; (4) C–C bond formation via nucleophilic attack of the enol/enolate onto the carbon of CO2; and (5) potential follow up reactions such as cleavage of the product from the cofactor.
 | ||
| Scheme 7 General mechanistic steps of enzymatic carboxylation reactions.50 | ||
Since carboxylation is a thermodynamically controlled reaction, enzymatic carboxylation (both nature and synthesis) is accomplished either by reducing the required reaction energy or by shifting the reaction equilibrium. Strategies to render carboxylation reactions more favourable include: (i) using high energy starting materials (electron-rich substrates); (ii) the activation of CO2 through interaction with the enzyme (carboxyphosphate formation, binding to biotin, and coordination); (iii) supply of external reducing equivalents, such as NADPH or ferredoxins; and (iv) formation of low energy products, such as in the case of the carboxylation of phenolic compounds.50
Recent reviews have detailed the most relevant enzymatic carboxylation reactions.50,51 Maybe the most promising biosynthetic carboxylation for implementation at a medium scale is the fermentation production of succinic acid,52 an important building block for the synthesis of polyesters, see Scheme 8. This route uses glycerol as a carbon source and fixes one CO2 molecule by the carboxylation of phosphoenolpyruvate.
 | ||
| Scheme 8 Fermentative production of succinic acid from glycerol and CO2. (PEPCK = phosphoenolpyruvate carboxykinase; MDH = malate deshydrogenase, FH = fumarate hydratase; FRD = fumarate reductase; QH = quinol).52 | ||
Enzymatic carboxylation reactions have relevant drawbacks that discourage their implementation at a large scale. On the one hand, their low productivity and incomplete conversion, are mainly due to the high amount of energy that is required. Moreover, they are sensitive since the operational window of enzymes is usually quite narrow (temperatures, solvent, pH, pressure, and inhibitors). This directly clashes with the variable conditions that might occur in an industrial process. Moreover, the cost of these enzymes is quite high, and the recovery and reuse of enzymes and cofactors are crucial to render the process economically feasible. Each individual enzyme would require its specific immobilization strategy (encapsulation, adsorption, and covalent immobilization) and, although some studies have evaluated the heterogenization of enzymes,48,53,54 these procedures are far from being developed for their scale-up.
Despite these limitations, biotechnology has several advantages concerning the chemo-catalytic industry, such as the use of non-toxic compounds, and the soft conditions in terms of pH, temperature, and pressure. Considering these strong points, several authors have made relevant efforts to overcome the inherent disadvantages of enzymatic routes. Most of these studies have the objective of increasing the yield, which is affected not only by the thermodynamics but also by the irreversible production of bicarbonates since reactions occur in aqueous media. Different strategies for pushing the equilibrium towards target compounds are developed,55 but most of these solutions are not applicable in the context of reaction engineering.
Modifying temperature or pressure to increase the availability of CO2 is difficult due to the irreversible deactivation that most carboxylases experience under these conditions.56 Few natural carboxylases can survive these conditions, but enzyme engineering has found an interesting research niche in generating variants of carboxylases with enhanced stability and activity.49,57 However, the current state of the art of these studies is far from being applicable at an industrial scale and the high costs of these mutations limit future applications to high-value fine chemicals, with the pharmaceutical sector being the main candidate. This fact reduces the applicability of enzymatic carboxylation as a large-scale capture and utilization (CCU) technology. An exception to this general situation is the pyrrole carboxylation, which has been already tested in continuous flow at high pressure (65 bar), achieving a space-time yield of 24 ± 7 μmol h−1 using immobilized B. megaterium (PYR2910).58
Another option proposed in the literature is product removal, with the subsequent displacement of the reaction equilibrium. This solution is useful when carboxylic acid is not the final product but an intermediate, such as in the synthesis of methionine (carboxylation + amination)59 or when carboxylic acid derivatives form insoluble salts, such as in the case of benzoic acid. An interesting increase in conversion (from 37% to 97%) was observed by adding 50 mM of tetrabutylammonium bromide to the reaction medium.60
Thermocatalytic carboxylation
The chemical approach of CO2 fixation is considered one of the most versatile strategies to produce carboxylic acids from quite different substrates, including alcohols, olefins, aromatics, and alkynes. The extensive bibliography on this topic has been analysed and discussed in several comprehensive reviews, analysing the main advances, with some of them structured based on the substrates,61–66 mechanisms,67,68 and catalysts.69–73The success of a carboxylation reaction depends on various factors, including temperature, pressure, and the need for a catalyst or a precursor. Different carboxylation reactions require specific conditions, mainly influenced by the reactivity of the organic compound (the acidity of the C–H bond). Thus, carboxylation can occur in the liquid phase, at soft temperatures using organometallic catalysts (homogeneous catalysis); or at higher temperatures, with a solvent-free configuration, using inorganic salts as precursors.
In general, thermocatalytic carboxylation can follow two opposite mechanisms, mainly depending on the specific reaction conditions and the nature of the organic compound involved: the nucleophilic addition and the electrophilic one. The first mechanism involves a nucleophilic attack on the organic compound by CO2. Common nucleophiles include organometallic reagents, such as Grignard reagents or organolithium compounds. In the second mechanism, CO2 is activated by a strong electrophile, often a metal catalyst. The metal site polarizes the CO2 molecule, making it more reactive and susceptible to electrophilic attack by the organic compound. The Kolbe–Schmitt reaction is a typical example of this type of carboxylation. An initial overview of these two reactions is needed to identify the aspects that have captured the attention of the researchers in the last few decades, trying to develop suitable reactions that can have a crucial role in a sustainable future industry.
| R−Mg−X + CO2 → R−COOH | (2) |
The high reduction potential of Grignard reagents in the presence of nucleophilic bases allows the formation of C–C bonds to spontaneously produce carboxylic acids from gaseous or solid-state CO2. The reaction at room temperature is one of the strongest points of this reaction. However, the limited availability of the parent reagents (these reactions are not compatible with sensitive functional groups, such as aldehydes, ketones, or nitriles, as they rapidly react with organolithium or Grignard reagents), as well as their chemical reactivity and complex manipulation makes CO2 fixation by this method unattractive for industrial implementation. Some authors consider this mechanism to study possible carboxylation reactions, mainly of aromatic compounds (Scheme 9), and relevant knowledge about mechanisms, conditions, and catalysts has been obtained from these studies.64 However, the relevance of these studies is limited to the academic interest.
