
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolvation and interfacial chemistry in ionic liquid based electrolytes toward rechargeable lithium-metal batteries
Haifeng
Tu†
ab,
Keyang
Peng†
ab,
Jiangyan
Xue†
ab,
Jingjing
Xu
 *ab,
Jiawei
Zhao
ab,
Yuyue
Guo
ab,
Suwan
Lu
ab,
Zhicheng
Wang
cd,
Liquan
Chen
cd,
Hong
Li
cd and
Xiaodong
Wu
*abc
*ab,
Jiawei
Zhao
ab,
Yuyue
Guo
ab,
Suwan
Lu
ab,
Zhicheng
Wang
cd,
Liquan
Chen
cd,
Hong
Li
cd and
Xiaodong
Wu
*abc
aSchool of Nano-Tech and Nano-Bionics, University of Science and Technology of China, Hefei 230026, China. E-mail: jjxu2011@sinano.ac.cn; xdwu2011@sinano.ac.cn
bi-Lab, Suzhou Institute of Nano-Tech and Nano-Bionics, Chinese Academy of Sciences, Suzhou 215123, China
cTianmu Lake Institute of Advanced Energy Storage Technologies Co., Ltd, Liyang 213300, China
dInstitute of Physics, Chinese Academy of Sciences, Beijing 100190, China
First published on 6th November 2024
Abstract
Rechargeable lithium metal batteries (LMBs) are a highly promising technology for high-energy-density storage systems due to the low electrochemical potential and high theoretical capacity of the lithium metal anode. The electrolyte plays a pivotal role among the critical components of LMBs. However, traditional organic electrolytes pose significant safety risks and shorten the battery life due to their electrochemical instability, volatility, and flammability. Alternatively, ionic liquids (ILs), composed of anions and cations, are room-temperature molten salts characterized by ultra-low volatility, high ionic conductivity, excellent thermal stability, low flammability, and wide electrochemical windows. Based on these properties, liquid IL electrolytes (ILEs) and polymeric IL electrolytes (PILEs) have shown immense potential in enhancing battery cycle stability, energy density, lifespan, and safety. This review aims to comprehensively explore and summarize recent applications of ILEs and PILEs in LMBs, including their use as liquid and solid-state electrolytes, as well as ILs serving as film-forming additives, interfacial wetting agents, and pretreatment reagents. Additionally, the review delves into the solvation structures of Li+ ions within different IL-based electrolytes and the resulting interfacial chemical characteristics. Finally, based on literature reports and our previous work, we identify current challenges and propose solutions for the future application of IL-based electrolytes in LMBs.
 Haifeng Tu | Haifeng Tu received his bachelors degree from the China University of Mining and Technology in 2020. He is currently a PhD candidate at the University of Science and Technology of China at the School of Nanotech and Nanobionics. His research is focused on lithium metal batteries, ionic liquid electrolytes and the design of electrolyte–electrode interfaces. |
 Keyang Peng | Keyang Peng is currently a graduate student at the School of Nano-Tech and Nano-Bionics, University of Science and Technology of China. His research interest mainly focuses on the structural design of high-performance polymer electrolytes for safe lithium metal batteries. |
 Jiangyan Xue | Jiangyan Xue obtained her PhD degree in 2022 from Soochow University. Now she is a postdoctoral fellow at Suzhou Institute of Nano-Tech and Nano-Bionics (SINANO), Chinese Academy of Sciences. Her research interest includes the development of functional nanomaterials for energy storage and transformation (including water splitting, lithium/sodium ion batteries, lithium/sodium–sulfur batteries). |
 Jingjing Xu | Jingjing Xu is currently a professor at the College of Materials Science and Engineering, Hohai University. Her research is focused on the fundamental study of electrolytes/interfaces and key materials for novel energy storage batteries, aiming to address issues such as safety performance and cycle life in the practical application of lithium secondary batteries. |
 Xiaodong Wu | Xiaodong Wu is a researcher in Suzhou Institute of Nano-Tech and Nano-Bionics, Chinese Academy of Sciences. As the project leader, he has undertaken national key research and development projects, pilot projects of the Chinese Academy of Sciences, and general projects of the National Natural Science Foundation of China. His research interests include high-capacity cathode and anode materials for lithium-ion batteries, electrolyte/electrode interfaces and Li–S batteries. |
1 Introduction
Thirty years ago, Sony commercialized the first lithium-ion batteries (LIBs) by using LiCoO2 (LCO) as the cathode, graphite (Gr) as the anode, and an ethylene carbonate (EC)-based electrolyte.1 With continuous technological advancements and iterations, LIBs have been widely used in modern electronic devices such as smartphones and laptops. However, the energy density of current LIBs is approaching its theoretical value due to the limited specific capacity of the Gr anode (374 mA h g−1), which cannot satisfy the market requirement.2–4 Alternatively, lithium metal batteries (LMBs), which utilize lithium metal as the anode (LMA), can theoretically provide a much higher specific capacity of up to 3860 mA h g−1, far exceeding that of the traditional Gr anode.5–7 Additionally, LMBs can be paired with high-capacity cathode materials to further boost the energy density, including nickel-rich, layered transition metal oxides (LiNixMnyCozO2, NCM), cobalt-free Li1.2Ni0.2Mn0.6O2 (LNM), spinel LiNi0.5Mn1.5O4 (LNMO), and sulfur (S).8–11However, traditional carbonate-based and ether-based electrolytes undergo uncontrollable side reactions with LMA, leading to anode pulverization and electrolyte depletion.12 Moreover, the electrode/electrolyte interfaces (EEIs), including the solid electrolyte interphase (SEI) and the cathode electrolyte interphase (CEI), are chemically unstable and mechanically fragile.13,14 The organic-rich SEI fails to suppress dendrite growth and continuous Li consumption, resulting in poor cycling stability.15 Moreover, the unstable CEI cannot effectively inhibit the catalytic decomposition of the electrolyte by the cathode material. For example, high-nickel NCM cathodes undergo irreversible phase transitions and metal ion dissolution during cycling, limiting the application of high-voltage LMBs.16 Among numerous efforts in the literature, innovative electrolyte designs offer a cost-effective and convenient solution for the current development of LMBs.17–19
Ionic liquids (ILs) are salts with melting points below 100 °C, composed of organic cations and organic or inorganic anions.20 Paul Walden first discovered and reported ethylammonium nitrate in 1914, marking the beginning of a surge in IL research.21 In recent years, the number of studies on ILs as electrolyte materials (ILEs) has significantly increased, as evidenced by the growing number of related articles published in academic journals (Fig. 1a), reflecting researchers' strong interest in this field. In general, as shown in Fig. 1b, several key parameters should be considered using ILs as electrolytes for rechargeable LMBs. They have low melting point, low viscosity, wide electrochemical window, low volatility and non-flammability, and high ionic conductivity, which are essential for advancing the cycle performance for LMBs.
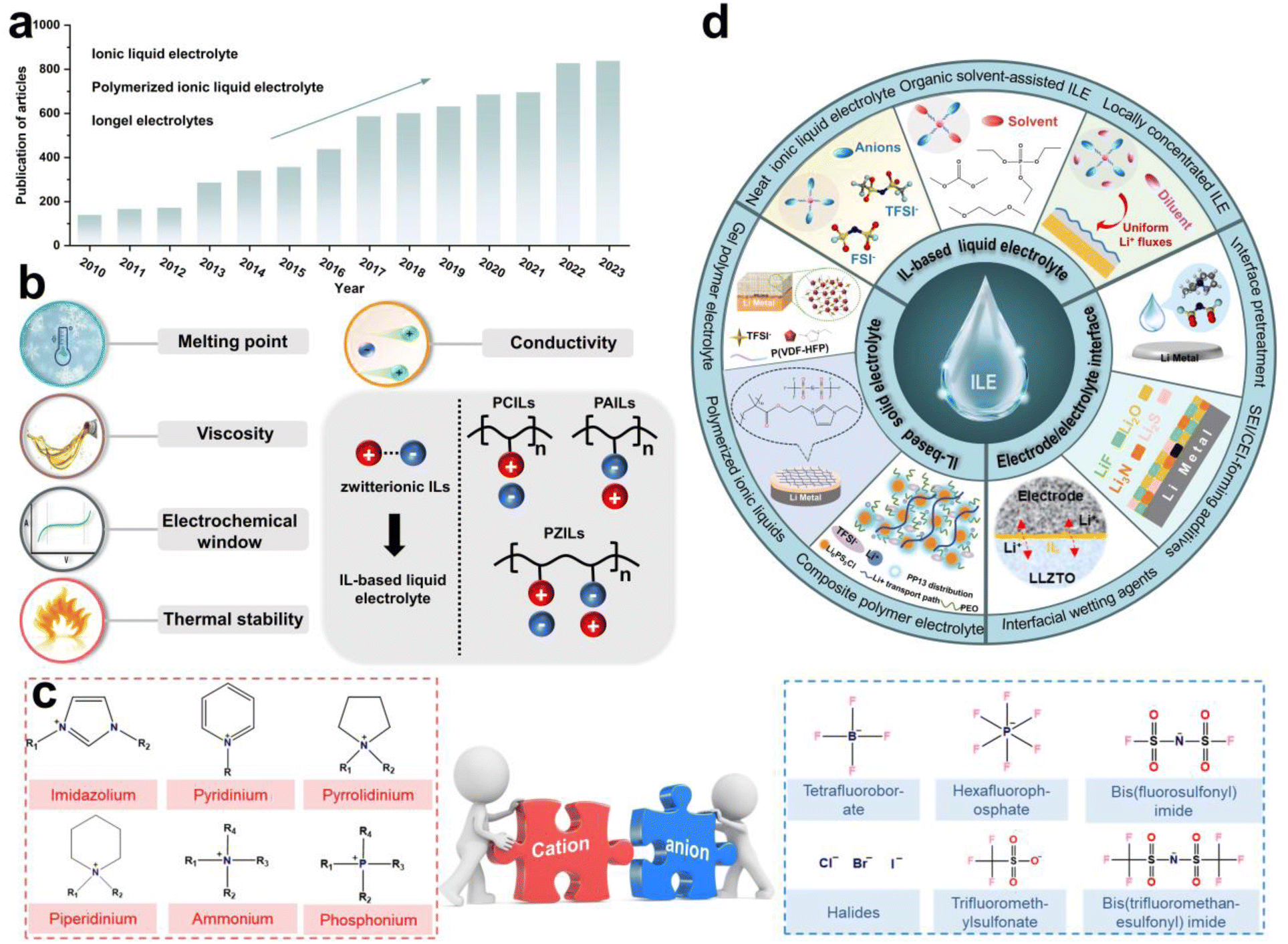 | ||
| Fig. 1 (a) Number of publications of ionic liquids applied in batteries. Data were obtained from Web of Science on the 30th May, 2024, via keywords such as ionic liquid electrolyte, polymerized ionic liquid electrolyte and iongel electrolyte. (b) The key parameters for evaluating ionic liquids as electrolytes. (c) Chemical structures of various ILs. (d) Summary of ionic liquid application. | ||
Based on molecular characteristics, ILs can be divided into small molecule zwitterionic ILs and poly(ionic liquids) (PILs).22 PILs are polymeric materials formed by the polymerization of IL monomers,23 combining the properties of polymers and ILs, typically exhibiting higher mechanical strength and stability while retaining the excellent ionic conductivity and chemical stability. The structure of PILs usually includes a monomer with positive and negative ions and a polymer backbone, which can further be categorized into polycation ILs (PCILs), polyanion ILs (PAILs), and poly(zwitterion) ILs (PZILs).24 In addition, by adjusting the type of monomers and polymerization methods, PILs with different physicochemical properties can be designed for solid-state LMBs. Fig. 1c presents common organic cations and inorganic or organic anions, including imidazolium, pyridinium, tetrafluoroborate, hexafluorophosphate and so on. Several reviews have summarized the application of ILs in batteries. For instance, Wu et al. reviewed the progress of IL-based electrolytes in non-aqueous and aqueous metal batteries, and Zhou et al. reported on the composition, classification, and application of common IL electrolytes in different valence state ion batteries.25,26 However, a systematic review focusing on the application of ILs in LMBs from the perspective of electrolyte solvation chemistry and the interfacial chemistry formed by ILs is still lacking.
Herein, based on our previous work and the recent reported literature on IL-based electrolytes in LMBs, we have systematically summarized the IL-based liquid and solid electrolytes, as well as the use of ILs as additives or wetting agents for EEI treatment (Fig. 1d). The solvation structure of Li+ ions and the unique interfacial chemical components formed are discussed in detail. We propose that the application of ILs and PILs in LMBs is still in its early stages, with a lack of fundamental understanding of the solvation chemistry and advanced in situ characterization techniques for interfacial chemical components. Furthermore, there remains significant potential for the molecular design of ILs. For example, machine learning and artificial intelligence (AI) big data screening can efficiently design target IL molecules. Finally, we propose a novel concept for the application of PILs in solid electrolytes-the solidification of electrodes, which expands the imaginative scope for designing solid-state electrolytes and non-flammable liquid ILEs. This timely and critical review provides an overview of the latest design strategies for ILs in LMB applications, offering insights for the design of next-generation ILEs based on solvation chemistry and interfacial chemistry modulation.
2 IL-based liquid electrolytes
Traditional carbonate- or ether-based electrolytes suffer from uncontrolled side reactions with electrodes, forming chemically unstable SEI and CEI, which in turn results in dendrite growth, dead Li, and continuous Li consumption.27,28 Alternatively, ILs based on cations such as imidazolium or pyrrolidinium exhibit excellent film-forming ability and a wide electrochemical window, making them suitable for LMBs operating under high voltages or other special environmental conditions.29 ILEs also possess low volatility and non-flammability, which can significantly enhance the battery safety.30 Compared to IL-based solid-state electrolytes, IL-based liquid electrolytes have numerous advantages in LMBs: (i) high ionic conductivity to facilitate more efficient Li+ ion transport, and enhance the rate performance of the battery (Fig. 2a); (ii) better wettability to reduce interfacial resistance, and improve overall battery performance (Fig. 2b); (iii) well-established preparation to enable easy large-scale production (Fig. 2c); (iv) significant flexibility in design and application to tune their performance (Fig. 2d). In summary, based on the types of electrolyte solvents, liquid ILEs can be classified into neat ILEs, organic solvent-assisted ILEs, and locally concentrated ILEs. | ||
| Fig. 2 The advantages of liquid ILEs. (a) Schematic of rapid Li+ ion transport in liquid ILEs. The river represents the liquid IL, and the sailboats represent Li+ ions. Schematic illustrations of liquid electrolytes (b) wetting electrodes, (c) large-scale production, and (d) design flexibility. | ||
2.1 Neat ionic liquid electrolytes
The advancement of neat ILEs can be categorized into two key areas: the regulation of solvation structures and the design of organic cations. A common approach to solvation structure regulation involves using dual-anion ILEs. In such systems, the competitive interactions between TFSI− and FSI− anions slow the reaction kinetics of FSI−, leading to the formation of a thin and dense SEI layer. Compared to single-anion ILEs, dual-anion ILEs show superior compatibility with lithium metal, resulting in more uniform, compact, and dendrite-free Li deposition. In high-concentration ILEs, the proportion of contact ion pairs (CIPs) and aggregates (AGGs) in the solvation structure increases, creating an anion-rich environment. This enhances the reduction of anions, increasing the LiF content at the interface and stabilizing both the anode and cathode. However, the redox activity of organic cations can lead to higher organic content at the EEI, reducing the interface stability and increasing the impedance. To address this, redesigning organic cations can significantly improve the performance of neat ILEs by introducing ether oxygen atoms into side chains, extending the chain length, and fluorinating the cations. This section provides a comprehensive overview of the mechanisms behind the design strategies for neat ILEs.Wu et al. reported an ionic liquid electrolyte containing both FSI− and TFSI− anions, denoted as 0.8Pyr14FSI–0.2LiTFSI (Fig. 3a).31 The synergistic effect of FSI− and TFSI− enabled NCM88 (LiNi0.88Co0.09Mn0.03O2) to exhibit excellent long-cycle stability. In this electrolyte, Li/NCM88 batteries showed 88% capacity retention after 1000 cycles with almost no voltage decay, and the average coulombic efficiency exceeded 99.94%. From the cross-section images of the cycled cathode, commercial electrolytes led to various cracks and particle fractures in electrodes at high states of charge (SOC) due to stress from expansion and the H2–H3 phase transition after repeated charge–discharge cycles. In contrast, the NCM88 cathode cycled in the ILE showed no severe cracking indicating that the CEI film is more stable. Neat ILEs can also form a stable SEI on LMA. Using reactive molecular dynamics simulations (MD), the atomic details of SEI formation at the LMA and ILE interface can be observed. Yang et al. developed an atomic model of SEI formation at the interface between LMA and the [BMIM][TFSI] (bis(trifluoromethylsulfonyl)-imide/1-butyl-3-methylimidazolium) ILE (Fig. 3b).32 Reactive force field (ReaxFF) molecular dynamics simulations revealed significant differences in the decomposition rates and extent of TFSI− anions and BMIM+ cations, with TFSI− decomposition products being predominant. At 300 K, the SEI exhibited a bilayer structure with a 2.5 nm ordered inorganic layer near the LMA and a 7.5 nm porous organic layer near the ILE. The excellent electrochemical stability of ILEs in batteries is attributed not only to the formation of a stable EEI but also to their intrinsically wide voltage window. Qi et al. systematically synthesized piperidinium- and pyrrolidinium-based ILs with various substituents, including cyanomethyl, benzyl, butyl, hexyl, and octyl, and investigated their electrochemical stability.33 As shown in Fig. 3c, introducing electron-donating substituents into the side chains can enhance the oxidative stability of ILs. For example, [C6Py][TFSI] has a decomposition voltage of 5.2 V, higher than that of [CCNPy][TFSI] (3.81 V). The order with respect to decomposition voltage for piperidinium-based ILs is similar to that of pyrrolidinium-based ILs, with [C6Pip][TFSI] exhibiting the highest decomposition voltage of 5.09 V. When the substituent changes from a hexyl group to an octyl group, the change of voltage stability is negligible due to the reduced electron-donating ability with the increased length of the carbon chain. Unlike the work contributed by Qi et al., Warrington et al. functionalized the side chains with ether groups to enhance the fluidity of ILs and reduce Li+–anion interactions in highly concentrated ionic liquid electrolytes (HCILEs), facilitating faster Li+ ion transport.34 As shown in Fig. 3d, three novel ILEs were compared, specifically [N111,1O1][FSI] (1-methoxymethyl-1,1,1-tri-methylammonium), [C1O1mpip][FSI] (N-methoxymethyl-N-methylpiperidinium), and [C1O1mmor][FSI] (N-methoxymethyl-N-methylmorpholinium). It was found that (LiFSI)0.5([N111,1O1][FSI])0.5 exhibited the lowest viscosity and the highest ionic conductivity. This work demonstrates the flexibility in the design and synthesis of ILEs.
