DOI:
10.1039/D3TB02834H
(Paper)
J. Mater. Chem. B, 2024,
12, 6577-6586
Administration sequence- and formation-dependent vaccination using acid-degradable polymeric nanoparticles with high antigen encapsulation capability†
Received
30th November 2023
, Accepted 30th May 2024
First published on 30th May 2024
Abstract
Vaccines aim to efficiently and specifically activate the immune system via a cascade of antigen uptake, processing, and presentation by antigen-presenting cells (APCs) to CD4 and CD8 T cells, which in turn drive humoral and cellular immune responses. The specific formulation of vaccine carriers can not only shield the antigens from premature sequestering before reaching APCs but also favorably promote intracellular antigen presentation and processing. This study compares two different acid-degradable polymeric nanoparticles that are capable of encapsulating a moderately immunogenic antigen, GFP, at nearly full efficacy via electrostatic interactions or molecular affinity between His tag and Ni–NTA-conjugated monomners. This resulted in GFP-encapsulating NPs composed of ketal monomers and crosslinkers (KMX/GFP NPs) and NTA-conjugated ketal monomers and crosslinkers (NKMX/GFP NPs), respectively. Encapsulated GFP was found to be released more rapidly from NKMX/GFP NPs (electrostatic encapsulation) than from KMX/GFP NPs (affinity-driven encapsulation). In vivo vaccination studies demonstrated that while repeated injections of either NP formulation resulted in poorer generation of anti-GFP antibodies than injections of the GFP antigen itself, sequential injections of NPs and GFP as prime and booster vaccines, respectively, restored the humoral response. We proposed that NPs primarily assist APCs in antigen presentation by T cells, and B cells need to be further stimulated by free protein antigens to produce antibodies. The findings of this study suggest that the immune response can be modulated by varying the chemistry of vaccine carriers and the sequences of vaccination with free antigens and antigen-encapsulating NPs.
Introduction
Since its discovery, vaccination has played a pivotal role in improving human health. The demand for vaccines that can efficiently activate the immune system for a desired response against a pathogenic target has become greater than ever with the potential emergence of serious health threats, such as the recent COVID-19 pandemic.1–5 Immune activation starts with introducing an antigen into antigen-presenting cells (APCs) that can trigger the cascade of adaptive immunity, including humoral and cellular responses against the antigen.6–10 Though many forms of antigenic proteins are poorly immunogenic upon administration, the design of efficient, safe, and versatile delivery carriers can overcome this limitation.11–15 CD4 and CD8 T cells are prime targets of vaccination for their central roles in initiating the cascade of immune response.16–18 Ideal vaccine carriers adeptly encapsulate antigens, effectively deliver and release them within APCs, and facilitate the presentation of the antigenic peptides to targeted CD4 or CD8 T cells.
The molecular tunability of polymeric nanoparticles (NPs) for physical (e.g., size) and chemical (e.g., degradability) properties makes them promising antigens and adjuvant carriers for versatile and efficient vaccination.19–23 Their multifaceted function of efficient and facile encapsulation of antigens and intracellular release within APCs have been successfully employed for successful vaccination.24–27 After uptake into APCs, exogenous protein antigens are degraded into peptides in endosomal compartments, which are in turn transported back to the cell surface by class II MHC molecules for recognition by antigen receptors of CD4 T cells. In contrast, the transport of peptides by class I MHC molecules for recognition by CD8 T cells requires entry or origination of the antigen in the cytoplasm. Thus, a potentially useful property of polymeric NPs is assisting the antigen to escape from the endosomal pathway to the cytoplasm to promote both CD4 and CD8 activation. However, accomplishing loading of both class I and II MHC pathways remains a major technical challenge for polymeric NP vaccines.28,29
Conventional vaccines are repeatedly administered in the same formulations. Considering the diverse and distinct antigen processing and presentation pathways in generating humoral versus cellular immunity, vaccination with varying formulations in different orders could provide versatility and targeted immune activation. This proof-of-concept study demonstrates the efficacy of acid-degradable polymeric NPs capable of efficiently encapsulating a broad range of recombinant protein antigens in their capacity of delivering the antigen payloads to APCs for antigen presentation to T cells toward immune modulations for targeted vaccination outcomes.
Results and discussion
Efficient encapsulation of GFP in relatively monodispersed KMX/GFP and NKMX/GFP NPs
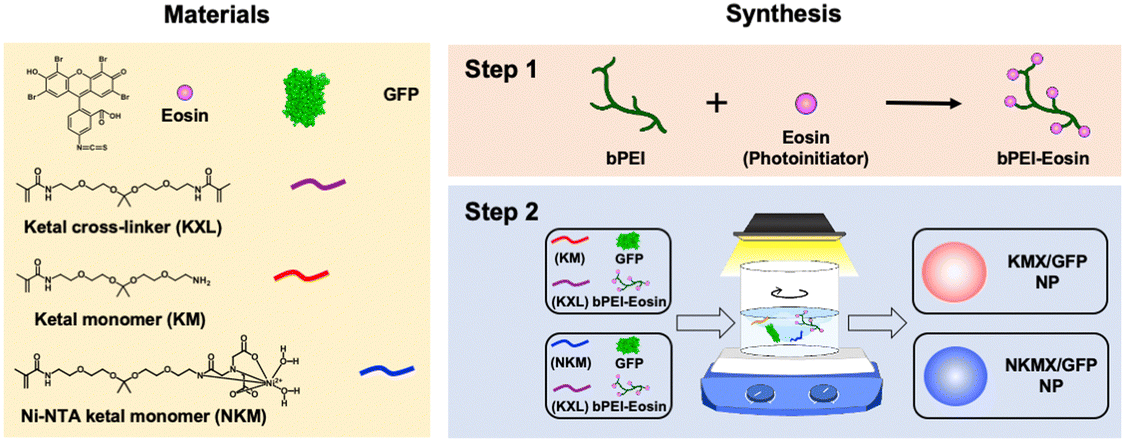
Efficient vaccination requires a formulation with an efficient and versatile antigen encapsulation. His-tagged GFP was encapsulated in KMX/GFP and NKMX/GFP NPs, synthesized from an acid-cleavable amino ketal methacrylamide monomer (KM), Ni–NTA ketal methacrylamide monomer (NKM), and ketal bismethacrylamide crosslinker (KXL), via attractive electrostatic interactions alone or in combination with the molecular affinity between His-tag and Ni–NTA, respectively (Fig. 1 and Fig. S1, ESI†). GFP is known to be anionic at neutral pH with a pKa of 6.030 and can be efficiently retained in a cationic molecular environment such as inside KMX NPs. Not all protein antigens are necessarily anionic, but many recombinant proteins are often His-tagged for separation and purification. Therefore, affinity-based protein encapsulation by Ni–NTA provides the opportunity to deliver a broad range of recombinant protein antigens. KM and NKM were pre-incubated with His-tagged GFP along with KXL prior to photopolymerization from the surface of eosin Y-conjugated PEI, as illustrated in Fig. 1. This surface-initiated photopolymerization enables the synthetic flexibility of controlled, relatively monodispersed size, differential protein loading, and differential structure construction, and freedom from GFP-free smaller particles, as previously validated.31 Finally, the monomers and crosslinkers contain an acid-cleavable ketal linkage used for intracellular delivery of drugs, nucleic acids, and proteins in various applications.
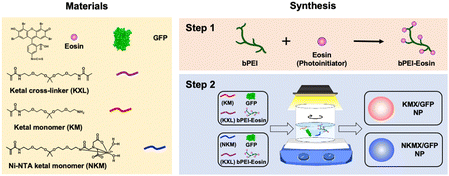 |
| | Fig. 1 Synthesis of KMX/GFP and NKMX/GFP NPs. | |
The sizes of KMX/GFP and NKMX/GFP NPs were approximately 170 and 230 nm in diameter with relatively narrow size distributions indicated by polydispersity indices of 0.26 and 0.22, respectively (Fig. 2A). The size of NKMX/GFP NPs was slightly larger than that of KMX/GFP, attributed to the relatively bulky Ni–NTA end of NKM in comparison to KM with the amino end of KM (Fig. 1). This observation also implies that the charge of KM is sufficient for electrostatic interaction with a counter-charged protein. TEM images showed that both NPs were spherical with no aggregation (Fig. 2A). As speculated, the incorporation of NKM into NPs decreased the surface charge (Fig. 2B) from 17.3 mV (KMX/GFP NPs) to 12.8 mV (NKMX/GFP NPs) (Fig. 2B). Regardless of the different molecular ratios of KM to NKM in both NPs, the majority of His-tagged GFP was encapsulated with efficiencies of approximately 93% and 96% in KMX/GFP and NKMX/GFP NPs, respectively. These results indicated that the incorporation of NKM into the NPs moderately improved GFP encapsulation with an increased size and lowered surface charge.
