
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Molecular insights into solid-state photochromism in bulk and confined N-salicylidenes†
Kieran
Griffiths
 ,
Harry
Brough
,
Ryan J.
Bragg
,
Nathan R.
Halcovitch
and
John M.
Griffin
*
,
Harry
Brough
,
Ryan J.
Bragg
,
Nathan R.
Halcovitch
and
John M.
Griffin
*
Department of Chemistry, Lancaster University, Lancaster LA1 4YB, UK. E-mail: j.griffin@lancaster.ac.uk
First published on 23rd October 2024
Abstract
N-Salicylidenes are well known to exhibit solid-state photochromism, but the precise structure–property relationships are not fully understood and it is difficult to control or impart specific photochromic properties through molecular design alone. In this study, we use solid-state NMR and DFT calculations to investigate the link between the solid-state structure and the photochromic properties. We show that the photochromic properties are highly dependent on the molecular geometry within the crystal structure, which itself is an indicator of the amount of free space available for light-induced tautomerisation. Specifically, 13C solid-state NMR experiments and DFT calculations reveal that the imine chemical shift of the ground-state enol isomer is highly dependent on the molecular geometry, and this helps to rationalise the known empirical torsion angle dependence of the photochromic properties. Upon inclusion within a metal–organic framework, the N-salicylidene molecules are found to adopt geometries close to their ground-state energy minima and this is coupled with the emergence of photochromism for molecules that are not photochromic in the bulk crystalline state. This work highlights that controlling the amount of available steric freedom is key to unlocking the photoactive conformation in the solid state.
Introduction
Molecular solar thermal (MOST) systems are gaining increasing interest as a potential technology for solar energy capture and storage.1–3 MOST systems are designed around photoresponsive moieties that undergo isomerisation to a higher energy metastable structure in response to a light stimulus. The light-induced structure change is typically accompanied by a change in absorbance, meaning that MOST systems are generally also photochromic. Photochromism is therefore a useful indicator of potential MOST materials, and can also find use in applications such as sensing or data storage.4,5 Well known examples of photochromic molecules of interest for MOST applications include azobenzenes6,7 and norbornadienes.8,9 While photoswitching in solution is facile for these classes of compounds, examples of solid-state photochromism are more limited. This is because there is limited space in a typical crystalline lattice to allow for the conformational rearrangement needed to undergo light-induced structure changes. Solid-state photochromism is of particular interest for high energy density MOST devices and other applications in e.g., coatings.10 Systems which do exhibit solid-state photochromism are often those where the light-induced isomerisation results in minimal change to the overall molecular geometry, for example diarylethenes and, in limited cases, spiropyrans.11–14One class of compounds which has been widely observed to exhibit solid-state photochromism is N-salicylidenes (also called anils or Schiff bases).15 The light-induced colour changes in N-salicylidenes have been proposed to occur due to a hydrogen atom transfer from the enol group to the imine-nitrogen to form a cis-keto tautomer. This is followed by subsequent rotation of the amine-bound arene to form a trans-keto tautomer (Fig. 1). The energy difference between the ground-state enol and the metastable trans-keto tautomers can be appreciable, making N-salicylidenes of potential interest for MOST applications. There are numerous examples of N-salicylidenes which display solid-state photochromism, but there are also many structurally similar examples which are not photochromic. The propensity for N-salicylidenes to display solid-state photochromism has been linked to the amount of free space or packing density in the crystal structure. In some cases, photochromism has been shown to be more likely when bulky substituents are used.16 However, other groups have argued that the torsion angle between the arene rings is of importance to allow the light-induced tautomerisation.17,18 Despite this, photoswitching within molecular crystals remains difficult to predict and control, thus complicating the molecular design of these photoswitches for solid-state MOST energy storage and other applications.
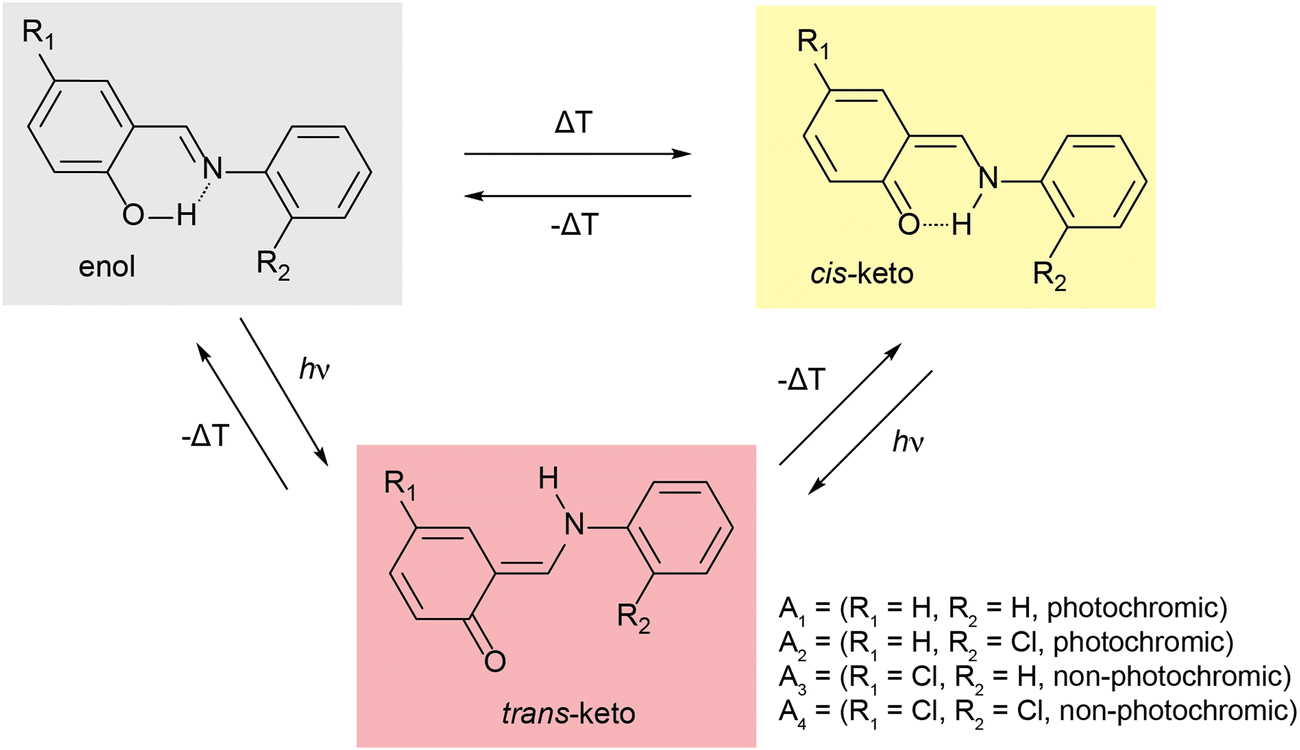 | ||
| Fig. 1 Scheme showing temperature- and light-induced structural changes in N-salicylidenes. A1–A4 denote molecules used in this study. | ||
One strategy to facilitate light-induced structural changes in the solid state is to incorporate photoswitches within the void spaces of porous materials. This has been demonstrated in several studies on photoswitches confined within the pores of metal–organic frameworks (MOFs),12,19,20 as well as with N-salicylidenes confined in the void spaces of crystalline clathrates and single guest molecular capsules.21,22 While light-induced structure changes may be restricted or prevented in the bulk crystalline structure, occlusion in a well-defined pore space can allow solid-state photoswitching of the guest up to quantitative levels.23–25 In this work, we study the solid-state photochromic properties of four structurally related N-salicylidenes (Fig. 1), and their incorporation within the well-known MOF MIL-53 [Al(OH)(C8O4H4), 1]. The molecules studied are chosen to offer a systematic variation in structure and photochromic properties. N-salicylideneaniline (A1) displays solid-state photochromism and was one of the first aniline derivatives to be studied for its photochromic properties at ambient temperature.26N-salicylidene-2-chloroaniline (A2) also displays solid-state photochromism, but N-(5-chlorosalidylidene)aniline (A3) is not photochromic. These confirm the known empirical trend of low-density crystal packing giving photochromic properties, and densely packed structures being non-photochromic.27 Finally, N-(5-chlorosalicylidene)-2-chloroaniline is also non-photochromic at ambient temperature. Here, we show that solid-state NMR is a sensitive probe of the torsion angle in N-salicylidenes, and comparison between DFT calculations on periodic crystal structures and isolated molecules helps to rationalise the known empirical torsion angle dependence of the photochromic properties. Using this information, we are able to identify clear changes in molecular geometry upon incorporation within the MOF architecture, and these changes are found to lead to the emergence of photochromism for the two molecules that are not photochromic in the bulk phase. In this way, MOF incorporation offers a transferrable strategy for imparting solid-state photochromism into N-salicylidenes that are photochemically inactive in the native state.