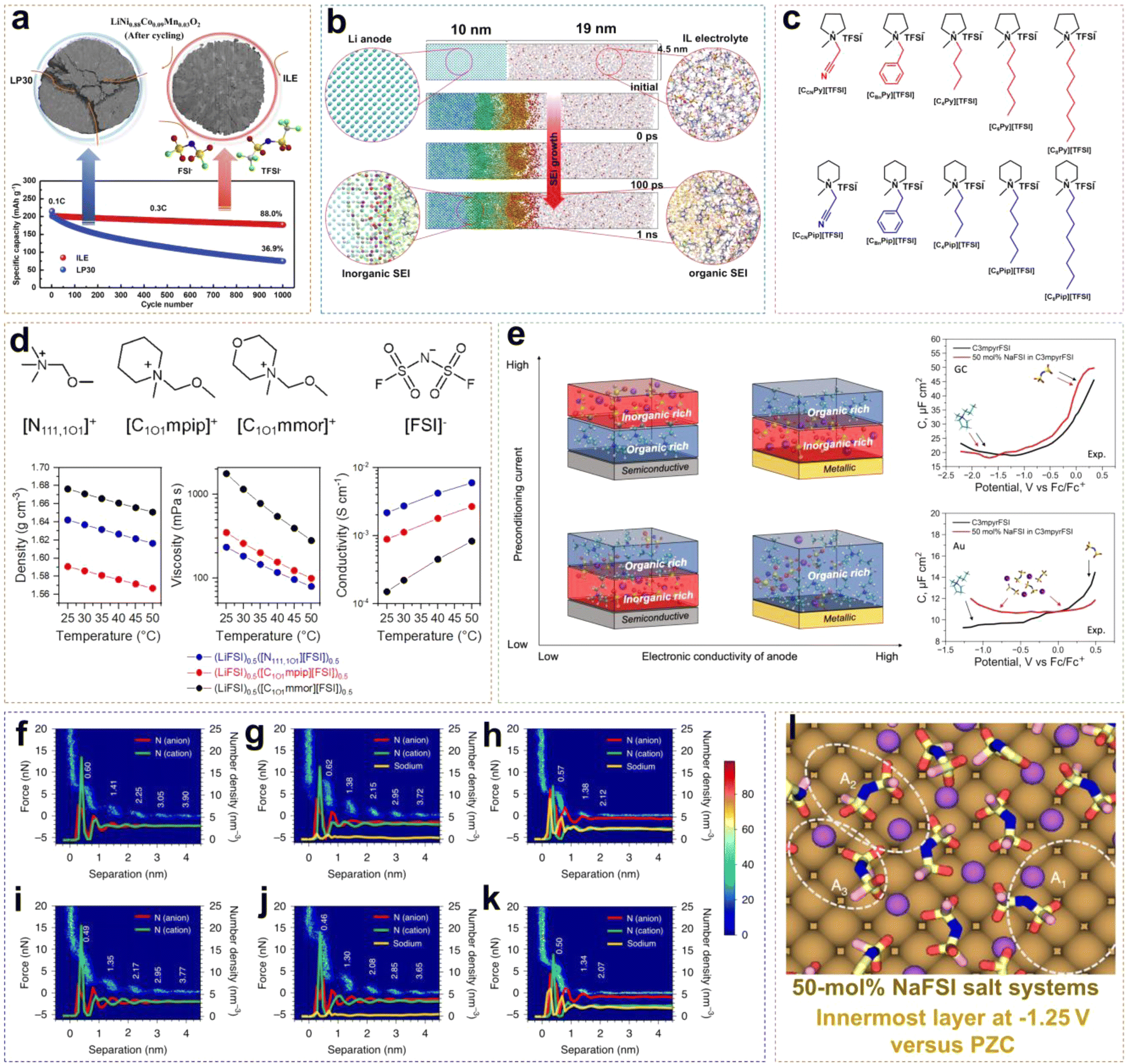 | ||
| Fig. 3 (a) Long-term cycling performance of an exemplary Li/NCM88 cell using ILE and LP30 electrolyte. Reprinted from ref. 31 with permission. Copyright 2021, Elsevier. (b) The formation of the SEI layer from the ReaxFF MD simulation at 300 K using a [BMIM] [TFSI] IL electrolyte. Reprinted from ref. 32 with permission. Copyright 2022, Wiley. (c) Chemical structures of pyrrolidinium- and piperidinium-based ILs. Reprinted from ref. 33 with permission. Copyright 2020, American Chemical Society. (d) Chemical structures and corresponding density, viscosity, and conductivity of different ether-functionalized ILEs. Reprinted from ref. 34 with permission. Copyright 2023, Elsevier. (e) Schematic relationships between the SEI composition, applied preconditioning current, and dielectric nature of the anode material. Reprinted from ref. 35 with permission. Copyright 2023, The Royal Society of Chemistry. AFM force–distance two-dimensional histograms and ion number density profiles of FSI−, C3mpyr+ and Na+ from MD simulations at OCP (AFM) and PZC (MD) (f–h) and OCP −0.5 V (AFM) and −0.5 V versus PZC (MD) (i–k) for C3mpyrFSI IL with different NaFSI concentrations of 0 mol% (f, and i), 10 mol% (g, and j), and 50 mol% (h, and k). (l) Typical Na–FSI coordination complexes in the innermost layer at −1.25 V versus PZC in the 50 mol% salt systems. Reprinted from ref. 36 with permission. Copyright 2020, Springer Nature. | ||
Forsyth et al. found that the composition of the EEI in ILEs is significantly influenced by the dielectric properties of the electrode material.35 As shown in Fig. 3e, ILEs form an organic–inorganic double layer SEI on semiconductor electrodes. At low current densities, the inner and outer layers are composed of inorganic and organic components, respectively; at high current densities, this order is reversed. This is attributed to the increased surface negative charge on the semiconductor electrode with rising current, which increases the number of IL cations. The metallic electrode surface, characterized by high electron polarizability and strong dispersion forces, requires higher current densities to form an anion-derived interface. Differential capacitance measurements, obtained by recording electrochemical impedance spectra (EIS) at various applied potentials in the non-faradaic region, reveal that on semiconductive glassy carbon (GC) electrodes, the C–E (differential capacitance–electrical potential) curve of pure C3mpyrFSI exhibits a descending shape from positive to negative potential regions. This behavior arises from the tighter packing of [FSI]− anions compared to cations at the interface, resulting in increased charge accumulation and higher capacitance. However, the shape of the C–E curve on Au electrodes is significantly influenced by the dominance of the NaxFSIy species. This also indicates a substantial difference in the composition of the electric double layer (EDL) across different electrode surfaces. Rakov et al. investigated the nanostructure of IL electrode surfaces at NaFSI concentrations and open circuit potential (OCP) using atomic force microscopy (AFM) and MD simulations.36 They found that at a distance of 4.5 nm from the electrode, there exists multiple nanostructured layers that decay into the bulk phase (Fig. 3f). By comparison, adding 10 mol% NaFSI salt enhances the rupture force required to disrupt each layer due to increased ion–ion association (Fig. 3g). In a system with 50 mol% NaFSI, the force significantly decreases after only two to three steps, indicating a change in the physical dimensions of interfacial ion packing in the HCILE system (Fig. 3h). When a potential of −0.5 V is applied to the working electrode, the impact on the number of ion layers becomes negligible (Fig. 3i–k). The ion number density profiles obtained from MD simulations align with AFM measurements, showing the formation of molten salt-like Nax(FSI)y ion aggregates at the electrode interface in HCILE systems (Fig. 3l). Rakov et al.'s work clearly elucidates the nanoscale solvation structure at charged alkali metal anodes in HCILE systems, shedding light on the mechanism by which these systems improve the cycling stability of alkali metal anodes.
Due to the positive charge dispersion around the imidazolium ring, which reduces electrostatic interactions between ion pairs, ILs based on Emim+ cations typically exhibit lower viscosity. Sun et al. developed an EM-5Li-Na ILE (5 M LiFSI/0.16 M NaTFSI/EmimFSI) with high ionic conductivity of 2.6 mS cm−1.37 The prepared Li/LCO cell exhibited great electrochemical reversibility and long-cycle stability, because the formation of an F-rich SEI effectively suppresses the reduction of Emim+ cations. Additionally, Na+ ions may provide positively charged electrostatic shielding around the initial Li dendrite growth. Li/LCO cell retained about 81% of initial capacity after 1200 cycles at a high rate of 0.7C, with an average coulombic efficiency (CE) of approximately 99.9% (Fig. 4a). Liang et al. combined the IL 4.5 FSI-TFSI ILE (4.5 M LiFSI in Py13FSI + 1 wt% LiTFSI) with the low loading anode material (Cu@Si-PAN), and the Cu@Si-PAN/LNMO battery exhibited a specific capacity of 120 mA h g−1 and retained 80% capacity after 120 cycles (Fig. 4b).38 Liu et al. synthesized PMpyrfFSI (1-methyl-1-propyl-3-fluoropyrrolidinium) via a one-step quaternization method, which can enter the inner-Helmholtz layer of electrodes with high Fermi levels, forming a stable EEI.39 As shown in Fig. 4c, compared to non-fluorinated PMpyr+, PMpyrf+ lowered the energy levels of the HOMO (Highest Occupied Molecular Orbital) and LUMO (Lowest Unoccupied Molecular Orbital), indicating that fluorinated cations are more resistant to oxidation and more readily reduced. The density functional theory (DFT) calculations showed that the C–F bond in PMpyrf+ is more prone to reductive decomposition than the C–H bond in PMpyr+, resulting in dendrite-free Li deposition. Additionally, on the NCM622 (LiNi0.6Co0.2Mn0.2O2) cathode side, the deprotonation and ring-opening potential of PMpyrf+ was −0.49 eV, compared to −0.03 eV for PMpyr+, indicating that the fluorinated cation is more oxidation-resistant, forming a stable and thin CEI that effectively suppresses electrolyte/cathode interfacial oxidation reactions (Fig. 4d).
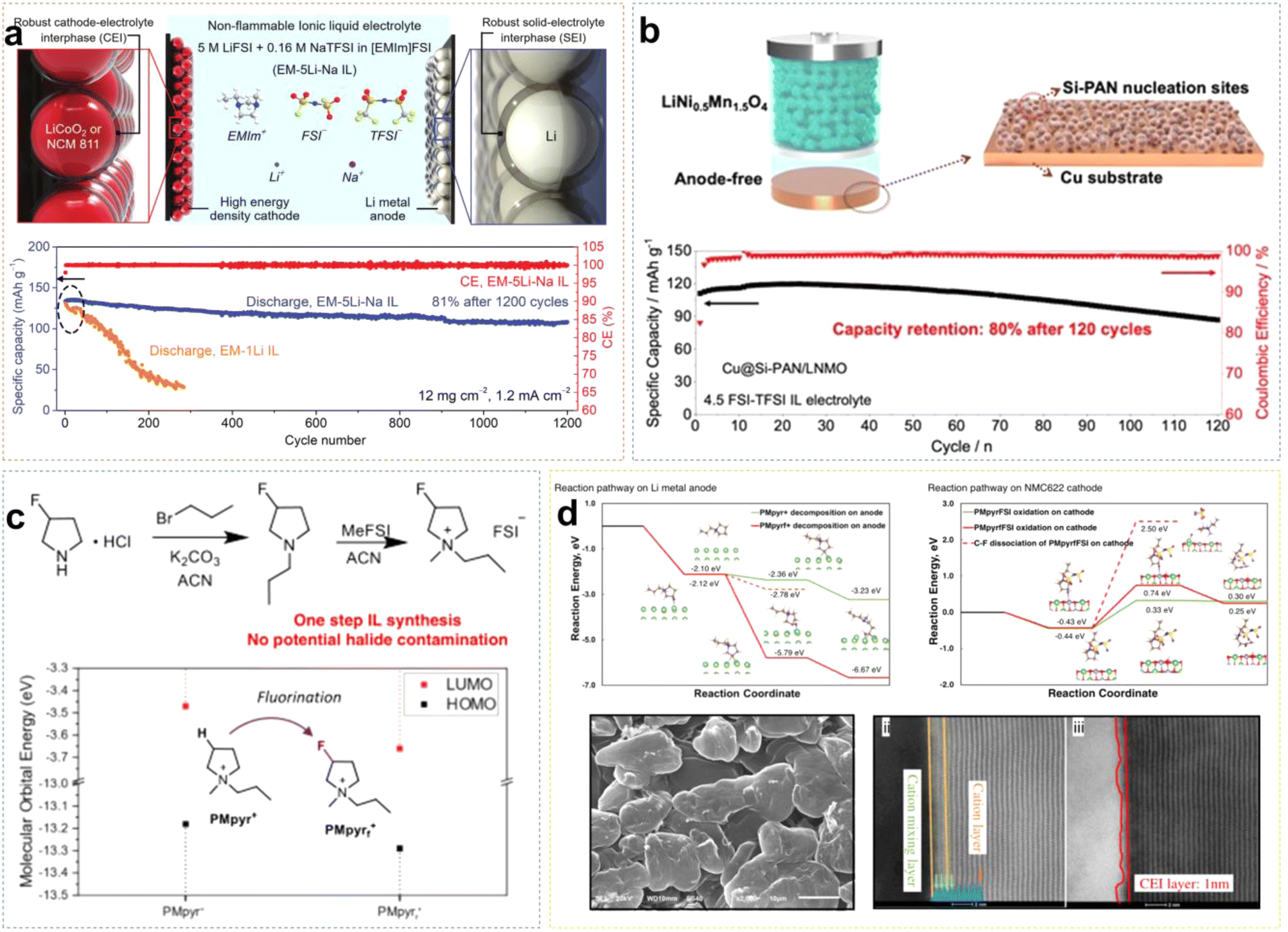 | ||
| Fig. 4 (a) Schematic diagram of robust EEI formed by EM-5Li-Na ILE and corresponding cycling performance in Li/LCO cells with the mass loading of 12 mg cm−2. Reprinted from ref. 37 with permission. Copyright 2020, Wiley. (b) Schematic illustrations of the high-voltage LNMO anode-free batteries using the Cu@Si-PAN current collector, and corresponding cycling performance of the LNMO anode-free batteries with 4.5 FSI-TFSI ILE. Reprinted from ref. 38 with permission. Copyright 2022, Wiley. (c) Synthesis route for PMpyrfFSI and comparison of HOMO/LUMO energy levels for PMpyrf+ and PMpyr+. (d) PMpyrf+ and PMpyr+ reduction and oxidation pathway on Li metal and NMC622 cathode, respectively; the morphology of plated Li on Cu foil and HAADF-STEM analysis of cycled NMC622 cathodes. Reprinted from ref. 39 with permission. Copyright 2023, Springer Nature. | ||
2.2 Organic solvent-assisted ILE
Neat ILEs in LMBs possess some inherent advantages compared with traditional electrolytes, but several challenges remain. (i) Neat ILEs exhibit relatively low ionic conductivity at room temperature, limiting their applicability across a wide temperature range to a certain extent. (ii) Li+ ions in neat ILEs with high viscosity show slow migration and poor rate performance. (iii) The reduction of IL organic cations can still form a potentially unstable EEI. (iv) The high preparation cost of ILs poses a challenge for large-scale commercial applications. Mixing ILs with organic solvents is a promising solution. For example, adding ester and ether solvents to ILEs can improve ionic conductivity, reduce viscosity and cost, while retaining the advantages of ionic liquids (Fig. 5a).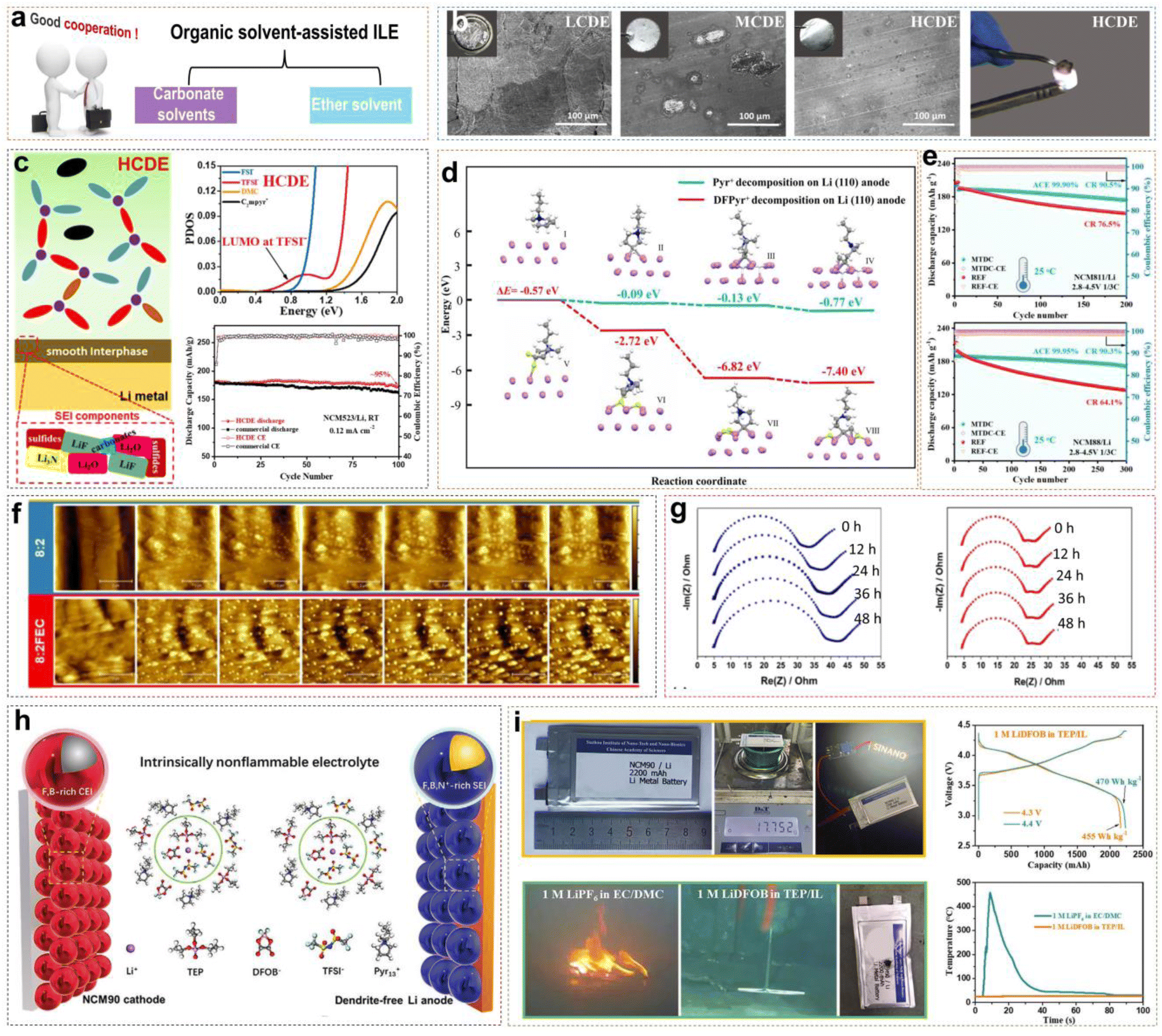 | ||
Fig. 5 (a) Classification of organic solvents for organic solvent-assisted ILEs. (b) Corrosion of Al foil in different electrolytes and combustion tests of HCDE. (c) Schematic of the SEI formed on the LMA surface and PDOS obtained by DFT calculations with HCDE; corresponding cycling performance of Li/NCM523 LMBs. Reprinted from ref. 40 with permission. Copyright 2020, Elsevier. (d) Reduction pathway of difluorinated IL on Li metal. (e) Cycling stability and coulombic efficiency of Li/NMC811 and Li/NMC88 cells under 2.8–4.5 V cutoff voltage at 25 °C. Reprinted from ref. 41 with permission. Copyright 2023, Wiley. (f) In situ AFM mappings (area 5 × 5 μm2) of lithium plated onto nickel current collectors at 80 μA cm−2 employing 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, and 8:2FEC as the electrolytes. (g) EIS performed on LiNi/Li cells using 8:2 and 8:2FEC during a two-day rest period with spectra recorded every 12 h. Reprinted from ref. 42 with permission. Copyright 2023, American Chemical Society. (h) Schematic diagram of the Li/NCM90 LMBs and solvation structure with the intrinsically non-flammable electrolyte. (i) Nail penetration test, charge/discharge curves and temperature versus time of the Li/NCM90 pouch cells. Reprinted from ref. 43 with permission. Copyright 2023, Wiley. :2, and 8:2FEC as the electrolytes. (g) EIS performed on LiNi/Li cells using 8:2 and 8:2FEC during a two-day rest period with spectra recorded every 12 h. Reprinted from ref. 42 with permission. Copyright 2023, American Chemical Society. (h) Schematic diagram of the Li/NCM90 LMBs and solvation structure with the intrinsically non-flammable electrolyte. (i) Nail penetration test, charge/discharge curves and temperature versus time of the Li/NCM90 pouch cells. Reprinted from ref. 43 with permission. Copyright 2023, Wiley. | ||
In our previous work, we explored the applicability of organic solvent-assisted ILEs in high-voltage LMBs at room temperature using N-ethyl-N-methyl-pyrrolidinium bis(fluorosulfonyl)imide ([C2mpyr][FSI]) IL as the solvent and dimethyl carbonate (DMC) as the co-solvent.40 In a low-concentration dual-anion electrolyte (LCDE) and moderate-concentration dual-anion electrolyte (MCDE), free DMC molecules and FSI− anions coordinate with Al3+, dissolving the Al2O3 layer and continuously corroding the Al metal (Fig. 5b). However, in highly concentrated dual-anion electrolyte (HCDE), DMC and FSI− anions coordinate with Li+, which can prevent the corrosion of the Al current collector. Compared to LCDE, HCDE exhibits excellent non-flammability. In addition, with the increase of LiTFSI salt concentration, free DMC solvent molecules coordinate with Li+ ions. This coordination increases the energy required to break the solvation structure, significantly reducing the volatility of the electrolyte and enhancing thermal stability. DFT calculations reveal that in the HCDE system, LUMO energy levels are as follows: TFSI− < FSI− < DMC < C2mpyr+. Consequently, the SEI is primarily formed from the reductive decomposition of TFSI− and FSI− anions, and the main products of Li3N, LiF, and Li2O. A 4.5 V Li/NCM523 (LiNi0.5Co0.2Mn0.3O2) cell retains 95% of the initial capacity after 100 cycles (Fig. 5c).
Li et al. presented the synthesis of DFPyrTFSI (3,3-difluoro-N-methyl-N-propylpyrrolidinium bis(fluorosulfonyl)imide) IL, by using gem-difluorinated pyrrolidinium as a precursor and through a series of reactions, including the Eschweiler–Clarke reaction, quaternization, and anion-exchange (Fig. 5d).41 DMC was mixed with DFPyrTFSI (mass ratio of 1:1) as a co-solvent, LiTFSI and LiDFOB (8:1 by mole) were added afterward to form a DMC-assisted ILE (2 M LiTFSI/LiDFOB in IL/DMC). Similar to the work by Liu et al., the difluorinated IL cation exhibited lower HOMO and LUMO energy levels compared with non-fluorinated cations. More interestingly, DFT calculations suggest that the DFPyr+ cation would be more prone to defluorination reduction at the anode, and could lead to the formation of a fluorine-rich SEI. A Li/NCM811 (LiNi0.8Co0.1Mn0.1O2) cell retained 90.5% of the initial capacity after 200 cycles, and an ultrahigh Ni-rich Li/NCM88 cell demonstrated excellent cycling stability, retaining 90.3% of the initial capacity after 300 cycles with a high average efficiency of 99.95%. Recently, Li et al. designed 4,4-difluoro-N-methyl-N-propylpiperidinium bis(fluorosulfonyl)imi-de (DFP13TFSI) by adopting a similar strategy, and the result shows that the 40 wt% LiTFSI/1 wt% LiDFOB/DFP13TFSI/DMC ILE can be used to assemble the Li/NCM88 cell to deliver a high-capacity retention of 91.4% after 300 cycles.44
Meanwhile, the nucleation efficiency of Li+ ions is usually accelerated after incorporating co-solvents into ILEs. Stępień et al. observed the actual Li+ nuclei growth on a nickel (Ni) current collector by in situ AFM mapping.42 They selected target electrolytes consisting of Pyr14FSI-LiTFSI with ratios of 8:2 and compared them with 5 wt% FEC added as a control (8:2FEC). As shown in Fig. 5f, both systems exhibited granular Li deposition. In the 8:2 system, the Li particles varied in size, leading to a rough surface with the continuous growth of new Li nuclei. Conversely, in the 8:2FEC system, the Li particles were uniformly distributed and consistent in size, with no new nuclei appearing after 10 minutes. The above results suggested that FEC restrained the reductive decomposition of the electrolyte and induced smoother and denser Li deposition, which could reduce and stabilize the lithium nucleation overpotential significantly. When Li was deposited on a Ni current collector to form a LiNi/Li battery, impedance measurements taken every 12 h showed that the cell impedance increased over time in the 8:2 system. In contrast, the system with FEC remained relatively stable, indicating a more stable interface after initial Li plating in the 8:2FEC system (Fig. 5g). Unfortunately, the presence of flammable organic solvents like DMC and FEC reduces the safety of ILEs. Designing organic solvent-assisted ILEs that are both high-voltage and safe presents a significant challenge. Recently, we developed a triethyl phosphate (TEP)-assisted ILE using 1 M lithium difluoro(oxalato)borate (LiDFOB) in TEP and [Pyr13][TFSI] IL.43 Benefitting from high ionic conductivity, good thermal stability, and excellent passivation ability of LiDFOB on Al current collectors, we chose it as the Li salt. Particularly, TEP solvent is non-flammable, low viscosity, low cost, but reacts severely with metallic Li during charge–discharge cycles. Non-flammable ILs bring a large amount of Pyr13+ cations and TFSI− anions into the electrolyte. The Pyr13+ cations can ensure a uniform Li+ ion flux distribution on the electrode surface by electrostatic shielding. Meanwhile, TFSI− and DFOB− anions would participate in the Li+ solvation structure to form F- and B-rich CEI/SEI layers on the high-nickel cathode or LMA by oxidizing or reducing preferentially. All the above suppress side reactions between the electrode and electrolyte effectively (Fig. 5h). A 2.2 Ah multi-layer Li/NCM90 pouch cell with non-flammable electrolytes has been achieved with high energy densities of 455 W h kg−1 at 4.3 V and 470 W h kg−1 at 4.4 V, besides passing the nail penetration test stably.
