
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unity fluorescent carbene–gold(I)–acetylide complexes with two-photon absorption and energy-efficient blue FOLEDs†
Alexander C.
Brannan
a,
Hwan-Hee
Cho
 b,
Jonathan
Daniel
c,
Amelia J.
Harvey
a,
Charles T.
Smith
d,
Nguyen
Le Phuoc
e,
Mikko
Linnolahti
*e,
Mireille
Blanchard-Desce
*c,
Neil C.
Greenham
*b and
Alexander S.
Romanov
*a
b,
Jonathan
Daniel
c,
Amelia J.
Harvey
a,
Charles T.
Smith
d,
Nguyen
Le Phuoc
e,
Mikko
Linnolahti
*e,
Mireille
Blanchard-Desce
*c,
Neil C.
Greenham
*b and
Alexander S.
Romanov
*a
aDepartment of Chemistry, The University of Manchester, Oxford Rd, Manchester M13 9PL, UK. E-mail: alexander.romanov@manchester.ac.uk
bDepartment of Physics, Cavendish Laboratory, Cambridge University, Cambridge, CB3 0HF, UK. E-mail: ncg11@cam.ac.uk
cInstitut des Sciences Moléculaires (ISM, UMR5255), University of Bordeaux, Centre National de la Recherche Scientifique, Institut Polytechnique de Bordeaux, Bat A12, 351 Cours de la Libération, 33405 Talence, France. E-mail: mireille.blanchard-desce@u-bordeaux.fr
dDepartment of Physics and Astronomy and the Photon Science Institute, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK
eDepartment of Chemistry, University of Eastern Finland, FI-80101 Joensuu, Finland. E-mail: mikko.linnolahti@uef.fi
First published on 19th July 2024
Abstract
Two fluorescent organometallic “carbene–gold(I)–acetylide” (CMAc) complexes based on bicyclic alkyl(amino) carbene (BIC), 4-ethynyl-2,1,3-benzothiadiazole (Au1) and 4,7-diethynyl-2,1,3-benzothiadiazole (Au2) have been synthesized and characterized. Compounds Au1 and Au2 exhibit blue to yellow fluorescence with up to unity fluorescence quantum yields and radiative rates exceeding 108 s−1. The dipolar Au1 complex exhibits a low two photon absorption cross-section (2PA σ2) of 0.8 GM, whereas the quadrupolar Au2 complex shows a high 2PA σ2 value of 90 GM. A fluorescent CMAc organic light emitting diode (OLED) emits blue electroluminescence with up to 4.7% external quantum efficiency.
Introduction
Materials with third-order nonlinear optical (NLO) properties, for instance two-photon absorption (2PA) and two-photon excited fluorescence (2PF), has attracted ever-increasing interest from academia and industry due to their promising applications as materials for nano- or microfabrication and 3D-optical memory storage,1 optical power limiting,2 imaging and multiphoton microscopy,3 and are particularly useful for biomedical applications such as photosensitizers for the photodynamic therapy of cancer4 and many others. During the 2PA event, the molecule absorbs simultaneously two photons having the same polarization, while their photon's energy is equal to or slightly above half the energy gap between the ground and first excited states.5 Therefore, 2PA molecular chromophores are characterized by the appearance of an absorption band at a lower energy than the corresponding S0 → S1 transition. The 2PA spectra exhibit a cross-section value, σ2, measured in Göppert-Mayer (GM) units, where 1 GM equals 10−50 cm4 s photon−1. Large 2PA cross-sections (over 100 GM) and photochemical stability are highly sought-after characteristics to unlock the whole portfolio of advanced applications offered by 2PA materials. The best performing 2PA materials are commonly large macromolecules (conjugated polymers and Ru-containing dendrimers, σ2 up to 104 GM),6 while gold nanorods7 are the champion inorganic materials with normalised σ2 values up to 108 GM, albeit reported for solutions only. By contrast, small molecule organic and organometallic 2PA materials suffer from low σ2 values (<1 GM) thus requiring a high laser power which may cause adverse effects in biomedical applications.Such a striking difference between small and macromolecules suggests that efficient 2PA materials should exhibit a large change in polarization upon photoexcitation.8 Therefore, the 2PA molecular design strategy pinpoints molecules with a large transition dipole moment (Δμ, difference between ground and excited states).9 Within the two-level approximation, the 2PA spectrum is represented by eqn (1):
| σ2(ν) = B|Δμ10|2|μ10|2g2(ν), | (1) |
Various molecular designs have been proposed based on combinations between the donor (D) and acceptor (A) moieties, connected with π-conjugated bridging units, to form extended π-conjugated systems to enhance the NLO properties of dipolar (D–π–A, D–π–D or A–π–A), quadrupolar (D–A–D or A–D–A), octupolar or even multipolar 2PA chromophores.9 Among these molecular designs, small molecule organometallic alkynyl complexes of gold(I), ruthenium(II), and platinum(II) demonstrated particularly strong 2PA cross-section values ranging from 10 to 104 GM, which is on par with those of organic macromolecules.10 Late heavy transition metals (4d and 5d) appeared to be particularly beneficial due to their electron-rich nature and greater polarizability compared with 3d metal alkynyl complexes.10 For instance, a common molecular design for neutral linear Au(I)–complexes involves an alkynyl moiety with optionally-substituted fluorenyl benzothiazole (BTD) ligands, where the Au-atom possesses only 14 valence electrons (ve) and may only have one alkynyl ligand.11 However, 2PA σ2 values may be enhanced by coordination with strong σ-donor ligands, for instance phosphines or N-heterocyclic carbenes (NHC).11 Such dipolar chromophores of the D–(Au)–A type gold complexes benefit from one-photon absorption (1PA) often happening in the UV-vis region, thus making them optically transparent for the desired applications in the visible wavelength range.10,11 Several new linear (L)Au(I)-complexes with directly coordinated 4-ethynylbenzothiadiazole (2PA-chromophores without a fluorenyl π-conjugated bridge) have been recently reported to have near unity fluorescence quantum yields with radiative rates of over 107 s−1.12 Regardless of such spectacular photophysical properties, there are no reports of their 2PA properties or tests of their performance in organic light-emitting diodes (OLEDs). Recently, we prepared a series of bright phosphorescent carbene–metal–acetylide (CMAc)13 complexes with cyclic alkyl(amino) carbene (CAAC) and cyclic alkyl(amino) carbene (BIC) ligands.14 The BIC-carbene ligand is a strong σ-donor, which increases the electron density on the Au-atom. In addition, the BIC-carbene shows greater π-back bonding with the Au atom, thanks to the empty pz-orbital on the carbene–carbon, thus providing greater delocalization of the electron density, making it a very promising building block for 2PA materials.
All these factors spurred our interest and motivated us to prepare new dipolar (Au1, Fig. 1) and quadrupolar (Au2) carbene–Au(I)–acetylide (CMAc) materials, with a benzothiadiazole core, to reveal a principle advancement in the materials design strategy for efficient 2PA. We probe the effect of the superior electronic properties (σ-donor and π-acceptor) of the BIC-carbene on the 2PA efficiency, while investigating the applied potential of the title materials in OLED devices.