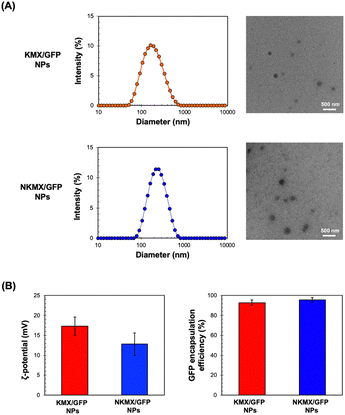 |
| | Fig. 2 Characterization of KMX/GFP and NKMX/GFP NPs: (A) size and morphology determined by DLS particle analysis and observed by TEM, and (B) zeta potential and GFP encapsulation efficiency (n = 5). | |
GFP release from KMX/GFP and NKMX/GFP NPs at endosomal pH
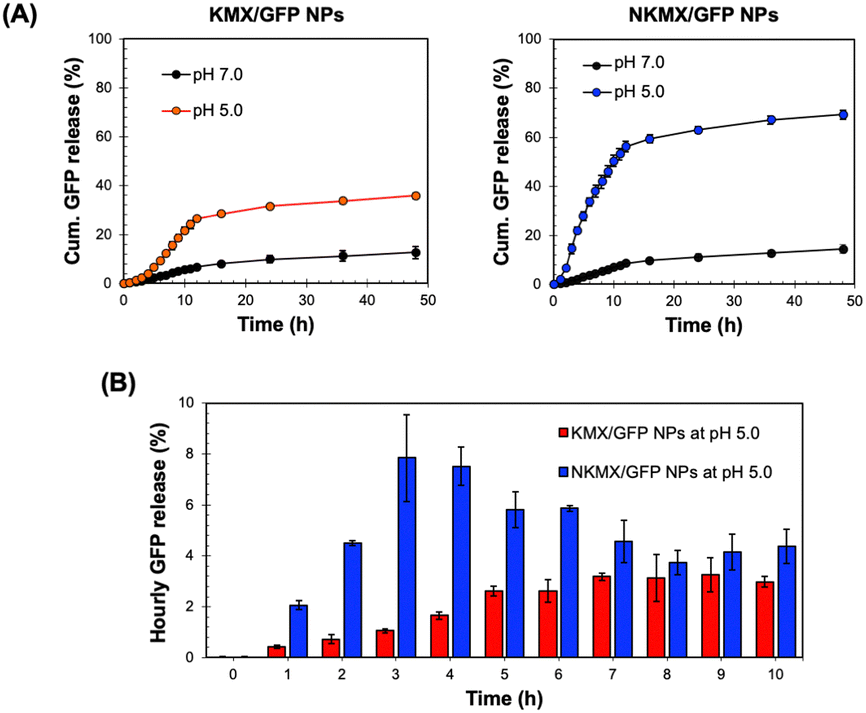
Antigens delivered to an antigen-presenting cell (APC) are processed via endogenous or exogenous pathways, depending on their intracellular localization.32 Therefore, the rapid release of antigens from NPs determines not only the overall antigen presentation efficacy but also whether it is presented by MHC I or MHC II on the surface. At a physiological pH of 7.0, only a small fraction (10–15%) of encapsulated GFP was released from the NPs after 48 h, while a mildly acidic endosomal pH triggered a much greater release of GFP (Fig. 3A). In contrast to the similar release at pH 7.0, a substantially higher amount of GFP was released from NKMX/GFP NPs (∼69%) than KMX/GFP NPs (∼36%) after 48 h. Notably, while the time required to reach a plateau of accumulated GFP release was almost identical for both NPs, there was an initial lag in GFP release from the KMX/GFP NPs (Fig. 3A). The almost 2-fold difference in the overall GFP release between NPs and the greater initial GFP release from NKMX/GFP NPs can be attributed to a stronger attractive electrostatic interaction between the protonated amino end of KM at acidic pH and GFP. Likewise, it was confirmed that approximately twice as much GFP was still retained in KMX/GFP NPs than in NKMX/GFP NPs when unreleased GFP was measured (data not shown). His-tagged GFP in NKMX/GFP NPs was released immediately upon hydrolysis of the ketal linkage, while the strengthened cationic environment KMX/GFP NPs retained GFP from rapid release. This outcome was further supported by the GFP detected per hour over the first 10 h (Fig. 3B). This result demonstrated that GFP was released from NKMX/GFP NPs in a burst-like manner while KMX/GFP NPs released GFP in a sustained manner. The results in Fig. 3 suggest a rapid release of GFP from NKMX/GFP NPs in mildly acidic endosomes, possibly leading to fast degradation in comparison to KMX/GFP NPs.
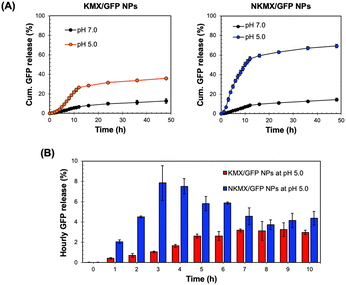 |
| | Fig. 3 Acid-triggered release of GFP from KMX/GFP and NKMX/GFP NPs in DI water at 37 °C in a shaking incubator: (A) accumulated release for 48 h and (B) hourly release for the initial 10 h (mean ± SD; n = 3). | |
Intracellular trafficking of GFP released from KMX/GFP and NKMX/GFP NPs
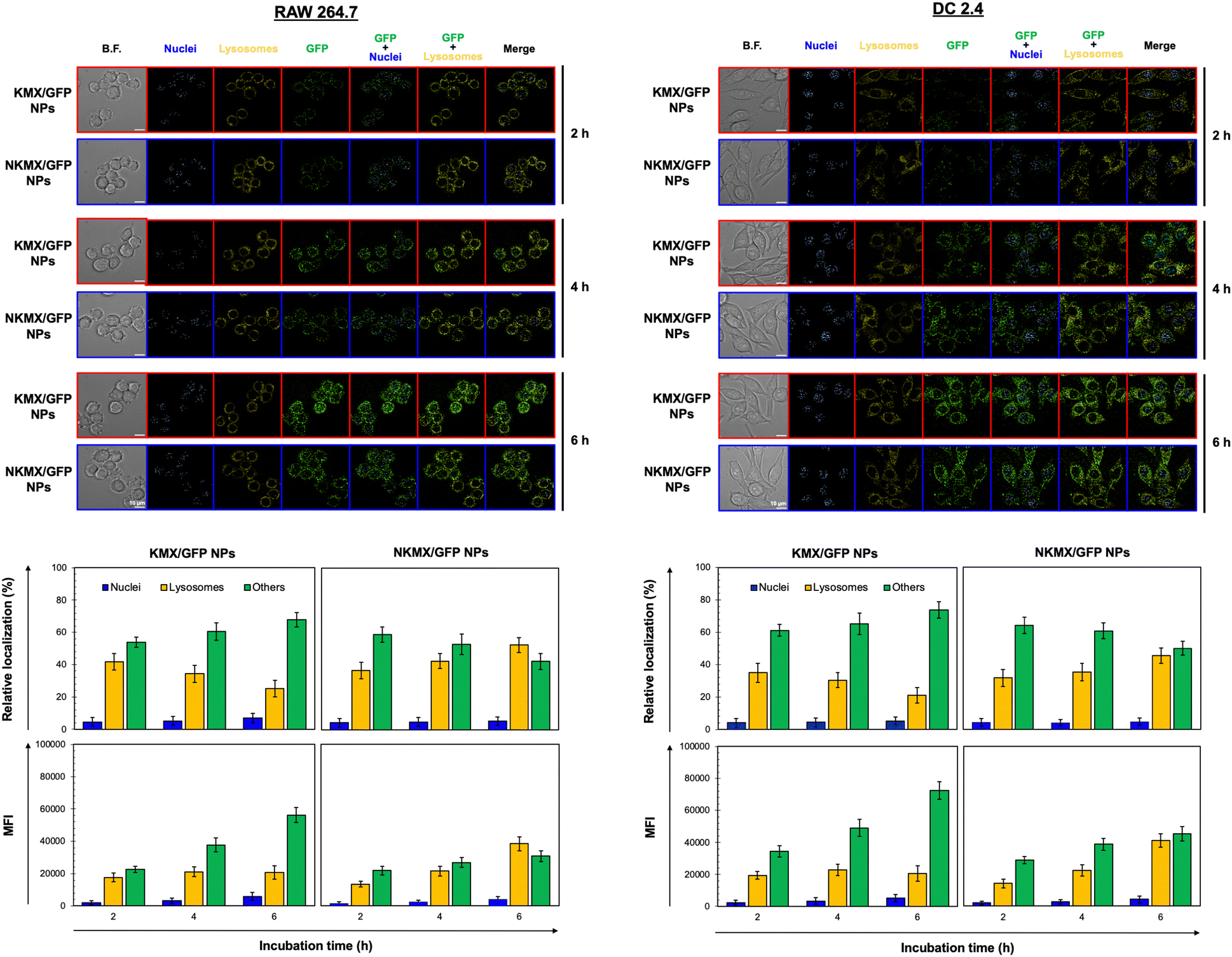
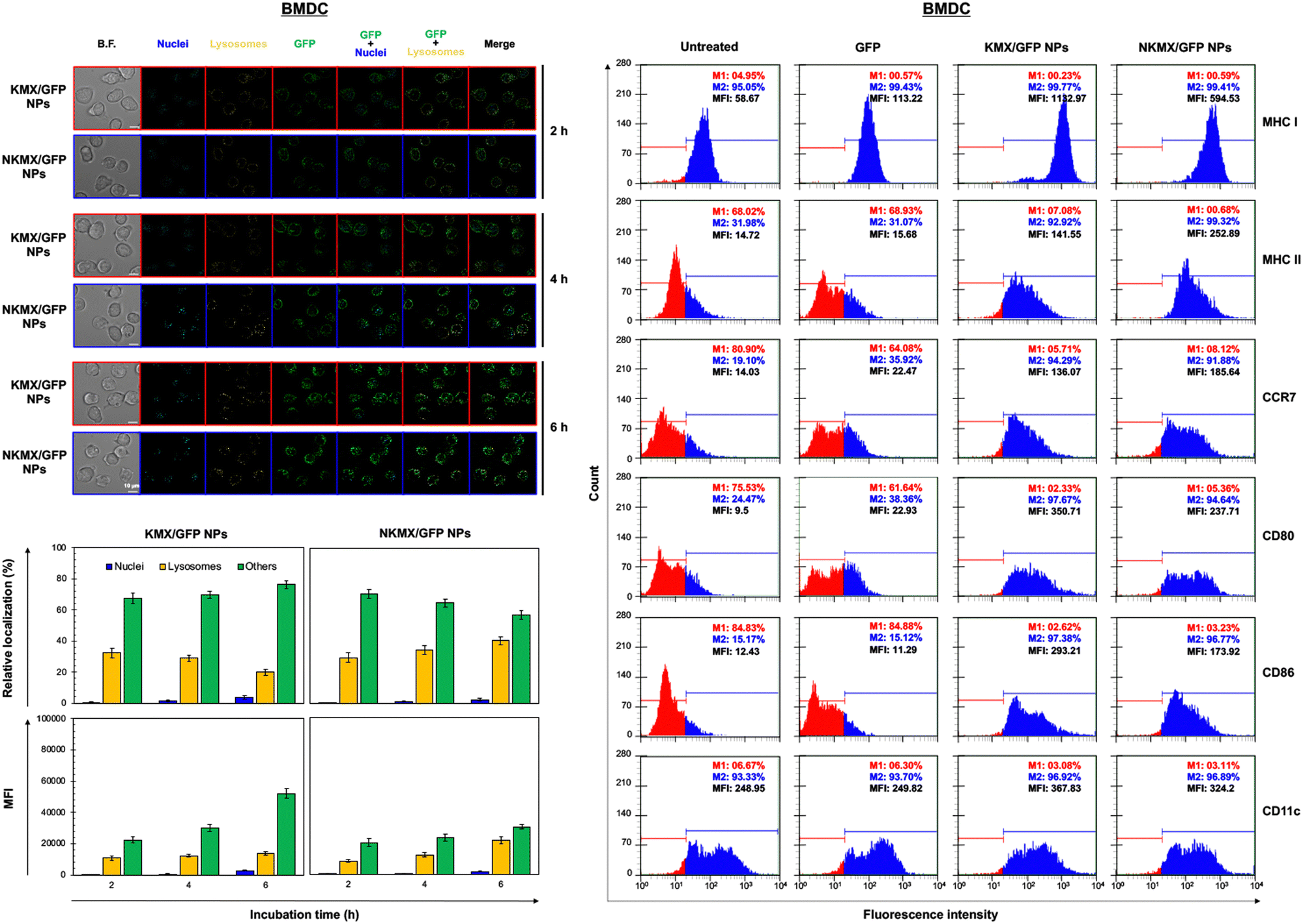
Upon administration, vaccines are taken up by APCs, including macrophages and dendritic cells (DCs). While macrophages and DCs are known to be effective in degrading and salvaging antigens, the delivery of antigenic proteins to them, especially in route to MHC I pathway, is challenging.33,34 The intracellular distributions of GFP released from NPs in the nucleus, lysosome, and/or other places (e.g., cytoplasm) were tracked using a superresolution fluorescence microscope, which is capable of imaging the nanoparticles with a resolution of ∼20 nm.35 The capability of