Ether-based electrolytes have greater resistance to reduction than ester-based electrolytes, so they have been able to form thinner SEI layers on the anode and possess higher initial coulombic efficiency. Using ether-based electrolytes as co-solvents with ILs improves Li+ ion transport kinetics in ILEs while overcoming their inherent oxidative instability. Pal et al. reported an ether-aided 80IL20DME electrolyte (3.2 mol kg−1 LiFSI in C3mpyrFSI:DME = 80:20 wt:wt), demonstrating excellent anode stability and high voltage tolerance.45Fig. 6a shows SEM (scanning electron microscopy) images of Li morphology after cycling in both 80IL20DME and 100IL (3.2 mol kg−1 LiFSI in C3mpyrFSI) systems. After 10 and 100 cycles, obviously larger and denser Li metal particles were deposited in the DME-assisted ILE, and verified by Li deposited on a Cu substrate simultaneously. However, the 100IL and 80IL20DME systems also possessed rod-like deposits and block-like deposits, respectively. Titration gas chromatography (TGC) quantification revealed that the SEI formation resulted in an 11% Li loss, with approximately 89% of the loss attributed to dead Li formation in the 100IL system. Meanwhile, the active Li loss was 43% and 57% in the 80IL20DME system, which were attributed to the SEI and dead Li, respectively (Fig. 6b). The results indicate that DME-assisted ILEs form a greater uniform and denser protective SEI than 100IL. In Li/Li symmetric cells, 80IL20DME exhibited lower polarization voltage compared to the 100IL. The Li/80IL20DME/NMC811 cell maintained an average coulombic efficiency of about 99.8% and a capacity retention of 81% after 300 cycles. Additionally, the Li|80IL20DME|NMC622 cell maintained an average coulombic efficiency of 99.23% after 100 cycles, based on the active material loading, which was as high as 22.4 mg cm−2 in the cathode. All the above results demonstrated the efficacy of ether-aided ILs in forming stable SEI layers and improving overall battery performance. Ding et al. dissolved 1 M LiFSI and 0.3 M LiNO3 in a solution of PP13TFSI and DME with a volume ratio of 1:4.46 Raman spectroscopy revealed that FSI−, TFSI−, and NO3− anions formed anion-rich solvation structures with Li+ ions and solvent molecules (Fig. 6c). Meanwhile, the reduction of free solvent molecules enhanced the oxidative stability of the electrolyte system and promoted the formation of an inorganic-rich EEI. Linear sweep voltammetry (LSV) results indicated that the electrochemical window of the ether-based ILE was extended to 4.5 V. Additionally, DME, LiNO3, and IL have higher HOMO energy levels which make it more likely to decompose on the NCM811 surface and form a CEI layer. The 4.3 V Li/NCM811 cells still exhibited stable cycling for 150 and 100 cycles, even though they had low N/P ratios of 2 and 1 (Fig. 6d). This demonstrates that the combination of LiFSI, LiNO3, PP13TFSI, and DME in the electrolyte formulation effectively enhances the stability and performance of LMBs even under challenging conditions.
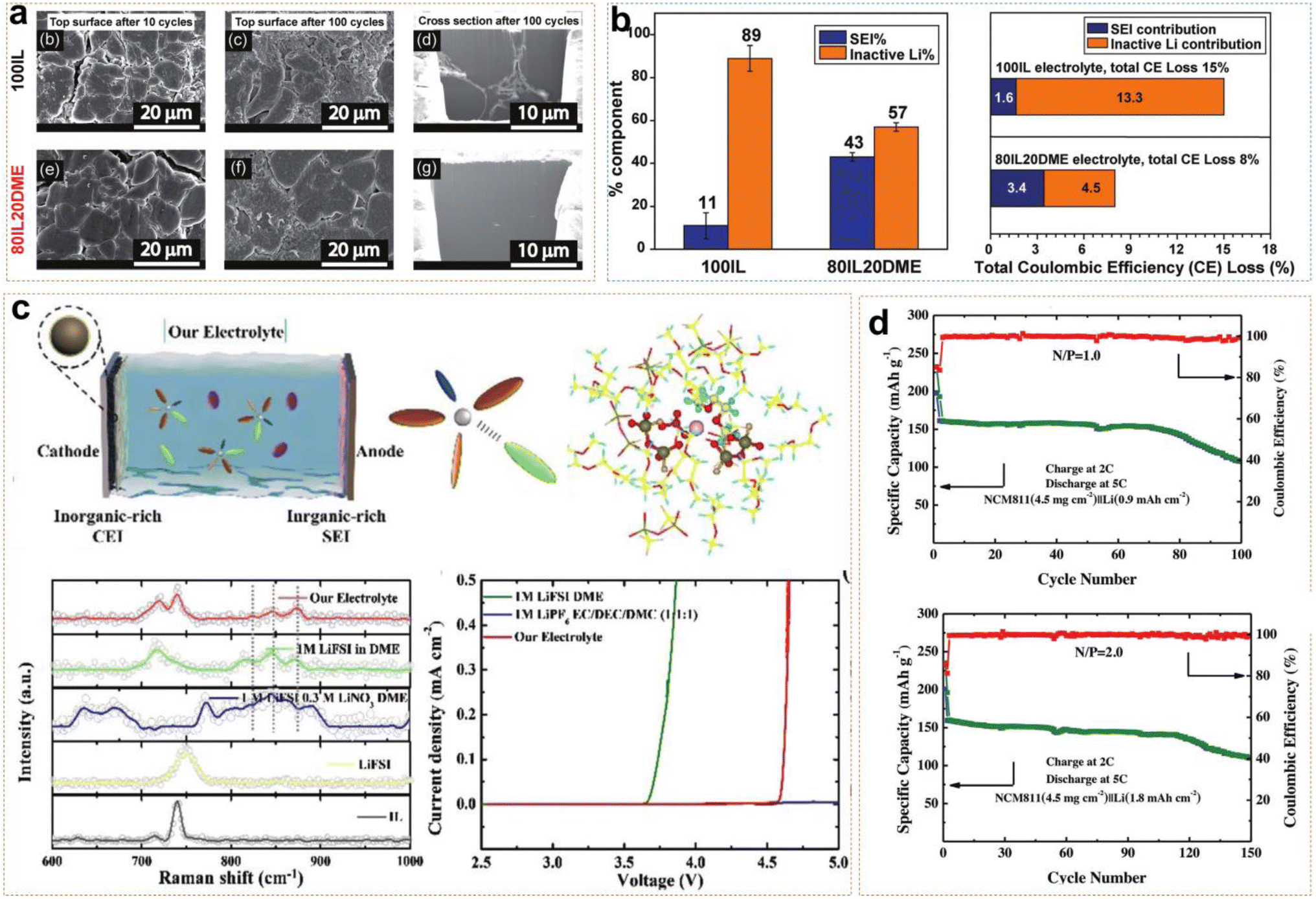 | ||
| Fig. 6 (a) Morphology of the top surface and cross-section using Cryo-FIB in 100IL and 80IL20DME electrolyte. (b) Quantitative analysis of Liinactive0 and LiSEI+ by the TGC quantification method. Reprinted from ref. 45 with permission. Copyright 2022, The Royal Society of Chemistry. (c) Schematic illustration of Li/NCM811 full cells in IL-based electrolytes; Raman spectra, and linear sweep voltammetry curves. (d) Cycling performance of the Li/NCM811 full cell under N/P ratios of 2 and 1. Reprinted from ref. 46 with permission. Copyright 2023, Wiley. | ||
2.3 Locally concentrated ILE
Although organic solvents (such as ethers and esters) can reduce the viscosity of ILs and improve ionic conductivity in batteries, they also introduce new challenges, such as poor thermal stability and safety, electrochemical instability, and strong side reactions. Ether and ester solvents usually evaporate or decompose under high-temperature conditions due to their low boiling points and high volatility, which increases the risk of thermal runaway. Additionally, organic solvents may lead to the formation of uneven and unstable SEI layers on the LMA surface. Thus, organic solvent-assisted ILE is in contrast to our original design concept of safety. In particular, for ester solvents, a thick and poorly conductive SEI formed from the decomposition products, namely, alkyl carbonates, which increase the interfacial impedance.47 Furthermore, ether solvents are prone to decomposition under high voltage conditions, which makes them incompatible with high-voltage cathode materials.48 The concept of locally concentrated ILE (LCILE) is similar to the classical LHCE (localized high concentration electrolyte), but both of them still have some subtle differences.49 For the LHCE, only a salt concentration above a threshold (typically >3 mol kg−1), can reduce the free solvent content and increase the content of contact ion pairs (CIPs) and aggregates (AGGs) significantly.50 In contrast, ILs contain abundant film-forming anions, so the Li salt concentration in concentrated ionic liquid electrolytes (CILEs) is usually less than 2 mol kg−1.The basic design principles of LCILE include the following aspects: (i) the CILEs are formed by dissolving 1.5–2 mol kg−1 of Li salts into neat ILs. This leads to the creation of abundant anionic aggregate structures, which significantly enhance the stability of the EEI. (ii) The selection of non-polar or weakly polar co-solvents ensures that the original solvation structure of the CILEs is not disrupted. Additionally, the co-solvents should possess a wide electrochemical window and high thermal stability to maintain the LCILE's stability under high-voltage and high-temperature conditions. (iii) Adjusting the component ratios and Li salt concentrations helps maintain appropriate electrolyte viscosity while improving ionic conductivity, ensuring that the Li+ ion kinetics in the LCILE system meet the requirements for its applications.
Fig. 7a shows the advantages and disadvantages of the traditional ether and carbonate cosolvents in CILEs. We can discover that LCILEs demonstrate significant advantages in terms of redox stability, viscosity, flammability, liquid range, and film-forming quality by comparison. Additionally, the composition of LCILEs is simple, usually consisting of a Li salt, IL, and cosolvent. Fig. 7b summarizes some recent reports about the compositions of LCILEs. LCILEs use LiFSI, LiTFSI, and LiDFOB as Li salts, sulfonamide-based ILs as solvents, and fluorinated ethers, aromatic molecules, and chlorinated molecules as cosolvents. In this section, we will explore the solvation structure and interfacial chemistry of LCILEs, and summarize their latest advancements in high-energy-density LMBs.
 | ||
| Fig. 7 (a) Comparison of the physicochemical and electrochemical properties of ILEs using an ether-based cosolvent, an ester-based cosolvent, and a locally concentrated cosolvent in a radar chart; and classical LCILE solvation structure. (b) Summary of the combination and collocation of LCILE composed of a lithium salt, cosolvent and ILs. | ||
Fluorinated ether cosolvents not only enhance Li+ ion conductivity and promote rapid charge–discharge performance by reducing the viscosity of the electrolyte, but also ensure a wide electrochemical window to make LCILEs operate at high voltages stably. In our previous work, we proposed an LCILE based on non-flammable PP13FSI and hydrofluoroether diluents (HFEs).51 HFEs address the inherent drawbacks of ILs, such as high viscosity, poor separate wettability, low ionic conductivity, and high cost, while maintaining the flame-retardant properties of the electrolyte and enhancing battery safety. In ILE systems, the strong coulombic interactions between cations and anions result in high viscosity and poor Li+ ion transport kinetics, leading to uneven Li deposition on the LMA surface. The addition of HFE accelerates Li+ ion transport while preserving the anion-rich solvation structure, ensuring the formation of a stable inorganic SEI layer (Fig. 8a). As a result, the assembled Li/Cu cell can cycle stably for 800 cycles with a coulombic efficiency of 99.4%. The Li/LFP cell can cycle for 1000 cycles at a high rate of 5C, with a capacity retention rate of 87%. Compared to single anions, the synergistic effect of FSI− and TFSI− dual anions further enhances SEI stability. Lee et al. designed an LCILE with LiTFSI as the lithium salt, Pyr13FSI IL as the solvent, and TTE as the cosolvent (1:2:2 by mol).56 The Li/LCO cell shows outstanding cycling performance with around 80% of capacity retention after 400 cycles. Meanwhile, we also found that LCILE based on LiDFOB and N-methyl-N-methoxyethyl-pyrrolidinium bis(trifluoromethylsulfonyl)imide (MEMPTFSI) IL could maintain the lithium salt concentration at an extremely low level of 0.1 mol L−1. This design of ultralow-concentration electrolyte (ULCE) employs LiDFOB as the lithium salt, which has excellent corrosion inhibition properties for aluminum foil and preferentially decomposes to form a stable, low-impedance SEI layer.57 The designed SEI effectively suppresses lithium dendrite growth and improves the high and low-temperature performances of lithium batteries. At −40 °C, Li/NCM622 batteries with commercial electrolytes fail to operate properly, while batteries with ULCE retain a discharge capacity of 115 mA h g−1. Even at −60 °C, the Li/NCM622 battery maintains 57% of room temperature capacity. Under high-temperature conditions of 70 °C, Li/NCM622 with ULCE can cycle for 150 cycles stably. In addition, organic cations play a significant role in LCILEs. Liu et al. discovered that Emim+ cations coordinate with FSI− less frequently compared to Pyr14+, resulting in lower viscosity and enhanced Li+ ion transport. Furthermore, the SEI formed with Emim+ is relatively more stable, due to its high nitrogen content.58
 | ||
| Fig. 8 (a) Schematic diagrams of Li plating on a Cu current collector and solvation structures of ILE and LHCE. Reprinted from ref. 51 with permission. Copyright 2021, Wiley. (b) Li+ ion solvation structure with mFBn and the corresponding Raman characterization and coordination number calculation. Reprinted from ref. 52 with permission. Copyright 2022, Wiley. (c) 1D 1H NMR spectra of the electrolytes and their constituents in the region of 6.25–7.05 ppm and 7.675–7.950 ppm. Reprinted from ref. 53 with permission. Copyright 2023, Wiley. (d) Schematic illustrations of the SEI/CEI components from LCM systems and Li+ diffusion path and migration energy barriers of Li+ along LiF–LiCl grain boundaries. Reprinted from ref. 54 with permission. Copyright 2024, American Chemical Society. (e) Schematic diagram of Li deposition behavior by regulating Tsand with OTE. Reprinted from ref. 55 with permission. Copyright 2021, Wiley. | ||
Fluorinated aromatic compounds are also excellent diluents for LCILEs, given their low affinity for Li+ and superior F-donating ability compared to HFE. 1,2-Difluorobenzene (1,2-dfBen) was selected as a diluent for ILE due to its extremely low dielectric constant and abundance of fluorinated functional groups. We found that the inclusion of 1,2-dfBen increases the AGG content in the ILE system, attributed to the enhanced interaction between Li+ and FSI− in the low dielectric environment.59 The abundance of AGG induces the formation of a robust SEI, significantly improving Li+ ion transport at the interface and stabilizing the LMA. The 1,2-dfBen diluent is also suitable for high-voltage cathode materials. Liu et al. designed an LCILE composed of LiFSI, EmimFSI, and 1,2-dfBen in a molar ratio of 1:2:2, which achieved stable cycling of high-voltage LiNi0.8Co0.15Al0.05O2 and LiNi0.8Mn0.1Co0.1O2 cathodes with a mass loading of 10 mg cm−2.60,61 Unlike 1,2-dfBen, monofluorobenzene (mFBn) possesses a single fluorine functional group, which can solvate Li+ ions partially. The self-diffusion coefficient of Li+ in the LCILE with mFBn (FEmF) is 3.5 × 10−11 m2 s−1, 2.5 times higher than in pure ILE (FE).52 As shown in Fig. 8b, the C–F stretching vibration of mFBn shifts from 241.9 cm−1 to 244.1 cm−1, indicating the involvement of mFBn in the solvation structure. MD simulations of FEmE show an additional peak at 2.22 Å in the Li–F (mFBn) curve, suggesting that mFBn enters the solvation sheath of Li+. Although the participation of mFBn in Li+ solvation is limited, it reduces the Li+–FSI− interaction, lowering the viscosity and enhancing the ionic conductivity. Fluorinated aromatic compounds can also influence the π–π interactions between the benzene ring and Emim+ cations and the coordination between Li+ and cosolvent F atoms, thereby affecting the physicochemical properties of the electrolyte system. Liu et al. compared monofluorobenzene (BnF), trifluoromethylbenzene (BnCF), and trifluoromethoxybenzene (BnOCF) as diluents, finding that BnOCF promotes near-complete decomposition of FSI− and Emim+, enhancing the reversibility of LMA.53 Raman spectroscopy analysis showed limited influence of BnOCF on Li+ and FSI− coordination. However, 1H NMR spectroscopy revealed that the peaks of Emim+ C5–H and C4–H shifted to lower frequencies, while the cosolvent peaks shifted to higher frequencies, indicating π–π interactions between Emim+ and the fluorinated aromatic cosolvent (Fig. 8c). However, the environmental and occupational safety of perfluoroalkyl and polyfluoroalkyl substances (PFAS) are of concern. Recently, Liu et al. reported LCILEs using anisole as a cosolvent.62 Anisole not only enhances ion transport by inducing nanophase-separated solvation structures but also regulates the deposition of organic cations and anions on LMA and the conversion of FSI− to LiF in the SEI. The coulombic efficiency improved from 99.19% in ILE to 99.71% in LCILE by optimizing the anisole content. This electrolyte enabled stable cycling for 400 cycles in Li/LiFePO4 cells and 350 cycles in Li/SPAN cells, with a capacity retention of 90% at an N/P ratio of 1.5.
Besides fluorinated diluents, chlorinated diluents have emerged as promising candidates for LCILEs and benefit from their lower cost and superior flame-retardant properties. We introduced dichloromethane (DCM) into the ILE system, formulating a 0.68 M LiFSI/C3mpyrFSI/DCM electrolyte (LCM).54 Our research demonstrated that chlorinated diluents effectively modulate the solvation structure of the ILE, reducing its viscosity and enhancing the bulk Li+ ion transport kinetics. Furthermore, the coordination between chlorinated diluents and IL induces the formation of a uniform dual-halide EEI. This EEI mitigates severe side reactions between the electrode and electrolyte under extreme conditions (high voltage, high temperature and low temperature) effectively, enabling stable operation of LMBs across a broad temperature range (−20 to 60 °C). Fig. 8d shows that unique dual-halide EEI not only inhibits the growth of lithium dendrites at the anode but also forms a stable CEI. Simulation results of Li migration paths indicate that the LiF–LiCl interface has a lower diffusion energy barrier than a single LiF interface. Zou et al. employed trichloromethane (TCM, CHCl3) as a diluent to develop an anion-strengthened solvation ILE (ASILEs) system, composed of 1 mol L−1 LiTFSI Pyr14FSI/TCM.63 TCM improved flame retardancy and enhanced the safety performance of LMBs. The assembled Li/NCM811 battery retained 81.6% of its initial capacity after 500 cycles at a 1C rate, and the Ah-level Li/NCM811 pouch cell achieved a high energy density of 386 W h kg−1. The results demonstrate that the chlorinated diluents are feasible diluents in LCILE systems. LCILEs also have extensive applications not only in LMBs with insertion-type cathode materials but also Li–O2 batteries. In Li–O2 batteries, LCILEs stabilize O2− intermediates, enhancing reaction kinetics and reducing overcharge potentials in the O2/O2− redox coupling. Although the use of N,N-diethyl-N-methyl-N-(2-methoxyethyl)ammonium bis(trifluoromethanesulfonyl)imide ([DEME][TFSI]) IL as a solvent in ILEs can lower the overpotential of Li–O2 batteries to 0.68 V, the limiting current density at the Li anode remains low. As shown in Fig. 8e, Cai et al. assessed Li+ transport capabilities using Sand's time (Tsand), which measures the duration during which Li+ ions are depleted near a flat electrode surface without replenishment. By incorporating 1H,1H,5H-octafluoropentyl 1,1,2,2-tetrafluoroethyl ether (OTE) and anisole (MOP) to create LCILEs, the system maintained a high initial Li+ ion concentration (C0) while simultaneously reducing viscosity and increasing the Li+ diffusion coefficient (D), thereby significantly extending Tsand.55,64 These studies provide important theoretical and practical foundations for the development of novel LCILEs. Table 1 summarizes the properties of IL-based electrolytes designed by using IL as a solvent in recent years.