Experimental details
Synthesis
MIL-53 (1) was synthesised following reported synthetic procedures.28 The initial as-synthesised product (1as) was refluxed in DMF for 3 days to remove excess benzene dicarboxylate (BDC) linkers from the pores. The resulting product, hydrated narrow-pore MIL-53 (1np), was characterised by X-ray power diffraction (XRPD) and 13C CPMAS NMR, giving results consistent with previously reported data.29N-salicylidenes A1–A4 were synthesised following reported procedures and characterised by 1H solution-state NMR. XRPD was used to identify the phase of the micro-crystalline powders, and these were assigned to reported structures.30–33
MOF-N-salicylidene host–guest complexes (1⊃An) were prepared by a gas phase loading procedure. 1np (50 mg) was heated to 140 °C under vacuum for 12 hours to remove guest water molecules. The relevant A1–A4 guest (50 mg) was then added and heated at 100 °C under vacuum for a further 6 hours. After this time all excess guest molecules had been removed which was confirmed by DSC and TGA (Fig. S1 and S2, ESI†). The MOF-N-salicylidene complexes were collected as pale-yellow powders.
Characterisation
13C cross-polarisation (CP) magic-angle spinning (MAS) NMR experiments were performed on Bruker Avance III HD spectrometer operating at magnetic field strength of 16.4 T, corresponding to 1H and 13C Larmor frequencies of 700 and 176 MHz, respectively. Spectra are referenced relative to tetramethylsilane using the CH3 (13C = 20.5 ppm) resonance of L-alanine as a secondary reference. 13C NMR spectra were recorded at a MAS rate of 16.0 kHz using CP to transfer magnetization from 1H with a contact time of 3 ms. The CP pulse was ramped linearly from 70–100% power. 1H heteronuclear decoupling using two-pulse phase modulation (TPPM)34 with a pulse length of 4.8 μs and a radiofrequency field strength of 100 kHz was applied during acquisition. Spectra are the sum of 1024 transients separated by a recycle interval of 3 s. The sample temperature in variable-temperature experiments was calibrated using Pb(NO3)2.35Solution-state NMR experiments were performed on a Bruker Avance III 400 NMR spectrometer with a 5 mm 1H-X broadband observe probe was used to collected 1H NMR data.
XRPD patterns were measured with a Rigaku SmartLab X-ray diffractometer with a 9 kW rotating anode Cu-source equipped with a high-resolution vertical θ/θ 4-circle goniometer and D/teX-ULTRA 250 high-speed position-sensitive detector system in reflectance mode. The system was configured with parallel-beam optics and a Ge(220) 2 bounce monochromator on the incident side. Powdered solid samples were prepared on glass slides. The measurements were performed as θ/2θ scans with a step size of 0.01 degrees.
Samples were irradiated with an OmniCure LX5 365 nm LED head with a power of 425 mW and a 3 mm focusing lens. 50 mg of finely ground 1⊃An was spread homogenously over a microscope slide. The powder was spread into a circle with a 1 cm radius which was approximately 0.5 mm thick so that irradiation was approximately uniform. The slide was placed under the light at a distance of 5 cm. The beam was set to 100% intensity and exposed for a fixed duration. The sample was periodically agitated to allow all particulates to be exposed to the beam.
UV-vis reflectance spectra were recorded with a Cary 5000 UV-Vis-NIR spectrometer between 200–800 nm with a mounted solid-state powder sample cell. The samples were spread homogenously within the solid-state powder sample cell and compressed by manual pressure with no additional matrix. For UV irradiated samples, the cell containing the powder was treated with 365 nm light prior to spectra being recorded.
First-principles calculations of NMR parameters were carried out under periodic boundary conditions using the CASTEP code employing the gauge-including projector augmented wave (GIPAW) algorithm.36 Prior to calculation of the NMR parameters, structures were fully geometry optimised, with all atomic positions allowed to vary. Calculations were performed using a planewave energy cut off of 60 Ry while reciprocal space was sampled via a Monkhorst–Pack grid with a k-point spacing of 0.05 A−1. A reference shielding of 168.5 ppm was used to convert calculated 13C chemical shieldings to chemical shifts. Optimisations were performed with the rSCAN functional, as well as with PBE with the D3 semi-empirical dispersion correction scheme. Calculations were performed both fixing the unit cell parameters at their experimental values, and allowing them to freely optimise. In all cases, all atoms were allowed to relax. Single molecule optimisations and energy calculations were performed within the Gaussian code using the B3LYP functional and 6-31G basis set. Torsion angles were fixed to specified values while all other atoms positions were allowed to vary. Single molecule NMR calculations were performed using the B3LYP functional and cc-pVDZ basis set.
Thermogravimetric analysis (TGA) traces were recorded on a TA instruments Q500 thermogravimetric analyser. Data was collected between 25–800 °C at a ramp rate of 10 °C min−1 held within platinum sample pans.
Results and discussion
Structural study of bulk crystalline N-salicylidenes
The N-salicylidenes A1–A4 have been reported in several different polymorphic phases and the reported photochromic or thermochromic properties are dependent on the crystal packing (Table 1 and Table S1, ESI†). Prior to analysis, XRPD was used to characterise the polymorphs of An compounds used in this study. A1 and A2 were found to crystallise in the orthorhombic space groups Fd2d and P212121, respectively, and both phases exhibit photochromism in response to 365 nm light. A3 and A4 were found to crystallise in the orthorhombic space groups Pbca and Pca21, respectively, and do not exhibit photochromism.| A1 | A2 | A3 | A4 | |
|---|---|---|---|---|
| Crystal system | Orthorhombic | Orthorhombic | Orthorhombic | Orthorhombic |
| Space group | Fd2d | P212121 | Pca21 | Pbca |
| Z | 16 | 4 | 4 | 8 |
| Experimental torsion angle/° | 47.1 | 47.0 | 3.5 | 5.7 |
| DFT-optimised torsion angle (change from experimental)/° | 43.5 (−3.6) | 43.1 (−3.9) | 3.8 (+0.3) | 6.9 (+1.2) |
| Photochromic | Yes | Yes | No | No |
One of the key structural indicators of photochromism in N-salicylidenes is the torsion angle, ϕ, around the imine linkage (Fig. 2a), whereby structures with ϕ between 35–50° typically exhibit photochromism, and structures with ϕ close to 0° are not photochromic. It has previously been posited that larger torsion angles are enabled by low density packing of molecules in the crystal structure, and this in turn is associated with solid-state photochromism.37,38 The torsion angles for An compounds follow this empirical trend, with A1 and A2 (photochromic) having large torsion angles of 47.7° and 47.0°, respectively, and A3 and A4 (non-photochromic) having small torsion angles of 3.5° and 5.7°, respectively. For the present study, we hypothesised that it should be possible to probe the imine torsion angle using solid-state NMR spectroscopy. To this end, 13C CPMAS NMR spectra were recorded for bulk crystalline samples of A1–A4 (Fig. 2b). For the photochromic N-salicylidenes A1 and A2, two resonances are seen above 160 ppm which is within the chemical shift range expected for an arene OH group and is also the upper boundary of the range for imine carbons. However, for the non-photochromic N-salicylidenes A3 and A4, one of these resonances is observed below 160 ppm. To help interpret the experimental 13C CPMAS NMR spectra, 13C chemical shifts were calculated through periodic DFT calculations on the published crystal structures (CCDC: A1 = 273062,32 A2 = 1124413,33 A3 = 1131744,31 A4 = 72291130) which show all molecules in the enol tautomeric form. The calculated 13C chemical shifts for the geometry optimised crystal structures show good overall agreement with the experimental chemical shifts, confirming the equilibrium in the ground state is very close to the pure enol tautomer. However, it is noteworthy that the calculated and experimental chemical shifts for the imine carbon C7 are split into two regions for the photochromic (160–168 ppm) and non-photochromic (152–154 ppm) N-salicylidenes (Table S2, ESI†). This helps spectral assignment, which would otherwise be ambiguous due to the switching of the relative shifts of the C1 and C7 resonances, but also points towards a marked dependence of the C7 chemical shift on the structure. Inspection of the geometry-optimised crystal structures for A1 and A2 revealed that the DFT optimisation decreased the torsion angle decreased by 3.6° and 3.9° respectively, relative to the experimental crystal structures; in contrast, the torsion angles for A3 and A4 increased, albeit by a much smaller amount of 0.3° and 1.2° respectively (Table 1). These values were found to be independent of the DFT methodology used (Table S3, ESI†). The free volume around each N-salicylidene molecule (Vfree) in the bulk crystal structures can be estimated through consideration of the unit cell volume, guest volume, and number of molecules in the unit cell (Z).39 The Vfree values for non-photochromic crystalline structures are lower than the photochromic crystal structures (Table S4, ESI†), indicating that the non-photochromic molecules A3 and A4 are more densely packed, with less free volume to allow light-induced tautomerisation to occur. This also helps to rationalise the larger change in torsion angle for the photochromic structures compared to the non-photochromic structures observed in the DFT geometry optimisations, whereby the larger free volume around the photochromic molecules A1 and A2 permits larger structural distortions.
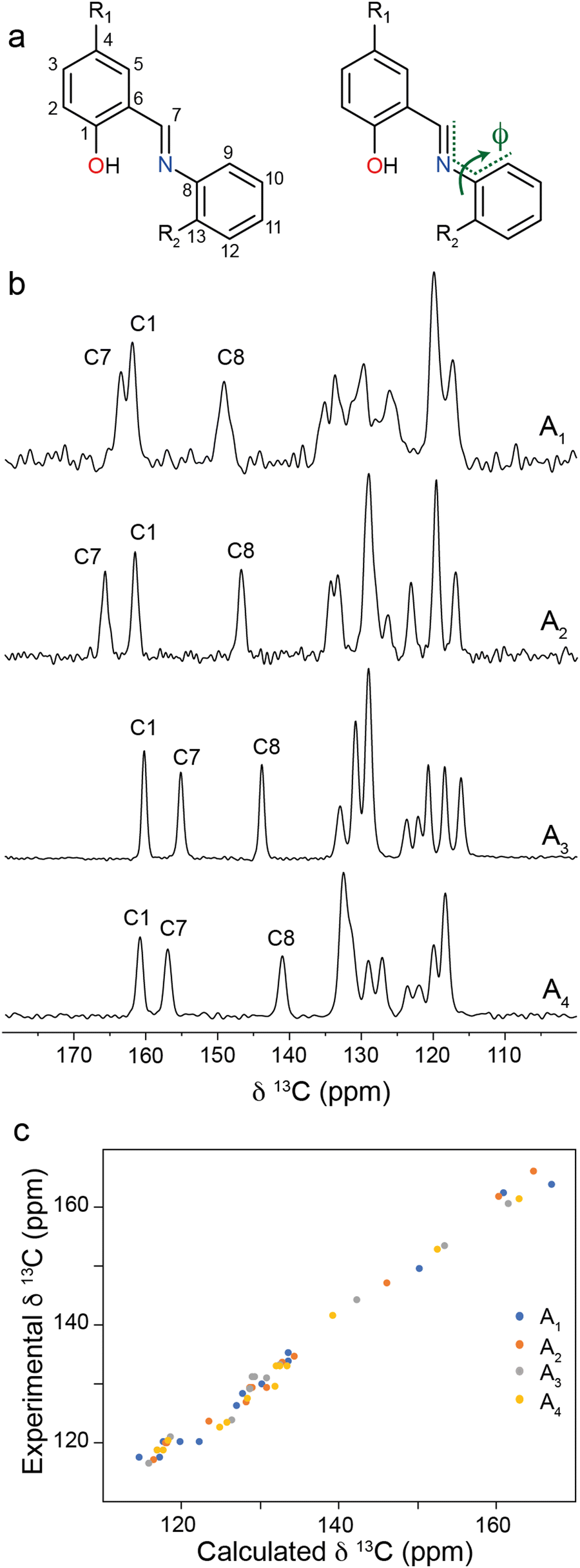 | ||
| Fig. 2 (a) Labelling scheme and depiction of the C–C–C–N imine torsion angle, ϕ, for A1–A4. (b) 13C CPMAS NMR spectra of bulk crystalline N-salicylidenes, A1–A4. (c) Comparison of DFT-calculated and experimental 13C chemical shifts for A1–A4. | ||
To gain further insight into the differences in torsion angle between the structures studied, single-molecule DFT calculations were performed on A1–A4 in each of the tautomeric forms. Geometry optimisations were performed with the imine torsion angles, ϕ, fixed in 10° increments between 0°–90° while all other atoms positions were allowed to vary. Total energies for A1–A4 are shown in Fig. 3. The data show that the enol tautomers are the lowest energy tautomers for molecules and all torsion angles, and an energy minimum is observed when ϕ is between 30°–40°. The cis-keto tautomers are intermediate in energy with minima observed at ϕ = 0°, while the trans-keto tautomers are up to 0.6 eV per molecule higher in energy, also with minima ϕ = 0°. This energy difference is equivalent to 294 J g−1, suggesting that N-salicylidenes could be competitive with other photochromic molecules such as azobenzene in terms of stored energy, depending on the photostationary state that can be achieved.
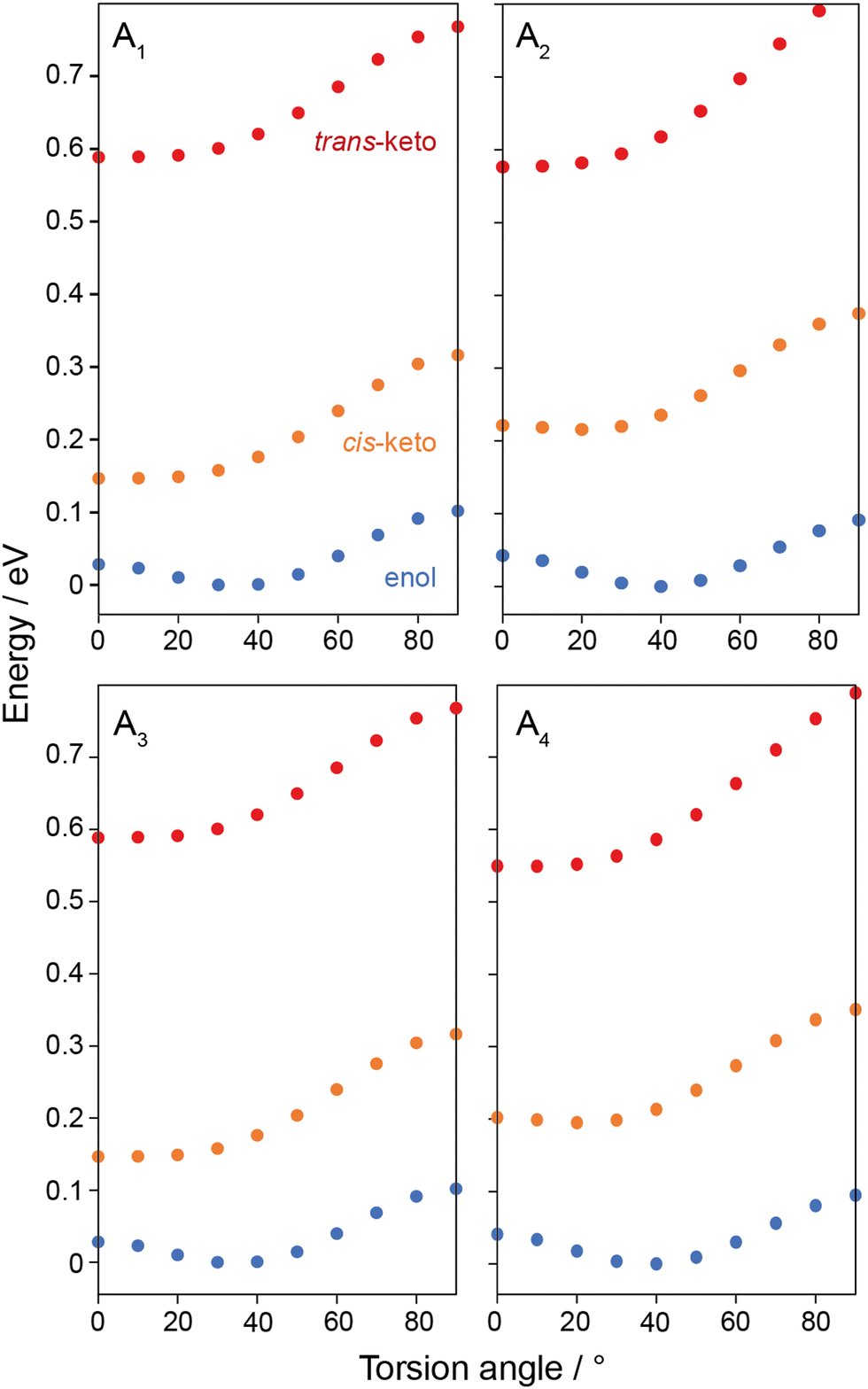 | ||
| Fig. 3 Plots of total energies of enol (blue), cis-keto (yellow) and trans-keto (red) tautomers of A1–A4 obtained from single molecule DFT calculations with imine torsion angles fixed between 0–90°. Energies for each molecule are normalised relative to the lowest energy conformer. | ||
The single-molecule energy profiles help to explain the change in torsion angles when the crystal structures are geometry optimised. For all structures, the geometry optimisation causes the torsion angle to deviate slightly towards the energy minimum of the enol tautomer at ϕ ∼ 35°. However, the magnitude of the change is variable: for photochromic structures A1 and A2 a substantial decrease in torsion angle is observed, which suggests there is a degree of structural freedom around the phenolic part of the molecule, consistent with the larger Vfree values for these structures. The precise difference in torsion angle between the experimental crystal structures and the geometry-optimised structures could result from thermal effects in the diffraction experiment, whereas the DFT calculation is carried out at an effective temperature of 0 K. Indeed, in a previous study investigating the photochromic N-salicylidene N-(3,5-di-t-butylsalicylidene)-4-amino-pyridine (with torsion angle ϕ = 42.3°), millisecond timescale rotational dynamics of the pyridyl group were observed at ambient temperature, confirming a significant degree of structural freedom around that end of the molecule.40 This structure also showed a significant reduction in torsion angle to ϕ = 38.5° upon DFT geometry optimisation. This is contrasted by the small increase in torsion angle during the geometry optimisation for the non-photochromic crystal structures A3 and A4. The fact that these crystal structures show almost planar geometries suggests that the crystal packing distorts the molecule away from the energy minimum geometry at ϕ ∼ 35°, and the dense molecular packing that is responsible for the imposed planar geometry (consistent with the smaller Vfree values) will also likely restrict the ability to further change geometry to the trans-keto tautomer. To probe the chemical effect of the R-group substituents on the electronic structure and molecular geometry, single-molecule calculations of electron density (Fig. S3, ESI†) and NMR parameters (Table S5, ESI†) were performed on fully-optimised structures for A1 and A3. The results show that the substitution of Cl for H at the R1 position has overall a very small effect, with very similar chemical shieldings, electron densities and ground-state molecular geometries observed for both molecules. This further supports that the dominant influence on the photochromic properties is the crystal packing rather than intrinsic differences in the molecular structure or geometry due to the different substituents used.
These results rationalise the empirical dependence of the photochromic properties of N-salicylidenes on the torsion angle. When the bulk crystal structure exhibits a torsion angle close to 35°, this indicates the structure is relatively free of packing-induced distortion, which then allows the molecules to adopt geometries close to their energetic minima. This also indicates low density packing, and suggests there is sufficient freedom for movement to convert between the enol, cis-keto and trans-keto tautomers upon UV irradiation. It is also noteworthy that the energy profile of the trans-keto tautomer is relatively flat from ϕ ∼ 0–30° (Fig. 3), so it may be possible for light-induced trans-keto tautomers to be accommodated within the crystal structure even if a small packing-imposed distortion away from the true minimum energy geometry is present. In contrast, crystal structures which show torsion angles of close to 0° must do so because of close molecular packing, which distorts the molecular geometries away from the energetic minima and will also act to restrict or inhibit the pedal motion and change in molecular geometry required for conversion to the trans-keto tautomer.
Structural analysis of MOF-N-salicylidenes complexes
MIL-53 (1) is constructed from chains of corner sharing Al4(OH)2 octahedra which are interconnected by the dicarboxylate groups of benzene dicarboxylate (BDC) linker units. In the guest-free dehydrated form, the three-dimensional microporous framework has orthorhombic symmetry (Imma) and forms a large-pore structure (1lp) characterised by one-dimensional diamond-shaped channels with an internal diameter of 0.85 nm.28 Under ambient conditions, 1 absorbs water molecules which hydrogen bond to hydroxide ions coordinated to the Al(III) octahedra. This causes a significant contraction of the pores and results in a narrow-pore phase (1np) with a monoclinic symmetry (P21/c). When heated under vacuum, the water molecules are removed and hydrogen bonds broken, which causes an opening of the pores back to the large-pore phase.The N-salicylidenes, A1–A4, were separately loaded into 1via a gas-phase procedure under vacuum which also removes any excess bulk N-salicylidene.19,41,42 Differential scanning calorimetry was performed to confirm removal of any excess via the absence of melting features (Fig. S1, ESI†). Thermogravimetric analysis showed that each N-salicylidene loads to 0.5 molecules per formula unit of the framework, which is consistent with previously reported loadings of azobenzene (AB) into the same framework and equivalent to 1.0 guest N-salicylidene per pore (Fig. S2, ESI†).43 Upon guest loading, for each complex 1⊃An there are changes in the intensities and positions of reflections in the XRPD patterns (Fig. S4a–e, ESI†) which is consistent with the deformation of the porous channels and guest-induced breathing behaviour observed in 1.44,45 Le Bail fits for all complexes show that they crystallise in the orthorhombic space group Imma which is consistent with the 1lp structure. Relevant unit cell parameters are summarised in Table 2. Additionally, the unit cell parameters for 1⊃A1 and 1⊃A3 are very similar to those previously reported for 1⊃AB by Hermann et al.43 Notably, 1⊃A2 and 1⊃A4, have an extended a dimension and a contracted b dimension; the significant lengthening of a dimension appears to be a consequence of a large R1 = Cl group of the guest molecule. The c dimension, which represents Al–O–Al bonds, remains relatively consistent throughout the series. For the N-salicylidene-loaded complexes, the observation of the large-pore framework indicates that there are no water molecules in the structures which would lead to the contraction of the channels by hydrogen bonding28 – this is also supported by TGA measurements (Fig. S2, ESI†). Indeed this is expected from the gas-phase loading procedure which simultaneously removes water molecules from the framework.
Guest conformation within 1
To gain further insight into the packing and orientation of the N-salicylidene guest molecules occluded in 1, 13C CPMAS NMR spectra were recorded (Fig. 4). For hydrated 1np, three resonances are observed corresponding to crystallographically distinct carbon sites within the BDC linkers. The chemical shifts are consistent with previous reports.29 As the structure is loaded with N-salicylidenes, additional resonances corresponding to the guest molecules emerge. Moreover, the BDC carbonyl resonance shifts from 175.3 ppm to 171.0 ppm for all 1⊃An complexes. This is consistent with the guest-induced expansion from the hydrated narrow-pore structure to the large-pore structure.29,46 The carbonyl chemical shift is particularly sensitive to hydration and the reduction in chemical shift is a further indication of the absence of water molecules.28,47 In contrast, the chemical shifts of the quaternary carbons (136.9–137.6 ppm) and ring carbons (128.8–130.4 ppm) of the BDC linker in the guest-loaded structures show little deviation from hydrated 1np.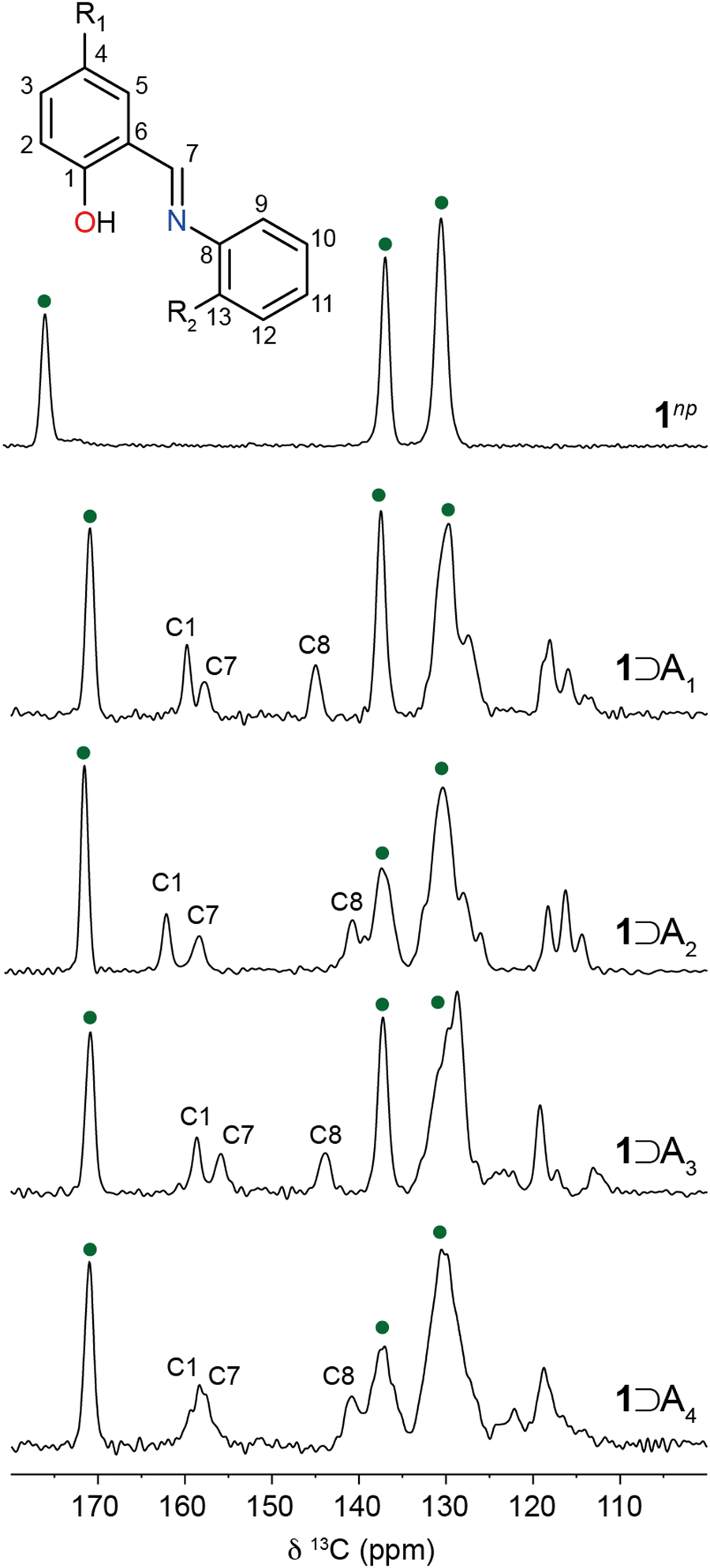 | ||
| Fig. 4 13C CPMAS NMR spectra of 1⊃An host–guest complexes. Resonances corresponding to 1 are denoted by green circles. | ||
1⊃A1 is presented as a model system to understand guest behaviour in 1. Considering the guest molecules within 1⊃A1, the experimental resonances are characteristic of the enol form with the C–OH (C1) resonance at 159.8 ppm (Fig. 4). However, this resonance is shifted from its chemical shift in the bulk crystal (162.2 ppm). Similarly, resonances corresponding to C7 (158.0 ppm) and C8 (145.2 ppm) have shifted considerably when compared to bulk A1 (Fig. 1, 162.3 ppm and 149.5 ppm respectively). This confirms A1 is incorporated into the pores of 1 with a different local environment or molecular geometry. Variable-temperature 13C CPMAS NMR spectra recorded between 248–360 K (Fig. S5, ESI†) show small changes in the positions and intensities of the enol ring resonances suggesting the possible onset of thermally induced ring dynamics, indicating that the A1 molecules have some degree of local mobility when confined within the MOF pore.
To investigate the relationship between torsion angle and 13C chemical shift, DFT calculations on isolated enol N-salicylidene molecules with torsion angles between 0–90° were performed. The calculated chemical shifts of the carbons adjacent to the imine linkage show systematic changes with respect to the torsion angle, varying between 153.0–164.0 ppm (C7, Fig. 5a) and 141.5–154.0 ppm (C8, Fig. 5b). Interestingly, the calculated 13C chemical shifts for the C7 imine carbons in isolated single molecules with torsion angles close to those in the experimental diffraction structures agree well with the experimental 13C chemical shifts for the respective crystalline N-salicylidenes (Table S2, ESI†). Additionally, for C8 there is a similar relationship (Fig. 5b); however, there is more deviation from the calculated values, suggesting that intermolecular effects or possibly ring dynamics may contribute to the chemical shift at this position. There is also unsurprisingly a clear dependence on the nature of the substituent at the R2 position. Therefore, while chemical shifts of the C7 imine carbons and the C8 carbons both provide sensitive probes of the torsion angle, C7 is largely independent of substituent effects from the R2 position, and is less influenced by secondary structural effects.
 | ||
| Fig. 5 DFT-calculated 13C chemical shifts of (a) C7 and (b) C8 in single molecules of A1–A4 compounds at fixed torsion angles. (c) Correlation of torsion angles in crystal structures of A1–A4 against experimental 13C chemical shift of C7 (black dots). Also shown are points representing predicted torsion angles for A1–A4 within 1⊃An host–guest complexes based on the calculated torsion angle dependence of the C7 chemical shift derived from the plot in (a) (coloured dots). | ||
To gain insight into the geometry of the guest molecules under confinement within 1, the experimental chemical shift of C7 was used to estimate the molecular torsion angle using the dependence in Fig. 5a. These values are plotted in Fig. 5c, together with the corresponding values for the bulk crystalline structures. The points for the bulk crystalline N-salicylidenes are split into two regions, where the non-photochromic structures (A3 and A4) have experimental chemical shifts between 152.0–153.0 ppm and are almost planar, while the photochromic structures (A1 and A2) have experimental resonances between 160.0–161.5 ppm and torsion angles between 45°–47°. Interestingly, the experimental resonances of C7 for 1⊃An complexes are grouped in an intermediate range between 156.0–158.0 ppm, indicating torsion angles between 21°–32°. This suggests that occlusion of guest molecules in the pores of 1 imparts freedom to allow the planar molecules A3 and A4 to adopt a geometry closer to the energetic minimum. However, we note that the torsion angles for all molecules are somewhat smaller than the true energetic minimum which is expected at ∼35°, suggesting that the structures still remain slightly constrained within the pore environment.
Photoswitching properties of 1⊃An
To determine whether occlusion of A1–A4 in the framework of 1 alters their photochromic properties, the bulk crystalline N-salicylidenes, and the host–guest complexes 1⊃An were each irradiated with 365 nm light for five minutes. Bulk crystalline A1 and A2 were irradiated as bright yellow powders, and over the course of the experiment the surface crystallites changed to bright red (Fig. 6a). Bulk crystalline A3 and A4 were irradiated as bright orange powders and no change in colour was observed over the course of the experiment (Fig. 6a). These results are consistent with the reported photochromic properties, i.e., A1 and A2 are photochromic whereas A3 and A4 are non-photochromic.15,27 UV-vis reflectance spectra for the photochromic A1 and A2 show that the unirradiated form reflectance plateaus of 3% from 200–400 nm, before steadily increasing up to 80% between 400–500 nm (Fig. 6b and Fig. S6, ESI†). The yellow colour observed is indicative of the presence of a minor component of the cis-keto tautomer; however, any cis-keto tautomer present appears to be below the level of detection in the 13C CPMAS NMR experiments discussed above, which is estimated to be approximately 3% based on the maximum signal-to-noise ratio obtained. After irradiation the low-reflectance plateau extends to 550 nm, before sharply increasing up to 81% reflectance at 550 nm. The red colour observed and extension of the plateau is indicative of the trans-keto tautomer. The red colouring persisted for 3–5 minutes for both samples before fading to the characteristic yellow colour of the cis-keto isomer. UV irradiation and decay cycles were repeated up to ten times without any changes to the observed properties. UV irradiation of A3 and A4 produced no change in the reflectance properties (Fig. S7, ESI†).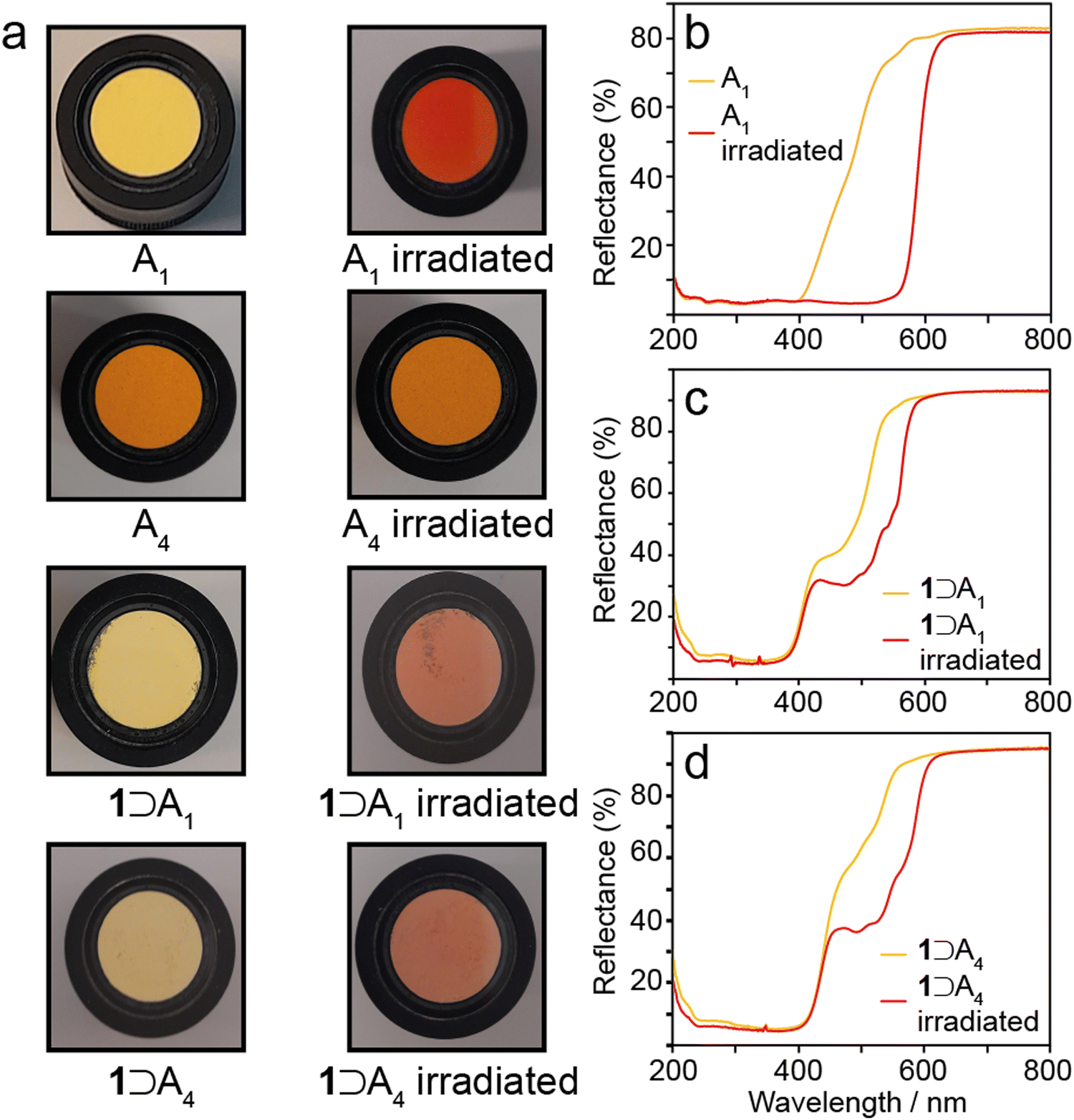 | ||
| Fig. 6 (a) Observed colour changes of A1 and A4, and 1⊃A1 and 1⊃A4 after 365 nm irradiation. UV-vis reflectance spectra of (b) A1, (c) 1⊃A1, and (d) 1⊃A4 before and after 365 nm irradiation. | ||
The 1⊃An complexes all appear pale yellow in colour (Fig. 6a) and have similar UV-vis reflectance spectra to bulk A1 and A2 (Fig. 6b–d and Fig. S8, ESI†), where there is low reflectance between 200–400 nm, with a sharp increase from 400–700 nm. Noticeably, this represents a marked change in the reflectance properties of 1⊃A3 and 1⊃A4 when compared to the bulk crystalline phases A3 and A4, where the absorbing plateaus extend to 520 nm and 525 nm respectively. Occlusion within the MOF structure changes the reflectance profiles to become very similar to A1 and A2. This mirrors the 13C chemical shifts which suggest convergence of the torsion angles to around 30° (Fig. 5c), representing large geometry changes for A3 and A4 but smaller changes for A1 and A2. When exposed to 365 nm light the pale yellow 1⊃An complexes gradually change, over a period of ten minutes, to pale red (Fig. 6). UV-vis reflectance measurements show a shoulder with 30–40% reflectance between 460–520 nm, which is increased relative to the unirradiated samples and within the reflectance range of the trans-keto tautomer (Fig. 6d and Fig. S8, ESI†). Over a period of five minutes the pale red colouring returns to pale yellow for each example. The UV irradiation and decay cycles were repeated ten times without any changes to the observed behaviour. Therefore, the occlusion of A3 and A4 within 1 imparts photochromic properties which are not present in the bulk crystalline structures. This is attributed to the increased free volume afforded to the guest N-salicylidene molecules, which we estimate increases by at least a factor of 3 compared to the bulk crystalline structures (Table S4, ESI†). The results mirror a previous study of a structurally similar N-salicylidene (E)-5-methyl-2-((p-tolylimino)-methyl)-phenol (Me2SA) occluded within a molecular capsule.22 In the bulk crystal structure, Me2SA adopts a planar structure and does not display photochromism, but the additional free volume afforded by encapsulation within a molecular cavity imparted photochromic properties in both the solution and solid-state. Together, these results highlight how controlled confinement within a suitable host can be used to tailor the photochromic properties of N-salicylidenes.
In addition to the photochromic properties, the thermochromic properties of A1–A4 were also investigated by cooling samples to approximately 210 K through immersion in acetone cooled with dry ice (Fig. S9, ESI†). For the photochromic N-salicylidenes A1 and A2, a colour change from pale yellow to colourless was observed upon cooling. The non-photochromic structures, A3 and A4, changed from a bright orange to yellow. This is consistent with a tautomeric equilibrium between cis-keto and enol isomers, and where the non-photochromic (planar) structures have a higher proportion of the cis-keto form resulting in a stronger colour at ambient temperature and a more pronounced colour change. Upon occlusion of A1–A4 within 1, the colour change upon cooling was less pronounced for all samples, transitioning from dull yellow to colourless. This further supports that the guest molecules of 1⊃A3 and 1⊃A4 adopt more twisted conformations when compared to their bulk crystal structures, which pushes the tautomeric equilibrium further towards the enol structure as the sample is cooled.
Conclusions
A structural investigation of a set of crystalline N-salicylidenes has revealed distinct differences in the geometries of their possible tautomeric forms, whereby the enol has an energetic minimum with a torsion angle of ∼35° and the cis- and trans-keto tautomers adopt planar structures at the energetic minima. Inspection of the crystal structures shows that photochromic properties are only observed when the molecules exhibit a torsion angle close to the energetic minimum of the enol tautomer in the native state. This is attributed to the crystal structure having a low enough packing density that intermolecular effects do not distort the geometry of the enol ring, and sufficient local void space is available to permit light-induced structural changes to the trans-keto tautomer. Conversely, structures which show a planar geometry in the native state are indicative of high packing density which exerts pressure on the molecules, forcing them to deviate from the energy minimum geometry. This will also act to restrict the ability of the molecules to undergo the light-induced structural change required to form the trans-keto tautomer, thereby inhibiting photochromism.13C CPMAS NMR experiments highlight that the C7 chemical shift is highly sensitive to the torsion angle of the enol isomer and is not significantly affect by intermolecular effects. Using this as a structural probe, the conformations of the enol isomers of all molecules occluded within the pores of 1 were found to be close to the calculated energetic minima with torsion angles between 21–32°. For A1 and A2, this does not represent a significant change in geometry from the bulk crystalline state; however, for A3 and A4, this represents a marked change in geometry which is indicative of increased local free volume. For these molecules, which do not exhibit photochromism in the bulk crystalline state, occlusion within 1 results in the emergence of photochromic properties. This is attributed to the internal void space of the MOF structure allowing sterically unhindered photoswitching between enol and trans-keto isomers. Further studies investigating the effect of the framework structure itself on photochromic behaviour, such as the inclusion of pendant groups on the linker to stabilise the trans-keto tautomer, are ongoing.
Author contributions
K. G.: conceptualization, synthesis, investigation, formal analysis, writing – original draft, writing – review and editing. H. B.: formal analysis, methodology, software. R. J. B.: formal analysis, methodology, software. N. H.: conceptualization, synthesis, investigation, formal analysis, writing – original draft, writing – review and editing, funding acquisition. J. M. G.: conceptualization, synthesis, investigation, formal analysis, writing – original draft, writing – review and editing, funding acquisition.Data availability
The data supporting this article is available at https://doi.org/10.17635/lancaster/researchdata/679.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Leverhulme Trust (Research Project Grant Number RPG-2018-395) and EPSRC (project EP/V05001X/1) for funding this work.References
- J. Orrego-Hernández, A. Dreos and K. Moth-Poulsen, Acc. Chem. Res., 2020, 53, 1478–1487 CrossRef PubMed.
- Q. Qiu, Y. Shi and G. G. D. Han, J. Mater. Chem. C, 2021, 9, 11444–11463 RSC.
- L. Dong, Y. Chen, F. Zhai, L. Tang, W. Gao, J. Tang, Y. Feng and W. Feng, J. Mater. Chem. A, 2020, 8, 18668–18676 RSC.
- S. Kawata and Y. Kawata, Chem. Rev., 2000, 100, 1777–1788 CrossRef CAS.
- X. Li, M. Liu, J. Shao, H. Sun, Q. Zhang, D. Peng and F. Liu, Adv. Funct. Mater., 2024, 2402603 CrossRef CAS.
- M. Poutanen, Z. Ahmed, L. Rautkari, O. Ikkala and A. Priimagi, ACS Macro Lett., 2018, 7, 381–386 CrossRef CAS PubMed.
- Y. Shi, M. A. Gerkman, Q. Qiu, S. Zhang and G. G. D. Han, J. Mater. Chem. A, 2021, 9, 9798–9808 RSC.
- J. Orrego-Hernández, A. Dreos and K. Moth-Poulsen, Acc. Chem. Res., 2020, 53, 1478–1487 CrossRef.
- R. J. Salthouse and K. Moth-Poulsen, J. Mater. Chem. A, 2024, 12, 3180–3208 RSC.
- C. Wallace, K. Griffiths, B. L. Dale, S. Roberts, J. Parsons, J. M. Griffin and V. Görtz, ACS Appl. Mater. Interfaces, 2023, 15, 31787–31794 CrossRef CAS.
- C. Bin Fan, Z. Q. Liu, L. Le Gong, A. M. Zheng, L. Zhang, C. S. Yan, H. Q. Wu, X. F. Feng and F. Luo, Chem. Commun., 2017, 53, 763–766 RSC.
- I. M. Walton, J. M. Cox, J. A. Coppin, C. M. Linderman, D. G. Patel and J. B. Benedict, Chem. Commun., 2013, 49, 8012–8014 RSC.
- H. A. Schwartz, S. Olthof, D. Schaniel, K. Meerholz and U. Ruschewitz, Inorg. Chem., 2017, 56, 13101–13110 CrossRef PubMed.
- P. Naumov, P. Yu and K. Sakurai, J. Phys. Chem. A, 2008, 112, 5810–5814 CrossRef CAS PubMed.
- E. Hadjoudis and I. M. Mavridis, Chem. Soc. Rev., 2004, 33, 579–588 CAS.
- S. Hasebe, K. Hatakeyama-Sato, K. Oyaizu, T. Asahi and H. Koshima, ACS Omega, 2024, 9, 1463–1471 CrossRef CAS PubMed.
- H. Sugiyama and H. Uekusa, CrystEngComm, 2018, 20, 2144–2151 RSC.
- K. Johmoto, T. Ishida, A. Sekine, H. Uekusa and Y. Ohashi, Acta Crystallogr., Sect. B: Struct. Sci., 2012, 68, 297–304 CrossRef CAS.
- K. Griffiths, N. R. Halcovitch and J. M. Griffin, Chem. Sci., 2022, 13, 3014–3019 RSC.
- D. Hermann, H. A. Schwartz, M. Werker, D. Schaniel and U. Ruschewitz, Chem. – Eur. J., 2019, 25, 3606–3616 CrossRef CAS.
- H. M. H. K. Toshio Kawato Kiichi Amimoto and H. Kanatomi, Molecular Crystals and Liquid Crystals Science and Technology. Section A, Mol. Cryst. Liq. Cryst., 2000, 345, 57–62 CrossRef.
- M. Sliwa, P. Naumov, H.-J. Choi, Q.-T. Nguyen, B. Debus, S. Delbaere and C. Ruckebusch, ChemPhysChem, 2011, 12, 1669–1672 CrossRef CAS.
- J. He, K. Aggarwal, N. Katyal, S. He, E. Chiang, S. G. Dunning, J. E. Reynolds, A. Steiner, G. Henkelman, E. L. Que and S. M. Humphrey, J. Am. Chem. Soc., 2020, 142, 6467–6471 CrossRef CAS PubMed.
- N. Yanai, T. Uemura, M. Inoue, R. Matsuda, T. Fukushima, M. Tsujimoto, S. Isoda and S. Kitagawa, J. Am. Chem. Soc., 2012, 134, 4501–4504 CrossRef CAS.
- H. A. Schwartz, M. Werker, C. Tobeck, R. Christoffels, D. Schaniel, S. Olthof, K. Meerholz, H. Kopacka, H. Huppertz and U. Ruschewitz, ChemPhotoChem, 2020, 4, 195–206 CrossRef CAS.
- M. D. Cohen, G. M. J. Schmidt and S. Flavian, J. Chem. Soc., 1964, 2041–2051 RSC.
- E. Hadjoudis, ChemInform, 2010, 22, 295 Search PubMed.
- T. Loiseau, C. Serre, C. Huguenard, G. Fink, F. Taulelle, M. Henry, T. Bataille and G. Férey, Chem. – Eur. J., 2004, 10, 1373–1382 CrossRef CAS.
- G. P. M. Bignami, Z. H. Davis, D. M. Dawson, S. A. Morris, S. E. Russell, D. McKay, R. E. Parke, D. Iuga, R. E. Morris and S. E. Ashbrook, Chem. Sci., 2018, 9, 850–859 RSC.
- X. Zhang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, o512 CrossRef CAS PubMed.
- J. Bregman, L. Leiserowitz and G. M. J. Schmidt, J. Chem. Soc., 1964, 2068–2085 RSC.
- F. Arod, M. Gardon, P. Pattison and G. Chapuis, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2005, 61, o317–o320 CrossRef.
- J. Bregman, L. Leiserowitz and K. Osaki, J. Chem. Soc., 1964, 2086–2100 RSC.
- A. E. Bennett, C. M. Rienstra, M. Auger, K. V. Lakshmi and R. G. Griffin, J. Chem. Phys., 1995, 103, 6951–6958 CrossRef CAS.
- A. Bielecki and D. P. Burum, J. Magn. Reson., Ser. A, 1995, 116, 215–220 CrossRef CAS.
- C. J. Pickard and F. Mauri, Phys. Rev. B, 2001, 63, 245101 CrossRef.
- K. Johmoto, A. Sekine and H. Uekusa, Cryst. Growth Des., 2012, 12, 4779–4786 CrossRef CAS.
- F. Robert, A. D. Naik, B. Tinant, R. Robiette and Y. Garcia, Chem. – Eur. J., 2009, 15, 4327–4342 CrossRef CAS.
- P.-L. Jacquemin, K. Robeyns, M. Devillers and Y. Garcia, Chem. – Eur. J., 2015, 21, 6832–6845 CrossRef CAS PubMed.
- B. L. Dale, N. R. Halcovitch, M. J. G. Peach and J. M. Griffin, Magn. Reson. Chem., 2019, 57, 230–242 CrossRef CAS PubMed.
- K. Griffiths, N. R. Halcovitch and J. M. Griffin, Inorg. Chem., 2021, 60, 12950–12960 CrossRef CAS.
- K. Griffiths, N. R. Halcovitch and J. M. Griffin, Chem. Mater., 2020, 32, 9925–9936 CrossRef CAS.
- D. Hermann, H. Emerich, R. Lepski, D. Schaniel and U. Ruschewitz, Inorg. Chem., 2013, 52, 2744–2749 CrossRef CAS.
- W. P. Mounfield and K. S. Walton, J. Colloid Interface Sci., 2015, 447, 33–39 CrossRef CAS PubMed.
- F. Nouar, T. Devic, H. Chevreau, N. Guillou, E. Gibson, G. Clet, M. Daturi, A. Vimont, J. M. Grenèche, M. I. Breeze, R. I. Walton, P. L. Llewellyn and C. Serre, Chem. Commun., 2012, 48, 10237–10239 RSC.
- C. M. Rice, Z. H. Davis, D. McKay, G. P. M. Bignami, R. G. Chitac, D. M. Dawson, R. E. Morris and S. E. Ashbrook, Phys. Chem. Chem. Phys., 2020, 22, 14514–14526 RSC.
- R. Giovine, C. Volkringer, J. Trébosc, J.-P. Amoureux, T. Loiseau, O. Lafon and F. Pourpoint, Acta Crystallogr., Sect. C: Struct. Chem., 2017, 73, 176–183 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tc02862g |
| This journal is © The Royal Society of Chemistry 2024 |