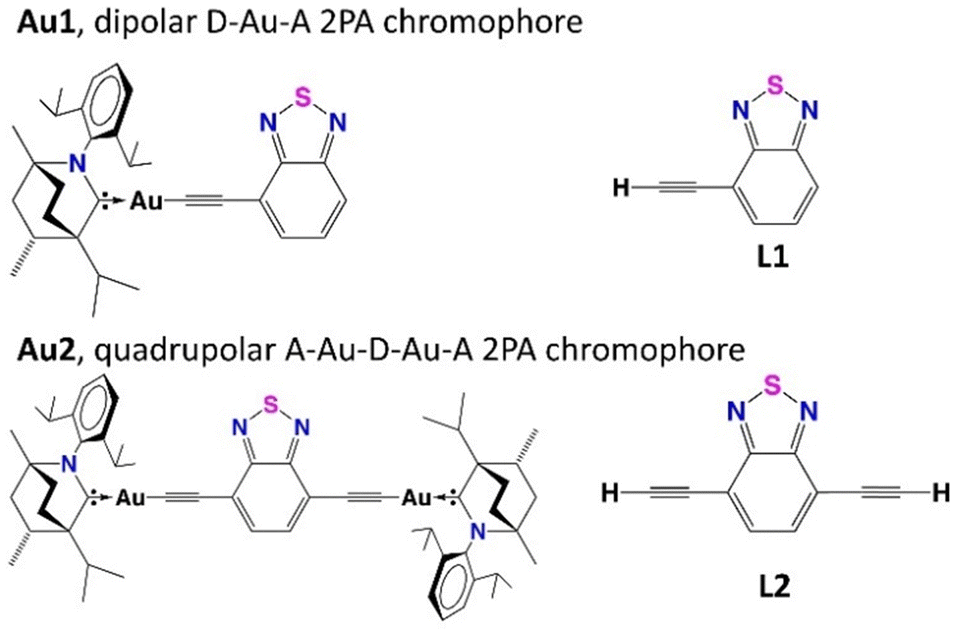 | ||
| Fig. 1 Structures of new dipolar Au1 and quadrupolar Au2 carbene complexes and corresponding ligands L1 and L2. | ||
Results and discussion
Synthesis and structure
The ligands 4-ethynylbenzothiadiazole (L1) and 4,7-diethynylbenzothiadiazole (L2) were prepared via Sonogashira cross-coupling between trimethylsilylacetylene and 4-bromo-2,1,3-benzothiadiazole or 4,7-dibromo-2,1,3-benzothiadiazole, respectively, followed by deprotection with tetra-N-butylammonium fluoride (TBAF). L1 and L2 were subsequently reacted with (BIC)AuCl under basic conditions to give carbene–metal–acetylide (CMAc) complexes Au1 and Au2, respectively. Both complexes are yellow solids, soluble in polar and non-polar aprotic solvents such as dichloromethane (DCM), tetrahydrofuran (THF), toluene and methylcyclohexane (MCH). The complexes were fully characterised by NMR, IR, HRMS and elemental analysis. Both, Au1 and Au2 show good thermal stability with decomposition temperature Td of 297 and 324 °C, respectively (Fig. S1, ESI†). The increase in thermal stability on increasing the number of (BIC)Au moieties concur with our previous work on gold(I) alkynyl complexes of the type (BIC)Au(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CPh).13cAu1 and Au2 exhibit carbene resonances at 261.09 and 261.20 ppm in 13C{1H} NMR which is similar to the previously reported 261.4 ppm for the (BIC)Au(CCPh) complex.13c BIC carbenes show greater σ-donating and π-accepting properties compared with NHC carbenes due to the absence of the second N atom adjacent to the carbene carbon. This results in the deshielding of 13C-carbene compared to similar NHC complexes, for instance, (NHC)Au(ethynylBTD)12 shows 13C-carbene resonances in the range of 187–189 ppm (up to 75 ppm upfield shift compared to complexes Au1 and Au2 with BIC-carbene). The IR spectra show triple CC bond stretching vibrations at 2105 and 2107 cm−1 for Au1 and Au2, which is ca. 12 cm−1 lower frequency compared to 2117 cm−1 for the (BIC)Au(CCPh) complex.13c This fact supports the stronger bonding of the ethynyl(BTD) ligand to the Au-atom that results in a weaker CC bond as a consequence of the lower CC bond frequency vibration compared to the (BIC)Au(CCPh) complex.
CPh).13cAu1 and Au2 exhibit carbene resonances at 261.09 and 261.20 ppm in 13C{1H} NMR which is similar to the previously reported 261.4 ppm for the (BIC)Au(CCPh) complex.13c BIC carbenes show greater σ-donating and π-accepting properties compared with NHC carbenes due to the absence of the second N atom adjacent to the carbene carbon. This results in the deshielding of 13C-carbene compared to similar NHC complexes, for instance, (NHC)Au(ethynylBTD)12 shows 13C-carbene resonances in the range of 187–189 ppm (up to 75 ppm upfield shift compared to complexes Au1 and Au2 with BIC-carbene). The IR spectra show triple CC bond stretching vibrations at 2105 and 2107 cm−1 for Au1 and Au2, which is ca. 12 cm−1 lower frequency compared to 2117 cm−1 for the (BIC)Au(CCPh) complex.13c This fact supports the stronger bonding of the ethynyl(BTD) ligand to the Au-atom that results in a weaker CC bond as a consequence of the lower CC bond frequency vibration compared to the (BIC)Au(CCPh) complex.
Cyclic voltammetry was performed for 1.4 mM THF solutions of Au1 and Au2 using a glassy carbon electrode and [n-Bu4N]PF6 as the supporting electrolyte (0.13 M) at a scan rate of 100 mV s−1 (Fig. 2, with data summarized in Table 1 and Table S1, ESI†). Both Au1 and Au2 show one quasi-reversible reduction with little variation in E1/2 values at −2.0 ± 0.02 V. The peak-to-peak separation (ΔEp) is 146 mV for Au1 and Au2 which is larger than the theoretical 59 mV value for the reversible redox process. The ratio between anodic and cathodic currents, ipa/ipc = 0.9 for both complexes, is close to unity for a fully reversible reduction process. Au1 and Au2 possess a similar current peak value for the reduction process, suggesting a one-electron reduction process for both complexes. Collectively, the experimental data suggest a quasi-reversible character for the reduction event, consistent with the analogous NHC complexes.12 We reported a similar (BIC)Au(CCPh) complex which shows a quasi-reversible reduction process at −2.90 ± 0.03 V and demonstrated that the LUMO is predominantly localised on the BIC carbene ligand.13c The 0.9 V difference in reduction potentials for Au1/Au2 compared to this (BIC)Au(CCPh) complex suggests a much greater delocalization of the LUMO. The theoretical DFT calculations reveal that the LUMO is broadly delocalised over the ethynylBTD, gold and BiC ligands (Fig. 2 and Table S4, ESI†).