| Electrolyte composition | Molar ratio or concentration | Battery type | Mass loading (mg cm−2) and N/P ratio | Cycling stability (voltage, CR, cycles, rate, CE) | Ref. |
|---|---|---|---|---|---|
| LiTFSI:Pyr14FSI |
1:4 |
Li/NCM88 | 2.8, excessive Li | 3.0 V–4.3 V, 88%, 1000 cycles, 0.3C, 99.94%, RT | 31 |
| LiTFSI/[C6Py][TFSI] | 1 mol L−1 | Li/LNMO | 2.7, excessive Li | 3.5 V–4.9 V, 70.2%, 55 cycles, 0.1C, 97%, 60 °C | 33 |
| LiFSI:[C1O1mpip][FSI] |
1:1 |
Li/LFP | 3.82, excessive Li | 2.5 V–3.8 V, 98.8%, 100 cycles, 0.2C, >99%, 50 °C | 34 |
| LiTFSI/NaTFSI/EMImFSI | 5 mol L−1 | Li/LCO | 10, 2 | 2.8 V–4.3 V, 90%, 140 cycles, 0.7C, 99.9%, RT | 37 |
| Li/NCM811 | 10, 1.8 | 2.8 V–4.4 V, 95%, 120 cycles, 0.5C, 99.8%, RT | |||
| LiFSI/Py13FSI + 1 wt% LiTFSI | 4.7 mol L−1 | Cu@SiPAN/LMNO | 10, anode-free | 3.0 V–4.85 V, 80%, 120 cycles, 50 mA g−1, 99%, RT | 38 |
| LiFSI:PMpyrfFSI |
1:1 |
Li/NCM622 | 8.8, 3 | 3.0 V–4.6 V, 98.5%, 100 cycles, 0.3C, 99%, RT | 39 |
| LiTFSI:[C2mpyr][FSI]:DMC |
13:4:13 |
Li/NCM523 | 4, excessive Li | 2.8 V–4.5 V, 95%, 100 cycles, 0.12 mA cm−2, 99%, RT | 40 |
| Li/LFP | 2, excessive Li | 2.5 V–4.2 V, 80%, 500 cycles, 0.15 mA cm−2, 99.9%, RT | |||
| LiDFOB:TEP:[Pyr13][TFSI] |
10:29:17 |
Li/NCM90 | 15, 3 | 2.8 V–4.4 V, 99%, 50 cycles, 0.1C, 99%, RT | 43 |
| LiFSI/LiNO3/PP13TFSI/DME | 1 mol L−1 | Li/NCM811 | 4.5, 1 | 3.0 V–4.3 V, 66%, 100 cycles, 2C, >99%, RT | 46 |
| LiFSI/C3mpyrFSI/DME | 3.2 mol kg−1 | Li/NCM811 | 6.4, 50 μm Li foil | 2.8 V–4.4 V, 81%, 300 cycles, 0.2C, 99.8%, RT | 45 |
| LiTFSI/LiDFOB/DMC/DFPyrTFSI | 8:1:20:4 |
Li/NCM811 | 5, excessive Li | 2.8 V–4.5 V, 90.5%, 200 cycles, 0.3C, 99.9%, RT | 41 |
| Li/NCM88 | 5, excessive Li | 2.8 V–4.5 V, 90.3%, 300 cycles, 0.3C, 99.95%, RT | |||
| LiTFSI/LiDFOB/DMC/DFPTFSI | 40:3:60:12 |
Li/NCM811 | 5, excessive Li | 2.8 V–4.5 V, 93.6%, 200 cycles, 0.2C, >99%, RT | 44 |
| Li/NCM88 | 5, excessive Li | 2.8 V–4.5 V, 91.4%, 300 cycles, 0.2C, >99%, RT | |||
| LiFSI/Pyr13FSI/1,2-dfBen | 8:18:15 |
Li/LFP | 9.5, excessive Li | 2.5 V–4.0 V, 96%, 250 cycles, 0.5C, >99.6%, RT | 59 |
| LiTFSI/PP13FSI/TTE | 1:2:2 |
Li/LCO | 8.0, excessive Li | 3.0 V–4.3 V, 80%, 350 cycles, 0.5C, >99.6%, RT | 56 |
| LiFSI/Pyr13FSI/DCM | 8:18:23 |
Li/LCO | 11.5, 5.28 | 2.8 V–4.4 V, 80.6%, 70 cycles, 0.3C, 99%, RT | 54 |
| LiTFSI/Pyr14FSI/TCM | 1 mol L−1 | Li/NCM811 | 5, excessive Li | 3.0 V–4.3 V, 81.6%, 500 cycles, 1.0C, 99.9%, RT | 63 |
| LiFSI/EmimFSI/BnOCF | 1:2:0.55 |
Li/NCA | 21, 1.2 | 2.8 V–4.4 V, 71%, 150 cycles, 0.1C, >99%, RT | 53 |
| LiFSI/EmimFSI/mFBn | 1:2:2 |
Li/SPAN | 2.7, 1.8 | 1.0 V–3.0 V, 71%, 250 cycles, 0.3C, >99%, RT | 52 |
| LiFSI/EmimFSI/dFBn | 1:2:2 |
Li/NCM811 | 10, 1 | 2.8 V–4.4 V, 62%, 100 cycles, 0.3C, >99%, RT | 60 |
| Li/NCM811 | 10, anode-free | 2.8 V–4.4 V, 76%, 250 cycles, 0.3C, >99.5%, RT | |||
| LiFSI/EmimFSI/dFBn | 1:2:2 |
Li/NCA | 10, excessive Li | 2.8 V–4.4 V, 92.8%, 100 cycles, 0.1C, >99.5%, −20 °C | 61 |
| Li/NCA | 10, 2.35 | 2.8 V–4.4 V, 70%, 100 cycles, 0.1C, >99%, −20 °C | |||
| LiFSI/PP13FSI/HFE | 1:2:4 |
Li/LFP | 5, excessive Li | 2.5 V–4.2 V, 87%, 1000 cycles, 5C, >99.5%, RT | 51 |
| LiDFOB/MEMPTFSI/HFE | 3:34:132 |
Li/NCM622 | 5, excessive Li | 2.8 V–4.5 V, 96%, 100 cycles, 0.2C, 99.9%, RT | 57 |
| LiFSI/EmimFSI/DCM | 5:11.73:117 |
Li/LFP | N/A, excessive Li | 2.5 V–4.0 V, 100%, 150 cycles, 0.1C, >99%, −40 °C | 65 |
| LiFSI/Pyr14FSI/BTFE | 3:4:4 |
Li/LFP | 6.3, excessive Li | 2.2 V–4.0 V, 94.6%, 400 cycles, 1C, >99.97%, RT | 66 |
| Li/NCM523 | 7.6, excessive Li | 2.8 V–4.3 V, 93.9%, 150 cycles, 0.3C, 99.91%, RT | |||
| LiFSI/EmimFSI/Anisole | 1:2:6 |
Li/LFP | 10, 1.5 | 2.4 V–3.6 V, 94%, 400 cycles, 0.3C, >99%, RT | 62 |
| Li/SPAN | 2.8 2.5 | 1.0 V–3.0 V, 90%, 350 cycles, 0.3C, >99%, RT | |||
| LiFSI/EmimFSI/BTFE | 1:2:2 |
Li/NCM811 | 10, excessive Li | 2.8 V–4.4 V, 96%, 200 cycles, 0.3C, 99.91%, RT | 58 |
| LiFSI/PP13FSI/HFE | 1:2:4 |
Li/Gr | 2, excessive Li | 0.01 V–1 V, 85%, 300 cycles, 3C, >99%, RT | 67 |
| LiTFSI/DEMETFSI/MOP | 0.95:1:3 |
Li/O2 | N/A, excessive Li | 4 V charging voltage, 120 cycles, 0.16 V overpotential | 64 |
| LiTFSI/DEMETFSI/OTE | 0.8:1:4 |
Li/O2 | N/A, excessive Li | 4 V charging voltage, 75 cycles, 0.064 V overpotential | 55 |
| NaTFSI/Py13FSI/TFEE | 1:3:1 |
Na/NFM | 1.3, excessive Na | 2.0 V–4.1 V, 92%, 150 cycles, 0.5C, >99%, RT | 68 |
| NaPF6/C4C1imBF4/diglyme | 1 mol L−1 | Na/NVP | 2, excessive Na | 2.5 V–4.0 V, 90.7%, 1000 cycles, 2C, 99.6%, RT | 69 |
3 IL-based solid electrolytes
Owing to their excellent ion conductivity across a broad temperature range, liquid electrolytes have been widely acknowledged for over two decades.70,71 Nevertheless, the risk of leakage and flammability along with the conventional organic liquid electrolytes also needs to be considered, so there is still a gap in the large-scale use of LMBs.72–75 Compared to liquid electrolytes, solid electrolytes exhibit higher energy density and greater safety, which could have tremendous potential in batteries and other electrochemical devices.76,77 Despite their numerous advantages, the main challenge faced by advanced application is the low ionic conductivity and slow Li+ transfer kinetics.78 Obviously, the large interfacial resistance originating from the insufficient solid–solid contact at the EEI always hinders the development of solid-state LMBs.70In addition to their prominent role in liquid–electrolyte battery systems, ILs have emerged to be pivotal for promoting the development of solid-state electrolytes. Their practical use can be broadly classified into three key areas: (i) PILs act directly as polymer matrices through polymerization initiated by an initiator; (ii) utilization of ILs as plasticizers and charge carriers to enhance ionic conductivity and other properties of polymer electrolytes; (iii) incorporation of ILs to improve the interfacial contact in composite solid-state polymer electrolytes (CSPEs). The subsequent sections will discuss these three applications and clarify how ILs facilitate the development and performance of solid-state electrolyte systems.
3.1 PIL-based solid electrolytes
The PILs are composed of distinct IL monomers that are covalently linked together through the identical repeating unit (Fig. 9a).79–82 The uniqueness of PILs lies in the integration of their exceptional properties (ILs) with the macromolecular structure of polymers.81,83–85 Consequently, PILs possess superior properties such as electrochemical stability, thermal stability, and excellent mechanical strength by regulating the backbone structure.86 As shown in Fig. 9b, we compared the main properties of solid-state electrolytes using PIL-based SPEs, oxide-based inorganic solid-state electrolytes (ISEs), and sulfide-based ISEs. Ideally, PILs would form self-supporting membranes with high conductivity and electrochemical stability that are able to dissolve substantial amounts of lithium salts. However, the polymerization of PIL monomers markedly elevates their glass transition temperature (Tg), consequently reducing their ionic conductivity by even several orders of magnitude.87,88 Researchers have primarily adopted two strategies to address this issue: (i) designing novel PILs with complex macromolecular structures; (ii) introduction of modifications such as copolymers or plasticizers. This section will discuss the direct polymerization of neat PIL monomers and other modification approaches.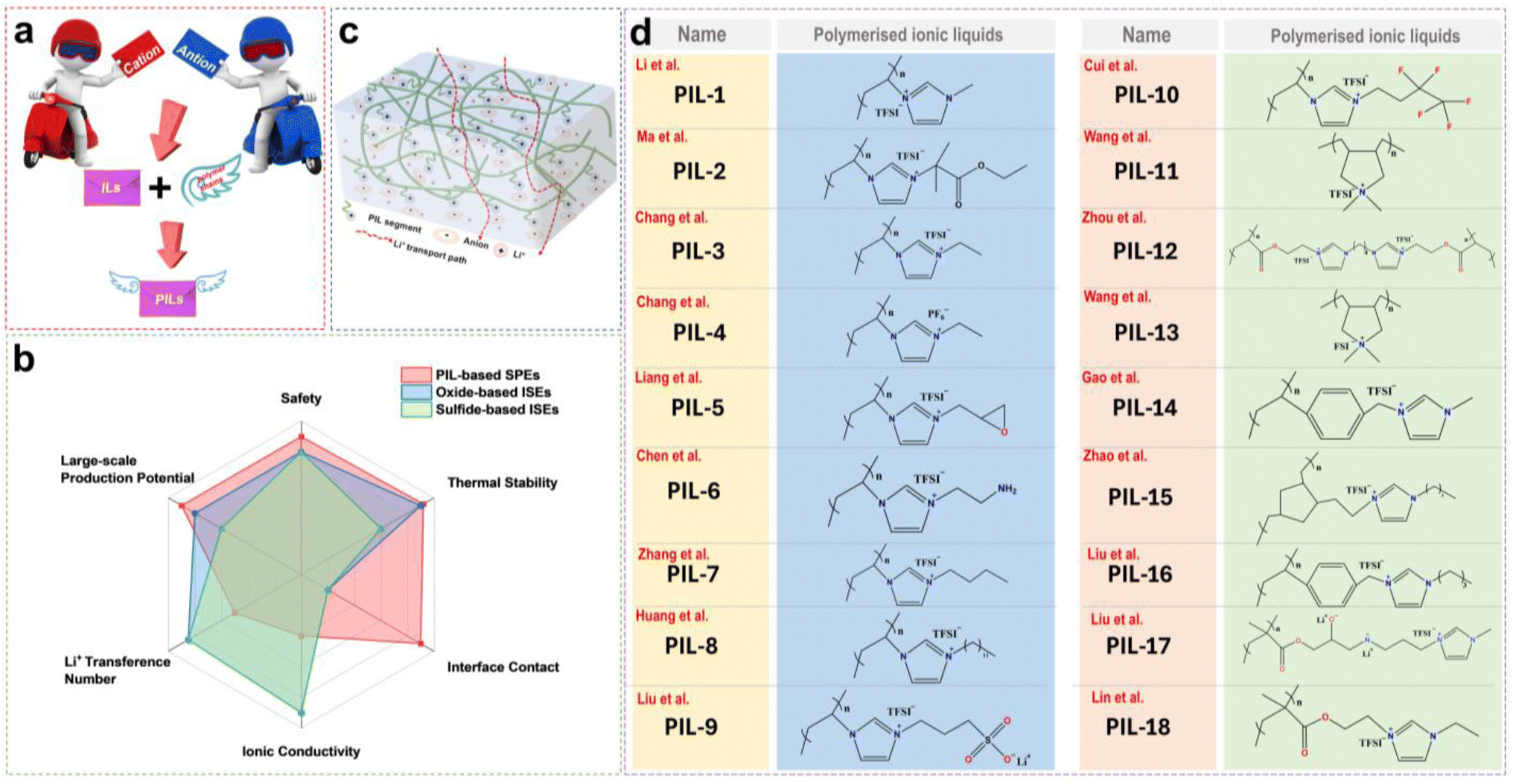 | ||
| Fig. 9 (a) Schematic illustration of the relationship between ILs and PILs. (b) Comparison of the main properties of solid-state electrolytes using PIL-based SPEs, oxide-based inorganic solid-state electrolytes (ISEs), and sulfide-based ISEs in a radar chart. (c) Schematic illustration of PIL-based solid-state electrolyte transport model. (d) Summary of the chemical structures of the PILs used for solid-state electrolytes. | ||
:1.5 of [PDADMA][FSI]:LiFSI (Fig. 10a). Corresponding experimental data also indicated that increasing the lithium salt content facilitates Li+ ion transport, as illustrated in Fig. 10b. When the salt content increases, more Li+ ions start to co-coordinate to those FSI− anions originally associated with the polycations only. This reduces the interactions between polycations and FSI−anions, allowing greater freedom of movement for the polymer backbone, to raise the ionic conductivity. However, excessively high salt content can give rise to phase segregation and the formation of crystalline phases, consequently decreasing the number of “free” charge carriers and making low ion conductivity. The optimized PIL electrolyte is used in lithium symmetrical batteries and LMBs, exhibiting long-term stability and consistent cycling characteristics across varying current densities, thereby ensuring better performance (Fig. 10c).
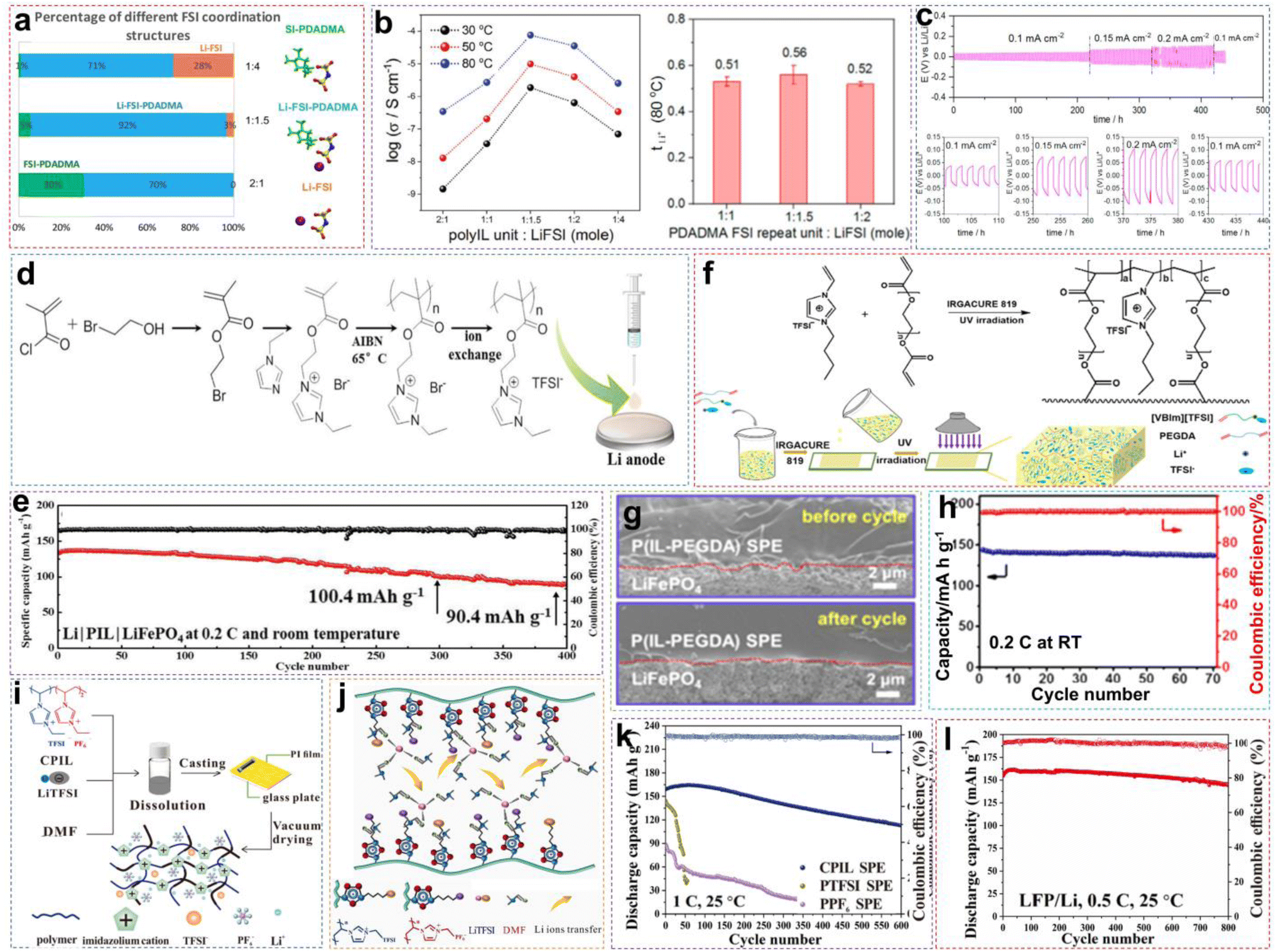 | ||
| Fig. 10 (a) Percentage of anions in three types of FSI− coordination environment calculated from MD simulations at 353 K. (b) The ion conductivities of different polyIL/salt systems at different temperatures and the Li+ transference number of solid polymer electrolytes with varying polymer-to-salt ratios. (c) The Li/Li symmetric battery cycling performances of the 1:1 polymer electrolyte at various current densities (i.e., 0.1, 0.15, and 0.2 mA cm−2). Reproduced from ref. 89 with permission. Copyright 2019, Elsevier. (d) Schematic representation of the synthesis of PIL. (e) Cycling performance of LFP/PIL/Li batteries at 0.2C and RT. Reproduced from ref. 90 with permission. Copyright 2023, The Royal Society of Chemistry. (f) Schematic diagram of the P(IL-PEGDA) SPE copolymerization reaction and preparation process. (g) SEM image of the interface between the LFP cathode and P(IL-PEGDA) SPE before and after 50 cycles. (h) Cycling performance of LFP/P(IL-PEGDA) SPE/Li batteries at 0.2C and RT. Reproduced from ref. 91 with permission. Copyright 2020, American Chemical Society. (i) Schematic illustration of the fabrication of solid polymer electrolytes. (j) Schematic representation of Li+ transfer in robust-flexible CPIL SPE. (k) Cycling performance of NCM811/CPIL SPE/Li batteries at 1C and RT. (l) Cycling performance of LFP/CPIL SPE/Li batteries at 0.5C and RT. Reproduced from ref. 84 with permission. Copyright 2024, Elsevier. | ||
Besides utilizing high-salt battery systems to achieve satisfactory performances, leveraging the structural adjustability of ILs and rationally designing the molecular structures of PIL monomers are other convenient and effective strategies.90,92 This approach not only renders certain good electrochemical performances but also imparts unique properties to the polymers, such as hydrophobicity and self-healing capability. As shown in Fig. 10d, Lin et al. synthesized a novel PIL polymer electrolyte by grafting IL chain units (EMIM+) into polymer backbones (poly(methyl methacrylate) (PMMA)).90 PMMA, which is chemically stable with lithium metal, served as the matrix for the electrolyte, while the H2, H4, and H5 protons in EMIM+ acted as hydrogen bond donors, interacting with oxygen-containing groups in PMMA to form hydrogen bonds. This interaction enhanced the Li+ transport and self-healing abilities of the polymer. Due to the external hydrogen bond interactions, the designed PIL could spontaneously heal cracks caused by dendrite growth at the EEI, resulting in uniform lithium deposition. As described in Fig. 10e, 91.2% of the LFP/PIL/Li battery capacity was retained after 206 cycles at RT (0.5C). When the temperature increased to 48 °C, 74.5% of the battery capacity was retained after 560 cycles, while CE stabilized near 100%. In summary, neat PILs have made significant strides as solid electrolytes. Nevertheless, the marked decrease in ionic conductivity following polymerization remains a pressing issue requiring resolution. Therefore, researchers must meticulously design PIL structures, incorporate additional functional groups for functionalization, and enhance their ionic conductivities while preserving their excellent mechanical properties.
Recently, Chang et al. produced a rigid–flexible PIL-based random copolymer by utilizing two PIL monomers with different anions.84 This was accomplished by precisely adjusting the ratio of the soft segment (1-vinyl-3-ethylimidazolium bis(trifluoromethylsulfonyl)imide) ([VEIM][TFSI]) to the hard segment (1-vinyl-3-ethylimidazolium hexafluorophosphate ([VEIM][PF6])) through making use of cross-linked copolymerization reactions (denoted as CPIL) (Fig. 10i). Notably, the large volume and more delocalized structure of the TFSI− anion in the soft segment facilitate fast ion migration, while the hard segment provides excellent mechanical strength and excellent antioxidative properties. Furthermore, the residual DMF molecules in the form of [Li(DMF)x]+ acted as a polar solvent, softening the PTFSI and PPF6 chains, which allowed the dissociated Li+ to be transported among the CPIL (Fig. 10j). The combination of the above factors led to the establishment of an effective Li+ transport pathway in SPE, which was made possible for the rational regulation of the ratio of the two components, so that an ionic conductivity of 1.06 × 10−4 S cm−1 and a stable electrochemical window up to 4.5 V (vs. Li+/Li) at RT could be satisfied. Based on the above advantages, the solid-state NCM811/CPIL SPE/Li batteries put up a stable cycling behavior, maintaining a high CR of 70.8% after 600 cycles at 1C (Fig. 10k). Besides, the LFP/CPIL SPE/Li batteries demonstrated a discharge capacity of 144.7 mA h g−1, with a CR of 89.6% after 800 cycles at 0.5C (Fig. 10l).