 | ||
| Scheme 9 2,6-Dimethylphenylmagnesium bromide carboxylation at room temperature, using different bases and THF saturated in CO2. A maximum yield of 77% is obtained after 30 min using DBU (1,8-diazabicycloundec-7-ene) 50 mol%.74 | ||
 | ||
| Scheme 10 Synthesis of salicylic acid by phenol carboxylation. | ||
One of the drawbacks of this method is that the reaction mixture is a waxy solid or a highly viscous liquid, which limits the contact surface between alkali metal phenoxide and CO2. Different studies have proposed conducting the Kolbe–Schmitt reaction in solution or suspension, proposing polar media such as phenol, ketones, dimethylsulfoxide (DMSO), or THF, among others.64 However, it was demonstrated that while solvents improved the reaction's rate constants by dissolving the reactants, they also allowed the reverse reaction to take place because the final product was retained in the reactive phase.76
Most of the current Kolbe–Schmitt reactions are based on the optimized process patented by Marassé in 1893: the reaction is performed at high temperature, with a mixture of free phenol and anhydrous potassium, rubidium, or cesium carbonate under CO2 pressure, with the best results achieved when using the caesium salt.77 This reaction is limited by the requirement of dry phenoxide since moisture can bind with the metal cation, thereby the coordination between CO2 and the alkali metal cation is prevented.78
Alternatively, the carboxylation of phenol with CO2 (1 atm) in the presence of NaOH and the additive 2,4,6-trimethylphenol (TMP) was reported by Larrosa and co-workers, see Scheme 11.82 The high yields obtained with several phenol derivatives (up to 90%) represent a significant improvement of the original Kolbe–Schmitt configuration allowing carboxylation at atmospheric pressure (easier to implement on a large scale in combination with CO2 capture techniques). Additionally, it offers other advantages since it does not generate water and the isolation of the phenoxide intermediate is not required. However, this reaction is ineffective in the presence of EWGs on the aromatic ring. The carboxylation of aromatic compounds was also proposed in the presence of organic bases, the diazabicyclo[5.4.0]undec-7-ene (DBU) being the most used one.83 Excellent yields of salicylic derivatives (>99%) are obtained under atmospheric CO2 pressure at soft temperatures.
 | ||
| Scheme 11 Phenol carboxylation using the TMP additive.82 | ||
All these studies consider reactions involving highly acidic C–H or N–H bonds of aromatic heterocycles, concluding that the presence of highly energetic substances (strong base or highly reducing metal) is imperative for the construction of nucleophilic carbons, a necessary step for reacting with CO2. Thus, to perform the carboxylation of less acidic C–H bond (pKa > 40) with CO2 is very challenging. This problem was solved in 2016 by using molten Cs2CO3 or K2CO3, which resulted in good yields of furan-2,5-dicarboxylic acid (FDCA) when furoic acid was used as the reactant.84 Considering the industrial relevance of this compound for the polymer industry, a separate section of this review is dedicated to this reaction.
After an extensive screening of different aromatics with several Lewis and Brønsted acid catalysts,64 only AlCl3 performed the carboxylation successfully. However, the low reactivity of CO2 and the highly reactive nature of organoaluminium (intermediate in the reaction) afforded low yields, with the formation of undesired products. This drawback was solved using an Al2Cl6/Al system.86 The role of additive Al was defined as very crucial to direct the reaction for the selective formation of the carboxylic acid by scavenging the HCl to produce AlCl3, which further participated in the reaction and assisted the carboxylation process. The effect of temperature, CO2 pressure, the amount of catalyst, and the reaction mechanisms were investigated thoroughly, resulting in milder conditions in all cases than those reported for basic systems. Experimental and theoretical studies based on density functional theory (DFT) concluded that the activation of CO2 takes place by super-electrophilic AlCl3 followed by a reaction with aromatic compounds. Based on these results, other authors proposed the use of organic catalysts with Lewis acidity, and good results were obtained with different dialkyl aluminium chlorides (Me2AlCl and Et2AlCl), at room temperature and medium CO2 pressure (30 bar).87,88 AlCl3-derived catalysts are the most studied ones, and only a few references to other Lewis acids are reported, as discussed by Rawal and co-workers.64
A common drawback of this approach is that carboxylation catalyzed by Lewis acids is moisture sensitive, so it must be performed in an inert atmosphere, reducing the potential for a possible scale-up.
The first-row transition metal ions are easily available and their price is quite low; 3d transition metal-based catalysts are the most active ones, but 4d and 5d can be used under milder conditions.
The earliest coupling reaction of CO2 and ethene (the 1980s) involved the use of nickel lactone, using donor ligands as potential activators of the β-hydride elimination reaction, see Scheme 12. This reaction has been the basis for an extensive bibliography on mechanisms, activators, and conditions to optimize the yields obtained. Other transition metal complexes have also been utilized for acrylate formation, highlighting molybdenum, tungsten, palladium, and platinum, with iron, and ruthenium being of significantly lower relevance. All these contributions are discussed in a very comprehensive review.69 Group VI metal centres induce more facile oxidative coupling of CO2 and ethene than Ni, which has a direct influence on the reaction conditions. Thus, carboxylation can occur at ambient temperature and pressure with Mo/W complexes, whereas high gas pressures are required with Ni.
 | ||
| Scheme 12 Ni mediated catalytic cycle as discussed by Fisher and co-workers.91 | ||
As for the carboxylation of aryl compounds, copper is one of the most used metals. In the initial studies, the in situ generation of copper complexes between CuCl and organic precursors was proposed, using tetrahydrofuran (THF) or dimethylfuran (DMF) solvents and mild temperatures.89,90 Other authors have demonstrated a high activity of gold(I) complexes containing strong donor ligands, for different aromatic systems, even when C–H bonds are not very acidic (pKa < 30.3).92 To a lesser extent, many other transition metals have been considered, including Ag, Zn, or Ru.70
The use of transition metal catalysts is an efficient strategy to achieve high activity for many different substrates (high functional group tolerance), providing high selectivity under mild conditions. However, organic solvents, basic salts, and organic precursors are needed to stabilize the complexes that activate CO2, resulting in difficult methodologies that are not easy to apply on a medium or large scale. Maybe the most promising future of this strategy is photocatalysis, i.e., using these transition metals but obtaining the required energy from a light source.
MOFs. One of the requirements of green chemistry is the use of heterogeneous catalysts instead of homogeneous ones. Working with solid catalysts has many advantages, including simple product purification, recyclability, etc. The development of optimal catalysts for carboxylation must consider not only the activity, selectivity, and stability, but also the cost and moisture sensitivity. The special characteristics of metal–organic frameworks (MOFs) are very interesting, and these catalysts have attracted a lot of attention.93,94 These materials have a well-structured organic framework (structural flexibility, porous nature, and tunable size) enriched with metal ions and ligands that introduce Lewis acid and basic characteristics.
Liu and colleagues reported the first study about the carboxylation of terminal alkynes with CO2 to propiolic acid by using Ag@MIL-101, achieving high efficiency as a bifunctional material, for the CO2 capture and the carboxylation at 100 °C and atmospheric pressure.95 The use of heterogeneous catalysts allowed their recovery and reuse. This study demonstrates that this MOF can be recycled up to five times without any significant change in the catalytic ability or the structure. The use of Cs2CO3 is required to abstract a proton from the C(sp)-H bond, leading to the formation of silver acetylide followed by the insertion of adsorbed CO2 into the Ag–C bond to form carboxylic acid. This study paves the way for other researchers, proposing the use of this MOF with Ag, Cu, and Pd@Cu nanocrystals.96–100 In all the cases, terminal alkynes (hetero aromatics and aromatic rings consisting of EWGs or EDGs) were used as substrates. Other MOFs, such as UiO67(dcppy) and ZIF-8 have been also proposed in preliminary approaches, using organic solvents and always at a very small scale.
The optimal situation involves the use of metal-modified MOFs that can be active, selective, and stable in aqueous-phase carboxylation reactions or under solvent-free conditions. These aspects are crucial for establishing carboxylation as a green route to obtain carboxylic acids at an industrial scale.
Main industrial processes involving carboxylation
Carboxylic acids and their derivatives are used in the formulation of solvents, surfactants, and emulsifying agents due to their amphiphilic nature. This property is crucial for the creation of stable emulsions and the development of cleaning products and cosmetics. Carboxylic groups are commonly incorporated into drug molecules because they enhance the drug's solubility, influence its adsorption and distribution in the body, and participate in specific interactions with target proteins. Aspirin (acetylsalicylic acid) is the most relevant example of a carboxylic-derived drug. Its production includes a key step of CO2 incorporation, serving as the reference for carboxylation at an industrial scale.For the near future, carboxylation reactions hold significant potential for reducing the environmental impact of chemical processes. The principles of green chemistry, which focus on designing sustainable and environmentally friendly reactions, can be applied to carboxylation: (1) atom economy: carboxylation reactions often lead to the incorporation of CO2 into valuable products, maximizing the use of starting materials and minimizing waste; (2) renewable feedstocks: CO2 is a renewable feedstock that can be harnessed to create valuable organic compounds, reducing the reliance on fossil-based resources; (3) reduced hazardous substances: by using CO2 as a feedstock and employing mild reaction conditions, carboxylation can reduce the generation of hazardous waste and by-products.
Although academic interest in carboxylation is very relevant, only a few chemicals have real potential to be suitable for scaling in the near future. This selected group includes pharmaceutical chemicals, such as ibuprofen and naproxen, which can be obtained through enzymatic routes, and green polymers that can be obtained by thermocatalytic strategies. This group includes polyacrylic acid and polyethylene furanoate (PEF). Polyacrylic acid and its derivates are used in a range of applications, from superabsorbent materials in diapers to drug delivery systems. Acrylic acid can be synthesized through carboxylation of propylene with CO2. PEF is the green substitute for polyethylene terephthalate (PET), one of the most used polymers, a basic material for plastic bottles and packaging. The last part of the review is dedicated to summarizing the main aspects of these processes from an engineering perspective.
According to the latest reports, the salicylic acid market was valued at $431.1 million in 2020 and is projected to reach almost $890 million by 2030.101 The large-scale use of this compound in the pharmaceutical sector is the major driver of this market in Asia–Pacific and Africa areas, whereas a more stable situation is considered for Europe and North America, where its modest growth is mainly due to the use of salicylic acid in the food & beverage industry for packaging.
This reaction was reported for the first time in 1987, using homogeneous iron salts as metal catalysts. This preliminary approach was performed at −78 °C, using THF solvent, obtaining a metal lactone complex that stabilizes ethylene, thus allowing the reaction with the CO2 stream (1 bar).102 This pioneering study was revisited in the first decade of the 21st century, with a relevant number of studies that deepen the reaction conditions, the mechanism, and possible catalysts. Mo and W complexes were identified as the optimal homogeneous catalysts, enabling carboxylation at room temperature and pressure,103,104 whereas more severe conditions are required with Ni.105 THF is the most studied solvent, and, in all the cases, a base is required to stabilize the intermediates since, due to thermodynamics, the formation of acrylic acid is unfavourable, and the equilibrium must be shifted,106 see eqn (3) and (4).
 | (3) |
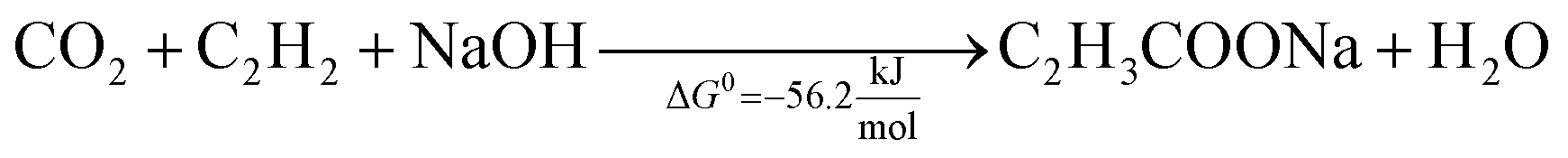 | (4) |
The use of NaOH itself is not recommended because of the formation of a stable carbonate (NaHCO3) that is significantly more favourable from a thermodynamic point of view (ΔG0 = −826.4 kJ mol−1). Therefore, this reaction requires sodium bases that are basic enough to produce sodium acrylate but not so basic to form irreversible carbonates. In addition, these bases should be capable of regeneration by using NaOH. Promising results are reported with certain sodium phenolates and alkoxides (NaO-t-Bu and NaO-i-Pr) using homogeneous nickel or palladium catalysts.62
Based on these results, Manzini and colleagues proposed the first continuous process using amide solvents, NaO-i-Pr, and Pd-catalysts at 145 °C, see Scheme 13.107 In this concept, water is added to a continuous stream from the reactor after depressurization to separate sodium acrylate from the catalyst. The alcohol formed in the reaction can be distilled from the product phase and recycled with NaOH, leaving behind pure sodium acrylate. The catalyst and the organic solvent can also be recycled, improving the sustainability of the process.
 | ||
| Scheme 13 : Process concept for the Pd-catalyzed synthesis of sodium acrylate from ethylene and Co2 using amide solvents and NaO-iPr.107 | ||
This route is an interesting alternative to propylene oxidation. From an economic perspective, the CO2 route could result in significant raw material savings, mainly because of the increase in ethane cracking that produces ethylene and only minor amounts of propylene. Ethylene can also be obtained by ethanol dehydration, and the production of ethanol by sugar fermentation is a well-known process, already established at an industrial scale. Thus, this route offers a double benefit from the sustainability point of view by capturing CO2 and using a biomass-derived compound as the raw material. However, the overall energy consumption must also be considered since the CO2 route is a net steam consumer (due to base regeneration) whereas propylene oxidation produces steam (highly exothermic reaction). Moreover, this approach still has significant drawbacks, including the low turnover numbers (<0.1 h−1), the stoichiometric amount of base required, and the use of solvents.108
One of the main candidates to lead this transition is polyethylene 2,5-furandicarboxylate (PEF), the renewable substitute for the most widely used plastic (polyethylene terephthalate, PEF). The most advanced route to produce FDCA involves converting edible fructose into 5-hydroxymethylfurfural (HMF) and oxidizing it to FDCA. This route is treated as a benchmark process, with plans for a 5 kt/a pilot plant to be operational in 2023.110 However, the future scale-up of this approach is restricted by the high price of HMF, which is influenced by the use of edible biomass and the purity requirements.
An alternative route to produce FDCA is based on furfural (FFL), a cyclic aldehyde obtained by the acidic hydrolysis of pentoses, i.e., with a non-edible origin. This route is a two-step process involving the partial oxidation of furfural yielding furoic acid followed by a second step that can involve three different catalytic routes: (1) disproportionation (Henkel reaction), (2) carbonylation, or (3) carboxylation. The first option has limited selectivity because of the simultaneous production of furan. The second one is a complex process using non sustainable chemicals. Thus, furoic acid carboxylation is the most promising approach according to the principles of green chemistry.
Considering the current state of the art, the acidic approach is disregarded since poor yields are obtained86 and, despite operating at ambient temperature, high CO2 pressures are needed, making it impossible to implement this reaction in a potential capture system. The complexity of the process also discourages the transition-metal strategy, although some studies have proposed the use of Cu and Pd salts.63
The base-mediated furoic acid carboxylation was first reported in 2016, as a particular application of the Kolbe–Schmitt reaction.84 The carboxylation was studied in a free-solvent configuration, using Cs2CO3 as a molten salt, as shown in Scheme 14. Further studies were focused on the study of the mechanism, mainly to understand the role of Cs+.65
 | ||
| Scheme 14 Overview of the reaction steps for the conversion of furfural to FDCA using the carboxylation route.65 | ||
Dick and co-workers conducted the first study in a fixed-bed flow reactor (mole-scale), yielding almost 90% FDCA working at 260–285 °C and 8 bar of CO2.111 To complete these studies, Wang and colleagues, in a comprehensive study, optimized the reaction conditions, concluding that maximum yield is obtained with a furoic acid![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :salt molar ratio of 3:5 (higher excess reduces the activity), and at 270 °C (higher temperatures promote decomposition).112
:salt molar ratio of 3:5 (higher excess reduces the activity), and at 270 °C (higher temperatures promote decomposition).112
The substitution of Cs2CO3 is of key interest because of economic reasons. Preliminary results suggest that, by modifying the conditions, this salt could be substituted with K2CO3.113 This strategy indicates the significant role of formates, also corroborated by DFT studies.114
Another strategy involves FDCA production by the simultaneous reaction of HMF and furfural, considering that both aldehydes are obtained during the same hydrolytic process. This approach enables the production of more than 71% FDCA after 36 h at 260 °C, using a bimetallic–lignin complex in the presence of Cs2CO3.115
All these studies demonstrate the promising future of this approach. However, relevant drawbacks must be overcome, including the narrow range of reaction conditions to produce FDCA, preventing decomposition, the development of effective heterogeneous catalysts, the optimization of reaction conditions, and a relevant cost reduction.
According to the latest data available, in 2022, the market volume of furfural amounted to over 365.24 metric tons worldwide.116 By the year 2030, it is projected to grow to nearly 505.41 metric tons. This global production capacity is still far from meeting the current PET demand, estimated at almost 35.3 million metric tons,117 but the development of an efficient furfural valorisation process can enhance its production. Increasing furfural production can have a direct impact on the process economy since, according to preliminary studies by Dubbink and co-workers, a cost reduction of 75% is required to meet the target FDCA selling price for a profitable design.118
Main challenges for future green carboxylation reactions
All advanced studies on carboxylation (already industrially implemented or with an expected near scale-up) use high-purity CO2 (specially manufactured for this purpose) and, in general, operate at high pressure. This makes the processes costly and environmentally unsustainable. Therefore, the greatest challenge is adapting these processes to use CO2 captured directly from the atmosphere (DAC) or from industrial processes that produce it (exhaust gases, CCU).The significant number of possible technologies for capturing CO2 from exhaust gas as well as their relevant technical and economic drawbacks that hinder their industrial implementation are well known, and their analysis is out of the scope of this work.119 Carboxylation stands out as one of the most promising alternatives among CO2-capture-and-utilization technologies (CCU). Although the capture technologies that are affected by technical limitations (very high flowrate, low concentration, and potential catalytic impurities) need to be optimized, the development of an efficient valorisation process could be the impulse required for some of these technologies to achieve the required economic feasibility.
Another option is direct air capture (DAC) of CO2, which involves extracting CO2 from the atmosphere.120 While this approach addresses CO2 directly relevant to the environment, it also presents significant barriers to large-scale implementation, mainly related to the low concentration of CO2 in the air, requiring highly efficient, advanced and costly capture equipment.
With this scenario, carboxylation using atmospheric or captured residual CO2 is far from becoming a reality, and there are no scientific articles that consider this configuration. Therefore, the analysis of the potential consequences or additional difficulties of using these streams remains incomplete.