acquiring the highest resolution images is traded off with the inability of confocal imaging, which results in overlaid but pinpointed fluorescence location of GFP in the nuclei even at the highest intensity settings. The analysis showed that GFP quickly accumulated in the lysosome and cytoplasm, with very little accumulation found in the nucleus (Fig. 4 and 5). The relative accumulation of GFP in the lysosome decreased with time, while its release into the cytoplasm increased when delivered by KMX/GFP NPs. In contrast, GFP found in the lysosome increased with the incubation time, and its release into the cytoplasm decreased when NKMX/GFP NPs were incubated with RAW 264.7 cells, DC2.4 cells, and BMDCs (Fig. 4 and 5). One possible explanation for this observation is stronger proton buffering and GFP retention by KMX NPs in the lysosome, resulting in a faster cytosolic release of intact proteins (GFP). When delivered by NKMX/GFP NPs, GFP is quickly released into the reducing environment in the lysosome with slower release into the cytoplasm due to limited proton buffering. Previous studies have also demonstrated that successful delivery of a therapeutic payload (e.g., proteins and nucleic acids) requires cytosolic release, avoiding premature degradation and inactivation.36,37 The relatively slower cellular uptake of NKMX/GFP NPs than KMX/GFP NPs, as indicated by mean fluorescence intensity (MFI), likely contributed to the slower cytosolic release (Fig. 4 and 5). Antigen delivery in carriers such as NPs could be beneficial for extended bioavailability, avoidance of immediate immune response, and targeted uptake by phagocytic cells, which together can improve both vaccination efficacy and safety.38,39 However, they would require utilizing an energy-dependent route (e.g., macropinocytosis) or via a structurally challenging uptake process (e.g., different membrane potential).40–42 In contrast to NPs that contain multiple GFP proteins inside, free GFP proteins parsley distributed in a cell were not highly visible (Fig. S2, ESI†). When incubated with RAW 264.7 cells, DC 2.4 cells, and BMDCs, KMX/GFP and NKXM/GFP NPs were found to be minimally toxic with relative viability of RAW 264.7 cells at ∼91 and ∼71%, DC 2.4 cells at ∼89 and ∼62%, and BMDCs at ∼94 and 86%, respectively, at a highest concentration of 200 μg mL−1 NPs (Fig. S3, ESI†). Despite the lower zeta potential than KMX/GFP NPs (Fig. 2B), Ni–NTA groups in NKMX/GFP NPs contributed to cytotoxicity.43 At a concentration of 100 μg mL−1, KMX/GFP and NKMX/GFP NPs showed tolerable cytotoxicity of 20% or lower, allowing vaccination at a dose of up to 0.3 μg mL−1 antigens. Incubation of BMDCs with KMX/GFP and NKMX/GFP NPs induced maturation, as indicated by the upregulated expression of MHC II, CCR7, CD80, and CD86 (Fig. 5). This was attributed to the elevated antigen processing and presentation activities when NP vaccines were taken up by BMDCs.44 In contrast, RAW 264.7 macrophages showed only a marginally upregulated CCR7, and DC 2.4 cells remained unchanged for the expression of surface markers (Fig. S4 and S5, ESI†). This observation implies efficient in vivo DC activation upon vaccination with KMX/GFP and NKMX/GFP NPs.
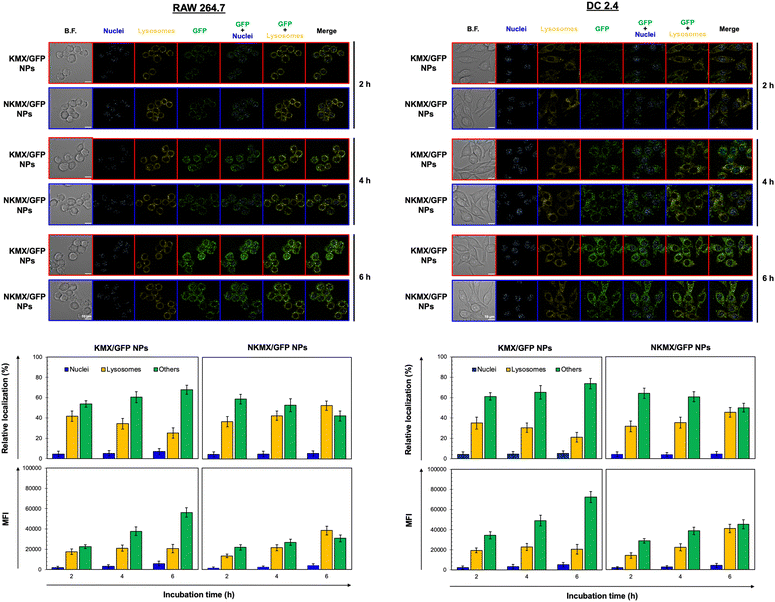 |
| | Fig. 4 Intracellular distribution and accumulation of GFP delivered by KMX/GFP and NKMX/GFP NPs in RAW 264.7 and DC 2.4 cells for 2, 4, and 6 h. The fluorescence of GFP in the cells was imaged by superresolution fluorescence microscopy (image n = 8–10), and the colocalized fluorescence of GFP and intracellular organelles was further quantified. In addition, the cellular uptake of KMX/GFP and NKMX/GFP NPs was quantified as mean fluorescence intensity (MFI). | |
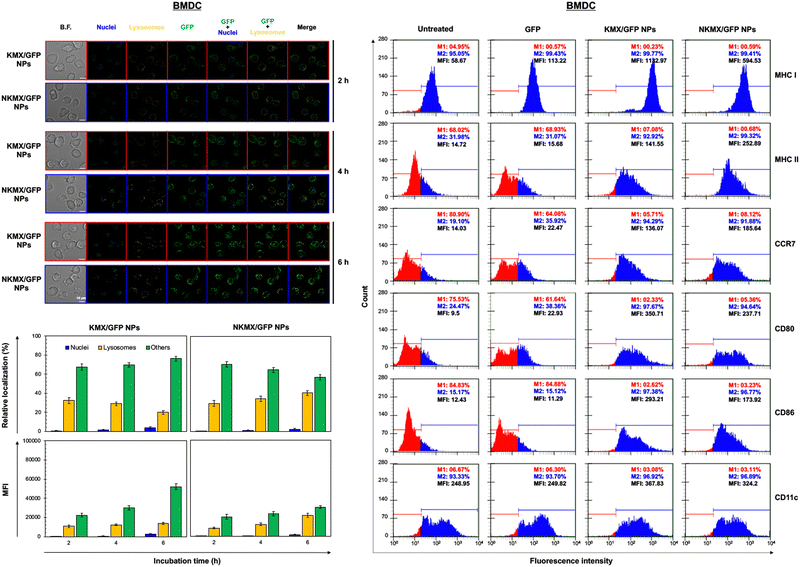 |
| | Fig. 5 Intracellular distribution and accumulation of GFP delivered by KMX/GFP and NKMX/GFP NPs in BMDCs for 2, 4, and 6 h, and the changes of cell surface markers on BMDCs after incubation with KMX/GFP and NKMX/GFP NPs. The fluorescence of GFP in the cells was imaged by superresolution fluorescence microscopy (image n = 7–9), and the colocalized fluorescence of GFP and intracellular organelles was further quantified. In addition, the cellular uptake of KMX/GFP and NKMX/GFP NPs was quantified as mean fluorescence intensity (MFI). Cell surface markers on BMDCs after incubation with free GFP, KMX/GFP NPs, and NKMX/GFP NPs for 6 h were analyzed by flow cytometry. | |
Immune response determined by the order of vaccination in varying formulations
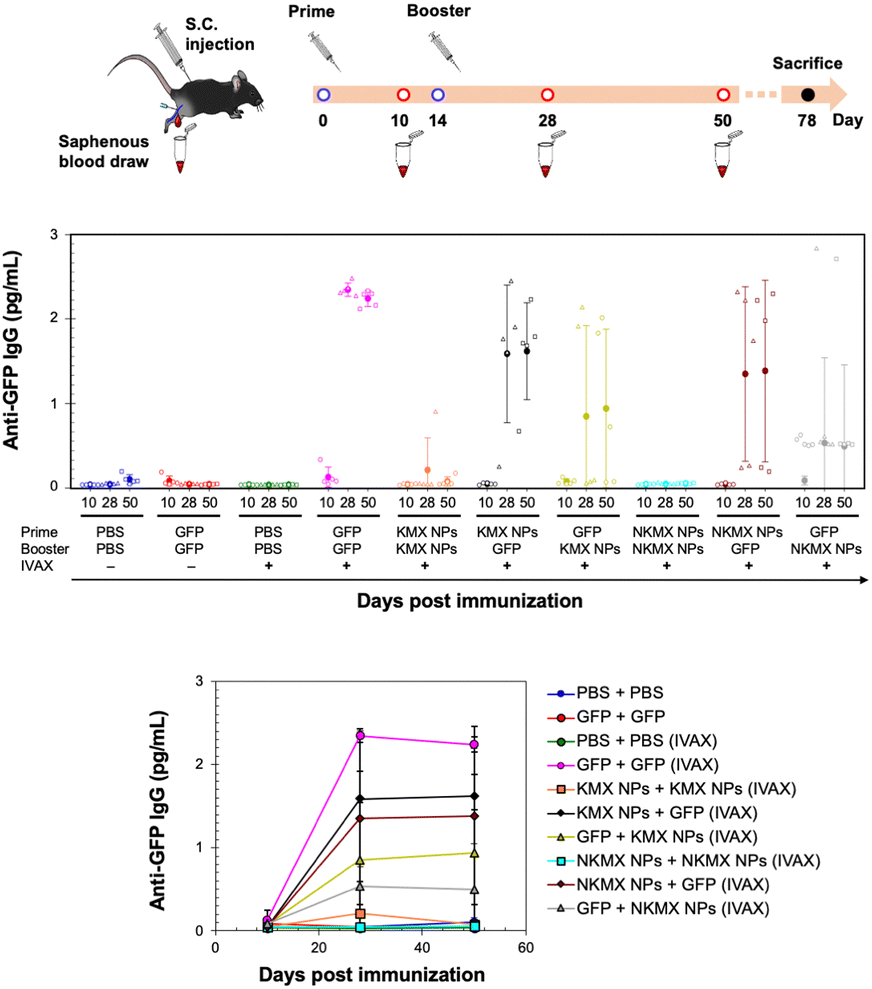
In vivo studies allow the assessment of humoral immunity initiated by antigen presentation to T cells. C57BL/6 mice, a common animal model in immunology, were vaccinated with 5 μg of GFP, KMX/GFP NPs, or NKMX/GFP NPs in PBS, in various combinations of GFP and NPs for prime (Day 0) and booster (Day 14) injections, with or without IVAX-1 adjuvant (CpG/MPLA/AddaVAX) (Fig. 6 and Fig. S6, ESI†). The animals were bled on Day 10 (after the prime but before the booster injection), Day 28 (2 weeks after the booster injection), and Day 50 (∼ a month after complete vaccination), followed by evaluation of the generation of antibodies against GFP by ELISA. Robust anti-GFP IgG responses were induced by vaccination with GFP–GFP, KMX/GFP NPs–GFP, and NKMX/GFP NPs–GFP (prime–booster) and the efficiently activated humoral immunity lasted about a month with no changes. No difference in the IgG2a/IgG1 ratio, observed between KMX/GFP NPs and NKMX/GFP NPs (Fig. S7, ESI†), confirmed their roles in the Th1- or Th2-biased polarization of humoral immunity. In contrast, administration of KMX/GFP NPs and NKMX/GFP NPs for both prime and boost injections did not yield measurable antibodies, consistent with the notion that the antigen is sequestered inside the NPs until it is endocytosed and intracellularly degraded in an APC. Processed peptides are presented in the context of MHC molecules on T cells at the APC surface. This finding emphasized the importance of administering soluble antigens (GFP), during the prime and/or boost injection to induce antibodies. It was demonstrated that the administration of NPs for prime followed by soluble GFP for boost induced higher IgG signals than groups in which soluble GFP was given first. This may indicate a role for NPs in modulating antibody response via prime injection. Overall, the production of anti-GFP antibodies by NPs was not as effective as that of free GFP. This might be attributed to the higher number of free proteins than NPs for the same amount of GFP, which is also attributed to the relatively low statistical differences among the treatment groups and warrants a subsequent study at a higher dose than that used in the current study. Regardless of the formulation, IVAX-1 was indispensable for efficient antibody generation against relatively weak antigens, such as GFP. Further studies are required to more precisely define the potential of these NPs to enhance or downregulate both T cell and antibody responses. In particular, conventional vaccination strategies repeatedly administer the same formulations in prime and boost(s). However, the results shown in Fig. 6 indicate the possibility of modulating the immune response by varying the immunization sequence with varying formulations.
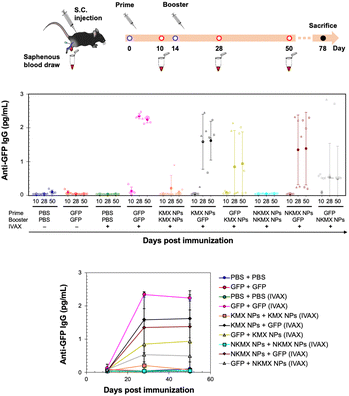 |
| | Fig. 6 Humoral response to GFP, KMX/GFP NPs, and NKMX/GFP NPs in varying combinations of prime and booster injections. C57BL/6 mice were vaccinated twice on Day 14, and their blood samples were collected on Days 10, 28, and 50 for the analysis of anti-GFP antibodies by ELISA (n = 5). | |
Hypothesis of humoral immune responses to antigen formulations in varying administration orders
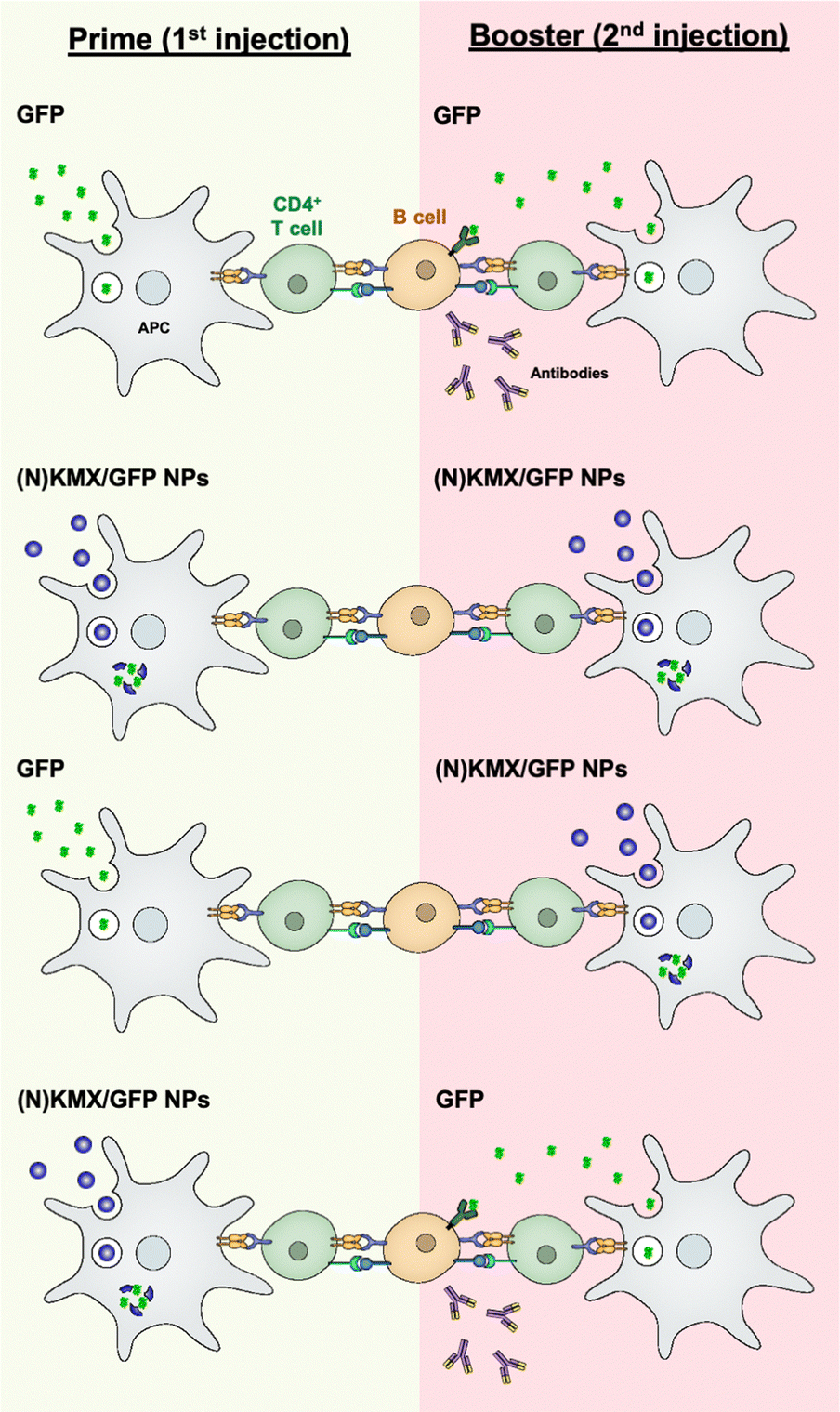
Antigen presentation by APC to T cells plays a pivotal role in orchestrating adaptive immunity, including humoral and cellular responses, which is the prime target for achieving efficient vaccination. The results of this study indicate that the magnitude and type of immune response can be modulated by antigen formulations (soluble vs. encapsulated) and the sequence of their administration (summarized in Fig. 7). Free proteins circulate upon injection before being internalized by an APC mainly via micropinocytosis,45 degraded in the lysosome into peptides, and loaded onto MHC II for antigen presentation46 that are required for B cell-mediated antibody production. We proposed that the KMX/GFP and NKMX/GFP NPs used in this study protect their cargo upon administration, rapidly degrade in the mildly acidic lysosome of an APC after endocytosis, and rapidly release their payload not only to the lysosome but also to the cytoplasm (Fig. 4 and 5). Free antigens are also available to bind to the corresponding antigen receptors on B cells, which differentiate into antibody-secreting plasma cells with cognate help from CD4 T cells. When GFP was delivered as a free protein, its antigenic peptides were simultaneously presented to CD4 T cells and to B cells. When administered again as a booster, it is processed for the same purpose and simultaneously binds to the B cells that are already primed from the prior vaccination, generating a strong humoral response, including secreting antibodies (the first scenario in Fig. 7). We propose that GFP delivered by acid-degradable NPs is processed in an APC, and processed GFP peptides are presented to CD4 cells, which are required for B cell differentiation. Repeated vaccination with GFP-encapsulating NPs does not provide free GFP to stimulate activated B cells (the second scenario in Fig. 7). When GFP is delivered to NPs as a booster, B cells that are activated upon prime injection of GFP are not activated to become antibody-producing plasma cells (the third scenario in Fig. 7). It has been proposed that GFP released from NPs in an APC is processed and presented as peptides to CD4 T cells. Booster injection of free GFP enables the activation of B cells to differentiate into plasma cells (the fourth scenario in Fig. 7). The results of this study allow us to develop vaccine strategies for the activation of humoral immunity. The findings presented in this study indicate that the administration of various antigen formulations for prime and booster injections could improve strategies for achieving efficient and targeted vaccination. Studies are underway to test whether this platform, when used to deliver immunodominant antigens from specific pathogens, can influence immunogenicity, particularly for broadening antibody profiles and improving efficacy against challenge. One of the areas to be investigated in a subsequent study is to determine whether KMX/GFP and NKMX/GFP NPs affect Th1- vs. Th2-biased polarization of humoral immunity, which may provide a new insight into the molecular modulation of the immune system to generate the desired modes of immune response.
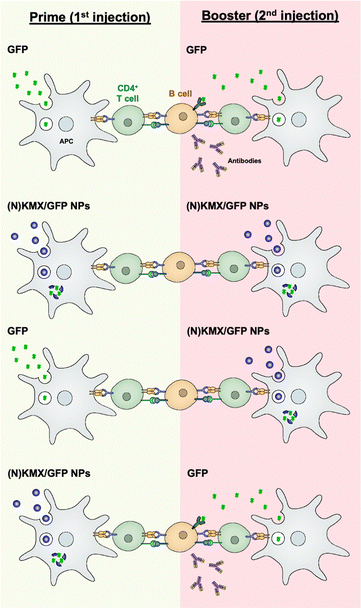 |
| | Fig. 7 Schematic showing hypothesized modulation of the humoral immune response by alternating administration of free antigens and antigen-encapsulating NPs. | |
Conclusions
Successful vaccination is measured by the magnitude and type of immune response to the target antigen. In this study, a model antigen (GFP) was encapsulated in acid-degradable polymeric NPs via electrostatic interactions and molecular affinity. The resulting vaccine carriers were shown in vitro to release the antigen payload once internalized by an APC in response to the mildly acidic lysosome. Interestingly, the magnitude and quality of the immune response in mice were greatly affected by antigen formulation and vaccination sequence. While repeated vaccination with antigenic proteins was most efficient in eliciting an immune response, only limited humoral immunity was observed when antigen-encapsulating NPs were used for both prime and booster injections. Alternating free antigens and antigen-encapsulating NPs for prime and booster injections generated qualitatively different antibody productions, which is indicative of the modulation of the immune response by this approach. The findings of this study suggest that the efficacy of a vaccine may be affected by the immunological nature of the antigen and its delivery carriers. For example, the GFP used in this study is weakly immunogenic, and the magnitude of the immune response observed in this study is likely affected by the immunogenic nature of different antigens. The current proof-of-concept study needs to be further extended to investigate the roles of more common antigens, such as ovalbumin, hemagglutinin, and viral surface antigens (e.g., gp120 on HIV and SARS-CoV-2 spike proteins). Likewise, the polymeric NPs used in this study intracellularly release GFP in APC. Vaccination with antigen-encapsulating NPs with different intracellular behaviors, such as stimuli-responsive degradability and localization, may show different traits of stimulating and modulating the immune response. Overall, this study suggests that versatile strategies, including formulations and sequences, need to be considered in order to accomplish efficient and targeted vaccination.
Experimental
Materials
Branched polyethylenimine (bPEI, 25 kDa) and Hoechst 33342 dye were purchased from Sigma-Aldrich (St. Louis, MO). Eosin-5-isothiocyanate and LysoTrackerTM Red DND-99 dye were purchased from Thermo Fisher Scientific (Waltham, MA). RAW 264.7 murine macrophage cells and DC 2.4 murine dendritic cells (ATCC, Rockville, MD) were cultured in Dulbecco's modified Eagle's medium (DMEM) and RPMI 1640 (MediaTech, Manassas, VA), respectively, with 10% fetal bovine serum (FBS) (Atlanta Biologicals, Flowery Branch, GA) and 1% antibiotics (100 units per mL penicillin; 100 μg per mL streptomycin) (Gibco, Grand Island, NY) at 37 °C, 5% CO2, and 95% humidity. AddaVAX™ (squalene oil-in-water emulsion) was purchased from InvivoGen (San Diego, CA). TLR9 agonist, CpG 1018-ODN, and TLR4 agonist, monophosphoryl lipid A (MPLA), were purchased from Integrated DNA Technologies (Coralville, Iowa) and Avanti Polar Lipids (Alabaster, AL), respectively. GFP with a 6-His tag was expressed in Escherichia coli BL21 and purified using His-Pur Ni–NTA resin chromatography and Triton-X114 endotoxin removal. Purified proteins were evaluated by SDS-PAGE and bicinchoninic acid (BCA) protein assays (Pierce Biotechnology, Rockford, IL).
Preparation and characterization of KMX/GFP and NKMX/GFP nanoparticles (NPs)
Acid-degradable NPs were synthesized via surface-initiated photopolymerization of acid-degradable amino-ketal methacrylamide (KM) and Ni–NTA-ketal methacrylamide (NKM) monomers (Fig. S1, ESI†), and ketal bismethacrylamide crosslinker (KXL), for electrostatic or affinitive encapsulation of GFP as an antigen (Fig. 1). as described in the ESI† in detail. For the synthesis of KMX/GFP NPs, 20 μL of 100 mg mL−1 KM (2 mg), 10 μL of 100 mg mL−1 KXL (1 mg), 10 μL of 1 mg mL−1 GFP (10 μg), and 50 μL of 200 mg mL−1 ascorbic acid (10 mg), all in deionized (DI) water, were mixed dropwise with 910 μL of 86.4 μg mL−1 PEI–eosin conjugate, a total mixing volume of 1 mL, with vigorous stirring for 10 min. For the synthesis of NKMX/GFP NPs, 20 μL of 100 mg mL−1 Ni–NTA-KM (2 mg), 10 μL of 100 mg mL−1 KXL (1 mg), 10 μL of 1 mg mL−1 GFP (10 μg), and 50 μL of 200 mg mL−1 ascorbic acid (10 mg) were also mixed with 910 μL of 86.4 μg mL−1 PEI–eosin conjugate in the same way. Then, the mixture was photopolymerized under a halogen lamp at 700 klux for 10 min with vigorous stirring, followed by additional stirring for 10 min without light. Unreacted monomers, crosslinkers, and other reagents were removed by centrifugal filtration (MWCO 100 kDa, Millipore, Bedford, MA) three times at 3000 g and 4 °C for 10 min. The resulting KMX/GFP and NKMX/GFP NPs were finally re-suspended in 1 mL of DI water and stored at 4 °C before being used for further studies.
The size and zeta potential of KMX/GFP and NKMX/GFP NPs were measured using a dynamic light scattering (DLS) particle analyzer (Zetasizer Nano ZS, Malvern Panalytical, Malvern, UK), with a refractive index of 1.59 and an absorption of 0.01 at 25 °C. For 1 mL of KMX/GFP and NKMX/GFP NPs at a concentration of 10 μg His-tagged GFP/mL, each size measurement was conducted with a series of 15 runs using disposable cuvettes (ZEN0040, Malvern Panalytical) and each zeta-potential analysis was conducted with a series of 100 runs using DTS1070 folded capillary cells (Malvern Panalytical). The morphologies of KMX/GFP and NKMX/GFP NPs were observed using transmission electron microscopy (TEM). Briefly, 10 μL of KMX/GFP or NKMX/GFP NPs at the concentration used for DLS and zeta-potential analysis were dropped on a carbon-coated copper grid (Thermo Fisher Scientific), air-dried for 30 min at room temperature, and observed under a JEOL 2100F transmission electron microscope (JEOL, Peabody, MA) at 200 kV. The encapsulation efficiency of His-tagged GFP in KMX/GFP and NKMX/GFP NPs was determined by measuring the excitation/emission (390 nm/510 nm) of the released GFP following hydrolysis using a fluorescence spectrometer (Synergy H1, BioTek, VT). KMX/GFP and NKMX/GFP NPs were hydrolyzed in a 100 mM acetate buffer at pH 5.0 with vigorous stirring at room temperature for 24 h. The released GFP was determined by comparing the hydrolyzed samples with a calibration curve of free His-tagged GFP in DIW at a concentration range of 0–20 μg mL−1.
pH-Triggered GFP release from KMX/GFP and NKMX/GFP NPs
The release of His-tagged GFP from KMX/GFP and NKMX/GFP NPs in a pH-dependent manner was evaluated by measuring the fluorescence of free GFP at the endosomal/phagolysosomal and physiological pH of 5.0 and 7.4, respectively. Briefly, a Spectra-Por® Float-A-Lyzer® G2 dialysis device (MWCO 100 kDa) (Sigma-Aldrich) containing KMX/GFP and NKMX/GFP NPs at a concentration of 5 μg mL−1 GFP in 100 mM acetate buffer (pH 5.0) was incubated on a shaker incubator at 37 °C. At different time points, 4 mL of dialyzed samples were collected while replenishing the buffers in the same volume and analyzed for fluorescence using a Synergy H1 Microplate Fluorescence Reader (Synergy H1, BioTek, VT) at excitation/emission wavelengths of 385 nm/524 nm (pH 5.0) or 390 nm/510 nm (pH 7.4), without further dilution or reconstitution. The amount of GFP released was calculated by comparing the fluorescence of GFP at known concentrations, as described above.
Cytotoxicity and intracellular distribution of KMX/GFP and NKMX/GFP NPs
The dose-dependent toxicity of KMX/GFP and NKMX/GFP NPs in vitro was assessed at concentrations of up to 200 μg mL−1 by the conventional MTT assay using RAW 264.7 cells, DC 2.4 cells, and BMDCs47 seeded at a density of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per well in a 96-well plate with 0.1 mL of medium, 24 h prior to the experiment. After the cells were incubated with KMX/GFP and NKMX/GFP NPs for 24 h, 10 μL of 5 mg mL−1 MTT in PBS was added to each well, followed by further incubation for 2 h. The MTT-containing medium was aspirated, 0.2 mL of DMSO was added to dissolve the formazan crystals produced by the live cells, and the absorbance of the formazan was measured at 570 nm using a SpectraMax Plus microplate reader (Molecular Devices, Sunnyvale, CA). The relative viability of the cells was determined by comparing the absorbance of treated and untreated cells.
000 cells per well in a 96-well plate with 0.1 mL of medium, 24 h prior to the experiment. After the cells were incubated with KMX/GFP and NKMX/GFP NPs for 24 h, 10 μL of 5 mg mL−1 MTT in PBS was added to each well, followed by further incubation for 2 h. The MTT-containing medium was aspirated, 0.2 mL of DMSO was added to dissolve the formazan crystals produced by the live cells, and the absorbance of the formazan was measured at 570 nm using a SpectraMax Plus microplate reader (Molecular Devices, Sunnyvale, CA). The relative viability of the cells was determined by comparing the absorbance of treated and untreated cells.
The intracellular distribution of His-tagged GFP delivered by KMX/GFP and NKMX/GFP NPs was evaluated using a NanoresoTM Super Resolution Fluorescence Microscope (Sysmex Corporation, Kobe, Japan). Briefly, 20000 RAW 264.7 and DC 2.4 cells were seeded in an 8-well chamber dish (Thermo Fisher Scientific, Waltham, MA) and incubated for 24 h. After incubation with KMX/GFP and NKMX/GFP NPs at a GFP concentration of 1 μg mL−1 for 2, 4, and 6 h, the cells were stained with LysoTracker™ Red DND-99 at a concentration of 100 nM for 20 min in order to locate acidic intracellular organelles, and their nuclei were stained with Hoechst 33342 at a concentration of 1 μg mL−1 for 10 min. The cells were rinsed twice with DPBS, and the GFP fluorescence of the cells was captured at 10000 frames per image. The location of intracellular GFP and other fluorophores was precisely determined by analyzing the obtained images using ThunderSTORM for image filtering with a wavelet filter (B-Spline) and result visualization using the normalized Gaussian method and colocalized fluorescence, located in the same location, and was then quantified (% volume colocalized) using Image J software (National Institutes of Health, Bethesda, MD; https://imagej.nih.gov/ij/.) in the analysis mode.
Analysis of cell surface molecules after incubation with KMX/GFP and NKMX/GFP NPs
The cell surface molecules after incubation with KMX/GFP and NKMX/GFP NPs were measured by flow cytometry. Briefly, RAW 264.7, DC2.4, and immature BMDCs were seeded at a density of 200000 per well in a 12-well plate 24 h prior to the experiment. After 6 h of incubation with GFP, KMXGFP, or NKMX/GFP NPs at a concentration of 1 μg mL−1 GFP or equivalent, the cells were trypsinized and stained with PE-Cy7 anti-mouse antibodies against MHCI, MHCII, CCR7, CD80, CD86 or CD11c (BioLegend, San Diego, CA) on ice for 20 min. The cells were then rinsed with 1× PBS three times and analyzed by flow cytometry using the unlabelled samples as controls.
Vaccine adjuvant preparation
To prepare IVAX-1, a customized adjuvant mixture used in this study, CpG1018-ODN,48,49 was dissolved in sterile water at 1 mM as a stock. To overcome this limited aqueous solubility, MPLA was incorporated into DOPG liposomes (an inert co-lipid). Briefly, MPLA and DOPG (both from Avanti Polar Lipids, Alabaster, AL) were dissolved at a molar ratio of 1:5 in chloroform, followed by evaporation under nitrogen and removal overnight under vacuum. The lipid film was then hydrated with 10 mM NaCl to a concentration of 5 mg mL−1 (2.835 mM) MPLA and sonicated in a Branson M1800 sonicating water bath (Branson Ultrasonics Corporation, Brookfield, CT) at room temperature for 15 min until the formulation was translucent with no large visible particles. The particle size distribution of the liposomes was determined to be approximately 100 nm using DLS. AddaVAX™ (squalene oil-in-water emulsion, Invitrogen Inc., San Diego, CA) was used at 50% of the dosing volume in adjuvanted formulations, as recommended by the manufacturer. In this study, mice received IVAX-1 comprising 1 nmol of CpG1018-ODN, 3 nmol of MPLA, and 25 μL of AddaVax as adjuvant. IVAX-1 was developed and well characterized at the UCI Vaccine R&D Center after performing systematic screens of different toll-like receptor (TLR) agonists and emulsions on the immunogenicity of recombinant hemagglutinin (HA) vaccines in mice.49
Vaccination with KMX/GFP and NKMX/GFP NPs
Female C57BL/6 mice (7–10 weeks of age), purchased from Charles River Laboratories (Wilmington, MA), received vaccines comprised of either free soluble His-tagged GFP or His-tagged GFP encapsulated in KMX/GFP or NKMX/GFP NPs, as prepared above. For immunization, 5 μg of His-tagged GFP or equivalent in 50 μL was administered in sterile PBS or IVAX-1 adjuvant via the subcutaneous route (base of tail) under brief anesthesia with inhaled 1% isoflurane/O2 mixture. The mice were weighed and monitored daily for 14 days post prime or boost for any changes in the behavior or development of lesions at the site of injection. At regular time points, plasma was collected into heparinized microcapillary tubes (Minicollect 0.8 mL Z Serum Sep Gold, Greiner Bio-One, Monroe, NC) by saphenous vein bleeding under anesthesia with inhaled 1% isoflurane/O2 and at the experimental endpoint by cardiac puncture under terminal anesthesia. All animal experiments were approved by the UCI Institutional Animal Care and Use Committee (IACUC protocol #AUP-18-096) and by the Animal Care and Use Review Office (ACURO) of the U.S. Army Medical Research and Material Command (USAMRMC). The laboratory animal resources at UCI are internationally accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC #000238).
Anti-GFP antibody quantification by ELISA
His-tagged GFP was used to coat Reacti-Bind microtiter plates (Thermo Fisher Scientific, Waltham, MA) at a concentration of 2 μg mL−1 in TBS (20 mM Tris/150 mM NaCl, pH 7.6; 100 μL per well) overnight at 4 °C. The plates were then washed four times with T-TBS, and TBS containing 0.05% Tween 20 (Thermo Fisher Scientific, Waltham, MA) and blocked with 300 μL per well of casein/TBS blocking buffer (Thermo Fisher Scientific, Waltham, MA) for 1–2 h. The blocking buffer was then aspirated, and the plates were air-dried and stored in desiccated foil pouches at 4 °C until use. For the ELISA assay, sera were diluted to 1/100 in casein/TBS blocking buffer containing E. coli lysate (GenScript, Piscataway, NJ) at 1.5 mg mL−1 final concentration and incubated for 30 min prior to addition to the plates. The plates were incubated for 45 min with gentle rocking at room temperature. After washing with T-TBS four times, 100 μL of goat anti-human IgG, IgG1 or IgG2c-HRP conjugates (Bethyl Laboratories, Montgomery, TX) diluted to 1/12500 in Guardian Stabilizer (Thermo Fisher Scientific, Waltham, MA) was added to wells and incubated for 45 min at room temperature. After washing with T-TBS four more times, plates were developed by adding 100 μL/per well SureBlue Reserve TMB developer (Kirkegaard and Perry Laboratories, Gaithersburg, MD) for 2–5 min in the dark. Development was stopped by the addition of 100 μL per well of 0.2 M H2SO4, and absorbance was measured at 450 nm using a FilterMax-F5 plate reader (Molecular Devices, San Jose, CA).
Statistical analysis
All data were analyzed for statistical significance using an unpaired Student's t-test for single comparisons at p < 0.05, p < 0.01, or p < 0.001.
Author contributions
Yeon Su Choi: conceptualization, methodology, validation, formal analysis, investigation, writing – drafting, review, and editing, and visualization; Jiin Felgner: validation, formal analysis, investigation, writing – drafting, review, and editing; Sharon Jan: methodology, validation, and formal analysis; Jenny E. Hernandez-Davies: methodology, validation, formal analysis, and investigation; D. Huw Davies: validation, formal analysis, investigation, writing – review and editing, and supervision; Young Jik Kwon: conceptualization, methodology, writing – original draft, writing – revised draft, writing – review and editing, supervision, visualization, and project administration.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Defense Threat Reduction Agency (DTRA) grant HDTRA1-18-1-0036 and a research gift from Pharma Research, Co., Ltd. The authors thank Olivia Ritchie and Rebecca Lee for proofreading the manuscript.
Notes and references
- F. P. Polack, S. J. Thomas, N. Kitchin, J. Absalon, A. Gurtman, S. Lockhart, J. L. Perez, G. Perez Marc, E. D. Moreira, C. Zerbini, R. Bailey, K. A. Swanson, S. Roychoudhury, K. Koury, P. Li, W. V. Kalina, D. Cooper, R. W. Frenck, Jr., L. L. Hammitt, O. Tureci, H. Nell, A. Schaefer, S. Unal, D. B. Tresnan, S. Mather, P. R. Dormitzer, U. Sahin, K. U. Jansen, W. C. Gruber and C. C. T. Group, N. Engl. J. Med., 2020, 383, 2603–2615 CrossRef CAS PubMed.
- J. Yang, W. Wang, Z. Chen, S. Lu, F. Yang, Z. Bi, L. Bao, F. Mo, X. Li, Y. Huang, W. Hong, Y. Yang, Y. Zhao, F. Ye, S. Lin, W. Deng, H. Chen, H. Lei, Z. Zhang, M. Luo, H. Gao, Y. Zheng, Y. Gong, X. Jiang, Y. Xu, Q. Lv, D. Li, M. Wang, F. Li, S. Wang, G. Wang, P. Yu, Y. Qu, L. Yang, H. Deng, A. Tong, J. Li, Z. Wang, J. Yang, G. Shen, Z. Zhao, Y. Li, J. Luo, H. Liu, W. Yu, M. Yang, J. Xu, J. Wang, H. Li, H. Wang, D. Kuang, P. Lin, Z. Hu, W. Guo, W. Cheng, Y. He, X. Song, C. Chen, Z. Xue, S. Yao, L. Chen, X. Ma, S. Chen, M. Gou, W. Huang, Y. Wang, C. Fan, Z. Tian, M. Shi, F. S. Wang, L. Dai, M. Wu, G. Li, G. Wang, Y. Peng, Z. Qian, C. Huang, J. Y. Lau, Z. Yang, Y. Wei, X. Cen, X. Peng, C. Qin, K. Zhang, G. Lu and X. Wei, Nature, 2020, 586, 572–577 CrossRef CAS PubMed.
- N. Vabret, G. J. Britton, C. Gruber, S. Hegde, J. Kim, M. Kuksin, R. Levantovsky, L. Malle, A. Moreira, M. D. Park, L. Pia, E. Risson, M. Saffern, B. Salome, M. Esai Selvan, M. P. Spindler, J. Tan, V. van der Heide, J. K. Gregory, K. Alexandropoulos, N. Bhardwaj, B. D. Brown, B. Greenbaum, Z. H. Gumus, D. Homann, A. Horowitz, A. O. Kamphorst, M. A. Curotto de Lafaille, S. Mehandru, M. Merad, R. M. Samstein and P. Sinai, Immunology Review Project, Immunity, 2020, 52, 910–941 CrossRef CAS PubMed.
- J. Y. Chung, M. N. Thone and Y. J. Kwon, Adv. Drug Delivery Rev., 2021, 170, 1–25 CrossRef CAS PubMed.
- A. Farlow, E. Torreele, G. Gray, K. Ruxrungtham, H. Rees, S. Prasad, C. Gomez, A. Sall, J. Magalhaes, P. Olliaro and P. Terblanche, Vaccines, 2023, 11, 690 CrossRef PubMed.
- W. Tai, S. Feng, B. Chai, S. Lu, G. Zhao, D. Chen, W. Yu, L. Ren, H. Shi, J. Lu, Z. Cai, M. Pang, X. Tan, P. Wang, J. Lin, Q. Sun, X. Peng and G. Cheng, Nat. Commun., 2023, 14, 2962 CrossRef CAS PubMed.
- J. Liu, A. Chandrashekar, D. Sellers, J. Barrett, C. Jacob-Dolan, M. Lifton, K. McMahan, M. Sciacca, H. VanWyk, C. Wu, J. Yu, A. Y. Collier and D. H. Barouch, Nature, 2022, 603, 493–496 CrossRef CAS PubMed.
- L. T. Gray, M. M. Raczy, P. S. Briquez, T. M. Marchell, A. T. Alpar, R. P. Wallace, L. R. Volpatti, M. S. Sasso, S. Cao, M. Nguyen, A. Mansurov, E. Budina, E. A. Watkins, A. Solanki, N. Mitrousis, J. W. Reda, S. S. Yu, A. C. Tremain, R. Wang, V. Nicolaescu, K. Furlong, S. Dvorkin, B. Manicassamy, G. Randall, D. S. Wilson, M. Kwissa, M. A. Swartz and J. A. Hubbell, Biomaterials, 2021, 278, 121159 CrossRef CAS PubMed.
- D. J. Irvine, M. A. Swartz and G. L. Szeto, Nat. Mater., 2013, 12, 978–990 CrossRef CAS PubMed.
- S. N. Mueller, S. Tian and J. M. DeSimone, Mol. Pharmaceutics, 2015, 12, 1356–1365 CrossRef CAS PubMed.
- J. F. Correia-Pinto, N. Csaba and M. J. Alonso, Int. J. Pharm., 2013, 440, 27–38 CrossRef CAS PubMed.
- J. W. Lim, W. Na, H. O. Kim, M. Yeom, A. Kang, G. Park, C. Park, J. Ki, S. Lee, B. Jung, H. H. Jeong, D. Park, D. Song and S. Haam, J. Mater. Chem. B, 2020, 8, 5620–5626 RSC.
- P. Li, G. Shi, X. Zhang, H. Song, C. Zhang, W. Wang, C. Li, B. Song, C. Wang and D. Kong, J. Mater. Chem. B, 2016, 4, 5608–5620 RSC.
- G. Liu, M. Zhu, X. Zhao and G. Nie, Adv. Drug Delivery Rev., 2021, 176, 113889 CrossRef CAS PubMed.
- M. F. Bachmann and G. T. Jennings, Nat. Rev. Immunol., 2010, 10, 787–796 CrossRef CAS PubMed.
- J. Ning, Q. Wang, Y. Chen, T. He, F. Zhang, X. Chen, L. Shi, A. Zhai, B. Li and C. Wu, J. Med. Virol., 2023, 95, e28743 CrossRef CAS PubMed.
- G. A. Koretzky, J. Immunol., 2010, 185, 2643–2644 CrossRef CAS PubMed.
- M. M. Painter, D. Mathew, R. R. Goel, S. A. Apostolidis, A. Pattekar, O. Kuthuru, A. E. Baxter, R. S. Herati, D. A. Oldridge, S. Gouma, P. Hicks, S. Dysinger, K. A. Lundgreen, L. Kuri-Cervantes, S. Adamski, A. Hicks, S. Korte, J. R. Giles, M. E. Weirick, C. M. McAllister, J. Dougherty, S. Long, K. D'Andrea, J. T. Hamilton, M. R. Betts, P. Bates, S. E. Hensley, A. Grifoni, D. Weiskopf, A. Sette, A. R. Greenplate and E. J. Wherry, Immunity, 2021, 54, 2133–2142.e3 CrossRef PubMed.
- C. Foged, B. Brodin, S. Frokjaer and A. Sundblad, Int. J. Pharm., 2005, 298, 315–322 CrossRef CAS PubMed.
- R. R. Shah, M. Taccone, E. Monaci, L. A. Brito, A. Bonci, D. T. O'Hagan, M. M. Amiji and A. Seubert, Sci. Rep., 2019, 9, 11520 CrossRef PubMed.
- G. P. Howard, G. Verma, X. Ke, W. M. Thayer, T. Hamerly, V. K. Baxter, J. E. Lee, R. R. Dinglasan and H. Q. Mao, Nano Res., 2019, 12, 837–844 CrossRef CAS PubMed.
- E. R. Steenblock and T. M. Fahmy, Mol. Ther., 2008, 16, 765–772 CrossRef CAS PubMed.
- J. A. Cohen, T. T. Beaudette, W. W. Tseng, E. M. Bachelder, I. Mende, E. G. Engleman and J. M. J. Fréchet, Bioconjugate Chem., 2009, 20, 111–119 CrossRef CAS PubMed.
- Q. Liu, X. Chen, J. Jia, W. Zhang, T. Yang, L. Wang and G. Ma, ACS Nano, 2015, 9, 4925–4938 CrossRef CAS PubMed.
- C. Pan, L. Wang, M. Zhang, J. Li, J. Liu and J. Liu, J. Am. Chem. Soc., 2023, 145, 13261–13272 CrossRef CAS PubMed.
- Y. Li, A. W. Frei, E. Y. Yang, I. Labrada-Miravet, C. Sun, Y. Rong, M. M. Samojlik, A. L. Bayer and C. L. Stabler, Biomaterials, 2020, 256, 120182 CrossRef CAS PubMed.
- P. O. Ilyinskii, C. J. Roy, C. P. O'Neil, E. A. Browning, L. A. Pittet, D. H. Altreuter, F. Alexis, E. Tonti, J. Shi, P. A. Basto, M. Iannacone, A. F. Radovic-Moreno, R. S. Langer, O. C. Farokhzad, U. H. von Andrian, L. P. Johnston and T. K. Kishimoto, Vaccine, 2014, 32, 2882–2895 CrossRef CAS PubMed.
- S. Foster, C. L. Duvall, E. F. Crownover, A. S. Hoffman and P. S. Stayton, Bioconjugate Chem., 2010, 21, 2205–2212 CrossRef CAS PubMed.
- R. Vyasamneni, V. Kohler, B. Karki, G. Mahimkar, E. Esaulova, J. McGee, D. Kallin, J. H. Sheen, D. Harjanto, M. Kirsch, A. Poran, J. Dong, L. Srinivasan, R. B. Gaynor, M. E. Bushway and J. R. Srouji, Cells Rep. Methods, 2023, 3, 100388 CrossRef CAS PubMed.
- B. C. Campbell, M. G. Paez-Segala, L. L. Looger, G. A. Petsko and C. F. Liu, Nat. Methods, 2022, 19, 1612–1621 CrossRef CAS PubMed.
- S. K. Cho, R. T. Lee, Y. H. Hwang and Y. J. Kwon, ChemMedChem, 2022, 17, e202100718 CrossRef CAS PubMed.
- J. S. Blum, P. A. Wearsch and P. Cresswell, Annu. Rev. Immunol., 2013, 31, 443–473 CrossRef CAS PubMed.
- E. M. Muntjewerff, L. D. Meesters and G. van den Bogaart, Front. Immunol., 2020, 11, 1276 CrossRef CAS PubMed.
- T. Zhang, A. Aipire, Y. Li, C. Guo and J. Li, Biomed. Pharmacother., 2023, 168, 115758 CrossRef CAS PubMed.
- D. Hara, S. N. Uno, T. Motoki, Y. Kazuta, Y. Norimine, M. Suganuma, S. Fujiyama, Y. Shimaoka, K. Yamashita, M. Okada, Y. Nishikawa, H. Amino and S. Iwanaga, J. Phys. Chem. B, 2021, 125, 8703–8711 CrossRef CAS PubMed.
- Y. J. Kwon, S. M. Standley, A. P. Goodwin, E. R. Gillies and J. M. J. Fréchet, Mol. Pharmaceutics, 2005, 2, 83–91 CrossRef CAS PubMed.
- M. S. Shim, X. Wang, R. Ragan and Y. J. Kwon, Microsc. Res. Tech., 2010, 73, 845–856 CrossRef CAS PubMed.
- P. Zamani, M. Mashreghi, M. Rezazade Bazaz, S. Zargari, F. Alizadeh, M. Dorrigiv, A. Abdoli, H. Aminianfar, M. Hatamipour, J. Zarqi, S. Behboodifar, Y. Samsami, S. Khorshid Sokhangouy, Y. Sefidbakht, V. Uskokovic, S. M. Rezayat, M. R. Jaafari and S. Mozaffari-Jovin, J. Control Release, 2023, 360, 316–334 CrossRef CAS PubMed.
- E. Yan Wang, M. Sarmadi, B. Ying, A. Jaklenec and R. Langer, Biomaterials, 2023, 303, 122345 CrossRef PubMed.
- C. C. Norbury, L. J. Hewlett, A. R. Prescott, N. Shastri and C. Watts, Immunity, 1995, 3, 783–791 CrossRef CAS PubMed.
- Y. Shi, J. Huang, Y. Liu, J. Liu, X. Guo, J. Li, L. Gong, X. Zhou, G. Cheng, Y. Qiu, J. You and Y. Lou, Sci. Adv., 2022, 8, eabo1827 CrossRef CAS PubMed.
- Z. Liu and P. A. Roche, Front. Physiol., 2015, 6, 1 Search PubMed.
- S. Latvala, J. Hedberg, S. Di Bucchianico, L. Moller, I. Odnevall Wallinder, K. Elihn and H. L. Karlsson, PLoS One, 2016, 11, e0159684 CrossRef PubMed.
- H. Zhao, Y. Li, B. Zhao, C. Zheng, M. Niu, Q. Song, X. Liu, Q. Feng, Z. Zhang and L. Wang, Acta Pharm. Sin. B, 2023, 13, 3892–3905 CrossRef PubMed.
- J. C. Charpentier and P. D. King, Cell Commun. Signaling, 2021, 19, 92 CrossRef CAS PubMed.
- P. A. Roche and K. Furuta, Nat. Rev. Immunol., 2015, 15, 203–217 CrossRef CAS PubMed.
- M. N. Thone, J. Y. Chung, D. Ingato, M. L. Lugin and Y. J. Kwon, Adv. Ther., 2023, 6, 2200125 CrossRef CAS PubMed.
- J. E. Hernandez-Davies, J. Felgner, S. Strohmeier, E. J. Pone, A. Jain, S. Jan, R. Nakajima, A. Jasinskas, E. Strahsburger, F. Krammer, P. L. Felgner and D. H. Davies, Front. Immunol., 2021, 12, 692151 CrossRef CAS PubMed.
- J. E. Hernandez-Davies, E. P. Dollinger, E. J. Pone, J. Felgner, L. Liang, S. Strohmeier, S. Jan, T. J. Albin, A. Jain, R. Nakajima, A. Jasinskas, F. Krammer, A. Esser-Kahn, P. L. Felgner, Q. Nie and D. H. Davies, Sci. Rep., 2022, 12, 9198 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *acde
*acde