For polymer electrolytes comprising PILs as the matrix, the most prevalent approach to enhancing the ionic conductivity is to select ILs devoid of polymerization sites as plasticizers to optimize the overall performance of the PILs, which will be formulated in conjunction with this topic in the subsequent section. Furthermore, Fig. 9d summarizes the structural types of PILs employed in PIL-based solid-state electrolytes in recent years, along with their corresponding performances as listed in Table 2.
| Electrolyte composition | Conductivity (S cm−1) | ESW (V) | Battery type | Cycling stability (voltage, CR, cycles, rate, CE, temperature) | Ref. |
|---|---|---|---|---|---|
| LiTFSI/PEGDA/PEO/PIL-1 (poly[1-vinyl-3-methylimidazolium bis(trifluoromethylsulfo-nyl)imide]) | 4.3 × 10−5 (RT) | 5.44 V | Li/LFP | 2.8 V–4.0 V, 77.6%, 50 cycles, 0.2C, >99%, 55 °C | 94 |
| 6.12 × 10−4 (55 °C) | |||||
| LiTFSI/PEGMEM/PEGDA/EC/PC/PIL-2 (poly[1-vinyl-3-isobutyrate ethylimidazole bis(trifluoromethylsulfonyl)imide]) | 2.15 × 10−4 (30 °C) | ∼4.8 V | Li/LFP | 2.7 V–4.0 V, 96%, 200 cycles, 0.1C, N/A, 60 °C | 80 |
| Li/LCO | 3 V–4.2 V, 92.1%, 100 cycles, 0.1C, N/A, RT | ||||
| LiTFSI/PIL-3 (poly[1-vinyl-3-ethylimidazolium bis(trifluoromethylsulfonyl)imide])/PIL-4 (poly[1-vinyl-3-ethylimidazolium hexafluorophosphate]) | 1.06 × 10−4 (RT) | 4.5 V | Li/NCM811 | 2.7 V–4.3 V, 70.8%, 600 cycles, 1C, N/A, RT | 84 |
| Li/LFP | 2 V–4 V, 89.6%, 800 cycles, 0.5C, N/A, RT | ||||
| LiTFSI/EMIMTFSI/PEI/PVDF-HFP/PIL-5 (poly[1-vinyl-3-(oxiran-2-ylmethyl)imidazolium bis(trifluoromethylsulfonyl)imide]) | 1.8 × 10−3 (RT) | 5 V | Li/LFP | 2.5 V–4.2 V, 98%, 200 cycles, 0.5C, 98%, RT | 86 |
| Li/NCM622 | 2.7 V–4.3 V, 98%, 50 cycles, 0.1C, N/A, RT | ||||
| LiTFSI/PEGDA/BA/EPPOSS/BMIMTFSI/PIL-6 (poly[amino-vinyl bifunctionalized imidazolium bis(trifluoromethylsulfonyl)imide]) | 2.5 × 10−4 (RT) | ∼5.1 V | Li/LFP | 2.5 V–4.2 V, ∼100%, 200 cycles, 0.2C, 98%, RT | 95 |
| LiTFSI/PEGDA/PIL-7 (poly[1-vinyl-3-butylimidazolium bis(trifluoromethylsulfonyl)imide]) | 1.4 × 10−4 (30 °C) | 5 V | Li/LFP | 2.5 V–4.2 V, 97%, 70 cycles, 0.2C, ∼100%, RT | 91 |
| LiTFSI/PEGDMA/BMIMTFSI/VC/PIL-8 (poly[1-vinyl-3-dodecylimidazolium bis(trifluoromethanesulfonyl)imide]) | 7 × 10−4 (RT) | 5 V | Li/LFP | 2.8 V–4.2 V, 98.9%, 100 cycles, 0.1C, N/A, RT | 82 |
| PEGMEA/PEGDA/PIL-9 (poly[1-vinyl-3-(propylsulphopropyl)imidazolium bis(trifluoro-methanesulfonyl)imide]) | 1.1 × 10−5 (30 °C) | 5.4 V | Li/LFP | 2.5 V–4 V, 90.16%, 100 cycles, 0.2C, 99%, RT | 96 |
| LiTFSI/PVDF-HFP/GO-g-PIL-10 (poly[3-(3,3,4,4,4-pentafluorobutyl)-1-vinyl-1H-imidazole-3-ium bis(trifluoromethanesulfonyl)imide]) | 3.24 × 10−4 (RT) | ∼4.75 V | Li/LFP | N/A, 82%, 350 cycles, 0.5C, N/A, 30 °C | 97 |
| LiFSI/Pyr13 FSI/PIL-11 (poly[(diallyldimethylammonium) bis(trifluoromethanesulfonyl)imide]) | 1.7 × 10−5 (RT) | N/A | Li/NCM | 3.0 V–4.3 V, 88.8%, 50 cycles, 0.05C, N/A, 50 °C | 98 |
| LiTFSI/EMITFSI/FEC/PIL-11/PIL-12 (poly-[1,4-bis[3-(2-acryloyloxyethyl)imidazolium-1-yl]butane bis[bis(trifluoromethanesulfonyl)imide]]) | 1.06 × 10−3 (RT) | ∼4.4 V | Li/LFP | 2.4 V–4.2 V, 97.7%, 100 cycles, 0.1C, ∼100%, RT | 85 |
| LiFSI/PIL-13 (poly[(diallyldimethylammonium) bis(fluorosulfonyl)imide]) | 7 × 10−5 (80 °C) | N/A | Li/LFP | 2.5 V–3.8 V, N/A, 30 cycles, 0.067C, 99.94%, 80 °C | 89 |
| Li/NCM | 3.0 V–4.3 V, 67.5%, 50 cycles, 0.1C, 99.95%, 80 °C | ||||
| LiTFSI/EMIMTFSI/TEOS/PVDF-HFP/PIL-11 | 5.3 × 10−4 (20 °C) | 4.9 V | Li/LFP | 2.5–4.0 V, 95.7%, 250 cycles, 3C, 99.1%, 100 °C | 99 |
| LiTFSI/PEO/PVDF-HFP/PIL-14 (poly[1-(4-vinylbenzyl)-3-methylimidazolium bis(trifluoromethylsulfonyl)imide]) | 3.7 × 10−4 (RT) | 5 V | Li/LFP | 2.5 V–3.8 V, 97.1%, 250 cycles, 0.5C, N/A, RT | 81 |
| LiTFSI/PEG/PIL-15 (poly[(1-ethyl-3-(2-3-vi-nylcyclopentyl)ethyl)-1H-imidazol-3-ium bis(trifluoromethylsulfonyl)imide]) | 1.5 × 10−5 (30 °C) | 4.6 V | Li/LFP | 2.5 V–4.0 V, 92%, 70 cycles, 0.2C, N/A, 50 °C | 100 |
| LiTFSI/PEGDA/POSS/PIL-16 (poly[(1-(4-vinylbenzyl)-3-butylimidazolium bis(trifluoromethanesulfonyl)imide]) | 1.8 × 10−4 (30 °C) | ∼5 V | Li/LFP | 2.5 V–4.0 V, 80%, 150 cycles, 0.5C, N/A, RT | 101 |
| LiTFSI/MEMPTFSI/PIL-17 (poly[dilithium mono(3-(methacryloyloxy)-2-oxidopropyl)-3-(1-methyl-1H-imidazol-3 ium-3-yl)prop-yl bis(trifluoromethylsulfonyl)imide]) | 4.3 × 10−4 (30 °C) | 4.3 V | Li/LFP | 2.5 V–4.0 V, 87.6%, 144 cycles, 0.2C, N/A, 30 °C | 92 |
| LiTFSI/PIL-18 (poly[(1-ethyl-3-(2-methacryloyloxy)ethyl)imidazolium bis(trifluoro methylsulphonyl)imide]) | 1.76 × 10−4 (25 °C) | 5.2 V | Li/LFP | 2.5 V–4.3 V, 91.2%, 206 cycles, 0.5C, N/A, RT | 90 |
3.2 IL as a plasticizer for gel polymer electrolytes
Among the various methods used to enhance the ionic conductivities of SPEs, incorporating plasticizers to prepare gel polymer electrolytes (GPEs) is one of the most effective and commonly used approaches.92,98 Compared with traditional organic solvents, ILs are among the best choices for plasticizers due to their unique advantages, such as extremely low vapor pressure, non-flammability, and good thermal stability.86,97,101 These GPEs are also known as ionogel electrolytes.86 On the one hand, ILs facilitate the dissociation of lithium salts through their interactions;102,103 on the other hand, they weaken the interactions between polymer chains and simultaneously increase the number of amorphous regions, thereby effectively increasing the ionic conductivity of the electrolyte.104,105 In addition, the special ion channels formed by the ionic liquid in conjunction with other components play a significant role in Li+ conduction.106,107 The combined effect of these components enables the electrolyte to exhibit excellent overall performance.For instance, Yuan et al. successfully developed a novel solvate ionic liquid (SIL)-based SPE by incorporating an SIL of [Li(G4)1][TFSI] containing the functional additive LiBOB into the PVDF-HFP polymer matrix (named PLGB) (Fig. 11a).107 The interaction between SIL and PVDF-HFP disrupted the regular arrangement of polymer chains, increasing the proportion of the amorphous phase, which lessens the difficulty of chain movement and ion migration in the PLGB. The addition of LiBOB not only further adjusted the Li+ coordination environment by competing with TFSI−, but also helped to form the rigid–flexible coupling interface chemistry that buffered the volumetric changes of Li metal during cycling, giving a uniform and dendrite-free Li deposition. Through the synergistic effects of SIL and LiBOB, the solid-state electrolyte displayed a unique solvated structure that promoted Li+ transport along the polymer matrix. The optimized electrolyte revealed high ionic conductivity (2.18 × 10−3 S cm−1) at RT, high Li+ transference number (0.86), and outstanding electrochemical stability (up to 5.7 V vs. Li/Li+) (Fig. 11b). The assembled solid-state LFP/PLGB/Li battery delivered a high capacity of 143.2 mA h g−1 and capacity retention of 95.9% after 500 cycles at 0.5C (Fig. 11c). Recently, Wang et al. reported a hyperbranched polyurethane electrolyte (HPU-IL) by reacting hyperbranched polyether (HPEG) with isophorone diisocyanate (IPDI) in the presence of LiTFSI and 1-n-butyl-1-methylpyrrolidinium bis(trifluoromethanesulfonyl)-imide ([Pry][TFSI]) (Fig. 11d).108 After adding the IL, the X-ray diffraction (XRD) results revealed a new diffuse diffraction peak at 12°, indicating that the IL significantly increased the mobility of the polymer chain segments and interacted with the polymer backbone. This interaction enhanced hydrogen bonding interactions between carbamate and caused microphase separation, which accelerated ion transport. As the battery charged and discharged repetitively, no obvious lithium dendrites were observed on the surface of the LMA. The surface remained smooth and dense, making it clear that the presence of numerous hydrogen bonds in the HPU-IL electrolyte also solves the electrode/electrolyte interfacial contact problem and promotes the formation of a stable SEI at the interface. Owing to the structural advantages of the hyperbranched polyurethane and the plasticizing effect of the IL, the lithium salt dissociation capability, electrochemical stability of the electrolyte, and Li+ migration capability were significantly improved. The LFP/HPU-IL/Li battery could maintain a discharge capacity of 118 mA h g−1 after 1000 stable cycles at 0.5C and RT (Fig. 11e).
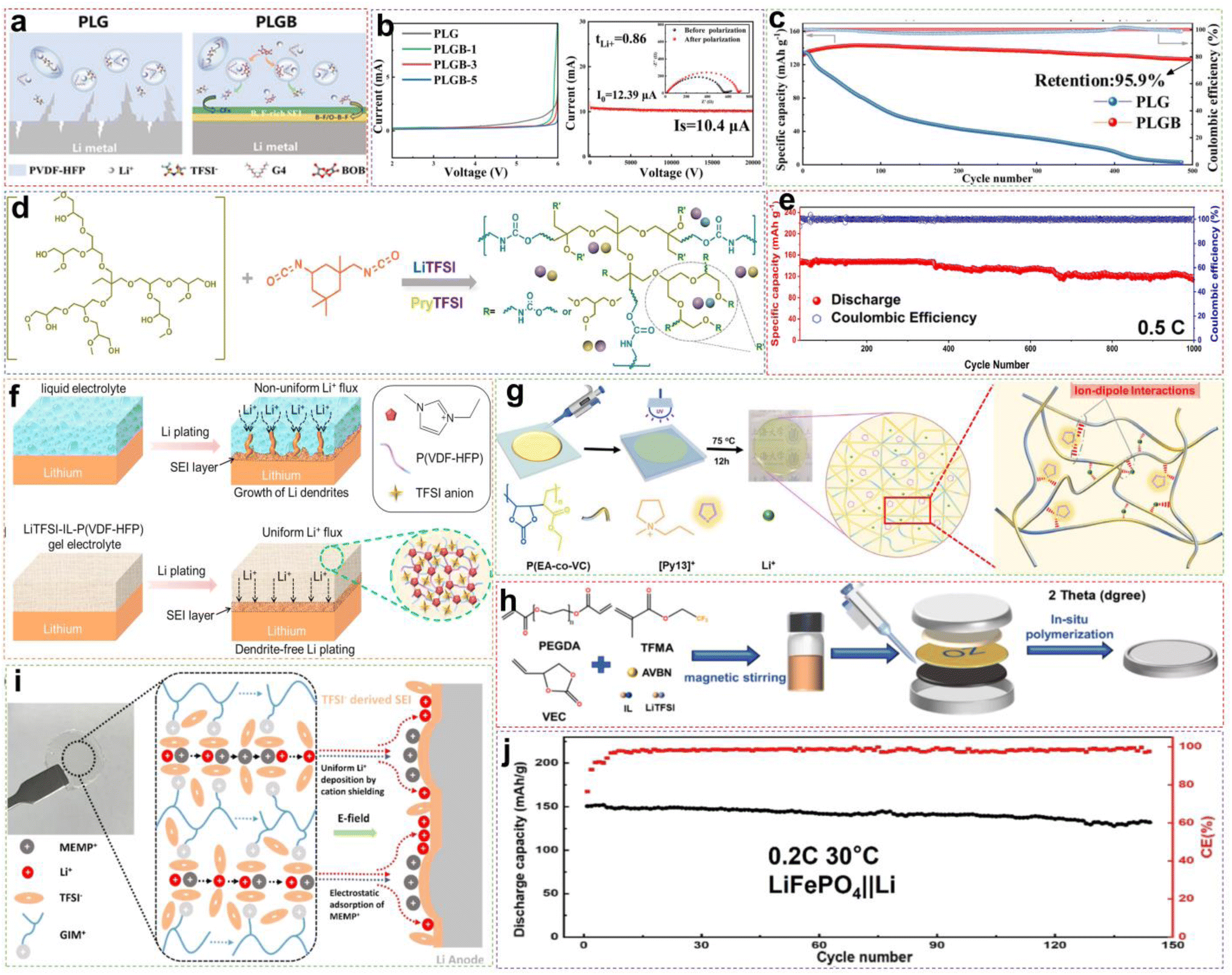 | ||
| Fig. 11 (a). Schematic representation of Li+ transport in PLG and PLGB GPE. (b) The LSV measurement of SS/GPE/Li batteries and DC polarization profiles of the symmetrical Li/PLGB/Li battery (the inset shows the impedance spectra before and after polarization). (c) A comparison of the long-term cycle performance between LFP/PLG/Li and LFP/PLGB/Li batteries at 0.5C. Reproduced from ref. 107 with permission. Copyright 2023, The Royal Society of Chemistry. (d) Schematic diagram of the HPU-based electrolyte. (e) Cycling performance of the LFP/HPU1.5-IL1.5/Li cell at 0.5C and RT. Reproduced from ref. 108 with permission. Copyright 2024, Elsevier. (f) Schematic illustration of the electrochemical deposition behavior of lithium metal anodes with liquid organic solution electrolyte and LiTFSI-IL-P(VDF-HFP) gel electrolyte. Reproduced from ref. 109 with permission. Copyright 2018, Elsevier. (g) Schematic diagram for the preparation of CLSPE-IL. Reproduced from ref. 110 with permission. Copyright 2022, Elsevier. (h) In situ thermal curing battery assembly flow chart. Reproduced from ref. 111 with permission. Copyright 2024, Elsevier. (i) Schematic illustration of GM-GPE that enhances ionic transport and stabilizes the GPE/Li anode interface. (j) Cycling performances of the LFP/GM-GPE/Li cell at 30 °C. Reproduced from ref. 92 with permission. Copyright 2024, American Chemical Society. | ||
Unfortunately, it is worth noting that the excessive introduction of ILs usually brings out an increase of ionic conductivity accompanied by a reduction in mechanical properties, which weakens their ability to resist lithium dendrites.112 Extensive studies have been conducted to reach a balance between the ionic conductivity and mechanical properties. Chen et al. reported the preparation of IL ([EMIM][TFSI]) immobilized GPEs, termed as IL-P(VDF-HFP).109 As shown in Fig. 11f, the ion–dipole interactions between the imidazolium cation in [EMIM][TFSI] and the polar groups −CFx in P(VDF-HFP) enable stable and dendrite-free Li+ plating/stripping. Additionally, a tightly crosslinked gel framework in the polymer matrix takes shape by the incorporation of [EMIM][TFSI] greatly strengthening the mechanical performance and thermal stability. The LiTFSI-IL-P(VDF-HFP) gel electrolyte, with excellent flexibility, also displayed close interfacial contact with electrodes and outstanding self-healing properties. Consequently, the LFP/LiTFSI-IL-P(VDF-HFP)/Li battery demonstrated superior cycling stability and rate performance. Wang et al. utilized ethyl acrylate (EA) and vinylene carbonate (VC) as the polymeric monomer, LiTFSI as the lithium salt, and N-methyl-N-propylpyrrolidine bis (trifluoromethylsulfonyl) imide ([Py13][TFSI]) as the additive to prepare a viscoelastic polymer electrolyte with both high ionic conductivity and favorable mechanical properties through UV polymerization (Fig. 11g).110 The study found that introducing the IL disrupted the crystallization of polymer chains, increasing chain mobility and facilitating Li+ migration in the electrolyte, which conveniently obtained an ionic conductivity of 2.77 × 10−4 S cm−1 at RT. On the other hand, the ion–dipole interactions between the cations in the IL and the oxygen atoms on the copolymer chains enhanced the mechanical properties. So by accommodating the IL content, the polymer electrolyte would possess high tensile strength up to 11.4 MPa and excellent stretchability elongation of 387% at break. Clearly, the resulting LFP/Li battery demonstrated excellent cycling stability at 0.2C, with a discharge capacity of 136.7 mA h g−1 after 500 cycles.
Except for utilizing intermolecular interactions to balance ionic conductivity and mechanical properties, forming a reinforcing framework into GPEs is another effective approach. Xu et al. successfully prepared a novel polycarbonate-fluorinated solid electrolyte by in situ thermal curing method.111 This involved the use of a polyester separator containing abundant polar functional groups as the reinforcing framework, in conjunction with vinylene carbonate (VEC) and trifluoroethyl methacrylate (TFEMA) as the polymer monomers, poly(ethylene glycol) diacrylate (PEGDA) with a flexible backbone as the flexible cross-linking point, and IL ([EMIM][TFSI]) as the plasticizer (Fig. 11h). The introduction of [EMIM][TFSI] promotes dissociation of the lithium salt, generating more mobile Li+ ions. The strong interaction between –CF3 in TFEMA and TFSI− competes with Li+, reducing the coordination ability of TFSI− and Li+. This facilitates the coordination and de-coordination of Li+ among the polymer chains, thereby increasing the Li+ transference number. Additionally, using a polyester membrane as a reinforcing framework reinforces the mechanical properties of the polymer and its ability to resist lithium dendrite penetration. Through the synergistic effects of these factors, the optimized electrolyte (denoted as 31VPIF/OZ), presented excellent ionic conductivity (3.58 × 10−4 S cm−1 at RT), high Li+ transference number (0.52), and wide electrochemical window (5.4 V). The assembled LFP/31VPIF/OZ/Li battery exhibited 91.5% capacity retention and 99.85% coulombic efficiency after 600 cycles at 0.5C and RT.
ILs play crucial roles as plasticizers in traditional polymer matrices. Because of the strong chemical affinity between PILs and ILs, ILs can be well confined within the PIL, increasing the ionic conductivity and decreasing the risk of leakage.112 Recently, our research group successfully synthesized a PIL monomer (denoted as [GIM][TFSI]), with polar charges far from the main chain through designing the monomer structure.92 This structure of the monomer was designed in a way that the imidazole cation away from the main chain increases the free volume of the PIL units, thus facilitating the Li+ transport rate. Subsequently, by combining this monomer with LiTFSI and the IL plasticizer N-methyl-N-methoxyethyl-pyrrolidinium bis(trifluoromethylsulfonyl)imide ([MEMP][TFSI]), PIL-based GPE (denoted as GM-GPE) could be obtained by UV light curing. The rich polar charges in GM-GPE effectively promote the dissociation of LiTFSI, and the cationic backbone also can anchor anions to the polymer chains through coulombic interactions (Fig. 11i). The long flexible side chain structure of [GIM][TFSI] and the introduction of the plasticizer will cause the ionic conductivity of GM-GPE to be as high as 4.3 × 10−4 S cm−1 at 30 °C. When GM-GPE is applied to LMBs, it is beneficial to generate an SEI film derived from TFSI− anions, thereby limiting the growth of lithium dendrites. The MEMP+ can migrate to the surface of the lithium metal anode under the influence of the electric field, giving rise to a cationic electrostatic shielding effect, further promoting uniform Li+ deposition. Thanks to the synergistic effects of these advantages, the LFP/GM-GPE/Li cell displayed a high discharge capacity of 150 mA h g−1 at 0.2C, and the capacity retention rate reached 87.6% after 144 cycles at 30 °C (Fig. 11j).
3.3 IL-based composite polymer electrolytes
In 1982, Weston and his colleague Steele first introduced inorganic particles, specifically aluminum oxide (Al2O3), into polyethylene oxide (PEO) to prepare solid electrolytes.113 They found that the conductivity and Li+ transference number of the electrolyte were slightly impacted with a small amount of Al2O3. However, the mechanical properties were significantly improved. Subsequently, various inorganic fillers have been incorporated into polymer systems, leading to the development of CSPEs. The inclusion of inorganic fillers would improve the thermal/electrochemical stability, mechanical properties, ionic conductivity, or ion transference number of polymer electrolytes.114 Despite these advantages in CSPEs, it's still challenging to modify the organic/inorganic phase interface contact.115,116 To overcome the interfacial issues, introducing a small amount of IL between the organic and inorganic phases has been considered a convenient and effective strategy. This section focuses on the recent advances in CSPEs with added ILs, emphasizing the role of ILs at the organic/inorganic interface.Similarly, Pan et al. prepared a series of CSPEs constructed from PVDF-HFP, [PP13][TFSI], LiTFSI and various inorganic fillers.118 They found that the mechanical properties of the CSPE were improved with the addition of 5 wt% TiO2 particles, which formed a dense electrolyte membrane by increasing the amorphous phase of PVDF-HFP and filling the pores of the CSPE. The mechanical properties would be improved by the addition of inert fillers; however, the ionic conductivity might be significantly reduced when large amounts of inert fillers were added, owing to the aggregation of nanoparticles. Therefore, the content of nanoparticles as inert fillers is typically limited to less than 10 wt% of the total CSPE in most previous studies. Recently, Kim et al. synthesized vinyl mesoporous silica nanoparticles (VMSNs) as reinforcing fillers by introducing vinyl groups on the surface of mesoporous silica nanoparticles (MSNs) via the surface grafting method.117 Compared to physically dispersed CSPEs containing MSNs, functionalized VMSNs can chemically crosslink with poly(ethylene glycol) (PEG) oligomers in the polymer matrix, enhancing the dispersion of mesoporous silica nanoparticles (Fig. 12a). This offers CSPEs higher mechanical properties (1.1 × 106 Pa) and higher ionic conductivity (1 × 10−4 S cm−1) at RT.