Conclusions and outlook
The demand for CO2 sequestration to produce value-added products is high, and carboxylation stands out as one of the most attractive options to obtain commodity chemicals. Carboxylic acids or their esters play a crucial role in the synthesis of relevant biologically active molecules, polymers, and natural products, many of which are currently produced using non-renewable resources.The high stability of the CO2 molecule, attributed to its highest oxidation state and linear configuration, introduces relevant thermodynamic and kinetic limitations. Activating CO2 is challenging, and various techniques have been developed to overcome this drawback. These include energy activation through electrochemical and photochemical approaches, as well as biological and chemical activation, using enzymes and catalysts, respectively; or a combination of both these approaches, utilizing chemical catalysts to improve the electrochemical or photochemical route. These routes have been well documented over the last few decades, proposing and optimizing procedures for the synthesis of hundreds of chemicals.
Despite the wealth of research studies, the large-scale transformation of CO2 into value-added products remains a significant challenge. While these studies are academically interesting, their limited industrial application is due to various reasons hindering economic feasibility. These include the difficult scaling of some configurations, the use of complex, non-commercial catalysts, the need for too harsh conditions and toxic compounds, and the low yields obtained, mainly for asymmetric carboxylation reactions.
From an industrial viewpoint, catalytic routes are the most promising ones. In fact, this is the only methodology already implemented on a large scale (production of salicylic acid) and it is expected that this approach will be extended to the sustainable production of other compounds, such as renewable polymers.
Despite notable recent advances, common drawbacks that must guide future research in this field include the development of cheap and sustainable (metal-free if possible) catalysts, which are active for carboxylation reactions under atmospheric pressure of CO2 and at mild temperatures. Sustainable methods, such as photocatalysis and electrocatalysis still have a long way to go before they can be scaled whereas, currently, enzymatic and transition metal-catalyzed carboxylation represent the most suitable configurations from an industrial viewpoint.
The most ambitious goal in this field is the development of industrially viable carboxylation processes that utilize CO2 captured either directly from the atmosphere or from waste streams.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study has been financially supported by the Spanish Ministry of Science and Innovation (PID2020-112587RB-I00).References
- O. Akeeb, L. Wang, W. G. Xie, R. Davis, M. Alkasrawi and S. Toan, J. Environ. Manage., 2022, 313, 115026, DOI:10.1016/j.jenvman.2022.115026.
- H. Li, J. Cleaner Prod., 2023, 414, 137679, DOI:10.1016/j.clepro.2023.137679.
- F. Nocito and A. Dibenedetto, Curr. Opin. Green Sustainable Chem., 2020, 21, 34–43, DOI:10.1016/j.cogsc.2019.10.002.
- K. Kula, J. J. Klemes, Y. Van Fan, P. S. Varbanov, G. K. Gaurav and R. Jasinski, Rev. Chem. Eng., 2023, 40, 457–480, DOI:10.1515/revce-2023-0010.
- H. Wang, Z. Xin and Y. H. Li, Top. Curr. Chem., 2017, 375, 49, DOI:10.1007/s41061-017-0137-4.
- M. Chehrazi and B. K. Moghadas, J. CO2 Util., 2022, 61, 102030, DOI:10.1016/j.jcou.2022.102030.
- H. Kolbe, Justus Liebigs Ann. Chem., 1860, 113, 125–127, DOI:10.1002/jlac.18601130120.
- T. Iijima and T. Yamaguchi, Appl. Catal., A, 2008, 345, 12–17, DOI:10.1016/j.apcata.2008.03.037.
- M. Tamura, M. Honda, Y. Nakagawa and K. Tomishig, J. Chem. Technol. Biotechnol., 2014, 89, 19–33, DOI:10.1002/jctb.4209.
- G. Centi and S. Perathoner, Catal. Today, 2009, 148, 191–205, DOI:10.1016/j.cattod.2009.07.075.
- L. Faba, P. Rapado and S. Ordóñez, Greenhouse Gases: Sci. Technol., 2023, 13, 227–244, DOI:10.1002/ghg.2175.
- A. S. Lindsey and H. Jeskey, Chem. Rev., 1957, 57, 583–620, DOI:10.1021/cr50016a001.
- G. Silvestri, S. Gambino, G. Filardo and A. Gulotta, Angew. Chem., Int. Ed., 1984, 23, 979–980, DOI:10.1002/anie.198409791.
- P.-L. Fabre, D. Chen, O. Reynes, C.-L. Nadia and V. J. Sartor, Electroanal. Chem., 2013, 711, 25–31, DOI:10.1016/j.elechem.2013.10.014.
- R. Matthessen, J. Fransaer, K. Binnemans and D. E. De Vos, Beilstein J. Org. Chem., 2014, 10, 2484–2500, DOI:10.3762/bjoc.10.260.
- H. Senboku, in Encyclopedia of Applied Chemistry, ed. G. Kreysa, K. Ota and R. Savinell, Springer, New York, 2014, pp. 469–474 Search PubMed.
- S. N. Steinmann, C. Michel, R. Schwiedernoch, M. Wu and P. J. Sautet, J. Catal., 2016, 343, 240–247, DOI:10.1016/j.jcata.2016.01.008.
- H. Semboku, K. Nagakura, T. Fukuhara and S. Hará, Tetrahedron, 2015, 71, 3850–3856, DOI:10.1016/j.tet.2015.04.020.
- H. Semboku and D. Katayama, Curr. Opin. Green Sustainable Chem., 2017, 3, 50–54, DOI:10.1016/j.cogsc.2016.10.003.
- H. Tateno, Y. Matsumura, K. Nakabayashi, H. Senboku and M. Atobe, RSC Adv., 2015, 5, 98721–98723, 10.1039/c5ra19289g.
- Z. Zhao, Y. Liu, S. Wang, S. Tang, D. Ma, Z. Zhu, C. Guo and Y. Qiu, Angew. Chem., Int. Ed., 2023, 62, e202214710, DOI:10.1002/anie.202214710.
- AHRQ (Agency of Healthcare Research and Quality), in Medical Expenditure Panel Survey (MEEPS) 2013-2020, 2022, Rockville, MD. https://clincalc.com/DrugStats/Drugs/Naproxen Search PubMed.
- D. Acetti, E. Brenna, G. Fronza and C. Fuganti, Talanta, 2008, 76, 651–655, DOI:10.10167/j.talanta.2008.04.009.
- J. G. Speight, Handbook of Industrial Hydrocarbon Processes, Gulf Professional Publishing, Houston TX, USA, 2019 Search PubMed.
- T. B. Poulsen and K. A. Jorgensen, Chem. Rev., 2008, 108, 2903–2915, DOI:10.1021/cr078372e.
- M. W. Ha and S.-M. Paek, Molecules, 2021, 26, 4792, DOI:10.3390/molecules26164792.
- D.-T. Yang, M. Zhu, Z. J. Schiffer, K. Williams, X. Song, X. Liu and K. Manthiram, ACS Catal., 2019, 9, 3764–4708, DOI:10.1021/acscatal.9b00818.
- B.-L. Chen, Z.-Y. Tu, H.-W. Zhu, W.-W. Sun, H. Wang and J.-X. Lu, Electrochim. Acta, 2014, 116, 475–483, DOI:10.1016/j.electacta.2013.11.001.
- X. Zhang, Z. H. Li, H. S. Chen, C. R. Shen, H. H. Wu and K. W. Dong, ChemSusChem, 2023, 16, e202300807, DOI:10.1002/cssc.202300807.
- Y. W. Wang, S. Y. Tang, G. Q. Yang, S. Y. Wang, D. K. Ma and Y. Qiu, Angew. Chem., Int. Ed., 2022, 61, e202207746, DOI:10.1002/anie.202207746.
- Y.-Y. Gui, W.-J. Zhou, J.-H. Ye and D.-G. Yu, ChemSusChem, 2017, 10, 1337–1340, DOI:10.1002/cssc.201700502.
- X. He, L.-Q. Qiu, W.-J. Wang, K.-H. Chen and N. He, Green Chem., 2020, 22, 7301–7320, 10.1039/d0gc02743j.
- S. Pradhan, S. Roy, B. Sahoo and I. Chatterjee, Chem.– Eur. J., 2021, 27, 2254–2269, DOI:10.1002/chem.202003685.
- Z. H. Wang, Y. Sun, L. Y. Shen, W. C. Yang, F. Meng and P. H. Li, Org. Chem. Front., 2022, 9, 853–873, 10.1039/d4qo01512e.
- J. Schneider, H. Jia, J. T. Muckerman and E. Fujita, Chem. Soc. Rev., 2012, 41, 2036–2051, 10.1039/c1cs15278e.
- M. D. Otero, B. Batanero and F. Barba, Tetrahedron Lett., 2006, 47, 2171–2173, DOI:10.1016/j.tetlet.2006.09.132.
- Y. Masuda, N. Ishida and M. Murakami, J. Am. Chem. Soc., 2015, 137, 14063–14066, DOI:10.1021/jacs.5b10032.
- N. Arora, P. Dhiman, S. Kumar, G. Sing and V. Monga, Bioorg. Chem., 2020, 79, 103668, DOI:10.1016/j.bioorg.2020.103668.
- DBMR (Data Bridge Market Research), Global anxiolytics market – Industry trends and forecast to 2009, 2022, available at: https://www.databridgemarketresearch.com/reports/global-benzodiazepine-drugs-market Search PubMed.
- N. Ishida, Y. Masuda, Y. Imamura, K. Yamazaki and M. Murakami, J. Am. Chem. Soc., 2019, 141, 19611–19615, DOI:10.1021/jacs.9b12529.
- H. Seo, M. H. Katcher and T. F. Jamison, Nat. Chem., 2017, 9, 453–456, DOI:10.1038/nchem.2690.
- Q. Y. Meng, T. E. Schirmer, A. L. Berger, K. Donabauer and B. König, J. Am. Chem. Soc., 2019, 141, 19611–19615, DOI:10.1021/jacs.9b12529.
- G. Burkhart and H. Hoberg, Angew. Chem., Int. Ed., 1982, 21, 76, DOI:10.1002/anie.198200762.
- J. Hou, A. Ea, W. Feng, J. H. Xu, Y. Zhao and J. Wu, J. Am. Chem. Soc., 2018, 140, 5257–5263, DOI:10.1021/jacs.8b01561.
- B. Wiltschi, T. Cernava, A. Dennig, M. Galindo Casas, M. Geier, S. Gruber, M. Haberbauers, P. Heidinger, E. Herrero Acero, R. Kratzer, C. Luley-Goedl, C. A. Müller, J. Pitzer, D. Ribitsch, M. Sauer, K. Schmölzer, W. Schnitzhofer, C. W. Sensen, J. Soh, K. Steiner, C. K. Winkler, M. Winkler and T. Wriessnegger, Biotechnol. Adv., 2020, 40, 107520, DOI:10.1016/j.biotechadv.2020.107520.
- C. K. Winkler, J. H. Schrittwieser and K. Kroutil, ACS Cent. Sci., 2021, 7, 55–71, DOI:10.1021/acscentsci.0c01496.
- S. Wu, R. Snajdrova, J. C. Moore, K. Baldenius and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2021, 60, 88–119, DOI:10.1002/anie.202006648.
- R. A. Sheldon, Adv. Synth. Catal., 2007, 349, 1289–1307, DOI:10.1002/adsc.200700082.
- I. Bernhardsgrutter, G. M. Stoffel, T. E. Miller and T. J. Erb, Curr. Opin. Biotechnol., 2021, 67, 80–87, DOI:10.1016/j.copbio.2021.01.003.
- S. Bierbaumer, M. Nattermann, I. Schulz, R. Zschoche, T. J. Erb, C. K. Winkler, M. Tinzl and S. M. Glueck, Chem. Rev., 2023, 123, 5702–5754, DOI:10.1021/acs.chemrev.2c00581.
- S. E. Payer, K. Faber and S. M. Glueck, Adv. Synth. Catal., 2019, 361, 2402–2420, DOI:10.1002/adsc.201900275.
- J. H. Ahn, Y.-S. Jang and S. Y. Lee, Curr. Opin. Biotechnol., 2016, 42, 54–66, DOI:10.1016/j.copbio.2016.02.034.
- A. Basso and S. Serban, Mol. Catal., 2019, 479, 110607, DOI:10.1016/j.mcat.2019.110607.
- M. Romero-Fernandez and F. Paradisi, Curr. Opin. Chem. Biol., 2020, 55, 1–8, DOI:10.1016/j.cbpa.2019.11.008.
- G. A. Aleku, G. W. Roberts, G. R. Titchiner and D. Leys, ChemSusChem, 2021, 14, 1781–1804, DOI:10.1002/cssc.202100159.
- K. Plasch, G. Hofer, W. Keller, S. Hay, D. J. Heyes, A. Dennig, S. M. Glueck and K. Faber, Green Chem., 2018, 20, 1754–1759, 10.1039/c8gc00008e.
- R. J. Spreitzer and M. E. Salvucci, Annu. Rev. Plant Biol., 2002, 53, 449–475, DOI:10.1146/annurev.arplant.53.100301.135233.
- T. Matsuda, R. Marukado, S. Koguchi, T. Nagasawa, M. Mukouyama, T. Harada and K. Nakamura, Tetrahedron Lett., 2008, 49, 6019–6020, DOI:10.1016/j.tetlet.2008.08.004.
- J. Martin, L. Eisoldt and A. Skerra, Nat. Catal., 2018, 1, 555–561, DOI:10.1038/s41929-018-0107-4.
- J. Ren, P. Yao, S. Yu, W. Dong, Q. Chen, J. Feng, Q. Wu and D. Zhu, ACS Catal., 2016, 6, 564–567, DOI:10.1021/acscatal.5b02529.
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742, DOI:10.1021/cr4002758.
- S. Dabral and T. Schaub, Adv. Synth. Catal., 2019, 361, 223–246, DOI:10.1002/adcs.201801215.
- R. Drault, Y. Snoussi, S. Paul, I. Itabaiana and R. Wojcieszak, ChemSusChem, 2020, 13, 5164–5172, DOI:10.1002/cssc.202001393.
- A. Rawat, S. Dhakla, P. Lama and T. K. Pal, J. CO2 Util., 2022, 59, 101939, DOI:10.1016/j.jcou.2022.101939.
- L. Faba, P. Rapado and S. Ordóñez, Greenhouse Gases: Sci. Technol., 2022, 13, 227–244, DOI:10.1002/ghg.2175.
- F. Behmagham, M. N. Abdullah, S. M. Saied, M. D. Azeez, R. R. Abbass, A. H. Adhab and E. Vessally, RSC Adv., 2023, 13, 32502–32517, 10.1039/d3ra04073a.
- T. Fujiharaj and Y. Tsuji, Front. Chem., 2019, 7, 430, DOI:10.3389/fchem.2019.00430.
- L. Wang, S. S. Que, Z. W. Ding and E. Vessally, RSC Adv., 2020, 10, 9103–9115, 10.1039/c9ra10755j.
- M. Hollering, B. Dutta and F. E. Kühn, Coord. Chem. Rev., 2016, 309, 51–67, DOI:10.1016/j.ccr.2015.10.002.
- S.-S. Yan, Q. Fu, L.-L. Liao, G.-Q. Sun, J.-H. Ye, L. Gong, Y.-Z. Bo-Xue, D.-G. Yu and D.-G. Coord, Chem. Rev., 2018, 374, 439–463, DOI:10.1016/j.ccr.2018.07.011.
- L. Veltri, R. Amuso, R. Mancuso and B. Gabriele, Molecules, 2022, 27, 262, DOI:10.3390/molecules27010262.
- S. Saini, P. K. Prajapati and S. L. Jain, Catal. Rev.: Sci. Eng., 2022, 64, 631–677, DOI:10.1080/01614940.2020.1831757.
- A. Kojcnovic, B. Likozar and M. Grilc, J. CO2 Util., 2022, 66, 102250, DOI:10.1016/j.jcou.2022.102250.
- J. M. V. Lauridsen, S. Y. Cho, H. Y. Baeand and J.-W. Lee, Organometallics, 2020, 39, 1652–1657, DOI:10.1021/acs.organomet.9b00838.
- R. Schmitt and J. für, Prakt. Chem., 1885, 31, 397 CrossRef.
- I. Stanescu and L. Achenie, Chem. Eng. Sci., 2006, 61, 6199–6212, DOI:10.1016/j.ces.2006.05.025.
- S. Marasée, US529182A, 1893.
- Z. Markovic, S. Markovic, N. Manojlovic and J. P. Simovic, J. Chem. Inf. Model., 2007, 47, 1520–1525, DOI:10.1021/ci700068b.
- O. Vechorkin, N. Hirt and X. Hu, Org. Lett., 2010, 12, 3567–3569, DOI:10.1021/ol101450u.
- K. F. Inamoto, N. Asano, Y. Nakamura, M. Yonemoto and Y. Kondo, Org. Lett., 2012, 14, 2622–2625, DOI:10.1021/ol300958c.
- W. J. Yoo, M. G. Capdevila, X. Du and S. Kobayashi, Org. Lett., 2012, 14, 5326–5329, DOI:10.1021/ol3025082.
- J. Luo, S. Preciado, P. Xie and I. Larrosa, Chem.–Eur. J., 2016, 22, 6798–6802, DOI:10.1002/chem.201601114.
- Y. Sadamitsu, A. Okumura, K. Saito and T. Yamada, Chem. Commun., 2019, 55, 9837–9840, 10.1039/c9cc04550c.
- A. I. Banerjee, G. R. Dick, T. Yoshino and M. W. Kanan, Nature, 2016, 531, 215–219, DOI:10.1038/nature17185.
- C. Friedel and J. M. Crafts, Compt. Rend, 1878, 86, 1368–1371 Search PubMed.
- G. A. Olah, B. Török, J. P. Joschek, I. Bucsi, P. M. Esteves, G. Rasul and G. K. S. Prakash, J. Am. Chem. Soc., 2002, 124, 11379–11391, DOI:10.1021/ja020787o.
- K. Nemoto, S. Onozawa, M. Konno, N. Morohashi and T. Hattori, Bull. Chem. Soc. Jpn., 2012, 85, 369–371, DOI:10.1246/bcsj.20110335.
- K. Nemoto, S. Tanaka, M. Konno, S. Onozawa, M. Chiba, Y. Tanaka, Y. Sasaki, R. Okubo and T. Hattori, Tetrahedron, 2016, 72, 734–745, DOI:10.1016/j.tet.2015.12.028.
- T. Ohishi, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2008, 47, 5792–5795, DOI:10.1002/anie.200801857.
- O. S. Nayal, J. Hong, Y. Yang and F. Mo, Org. Chem. Front., 2019, 6, 3673–3677, 10.1039/c9qo01023h.
- R. Fisher, J. Langer, A. Malassa, D. Walther, H. Gorls and G. Vaughan, Chem. Commun., 2006, 23, 2510–2512, 10.1039/b603540j.
- P. Lu, T. C. Boorman, A. M. Z. Slawin and I. Larrosa, J. Am. Chem. Soc., 2010, 132, 5580–5581, DOI:10.1021/ja101525w.
- S. Chaemchuen, N. A. Kabir, K. Zhou and F. Verpoort, Chem. Soc. Rev., 2013, 42, 9304–9332, 10.1039/c3cs60244c.
- D. De, T. K. Pal, S. Negoi, S. Senthilkumar, D. Das, S. S. Gupta and P. K. Bharadwaj, Chem. Eng. J., 2016, 22, 3387–3396, DOI:10.1002/chem.201504747.
- X. H. Liu, J. F. Ma, Z. Niu, G. M. Yang and P. Cheng, Angew. Chem., Int. Ed., 2015, 54, 988–991, DOI:10.1002/anie.201409103.
- R. A. Molla, K. Ghosh, B. Banerjee, M. A. Iqubal, S. K. Kundu, S. M. Islam and A. Bhaumik, J. Colloid Interface Sci., 2016, 477, 220–229, DOI:10.1016/j.jcis.2016.05.037.
- N. N. Zhu, X. H. Liu, T. Li, J. G. Ma, P. Cheng and G. M. Yang, Inorg. Chem., 2017, 56, 3414–3420, DOI:10.1021/acs.inorgchem.6b02855.
- M. Trivedia, K. Bhaskarana, A. Kumara, G. Singha, A. Kumarb and N. P. Rath, New J. Chem., 2016, 40, 3109–3118, 10.1039/c5nj02630j.
- Y. B. N. Tran, P. T. K. Nguyen, V. A. Dao and V. D. Le, New J. Chem., 2024, 48, 5300–5310, 10.1039/d4nj00076e.
- Z. Y. Gao, H. D. Wang, Y. C. Hu and J. M. Sun, J. Colloid Interface Sci., 2024, 671, 232–247, DOI:10.1016/j.jcis.2024.05.104.
- AMR (Allied Market Research), Salicylic Acid Market. Global Opportunity Analysis and Industry Forecast, 2022, pp. 2021–2030 Search PubMed.
- B. Yu, Z. F. Diao, C. X. Guo and L. N. He, J. CO2 Util., 2013, 1, 60–68, DOI:10.1016/j.jcou.2013.01.001.
- R. Alvarez, E. Carmona, A. Galindo, E. Gutierrez, J. M. Marin, A. Monge, M. L. Poveda, C. Ruiz and J. M. Savariault, Organometallics, 1989, 8, 2430–2439, DOI:10.1021/om00112a026.
- A. Galindo, A. Pastor, P. J. Perez and E. Carmona, Organometallics, 1993, 12, 4443–4451, DOI:10.1021/om00035a031.
- M. L. Lejkowski, R. Lindner, T. Kageyama, G. E. Bodizs, P. N. Plessow, I. B. Müller, A. Schäfer, F. Rominger, P. Hofmann, C. Futter, S. A. Schunk and M. Limbach, Chem. - Eur. J., 2012, 18, 14017–14025, DOI:10.1002/chem.201201757.
- S. Manzini, N. Huget, O. Trapp and T. Schaub, Eur. J. Org Chem., 2015, 7122–7130, DOI:10.1002/ejoc.201501113.
- S. Manzini, A. Cadu, A.-C. Schmidt, N. Huguet, O. Trapp, R. Paciello and T. Schaub, ChemCatChem, 2017, 9, 2269–2274, DOI:10.1002/cctc.201601150.
- M. D. Burkart, N. Harazi, C. L. Tway and E. L. Zeitler, ACS Catal., 2019, 9, 7937–7356, DOI:10.1021/acscatal.9b02113.
- D. Fabien, Y. Snoussi, I. Itabaiana and R. Wojcieszak, Sustainability, 2021, 13, 11278, DOI:10.3390/su132011278.
- N. V. Avantium, Atlantium to build FDCA flagship plant at CHemie Park Delfzijl, Netherlands, 2020, https://www.avantium.com/2020/avantium-to-build-fdca-flagship-plant-at-chemie-park-delfzijl-netherlands/ Search PubMed.
- G. R. Dick, A. D. Frankhouser, A. Banerjee and M. W. Kanan, Green Chem., 2017, 19, 2966–2972, 10.1039/c7gc01059a.
- Y. Wang, C.-Y. Guo, J. Shen, Y.-Q. Sun, Y.-X. Niu, P. Li, G. Liu and X.-Y. Wei, J. CO2 Util., 2021, 48, 101524, DOI:10.1016/j.jcou.2021.101524.
- X. Han, Y. Wang, G. Liu, M. Wang, C. Guo and J. Shen, J. CO2 Util., 2023, 75, 102572, DOI:10.1016/j.jcou.2023.102572.
- X. Han, C. Guo, Y. Wang, G. Liu and J. Shen, J. Mol. Struct., 2024, 1295, 136674, DOI:10.1016/j.molstruc.2023.136674.
- H. Zhou, H. Xu, X. Wang and Y. Liu, Green Chem., 2019, 21, 2966–2972, 10.1039/c9gc00869a.
- Statista Research Department, Global furfural market volume 2015-2030, available at: https://www.statista.com/statistics/1310459/furfural-market-volume-worldwide/ Search PubMed.
- Statista Research Department, Global production capacity of polyethylene terephthalate 2014-2024, 2023, available at: https://www.statista.com/statistics/242764/global-polyethylene-terephthalate-production-capacity/.
- G. H. C. Dubbink, T. R. J. Geverink, B. Haar, H. W. Koets, A. Kumar, H. Berg, A. G. J. Ham and J.-P. Lange, Biofuels, Bioprod. Biorefin., 2021, 15, 1021–1030, DOI:10.1002/bbb.2204.
- A. Dubey and A. Arora, J. Cleaner Prod., 2022, 373, 133932, DOI:10.1016/j.jclepro.2022.133932.
- E. E. Sanz-Pérez, C. R. Murdock, S. A. Didas and C. W. Jones, Chem. Rev., 2016, 116, 11840–11876, DOI:10.1021/acs.chemrev.6b00173.
| This journal is © The Royal Society of Chemistry 2024 |