
Preparation and characterization of macrophage membrane camouflaged cubosomes as a stabilized and immune evasive biomimetic nano-DDS†
Xuehui
Rui
 *a,
Yukihiro
Okamoto
a,
Nozomi Morishita
Watanabe
a,
Taro
Shimizu
b,
Ward
Wakileh
a,
Naoko
Kajimura
c and
Hiroshi
Umakoshi
*a
*a,
Yukihiro
Okamoto
a,
Nozomi Morishita
Watanabe
a,
Taro
Shimizu
b,
Ward
Wakileh
a,
Naoko
Kajimura
c and
Hiroshi
Umakoshi
*a
aDivision of Chemical Engineering, Graduate School of Engineering Science, Osaka University, 1-3 Machikaneyamacho, Toyonaka, Osaka 560-8531, Japan. E-mail: umakoshi.hiroshi.es@osaka-u.ac.jp; xuehui.rui@cheng.es.osaka-u.ac.jp
bResearch Institute for Microbial Diseases, Osaka University, 3-1 Yamadaoka, Suita, Osaka 565-0871, Japan
cResearch Center for Ultra-High Voltage Electron Microscopy, Osaka University, 7-1, Mihogaoka, Ibaraki, Osaka 567-0047, Japan
First published on 7th August 2024
Abstract
This study aims to develop a biomimetic nano-drug delivery system (nano-DDS) by employing a macrophage cell membrane camouflaging strategy to modify lyotropic liquid crystal nanoparticles (LLC-NPs). The cubic-structured LLC-NPs (Cubosomes, CBs) were prepared via a top-down approach (ultra-sonification) using monoolein (MO) and doped with the cationic lipid, DOTAP. The cell membrane camouflaging procedure induced changes in the cubic lipid phase from primitive cubic phase (QIIP) to a coexistence of QIIP and diamond cubic phase (QIID). The macrophage membrane camouflaging strategy protected CB cores from the destabilization by blood plasma and enhanced the stability of CBs. The in vitro experiment results revealed that the macrophage cell membrane coating significantly reduced macrophage uptake efficacy within 8 h of incubation compared to the non-camouflaged CBs, while it had minimal impact on cancer cell uptake efficacy. The macrophage membrane coated CBs showed lower accumulation in the heart, kidney and lungs in vivo. This study demonstrated the feasibility of employing cell membrane camouflaging on CBs and confirmed that the bio-functionalities of the CBs-based biomimetic nano-DDS were retained from the membrane source cells, and opened up promising possibilities for developing an efficient and safe drug delivery system based on the biomimetic approach.
1. Introduction
Non-lamellar lyotropic liquid crystalline nanoparticles (LLC-NPs) are self-assembled nanostructures formed by amphiphilic molecules, characterized by a unique, non-lamellar, three-dimensional, self-organizing structure with a high internal interfacial surface area.1,2 LLC-NPs have demonstrated remarkable dispersibility and stability surpassing that of conventional lipid-based nanoparticles, and they have great potential in drug delivery and cosmetics.3 In particular, LLC-NPs have been used for encapsulating hydrophobic drugs and nucleic acids to increase their bioavailability for DDS.4–6Different types of non-lamellar LLC-NPs including cubosomes (cubic structure) and hexosomes (hexagonal structure) have different geometries and properties. Since the discovery and investigation of cubosomes (CBs) by Kåre Larsson in 1989, CBs have gained attention as potential nano-DDS.7 CBs are square or spherical nanoparticles consisting of cavernous structures separating the internal aqueous channels and a large hydrophilic interface. The special structure endows CBs with good controlled-release properties and high encapsulation capacities of drug molecules.2,8,9 Also, C. Leal's previous research elucidated that, after the doping of a small amount of cationic lipid, the CBs could still form a stable and positively charged cubic phase.10 The cationic lipid doping in CBs provides the CBs with nucleic acid loading ability and allows for further surface modification.
Nanoparticle recognition by the mononuclear phagocyte system poses a challenge for the effective use of nano-DDS.11 To overcome this challenge, surface modification of CBs is necessary to extend their circulation time and reduce their immunogenicity. Table S1 (ESI†) summarizes some representative studies on CBs surface modification. To date, PEGylation is the most widely used strategy for surface modification of NPs, as PEG coatings on NPs shield the surface from aggregation, opsonization, and phagocytosis, prolonging systemic circulation time in vivo.12 However, some studies have reported that PEGylation can also induce immune responses in animals, indicating the need for alternative surface modification strategies that could improve the efficacy and safety of CBs-based nano-DDS.13,14 An additional crucial factor concerning CBs in drug delivery is their structural instability. Upon injection in vivo, CBs will quickly interact with plasma, leading to their collapse and consequent burst release of drug payloads.15,16 This destabilization significantly impedes the effective application of CBs for drug delivery purposes.
In recent years, the cell membrane coating strategy was established as a promising surface modification platform for nano-DDS. The long-circulation nature of red blood cells (RBC) granted the RBC membrane-camouflaged nanoparticles a superior circulation half-life and lower immunogenicity comparing to the traditional PEGylated nanoparticles.17 Furthermore, the macrophage membrane coating strategy has also been extensively utilized in the development of nano-DDS, as they can prolong the circulation, control the release of drugs, realize immune escape, and reduce immunogenicity due to the membrane proteins inherited from macrophages.18 Also, the application of a cell membrane layer coating to nanoparticles has been shown to enhance their stability over time and improve their safety profile.19
Given the tunable surface of CBs and the multifunctionality of macrophage cell membranes, we hypothesize that coating CBs with macrophage cell membranes presents a promising method for enhancing drug delivery performance. The cell membrane coating not only stabilizes the CBs in vivo, but also provides stealth properties to evade immune detection, potentially leading to improved therapeutic efficacy and reduced side effects. In this work, we first fabricated the cationic CBs by doping 1 mol% DOTAP into monoolein (MO). The positively charged surface of the CBs would facilitate the coating of the negatively charged cell membrane via electrostatic interaction. After the macrophage membrane coating, this nano-DDS was systematically characterized using dynamic light scattering (DLS), cryo-transmission electron microscopy (cryo-TEM), and small-angle X-ray scattering (SAXS). The cell internalization efficacy was investigated using flow cytometry (FCM) and confocal laser scanning fluorescence microscopy (CLSM). The in vivo distributions of CBs were investigated. Doxorubicin (DOX) was harnessed as model drug and the anti-cancer efficacy was investigated via apoptosis assay. Our outcomes demonstrated that the prepared cationic CBs could successfully be camouflaged by macrophage membrane and inherit the membrane proteins from macrophages to realize an immune escape without hindering cancer cell uptake efficacy. Furthermore, the macrophage membrane stabilized the CBs in vivo and exhibited reduced accumulations in mice organs including heart, kidney and lungs, indicating a prolonged circulation time span in vivo.
2. Experimental
2.1 Materials
Monoolein (≥99%, 1-oleoyl-rac-glycerol), Pluronic F127 (poly(ethylene oxide)–poly-(propylene oxide)–poly(ethylene oxide)) were purchased from Sigma-Aldrich. DOTAP (1,2-dipalmitoyl-3-trimethylammonium-propane (chloride salt)), 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 NBD-PE (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2-1,3-benzoxadiazol-4-yl) (ammonium salt)) were purchased from Avanti Polar Lipids. Doxorubicin Hydrochloride (DOX), D-PBS, D-MEM (with L-glutamine and phenol red), RPMI-1640 cell culture media (with L-glutamine and phenol red), RPMI-1640 (with L-glutamine, without phenol red), trypsin (0.25 w/v%, EDTA solution with phenol red), methanol (99.7%, methyl alcohol), MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide), penicillin–streptomycin solution, protease inhibitor cocktail set V (EDTA free), and LabAssay™ Phospholipid Kit were purchased from Fujifilm. FITC Annexin V Apoptosis Detection Kit I was purchased from BD Pharmingen. Pierce® BCA (Bicinchoninic acid) Protein Assay Kit was purchased from Thermo Fisher Scientific. DiR near-IR membrane probe (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine iodide) was purchased from Abcam.
:1 NBD-PE (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2-1,3-benzoxadiazol-4-yl) (ammonium salt)) were purchased from Avanti Polar Lipids. Doxorubicin Hydrochloride (DOX), D-PBS, D-MEM (with L-glutamine and phenol red), RPMI-1640 cell culture media (with L-glutamine and phenol red), RPMI-1640 (with L-glutamine, without phenol red), trypsin (0.25 w/v%, EDTA solution with phenol red), methanol (99.7%, methyl alcohol), MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide), penicillin–streptomycin solution, protease inhibitor cocktail set V (EDTA free), and LabAssay™ Phospholipid Kit were purchased from Fujifilm. FITC Annexin V Apoptosis Detection Kit I was purchased from BD Pharmingen. Pierce® BCA (Bicinchoninic acid) Protein Assay Kit was purchased from Thermo Fisher Scientific. DiR near-IR membrane probe (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine iodide) was purchased from Abcam.
2.2 Cell line and cell culture
J774.1 (a BALB/c mouse derived macrophage cell line), Colon26 (a BALB/c mouse derived colon cancer cell line) and HEK293 cells (a human fetal kidney-derived cell line) were obtained from JCRB Cell Bank. J774.1 and Colon26 cells were cultured using RPMI 1640 cell culture media, HEK293 cells were cultured in D-MEM media. All cell culture media were supplemented with 10% (v/v) fetal bovine serum and 1% (v/v) of penicillin/streptomycin. Cells were maintained at 37 °C in a humidified atmosphere with 5% CO2.2.3 Cubosomes preparation and macrophage membrane camouflaging
MO with DOTAP (1 mol%) or without DOTAP was dissolved in chloroform in a predetermined amount in Eppendorf tubes. The solutions were then vacuumed for 48 h to remove the organic solvent. The dried samples were then melted at 70 °C and hydrated using a preheated F127 solution (final F127 concentration: 2 mg mL−1) at a final MO concentration of 20 mg mL−1. The resulting mixtures were vortexed for 10 min and exposed to probe-type ultra-sonification (Ultrasonic Disruptor, UD-200, TOMY) on ice for 20 min at 100 W.The macrophage cell membrane vesicles were obtained from J774.1 cells through a sucrose gradient ultracentrifugation method. Briefly, J774.1 cells were cultured in 100 mm cell culture dishes. Once the cells reached 90% confluency, they were detached using pipetting and collected in D-PBS without using trypsin. After washing using D-PBS and centrifugation, the cell sediment was resuspended in cold tris-magnesium buffer (TM buffer, pH 7.4, 0.01 M tris and 0.001 M MgCl2) containing protease inhibitor and incubated for 15 min at 4 °C. The cell-TM buffer suspension was then extruded 20 times through a mini-extruder without polycarbonate membrane to rupture the cells. Sucrose solution was added to the cell homogenate to a final concentration of 0.25 M and the homogenate was centrifuged at 2000 g and 4 °C for 15 min. The resulting supernatant was collected and extruded through 200 nm and 100 nm pore-sized polycarbonate membranes in sequence followed by ultra-centrifugation (50000 rpm, 4 °C) for 120 min to collect the cell membrane vesicles. The extracted cell membrane vesicles were stored at 4 °C for immediate use and at −80 °C for long-term storage.
Sonification method was utilized for the cell membrane coating on CBs. The extracted cell membranes were resuspended in D-PBS and sonicated to disperse the cell membrane vesicles, The protein content and phospholipid concentration in the extracted macrophage membrane were determined using a BCA assay kit and a LabAssay phospholipid kit, respectively. Then, the cell membrane vesicles were mixed with CBs at different ratios (MO to protein weight ratio). The mixed samples were shaken and then vortexed for 5 min. After the vortex, samples were sonicated on ice in a sonification bath for 15 min.
For the cell internalization efficacy assay, 0.5 wt% of 18:1 NBD-PE (dye to MO ratio) was added to the MO or MO/DOTAP solution before vacuuming to prepare fluorescence dye-labeled CBs. For DOX-loaded CBs preparation, DOX was dissolved in methanol and then added to MO/DOTAP solution at a final ratio of 0.5 wt% (DOX to MO ratio) before vacuuming. For in vivo imaging research, 0.1 mol% of DiR was added to the MO/DOTAP solution before vacuuming to prepare DiR-labelled CBs.
In the following sections, cubosomes prepared solely from monoolein are referred as MO- only CBs; the DOTAP-doped cubosomes as cationic CBs; the extracted macrophage membrane vesicles as MMVs; and the macrophage membrane camouflaged CBs as MM@CBs.
2.4 Particle size, polydispersity index, and ζ potential
The hydrodynamic particle size, polydispersity index (PDI), and ζ potential of all the nanoparticle samples were characterized using ZEN5600-Zetasizer (Malvern, UK). 50 μL of CB sample was diluted in 1 mL ultrapure water (final MO concentration was 1 mg mL−1) before measurement. Cell membrane vesicles were diluted into 0.1 mg mL−1 (protein concentration) using ultrapure water before measurement. For investigating the CB stability in vitro, MO only CB, cationic CB and MM@CBs were added to D-PBS, RPMI cell culture media (without Phenol Red) and mouse blood plasma (collecting procedure is described in ESI†) respectively to a final MO concentration of 5 mg mL−1. After specific incubation time (0.5, 1, 2, 4, 6, 8, 10, 12, 16, 20, 24 h) at 37 °C, 50 μL of CB sample was diluted in 1 mL ultrapure water and the particle size were measured. All the samples were measured in triplicate at 25 °C.2.5 Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE)
SDS-PAGE samples (fresh J774.1 macrophage, extracted macrophage membrane vesicles and MM@CBs) were prepared in an SDS sample loading buffer and heated at 95 °C for 5 min. Then, 20 μL of the sample was loaded into each well of a 10% SDS-PAGE and run at 10 V cm−1 for 1 h. The resulting gel was stained with Coomassie Brilliant Blue for 2 hours and washed in an acetic acid solution overnight for subsequent visualization in ChemiDoc XRS Gel Imaging System (Bio-Red, USA).2.6 DOX loading efficacy and drug release pattern
The DOX drug encapsulation efficacy investigation was carried out using ultrafiltration units (USY-5, MWCO 50 kD, Advantec, Japan). To determine the amount of free DOX, 1 mL of DOX-loaded CBs was added to the ultrafiltration tube and pressurized with air for 10 min. The eluted solution was collected and analyzed using a fluorescence spectrometer (FP-8500 Spectrofluorometer, JASCO, Japan) at 25 °C, with an excitation wavelength of 485 nm and emission wavelength of 556.5 nm.The DOX encapsulation efficacy (EE) was calculated according to the following equation:
 | (1) |
The dialysis method was employed to investigate the in vitro release of DOX. Specifically, 1 mL of DOX-loaded CBs was added to a dialysis tubing with a molecular weight cut-off of 12–14 kD and dialyzed against 20 mL of D-PBS in a brown vial at room temperature. At predetermined time intervals, 1 mL of the dialysis buffer was withdrawn from the vial and replaced with 1 mL of fresh buffer. The DOX concentrations in the collected samples were determined using a spectrofluorometer as previously described.
2.7 Small-angle X-ray scattering (SAXS) investigation
SAXS investigation was carried out using BL19B2 beamline at Spring-8 (Super Photon ring-8 GeV radiation faculty). The experiment conditions were confirmed via trial experiments. The scattering exposure time was 300 s for each sample. The detector was a two-dimensional pixel detector PILATUS-2M (camera length is 3 m). The beam energy was set at 18 keV; X-ray wavelength (λ) was about 0.69 Å, q value range was set from 0.06 to 3 nm−1 and the sample-to-detector distance was calibrated using silver behenate.The lattice parameters were calculated using the eqn (2).
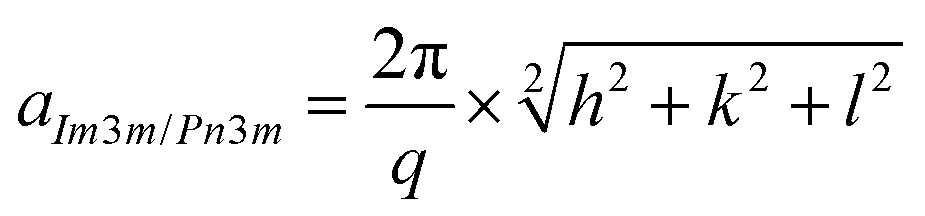 | (2) |
The water channel radius (rw) is calculated according to the following equations:20
For QIIP (Im3m) phase:
| rw = 0.306 × a − l | (3.1) |
For QIID (Pn3m) phase:
| rw = 0.391 × a − l | (3.2) |
For the biphasic CBs system (QIIP and QIID), the Bonnet ratio was calculated according to the equation:
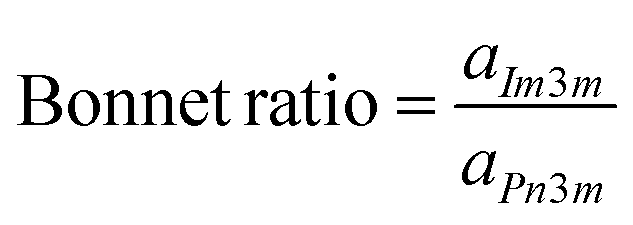 | (4) |
2.8 Cryogenic transmission electron microscopy (cryo-TEM)
Morphological characterization of different particles was performed using cryo-TEM. Vitrobot mark IV (Thermo/FEI, USA) was used for specimen preparation. Briefly, 2.4 μL of the sample was deposited onto Cu QUANTIFOIL grids (R1.2/1.3, 300 mesh) that were freshly glow discharged. The carbon-coated grids were then glow discharged at 10 mA for 20 s and blotted for 3 s (blotting force of 7 at 25 °C under 100% relative humidity) before being plunged into liquid ethane. CBs and macrophage membrane vesicles were imaged using a Titan KRIOS microscope (Thermo Fisher Scientific, USA) with an accelerating voltage of 300 kV and a defocus value of −4 μm at a magnification of 59000 which has a pixel size of 0.14 nm. Image processing and analysis were performed using ImageJ (Version 1.53t).
2.9 Cellular internalization assay
J774.1 and Colon26 cells were initially seeded in 6-well plates for flow cytometry investigation and in glass bottom dishes (35 mm diameter, Matsunami) for confocal laser scanning microscopy investigation (Eclipse TE2000-U, 518 Nikon, Japan). After incubation until they reached ∼70% confluency, the old cell culture medium was removed, and cells were rinsed three times with D-PBS. Fresh cell culture medium containing different CBs (doped with 0.5 wt% of NBD-PE) was added to each well at a final concentration of 0.15 mg mL−1 (calculated according to MO weight). After different periods of incubation, cells were washed with D-PBS and then collected for flow cytometer investigation and confocal laser scanning microscopy investigation. The cell uptake efficiency was evaluated by measuring the NBD fluorescence intensity in cells using flow cytometer (Applied Biosystems Acoustic focusing cytometer, Attune, USA).2.10 Annexin V/propidium iodide (PI) assay
Colon26 cells were seeded in 6-well plates and incubated. After the cells reached ∼70% confluency, cationic CBs (0.2 mg mL−1 MO), MM@CBs (0.2 mg mL−1 MO), free DOX (1 μg mL−1), DOX-loaded cationic CBs (1 μg mL−1 DOX, 0.2 mg mL−1 MO) and DOX-loaded MM@CB (1 μg mL−1 DOX, 0.2 mg mL−1 MO) were added to each well. After 12/24 h incubation period, cells were collected and investigated using an apoptosis assay kit according to the protocol (FITC Annexin V Apoptosis Detection Kit I, BD Pharmingen, USA).2.11 Biodistribution investigation
CBs were labelled with 0.1 mol% of DiR during preparation procedure for in vivo biodistribution investigation. BALB/c mice (male, 6 weeks old) were purchased from Japan SLC (Shizuoka, Japan). The experimental animals had unrestricted access to water and mouse chow and were kept under controlled environmental conditions, including constant temperature, humidity, and a 12-hour dark-light cycle. All animal experiments were approved in advance by Ethics Review Committee for Animal Experimentation of Osaka University.To study the in vivo distribution, two samples (DiR-MM@CB, DiR-CB) were injected in tail vein: concentration, 2 mg mL−1 (calculated according to MO amount); injection volume, 100 μL. For control group, 100 μL of PBS was injected in tail vein. Whole body and organ fluorescence images was recorded with IVIS (excitation: 720 nm; emission: 790 nm) under 5 s exposure time. At selected time (0.5, 1, 3, 6, 9 and 24 h), the whole-body images were obtained. After the final imaging (9 and 24 h), the mice were anesthetized with isoflurane inhalation and euthanized. The organs (liver, lungs, spleen, heart, kidneys) were collected and imaged using IVIS.
2.12 Data statistical analysis
All experimental data are presented as the mean value ± standard deviation (s.d). Statistically significant difference analysis was performed using Student's t-test for paired sample sets. For all analyses, p-values greater than 0.05 were considered as not statistically significant, p-values less than 0.05 were considered statistically significant. For all analyses, GraphPad Prism (version 9.0.0) software was used.3. Result and discussion
3.1 Cubosome preparation and characterization
For the sake of brevity, the monoolein-only CBs, DOTAP-doped CBs and macrophage membrane camouflaged cationic CBs fabricated for this study are referred to as MO-only CBs, cationic CBs and MM@CBs respectively. The MM@CBs preparation procedure is illustrated in Fig. 1(A). Hydrodynamic diameter and ζ-potential of different CBs were investigated, and the results are shown in Fig. 1(B). The average hydrodynamic diameters of fabricated MO-only CBs and cationic CBs are 205.4 ± 1.8 nm and 200.9 ± 2.2 nm, respectively. It should be noted that, the size of CBs is very dependent on the preparation procedure, especially the F127 concentration, water content and ultra-sonification power and timespan. In this study, the F127 concentration was set as 2 mg mL−1, and the ultra-sonification was conducted at 100 W for 20 min. In addition, heat treatment was utilized to promote the stability of the CBs’ structure.21 The coating efficacy of cell membrane vesicles on core-nanoparticles (core-NPs) is influenced by the curvature and size differences between cell membrane vesicles and core-NPs.22 Therefore, the size difference between cell membrane vesicles and core materials is an important parameter in determining the integrity of cell membrane camouflaging on CBs. In order to reduce the particle size, the extracted cell membrane vesicles were extruded through 100 nm pore-sized polycarbonate membrane. The average hydrodynamic particle size of MM@CBs (1:1 MO to protein weight ratio) was measured as 256.4 ± 16.6 nm, which was approximately 27.6% greater compared to the non-coated cationic CBs.
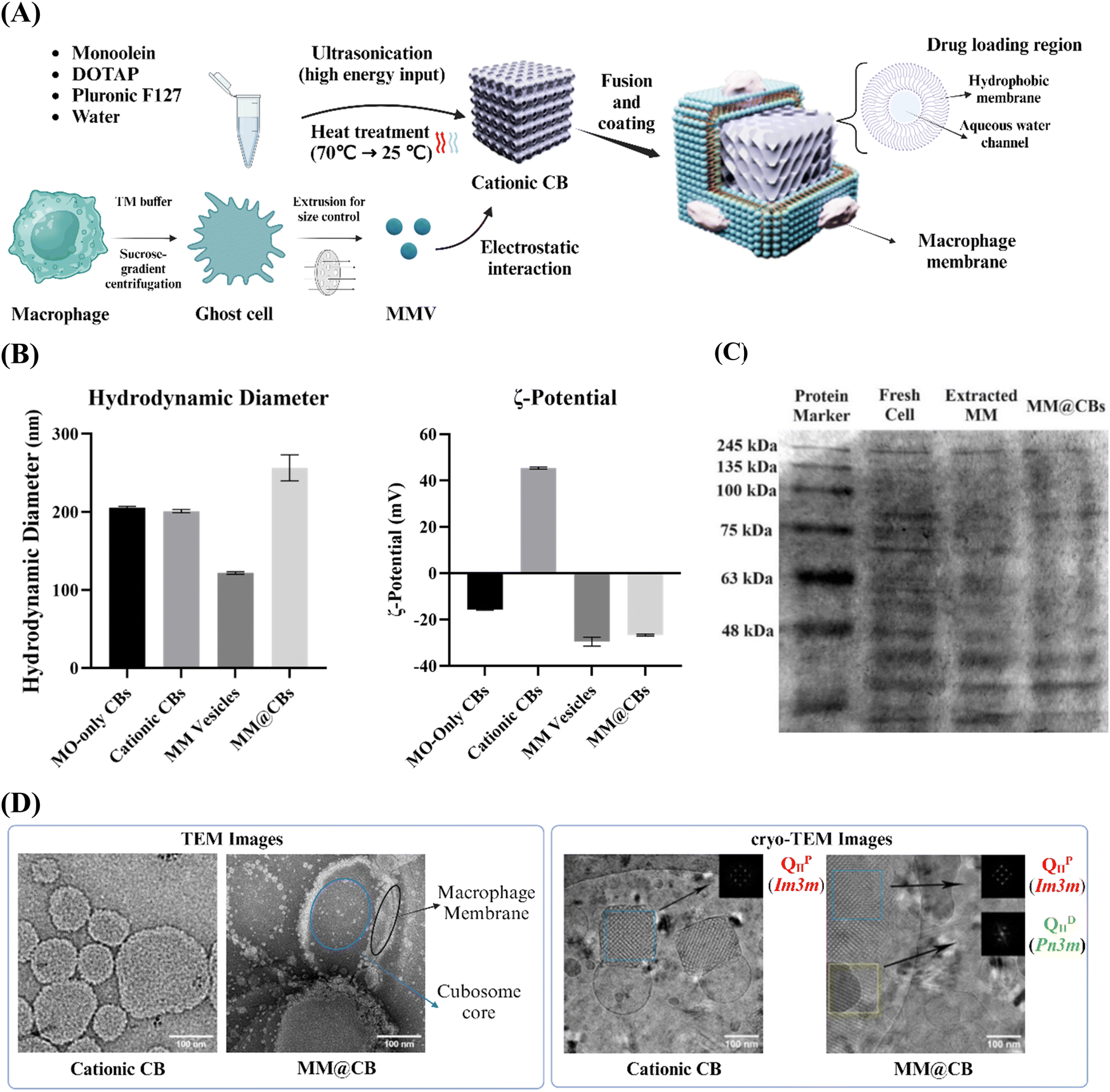 | ||
| Fig. 1 Schematic illustration of MM@CBs preparation procedure and results of characterizations. (A) Preparation procedure of MM@CBs. (B) Hydrodynamic particle size and ζ-potential of fabricated MO-only CBs, cationic CBs, extracted macrophage membrane vesicles (MM vesicles) and MM@CBs (MO to cell membrane protein = 1:1 weight ratio) in ultrapure water. All measurements were carried out at 25 °C. Error bars represent ± s.d. (n = 3). (C) SDS-PAGE result of fresh J774.1 cell lysis, extracted J774.1 membrane vesicles and the MM@CBs. (D) TEM and cryo-TEM images of cationic CBs and MM@CBs. The upper-right corner of cryo-TEM images show the fast Fourier transform (FFT) of the corresponding squared regions. Scale bar = 100 nm. Abbreviations: CB, cubosome; MMV, macrophage membrane vesicle; MM@CB, macrophage membrane camouflaged cubosome. | ||
Regarding the ζ-potential in ultrapure water, the MO-only CBs exhibited a negative ζ-potential (−15.7 ± 0.3 mV), while the cationic CBs displayed a high positive ζ-potential (45.4 ± 0.5 mV). After the cell membrane coating (1:1 MO to protein weight ratio), the ζ-potential shifted to −26.7 ± 0.4 mV, which closely resembled the ζ-potential of the extracted macrophage membrane vesicles (−29.5 ± 1.9 mV). The DLS and ζ-potential investigations revealed that the process of cell membrane camouflaging led to an increase in the particle size of the CBs and a significant shift in ζ-potential. These observations indicated a successful coating of the macrophage membranes on cationic CBs.
SDS-PAGE was employed to assess the presence of macrophage membrane proteins on MM@CBs. Fig. 1(C) displays distinct and consistent protein bands from MM@CBs comparable to those observed in macrophages. This indicated that MM@CBs have inherited the membrane proteins from macrophages.
TEM and cryo-TEM investigations were carried out to provide additional verification of the changes in CB morphology before and after cell membrane camouflaging. As shown in Fig. 1(D), the TEM images depict that the cationic CBs exhibited a round or square morphologies. After the cell membrane camouflaging, clear evidence of the surface changes on the cationic CBs was observed in the images. These surface changes are considered to be associated with the cell membrane that attached or fused onto the surface of cationic CBs. The fast Fourier transforms (FFT) acquired from the cryo-TEM images confirmed the existence of internal cubic phases. In the case of cationic CBs, the characteristic motifs and reflections of QIIP phase were confirmed in the FFT analysis. For the MM@CBs (1:1 MO to protein weight ratio), a coexistence of QIID and QIIP was observed.
The observations from the DLS, SDS-PAGE, TEM as well as the cryo-TEM results confirmed the successful coating of the cell membrane onto the cationic CBs, thereby solidifying the feasibility of the camouflaging strategy.
3.2 Cubosome phase changes after cell membrane camouflaging
As illustrated in Fig. 2(A), MO-based CBs could have different cubic phases, including primitive cubic phase (QIIP), diamond cubic phase (QIID) and gyroid cubic phase (QIIG). A phase transition of CBs was observed from the cryo-TEM images (Fig. 1(D)) after the cell membrane coating. For the purpose of investigating the influence of cell membrane camouflaging on the structure of CBs, a small-angle X-ray scattering (SAXS) investigation was conducted to assess the internal nanostructure of the CBs before and after cell membrane coating. MM@CBs were fabricated at different MO to cell membrane ratios. The representative SAXS patterns are presented in Fig. 2(B). The calculated lattice parameters (a) and the water channel radius (rw) of different CBs are presented in Table 1. | ||
| Fig. 2 Illustration of different phases of cubosomes and SAXS investigation results. (A) Different phase structures of cubosomes. (i) QIIP; (ii) QIID and (iii) QIIG. (B) The one-dimensional SAXS patterns of different CBs and the cubic phase transition illustration. From bottom to top: MO-only CBs (20 mg mL−1 MO), cationic CBs (20 mg mL−1 MO + 1 mol% DOTAP), and MM@CBs with varying compositions (10 mg mL−1 MO + 1 mol% DOTAP; MO to cell membrane protein weight ratios are 10:1, 7:1, 4:1, 3:1, 2:1 and 1:1). All measurements were conducted at 37 °C. Bragg peak spacing ratios identifying Im3m and Pn3m space group are represented in black and red, respectively. | ||
| CB type | Formulation | Space group | Lattice parameter (a) [nm] | Water Channel radius (rw) [nm] |
|---|---|---|---|---|
| The ratios represent MO: cell membrane protein weight ratio. | ||||
| MO-only CBs | MO | Im3m | 13.05 | 2.72 |
| Cationic CBs | MO + 1 mol% DOTAP | Im3m | 14.46 | 2.29 |
| MM@CBs | MO + 1 mol% DOTAP + MM (10:1 wt ratio) |
Im3m/Pn3m | 12.53/9.57 | 2.13/2.04 |
| MO + 1 mol% DOTAP + MM (7:1 wt ratio) |
Im3m/Pn3m | 12.40/9.35 | 2.09/1.96 | |
| MO + 1 mol% DOTAP + MM (4:1 wt ratio) |
Im3m/Pn3m | 12.03/8.94 | 1.98/1.80 | |
| MO + 1 mol% DOTAP + MM (3:1 wt ratio) |
Im3m/Pn3m | 11.68/8.81 | 1.88/1.75 | |
| MO + 1 mol% DOTAP + MM (2:1 wt ratio) |
Im3m/Pn3m | 11.57/8.75 | 1.84/1.72 | |
| MO + 1 mol% DOTAP + MM (1:1 wt ratio) |
Im3m/Pn3m | 11.57/8.69 | 1.84/1.70 | |
At a temperature of 37 °C, the SAXS patterns of MO-only CBs displayed prominent peaks at ∼0.681, 0.965, and 1.176 nm−1. On the other hand, the SAXS pattern of cationic CBs exhibited prominent peaks at ∼0.615, 0.870, and 1.074 nm−1. The SAXS patterns of both MO-only CB and cationic CBs exhibited distinct Bragg peaks at the space ratio of √2:√4:√6, corresponding to Miller indices [hkl]: [110], [200], and [211], respectively. These patterns are indicative of a primitive cubic phase (QIIP), belonging to the space group Im3m. The lattice parameters of the MO-only CBs and cationic CBs were measured to be 13.05 nm and 14.46 nm, respectively. The doping of the DOTAP cationic lipid resulted in a slight increase of lattice parameter. However, the internal QIIP nanostructure was retained, which is consistent to the previous reported results that the interplay between charge repulsion of the lipid membrane interfaces induced larger water channels in CBs.23
Following the macrophage membrane coating, the scattering intensity of the peaks in the CBs dispersions slightly decreased. This reduction can be attributed to factors such as dilution or changes in ionic strength.24 To minimize the influence of phase changes resulting from dilution and variations in ionic strength, we maintained a consistent dilution factor and ionic strength (same PBS volume) for different MM@CBs formulations. This approach allowed us to standardize the impact of dilution and ionic strength on the phases of CBs at a consistent level. For the MM@CBs with a 1:1 ratio (MO:cell membrane protein weight ratio) formulation, the Bragg peaks appeared at ∼ 0.775, 1.088, and 1.329 nm−1 represented a space ratio of √2:√4:√6, characterizing the retained QIIP phase. Meanwhile, the presence of peaks at ∼1.023, 1.220, and 1.424 nm−1 featured a space ratio of √2:√3:√4, characterizing the diamond cubic phase QIID with the Pn3m space group, indicating the coexistence of QIID and QIIP phases. Notably, the presence of QIID was observed even at a low cell membrane protein ratio (10:1 MO to protein weight ratio), and the lattice parameters of both QIID and QIIP phases decreased as the ratio of cell membrane vesicles increased. Both the phase transition from QIIP → QIID and the reduction of lattice parameter indicate an increase in the negative membrane curvature following cell membrane coating.25,26 As the cell membrane protein ratio increased, the relative intensity of Bragg peaks corresponding to QIID increased, which implies that the cell membrane coating promoted the phase change of cationic CBs. These findings highlight the sensitivity of the CBs’ structure to the cell membrane protein ratios, revealing the intriguing possibility of obtaining diverse nanoscale structures through the controlled manipulation of the membrane protein content.
There is a mathematical relationship between the coexisting QIID and QIIP surfaces, defined by the so-called Bonnet transformation.27 In previous research, it was established that the bicontinuous phases QIIP and QIID both consist of minimal surfaces. Under equilibrium conditions, the average Gaussian curvatures of these coexisting bicontinuous cubic phases (QIIP and QIID) are expected to be identical. It can be demonstrated that the ratio of the lattice parameters between QIIP and QIID phases (aIm3m/aPn3m) should be close to the theoretical value of 1.28, which is commonly referred to as the Bonnet relation27,28 To examine the relationship between the coexisting QIID and QIIP phases in MM@CBs, the ratio of the lattice parameters between QIIP and QIID phases (aIm3m/aPn3m) was calculated to examine the Bonnet relation between the coexisting QIIP and QIID phases. The results of this calculation are listed in Table 2. The lattice parameter ratios of various MM@CBs formulations fall within the range of 1.31–1.35 when measured at a temperature of 37 °C. These values closely align with the theoretical prediction and are consistent to the previously reported ratio of 1.33 for MO-based CB particles exhibiting coexisting QIID and QIIP phases.29 The Bonnet relationship indicates that QIIP and QIID phases can be interchanged through bending while maintaining a constant Gaussian curvature.
| MO to cell membrane protein weight ratio | QIIP lattice parameter (aIm3m) [nm] | QIID lattice parameter (aPn3m) [nm] | Bonnet ratio (aIm3m/aPn3m) |
|---|---|---|---|
| 10:1 |
12.53 | 9.57 | 1.31 |
| 7:1 |
12.40 | 9.37 | 1.35 |
| 4:1 |
12.03 | 8.94 | 1.35 |
| 3:1 |
11.68 | 8.81 | 1.32 |
| 2:1 |
11.57 | 8.75 | 1.32 |
| 1:1 |
11.57 | 8.69 | 1.33 |
The cubic phase of the CBs undergoing changes from QIIP to QIID eventually form a biphasic system. This could be attributed to the proteins and lipids present in the cell membrane vesicles fusing with the CBs’ surface, inducing a cubic phase transition. The various molecules existing in the cell membrane such as protein, peptides, phospholipids, and cholesterol are assumed to play roles in the CBs phase changes.30 Thomas G. Meikle et.al fabricated CBs for antimicrobial peptides (AMPs) delivery.25 Their research elucidated that, in the absence of peptides MO-based CBs exhibited QIIP (Im3m) phase. Whereas, after the loading of gramicidin A’ peptides, a sharp decrease in the lattice parameter was observed. From 3 to 5 mol% peptides, MO-based CBs underwent phase transition from QIIP (Im3m) to coexistence of QIIP and QIID (Pn3m) phases. On the other hand, Sampa Sarkar et al. carried out a systematic investigation on phase behavior of the quaternary lipid–water systems consisting of three different lipid species (monoolein, cholesterol, and various phospholipids) and water.26 The doping of cholesterol, phosphatidylcholine (PC), and saturated phosphatidylethanolamine (PE) in MO-based CBs drive a reduction in interfacial curvature and resulting in phase transitions in the sequence of QIID → QIIP →Lα, which is opposite to the phenomenon observed in this study. Additionally, the protein to phospholipid weight ratio of the extracted macrophage membrane vesicles was measured to be ∼7.29:1 (wt/wt) (ESI†). Therefore, it is rational to conclude that, the phase change undergone by the CBs (QIIP → QIID) after macrophage membrane coating was mainly attributed to the fusion of protein/peptides with CBs. A high coverage of binding protein on CBs interfacial membranes is believed to induce an incased CBs’ interfacial membrane curvature.31 In this study, the high positive charge density of cationic CBs, facilitated the recruitment and fusion of negatively charged macrophage membranes. The extensive coverage of macrophage membrane on cationic CBs induced a higher interfacial membrane curvature, resulting in the formation of QIID phases. Additionally, a higher membrane protein-to-MO ratio appeared to intensify this phase transition, as evidenced by increased QIID phases Brag peak intensities in SAXS pattern (Fig. 2(B)). Given that a low protein ratio may not achieve sufficient camouflaging on CBs, the 1:1 MO to protein weight ratio was chosen as the optimal ratio for the subsequent studies. This ratio was deemed suitable to ensure effective cell membrane camouflaging on the CBs.
3.3 Stability of cubosomes in vitro
To confirm the stability of the prepared CBs, different CBs were incubated with PBS, RPMI cell culture media, and mouse blood plasma, respectively. The procedure for mouse plasma extraction is detailed in the ESI.† The ζ-potential of the CBs was measured after 0.5 h of incubation, and the mean hydrodynamic diameter of the CBs was monitored over a 24 h incubation period.The ζ-potential results of different CBs after 0.5 h of incubation at 37 °C are shown in Fig. 3(A). Both MO-only CBs and MM@CBs exhibited negative ζ-potentials after incubation with PBS, RPMI media, and mouse plasma. For cationic CBs, a positive ζ-potential was observed after incubation with PBS. However, after incubation with RPMI media and mouse plasma, the ζ-potential of cationic CBs dramatically switched from positive to negative. At physiological pH, the acidic amino acids bearing negative charge in the cell culture media would attach to the positively charged surface of the cationic CBs, resulting in a net negative ζ-potential. In mouse plasma, serum proteins such as albumin and globulin would attach to or fuse with the CBs, leading to a negative ζ-potential.32
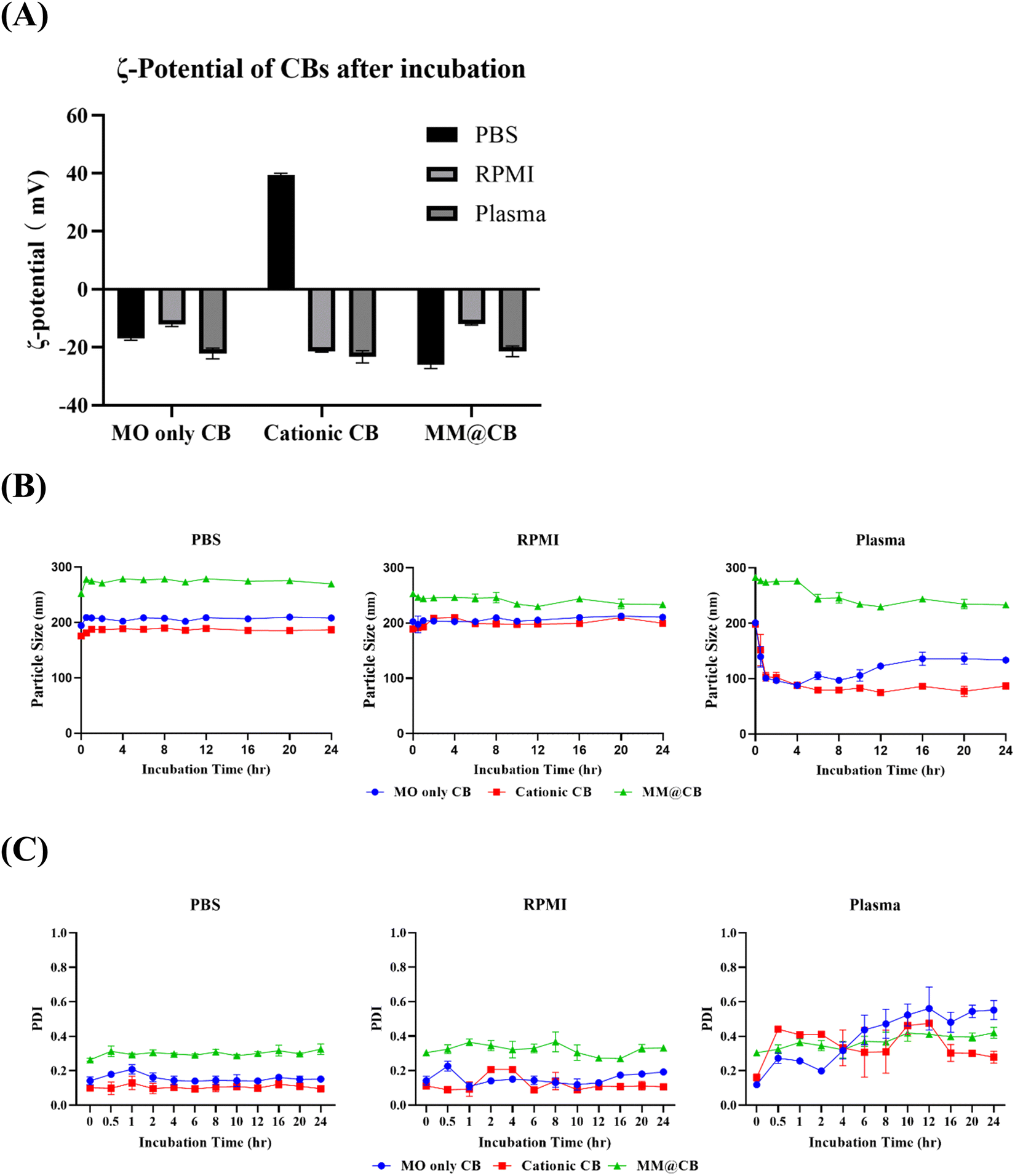 | ||
| Fig. 3 Cubosome stability characterization. (A) ζ-potential of different CBs after 0.5 h incubation with PBS, RPMI media and mouse plasma respectively. (B) Mean hydrodynamic diameters of different CBs during 24 h incubation with PBS, RPMI media and mouse plasma. (C) Polydispersity index (PDI) of different CBs during 24 h incubation with PBS, RPMI media and mouse plasma. Different CB samples were added into PBS, RPMI media and mouse plasma respectively to a final MO concentration at 5 mg mL−1 and incubated at 37 °C. For DLS and ζ-potential measurements, 50 μL of samples were diluted in 1 mL ultrapure water and measured. All measurements were carried out at 25 °C in triplicate. Error bars represent ± s.d. (n = 3). | ||
The variations in mean hydrodynamic diameter and polydispersity index (PDI) of different CBs after incubation in PBS, RPMI media, and mouse plasma are shown in Fig. 3(B) and (C). During incubation with PBS and RPMI media, all CBs exhibited stable particle sizes without significant variations. However, after incubation in mouse plasma, the size of MO-only CBs and cationic CBs immediately decreased from ∼200 nm to ∼100 nm, accompanied by increased PDI. These results imply a collapse of non-camouflaged CBs in plasma. In contrast, the size and PDI of MM@CBs remained stable during the first 4 h of incubation in mouse plasma, with only a slight decrease in particle size observed after 6 h incubation. These results demonstrate the enhanced stability of CBs after cell membrane camouflaging.
Previous studies have reported that MO-based CBs interact rapidly with plasma upon contact, resulting in partial destabilization and collapse of the CBs. Warunee et al.15 investigated the disintegration process of MO-based CBs in plasma. They incubated CBs with whole plasma and specific plasma components such as HDL (high-density lipoprotein), LDL (low-density lipoprotein), and albumin. Their study revealed that HDL affected CBs’ integrity, leading to the formation of smaller particles containing components from both CBs and HDL. When incubated with LDL, CBs fused with LDL. Albumin was shown to extract monoolein from the CB particles. J.C. Bode et al.16 also carried out an investigation on the interaction between MO-based CBs and blood components. Cryo-TEM investigation revealed that, when incubated in plasma, the CB particle surface was decomposed first, accompanied with a decreased particle size. Additionally, the F127 stabilizer was not able to protect the CBs from the described interactions with blood compounds.
Our results, along with previous research, reveal that MO-based CBs interact with blood plasma and disintegrated immediately after contact. The camouflaging of macrophage membrane enhanced the stability of CBs during incubation with plasma. The core–shell structure of MM@CBs is believed to prevent direct interaction between plasma components and CB cores, inhibiting CB disintegration.
3.4 Macrophage evasion and organ accumulation reduction of MM@CBs
Macrophage cell membrane proteins play a vital role in preventing the engulfment of foreign nanoparticles by macrophages and other mononuclear phagocytes.33,34 Phagocytosis is the primary mechanism for nanoparticle uptake by macrophages.35,36 Macrophage can easily phagocytize noncoated nanoparticles, whereas macrophage membrane coated nanoparticles can significantly evade recognition by macrophage cells.37 Functional membrane proteins from macrophages would inhibit the uptake by macrophages themselves. To examine the in vitro macrophage evasion performance of MM@CBs, a cellular internalization investigation was conducted on J774.1 cells and Colon26 cells using flow cytometry (FCM) and confocal laser microscopy (CLMS). CBs were doped with 0.5 wt% NBD-PE during preparation procedures to facilitate investigation by FCM and CLMS. According to the SAXS results (Fig. S5, ESI†), the doping of NBD-PE did not induce phase changes on the CBs, except for a slight increase in lattice parameter compared to the non-doped CBs.The cellular internalization efficacy after different incubation time spans (2, 4, 6, 8 and 12 h) was investigated using FCM. It was noted that individual macrophage in a population shows heterogeneity in the phagocytosis capacity.38 As shown in FCM dot plots (Fig. 4(A)), after a 4 h treatment with cationic CBs, the J774.1 macrophages were separated into two populations, including a low NBD fluorescence population (up left) and a high NBD fluorescence population (low right). This justified the heterogeneity in the macrophage internalization capacity. However, after a 4 h treatment with MM@CB, the cell population did not show clear separation. The NBD fluorescence intensity of MM@CBs treated J774.1 cells was generally lower than that of the cationic CBs treated cells, indicating a general inhibition of nanoparticles internalized by macrophages, solidifying the macrophage evasion capacity of MM@CBs. The internalization efficacy was monitored for 12 h using FCM. The results (Fig. 4(B)) revealed that, during 2 to 6 h treatment, the MM@CBs internalization efficacy was significantly lower than the cationic CBs. Compared to the cationic CB group, the cellular internalization efficacy, as calculated by the mean fluorescence intensity (MFI) increase, was reduced by 40.1 ± 13.8% (2 h incubation), 43.8 ± 12.1% (4 h incubation) and 36.6 ± 3.8% (6 h incubation) in the MM@CB group. After 8 h treatment, the internalization efficacies of cationic CBs and MM@CBs gradually converged. The CLSM images of J774.1 cells after CBs treatments were illustrated in Fig. 4(C). In comparison to the cationic CBs group, J774.1 cells treated with MM@CBs showed a lower NBD fluorescence intensity after 4 h incubation. Both FCM and CLSM results showed a reduction in the internalization efficacy of MM@CBs by the membrane source macrophage cell (J774.1), suggesting an in vitro immune escape capability of MM@CBs. However, according to the FCM results (Fig. 4(B)) and CLSM (Fig. S3, ESI†), no significant difference in internalization efficacy was observed between MM@CBs and cationic CBs when interacting with Colon26 cancer cells. Considering that both cationic CBs and MM@CBs exhibited negative ζ-potential upon contact with cell culture media (Fig. 3(A)), it appears that surface charge is not the primary factor influencing cellular internalization efficacy. In this study, the membrane source macrophages (J774.1) are non-polarized (M0) phenotype, which may display negligible selective internalization ability towards other cells.37,39,40 Generally, macrophages can be influenced by various factors to change their phenotype into two categories: M1 phenotype (pro-inflammatory) and M2 phenotype (anti-inflammatory). A previous study by C. Hu et al.41 constructed macrophage membrane coated nanoparticles for anti-tumor applications. Their findings revealed that both M0 and M1 macrophage membrane-coated nanoparticles demonstrated macrophage evasion performance in vitro, while their cancer cell internalization efficacies were comparable. Other research has indicated that tumor-associated macrophage (TAM) membrane coated nanoparticles exhibited better tumor targeting performance.42,43 TAM is a special class of macrophage present in the microenvironment of solid tumors and demonstrate tumor-homing capabilities.44 This enhanced cancer targeting is likely due to the presence of specific surface markers on TAMs that facilitate their interaction with cancer cells. However, TAM involves both M1 and M2 phenotypes, and the mechanisms of macrophage polarization and their cancer-targeting performance require further investigation. Overall, MM@CBs demonstrated notable macrophage evasion compared to non-coated CBs, though they did not significantly alter the internalization efficacy in Colon26 cancer cells.
 | ||
Fig. 4
In vitro immune-escape performance and in vivo distribution investigation results. (A) Dot plot of J774.1 internalization efficacy after 4 h treatment with 0.15 mg mL−1 of NBD-PE doped cationic CBs and MM@CBs (calculated according to MO concentration). (B) J774.1 and Colon26 cell internalization efficacies on cationic CBs and MM@CBs after different incubation time periods. Cells were incubated with 0.15 mg mL−1 CBs (calculated according to MO concentration) doped with NBD-PE. The black bars represent the cationic CBs group and the gray bars represent the MM@CBs group. Error bars represent ± s.d. n = 3. (C) Confocal laser scanning microscopy (CLSM) images of J774.1 cells after 4 h treatment with NBD-PE doped cationic CBs and MM@CBs (0.15 mg mL−1 CBs, calculated according to MO). The three columns are corresponding to DAPI channel, NBD channel and merged pictures respectively. Scale bar = 40 μm. (D) Whole body fluorescence imaging of mice after i.v injection of DiR-labeled cubosome samples for 9 and 24 h. Color scale ranges from 3 × 107 to 3 × 108 (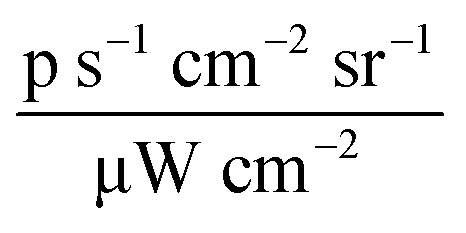 ). (E) DiR radiance efficiency in the main organs after i.v injection. Error bars = ± s.d, n = 3. ns: not significant; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. ). (E) DiR radiance efficiency in the main organs after i.v injection. Error bars = ± s.d, n = 3. ns: not significant; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. | ||
Based on the high stability and macrophage evasion capabilities of MM@CBs observed in vitro, we further investigated their biodistribution in mice. Fig. S4 (ESI†) summarized the biodistribution results for 24 h post-injection. Fig. 4(D) depicts the in vivo biodistribution images of BALB/c mice following intravenous (i.v) injection of different CBs for 9 h and 24 h. The cationic CBs showed a wide distribution in whole mouse body, whereas MM@CBs did not. As mentioned previously, the MO-based CBs interact with plasma rapidly upon contact, leading to a fraction of CBs destabilization and collapsing (Fig. 3(B)), resulting in a widespread of DiR in mice bodies. In the case of MM@CBs, the nanoparticles appear to be stabilized, leading to moderate interactions between CB cores and plasma, thereby reducing CB collapse. The CBs accumulation in main organs (liver, spleen, kidneys, heart and lungs) were investigated at 9 h and 24 h post-injection. Cationic CBs and MM@CBs exhibited comparable accumulation in liver and spleen which is due to the liver/spleen filtration.45 However, MM@CBs exhibited lower accumulation in organs including lungs, kidneys, and heart. Especially, MM@CBs showed a marked reduced accumulation in lungs (9 h and 24 h post-injection, p < 0.0001). Previous research demonstrated a similarly high accumulation of MO-based CBs in heart, kidneys and lungs.46 Firstly, the cationic CBs partially collapsed upon contacting with plasma (Fig. 3(B)), resulting in a widespread of DiR signals in main organs. Another factor is that the cationic CBs was masked with a layer of serum protein corona upon contacting with plasma. Which is evidenced by the ζ-potential switch of cationic CBs (Fig. 3(A)) upon contacting with plasma. The serum protein corona masking on cationic CBs would trigger the recognition of nanoparticles by macrophages,47 and could also determine their in vivo fate.48,49 Certain distinct serum protein species formed on to the nanoparticles’ surface could promote their accumulation in lungs.48 Coating nanoparticles with cell membrane could reduce the formation of serum protein corona and reduce their accumulation in certain organs.50,51 Generally, this reduced accumulation in heart, kidneys and lungs would likely prolong CB circulation in vivo, moderate the side effect of cargo drug molecules on those organs, reduce the off-target effects.
3.5 DOX loading and anticancer performance in vitro
DOX was selected as the model drug to evaluate the drug loading capacity and anti-cancer performance of cationic CBs and MM@CBs in this study. During the preparation process, 0.5 wt% of DOX was loaded into the cationic CBs. Characterization results of DOX-loaded cationic CBs and MM@CBs are shown in Table 3. The drug encapsulation efficacy (EE) results (Table S2, ESI†) revealed a high EE of DOX of approximately 91.93%. This high EE can be attributed to the presence of a hydrophilic inner water channel and the large surface area between the bilayer and internal water channels in CBs, facilitating the embedding of DOX. The macrophage membrane camouflaging process slightly reduced the DOX encapsulation efficacy to approximately 86.29%. The biocompatibilities of prepared CBs were investigated on HEK293 cells via MTT assay. The MTT results (Fig. S2, ESI†) demonstrate that 0.2 mg mL−1 of MO did not induce significant inhibition of cellular proliferation after 24 h of incubation.| CB formulation | Hydrodynamic diameter [nm] | PDI | Zeta potential [nm] | Space group | Lattice parameter [nm] |
|---|---|---|---|---|---|
| Cationic CBs + DOX | 228.2 ± 3.5 | 0.14 ± 0.03 | 29.9 ± 1.5 | Im3m | 14.46 |
| MM@CBs + DOX | 270.7 ± 5.9 | 0.28 ± 0.01 | −27.5 ± 1.9 | Im3m/Pn3m | 14.28/10.95 |
The DOX release pattern in vitro results are shown in Fig. 5(A). Both the cationic CBs and the MM@CBs demonstrated comparable drug release patterns. However, it was observed that the general release ratio from MM@CBs was slightly higher than that from cationic CBs. The reduction of DOX EE and the difference in drug release rates can be attributed to the phase and structure changes of CBs after macrophage camouflaging. During the cell membrane coating process, the cubic lipid phase change (from Im3m to Pn3m), as well as the reduction of the water channel radius result in partial DOX leakage from the CBs, and induced a relatively higher DOX release rate from MM@CBs. These results highlight the importance of carefully considering the impact of the cell membrane coating and phase changes on drug release behavior in CBs. Understanding these factors can aid in developing a controlled drug release pattern. Further investigations are warranted to delve deeper into the underlying mechanisms and to fine-tune the drug-release properties of MM@CBs for specific therapeutic purposes.
 | ||
| Fig. 5 DOX release pattern and Colon26 Annexin-V/PI assay results. (A) Release pattern of DOX-loaded cationic CBs (blue) and MM@CBs (red) in vitro. (B) The stack bar graphs showing the percentage of living cells, early apoptotic cells and late apoptotic Colon26 cells (C) the dot graph of the Colon26 cells Annexin-V/PI assay results after cationic CBs, MM@CBs, free DOX, DOX-loaded cationic CBs and DOX-loaded MM@CBs treatments for 12/24 h. The operation is detailed in Experimental Section 2.10. Error bars represent ± s.d. (n = 3). | ||
The anti-cancer performance was assessed using Colon26 cell lines. Free DOX and different CBs containing DOX were introduced to Colon26 cells at a final DOX concentration of 1 μg mL−1 (MO concentration was 0.2 mg mL−1) and subjected to 12/24 h treatment. For comparation, cationic CB (without DOX) and MM@CB (without DOX) were introduced to Colon26 cells at a final concentration of 0.2 mg mL−1 (based on MO concentration). As shown in Fig. 5(B) and (C), cationic CBs and MM@CBs induced mild and comparable apoptosis on Colon26 cells. The cationic CBs + DOX and MM@CBs + DOX groups exhibited slightly enhanced anti-cancer performance compared to the free DOX group after 12 h treatment, with no remarkable difference between the cationic CBs + DOX group and MM@CBs + DOX group. However, after 24 h, both the cationic CBs + DOX group and MM@CBs + DOX groups notably outperformed the free DOX group. The main difference was observed in the late apoptotic cell ratio in which the MM@CBs + DOX group is especially higher than that of the free DOX group as well as the cationic CBs + DOX group. This can be attributed to the higher release rate of DOX from MM@CBs upon cellular uptake. Given that, cationic CBs and MM@CBs showed comparable internalization efficacy by Colon26 cells. MM@CBs with faster DOX release kinetics might lead to a higher concentration of free DOX inside the cells, potentially triggering earlier apoptosis and accelerate the late apoptotic ratios in Colon26 cells.
4. Conclusion
The objective of this study was to explore a method for stabilizing and improving the immune escape capabilities of cubosomes (CBs) by utilizing macrophage cell membranes as a surface modification strategy, instead of the conventional PEGylation approach. Our systematic characterization results revealed that, the CBs structure slightly changed from QIIP to a coexistence of QIIP and QIID after the macrophage membrane camouflaging. The MM@CBs shows delayed internalization by macrophages (J774.1), while there was no notable impact on cancer cell uptake efficacy. Also, the macrophage membrane camouflaging stabilized the CBs in vivo and reduced their accumulation in heart, kidneys and lungs. These results suggest that the cell membrane camouflaging strategy presents a promising approach for surface modification of CBs, effectively combining the physical properties of CBs with the bio-functionalities of cell membranes. Taken together, our results indicate that this approach holds potential for enhancing the performance of CBs-based nanomedicines delivery, leveraging the immune escape benefits of macrophage cell membrane coating and the ability to deliver a wide range of therapeutic drug molecules. Additionally, further engineering of membrane source cells, would likely promote their cancer-targeting performance,In conclusion, the study's outcomes highlight the feasibility of employing macrophage cell membrane camouflaging as a surface modification strategy for CBs. The successful integration of physical and bio-functional properties in this approach shows promise for its potential applications in future drug delivery research and development.
Author contributions
X. R.: conceptualization, experiment, data analysis, data validation, data curation, writing – original draft preparation; Y. O.: supervision, conceptualization, in vivo experiment, data analysis, data validation, writing – review and editing. N. M. W.: supervision, conceptualization, SAXS and cryo-TEM experiment, data validation, writing – review and editing. T. S.: In vivo experiment, data analysis, data validation. W. W.: SAXS experiment, data analysis, writing – review. N. K.: cryo-TEM experiments, data analysis, writing – review. H. U.: supervision, conceptualization, writing – review and editing. All authors (X. R., Y. O., N. M. W., T. S., W. W. and H. U.) reviewed the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The author X. R., expresses gratitude to the Japanese Government for the MEXT scholarship that supported this research. This work received financial support from JSPS KAKENHI (23K17862, 21H04628) and the Grant in-Aid for Transformative Research Areas (A) on “Material Symbiosis” (23H04075) from MEXT, Japan. One of the authors, Y. O., thanks the Multidisciplinary Research Laboratory System in Osaka University, JKA, and its promotion funds from KEIRIN RACE, Tateisi Science and Technology Foundation, Mukai Science and Technology Foundation, and Toyota Physical and Chemical Research Institute for their valuable financial support in facilitating this research. All the coauthors thank to SPring-8 for conducting the SAXS measurements in this study. The synchrotron radiation experiments were performed at the BL19B2 beamline of SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (proposal no. 2023B1739). Appreciation is also extended to Prof. Kaoru Mitsuoka from Osaka University Research Center for Ultra-High Voltage Electron Microscopy for generously providing TEM and cryo-TEM equipment, and to Assistant Prof. Shuichiro Fukushima from the R3 Institute for Newly-Emerging Science Design, Osaka University for the technical assistant of confocal microscope experiments.References
- S. Murgia, S. Biffi and R. Mezzenga, Curr. Opin. Colloid Interface Sci., 2020, 48, 28–39 CrossRef CAS.
- J. Zhai, C. Fong, N. Tran and C. J. Drummond, ACS Nano, 2019, 13, 6178–6206 CrossRef CAS PubMed.
- R. Mezzenga, J. M. Seddon, C. J. Drummond, B. J. Boyd, G. E. Schröder-Turk and L. Sagalowicz, Adv. Mater., 2019, 31(35), 1900818 CrossRef PubMed.
- B. Boyd, S. Khoo, D. Whittaker, G. Davey and C. Porter, Int. J. Pharm., 2007, 340, 52–60 CrossRef CAS PubMed.
- H. Kim and C. Leal, ACS Nano, 2015, 9, 10214–10226 CrossRef CAS PubMed.
- S. Sarkar, N. Tran, S. K. Soni, Z. Nasa, C. J. Drummond and C. E. Conn, ACS Appl. Mater. Interfaces, 2021, 13, 2336–2345 CrossRef CAS PubMed.
- A. Angelova, B. Angelov, R. Mutafchieva, S. Lesieur and P. Couvreur, Acc. Chem. Res., 2010, 44, 147–156 CrossRef PubMed.
- X. Mulet, B. J. Boyd and C. J. Drummond, J. Colloid Interface Sci., 2013, 393, 1–20 CrossRef CAS PubMed.
- H. M. Barriga, M. N. Holme and M. M. Stevens, Angew. Chem., Int. Ed., 2018, 58, 2958–2978 CrossRef PubMed.
- C. Leal, N. F. Bouxsein, K. K. Ewert and C. R. Safinya, J. Am. Chem. Soc., 2010, 132, 16841–16847 CrossRef CAS PubMed.
- J. A. Mills, F. Liu, T. R. Jarrett, N. L. Fletcher and K. J. Thurecht, Biomater. Sci., 2022, 10, 3029–3053 RSC.
- J. S. Suk, Q. Xu, N. Kim, J. Hanes and L. M. Ensign, Adv. Drug Delivery Rev., 2016, 99, 28–51 CrossRef CAS PubMed.
- K. Shiraishi and M. Yokoyama, Sci. Technol. Adv. Mater., 2019, 20, 324–336 CrossRef CAS PubMed.
- M. Mohamed, A. S. Abu Lila, T. Shimizu, E. Alaaeldin, A. Hussein, H. A. Sarhan, J. Szebeni and T. Ishida, Sci. Technol. Adv. Mater., 2019, 20, 710–724 CrossRef CAS PubMed.
- W. Leesajakul, M. Nakano, A. Taniguchi and T. Handa, Colloids Surf., B, 2004, 34, 253–258 CrossRef CAS PubMed.
- J. C. Bode, J. Kuntsche, S. S. Funari and H. Bunjes, Int. J. Pharm., 2013, 448, 87–95 CrossRef CAS PubMed.
- C. M. J. Hu, L. Zhang, S. Aryal, C. Cheung, R. H. Fang and L. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 10980–10985 CrossRef CAS PubMed.
- D. Wang, S. Wang, Z. Zhou, D. Bai, Q. Zhang, X. Ai, W. Gao and L. Zhang, Adv. Healthcare Mater., 2021, 11(7), 2101349 CrossRef PubMed.
- W. Chen, K. Zeng, H. Liu, J. Ouyang, L. Wang, Y. Liu, H. Wang, L. Deng and Y. Liu, Adv. Funct. Mater., 2017, 27(11), 1605795 CrossRef.
- C. V. Kulkarni, W. Wachter, G. Iglesias-Salto, S. Engelskirchen and S. Ahualli, Phys. Chem. Chem. Phys., 2011, 13, 3004–3021 RSC.
- J. Barauskas, M. Johnsson, F. Joabsson and F. Tiberg, Langmuir, 2005, 21, 2569–2577 CrossRef CAS PubMed.
- L. Liu, X. Bai, M.-V. Martikainen, A. Kårlund, M. Roponen, W. Xu, G. Hu, E. Tasciotti and V.-P. Lehto, Nat. Commun., 2021, 12, 5726 CrossRef CAS PubMed.
- B. Angelov, A. Angelova, M. Drechsler, V. M. Garamus, R. Mutafchieva and S. Lesieur, Soft Matter, 2015, 11, 3686–3692 RSC.
- A. Yaghmur, P. Laggner, M. Almgren and M. Rappolt, PLoS One, 2008, 3(11), e3747 CrossRef PubMed.
- T. G. Meikle, A. Zabara, L. J. Waddington, F. Separovic, C. J. Drummond and C. E. Conn, Colloids Surf., B, 2017, 152, 143–151 CrossRef CAS PubMed.
- S. Sarkar, N. Tran, M. H. Rashid, T. C. Le, I. Yarovsky, C. E. Conn and C. J. Drummond, ACS Appl. Bio Mater., 2018, 2, 182–195 CrossRef PubMed.
- L. Han and S. Che, Adv. Mater., 2018, 30, 1705708 CrossRef PubMed.
- K. Larsson and F. Tiberg, Curr. Opin. Colloid Interface Sci., 2005, 9, 365–369 CrossRef CAS.
- T. Abraham, M. Hato and M. Hirai, Biotechnol. Prog., 2008, 21, 255–262 CrossRef PubMed.
- C. E. Conn and C. J. Drummond, Soft Matter, 2013, 9, 3449–3464 RSC.
- B. Angelov, A. Angelova, S. K. Filippov, M. Drechsler, P. Štěpánek and S. Lesieur, ACS Nano, 2014, 8, 5216–5226 CrossRef CAS PubMed.
- C. C. Fleischer and C. K. Payne, J. Phys. Chem. B, 2012, 116, 8901–8907 CrossRef CAS PubMed.
- Y. Wu, S. Wan, S. Yang, H. Hu, C. Zhang, J. Lai, J. Zhou, W. Chen, X. Tang, J. Luo, X. Zhou, L. Yu, L. Wang, A. Wu, Q. Fan and J. Wu, J. Nanobiotechnol., 2022, 20, 542 CrossRef CAS PubMed.
- N. G. Sosale, K. R. Spinler, C. Alvey and D. E. Discher, Curr. Opin. Immunol., 2015, 35, 107–112 CrossRef CAS PubMed.
- H. H. Gustafson, D. Holt-Casper, D. W. Grainger and H. Ghandehari, Nano Today, 2015, 10, 487–510 CrossRef CAS PubMed.
- N. Khatoon, Z. Zhang, C. Zhou and M. Chu, Biomater. Sci., 2022, 10, 1193–1208 RSC.
- A. Parodi, N. Quattrocchi, A. L. van de Ven, C. Chiappini, M. Evangelopoulos, J. O. Martinez, B. S. Brown, S. Z. Khaled, I. K. Yazdi, M. V. Enzo, L. Isenhart, M. Ferrari and E. Tasciotti, Nat. Nanotechnol., 2012, 8, 61–68 CrossRef PubMed.
- K. Sachdeva, M. Goel and V. Sundaramurthy, Traffic, 2020, 21, 522–533 CrossRef CAS PubMed.
- K. Ling, Z. Zhao, R. Wu, C. Tao, S. Liu, T. Yu, Q. Cao, J. Yan, T. Ge, M. Shariati, M. Sadeghi and J. Liu, Nanoscale, 2024, 16, 1673–1684 RSC.
- K. Y. Teo, C. Sevencan, Y. A. Cheow, S. Zhang, D. T. Leong and W. S. Toh, Small Sci., 2022, 2(4), 2100116 CrossRef CAS.
- C. Hu, T. Lei, Y. Wang, J. Cao, X. Yang, L. Qin, R. Liu, Y. Zhou, F. Tong, C. S. Umeshappa and H. Gao, Biomaterials, 2020, 255, 120159 CrossRef CAS PubMed.
- C. Chen, M. Song, Y. Du, Y. Yu, C. Li, Y. Han, F. Yan, Z. Shi and S. Feng, Nano Lett., 2021, 21, 5522–5531 CrossRef CAS PubMed.
- T. Du, Y. Wang, Z. Luan, C. Zhao and K. Yang, Int. J. Pharm., 2022, 624, 121911 CrossRef CAS PubMed.
- R. Noy and J. W. Pollard, Immunity, 2014, 41, 49–61 CrossRef CAS PubMed.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS PubMed.
- M. Dawoud, M. Mojally, R. Abdou and H. G. Attia, Pharm. Dev. Technol., 2023, 28, 277–287 CrossRef CAS PubMed.
- K. Saha, M. Rahimi, M. Yazdani, S. T. Kim, D. F. Moyano, S. Hou, R. Das, R. Mout, F. Rezaee, M. Mahmoudi and V. M. Rotello, ACS Nano, 2016, 10, 4421–4430 CrossRef CAS PubMed.
- M. Qiu, Y. Tang, J. Chen, R. Muriph, Z. Ye, C. Huang, J. Evans, E. P. Henske and Q. Xu, Proc. Natl. Acad. Sci. U. S. A., 2022, 119(8), e2116271119 CrossRef CAS PubMed.
- M. Qiu, Z. Glass, J. Chen, M. Haas, X. Jin, X. Zhao, X. Rui, Z. Ye, Y. Li, F. Zhang and Q. Xu, Proc. Natl. Acad. Sci. U. S. A., 2021, 118(10), e2020401118 CrossRef CAS PubMed.
- D. Zou, Z. Wu, X. Yi, Y. Hui, G. Yang, Y. Liu, J. Teng, H. Wang, A. Brooks, H. Wang, X. Liu, Z. P. Xu, M. S. Roberts, H. Gao and C.-X. Zhao, Proc. Natl. Acad. Sci. U. S. A., 2022, 120(1), e2214757120 CrossRef PubMed.
- C. Corbo, R. Molinaro, F. Taraballi, N. E. Toledano Furman, K. A. Hartman, M. B. Sherman, E. De Rosa, D. K. Kirui, F. Salvatore and E. Tasciotti, ACS Nano, 2017, 11, 3262–3273 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb01063a |
| This journal is © The Royal Society of Chemistry 2024 |