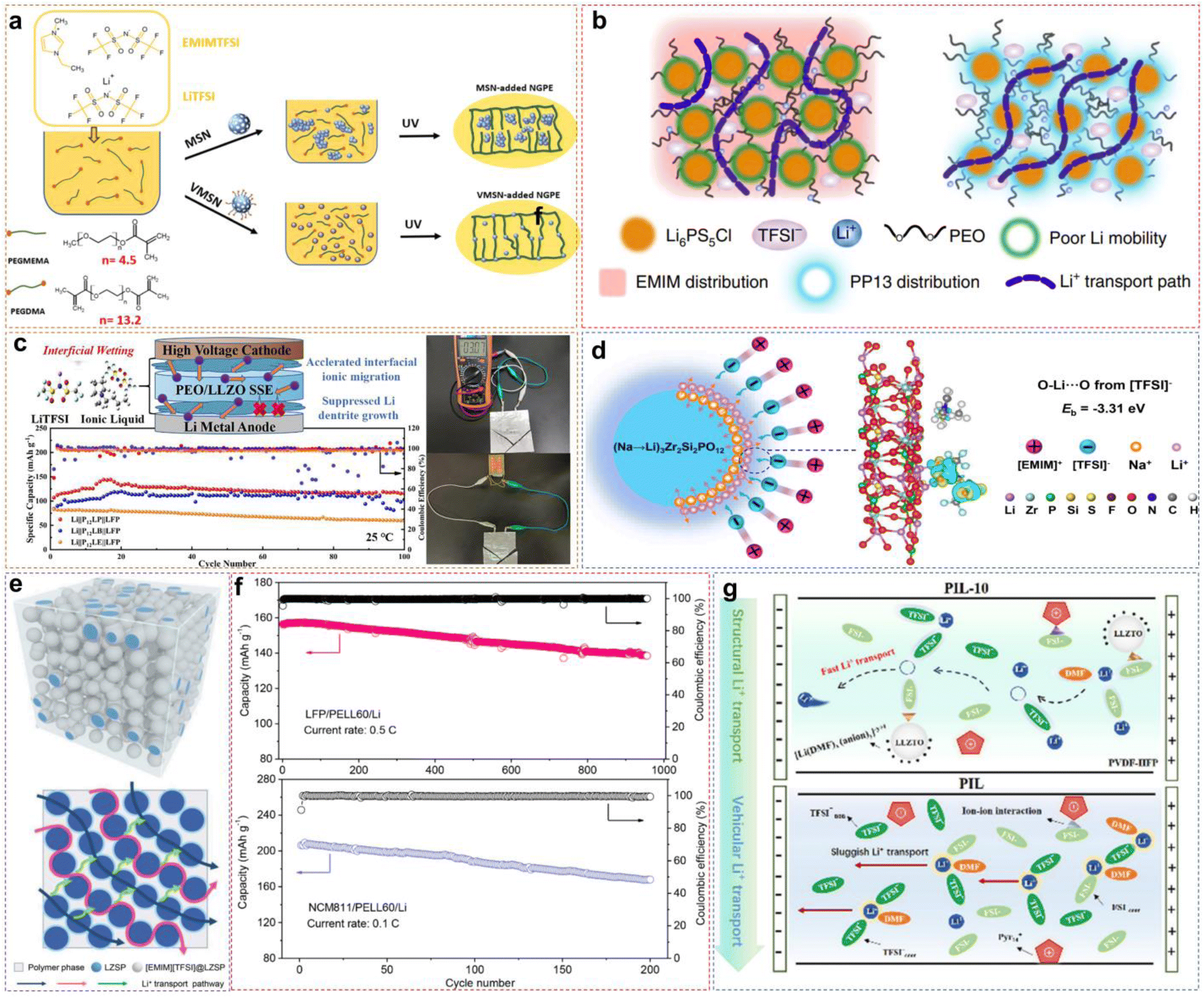 | ||
| Fig. 12 (a) Synthetic schematic diagram of nanohybrid gel polymer electrolytes (MSN-based NGPE and VMSN-based NGPE). Reproduced from ref. 117 with permission. Copyright 2023, American Chemical Society. (b) Proposed mechanism for Li+ diffusion in CSPEs with [EMIM][TFSI] and [PP13][TFSI] IL additives. Reproduced from ref. 120 with permission. Copyright 2022, Springer Nature. (c) Schematic diagram of CSPEs and pouch-type Li/PEO/LLZO@IL/LFP cells at 0.1C at RT. Reproduced from ref. 116 with permission. Copyright 2022, Elsevier. (d) Schematic diagram and DFT calculations on the interactions between LZSP surfaces and the [EMIM][TFSI] PCIL. (e) Schematic of the Li+ diffusion mechanism for PELL60. (f) Long-time cycling performance of LFP/PELL60/Li batteries at 0.5C and long-term cycling performance of NCM811/PELL60/Li batteries at 0.1C and RT. Reproduced from ref. 121 with permission. Copyright 2024, American Chemical Society. (g) Schematic illustrations of Li+ solvation structure and transformed transport mechanism in PIL-10 and PIL. Reproduced from ref. 122 with permission. Copyright 2024, Elsevier. | ||
The introduction of ILs can result in higher ionic conductivity and more thorough interfacial contact. However, excessive amounts of ILs can pose safety issues and increase the overall cost of the batteries. As shown in Fig. 12c, Yu et al. optimized the IL content in PEO/Li7La3Zr2O12 (LLZO) CSPEs and systematically studied the impact of IL cations on the interfacial behavior at the lithium anode side.116 With increasing IL content, the ionic conductivity and thermal stability of the CSPE were enhanced, and the EEI contact was simultaneously increased. However, when the IL content exceeded 17.5 wt%, the leakage of IL from the CSPE was observed, posing safety risks. Additionally, ILs directly participated in the SEI formation at the CSPE/Li interface, where ILs based on pyrrolidinium cations ([Py14]+) showed significantly more compatibility with the LMA, compared to ILs based on imidazolium cations ([BMIM]+ and [EMIM]+). The addition of [Py14][TFSI] resulted in the LiF and Li3N-riched SEI layer at the CSPE/Li interface, which facilitated Li+ conduction and suppressed lithium dendrite growth. The assembled pouch-type solid-state LFP/CSPE/Li cell achieved a specific capacity of 120 mA h g−1 and CE of more than 99% after 100 cycles at 0.1C.
The uniform dispersion of inorganic particles within the polymer matrix is crucial to achieve excellent overall performance of CSPEs. Like inorganic inert fillers, it is difficult to uniformly disperse the fillers within the polymer matrix when the content of the active filler is increased (beyond 10 wt%). Zhu et al. proposed using polymer-compatible ionic liquids (PCILs) to address the interfacial issues between the fillers and the polymer matrix.121 Using [EMIM][TFSI] as the liquid carrier, the Li3Zr2Si2PO12 (LZSP) was synthesized via the Na+/Li+ cationic exchange method. During the process, the cations concentrated on the filler surface and interacted with the adjacent PCIL, increasing the repulsive force and distance between particles and effectively solving the aggregation problem (Fig. 12d). The approximately 10 nm thick [EMIM][TFSI] coating uniformly covered the LZSP particle surfaces. The prepared PELL60 CSPE consisted of 30 wt% PVDF, 60 wt% [EMIM][TFSI]@LZSP, and 10 wt% LiTFSI. Subsequently, solid-state NMR revealed that in the CSPE composed of unmodified LZSP phases, the main conduction pathway for Li+ was through the LZSP phase. In contrast, PELL60 CSPE not only contributes to the highly dispersed LZSP powder, which builds up the Li+ conduction pathways in the native LZSP, but also facilitates pathways at the [EMIM][TFSI]@LZSP-PVDF interface and the intermediate spaces in PELL60 CSPE (Fig. 12e). As a result of the interconnected Li+ transport pathways established by [EMIM][TFSI] PCIL, the resulting PELL60 CSPE achieved a perfect combination of high ionic conductivity (8.3 × 10−4S cm−1), high Li+ transference number (0.81), excellent flexibility, and strong mechanical strength. The PELL60 CSPE exhibits decent cycling performance in both LFP and NCM811 batteries (Fig. 12f), as well as a high safety and high energy density of 424.9 W h kg−1 (excluding packing materials) in NCM811/PELL60/Li solid-state Li metal pouch batteries.
Recently, Lin et al. were the first to reveal the influence of inorganic active fillers on the solvation structure in IL-based CSPEs and their role in forming SEI layers on Li metal.122 IL containing FSI−/TFSI− anions and the LLZTO active filler were added to the PVDF-HFP matrix to prepare the CSPE. Detailed studies on the Li+ transport mechanism showed that LLZTO selectively anchored the FSI−/TFSI− anions, thereby altering the migration behavior of Li+ and the ratio of TFSI−/FSI− anions in the solvation shell. Adjusting the local environment aids in competing for coordination with TFSI− anions in the solvent structure dominated by FSI− anions, thereby forming a more stable interfacial chemistry (Fig. 12g). In the PIL-10 CSPE, the change in ionic environment enhances conductivity (1.24 × 10−3 S cm−1) and Li+ transference number (0.42) because of the preferable shift from the vehicular to structural Li+ transport. Additionally, the synergistic coordination of the solvent and the adjustment of the EEI offer the LFP/CSPE/Li stable battery cycling, achieving 95.4% CR after 500 cycles at 1C and RT. In the future, the design of composite electrolytes with high ionic conductivity, wide electrochemical window, high mechanical strength, and good interfacial contact and compatibility is a major research focus.
4 Electrolyte/electrode interface
The interface consists of two parallel sheets of excess charge, equal in magnitude but opposite in sign, commonly referred to as an “electric double layer”. Ideally, this interface would have zero thickness, and no irreversible reactions would occur between the electrode and the electrolyte (Fig. 13a).124 In reality, metallic Li, with its highly negative potential of −3.04 V vs. the standard hydrogen electrode, is extremely reactive and interacts with nearly any substance it contacts. When the electrode potential exceeds the electrochemical stability window of the electrolyte, a stable new layer forms on the electrode surface, known as the “interphase”. This is termed the SEI and CEI on the anode and cathode side, respectively (Fig. 13b).125 Both SEI and CEI are crucial for maintaining stable battery cycling. Ideally, SEI and CEI are electronic insulators and ionic conductors, preventing electrons from leaking out of the electrode and reacting with the electrolyte while allowing efficient lithium-ion transport. However, natural SEI and CEI typically exhibit low ionic conductivity and poor mechanical strength, leading to electrolyte penetration through the electrode/electrolyte interface (EEI) and continuous severe side reactions. | ||
| Fig. 13 (a) Schematic of electric double layer. (b) Illustration of the interphase formation and Li ion charge transfer process. | ||
Although ionic liquids (ILs), as either liquid or solid electrolytes, can stabilize LMBs during cycling, their high cost remains a significant limitation. Therefore, researchers are increasingly focused on how to achieve substantial performance improvements in LMBs with low doses of ILs. This section will explore three main approaches: using ILs as additives, pre-treating electrodes with ILs, and employing ILs as interface wetting agents. The low-dose ILs can introduce a rich variety of organic cations and inorganic anions into ether- or ester-based electrolytes. The Li dendrite protrusions typically exhibit higher electric potential, attracting organic cations that adsorb onto these sites, creating an electrostatic shielding effect to prevent excessive dendrite growth. Inorganic anions, on the other hand, become part of the Li+ ion solvation structure and decompose at the electrode surface, introducing inorganic components that stabilize the EEI. Moreover, using ILs for pre-treating electrodes is a cost-effective and scalable strategy. This process essentially constructs an artificial EEI. For instance, PILs can be applied to coat graphite or LMAs, reducing side reactions at the interface. In solid-state electrolyte applications, low-dose ionic liquids can enhance ionic conductivity at the interface, effectively lowering interfacial impedance. In summary, using the non-electrolytic functions of ILs to improve EEI stability marks a significant advancement in the application of ILs in LMBs.
4.1 SEI/CEI-forming additives
Lithium nitrate (LiNO3) has proven to be an effective additive for forming a stable SEI, but it has poor solubility in carbonate-based electrolytes. This solubility can be enhanced using cosolvents like dimethyl sulfoxide (DMSO) and sulfolane (SL).126,127 However, these cosolvents can compromise SEI stability, negatively impacting battery cycling performance. Therefore, Ma et al. designed 1-decyl-1-methylpyrrolidinium nitrate (Py110+NO3−), with weaker binding energy as an electrolyte additive. NO3− can incorporate into the Li+ solvation structure, and the low LUMO orbital of Li+–NO3− facilitates its reduction on LMA surfaces, forming a stable SEI layer (Fig. 14a).128 Using 2% IL-NO3−, Li/NCM811 cells retained 97.27% capacity after 200 cycles at 1C rate, and Li/LiCoO2 cells retained 80% capacity retention after 600 cycles. Huang et al. employed [EMIm][NO3] IL as a solvent component, achieving dense and smooth lithium deposition with Li+-coordinated NO3− solvation structures (Fig. 14b).129 With a mass loading of 20 mg cm−2, Li/NCM811 cells retained 70% capacity retention after 400 cycles at the 0.5C rate. Fang et al. introduced pyridinium trifluoroacetate (PyTFA) as an additive into commercial carbonate electrolytes.130 The strong coordination between the carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) in trifluoroacetate anions (TFA−) and Li+ facilitated the dissolution of LiNO3 in the carbonate electrolyte, creating an anion-rich formation of an inorganic-rich SEI, inducing uniform Li+ deposition (Fig. 14c). Li/NCM523 full cells using BEF-PyF/LN demonstrated excellent cycling stability with 92.9% CR after 100 cycles and a CE of 99.3%, while maintaining a capacity of 133.5 mA h g−1 at 3C. Single additives often struggle to meet the diverse electrochemical performance requirements of LMBs. Therefore, the synthesis and introduction of multifunctional additives are urgently needed for high-performance batteries. Gao et al. discovered that 1,3-dimethylimidazole dimethylphosphate (DIDP) and trimethylsilyl trifluoroacetate (TMSF) can undergo in situ transesterification in carbonate electrolytes to generate dimethyltrimethylsilyl phosphate (DTMSP) and 1,3-dimethylimidazole trifluoroacetate (DITFA) as multifunctional additives for LMBs (Fig. 14d).131 Due to the Si–O groups, DTMSP effectively removes H2O and HF, enhancing the moisture resistance of the electrolyte and contributing to the stability of the cathode.
O) in trifluoroacetate anions (TFA−) and Li+ facilitated the dissolution of LiNO3 in the carbonate electrolyte, creating an anion-rich formation of an inorganic-rich SEI, inducing uniform Li+ deposition (Fig. 14c). Li/NCM523 full cells using BEF-PyF/LN demonstrated excellent cycling stability with 92.9% CR after 100 cycles and a CE of 99.3%, while maintaining a capacity of 133.5 mA h g−1 at 3C. Single additives often struggle to meet the diverse electrochemical performance requirements of LMBs. Therefore, the synthesis and introduction of multifunctional additives are urgently needed for high-performance batteries. Gao et al. discovered that 1,3-dimethylimidazole dimethylphosphate (DIDP) and trimethylsilyl trifluoroacetate (TMSF) can undergo in situ transesterification in carbonate electrolytes to generate dimethyltrimethylsilyl phosphate (DTMSP) and 1,3-dimethylimidazole trifluoroacetate (DITFA) as multifunctional additives for LMBs (Fig. 14d).131 Due to the Si–O groups, DTMSP effectively removes H2O and HF, enhancing the moisture resistance of the electrolyte and contributing to the stability of the cathode.
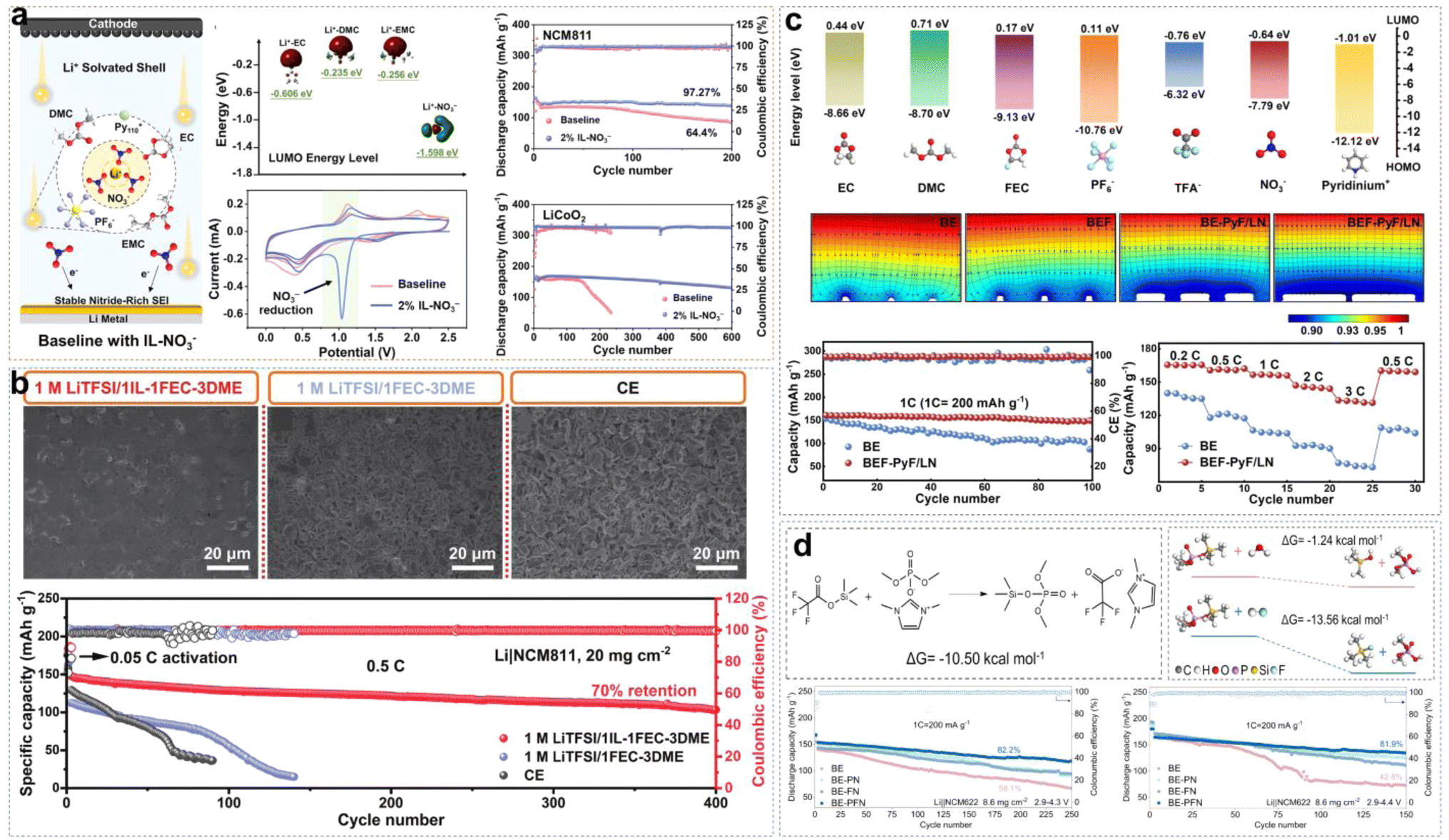 | ||
| Fig. 14 (a) Solvation structure of 2% IL-NO3− electrolyte; LUMO energy level of Li+-complexes; CV curves of Li/Cu cells for the baseline electrolyte and 2% IL-NO3− electrolyte; cycling performance of Li/NCM811 and Li/LCO cells. Reprinted from ref. 128 with permission. Copyright 2023, Elsevier. (b) SEM images of the cycled LMA in different electrolytes and corresponding cycling performance of Li/NCM811 cells. Reprinted from ref. 129 with permission. Copyright 2022, Wiley. (c) Molecular orbital energies of solvents and additives calculated by DFT; finite element analysis of Li deposition morphology simulated by COMSOL Multiphysics; cycling and rate performance of Li/LCO cells. Reprinted from ref. 130 with permission. Copyright 2022, Elsevier. (d) The reaction between DIDP and TMSF and the reaction paths of DTMSP and H2O/HF; long-term cycling performance in Li/NCM622 cells. Reprinted from ref. 131 with permission. Copyright 2024, Wiley. | ||
4.2 Interface pretreatment and wetting
Utilizing ILs to spontaneously form an SEI on electrode surfaces can significantly enhance electrode cycling stability. Wang et al. demonstrated that immersing LMA in Pyr13FSI leads to the in situ growth of an ordered organic/inorganic hybrid SEI layer, where the reactive FSI− anions reacts with metallic Li to form LiF.132 XPS and sum frequency generation (SFG) spectroscopy confirmed the dynamic formation process of LiF. The force curves of the atomic force microscopy (AFM) images showed a two-stage change: an initial slow increase followed by a plateau and then a sharp increase, indicating the mechanical differences between the organic and inorganic layers in the SEI (Fig. 15a). In addition to spontaneous formation, SEI can also be artificially constructed. In our previous work, we designed a cationic polymeric liquid (PIL) as an artificial SEI for high-voltage NCM622 cathodes and graphite anodes, aiming to suppress electrolyte decomposition on electrode surfaces.133 This PIL film, with its high mechanical modulus and excellent electrochemical and chemical stability, effectively inhibited interfacial side reactions. Its abundant cationic groups had strong coulombic interactions with PF6− anions, promoting lithium-ion conduction within the SEI. As shown in Fig. 15b, the PIL coating layer significantly improved the cycling stability of Li/Gr and Li/NCM622 cells. SEM images of cycled electrodes revealed that unmodified graphite particles had fuzzy boundaries and were covered by a thick coating due to severe EEI side reactions, while the morphology of PIL-coated graphite particles remained largely unchanged. Additionally, unmodified NCM622 particles exhibited a thick layer of degradation products on their surface after high temperature cycling. In contrast, the surface of the PIL-coated NCM622 particles remained uniformly smooth. Yu et al. developed a high-charge-density cationic polymer, poly(octaallyltetraazacyclodecane nitrate) (POTA-NO3), as an artificial SEI (Fig. 15c).134 The electrostatic shielding effect of OTA4+ effectively regulated lithium-ion deposition paths and facilitated the desolvation process. NO3− generated a stable inorganic SEI layer, preventing the decomposition of the polymer cation layer and electrolyte. POTA@Li/LCO cells demonstrated over 80% CR after 1400 cycles with a CE exceeding 98%. Qin et al. cross-linked ionic liquid 1-vinyl-3-methylimidazolium bis(fluorosulfonyl)imide (VMI-FSI) with polyethylene oxide (PEO) to form a self-healing membrane.135 FSI− was chemically decomposed into LiF and Li3N, aiding SEI formation on lithium and repairing SEI film cracks caused by lithium dendrite tearing (Fig. 15d).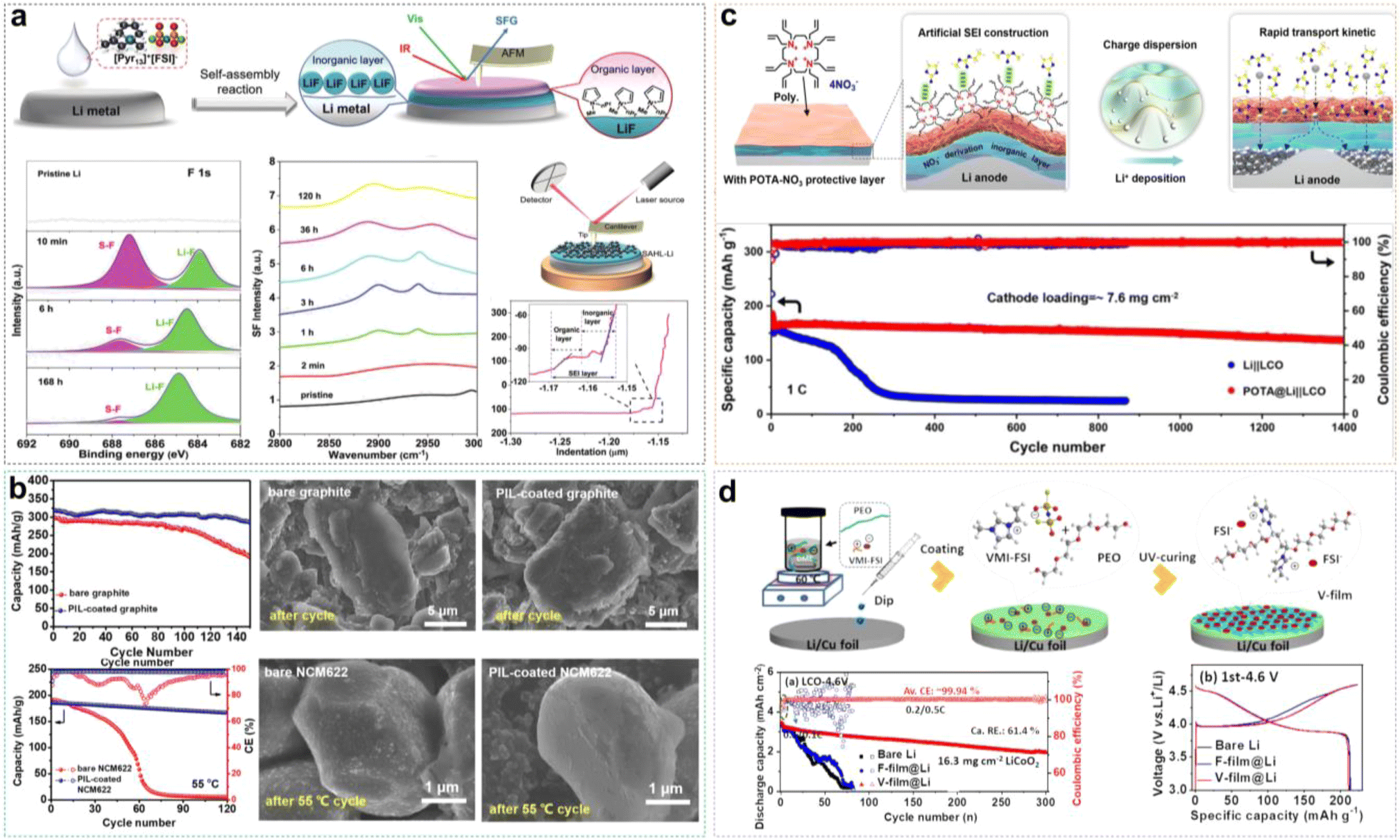 | ||
| Fig. 15 (a) Schematic illustration of artificial SEI formation on LMAs and corresponding evolution process probed by XPS, SFG and AFM. Reprinted from ref. 132 with permission. Copyright 2020, Wiley. (b) Cycling performance of Li/Gr and Li/NCM622 cells with PIL coating layer, and corresponding SEM images of cycled electrodes. Reprinted from ref. 133 with permission. Copyright 2022, Elsevier. (c) Schematic illustration of Li+ plating modulation by the POTA-NO3 protective layer and Li/LCO cycling performance. Reprinted from ref. 134 with permission. Copyright 2024, Elsevier. (d) Schematic illustrating the preparation of V-film, and cycling performance of Li/LiCoO2 full cells at cutoff voltages of 4.6 V. Reprinted from ref. 135 with permission. Copyright 2024, Wiley. | ||
ILs can also be used to wet solid-state interfaces and reduce interfacial resistance between solid electrolytes and cathodes. Im et al. prepared quasi-solid-state LMBs using Ag-coated Li6.4La3Zr1.7Ta0.3O12 powder (LLZTO), an Ag/C composite interlayer, and NCM333 cathodes, wetting the cathode and LLZTO@Ag/C with a 2 M LiFSI/Pyr13FSI additive.136 Shen et al. significantly enhanced the room-temperature performance of solid-state LMBs using a pyrrole-based IL (1-butyl-1-methyl pyrrolidinium bis(trifluoromethanesulfonyl)-imide) (BMP-IL) interlayer.137 The IL interlayer greatly facilitated lithium-ion conduction and reduced interfacial impedance by 400 times. The Li/LCO cell with the BMP-IL interlayer showed a high specific capacity of 122 mA h g−1 and an ultrahigh CR of 96% after 100 cycles.