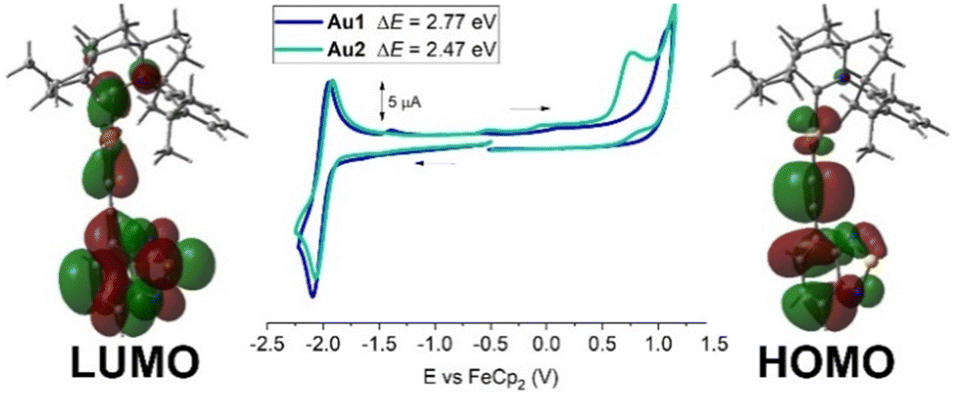 | ||
| Fig. 2 Full range cyclic voltammograms for Au1 and Au2 with frontier molecular orbitals associated with the redox processes. | ||
| λ em (nm) | τ (ns, 295 K) | Φ PL (%) | k r (107 s−1) | k nr (107 s−1) | S1e (eV) | λ em (nm) (τ, ns) 77 K | λ abs, (nm)f (ε/M−1 cm−1) | E red, (V) | LUMO (eV) | E ox, (V) | HOMO (eV) | λ 2PA, (nm) | σ 2, (GM) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Luminescence quantum yield measured with fluorescein in NaOHaq. (0.1 M, ΦPL = 0.9) as a reference. b Quantum yields determined using an integrating sphere. c Radiative rate constant kr = Φ/τ. d Nonradiative constant knr = (1 − Φ)/τ. e Singlet energy levels based on the onset of the emission spectra blue edge at 77 K in MCH. f UV-vis for CMAc solution in methycyclohexane. | ||||||||||||||
| Toluene solution | ||||||||||||||
| Au1 | 458 | 10.1 | >99a | 9.8 | 0.01 | 3.00 | 440 (8.2) | 391 (5070) | −2.02 | −3.46 | +1.06 | −6.23 | 800 | 0.8 |
| Au2 | 516 | 7.9 | >99a | 12.5 | 0.01 | 2.87 | 483 (6.9) | 356 (7680) | −1.99 | −3.48 | +0.76 | −5.95 | 720 | 90 |
445 (12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300) 300) |
820 | 2 | ||||||||||||
| 1 wt% Zeonex Film | ||||||||||||||
| Au1 | 460 | 9.4 | 81b | 8.6 | 2.2 | 2.98 | 461 (9.2) | — | — | — | — | — | — | — |
| Au2 | 534 | 6.1 | 52b | 8.5 | 7.9 | 2.58 | 529 (5.7) | — | — | — | — | — | — | — |
Both Au1 and Au2 show an irreversible oxidation process with no reduction back-peak (Fig. 2). The oxidation peak potential for Au1 (+1.06 V) is 0.3 V anodically shifted compared to Au2 (+0.76 V), suggesting an easier oxidation event for the π-extended and conjugated Au2 complex, which indicates greater delocalization of the HOMO. Theoretical calculations confirm this (Fig. 2 and Table S4, ESI†) where the HOMO is exclusively localised over the Au(ethynylBTD) moiety. Analogous NHC complexes12 were demonstrated to have similar trends and characteristics for the frontier orbitals. The calculations also indicate that there is only a 3–5% contribution of the gold atom to the HOMO and LUMO, suggesting that the redox events are predominantly localised on the BIC and ethynylBTD ligands. The energies of the HOMO and LUMO for Au1 and Au2 were estimated from the onset potential values for oxidation and reduction processes, see Table 1.
Single crystals for X-ray diffraction study were obtained by layer diffusion of hexane into DCM solution of Au1 at room temperature. The molecular structure of Au1 is shown in Fig. 3. Unlike analogous (NHC)Au(ethynylBTD) complexes with short aurophilic interactions,12Au1 is monomeric with no Au⋯Au intermolecular interactions. The Au1 molecules are arranged in a 3D-network via weak C–H⋯S and C–H⋯N intermolecular hydrogen bonds between BIC-carbene and BTD-ethynyl moieties. Complex Au1 exhibits a near linear geometry around the gold atom with a C1–Au–C25 angle of 175.7(4)°, whereas the Au–CC moiety experiences a ca. 16° bending distortion, with an Au1–C25–C26 angle of 164.4°. The torsion angle between the BIC and BTD ligand planes is 69.4(2)°, demonstrating a highly twisted geometry between the donor and the acceptor. The gold–carbene(C1) bond length is closely similar to that in analogous (NHC)Au(ethynylBTD) complexes12 whereas the gold–acetylide(C25) bond length of Au1 is longer by ca. 0.07 Å. Such a strong deviation is likely due to static disorder of the ethynylBTD moiety which limits detailed discussion of the Au1 structure.
 | ||
| Fig. 3 Crystal structure of complex Au1. Ellipsoids are shown at the 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) an angles (°): complex 7: Au–C1 2.020(4), Au–C25 2.073(12), C1–N1 1.308(6), C25–C26 1.188(17), N2–S1 1.552(13), N3–S1 1.653(14), C1–Au–C25 175.7(4)°, Au1–C25–C26 164.4°, and C1–N1⋯C27–C28 69.4(2)°. | ||
Photophysical properties and theoretical considerations
The UV-visible absorption spectra (UV-vis) for complexes Au1 and Au2 (Fig. 4(a), (d) and Table 1) show well-resolved and high-energy absorption bands up to 340 nm for Au1 and 375 nm for Au2. These are assigned to π–π* transitions for the core ethynylBTD ligand based on the high extinction coefficients (ε > 1 × 104 M−1 cm−1). Both, Au1 and Au2 exhibit a broad unstructured lower-energy absorption band, which suggests a charge transfer (CT) character of this transition. The CT-band for Au2 experiences a 52 nm red-shift compared with Au1 which is associated with greater π-delocalization for Au2. Complexes Au1 and Au2 display a weak negative solvatochromism for the CT-band, i.e. up to 12 nm blue-shift upon increasing solvent polarity (Fig. 4(a) and (d)). The extinction coefficients of Au2 for the CT band (Table 1) are twice as high as those of Au1, indicating a higher degree of allowedness for the CT transition with a greater number of (BIC)Au moieties. This correlates with the two-fold higher oscillator strength coefficient for Au2 (f = 0.9733, Table S6, ESI†) compared with Au1 (f = 0.3963). Comparison of the UV-vis spectra of the unmetallated ligands L1 and L2 (Fig. S3, ESI†) with their aurated complexes Au1 and Au2 reveals over 50 nm red-shift for the CT band upon coordination of the BIC-gold moiety to L1 and L2. Analogous phosphine and NHC-carbene gold complexes show similar red-shifts of their CT bands,12e which is due to an increase in conjugation and mixing of the 5d orbitals of the Au(I) centre with the π-system of the ethynylBTD ligand.12,15 The optical energy gap for Au1 (2.85 eV) and Au2 (2.49 eV) was calculated from the onset of the red edge of the CT band in the THF absorption spectrum. It is fully consistent with the electronic energy gap measured by cyclic voltammetry in THF solution for Au1 (2.77 eV) and Au2 (2.47 eV). This suggests that CT transition is predominantly HOMO → LUMO in nature. Our theoretical calculations suggest that the vertical S0 → S1 transitions (Table S6, ESI†) are 94% and 91% HOMO → LUMO for Au1 and Au2, respectively.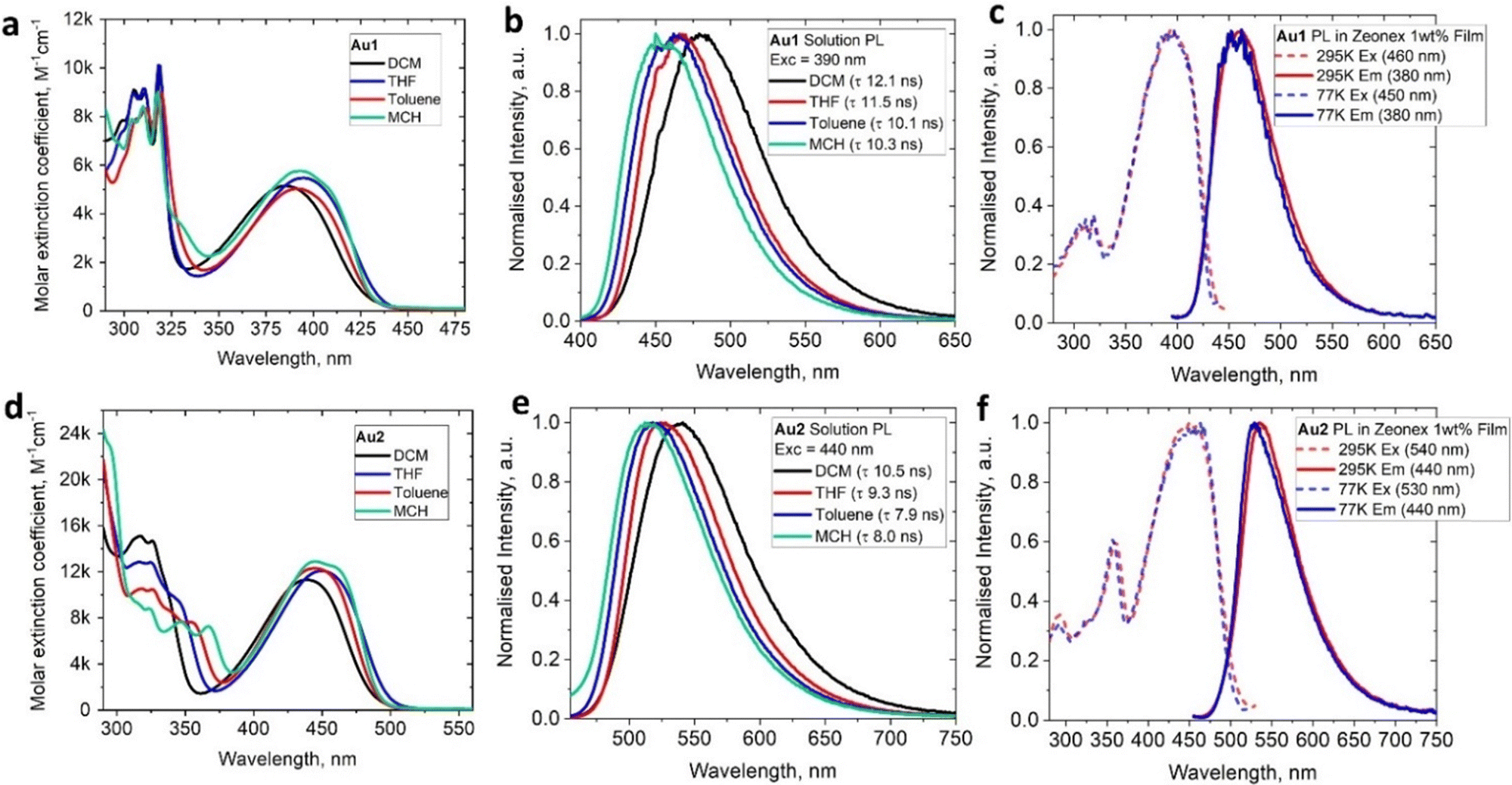 | ||
| Fig. 4 UV-vis spectra in various solvents for Au1 (a) and Au2 (d); emission spectra in various solvents for Au1 (b) and Au2 (e); emission (solid lines) and excitation (dashed line) spectra of Au1 (c) and Au2 (f) in Zeonex matrices with 1 wt% dopant at 298 K and 77 K. | ||
The photoluminescence (PL) spectra of Au1 and Au2 were measured in various solvents and 1%-doped Zeonex films at 298 and 77 K (Table 1 and Fig. 4, Fig. S4–S6, ESI†). Both complexes show broad, unstructured emission profiles in all media with positive solvatochromism in solutions (up to 25 nm red-shift MCH to DCM, Fig. 4(b) and (e)). This suggests a CT character of the emissive excited state. All complexes possess fast fluorescence excited state lifetimes in the range of 8–12 ns in all media (Table 1). These increase slightly with increasing solvent polarity (Fig. 4(b) and (e)). Both Au1 and Au2 show near-unity PL quantum yields (PLQY) in MCH and toluene solutions. Such bright and fast fluorescence results in radiative rates exceeding 1 × 108 s−1 (Table 1) for Au1 and Au2. This experimental fact parallels the theoretical calculations which predict high oscillator strength coefficients (Tables S6, ESI†) for both complexes, vide supra.
On cooling to 77 K, Zeonex films of Au1 and Au2 show nearly identical photophysical characteristics to those measured at 295 K. However, in frozen MCH glass Au1 and Au2 show a blue-shift of the emission peak of up to 30 nm (Fig. S5, ESI†). We did not detect phosphorescence or delayed emission in all media at 77 K. All these facts confirm a fluorescence emission mechanism for the complexes Au1 and Au2. Analogous (NHC)Au(ethynylBTD)12 complexes show similar PL performance in solution: green or yellow near-unity fluorescence with up to 11 ns excited state lifetime, albeit with much greater variations for PLQY values in Zeonex films (31–83%).
Theoretical calculations were performed for complexes Au1 and Au2 to reveal the non-emissive (dark) triplet excited state. The first triplet excited state T1 possesses HONTO and LUNTO orbitals predominantly localised over the ethynylBTD ligand (Tables S6 and S7, ESI†). Therefore, we ascribe a locally excited (3LE) character to the T1-state. It has a large HOMO/LUMO overlap integral that stabilizes the T1 state energy by ca. 1 eV for Au1 and 2 eV for Au2 compared to the S1 state energy. We calculate the spin–orbit coupling matrix elements (SOCME, Tables S8 and S9, ESI†) between the triplet and singlet excited states for Au1 and Au2 to support a very weak coupling between S1 and T1 states. This suggests a weak intersystem crossing (ISC), i.e. no phosphorescence for Au1 and Au2, consistent with the previous reports on fluorescent gold alkynyl complexes.12
Fluorescence from Au1 and Au2 is in stark contrast with our previously reported phosphorescent (BIC)Au(CCPh) complex having only ca. 0.6 eV energy difference between S1 and T1 states. Unlike Au1 and Au2, this complex emits fast prompt fluorescence from a high lying CT state and a dominant phosphorescence from the 3LE excited state localised at the phenylacetylide ligand thanks to high contribution of the gold orbitals (ca. 16%) and fast ISC rate of 9 ps.13c Collectively, the small contribution of the gold orbitals to HOMO and LUMO (3–5%) and the large energy gap between the S1 and T1 states for Au1 and Au2 suggests an inefficient ISC, thus explaining the dark nature of the T1 state.
 | ||
| Fig. 5 Two-photon cross-section (red: experimental, black: smooth) and molar extinction of complexes Au1 (a) and Au2 (d) in toluene saturated with argon (blue). Overlay of the one-photon fluorescence (blue) and two-photon fluorescence (green) for complexes Au1 (b) and Au2 (e) in MCH solution under argon. Optimized molecular structure of complexes Au1 (c) and Au2 (f) with orientation and value for the transition dipole moment, Δμ10. | ||
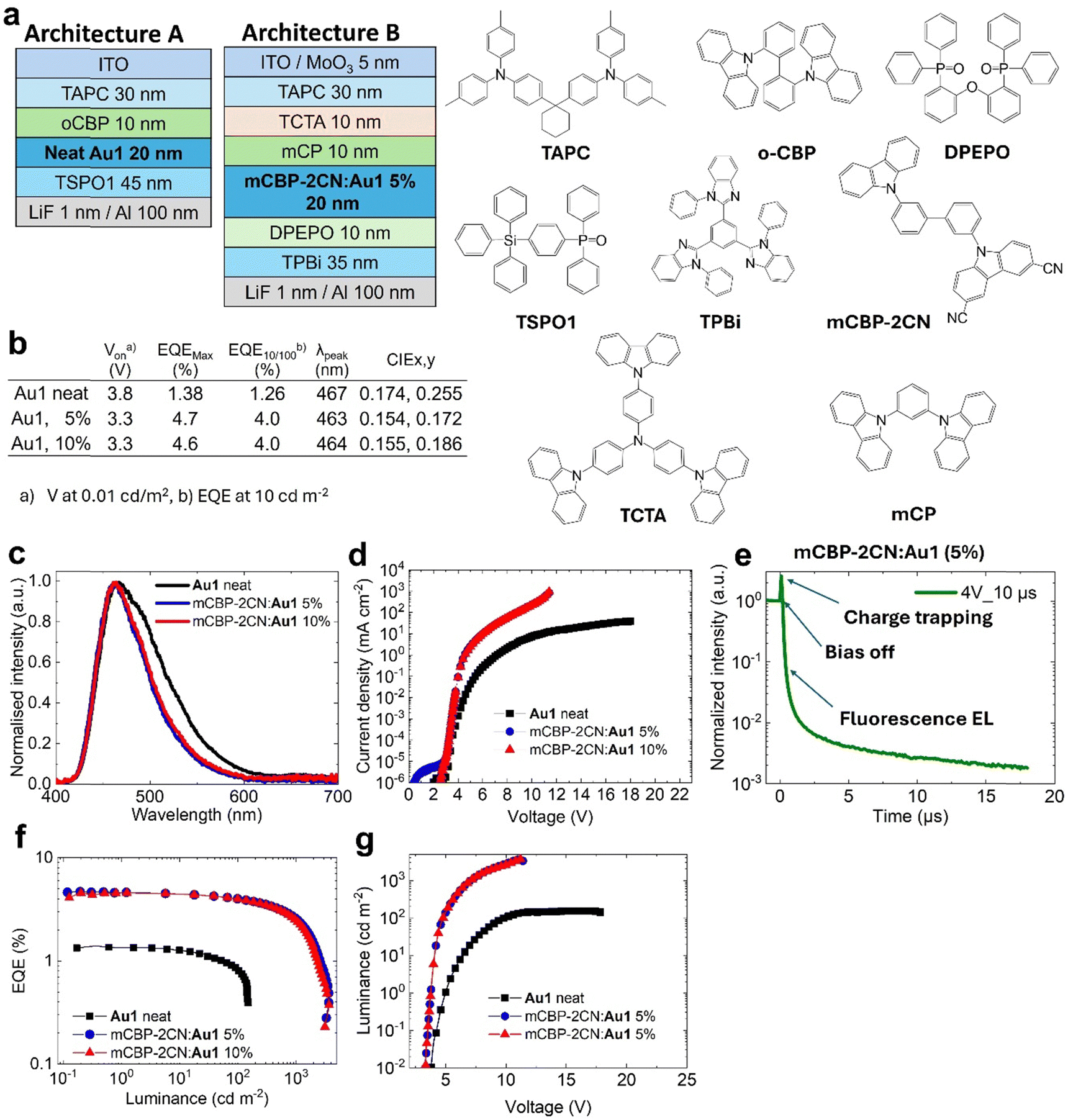 | ||
| Fig. 6 Vapor-deposited OLED device architectures. (a) OLED architectures A for neat Au1 and B for Au1 doped with mCBP-2CN at 5 and 10% by weight and chemical structures of the materials used; (b) performance data of vapor-deposited Au1 OLED; (c) normalised electroluminescence spectra from devices incorporating Au1 neat and in host–guest structures; (d) current density–voltage plot; (e) transient electroluminescence profiles for Au1 in mCBP-2CN at 5% by weight showing strong prompt fluorescence and weak phosphorescence profile after 1 μs delay; (f) EQE versus luminance plot; (g) luminance–voltage plot; and (h) excited state energy diagram for complex Au1 with radiative EL pathways. | ||
To enhance the performance of Au1 in OLED by increasing the probability of π⋯π stacking interactions, we attempted to prepare a host–guest OLED devices where Au1 was co-evaporated with the mCBP-2CN host at 5 and 10% concentration as shown in Fig. 6(a) (architecture B). mCBP-2CN is an ambipolar host with a high triplet energy of 2.77 eV and a well-stabilised HOMO energy level (−6.1 eV), which gives an acceptable energy alignment for complex Au1.17 Electroluminescence (EL) spectra for 5 and 10% Au1 OLED devices are shown in Fig. 6. The EL profile peaks at λmax = 467 nm perfectly matching with the fluorescence PL profile of complex Au1. The EL profile of the host–guest OLEDs is slightly narrower compared to the neat OLED, thus explaining the more blue CIE colour space coordinates for the former (0.15, 0.17, see Fig. 6(b)). Host–guest OLED devices show a low turn-on voltage down to VOn = 3.3 ± 0.3V (when luminance equals 0.1 cd m−2 at the applied bias). Fig. 6 shows the current density–voltage, luminance–voltage, EQE versus luminance plots for the best blue Au1 OLED in a host–guest environments. The OLEDs demonstrate a four-fold increase in peak EQEs of up to 4.7% compared to the host-free Au1 OLED. The Au1 devices experience a small EQE efficiency roll-off to 4% at a practical brightness of 100 cd m−2.
We measured the transient-EL for the mCBP-2CN:Au1(5%) OLED device (Fig. 6(e)) to reveal the role of the triplet state and the delayed-fluorescent component in OLEDs. The EL shows a biexponential decay with up to 10 ns fluorescence and microsecond delayed fluorescence in the OLED device held at a practical brightness of 100 cd m−2. However, the transient electroluminescence trace (Fig. 6(e)) shows a clear spike after “bias-off” which is characteristic of the delayed electroluminescence originating from the recombination of trapped charge carriers. Therefore, the similarity between PL and EL profiles with a dominant nanosecond range kinetics suggests the fluorescence electroluminescence in the OLED device.
Conclusions
We demonstrated a molecular design strategy towards thermally stable gold carbene–metal(I)–acetylide (CMAc) materials with unity fluorescence and high radiative rates exceeding 1 × 108 s−1. The bulky nature of the BIC-carbene ligand prevents aggregation in the solid state. Cyclic voltammetry data show that the redox processes for Au1 and Au2 are largely localised over the ethynyl-benzothiazole (ethynylBTD) ligand, resulting in greater stabilization of the HOMO energy levels down to −6.23 eV for Au2 due to greater π-conjugation. Absorption spectra and theoretical calculations confirm that the lowest energy vertical transition has over 90% HOMO → LUMO charge transfer (CT) character. Both Au1 and Au2 show a large HOMO/LUMO overlap integral (0.69) resulting in a significant stabilisation of the triplet excited state T1 energy. The low contribution of the gold atom (up to 5%) in the HOMO and LUMO coupled with a weak spin–orbit coupling between S1 and T1 states suggests a weak ISC process for Au1 and Au2, which explains the exclusive and bright fluorescence. We demonstrate an auration effect to enhance the two-photon absorption cross-sections (2PA σ2) for the quadrupolar chromophore Au2 (90 GM), i.e. a four-fold increase compared with the starting organic ligand L2 (23 GM) and a 100-fold increase compared to the dipolar chromophore Au1 (0.8 GM). The mutual orthogonality of the transition dipole vector between Au1 and Au2 is likely responsible for such a stark difference in 2PA σ2 values. Proof-of-concept fluorescent OLED devices have been fabricated with a maximum EQE of 4.7% approaching the theoretical maximum EQE of 5% thus indicating the excellent performance of the Au1 complex under electrical excitation. Overall, this work directs future CMAc material design to use ancillary carbene ligands with superior electronic properties (CAAC and BIC-carbenes) and π-conjugated alkynyl ligands to obtain quadrupolar type chromophores with large 2PA absorption cross-sections, bright 2PEF and energy-efficient CMAc OLEDs.Experimental section
General considerations
All reactions were performed under a N2 atmosphere. Solvents were dried as required. Reagents and catalysts were purchased from commercial vendors and used as received. (BIC)AuCl14b,c was prepared according to the literature procedure, see the ESI.† Synthetic procedures for L1 and L2 are found in the ESI.†1H and 13C{1H} NMR spectra were recorded using a Bruker AVIII HD 500 MHz NMR spectrometer. 1H NMR spectra (500.19 MHz) and 13C{1H} (125.79 MHz) were referenced to CD2Cl2 at δ 5.32 (13C, δ 53.84) and CDCl3 at δ 7.26 (13C, δ 77.16). IR spectra were recorded using a Bruker ALPHA FT-IR spectrometer equipped with a platinum ATR attachment. All electrochemical experiments were performed using an Autolab PGSTAT 302N computer-controlled potentiostat. Cyclic voltammetry (CV) was performed using a three-electrode configuration consisting of a glassy carbon macrodisk working electrode (GCE) (diameter of 3 mm; BASi, Indiana, USA) combined with a Pt wire counter electrode (99.99%; GoodFellow, Cambridge, UK) and an Ag wire pseudoreference electrode (99.99%; GoodFellow, Cambridge, UK). The GCE was polished between experiments using alumina slurry (0.3 μm), rinsed in distilled water and subjected to brief sonication to remove any adhering alumina microparticles. The metal electrodes were then dried in an oven at 100 °C to remove residual traces of water, the GCE was left to air dry and residual traces of water were removed under vacuum. The Ag wire pseudoreference electrodes were calibrated to the ferrocene/ferrocenium couple in THF at the end of each run to allow for any drift in potential, following IUPAC recommendations. All electrochemical measurements were performed at ambient temperatures under an inert N2 atmosphere in THF containing the complex under study (1.4 mM) and the supporting electrolyte [n-Bu4N][PF6] (0.13 M). Data were recorded using Autolab NOVA software (v. 1.11). Thermogravimetric analysis and elemental analyses were performed by the Microanalysis Laboratory at the University of Manchester. Mass spectrometry data were obtained by the Mass Spectrometry Laboratory at the University of Manchester. Thermogravimetric analysis was performed with a TA Instruments SDT650 simultaneous thermal analyser under a stream of nitrogen.:1 hexane:EtOAc) to give the pure product as a pale green crystalline solid in 64% yield (279 mg, 402 μmol). 1H NMR (500 MHz, CD2Cl2) δ 7.74 (d, J = 8.7 Hz, 1H, CH BTD), 7.52–7.46 (m, 2H, p-CH Dipp BIC and CH BTD), 7.43 (dd, J = 8.6, 7.1 Hz, 1H, CH BTD), 7.35–7.31 (m, 2H, m-CH Dipp BIC), 3.24–3.15 (m, 1H, CH iPr BIC), 3.07–2.98 (m, 1H, CH iPr Dipp BIC), 2.65–2.56 (m, 1H, CH iPr Dipp BIC), 2.31–2.22 (m, 1H, C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) (CH3) BIC), 2.13 (dd, J = 13.6, 10.5 Hz, 1H, CH BiCAAC), 1.84–1.70 (m, 3H, CH BIC), 1.64–1.58 (m, 1H, CH BIC), 1.56–1.52 (m, 1H, CH BIC overlap with water), 1.50–1.46 (m, 6H, 2xCH3 iPr Dipp BIC), 1.41 (d, J = 6.7 Hz, 3H, iPr Dipp BIC), 1.35 (d, J = 6.9 Hz, 3H, iPr Dipp BIC), 1.29 (d, J = 6.9 Hz, 3H, CH3 iPr BIC), 1.08 (t, J = 7.2 Hz, 6H, CH(C3) BIC and iPrCH3 BIC), 1.02 (s, 3H, CH3 BIC). 13C NMR (126 MHz, CD2Cl2) δ 261.09 (C: BIC), 156.12 (
(CH3) BIC), 2.13 (dd, J = 13.6, 10.5 Hz, 1H, CH BiCAAC), 1.84–1.70 (m, 3H, CH BIC), 1.64–1.58 (m, 1H, CH BIC), 1.56–1.52 (m, 1H, CH BIC overlap with water), 1.50–1.46 (m, 6H, 2xCH3 iPr Dipp BIC), 1.41 (d, J = 6.7 Hz, 3H, iPr Dipp BIC), 1.35 (d, J = 6.9 Hz, 3H, iPr Dipp BIC), 1.29 (d, J = 6.9 Hz, 3H, CH3 iPr BIC), 1.08 (t, J = 7.2 Hz, 6H, CH(C3) BIC and iPrCH3 BIC), 1.02 (s, 3H, CH3 BIC). 13C NMR (126 MHz, CD2Cl2) δ 261.09 (C: BIC), 156.12 (![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) = N BTD), 155.14 ( = N BTD), 145.08 (o-C Dipp BIC), 144.43 (o-C Dipp BIC), 141.21 (i-C Dipp BIC), 139.17 (BTD), 132.15 (BTD), 129.88 (BTD), 129.85 (p-C Dipp BIC), 125.27 (m-C Dipp BIC), 120.39 (BTD), 119.38 (m-C Dipp BIC), 115.40 (C ≡ C), 101.74 (CC), 64.44 (qCH3 BIC), 55.90 (qiPr BIC), 44.60 (CH(CH3)H2 BIC), 34.61 (H(CH3) BIC), 32.89 (CH2CH2 BIC), 31.72 (CH iPr BIC), 29.49 (CH iPr Dipp), 28.89 (CH iPr Dipp), 25.61 (CH3 iPr carbene), 25.39 (CH3 iPr Dipp), 24.05 (CqH3 carbene), 23.73 (CH3 iPr Dipp), 23.40 (CH3 iPr Dipp), 21.56 (CH BIC), 20.32 (CH2CH2 BIC), 19.69 (CH(H3) BIC), 16.23 (CH3 iPr BIC). IR (ATR, cm−1): 2105 (CC). Anal. calcd for C32H40AuN3S: C, 55.25; H, 5.80; N, 6.04; S, 4.61; found = C, 53.83; H, 6.46; N, 4.81; S, 3.02. HRMS C32H40AuN3S theoretical [M − H]+ = 696.2681, HMRS APCI(ASAP) = 696. 2680. TGA C32H40AuN3S Td = 197.2 °C.
:1 hexane:EtOAc) to give the pure product as a yellow powder in 55% yield (224 mg, 179 μmol). 1H NMR (500 MHz, CD2Cl2) δ 7.51–7.42 (m, 2H, BTD), 7.35–7.24 (m, 6H, Dipp BIC), 3.22–3.11 (m, 2H, CH iPr BIC), 3.04–2.93 (m, 2H, CH iPr Dipp BIC), 2.62–2.52 (m, 2H, CH iPr Dipp BIC), 2.30–2.19 (m, 2H, C(CH3) BIC), 2.15–2.05 (m, 2H, CH BIC), 1.84–1.67 (m, 8H, CH BIC), 1.62–1.50 (2H, CH BIC overlap with water), 1.49–1.41 (m, 12H, iPr Dipp BIC), 1.40–1.22 (m, 18H, 12H iPr Dipp BIC and 6H CH3 iPr BIC), 1.09–0.96 (m, 18H, 6H CH(C3) BIC, 6H iPrCH3 BIC and 6H CH3 BIC). 13C NMR (126 MHz, CD2Cl2) δ 261.20 (C: BIC), 155.84 (
= N BTD), 155.14 ( = N BTD), 145.08 (o-C Dipp BIC), 144.43 (o-C Dipp BIC), 141.21 (i-C Dipp BIC), 139.17 (BTD), 132.15 (BTD), 129.88 (BTD), 129.85 (p-C Dipp BIC), 125.27 (m-C Dipp BIC), 120.39 (BTD), 119.38 (m-C Dipp BIC), 115.40 (C ≡ C), 101.74 (CC), 64.44 (qCH3 BIC), 55.90 (qiPr BIC), 44.60 (CH(CH3)H2 BIC), 34.61 (H(CH3) BIC), 32.89 (CH2CH2 BIC), 31.72 (CH iPr BIC), 29.49 (CH iPr Dipp), 28.89 (CH iPr Dipp), 25.61 (CH3 iPr carbene), 25.39 (CH3 iPr Dipp), 24.05 (CqH3 carbene), 23.73 (CH3 iPr Dipp), 23.40 (CH3 iPr Dipp), 21.56 (CH BIC), 20.32 (CH2CH2 BIC), 19.69 (CH(H3) BIC), 16.23 (CH3 iPr BIC). IR (ATR, cm−1): 2105 (CC). Anal. calcd for C32H40AuN3S: C, 55.25; H, 5.80; N, 6.04; S, 4.61; found = C, 53.83; H, 6.46; N, 4.81; S, 3.02. HRMS C32H40AuN3S theoretical [M − H]+ = 696.2681, HMRS APCI(ASAP) = 696. 2680. TGA C32H40AuN3S Td = 197.2 °C.
:1 hexane:EtOAc) to give the pure product as a yellow powder in 55% yield (224 mg, 179 μmol). 1H NMR (500 MHz, CD2Cl2) δ 7.51–7.42 (m, 2H, BTD), 7.35–7.24 (m, 6H, Dipp BIC), 3.22–3.11 (m, 2H, CH iPr BIC), 3.04–2.93 (m, 2H, CH iPr Dipp BIC), 2.62–2.52 (m, 2H, CH iPr Dipp BIC), 2.30–2.19 (m, 2H, C(CH3) BIC), 2.15–2.05 (m, 2H, CH BIC), 1.84–1.67 (m, 8H, CH BIC), 1.62–1.50 (2H, CH BIC overlap with water), 1.49–1.41 (m, 12H, iPr Dipp BIC), 1.40–1.22 (m, 18H, 12H iPr Dipp BIC and 6H CH3 iPr BIC), 1.09–0.96 (m, 18H, 6H CH(C3) BIC, 6H iPrCH3 BIC and 6H CH3 BIC). 13C NMR (126 MHz, CD2Cl2) δ 261.20 (C: BIC), 155.84 (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N BTD), 145.04 (o-C Dipp BIC), 144.41 (o-C Dipp BIC), 141.19 (i-C Dipp BIC), 139.55 (BTD), 132.44 (BTD), 129.83 (p-C Dipp BIC), 125.26 (m-C Dipp BIC), 125.16 (m-C Dipp BIC), 117.90 (CC), 102.38 (CC), 64.35 (qCH3 BIC), 55.86 (qiPr BIC), 44.60 (CH(CH3)H2 BIC), 34.59 (H(CH3) BIC), 32.89 (CH2CH2 BIC), 31.69 (CH iPr BIC), 29.47 (CH iPr Dipp), 28.87 (CH iPr Dipp), 25.98 (CH3 iPr Carbene), 25.59 (CH3 iPr Dipp), 24.05 (CqH3 Carbene), 23.72 (CH3 iPr Dipp), 23.40 (CH3 iPr Dipp), 21.55 (CH BIC), 20.30 (CH2CH2 BIC), 19.68 (CH(H3) BIC), 19.64, 16.22 (CH3 iPr BIC). IR (ATR, cm−1): 2107 (CC). Anal. calcd for C58H76Au2N4S: C, 55.50; H, 6.10; N, 4.46; S, 2.55; found = C, 54.44; H, 6.13; N, 4.23; S, 2.34. HRMS C58H76Au2N4S theoretical [M − H]+ = 1255.5194; HRMS APCI(ASAP) = 1255.5191. TGA C58H76Au2N4S: Td = 321.8 °C.
000 Hz frequency and 10 μs pulse width) to collect transient electroluminescence.
N BTD), 145.04 (o-C Dipp BIC), 144.41 (o-C Dipp BIC), 141.19 (i-C Dipp BIC), 139.55 (BTD), 132.44 (BTD), 129.83 (p-C Dipp BIC), 125.26 (m-C Dipp BIC), 125.16 (m-C Dipp BIC), 117.90 (CC), 102.38 (CC), 64.35 (qCH3 BIC), 55.86 (qiPr BIC), 44.60 (CH(CH3)H2 BIC), 34.59 (H(CH3) BIC), 32.89 (CH2CH2 BIC), 31.69 (CH iPr BIC), 29.47 (CH iPr Dipp), 28.87 (CH iPr Dipp), 25.98 (CH3 iPr Carbene), 25.59 (CH3 iPr Dipp), 24.05 (CqH3 Carbene), 23.72 (CH3 iPr Dipp), 23.40 (CH3 iPr Dipp), 21.55 (CH BIC), 20.30 (CH2CH2 BIC), 19.68 (CH(H3) BIC), 19.64, 16.22 (CH3 iPr BIC). IR (ATR, cm−1): 2107 (CC). Anal. calcd for C58H76Au2N4S: C, 55.50; H, 6.10; N, 4.46; S, 2.55; found = C, 54.44; H, 6.13; N, 4.23; S, 2.34. HRMS C58H76Au2N4S theoretical [M − H]+ = 1255.5194; HRMS APCI(ASAP) = 1255.5191. TGA C58H76Au2N4S: Td = 321.8 °C.
000 Hz frequency and 10 μs pulse width) to collect transient electroluminescence.
Author contributions
H. H. C. developed and characterized the OLED devices and performed the transient electroluminescence study. A. S. R., A. H. and A. C. B. performed the molecular design, synthesis, purification, and characterization. A. S. R. performed X-ray crystallography. A. S.R., A. H. and A. C. B. performed steady-state photoluminescence, UV-vis, and electrochemistry studies. C. T. S. assisted with the acquisition of the two-photon photoluminescence. J. D. and M. B. D. carried out photophysical measurements, two-photon absorption and photoluminescence studies. M. B. D., N. G. and A. S. R. planned the project and designed the experiments. N. L. P. and M. L. performed theoretical calculations and analyzed theoretical data. A. S. R. wrote the manuscript. All authors contributed to the discussion of the results and analysis of the data, and reviewed and commented on the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. S. R. acknowledges the support from the Royal Society (grant no. URF\R1\180288, RGF\EA\181008, URF\R\231014), EPSRC (grant code EP/K039547/1). M. L. acknowledges the Academy of Finland Flagship Programme, Photonics Research and Innovation (PREIN), decision 320166, the Finnish Grid and Cloud Infrastructure resources (urn:nbn:fi:research-infras-2016072533). N. L. P. acknowledges the Doctoral Programme in Science, Forestry and Technology (Lumeto, University of Eastern Finland). The European Union's Horizon 2020 research and innovation programme grant agreement no. 101020167 (H.-H. C.). The authors thank Dr Louise Natrajan, EPSRC and the University of Manchester for access to the Centre for Radiochemistry Research National Nuclear User's Facility (NNUF, EP/T011289/1) to use FLS-920 fluorometer. M. B. D. and J. D. acknowledge the support from CNRS and University of Bordeaux.Notes and references
- (a) D. A. Parthenopoulos and P. M. Rentzepis, Science, 1989, 249, 843 CrossRef PubMed; (b) J. H. Strickler and W. W. Webb, Opt. Lett., 1991, 16, 1780 CrossRef CAS PubMed; (c) B. H. Cumpston, S. P. Ananthavel, S. Barlow, D. L. Dyer, J. E. Ehrlich, L. L. Erskine, A. A. Heikal, S. M. Kuebler, I. Y. S. Lee, D. McCord-Maughon, J. Qin, H. Röckel, M. Rumi, X. L. Wu, S. R. Marder and J. W. Perry, Nature, 1999, 398(6722), 51 CrossRef CAS; (d) Y. Gao, Y. Qu, T. Jiang, H. Zhang, N. He, B. Li, J. Wu and J. Hua, J. Mater. Chem. C, 2014, 2, 6353 RSC.
- (a) M. Charlot, N. Izard, O. Mongin, D. Riehl and M. Blanchard-Desce, Chem. Phys. Lett., 2006, 417(4–6), 297 CrossRef CAS; (b) R. Westlund, E. Malmström, C. Lopes, J. Öhgren, T. Rodgers, Y. Saito, S. Kawata, E. Glimsdal and M. Lindgren, Adv. Funct. Mater., 2008, 18, 1939 CrossRef CAS; (c) A. Purc, K. Sobczyk, Y. Sakagami, A. Ando, K. Kamada and D. T. Gryko, J. Mater. Chem. C, 2015, 3, 742 RSC; (d) S. Pascal, S. David, C. Andraud and O. Maury, Chem. Soc. Rev., 2021, 50, 6613 RSC.
- (a) W. Denk, J. J. Strickler and W. W. Webb, Science, 1990, 248, 73 CrossRef CAS PubMed; (b) A. Vaziri, J. Tang, H. Shroff and C. V. Shank, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 20221 CrossRef CAS PubMed; (c) Y. Chen, R. Guan, C. Zhang, J. Huang, L. Ji and H. Chao, Coord. Chem. Rev., 2016, 310, 16 CrossRef CAS; (d) C. Jin, F. Liang, J. Wang, L. Wang, J. Liu, X. Liao, T. W. Rees, B. Yuan, H. Wang, Y. Shen, Z. Pei, L. Ji and H. Chao, Angew. Chem., Int. Ed., 2020, 59, 15987 CrossRef CAS PubMed; (e) G. Prévot, T. Bsaibess, J. Daniel, C. Genevois, G. Clermont, I. Sasaki, S. Marais, F. Couillaud, S. Crauste-Manciet and M. Blanchard-Desce, Nanoscale Adv., 2020, 2(4), 1590 RSC; (f) M. Rosendale, J. Flores, C. Paviolo, P. Pagano, J. Daniel, J. Ferreira, J. B. Verlhac, L. Groc, L. Cognet and M. Blanchard-Desce, Adv. Mater., 2021, 33(22), 2006644 CrossRef CAS PubMed.
- (a) J. D. Bhawalkar, N. D. Kumar, C. F. Zhao and P. N. Prasad, J. Clin. Laser Med. Surg., 1997, 15, 201 CrossRef CAS PubMed; (b) M. Drobizhev, N. S. Makarov, S. E. Tillo, T. E. Hughes and A. Rebane, Nat. Methods, 2011, 8(5), 393 CrossRef CAS PubMed; (c) J. Schmitt, V. Heitz, A. Sour, F. Bolze, H. Ftouni, J.-F. Nicoud, L. Flamigni and B. Ventura, Angew. Chem., Int. Ed., 2015, 54, 169 CrossRef CAS PubMed; (d) Y. Wang, X. Shi, H. Fang, Z. Han, H. Yuan, Z. Zhu, L. Dong, Z. Guo and X. Wang, J. Med. Chem., 2022, 65(11), 7786 CrossRef CAS PubMed; (e) I. Sasaki, F. Brégier, G. Chemin, J. Daniel, J. Couvez, R. Chkair, M. Vaultier, V. Sol and M. Blanchard-Desce, Nanomaterials, 2024, 14(2), 216 CrossRef CAS PubMed.
- M. Göpper-Maier, Ann. Phys., 1931, 9(3), 273 CrossRef.
- (a) M. Rumi, S. Barlow, J. Wang, J. W. Perry and S. R. Marder, Adv. Polym. Sci., 2008, 213(1), 1 CAS; (b) R. L. Roberts, T. Schwich, T. C. Corkery, M. P. Cifuentes, K. A. Green, J. D. Farmer, P. J. Low, T. B. Marder, M. Samoc and M. G. Humphrey, Adv. Mater., 2009, 21, 2318 CrossRef CAS.
- (a) J. Olesiak-Banska, M. Gordel, R. Kolkowski, K. Matczyszyn and M. Samoc, J. Phys. Chem. C, 2012, 116, 13731 CrossRef CAS; (b) J. Olesiak-Banska, M. Waszkielewicz, P. Obstarczyk and M. Samoc, Chem. Soc. Rev., 2019, 48, 4087 RSC.
- S. R. Marder, C. B. Gorman, F. Meyers, J. Perry, G. Bourhill, J.-L. Bredas and B. M. Pierce, Science, 1994, 265, 632 CrossRef CAS PubMed.
- (a) A. Rebane, M. Drobizhev, N. S. Makarov, E. Beuerman, J. E. Haley, D. M. Krein, A. R. Burke, J. L. Flikkema and T. M. Cooper, J. Phys. Chem. A, 2011, 115, 4255 CrossRef CAS PubMed; (b) G. S. He, L.-S. Tan, Q. Zheng and P. N. Prasad, Chem. Rev., 2008, 108, 1245 CrossRef CAS PubMed.
- L. Zhang and M. G. Humphrey, Coord. Chem. Rev., 2022, 473, 214820 CrossRef CAS.
- (a) S. K. Hurst, M. P. Cifuentes, A. M. McDonagh, M. G. Humphrey, M. Samoc, B. Luther-Davies, I. Asselberghs and A. Persoons, J. Organomet. Chem., 2002, 642, 259 CrossRef CAS; (b) S. Goswami, R. W. Winkel and K. S. Schanze, Inorg. Chem., 2015, 54, 10007 CrossRef CAS PubMed; (c) T. R. Ensley, R. M. O’Donnell, J. J. Mihaly, J. E. Haley, T. A. Grusenmeyer and T. G. Gray, Appl. Opt., 2021, 60, G199 CrossRef PubMed.
- (a) I. R. Whittall, M. G. Humphrey, M. Samoc and B. Luther-Davies, Angew. Chem., Int. Ed. Engl., 1997, 36(4), 370 CrossRef CAS; (b) A. Moller, P. Bleckenwegner, U. Monkowius and F. Mohr, J. Organomet. Chem., 2016, 813, 1 CrossRef; (c) K. T. Chan, G. S. M. Tong, W.-P. To, C. Yang, L. Du, D. L. Phillips and C.-M. Che, Chem. Sci., 2017, 8, 2352 RSC; (d) A. Pinto, M. Echeverri, B. Gomez-Lor and L. Rodríguez, Dyes Pigm., 2022, 202, 110308 CrossRef CAS; (e) A. Pinto, M. Echeverri, B. Gomez-Lor and L. Rodríguez, Dalton Trans., 2022, 51, 8340 RSC.
- (a) A. S. Romanov and M. Bochmann, Organometallics, 2015, 34, 2439 CrossRef CAS PubMed; (b) A. S. Romanov, C. R. Becker, C. E. James, D. Di, D. Credgington, M. Linnolahti and M. Bochmann, Chem. – Eur. J., 2017, 23(19), 4625 CrossRef CAS PubMed; (c) A. C. Brannan, H.-H. Cho, A.-P. M. Reponen, M. Linnolahti, M. Bochmann, N. C. Greenham and A. S. Romanov., Adv. Mater., 2024, 36, 2306249 CrossRef CAS PubMed.
- (a) V. Lavallo, Y. Canac, A. DeHope, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2005, 44, 7236 CrossRef CAS PubMed; (b) E. Tomás-Mendivil, M. M. Hansmann, C. M. Weinstein, R. Jazzar, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2017, 139, 7753 CrossRef PubMed; (c) F. Chotard, V. Sivchik, M. Linnolahti, M. Bochman and A. S. Romanov, Chem. Mater., 2020, 32(14), 6114 CrossRef CAS.
- (a) G. Frapper and M. Kertesz, Inorg. Chem., 1993, 32, 732 CrossRef CAS; (b) M. A. Peay, J. E. Heckler, N. Deligonul and T. G. Gray, Organometallics, 2011, 30, 5071 CrossRef CAS.
- (a) D. Y. Kondakov, Philos. Trans. R. Soc., A, 2015, 373, 20140321 CrossRef PubMed; (b) D. Di, L. Yang, J. M. Richter, L. Meraldi, R. M. Altamimi, A. Y. Alyamani, D. Credgington, K. P. Musselman, J. L. MacManus-Driscoll and R. H. Friend, Adv. Mater., 2017, 29, 1605987 CrossRef PubMed; (c) J.-H. Lee, C.-H. Chen, P.-H. Lee, H.-Y. Lin, M.-K. Leung, T.-L. Chiu and C.-F. Lin, J. Mater. Chem. C, 2019, 7, 5874 RSC; (d) P. Han, C. Lin, E. Xia, J. Cheng, Q. Xia, D. Yang, A. Qin, D. Ma and B. Z. Tang, Angew. Chem., Int. Ed., 2023, 62, e2023103 Search PubMed.
- (a) C. S. Oh, J. Y. Lee, C. H. Noh and S. H. Kim, J. Mater. Chem. C, 2016, 4, 3792 RSC; (b) S. K. Shin, S. H. Hana and J. Y. Lee, J. Mater. Chem. C, 2018, 6, 10308 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2351761. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4tc02845g |
| This journal is © The Royal Society of Chemistry 2024 |