5 Summary and perspectives
The lithium metal anode is fundamental to achieving high-energy-density lithium batteries. Ionic liquids, with their unique properties, such as excellent thermal stability, high ionic conductivity, and low flammability have further propelled the development of LMBs. In this review, we have systematically discussed the latest developments of ILEs and PILEs in LMBs and summarized strategies for achieving stable cycling by regulating solvation and interfacial chemistry in different IL-based electrolytes. From the above directions, we can conclude that the structural diversity and adjustability of ILs allow the precise molecular design and facilitate collaboration with other components to modulate solvation structures and interfacial chemistry, thereby adapting to various application scenarios. Although certain progress in IL-based electrolytes has been achieved, there remain many challenges in the following aspects.Firstly, optimizing the synthesis processes and designing stable and cost-effective ILs are the primary challenges facing the commercialization of IL-based electrolytes. From an economic perspective, directly replacing existing electrolytes by IL-based ones is expected to significantly increase battery costs. This is because the preparation of ILs typically requires large amounts of organic solvents, and the purification processes are complex. Besides, the current design process for ILs resembles the mixing of cocktails, necessitating tedious and extensive experimental screening, which limits the rapid development of new ILs.
Secondly, the underlying mechanisms of ILs in LMBs require further investigation, with complexity research involving electrochemistry and interfacial chemistry. In IL-based electrolytes, the solvation structure of Li+ significantly influences the properties of the EEI, which is directly associated with the electrochemical performance of LMBs. Currently, characterization techniques are widely applied, including Raman spectroscopy, Fourier-transform infrared spectroscopy (FTIR), and nuclear magnetic resonance (NMR). However, it remains challenging to directly determine the influence of solvent molecules on solvation chemistry, the impact of solvation chemistry on interfacial chemistry, and ultimately, the effect of these factors on electrochemical performance.
Finally, to broaden the practical applications of ILs, further development at the cell device level is needed to address issues such as overall ion conduction and interfacial contact. Additionally, most evaluations of IL-based electrolytes are conducted in coin cells with excessive lithium (e.g., using lithium foil thickness >300 μm). Therefore, the reliability of IL-based electrolytes under practical conditions still requires further assessment.
For facilitating the practical development of IL-based electrolytes, our views on future research directions are listed below (Fig. 16).
 | ||
| Fig. 16 Prospects of ILs for future development. | ||
5.1 Rational design of ILs
In the synthesis and purification of ILs, more streamlined experimental routes are required to reduce production costs. Emerging technologies such as microwave irradiation and ultrasound-assisted reaction techniques present promising solutions. Besides, the development of efficient new catalysts can significantly improve reaction rates and selectivity, thereby simplifying synthesis procedures and minimizing byproduct formation.In terms of IL design, leveraging computer science can reduce the trial-and-error costs associated with traditional design methods. By extracting useful patterns from large datasets, computer science enables the rapid design of new ILs, significantly accelerating the development cycle. For instance, computers can be used to combine various types of cations and anions, with machine learning (ML) in artificial intelligence (AI) to predict IL properties such as melting point, viscosity, and conductivity. Deep learning (a subset of ML) can be performed to screen and evaluate ILs with the liquid phase at room temperature and ionic conductivities larger than 5 mS cm−1. The electrochemical window values calculated from the energy levels of ILs can serve as screening criteria to identify ILEs suitable for different high-voltage cathode materials. All results from this process are stored in a database, providing sufficient data support for ML to continue optimizing design schemes. Additionally, different functional groups can be introduced into cations or anions during the screening process to functionalize ILs for better performance. Furthermore, by screening, identifying, and simulating the interactions of various ILs and other electrolyte components (including salts, solvents, and diluents), the synergistic effects of multiple components can be effectively balanced, and electrolyte formulations can be optimized. This approach offers both theoretical and practical guidance for the efficient and intelligent development of new IL-based electrolytes in the future.
5.2 Profound comprehension of mechanisms
Developing multi-scale in situ characterization techniques is essential for a comprehensive understanding of actual electrolyte behaviors about solvation/desolvation and interfacial interactions during charge/discharge. For example, in situ magnetic resonance imaging (MRI), in situ FTIR, and super-resolution electrochemical microscopy can provide insights into the dynamic evolution of Li+-solvent/anion solvation environments and interfacial structures. This further elucidates the structure–property relationships of ILs and interfacial microchemical structures, thereby offering valuable guidance to design and synthesize more effective ILs. In addition to employing advanced characterization techniques, computational methods including DFT and MD have been utilized to study the solvation structures and interfacial chemistry of electrolytes. However, simplified models or approximate algorithms may not accurately describe the properties of different electrolytes. Therefore, precise and efficient new computational methods must be developed for the dynamic structures in complex systems, constructing cross-scale theoretical models that span from microscopic to mesoscopic levels. Researchers must also establish a relationship between theoretical calculations and experimental characterization results to ensure the reliability and validation of theoretical models.5.3 Practical application
Based on the unique physicochemical properties of ILs and PILs, we propose a novel concept of solid-state electrodes. In particular, as illustrated in Fig. 17, these solid-state electrodes are composed of active materials, inorganic nanoparticles, PILs, and ILEs. The structural characteristic of solid-state electrodes is that PILs serve as the main filling matrix, while ILEs facilitate wetting the polymer/electrode interface and act as a bridge for Li+ transport. To further enhance its performance, nano-inorganic fillers can also be incorporated into polymer electrolytes to construct additional ion transport pathways. The solid-state electrodes can: (i) be paired with traditional separators and liquid electrolytes to achieve high specific energy batteries with a wide temperature range and high rate capabilities; (ii) be combined with self-supporting solid electrolyte film, with a minimal amount of liquid electrolyte added to aid interfacial wetting, to realize pseudo-solid-state batteries; and (iii) ultimately achieve true all-solid-state batteries through in situ solidification techniques such as heating or lighting exposure in conjunction with solid electrolytes. This innovative solution provides greater flexibility in the electrochemical system design. The simple and convenient preparation process is compatible with existing lithium-ion battery production systems, reducing system transition costs and providing a new strategy for advancing high-energy-density batteries. | ||
| Fig. 17 Schematic illustration of solidified electrodes for applications in liquid and solid-state electrolytes. | ||
Additionally, to facilitate the commercialization of IL-based electrolytes, it is essential to evaluate these electrolytes under conditions involving high current stripping/plating processes or within high-loading pouch cells. For example, to achieve higher energy densities, such as 500 W h kg−1, the areal stripping/plating capacity must exceed 3 mA h cm−2, and the charging current density should increase to 3.0 mA cm−2. This requirement implies that high-energy-density batteries need to be fully charged within one hour.138 Consequently, many reported strategies must be re-evaluated to determine whether IL-based electrolytes can meet these performance thresholds and achieve satisfactory cycling performance. Furthermore, energy density should be assessed based on cell-level packaging rather than relying on rough estimates of the active materials. Due to complex interfacial interaction, the transition from laboratory-grade batteries to applications will be a huge challenge. Therefore, it is crucial to establish the targeted practical evaluation system and assessment standards to support the design of IL-based electrolytes that are suitable for high-energy-density battery applications.
In summary, future research should focus on IL-based electrolytes, including structural design, profound understanding of mechanisms, and practical applications to promote their widespread development in high-energy-density LMBs, establishing a solid foundation for the next generation of efficient and environmentally friendly battery technologies.
Data availability
Data availability is not applicable to this article as no new data were created or analyzed in this review.Author contributions
X. Wu and J. Xu conceived the idea and theme. H. Tu, K. Peng and J. Xue wrote the draft manuscript and contributed equally to this work. J. Zhao, Y. Guo, S. Lu, Z. Wang, L. Chen, H. Li, X. Wu and J. Xu edited and polished the manuscript. J. Xu and X. Wu supervised the project. All the authors contributed to the discussion and writing of this review.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
H. T., K. P. and J. X contributed equally to this work. This work was supported by the National Key R&D Program of China (Grant No. 2022YFE0207300) and the National Natural Science Foundation of China (Grant No. 22179142 and 22075314).Notes and references
- M. V. Reddy, A. Mauger, C. M. Julien, A. Paolella and K. Zaghib, Materials, 2020, 13, 1884 CrossRef CAS.
- J. Zhang, H. Zhang, S. Weng, R. Li, D. Lu, T. Deng, S. Zhang, L. Lv, J. Qi, X. Xiao, L. Fan, S. Geng, F. Wang, L. Chen, M. Noked, X. Wang and X. Fan, Nat. Commun., 2023, 14, 2211 CrossRef CAS.
- D. Lu, R. Li, M. M. Rahman, P. Yu, L. Lv, S. Yang, Y. Huang, C. Sun, S. Zhang, H. Zhang, J. Zhang, X. Xiao, T. Deng, L. Fan, L. Chen, J. Wang, E. Hu, C. Wang and X. Fan, Nature, 2024, 627, 101–107 CrossRef CAS.
- X. Huang, R. Li, C. Sun, H. Zhang, S. Zhang, L. Lv, Y. Huang, L. Fan, L. Chen, M. Noked and X. Fan, ACS Energy Lett., 2022, 7, 3947–3957 CrossRef CAS.
- L. Li, M. Wang, J. Wang, F. Ye, S. Wang, Y. Xu, J. Liu, G. Xu, Y. Zhang, Y. Zhang, C. Yan, N. V. Medhekar, M. Liu and Y. Zhang, J. Mater. Chem. A, 2020, 8, 8033–8040 RSC.
- H. Tu, L. Li, Y. Hu, Y. Zhang, Y. Wang, W. Huang, Z. Ren, H. Lin and M. Liu, Chem. Eng. J., 2022, 434, 134647 CrossRef CAS.
- L. Li, H. Tu, J. Wang, M. Wang, W. Li, X. Li, F. Ye, Q. Guan, F. Zhu, Y. Zhang, Y. Hu, C. Yan, H. Lin and M. Liu, Adv. Funct. Mater., 2023, 33, 2212499 CrossRef.
- H. Tu, Z. He, A. Sun, F. Mushtaq, L. Li, Z. Wang, Y. Kong, R. Huang, H. Lin, W. Li, F. Ye, P. Xue and M. Liu, Nano Lett., 2024, 24, 5714–5721 CrossRef CAS.
- J. Zhou, B. Hao, M. Peng, L. Zhang, H. Ji, J. Liu, W. Ling, C. Yan and T. Qian, Adv. Energy Mater., 2023, 13, 2204174 CrossRef CAS.
- H. Ji, Z. Wang, Y. Sun, Y. Zhou, S. Li, J. Zhou, T. Qian and C. Yan, Adv. Mater., 2023, 35, 2208590 CrossRef CAS.
- G. Liang, V. K. Peterson, K. W. See, Z. Guo and W. K. Pang, J. Mater. Chem. A, 2020, 8, 15373–15398 RSC.
- M. S. Kim, Z. Zhang, P. E. Rudnicki, Z. Yu, J. Wang, H. Wang, S. T. Oyakhire, Y. Chen, S. C. Kim, W. Zhang, D. T. Boyle, X. Kong, R. Xu, Z. Huang, W. Huang, S. F. Bent, L. W. Wang, J. Qin, Z. Bao and Y. Cui, Nat. Mater., 2022, 21, 445–454 CrossRef CAS PubMed.
- M. Ma, R. Huang, M. Ling, Y. Hu and H. Pan, Interdiscip. Mater., 2023, 2, 833–854 CAS.
- C. Zhu, C. Sun, R. Li, S. Weng, L. Fan, X. Wang, L. Chen, M. Noked and X. Fan, ACS Energy Lett., 2022, 7, 1338–1347 CrossRef CAS.
- T. Zhou, Y. Zhao, M. El Kazzi, J. W. Choi and A. Coskun, Angew. Chem., Int. Ed., 2022, 61, e202115884 CrossRef CAS.
- X. Ren, L. Zou, X. Cao, M. H. Engelhard, W. Liu, S. D. Burton, H. Lee, C. Niu, B. E. Matthews, Z. Zhu, C. Wang, B. W. Arey, J. Xiao, J. Liu, J. G. Zhang and W. Xu, Joule, 2019, 3, 1662–1676 CrossRef CAS.
- S. Chen, J. Zheng, L. Yu, X. Ren, M. H. Engelhard, C. Niu, H. Lee, W. Xu, J. Xiao, J. Liu and J. G. Zhang, Joule, 2018, 2, 1548–1558 CrossRef CAS.
- Q. K. Zhang, X. Q. Zhang, L. P. Hou, S. Y. Sun, Y. X. Zhan, J. L. Liang, F. S. Zhang, X. N. Feng, B. Q. Li and J. Q. Huang, Adv. Energy Mater., 2022, 12, 2200139 CrossRef CAS.
- S. Tan, Z. Shadike, J. Li, X. Wang, Y. Yang, R. Lin, A. Cresce, J. Hu, A. Hunt, I. Waluyo, L. Ma, F. Monaco, P. Cloetens, J. Xiao, Y. Liu, X. Q. Yang, K. Xu and E. Hu, Nat. Energy, 2022, 7, 484–494 CrossRef CAS.
- M. Palluzzi, A. Tsurumaki, H. Adenusi, M. A. Navarra and S. Passerini, Energy Mater., 2023, 3, 300049 CrossRef CAS.
- P. Walden, Acad. Imp. Sci. Saint-Pétersbourg, 1914, 85, 1800–1801 Search PubMed.
- J. Hwang, K. Matsumoto, C. Y. Chen and R. Hagiwara, Energy Environ. Sci., 2021, 14, 5834–5863 RSC.
- X. Tang, S. Lv, K. Jiang, G. Zhou and X. Liu, J. Power Sources, 2022, 542, 231792 CrossRef CAS.
- X. Wang, L. Jin, W. Feng, Z. Zhou and H. Zhang, Sci. China: Chem., 2023, 66, 3443–3466 CrossRef CAS.
- X. Wu, Y. Dai, N. W. Li, X. C. Chen and L. Yu, eScience, 2024, 4, 100173 CrossRef.
- W. Zhou, M. Zhang, X. Kong, W. Huang and Q. Zhang, Adv. Sci., 2021, 8, 2004490 CrossRef CAS.
- G. Zhang, X. Deng, J. Li, J. Wang, G. Shi, Y. Yang, J. Chang, K. Yu, S. Sen Chi, H. Wang, P. Wang, Z. Liu, Y. Gao, Z. Zheng, Y. Deng and C. Wang, Nano Energy, 2022, 95, 107014 CrossRef CAS.
- R. Han, Z. Wang, D. Huang, F. Zhang, A. Pan, H. Song, Y. Wei, Y. Liu, L. Wang, Y. Li, J. Xu, J. Hu and X. Wu, Small, 2023, 19, 2300571 CrossRef CAS.
- K. Matuszek, S. L. Piper, A. Brzęczek-Szafran, B. Roy, S. Saher, J. M. Pringle and D. R. MacFarlane, Adv. Mater., 2024, 36, 2313023 CrossRef CAS.
- P. Meng, Z. Yang, J. Zhang, M. Jiang, Y. Wang, X. Zhang, J. Luo and C. Fu, Energy Storage Mater., 2023, 63, 102953 CrossRef.
- F. Wu, S. Fang, M. Kuenzel, A. Mullaliu, J. K. Kim, X. Gao, T. Diemant, G. T. Kim and S. Passerini, Joule, 2021, 5, 2177–2194 CrossRef CAS.
- M. Y. Yang, S. V. Zybin, T. Das, B. V. Merinov, W. A. Goddard, E. K. Mok, H. J. Hah, H. E. Han, Y. C. Choi and S. H. Kim, Adv. Energy Mater., 2023, 13, 2202949 CrossRef CAS.
- H. Qi, Y. Ren, S. Guo, Y. Wang, S. Li, Y. Hu and F. Yan, ACS Appl. Mater. Interfaces, 2020, 12, 591–600 CrossRef CAS PubMed.
- A. Warrington, M. Hasanpoor, A. Balkis, P. C. Howlett, O. E. Hutt, M. Forsyth and J. M. Pringle, Energy Storage Mater., 2023, 63, 102984 CrossRef.
- D. A. Rakov, J. Sun, P. V. Cherepanov, K. Arano, P. C. Howlett, A. N. Simonov, F. Chen and M. Forsyth, Energy Environ. Sci., 2023, 16, 3919–3931 RSC.
- D. A. Rakov, F. Chen, S. A. Ferdousi, H. Li, T. Pathirana, A. N. Simonov, P. C. Howlett, R. Atkin and M. Forsyth, Nat. Mater., 2020, 19, 1096–1101 CrossRef CAS PubMed.
- H. Sun, G. Zhu, Y. Zhu, M. C. Lin, H. Chen, Y. Y. Li, W. H. Hung, B. Zhou, X. Wang, Y. Bai, M. Gu, C. L. Huang, H. C. Tai, X. Xu, M. Angell, J. J. Shyue and H. Dai, Adv. Mater., 2020, 32, 2001741 CrossRef CAS.
- P. Liang, H. Sun, C. L. Huang, G. Zhu, H. C. Tai, J. Li, F. Wang, Y. Wang, C. J. Huang, S. K. Jiang, M. C. Lin, Y. Y. Li, B. J. Hwang, C. A. Wang and H. Dai, Adv. Mater., 2022, 34, 2207361 CrossRef CAS.
- Q. Liu, W. Jiang, J. Xu, Y. Xu, Z. Yang, D. J. Yoo, K. Z. Pupek, C. Wang, C. Liu, K. Xu and Z. Zhang, Nat. Commun., 2023, 14, 3678 CrossRef CAS.
- Z. Wang, Y. Sun, Y. Mao, F. Zhang, L. Zheng, D. Fu, Y. Shen, J. Hu, H. Dong, J. Xu and X. Wu, Energy Storage Mater., 2020, 30, 228–237 CrossRef.
- Y. Li, F. Ding, Y. Shao, H. Wang, X. Guo, C. Liu, X. Sui, G. Sun, J. Zhou and Z. Wang, Angew. Chem., Int. Ed., 2024, 63, e202317148 CrossRef CAS PubMed.
- D. Stępień, B. Wolff, T. Diemant, G. T. Kim, F. Hausen, D. Bresser and S. Passerini, ACS Appl. Mater. Interfaces, 2023, 15, 25462–25472 CrossRef.
- Z. Wang, R. Han, H. Zhang, D. Huang, F. Zhang, D. Fu, Y. Liu, Y. Wei, H. Song, Y. Shen, J. Xu, J. Zheng, X. Wu and H. Li, Adv. Funct. Mater., 2023, 33, 2215065 CrossRef CAS.
- F. Ding, Y. Li, G. Zhang, H. Wang, B. Liu, C. Liu, L. Jiang, X. Sui and Z. Wang, Adv. Mater., 2024, 36, 2400177 CrossRef CAS.
- U. Pal, D. Rakov, B. Lu, B. Sayahpour, F. Chen, B. Roy, D. R. MacFarlane, M. Armand, P. C. Howlett, Y. S. Meng and M. Forsyth, Energy Environ. Sci., 2022, 15, 1907–1919 RSC.
- K. Ding, E. J. Begin, S. Yuan, M. Zhong, Y. Wang, Y. Zhang, X. Zeng, J. L. Bao and Y. Wang, Adv. Energy Mater., 2023, 13, 2302443 CrossRef CAS.
- S. Chen, J. J. Fan, Z. Cui, L. Tan, D. Ruan, X. Zhao, J. Jiang, S. Jiao and X. Ren, Angew. Chem., Int. Ed., 2023, 62, e202219310 CrossRef CAS PubMed.
- Y. Zou, Z. Ma, G. Liu, Q. Li, D. Yin, X. Shi, Z. Cao, Z. Tian, H. Kim, Y. Guo, C. Sun, L. Cavallo, L. Wang, H. N. Alshareef, Y. K. Sun and J. Ming, Angew. Chem., Int. Ed., 2023, 62, e202216189 CrossRef CAS.
- X. Liu, A. Mariani, H. Adenusi and S. Passerini, Angew. Chem., Int. Ed., 2023, 62, e202219318 CrossRef CAS.
- H. Jia, J. M. Kim, P. Gao, Y. Xu, M. H. Engelhard, B. E. Matthews, C. Wang and W. Xu, Angew. Chem., Int. Ed., 2023, 62, e202218005 CrossRef CAS PubMed.
- Z. Wang, F. Zhang, Y. Sun, L. Zheng, Y. Shen, D. Fu, W. Li, A. Pan, L. Wang, J. Xu, J. Hu and X. Wu, Adv. Energy Mater., 2021, 11, 2003752 CrossRef CAS.
- X. Liu, T. Diemant, A. Mariani, X. Dong, M. E. Di Pietro, A. Mele and S. Passerini, Adv. Mater., 2022, 34, 2207155 CrossRef CAS.
- X. Liu, A. Mariani, T. Diemant, M. E. Di Pietro, X. Dong, A. Mele and S. Passerini, Adv. Mater., 2024, 36, 2309062 CrossRef CAS.
- A. Sun, H. Tu, Z. Sun, Z. He, Y. Wang, J. Wang, Y. Zheng, F. Zhu, L. Wang, F. Mushtaq, P. Xue, J. Liu and M. Liu, ACS Energy Lett., 2024, 9, 2545–2553 CrossRef CAS.
- Y. Cai, Q. Zhang, Y. Lu, Z. Hao, Y. Ni and J. Chen, Angew. Chem., Int. Ed., 2021, 60, 25973–25980 CrossRef CAS PubMed.
- S. Lee, K. Park, B. Koo, C. Park, M. Jang, H. Lee and H. Lee, Adv. Funct. Mater., 2020, 30, 2003132 CrossRef CAS.
- Z. Wang, H. Zhang, J. Xu, A. Pan, F. Zhang, L. Wang, R. Han, J. Hu, M. Liu and X. Wu, Adv. Funct. Mater., 2022, 32, 2112598 CrossRef CAS.
- X. Liu, A. Mariani, M. Zarrabeitia, M. Enrica, D. Pietro, X. Dong, G. Antonio, A. Mele and S. Passerini, Energy Storage Mater., 2022, 44, 370–378 CrossRef.
- H. Tu, L. Li, Z. Wang, J. Wang, H. Lin, M. Wang, C. Yan and M. Liu, ACS Nano, 2022, 16, 16898–16908 CrossRef CAS.
- X. Liu, A. Mariani, T. Diemant, M. E. Di Pietro, X. Dong, M. Kuenzel, A. Mele and S. Passerini, Adv. Energy Mater., 2022, 12, 2200862 CrossRef CAS.
- X. Liu, A. Mariani, T. Diemant, X. Dong, P. H. Su and S. Passerini, Angew. Chem., Int. Ed., 2023, 62, e202305840 CrossRef PubMed.
- X. Liu, A. Mariani, T. Diemant, M. E. Di Pietro, X. Dong, P. H. Su, A. Mele and S. Passerini, ACS Energy Lett., 2024, 9, 3049–3057 CrossRef CAS.
- W. Zou, J. Zhang, M. Liu, J. Li, Z. Ren, W. Zhao, Y. Zhang, Y. Shen and Y. Tang, Adv. Mater., 2024, 36, 2400537 CrossRef CAS PubMed.
- Y. Cai, Y. Hou, Y. Lu, Q. Zhang, Z. Yan and J. Chen, Angew. Chem., Int. Ed., 2023, 62, e202218014 CrossRef CAS.
- Y. Zheng, H. Ji, T. Qian, S. Li, J. Liu, J. Zhou, Z. Wang, Y. Li and C. Yan, Nano Lett., 2023, 23, 3181–3188 CrossRef CAS.
- X. Liu, M. Zarrabeitia, A. Mariani, X. Gao, H. M. Schütz, S. Fang, T. Bizien, G. A. Elia and S. Passerini, Small Methods, 2021, 5, 2100168 CrossRef CAS PubMed.
- Z. Wang, H. Zhang, R. Han, J. Xu, A. Pan, F. Zhang, D. Huang, Y. Wei, L. Wang, H. Song, Y. Liu, Y. Shen, J. Hu and X. Wu, ACS Sustain. Chem. Eng., 2022, 10, 12023–12029 CrossRef CAS.
- Y. Liu, S. Lu, Z. Wang, J. Xu, S. Weng, J. Xue, H. Tu, F. Zhang, L. Liu, Y. Gao, H. Li, J. Zheng and X. Wu, Adv. Funct. Mater., 2024, 34, 2312295 CrossRef CAS.
- X. Hu, E. Matios, Y. Zhang, C. Wang, J. Luo and W. Li, Angew. Chem., Int. Ed., 2021, 133, 6043–6048 CrossRef.
- Y. Huang, B. Cao, Z. Geng and H. Li, Acc. Mater. Res., 2024, 5, 184–193 CrossRef CAS.
- H. Yang, J. Li, Z. Sun, R. Fang, D. Wang, K. He, H. Cheng and F. Li, Energy Storage Mater., 2020, 30, 113–129 CrossRef.
- Z. Li, S. Wang, J. Shi, Y. Liu, S. Zheng, H. Zou, Y. Chen, W. Kuang, K. Ding, L. Chen, Y. Lan, Y. Cai and Q. Zheng, Energy Storage Mater., 2022, 47, 262–270 CrossRef.
- J. Wan, J. Xie, X. Kong, Z. Liu, K. Liu, F. Shi, A. Pei, H. Chen, W. Chen, J. Chen, X. Zhang, L. Zong, J. Wang, L.-Q. Chen, J. Qin and Y. Cui, Nat. Nanotechnol., 2019, 14, 705–711 CrossRef CAS PubMed.
- M. Martinez-Ibañez, N. Boaretto, A. Santiago, L. Meabe, X. Wang, O. Zugazua, I. Raposo, M. Forsyth, M. Armand and H. Zhang, J. Power Sources, 2023, 557, 232554 CrossRef.
- X. Ma, J. Yu, Y. Hu, J. Texter and F. Yan, Ind. Chem. Mater., 2023, 1, 39–59 RSC.
- B. Tong, Z. Song, W. Feng, J. Zhu, H. Yu, X. Huang, M. Armand, Z. Zhou and H. Zhang, Adv. Energy Mater., 2023, 13, 2204085 CrossRef CAS.
- C. Heubner, S. Maletti, H. Auer, J. Hüttl, K. Voigt, O. Lohrberg, K. Nikolowski, M. Partsch and A. Michaelis, Adv. Funct. Mater., 2021, 31, 2106608 CrossRef CAS.
- N. W. Utomo, S. Hong, R. Sinha, K. Kim, Y. Deng, P. Ochonma, M. G. Kitahata, R. Garcia-Mendez, Y. L. Joo and L. A. Archer, Sci. Adv., 2024, 10, eado4719 CrossRef CAS PubMed.
- G. Eshetu, D. Mecerreyes, M. Forsyth, H. Zhang and M. Armand, Mol. Syst. Des. Eng., 2019, 4, 294–309 RSC.
- J. Ma, X. Ma, H. Zhang, F. Chen, X. Guan, J. Niu and X. Hu, J. Membr. Sci., 2022, 659, 120811 CrossRef CAS.
- L. Gao, W. Jiang, X. Zhang, Y. Sun, K. Chen, W. Li, H. Xie and J. Liu, Chem. Eng. J., 2024, 479, 147822 CrossRef CAS.
- T. Huang, M. Long, X. Wang, G. Wu and Y. Wang, Chem. Eng. J., 2019, 375, 122062 CrossRef CAS.
- A. Shaplov, R. Marcilla and D. Mecerreyes, Electrochim. Acta, 2015, 175, 18–34 CrossRef CAS.
- H. Chang, W. Li, H. Liu, H. Hu, W. Liu, Y. Jin and G. Cui, Chem. Eng. J., 2024, 481, 148602 CrossRef CAS.
- D. Zhou, R. Liu, J. Zhang, X. Qi, Y. He, B. Li, Q. Yang, Y. Hu and F. Kang, Nano Energy, 2017, 33, 45–54 CrossRef CAS.
- L. Liang, X. Chen, W. Yuan, H. Chen, H. Liao and Y. Zhang, ACS Appl. Mater. Interfaces, 2021, 13, 25410–25420 CrossRef CAS.
- V. Delhorbe, D. Bresser, H. Mendil-Jakani, P. Rannou, L. Bernard, T. Gutel, S. Lyonnard and L. Picard, Macromolecules, 2017, 50, 4309–4321 CrossRef CAS.
- F. Makhlooghiazad, L. Miguel Guerrero Mejía, G. Rollo-Walker, D. Kourati, M. Galceran, F. Chen, M. Deschamps, P. Howlett, L. A. O'Dell and M. Forsyth, J. Am. Chem. Soc., 2024, 146, 1992–2004 CrossRef CAS.
- X. Wang, F. Chen, G. M. A. Girard, H. Zhu, D. R. MacFarlane, D. Mecerreyes, M. Armand, P. C. Howlett and M. Forsyth, Joule, 2019, 3, 2687–2702 CrossRef CAS.
- X. Lin, S. Xu, Y. Tong, X. Liu, Z. Liu, P. Li, R. Liu, X. Feng, L. Shi and Y. Ma, Mater. Horiz., 2023, 10, 859–868 RSC.
- F. Zhang, Y. Sun, Z. Wang, D. Fu, J. Li, J. Hu, J. Xu and X. Wu, ACS Appl. Mater. Interfaces, 2020, 12, 23774–23780 CrossRef CAS.
- L. Liu, J. Xue, Y. Liu, S. Lu, S. Weng, Z. Wang, F. Zhang, D. Fu, J. Xu and X. Wu, ACS Appl. Mater. Interfaces, 2024, 16, 8895–8902 CrossRef CAS PubMed.
- N. Kiriy, S. Özenler, P. Voigt, O. Kobsch, J. Meier-Haack, K. Arnhold, A. Janke, U. L. Muza, M. Geisler and A. Lederer, Int. J. Mol. Sci., 2024, 25, 1595 CrossRef CAS.
- Y. Li, Z. Sun, L. Shi, S. Lu, Z. Sun, Y. Shi, H. Wu, Y. Zhang and S. Ding, Chem. Eng. J., 2019, 375, 121925 CrossRef CAS.
- X. Chen, L. Liang, W. Hu, H. Liao and Y. Zhang, J. Power Sources, 2022, 542, 231766 CrossRef CAS.
- J. Liu, Y. Xu, F. Xu, J. Li, Y. Chen, J. Qiao, Y. Han, Y. Ren and B. Lin, Ionics, 2023, 29, 2249–2259 CrossRef CAS.
- Y. Cui, G. Yu, R. Liu, D. Miao and D. Wu, Chin. J. Chem., 2023, 41, 2848–2854 CrossRef CAS.
- X. Wang, G. M. Girard, H. Zhu, R. Yunis, D. R. MacFarlane, D. Mecerreyes, A. J. Bhattacharyya, P. C. Howlett and M. Forsyth, ACS Appl. Energy Mater., 2019, 2, 6237–6245 CrossRef CAS.
- M. Yao, Q. Ruan, Y. Wang, L. Du, Q. Li, L. Xu, R. Wang and H. Zhang, Adv. Funct. Mater., 2023, 33, 2213702 CrossRef CAS.
- R. Zhao, J. Yang, B. Wang, Z. Ma, L. Pan and Y. Li, Chin. J. Chem., 2023, 41, 2493–2501 CrossRef CAS.
- Q. Liu, L. Li, G. Liu, X. He, Y. Niu and G. Li, J. Power Sources, 2024, 592, 233897 CrossRef CAS.
- A. J. D'Angelo and M. J. Panzer, Adv. Energy Mater., 2018, 8, 1801646 CrossRef.
- J. H. Shin, W. A. Henderson and S. Passerini, J. Electrochem. Soc., 2005, 152, A978 CrossRef CAS.
- M. Zhu, J. Wu, Y. Wang, M. Song, L. Long, S. H. Siyal, X. Yang and G. Sui, J. Energy Chem., 2019, 37, 126–142 CrossRef.
- J. Qian, B. Jin, Y. Li, X. Zhan, Y. Hou and Q. Zhang, J. Energy Chem., 2021, 56, 420–437 CrossRef CAS.
- L. Anh and D. Kim, ACS Appl. Energy Mater., 2019, 2, 2585–2595 CrossRef.
- Y. Yuan, X. Peng, B. Wang, K. Xue, Z. Li, Y. Ma, B. Zheng, Y. Song and H. Lu, J. Mater. Chem. A, 2023, 11, 1301–1311 RSC.
- H. Wang, X. Li, Q. Zeng, Z. Li, Y. Liu, J. Guan, Y. Jiang, L. Chen, Y. Cao and R. Li, Energy Storage Mater., 2024, 66, 103188 CrossRef.
- T. Chen, W. Kong, Z. Zhang, L. Wang, Y. Hu, G. Zhu, R. Chen, L. Ma, W. Yan, Y. Wang, J. Liu and Z. Jin, Nano Energy, 2018, 54, 17–25 CrossRef CAS.
- Z. Wang, Y. Wang, P. Zhai, P. Poldorn, S. Jungsuttiwong and S. Yuan, J. Energy Chem., 2022, 75, 340–348 CrossRef CAS.
- D. Xu, D. Zhao, X. Niu, T. Wang and Z. Yang, Chem. Eng. J., 2024, 490, 151780 CrossRef CAS.
- Q. Yang, Z. Zhang, X. Sun, Y. Hu, H. Xing and S. Dai, Chem. Soc. Rev., 2018, 47, 2020–2064 RSC.
- J. Weston and B. Steele, Solid State Ionics, 1982, 7, 75–79 CrossRef CAS.
- M. Gandolfo, D. Versaci, C. Francia, S. Bodoardo and J. Amici, Electrochim. Acta, 2023, 463, 142857 CrossRef CAS.
- D. Zhang, X. Xu, X. Huang, Z. Shi, Z. Wang, Z. Liu, R. Hu, J. Liu and M. Zhu, J. Mater. Chem. A, 2020, 8, 18043–18054 RSC.
- D. Yu, Z. Ma, Z. Liu, X. Jiang, H. A. Younus, X. Wang and S. Zhang, Chem. Eng. J., 2023, 457, 141043 CrossRef CAS.
- T. Kim, S. Yun, J. Chae, H. Kim and U. Choi, ACS Appl. Energy Mater., 2023, 7, 48–60 CrossRef.
- X. Pan, Q. Hou, L. Liu, J. Zhang, M. An and P. Yang, Ionics, 2021, 27, 2045–2051 CrossRef CAS.
- Z. Shen, Y. Cheng, S. Sun, X. Ke, L. Liu and Z. Shi, Carbon Energy, 2021, 3, 482–508 CrossRef CAS.
- M. Liu, S. Zhang, E. van Eck, C. Wang, S. Ganapathy and M. Wagemaker, Nat. Nanotechnol., 2022, 17, 959–967 CrossRef CAS PubMed.
- L. Zhu, J. Chen, Y. Wang, W. Feng, Y. Zhu, S. Lambregts, Y. Wu, C. Yang, E. van Eck, L. Peng, A. Kentgens, W. Tang and Y. Xia, J. Am. Chem. Soc., 2024, 146, 6591–6603 CrossRef CAS.
- W. Lin, H. Yuan, C. Tian, M. Song, T. Huang and A. Yu, Energy Storage Mater., 2024, 70, 103472 CrossRef.
- S. Xue, S. Chen, Y. Fu, H. Zhu, Y. Ji, Y. Song, F. Pan and L. Yang, Small, 2023, 19, 2305326 CrossRef CAS.
- R. Xu, X. Shen, X. X. Ma, C. Yan, X. Q. Zhang, X. Chen, J. F. Ding and J. Q. Huang, Angew. Chem., Int. Ed., 2021, 60, 4215–4220 CrossRef CAS.
- K. Xu, J. Power Sources, 2023, 559, 232652 CrossRef CAS.
- S. Liu, X. Ji, N. Piao, J. Chen, N. Eidson, J. Xu, P. Wang, L. Chen, J. Zhang, T. Deng, S. Hou, T. Jin, H. Wan, J. Li, J. Tu and C. Wang, Angew. Chem., Int. Ed., 2021, 60, 3661–3671 CrossRef CAS PubMed.
- N. Piao, S. Liu, B. Zhang, X. Ji, X. Fan, L. Wang, P. F. Wang, T. Jin, S. C. Liou, H. Yang, J. Jiang, K. Xu, M. A. Schroeder, X. He and C. Wang, ACS Energy Lett., 2021, 6, 1839–1848 CrossRef CAS.
- X. Ma, J. Yu, Y. Hu, J. Texter and F. Yan, Ind. Chem. Mater., 2023, 4, 101379 CAS.
- G. Huang, Y. Liao, X. Zhao, X. Jin, Z. Zhu, M. Guan and Y. Li, Adv. Funct. Mater., 2022, 33, 2211364 CrossRef.
- W. Fang, Z. Wen, L. Chen, Z. Qin, J. Li, Z. Zheng, Z. Weng, G. Wu, N. Zhang, X. Liu, X. Yuan and G. Chen, Nano Energy, 2022, 104, 107881 CrossRef CAS.
- Y. Gao, G. Wu, W. Fang, Z. Qin, T. Zhang, J. Yan, Y. Zhong, N. Zhang and G. Chen, Angew. Chem., Int. Ed., 2024, 63, e202403668 CrossRef CAS.
- J. Wang, J. Yang, Q. Xiao, J. Zhang, T. Li, L. Jia, Z. Wang, S. Cheng, L. Li, M. Liu, H. Liu, H. Lin and Y. Zhang, Adv. Funct. Mater., 2021, 31, 2007434 CrossRef CAS.
- F. Zhang, Z. Wang, L. Wang, W. Li, A. Pan, H. Song, J. Xu, J. Hu and X. Wu, Chem. Eng. J., 2022, 435, 135101 CrossRef CAS.
- J. Yu, X. Ma, X. Zou, Y. Hu, M. Yang, J. Yang, S. Sun and F. Yan, Energy Environ. Sci., 2024, 17, 4519–4530 RSC.
- Y. Qin, H. Wang, J. Zhou, R. Li, C. Jiang, Y. Wan, X. Wang, Z. Chen, X. Wang, Y. Liu, B. Guo and D. Wang, Angew. Chem., Int. Ed., 2024, 63, e202402456 Search PubMed.
- J. S. Kim, G. Yoon, S. Kim, S. Sugata, N. Yashiro, S. Suzuki, M. J. Lee, R. Kim, M. Badding, Z. Song, J. M. Chang and D. Im, Nat. Commun., 2023, 14, 782 CrossRef CAS.
- M. Shen, L. Zhang, C. Li, X. Feng, R. Zheng, H. Sun, Z. Wang and Y. Liu, J. Electroanal. Chem., 2024, 957, 118126 CrossRef CAS.
- J. Wang, L. Li, H. Hu, H. Hu, Q. Guan, M. Huang, L. Jia, H. Adenusi, K. V. Tian and J. Zhang, ACS Nano, 2022, 16, 17729–17760 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |