
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulti-metal (Fe, Cu, and Zn) coordinated hollow porous dodecahedron nanocage catalyst for oxygen reduction in Zn–air batteries†
Yanan
Pan‡
a,
Qi
Yang‡
a,
Xiaoying
Liu
a,
Fan
Qiu
a,
Junjie
Chen
a,
Mengdie
Yang
a,
Yang
Fan
a,
Haiou
Song
b and
Shupeng
Zhang
 *a
*a
aSchool of Chemistry and Chemical Engineering, Nanjing University of Science and Technology, Nanjing, 210094, PR China. E-mail: shupeng_2006@126.com; Fax: +86 25 84315519; Tel: +86 25 84315519
bSchool of Environment, Nanjing Normal University, Nanjing, 210097, PR China
First published on 5th September 2024
Abstract
The coupling of multiple low-cost metals and porous nanocarbon materials aimed at replacing precious metals to enhance electrocatalytic oxygen reduction is a critical challenge in some crucial research areas. In the present study, a hollow dodecahedron nanocage catalyst (Fe3O4/CuNCs/ZnNx-PHNC) was constructed by supporting copper nanoclusters, Fe3O4 nanoparticles, and Zn–Nx after sintering and annealing through the coordination of ZIF-8 and by doping copper and iron ions. We observed that the synergy of the multi-metals in the magnetically separable heterojunction catalyst induced electron transfer and inhibited hydrogen peroxide formation, thus improving its catalytic performance for the oxygen-reduction reaction. The catalyst demonstrated a half-wave potential as high as 0.832 V and a Tafel slope of 54 mV decade−1, superior to many non-precious metal catalysts reported in the literature. The assembled Zn–air battery (ZAB) exhibited a maximum power density of 162 mW cm−2 and ultrahigh stability of >500 h at 5 mA cm−2 current density. The ZAB's excellent performance indicates its high development and practical application prospects.
1. Introduction
The current global energy shortage has arisen because of the ever-increasing human demand for energy and the associated depletion of available energy resources.1,2 Human survival, economic status, and sustenance are highly dependent on energy availability, while the exploitation and use of other resources are also energy dependent.3Currently, the rapid development of the electronics industry requires a safe and stable energy supply system.4 Zn–air batteries (ZABs) are one of the most promising battery candidates because of their high theoretical energy density (1086 W h kg−1), low cost, high safety, and environmental friendliness.5 However, ZABs face some severe setbacks,6 with the lack of reliable and efficient bifunctional air electrodes as the biggest obstacle to their practical applications.7 Generally, ZABs’ efficiency depends on the catalytic performance of oxygen electrocatalysts on air electrodes, where the discharge process occurs in a kinetically retarded oxygen-reduction reaction (ORR). Therefore, ORR-efficient electrocatalysts are crucial for improving the performance of rechargeable ZABs. Moreover, platinum-based catalysts are typical commercial electrocatalysts for the ORR with excellent activity. However, their high cost, the natural scarcity of platinum, and their unsatisfactory durability limit their large-scale industrial applications.8,9 Therefore, there is an urgent need to develop highly active, durable, and cost-effective ORR catalysts.
Transition metals and their derivatives (bearing aluminum, oxygen, nitrogen, sulfur, selenium, carbides and hydroxides and their corresponding oxides, nitrides, sulfides, etc.) have attracted huge interest owing to their earth-abundance and high catalytic properties.10–13 Among them, Fe- and Cu-based nanoparticles are of immense interest because of their high electrical conductivity, excellent mechanical strength, and flexible electronic structure.14–16 However, finding a suitable catalyst substrate to load active nanoparticles is essential to expose the active sites more entirely for improved activity.
Zeolitic imidazolate frameworks (ZIFs) constructed from coordination networks between specific metal nodes and organic ligands are attractive substrates considering their unique properties of a high specific surface area, large pore volume, and multiple organometallic nature.17 Given these properties, ZIF-based materials have been widely applied in adsorption and purification.18 Introducing metallic species into ZIF precursors can inhibit the aggregation of nanoparticles using a strong anchoring effect, thereby leading to highly dispersed active sites. Additionally, calcination at high temperatures ensures a high graphitization of carbon precursors, which can improve the corrosion resistance and electrical conductivity.19 Due to their relatively high activity, Fe-doped ZIF materials are considered among the most promising oxygen-reduction electrocatalysts to replace the noble metal Pt.20–22 However, the H2O2 produced by the Fe–N–C catalyst formed by single-metal Fe doping in the ORR severely hinders the four-electron-transfer process in the ORR. As a result, the Fe active sites easily undergo the Fenton reaction with H2O2, potentially affecting the ORR activity.23,24
To overcome this drawback, other metals can be doped to induce electron transfer and inhibit H2O2 production. The significant synergistic effect between Fe3O4 nanoparticles and Cu nanoclusters’ catalytic active sites and carbon carriers effectively improves the electrocatalytic activity.25,26 In addition, Zn has less electronegativity (c = 1.65), making it easier to contribute its most valent electrons to Cu (c = 1.90) and Fe (c = 1.83). Therefore, Zn is compatible with Cu and Fe, and can optimize the electronic structure to produce synergy, promote the adsorption of reaction molecules and desorption of reaction intermediate, and improve the catalytic performance.27 Therefore, in the present study we employed ZIF-8 with a Zn atom as the substrate and introduced Fe and Cu atoms to construct composite catalysts.
The high surface area and short diffusion path of hollow nanocages provide more active sites, a larger liquid–solid contact area, and a relatively large functional surface for the adsorption–desorption of the substrate molecules themselves to promote the adsorption–desorption rate.28,29 Meanwhile, the hollow interior can prevent the encapsulated transition metal oxide nanoparticles from aggregating. The high dispersion of metal nanoparticles on the carbon skeleton can expose more active sites, providing excellent electrical conductivity and allowing for fast mass transfer. In addition, anisotropic nanocages have the characteristics of a uniform size, low density, large void space, and strong shell permeability, etc., which have strong utilization value for electrocatalysis.30 The synergetic enhancement of the interface structure and active sites on the catalyst utilizes the inherent advantages of the interface structure.31 Therefore, distributing the various nanoparticles entirely on the graded porous carbon and leveraging their synergistic advantages to promote catalytic performance has become a major challenge. Therefore, it is imperative to develop a transition metal-regulated skeletonization gold nanocage structure with a controllable microstructure to reveal changes in the electronic and environmental properties at the coupling interface, clarify the structural performance of the synergistic effect, and improve the ORR performance.32
The current research prepared a hollow-graded, porous carbon-based, cage-like catalyst co-regulated by multiple metals, and successfully applied it to ORR reactions and zinc–air batteries. We used ZIF-8 containing zinc, nitrogen, carbon, and oxygen as the precursor, which was transformable into a material with porous carbon-containing Zn–Nx active sites after high-temperature pyrolysis. Fe and Cu ions were introduced through coordination during ZIF-8 synthesis, and high-temperature sintering synchronously generated a hollow carbon-based catalyst controlled by the multiple metals. Further, salt melting and high-temperature annealing resulted in pore formation, and a hollow-graded porous carbon-based catalyst (named Fe3O4/CuNCs/ZnNx-PHNC) was obtained. The magnetically separable catalyst had a high specific surface area and high metal loading capacity. Moreover, Fe3O4, composed of Fe species with various valence states, could provide active sites for electron acquisition and loss, ensuring a high ORR activity. Second, the Cu nanoclusters have excellent stability, conductivity, and abundant oxygen defects, which can accelerate the conductivity of O-containing intermediates, thereby extending the lifespan of the Fe3O4/CuNCs/ZnNx-PHNC catalysts.
This study provides an effective strategy for synthesizing non-noble metal catalysts for use in ZABs. The assembled ZABs and flexible zinc–air batteries (FZABs) exhibited excellent discharge performances and strong stability, reflecting their optimistic commercial prospects and application value as air cathode catalysts.
2. Experimental section
2.1 Synthesis of ZIF-8
The production method for ZIF-8 refers followed the literature with some improvements.33,34 First, zinc nitrate hexahydrate (1.16 g) and dimethylimidazole (2.46 g) were dissolved in 40 ml of methanol, sonicated for 10 min, mixed and stirred for 12 h, and then rested at room temperature for 12 h. The supernatant after resting was poured out, then washed three times each with ultrapure water and ethanol, and dried in a vacuum oven at 60 °C to obtain ZIF-8 white crystals.2.2 Synthesis of Fe-ZIF-8 and Cu-ZIF-8
Following the same procedure used for the synthesis of ZIF-8, zinc nitrate hexahydrate (1.16 g) and iron acetylacetonate (0.35 g) were dissolved in 40 ml of methanol, and dimethylimidazole (2.46 g) was dissolved in 40 ml of methanol, sonicated for 10 min, mixed and stirred for 12 h, and then rested at room temperature for 12 h. The supernatant after resting was poured out, then washed three times each with ultrapure water and ethanol, and dried in a vacuum oven at 60 °C and the final product was named Fe-ZIF-8. The synthesis process was the same as for Fe-ZIF-8 except that iron acetylacetonate was replaced by copper nitrate (0.1 g), and the resulting product was named Cu-ZIF-8.2.3 Synthesis of Fe/Cu-ZIF-8
Iron acetylacetonate (0.35 g), copper nitrate (0.1 g), and zinc nitrate hexahydrate (1.16 g) were dissolved simultaneously in 40 ml of methanol, and the subsequent treatment was the same as that for the synthesis of ZIF-8.2.4 Synthesis of Fe3O4/CuNCs/ZnNx-PHNC
The dried product was mixed with NaCl and KCl (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4:1). After sintering at 925 °C for 2 h, the calcined products of ZIF-8, Cu-ZIF-8, Fe-ZIF-8, and Fe/Cu-ZIF-8 were extracted and filtered. The final catalysts were named ZnNx-PNC, CuNCs/ZnNx-PNC, Fe3O4/ZnNx-PHNC, and Fe3O4/CuNCs/ZnNx-PHNC. For the convenience of readers, we list the raw materials and the corresponding obtained materials in Table S1 (ESI†).
:4:1). After sintering at 925 °C for 2 h, the calcined products of ZIF-8, Cu-ZIF-8, Fe-ZIF-8, and Fe/Cu-ZIF-8 were extracted and filtered. The final catalysts were named ZnNx-PNC, CuNCs/ZnNx-PNC, Fe3O4/ZnNx-PHNC, and Fe3O4/CuNCs/ZnNx-PHNC. For the convenience of readers, we list the raw materials and the corresponding obtained materials in Table S1 (ESI†).
3. Results and discussion
3.1. Structure and characterization of the catalysts
The preparation procedure of Fe3O4/CuNCs/ZnNx-PHNC is shown in Fig. 1a. To synthesize Fe–Cu-ZIF-8, the precursors were first synthesized by dissolving a mixture of iron acetylacetonate, copper nitrate, and zinc nitrate hexahydrate in methanol before introducing imidazole dialdehyde dissolved in methanol with stirring at room temperature. Calcining Fe3O4/CuNCs/ZnNx-PHNC in the presence of NaCl and KCl under an Ar atmosphere formed the hollow-structured Fe3O4/CuNCs/ZnNx-PHNC.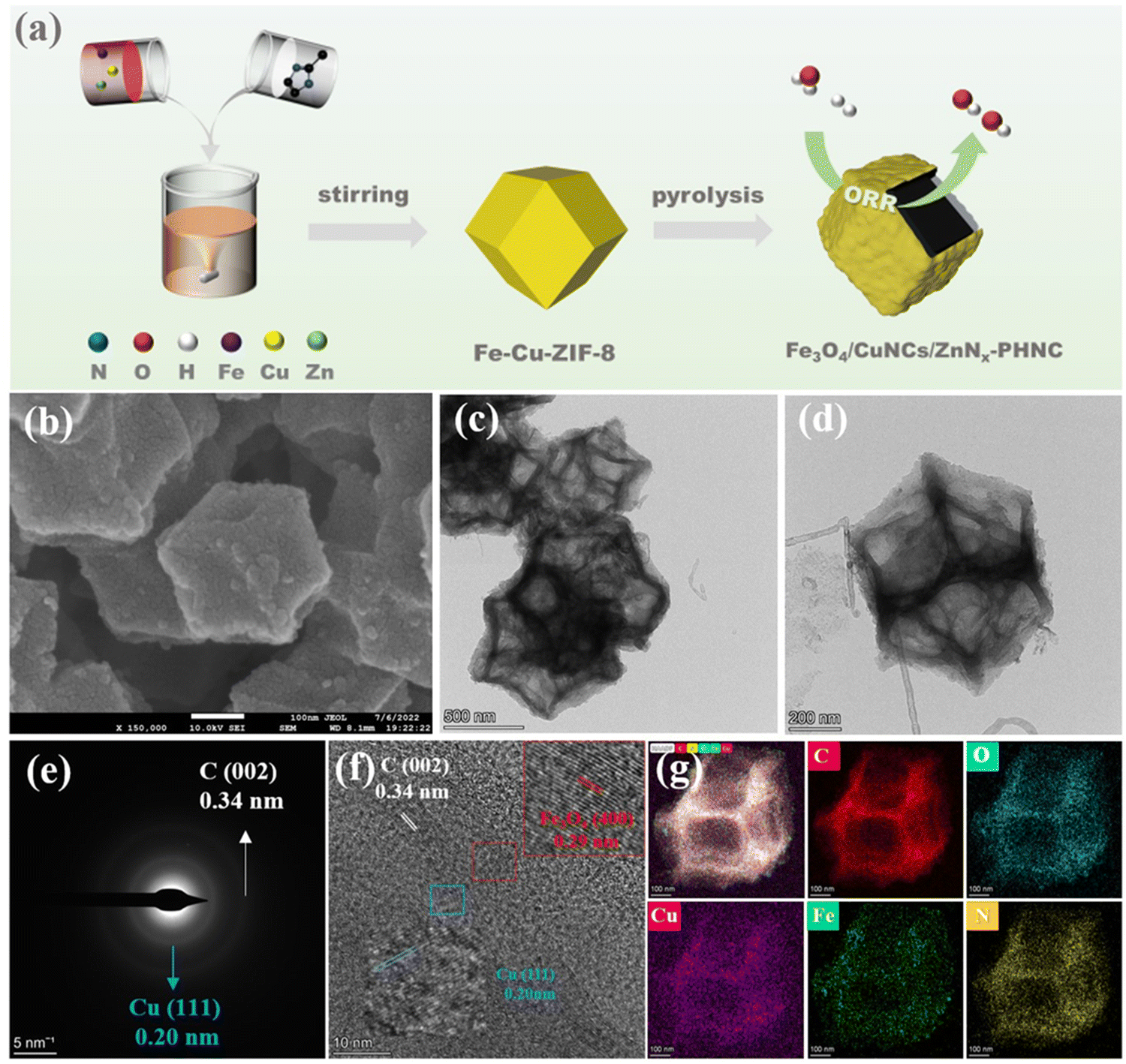 | ||
| Fig. 1 (a) Flow chart for the preparation of Fe3O4/CuNCs/ZnNx-PHNC; (b) SEM images of Fe3O4/CuNCs/ZnNx-PHNC at different resolutions; (c) and (d) TEM images of Fe3O4/CuNCs/ZnNx-PHNC at different resolutions; (e) selected area electron diffraction images of Fe3O4/CuNCs/ZnNx-PHNC; (f) high-resolution TEM images of Fe3O4/CuNCs/ZnNx-PHNC; (g) HAADF-STEM images of Fe3O4/CuNCs/ZnNx-PHNC and its elemental distribution images. | ||
Metal doping was determined from the X-ray diffraction (XRD) images of all the materials before sintering. As shown in Fig. S1 (ESI†), all the synthesized precursors conformed with the theoretical ZIF-8 peak structure. However, spurious peaks appeared at individual locations, attributable to the metal doping. Fig. 2a shows the XRD diffraction images of the four materials after sintering. The graphitic structure formed after calcination evinced the (002) diffraction peak centered at 25.4° and the (101) diffraction peak at 43.2° as the major crystallographic planes of C. For Fe3O4/CuNCs/ZnNx-PHNC, the 19.7° and 43.1° spikes corresponded to the crystallographic planes (212) and (440) of Fe3O4 (PDF#76-0956), respectively. Combined with the standard PDF card for Cu (PDF#04-0836), peaks for the Cu nanoclusters were also evident in the XRD pattern of Fe3O4/CuNCs/ZnNx-PHNC. The uniform embedding of Cu nanoclusters in the carbon layer as active sites is crucial for promoting the ORR performance. The XRD pattern of Fe3O4/ZnNx-PHNC only showed a weak Fe3O4 characteristic peak at 15.3°, likely due to the lack of Cu regulation and the minute Fe3O4 produced, mainly responsible for the lack of expected catalytic effect. Also, the XRD pattern of CuNCs/ZnNx-PNC did not show any Cu peak, indicating that single-metal doping could not stably form metal nanoclusters. In addition, only carbon peaks were observed in ZnNx-PNC.
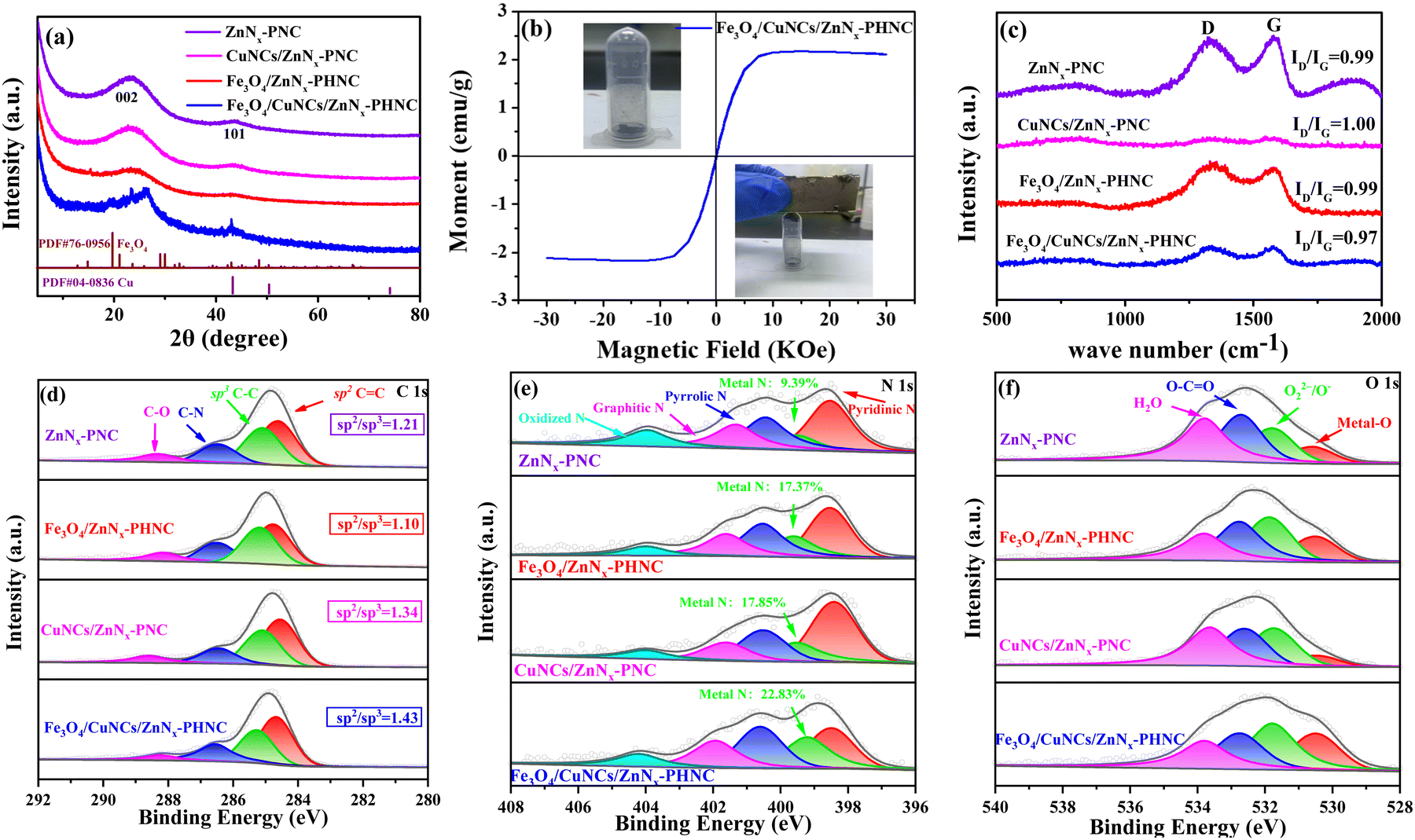 | ||
| Fig. 2 (a) XRD spectra of Fe3O4/CuNCs/ZnNx-PHNC, Fe3O4/ZnNx-PHNC, CuNCs/ZnNx-PNC and ZnNx-PNC; (b) VSM curves of Fe3O4/CuNCs/ZnNx-PHNC; (c) Raman spectra of Fe3O4/CuNCs/ZnNx-PHNC, Fe3O4/ZnNx-PHNC, CuNCs/ZnNx-PNC and ZnNx-PNC; (d) C 1s, (e) N 1s and (f) O 1s XPS spectra of Fe3O4/CuNCs/ZnNx-PHNC, Fe3O4/ZnNx-PHNC, CuNCs/ZnNx-PNC and ZnNx-PNC. | ||
The hysteresis curve (VSM) represents the magnetization of a ferromagnetic substance under alternating external magnetic field strengths. When the magnetization starts (Fig. 2b), the magnetic induction strength of the ferromagnet increases with the external magnetic field strength and the unit moment. At a certain magnetic field strength, the magnetic induction strength of the ferromagnet is optimized, and the unit moment plateaus. The inset shows the Fe3O4/CuNCs/ZnNx-PHNC catalyst powder, which was suspended when magnetized, indicating its strong magnetic property. This observation was consistent with the peak structure of Fe3O4 nanoparticles from the XRD spectra.
To investigate the structural defects and the degree of graphitization in the carbon materials, the Raman spectra of Fe3O4/CuNCs/ZnNx-PHNC, Fe3O4/ZnNx-PHNC, CuNCs/ZnNx-PNC, and ZnNx-PNC were examined (Fig. 2c). The D-band (1350 cm−1) was the vibration caused by lattice defects in the carbon matrix, corresponding to the defect position of sp3, while the G-band (1580 cm−1) was caused by vibrations belonging to the sp2 bonded graphitic carbon. The samples’ intensity ratios (ID/IG) of the D-band to G-band were 0.97, 0.99, 1.00, and 0.99, respectively. The intensity ratio for Fe3O4/CuNCs/ZnNx-PHNC was 0.97, indicating the presence of many defects in the carbon matrix. These defects in the lattice are essential sites for enhancing the electrocatalytic performance of the catalyst.35 Comparing the other three materials, we observed that the materials showed a large number of carbon defects and increased graphitization when calcined with added protective and stripping agents. This result shows that sintering and activators are crucial for preparing nanocarbon materials.36,37
N2 adsorption–desorption experiments were performed to further investigate the pore structures and specific surface areas of Fe3O4/CuNCs/ZnNx-PHNC, Fe3O4/ZnNx-PHNC, CuNCs/ZnNx-PNC, and ZnNx-PNC. The adsorption isotherms (Fig. S2a, ESI†) illustrated that the materials were characterized by hysteresis-return type IV profiles. Also, the samples had high specific surface areas and extensive porosity. From Fig. S2b (ESI†), the specific surface areas of Fe3O4/CuNCs/ZnNx-PHNC, Fe3O4/ZnNx-PHNC, CuNCs/ZnNx-PNC, and ZnNx-PNC were derived as 702, 773, 853, and 894 m2 g−1, respectively. Their pore size distribution (Fig. S2c, ESI†) was mainly dominated by micropores and mesopores, in which the hierarchical porous structure can shorten the distance for O2, thus accelerating the electron transport and mass transfer.38 Table S2 (ESI†) suggests that introducing Fe and Cu decreased the microporosity of Fe3O4/CuNCs/ZnNx-PHNC, whereas the average pore size (calculated by BJH) was the largest. This was related to the fact that the total BET, micropore area, and pore capacity ratio were reduced correspondingly with the magnitude of metal doping. CuNCs/ZnNx-PNC and ZnNx-PNC showed some folds spreading and may also exfoliate some graphitized lamellae, thus significantly altering their pore structure.
Furthermore, X-ray photoelectron spectroscopy (XPS) was used to assess the elemental composition of the catalysts. The samples’ surface elemental contents are listed in Table S3 (ESI†). Fig. S3a (ESI†) illustrates the full spectra of Fe3O4/CuNCs/ZnNx-PHNC, Fe3O4/ZnNx-PHNC, CuNCs/ZnNx-PNC, and ZnNx-PNC, showing the peaks of the elements in the various speciations. From the high-resolution spectrum of C 1s (Fig. 2d), the C peaks could be divided into four peaks: 284.7 ± 0.1, 285.2 ± 0.1, 286.5 ± 0.1, and 288.4 ± 0.2 eV, corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–C, C–N, and C–O bonds, respectively. Among these, the C–N bond indicated the successful doping of N atoms into the carbon nanostructures. The main peak at 284.7 ± 0.1 eV was ascribed to sp2 hybridized carbon, while that at 285.2 ± 0.1 eV belonged to sp3 hybridized carbon. The sp2 hybridized carbon significantly improves the conductivity of materials.39 Conversely, the conductivity and catalytic activity of sp3-hybridized carbon are relatively low.40
C, C–C, C–N, and C–O bonds, respectively. Among these, the C–N bond indicated the successful doping of N atoms into the carbon nanostructures. The main peak at 284.7 ± 0.1 eV was ascribed to sp2 hybridized carbon, while that at 285.2 ± 0.1 eV belonged to sp3 hybridized carbon. The sp2 hybridized carbon significantly improves the conductivity of materials.39 Conversely, the conductivity and catalytic activity of sp3-hybridized carbon are relatively low.40
Here, we compared the corresponding peak areas of the three metal-doped materials with the total peak area of C 1s (Table S4, ESI†) and calculated the relative contents of sp2 and sp3 based on the area ratio. The sp2/sp3 ratio values of ZnNx-PNC, Fe3O4/ZnNx-PHNC, CuNCs/ZnNx-PNC, and Fe3O4/CuNCs/ZnNx-PHNC were 1.21, 1.10, 1.34, and 1.43, respectively. Fe3O4/CuNCs/ZnNx-PHNC's surface has a relatively high sp2 hybrid carbon content and corresponding high electrical conductivity. The N 1s spectrum could be deconvoluted into peaks located at 398.5 ± 0.1, 399.4 ± 0.2, 400.5 ± 0.1, 401.6 ± 0.3, and 404.0 eV, ascribed to pyridinic nitrogen, metal–N, pyrrolic nitrogen, graphitic nitrogen, and nitrogen oxide species, respectively (Fig. 2e). The metal–N bonding pattern indicated that metals and N combined to form metal–N active sites between the carbon skeleton, contributing significantly to the ORR activity enhancement. By calculating the ratio of each peak area to the total peak area, the metal–N active site content was obtained (Table S5, ESI†). The metal–N content of Fe3O4/CuNCs/ZnNx-PHNC was 22.83%, evincing the sample's best ORR performance.
The XPS high-resolution spectra of O 1s exhibited main peaks at 530.5 ± 0.1, 531.8 ± 0.1, 532.7 ± 0.1, and 533.7 ± 0.1 eV, corresponding to metal–O, O22−/O−, C–CO, and H2O, respectively (Fig. 2f). O22−/O− is the chemisorbed oxygen, which is closely related to oxygen vacancies. Oxygen vacancies are crucial in regulating the surface electronic state, accelerating the oxygen-containing intermediate transfer, and enhancing the interaction between transition metal nanoparticles and carbon carriers; thus, enriching the electrochemical activity and enhancing the catalyst stability.41 Based on the ratio of the corresponding peak area to the total peak area, we could deduce the oxygen-vacancy content in the different substances. According to the peak proportion of the metal-doped materials (Table S6, ESI†), Fe3O4/CuNCs/ZnNx-PHNC had the highest surface oxygen-vacancy content, accounting for 33.75% of the total area. The XPS high-resolution spectrum of Fe 2p showed that Fe was primarily present as Fe2+ and Fe3+, as evidenced by the Fe 2p1/2 and Fe 2p3/2 orbital peaks, respectively. The peaks could be further deconvoluted into four peaks (Fig. S3b, ESI†),42 whereby the two peaks at 711.0 ± 0.2 and 712.8 ± 0.1 eV corresponded to the Fe 2p3/2 orbital, while the two peaks at 724.2 ± 0.2 and 726.0 ± 0.1 eV were designated as Fe 2p1/2 spin–orbit peaks. The absence of satellite peaks also corroborated the assignment of the final product to Fe3O4 rather than Fe2O3.43,44
Furthermore, the XPS high-resolution spectra of Cu 2p were dominated by Cu at peak positions of 934.1 ± 0.4 and 955.0 ± 0.6 eV, with Cu 2p3/2 and Cu 2p1/2 present (Fig. S3c, ESI†). These were consistent with the XPS images of Cu 2p in copper nanoclusters documented in other literature.45 In the XPS spectra of Zn 2p (Fig. S3d, ESI†), peaks were observed at 1021.9 ± 0.1 and 1045.2 ± 0.2 eV, corresponding to Zn2+.46 Because the boiling point of zinc is 907 °C, many experiments ignore Zn's presence after high-temperature calcination. However, while most zinc particles evaporate to form defects, the remaining atoms form Zn–N bonds, preventing them from evaporating.47 This result was also consistent with that found with metal–N bonds in the N 1s map of ZnNx-PNC, which was without transition metal doping.
The content ratio of metals in the materials was estimated using inductively coupled plasma mass spectrometry (ICP-MS). The concentrations of Fe, Cu, and Zn in the final product were 179.08, 17.06, and 10.80 ppb, respectively, with corresponding content ratios of 3.6%, 0.34%, and 0.21%.
The morphology and composition of the materials’ surface ultrastructures were examined by scanning electron microscopy (SEM). From the SEM image of the Fe/Cu-ZIF-8 precursor (Fig. S4a–c, ESI†), we observed that the ZIF-8 surface was wrinkled by Fe and Cu. However, the SEM micrographs of Fe-ZIF-8 (Fig. S4d–f, ESI†) and Cu-ZIF-8 (Fig. S4g–i, ESI†) showed no significant changes in morphology, indicating that single-metal doping did not significantly alter the dodecahedral structure of ZIF-8 when the precursor was synthesized. To further describe the basic three-dimensional and surface microscopic morphologies of the materials, we studied the SEM images of the materials after calcination. Fig. 1b and Fig. S5a–c (ESI†) show that Fe3O4/CuNCs/ZnNx-PHNC retained a complete dodecahedral structure, presenting some grain-like substances on the surface, preliminarily assumed as nanoparticles and nanoclusters formed by the metals. After calcination, the degree of graphitization and carbon defects of the material were significantly increased, which are crucial to increasing the conductivity and catalytic activity of the material. The basic morphology maintained by Fe3O4/ZnNx-PHNC (Fig. S5d–f, ESI†) was similar to that of Fe3O4/CuNCs/ZnNx-PHNC. However, the CuNCs/ZnNx-PNC surface (viewed at various resolutions) was smooth (Fig. S5g–i, ESI†), and wrinkled nanosheets appeared on the surface in the presence of activators and stripping agents. SEM proved that Fe contributes significantly to inducing the hollow nanocages. The SEM image of ZnNx-PNC (Fig. S5j–l, ESI†) confirmed that without metal doping, the complete dodecahedral structure could no longer be retained after high-temperature calcination, also proving that transition metal doping was essential to the structural stability of the material.
Further morphological observations were done using transmission electron microscopy (TEM). Fig. 1c depicts the TEM images of Fe3O4/CuNCs/ZnNx-PHNC at 500 nm resolution, showing that the sintered ZIF-8 did not agglomerate, retaining the dodecahedral structure.48Fig. 1d shows that the Fe3O4/CuNCs/ZnNx-PHNC dodecahedra had a laminar wrinkled nanosheet structure in the outer layer and distinct skeletonization in the inner layer, showing several dispersed skeletonized nanocage structures overall. The large number of defects formed in the outer and inner layers after calcination provided abundant electron-transport channels that enhanced O2 adsorption and shortened the mass-transfer distance, which were primarily responsible for the enhanced activity of Fe3O4/CuNCs/ZnNx-PHNC.49 Fig. S6a–c (ESI†) show the TEM images of Fe3O4/ZnNx-PHNC at different resolutions. The sample maintained the basic dodecahedral structure, with folded nanosheets on the outer layer and skeletonized porous layers on the inner layer. These structural forms were similar to those of Fe3O4/CuNCs/ZnNx-PHNC. In contrast, CuNCs/ZnNx-PNC (Fig. S6d–f, ESI†) showed no skeletonization despite the folded nanosheets on the surface, consistent with the SEM imagery. Moreover, ZnNx-PNC (representing the undoped transition metal) showed an irregular nanobulk after calcination (Fig. S6g–i, ESI†), with a rough surface and no skeletalization. This indicated that the basic skeleton of the material was destroyed by the high-temperature calcination via its collapse and splitting. The analysis showed that the Fe-doped carbon precursor could form skeletonized nanocages during calcination, while the non-doped metal precursor could not maintain a complete dodecahedral structure after sintering. The reason may be that in the early stages of calcination, Fe3+ forms an oxide of Fe on the surface of ZIF-8, and as calcination continues, the Kirkendall effect occurs between Zn2+ and the oxide of Fe to form ZnO. The continuous inhomogeneous interdiffusion of O2− ions and Zn2+ ions on ZIF-8 leads to the emergence of Kirkendall holes.50 The loss of Zn2+ destroys the crystal structure of ZIF-8, and the organic ligand dimethylimidazole evaporates from the interior due to the lack of coordination, causing ZIF-8 to gradually become hollow.51 Thus, metal doping is crucial for stabilizing the transition material morphology during calcination.
The selected electron diffraction (SAED) pattern of Fe3O4/CuNCs/ZnNx-PHNC (Fig. 1e) showed two diffracted circles. The radii of the outermost and innermost circles were 0.34 and 0.20 nm, respectively, corresponding to the (002) crystal plane of C and (111) crystal plane of Cu nanoclusters.52Fig. 1f depicts a high-resolution transmission electron microscopy (HRTEM) image of Fe3O4/CuNCs/ZnNx-PHNC. It could be observed that the metal nanoparticles were embedded in the graphitic carbon layer, inducing a polycrystalline structure, which improved the catalyst's stability. Further, measuring the lattice stripe spacing revealed that the 0.34 nm lattice spacing corresponded to the (002) crystalline plane of C. Compared with the standard Fe3O4 nanoparticle (PDF#76-0956), the 0.29 nm spacing corresponded to the (400) crystal plane of Fe3O4, while the 0.20 nm spacing was attributed to the (111) crystal plane (when compared with the standard Cu nanocluster, PDF#04-0836).53 These results were consistent with those from the XRD analysis.
Additional elemental mapping images of Fe3O4/CuNCs/ZnNx-PHNC using energy dispersive spectroscopy (EDS) (Fig. 1g) revealed that C, N, Fe, and Cu were uniformly dispersed on the skeletonized nanocages, and Fe3O4 nanoparticles were formed from the combination of Fe and O. The Cu nanoclusters were uniformly distributed in the nitrogen-doped carbon, forming Fe3O4 nanoparticles and Cu nanocluster active sites, which enabled efficient electron transfer during the ORR and enhanced the ORR catalytic activity.
3.2. Electrocatalytic performances of the catalysts
Next, cyclic voltammetry (CV) and linear scanning voltammetry (LSV) were performed to determine the electrocatalytic activity of the prepared catalysts for the ORR. The ORR performance of each catalyst was evaluated using commercial 20 wt% Pt/C. Fig. S7 (ESI†) shows the CV curves of the samples. In 0.1 M KOH electrolyte saturated with N2, no obvious redox peak was observed for the four materials, indicating there were no other side reactions on the materials. Conversely, in the O2-saturated electrolyte, oxygen-reduction peaks ensued, indicating oxygen electric reduction in the electrolyte. To investigate the ORR electrocatalytic activity of the synthesized catalyst materials, the catalytic performances of the materials were analyzed by testing the LSV curves with an electrochemical workstation.54 From the LSV curves (Fig. 3a), we observed that Fe3O4/CuNCs/ZnNx-PHNC outperformed the commercial 20 wt% Pt/C catalyst, with respective limiting currents of 5.69 and 5.63 mA cm−2, indicating the enhanced conductivity of the Fe3O4/CuNCs/ZnNx-PHNC catalyst for the ORR. The half-wave potential of Fe3O4/CuNCs/ZnNx-PHNC was 0.832 V, better than the 0.814 V of 20 wt% Pt/C, indicating that Fe3O4/CuNCs/ZnNx-PHNC reacted faster and had enhanced electrocatalytic activity. From the onset, Fe3O4/CuNCs/ZnNx-PHNC evinced a superior half-wave potential and limiting current than those of Fe3O4/ZnNx-PHNC, CuNCs/ZnNx-PNC, and ZnNx-PNC. Moreover, metal doping enhanced the conductivity and electron-donation ability of the material, proving that the abundant active sites of Fe3O4 nanoparticles and Cu nanogroups co-promoted the improved ORR activity.55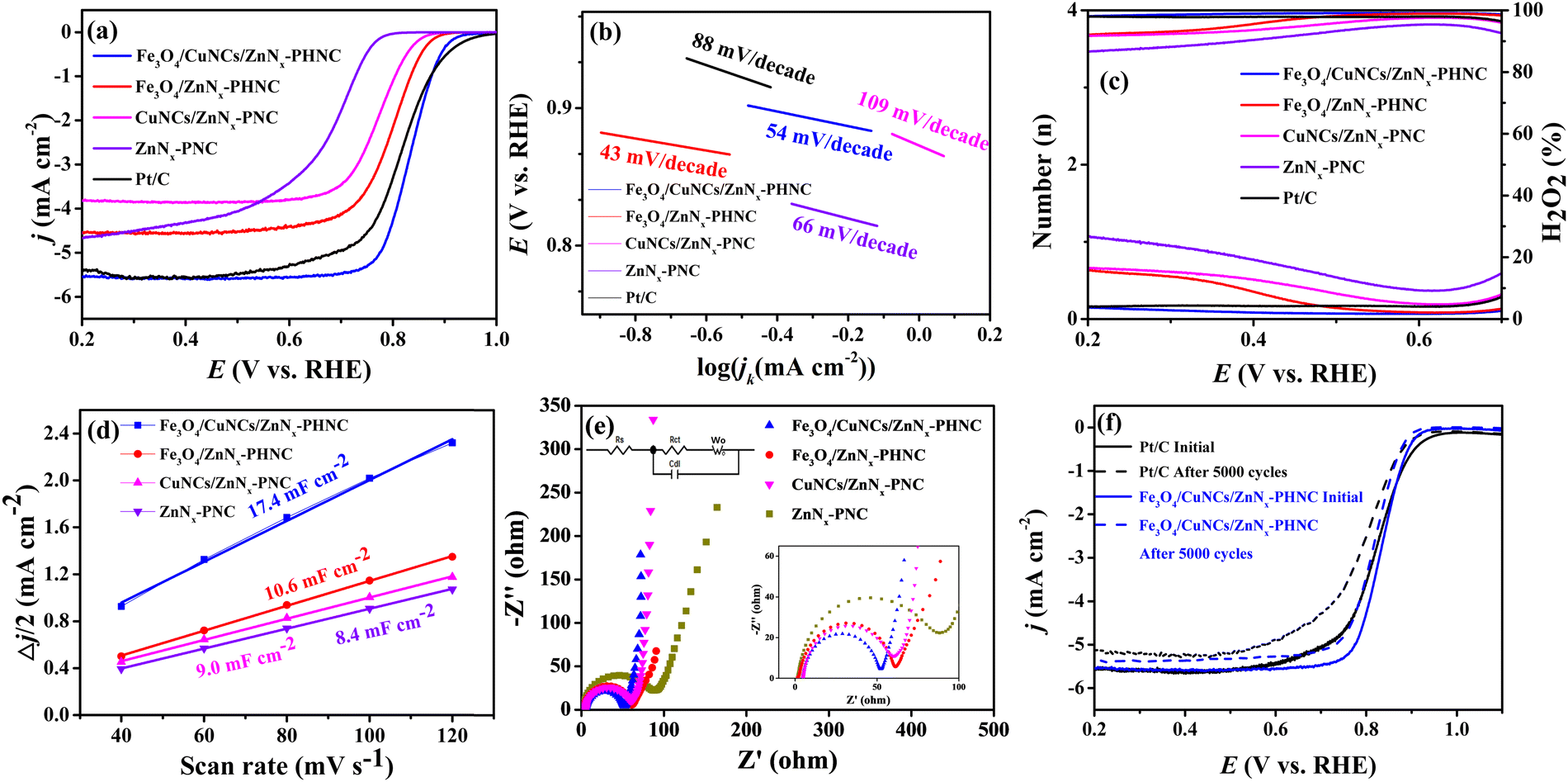 | ||
| Fig. 3 (a) LSV curves for Fe3O4/CuNCs/ZnNx-PHNC, Fe3O4/ZnNx-PHNC, CuNCs/ZnNx-PNC, ZnNx-PNC and Pt/C; (b) Tafel curves; (c) hydrogen peroxide yield and average number of electrons transferred; (d) double-layer capacitance (Cdl); (e) impedance diagrams of Fe3O4/CuNCs/ZnNx-PHNC, Fe3O4/ZnNx-PHNC, CuNCs/ZnNx-PNC and ZnNx-PNC (insets show partial enlargements); (f) stability experiments of Fe3O4/CuNCs/ZnNx-PHNC and Pt/C. | ||
Meanwhile, the Tafel slope values of Fe3O4/CuNCs/ZnNx-PHNC, Fe3O4/ZnNx-PHNC, CuNCs/ZnNx-PNC, ZnNx-PNC, and Pt/C were obtained as 54, 43, 109, 66, and 88 mV decade−1, respectively (Fig. 3b). The Tafel slopes of CuNCs/ZnNx-PNC and ZnNx-PNC were large, reflecting their poor ORR kinetic performance.56 For the mono-metallic catalyst samples, the lack of active sites and PHNC structure led to their failure to achieve the expected ORR performance. Fe3O4/CuNCs/ZnNx-PHNC and Fe3O4/ZnNx-PHNC had lower Tafel slopes and higher reaction kinetics than Pt/C, indicating that the active site and hollow nanocage structure promoted the ORR activity. On the other hand, it also indicated that the nanocage structure was essential for enhancing electron transport and improving the reaction kinetics. Further, the ORR properties of Fe3O4/CuNCs/ZnNx-PHNC and some recently published Fe-based catalysts were compared (Table S7, ESI†) and competitive ORR catalytic performance was demonstrated.
In addition, the K–L equation is essential for analyzing the ORR kinetics. As shown in Fig. S8 (ESI†), the K–L curves of the four materials displayed a good linear relationship, suggesting that their ORR reactions followed first-order kinetics. Meanwhile, the smaller the interval between the K–L curves, the more complete their ORR kinetics. The average numbers of transferred electrons for Fe3O4/CuNCs/ZnNx-PHNC, Fe3O4/ZnNx-PHNC, CuNCs/ZnNx-PNC, and ZnNx-PNC were 4.0, 3.6, 2.4, and 3.7, respectively. These values indicate that the reaction for Fe3O4/CuNCs/ZnNx-PHNC followed the four-electron reaction pathway entirely, whereas the other three materials also had incomplete side reactions.17
To further explore the actual catalytic pathway of each catalyst in the ORR reaction, RRDE test materials can be used to calculate the respective electron-transfer number (n) and the corresponding hydrogen peroxide yield (H2O2%) according to the measured ring and disk currents. Fig. 3c illustrates that the electron-transfer numbers of Fe3O4/CuNCs/ZnNx-PHNC and Pt/C were close to four, indicating that they were mainly involved in a four-electron transfer in the ORR reaction. Therefore, H2O2, as a product in the path of a two-electron reaction, was meager. At the same time, the electron-transfer numbers of Fe3O4/ZnNx-PHNC, CuNCs/ZnNx-PNC, and ZnNx-PNC were between three and four, suggesting a large number of two-electron-transfer pathways in their ORR reactions, generating a large amount of H2O2, thereby resulting in a relatively poor ORR activity. Also, it indicates that the synergistic effect of bimetals reduced the H2O2 production rate and accelerated the four-electron-transfer process during the reaction.
The magnitude of the electrochemical double-layer capacitance (Cdl) of an electrode is related linearly to the active specific surface area of the catalyst, with a larger Cdl resulting in a larger active specific surface area of the catalyst. The CV curves of Fe3O4/CuNCs/ZnNx-PHNC were tested under various scan rates at 1.106–1.175 V (vs. RHE) (Fig. S9, ESI†). At different scan rates, half of the sum of the absolute values of the current densities of the cathode and anode at 1.15 V were taken, and the slopes obtained were plotted and fitted to the Cdl of the electrodes of 17.4, 10.6, 9.0, and 8.4 mF cm−2 (Fig. 3d). We noticed that the catalytic active sites significantly affected the double-layer capacitance of the catalyst. As the material underwent catalysis, PHNC increased the active specific surface area and the exposure of active sites, thus exhibiting higher catalysis.
Electrochemical impedance spectroscopy (EIS) was next performed focused on the electrical conductivity and charge-transfer kinetics of the electrodes. For the collection of the EIS spectrum, it was obtained at a potential of −0.15 V, amplitude of 0.005 V, and frequency of 0.005 Hz to 1000 Hz. Fig. 3e shows the EIS measurements of Fe3O4/CuNCs/ZnNx-PHNC, Fe3O4/ZnNx-PHNC, CuNCs/ZnNx-PNC, and ZnNx-PNC fitted to the simulated equivalent circuit diagram based on the shape of the curve. The curves had semicircular and linear parts, so the simulation yielded the main components of the circuit consisting of Rs, Rct, Cdl, and Wo, where Rs is the electrolyte internal resistance, which is the sum of the material's internal resistance and the contact resistance between the material and the electrolyte surface, while Rct is the charge-transfer resistance, expressed by the radius of the fitted semicircle. According to the fitted impedance values of the circuit diagram (Table S8, ESI†), Fe3O4/CuNCs/ZnNx-PHNC had the least resistance and the fastest charge transfer during the charge and material transfers. The accelerated charge transfer could be attributed to the following: (1) the doping of Fe3O4 and Cu nanoparticles increased the material's conductivity; (2) the skeletonized nanocage structure accelerated O2 diffusion, thereby shortening the electron-transport distance; (3) the graphitized carbon and abundant carbon defects after sintering further accelerated the electron transport.57
Methanol tolerance and stability are important indicators for evaluating the catalytic activity and reproducibility of materials. Fig. S10a–c (ESI†) show that the current of Fe3O4/CuNCs/ZnNx-PHNC decreased by 2% after adding different molar concentrations of methanol, albeit the current normalized quickly. The curves of Pt/C further indicated that the current did not return to the original current after a 5% drop. Such a high tolerance and stability of Fe3O4/CuNCs/ZnNx-PHNC was mainly attributed to the stable skeletonized nanocages and the increased graphitization. Fig. 3f shows the durability of Fe3O4/CuNCs/ZnNx-PHNC and Pt/C. After 5000 CV cycles, the ultimate current density decreased by 5% and 6.9% for Fe3O4/CuNCs/ZnNx-PHNC and Pt/C, respectively. Accordingly, the onset electricity of Fe3O4/CuNCs/ZnNx-PHNC and Pt/C decreased by 6 and 20 mV, respectively, and the half-wave potential depreciated by 8 and 24 mV, respectively. These values proved that the skeletonized nanocage structure and the degree of graphitization in Fe3O4/CuNCs/ZnNx-PHNC were essential for enhancing the stability.58
3.3. Liquid and solid-state Zn–air battery performances of the catalysts
To verify the practical application of the material, we constructed a ZAB with a polished zinc plate as the anode and Fe3O4/CuNCs/ZnNx-PHNC material-coated nickel foam as the cathode (Fig. 4a). The alkaline electrolyte was 6 M KOH + 0.2 M Zn(CH3COO)2. The Fe3O4/CuNCs/ZnNx-PHNC assembled cell showed relatively strong stability. The inset in Fig. 4b depicts the actual test of the open-circuit voltage of the cell using a multimeter (OCV = 1.45 V). The multiplier performance of the battery was tested in the multi-current steps mode. Fig. S11 (ESI†) shows that the voltage changed at the beginning when various current densities were applied. However, the voltage was normal at the same current density, indicating that the material had excellent stability and multiplier performance.59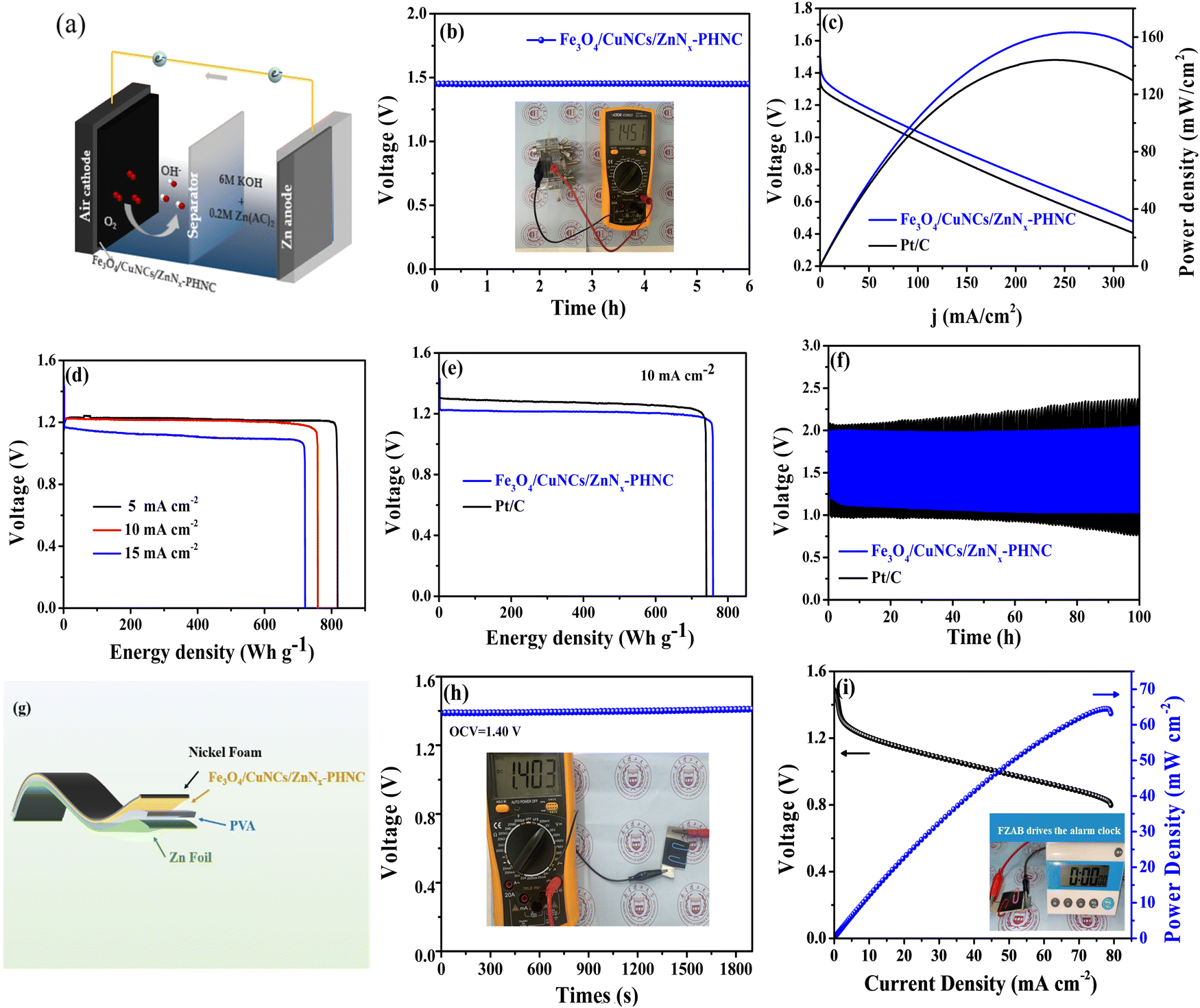 | ||
| Fig. 4 (a) Schematic of the assembled ZAB; (b) open-circuit voltage of the Fe3O4/CuNCs/ZnNx-PHNC-based ZAB (inset is the actual measurement using the universal meter). (c) Polarization curves and power density curves of Fe3O4/CuNCs/ZnNx-PHNC and Pt/C-based ZABs. (d) Specific capacity curves of Fe3O4/CuNCs/ZnNx-PHNC-based ZABs at different current densities. (e) Specific capacity curves of Fe3O4/CuNCs/ZnNx-PHNC and Pt/C-based ZABs at a current density of 10 mA cm−2. (f) Charge/discharge curves of Fe3O4/CuNCs/ZnNx-PHNC and Pt/C-based ZABs at a current density of 10 mA cm−2. (g) Schematic of a FZAB. (h) OCV plots (inset: multimeter measurement picture). (i) Polarization and power density curves (inset: FZAB powering an alarm clock). | ||
The power densities of the Fe3O4/CuNCs/ZnNx-PHNC and Pt/C-based ZABs were 162 and 144 mW cm−2, respectively (Fig. 4c). Comparing the power densities suggests that the Fe3O4/CuNCs/ZnNx-PHNC-based assembled cells were significantly stronger than the Pt/C-based ones in terms of the discharge. The discrepancy could be ascribed to the superior ORR from the Fe3O4/CuNCs/ZnNx-PHNC catalytic activity.60Fig. 4d illustrates the specific capacities of Fe3O4/CuNCs/ZnNx-PHNC at 5, 10, and 15 mA cm−2 as 818, 760, and 721 mA h g−1, respectively. Comparing the specific capacities of Fe3O4/CuNCs/ZnNx-PHNC and Pt/C at a 10 mA cm−2 discharge density (Fig. 4e) showed that Fe3O4/CuNCs/ZnNx-PHNC had superior ORR activity over Pt/C. Further, Fig. 4f depicts a 30 min discharge and 30 min charge cycle test for Fe3O4/CuNCs/ZnNx-PHNC and Pt/C-based catalyst cells at 10 mA cm−2. After 100 h of cycling, the Pt/C-based ZAB showed an unstable charging and discharging voltage plateau, while Fe3O4/CuNCs/ZnNx-PHNC maintained a stable charging and discharging voltage. Fig. S12a (ESI†) shows a stable charge/discharge plateau of the cell for 500 h at a current density of 5 mA cm−2. A stable charge/discharge was also maintained at 15 mA cm−2 for up to 200 h (Fig. S12b, ESI†). When the current density is too high, the durability of the battery will decrease, and the charging and discharging platforms will increase. Therefore, when the current density reached up to 20 mA cm−2, the charge/discharge test was conducted for 10 min each, and the battery maintained an excellent plateau for 500 cycles (Fig. S12c, ESI†). The combined charge/discharge tests at various current densities revealed that the Fe3O4/CuNCs/ZnNx-PHNC-based battery had excellent stability, mainly due to the skeletonized nanocage structure and abundant active sites of the material.61 By comparing recent articles (Table S9, ESI†), the Fe3O4/CuNCs/ZnNx-PHNC-based ZABs evinced a relatively impressive performance compared to that of other reported systems.
To further research into the potential of the rechargeable ZABs and their application in the increasingly attractive wearable electronic devices, we assembled FZABs and tested their performance. Among them, a 0.1 mm zinc plate was used as the anode, a PVA gel polymer as the electrolyte, and a nickel foam-loaded Fe3O4/CuNCs/ZnNx-PHNC as the air cathode (Fig. 4g). After a long-time open-circuit voltage test, we observed that the open-circuit voltage of the assembled FZAB was stable at 1.40 V (Fig. 4h), as proven by the attached multimeter picture. Fig. 4i illustrates that it demonstrated an excellent peak power density of 64.5 mW cm−2. To ascertain its practical usability, despite the adequate data support, we assembled a battery-driven LED electronic screen alarm clock. Here, we found that it could be used seamlessly, showing its future commercial prospects and practical applicability (video 1, ESI†).
4. Conclusions
A heterogeneous skeletonized nanocage catalyst Fe3O4/CuNCs/ZnNx-PHNC loaded with bimetals was prepared by stirring at room temperature and annealing at high temperature with NaCl and KCl as protectants and stripping agents, respectively. The rich active sites produced by the synergistic action of various metals and hollow nanostructures enabled the catalysts to show excellent ORR performance. When the prepared materials were used as liquid ZAB cathode catalysts, the ZABs exhibited excellent performance with a high OCV (1.45 V) and peak power density (162 mW cm−2). In addition, the ultrahigh specific capacity (760 mA h g−2) at a 10 mA cm−2 current density was far superior to that of the benchmark Pt/C-based catalyst. In addition, the Fe3O4/CuNCs/ZnNx-PHNC-based ZAB could be charged and discharged for more than 500 h at a 5 mA cm−2 current density, thus demonstrating high stability and high cycling performance. The excellent performance of the assembled FAZBs also proved their practical application potential as wearable flexible batteries. Overall, this study provides a feasible way to prepare efficient catalysts for ZAB cathodes to replace noble metal catalysts in practical applications.Data availability
The data supporting this article have been included as part of the ESI,† and in the Experimental section of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The Jiangsu Provincial Key Research and Development Program (BE2023820, BE2021720), Qinglan Project of Jiangsu Province of China, State Key Laboratory of Pollution Control and Resource Reuse Foundation (PCRRF19032) are greatly acknowledged.References
- Y. Jiao, X. Gu, P. Zhai, Y. Wei, W. Liu, Q. Chen, Z. Yang, J. Zuo, L. Wang, T. Xu and Y. Gong, Nano Lett., 2022, 22, 7386–7393 CrossRef CAS PubMed
.
- N. Shang, K. Wang, M. Wei, Y. Zuo, P. Zhang, H. Wang, Z. Chen and P. Pei, J. Mater. Chem. A, 2022, 10, 16369–16389 RSC
- M. Katsaiti, E. Papadogiannis, V. Dracopoulos, A. Keramidas and P. Lianos, J. Power Sources, 2023, 555, 232384 CrossRef CAS
- A. Wang, X. Zhang, S. Gao, C. Zhao, S. Kuang, S. Lu, J. Niu, G. Wang, W. Li, D. Chen, H. Zhang, X. Zhou, S. Zhang, B. Zhang and W. Wang, Adv. Mater., 2022, n/a, 2204247 CrossRef PubMed
- X. Chen, J. Pu, X. Hu, Y. Yao, Y. Dou, J. Jiang and W. Zhang, Small, 2022, 18, 2200578 CrossRef CAS PubMed
- W. Zhu, H. Liu, R. Yue, Y. Pei, J. Zhang, X. Liu, R. Li, Y. Yin and M. D. Guiver, Small, 2022, 18, 2202660 CrossRef CAS PubMed
- Y. Rao, W. Li, S. Chen, Q. Yue, Y. Zhang and Y. Kang, Small, 2022, 18, 2104411 CrossRef CAS PubMed
- A. Yu, Z. Peng, Y. Li, L. Zhu, P. Peng and F.-F. Li, ACS Appl. Mater. Interfaces, 2022, 14, 42337–42346 CrossRef CAS PubMed
- S. Huang, H. Zhang, J. Zhuang, M. Zhou, M. Gao, F. Zhang and Q. Wang, Adv. Energy Mater., 2022, 12, 2103622 CrossRef CAS
- S. Wu, D. Deng, E. Zhang, H. Li and L. Xu, Carbon, 2022, 196, 347–353 CrossRef CAS
- L. Xu, S. Wu, X. He, H. Wang, D. Deng, J. Wu and H. Li, Chem. Eng. J., 2022, 437, 135291 CrossRef CAS
- D. Deng, H. Ma, S. Wu, H. Wang, J. Qian, J. Wu, H. Li, C. Yan, H. Li and L. Xu, Renewables, 2023, 1, 362–372 CrossRef
- Y. Sun, X. Liu, M. Zhu, Z. Zhang, Z. Chen, S. Wang, Z. Ji, H. Yang and X. Wang, DeCarbon, 2023, 2, 100018 CrossRef
- H. Liu, F. Yu, K. Wu, G. Xu, C. Wu, H.-K. Liu and S.-X. Dou, Small, 2022, 18, 2106635 CrossRef CAS PubMed
- H. Yang, H. Huang, Q. Wang, L. Shang, T. Zhang and S. Wang, J. Mater. Chem. A, 2023, 11, 6191–6197 RSC
- D. Deng, J. Qian, X. Liu, H. Li, D. Su, H. Li, H. Li and L. Xu, Adv. Funct. Mater., 2022, 32, 2203471 CrossRef CAS
- W. Li, J. Wang, J. Chen, K. Chen, Z. Wen and A. Huang, Small, 2022, 18, 2202018 CrossRef CAS PubMed
- Y. Arafat, M. R. Azhar, Y. Zhong, H. R. Abid, M. O. Tadé and Z. Shao, Adv. Energy Mater., 2021, 11, 2100514 CrossRef CAS
- Q. Yu, J. Lv, J. Li, R. Yu, J. Wu, S. Xi, X. Li, N. Xu, L. Zhou and L. Mai, Energy Environ. Mater., 2023, 6, e12389 CrossRef CAS
- Y. Pan, Q. Yang, F. Qiu, X. Liu, C. Shen, Y. Fan, H. Song and S. Zhang, Mater. Today Chem., 2023, 34, 101787 CrossRef CAS
- Y. Pan, X. Ma, M. Wang, X. Yang, S. Liu, H.-C. Chen, Z. Zhuang, Y. Zhang, W.-C. Cheong, C. Zhang, X. Cao, R. Shen, Q. Xu, W. Zhu, Y. Liu, X. Wang, X. Zhang, W. Yan, J. Li, H. M. Chen, C. Chen and Y. Li, Adv. Mater., 2022, 34, 2203621 CrossRef CAS PubMed
- L. Deng, L. Qiu, R. Hu, L. Yao, Z. Zheng, X. Ren, Y. Li and C. He, Appl. Catal., B, 2022, 305, 121058 CrossRef CAS
- W. Wang, Q. Jia, S. Mukerjee and S. Chen, ACS Catal., 2019, 9, 10126–10141 CrossRef CAS
- T. Sun, S. Mitchell, J. Li, P. Lyu, X. Wu, J. Perez-Ramirez and J. Lu, Adv. Mater., 2021, 33, e2003075 CrossRef PubMed
- Y. Cheng, M. Wang, S. Lu, C. Tang, X. Wu, J.-P. Veder, B. Johannessen, L. Thomsen, J. Zhang, S.-Z. Yang, S. Wang and S. P. Jiang, Appl. Catal., B, 2021, 284, 119717 CrossRef CAS
- Y. Shen, Y. Xiao, H. Zhang, H. Fan, Y. Li, Z. Yan and W.-H. Zhang, Chem. Eng. J., 2023, 477, 146823 CrossRef CAS
- M. Tong, F. Sun, Y. Xie, Y. Wang, Y. Yang, C. Tian, L. Wang and H. Fu, Angew. Chem., Int. Ed., 2021, 60, 14005–14012 CrossRef CAS PubMed
- G. Yang, P. Hu, Y. Cao, F. Yuan and R. Xu, Nanoscale Res. Lett., 2010, 5, 1437 CrossRef CAS PubMed
- Y. Liu, H. Jiang, J. Hao, Y. Liu, H. Shen, W. Li and J. Li, ACS Appl. Mater. Interfaces, 2017, 9, 31841–31852 CrossRef CAS PubMed
- H. Hu, B. Guan, B. Xia and X. W. Lou, J. Am. Chem. Soc., 2015, 137, 5590–5595 CrossRef CAS PubMed
- K. Ding, J. Hu, J. Luo, L. Zhao, W. Jin, Y. Liu, Z. Wu, G. Zou, H. Hou and X. Ji, Adv. Funct. Mater., 2022, n/a, 2207331 CrossRef
- A. R. C. Bredar, M. D. Blanchet, A. R. Burton, B. E. Matthews, S. R. Spurgeon, R. B. Comes and B. H. Farnum, ACS Catal., 2022, 12, 3577–3588 CrossRef CAS
- W. da Silva Freitas, A. D’Epifanio, C. Lo Vecchio, I. Gatto, V. Baglio, V. C. A. Ficca, E. Placidi and B. Mecheri, Chem. Eng. J., 2023, 465, 142987 CrossRef CAS
- C. Xuan, B. Hou, W. Xia, Z. Peng, T. Shen, H. L. Xin, G. Zhang and D. Wang, J. Mater. Chem. A, 2018, 6, 10731–10739 RSC
- W. Wang, J. Si, J. Li, Q. Wang and S. Chen, Int. J. Hydrogen Energy, 2016, 41, 16858–16864 CrossRef CAS
- Q. Lv, N. Wang, W. Si, Z. Hou, X. Li, X. Wang, F. Zhao, Z. Yang, Y. Zhang and C. Huang, Appl. Catal., B, 2020, 261, 118234 CrossRef CAS
- K. Yang, Y. Cheng, Y. Lei, Q. Yang, X. Liu, T. Lu, H. Xie, J. Li, T. Zan and H. Wang, J. Solid State Chem., 2024, 329, 124401 CrossRef CAS
- K. Sheng, Q. Yi, L. Hou and A. Chen, J. Electrochem. Soc., 2020, 167, 070560 CrossRef
- J. Gao, Y. Wang, H. Wu, X. Liu, L. Wang, Q. Yu, A. Li, H. Wang, C. Song, Z. Gao, M. Peng, M. Zhang, N. Ma, J. Wang, W. Zhou, G. Wang, Z. Yin and D. Ma, Angew. Chem., Int. Ed., 2019, 58, 15089–15097 CrossRef CAS PubMed
- J. Zemek, J. Houdkova, P. Jiricek and M. Jelinek, Carbon, 2018, 134, 71–79 CrossRef CAS
- Y. Yang, X. Xu, P. Sun, H. Xu, L. Yang, X. Zeng, Y. Huang, S. Wang and D. Cao, Nano Energy, 2022, 100, 107466 CrossRef CAS
- C. Chen, J. Long, K. Shen, X. Liu and W. Zhang, ACS Appl. Mater. Interfaces, 2022, 14, 38677–38688 CrossRef CAS PubMed
- Y. Zuo, G. Wang, J. Peng, G. Li, Y. Ma, F. Yu, B. Dai, X. Guo and C.-P. Wong, J. Mater. Chem. A, 2016, 4, 2453–2460 RSC
- Y. Li, Y. Fu, S. Chen, Z. Huang, L. Wang and Y. Song, Composites, Part B, 2019, 171, 130–137 CrossRef CAS
- P. Sarkar, N. Nandi, N. Barnwal and K. Sahu, Mater. Today Chem., 2023, 27, 101341 CrossRef CAS
- Q. Wang, T. Ina, W.-T. Chen, L. Shang, F. Sun, S. Wei, D. Sun-Waterhouse, S. G. Telfer, T. Zhang and G. I. N. Waterhouse, Sci. Bull., 2020, 65, 1743–1751 CrossRef CAS PubMed
- J. Xu, S. Lai, D. Qi, M. Hu, X. Peng, Y. Liu, W. Liu, G. Hu, H. Xu, F. Li, C. Li, J. He, L. Zhuo, J. Sun, Y. Qiu, S. Zhang, J. Luo and X. Liu, Nano Res., 2020, 14, 1374–1381 CrossRef
- Q. Yang, R. Liu, Y. Pan, Z. Cao, Y. Liu, L. Wang, J. Yu, H. Song, Z. Ye and S. Zhang, J. Colloid Interface Sci., 2022, 628, 691–700 CrossRef CAS PubMed
- H. Yin, H. Xia, S. Zhao, K. Li, J. Zhang and S. Mu, Energy Environ. Mater., 2021, 4, 5–18 CrossRef CAS
- Y. Chen, S. Ji, S. Zhao, W. Chen, J. Dong, W.-C. Cheong, R. Shen, X. Wen, L. Zheng, A. I. Rykov, S. Cai, H. Tang, Z. Zhuang, C. Chen, Q. Peng, D. Wang and Y. Li, Nat. Commun., 2018, 9, 5422 CrossRef CAS PubMed
- K. Yu, K. Sun, W. C. Cheong, X. Tan, C. He, J. Zhang, J. Li and C. Chen, Small, 2023, 19, 2302611 CrossRef CAS PubMed
- B. Eren, D. Zherebetskyy, L. L. Patera, C. H. Wu, H. Bluhm, C. Africh, L.-W. Wang, G. A. Somorjai and M. Salmeron, Science, 2016, 351, 475–478 CrossRef CAS PubMed
- Y. Song, T. Zhang, G. Zhou, P. Liu, X. Yan, B. Xu and J. Guo, Appl. Surf. Sci., 2022, 589, 153022 CrossRef CAS
- K. Yuan, S. Sfaelou, M. Qiu, D. Lützenkirchen-Hecht, X. Zhuang, Y. Chen, C. Yuan, X. Feng and U. Scherf, ACS Energy Lett., 2018, 3, 252–260 CrossRef CAS
- J. Zhang, J. Chen, Y. Luo, Y. Chen, Y. Luo, C. Zhang, Y. Xue, H. Liu, G. Wang and R. Wang, J. Mater. Chem. A, 2021, 9, 5556–5565 RSC
- J. H. Lee, J.-h Jang, J. Kim and S. J. Yoo, J. Ind. Eng. Chem., 2021, 97, 466–475 CrossRef CAS
- Q. Yang, R. Liu, Y. Pan, Z. Cao, J. Zuo, F. Qiu, J. Yu, H. Song, Z. Ye and S. Zhang, ACS Appl. Mater. Interfaces, 2023, 15, 5720–5731 CrossRef CAS PubMed
- L. Li, Y. Li, Y. Xiao, R. Zeng, X. Tang, W. Yang, J. Huang, K. Yuan and Y. Chen, Chem. Commun., 2019, 55, 7538–7541 RSC
- Q. Yang, R. Liu, J. Gao, Y. Liu, S. Hu, H. Song and S. Zhang, ACS Appl. Energy Mater., 2022, 5, 1041–1051 CrossRef CAS
- H. Jiang, J. Gu, X. Zheng, M. Liu, X. Qiu, L. Wang, W. Li, Z. Chen, X. Ji and J. Li, Energy Environ. Sci., 2019, 12, 322–333 RSC
- Y. Li, H. Huang, S. Chen, X. Yu, C. Wang and T. Ma, Nano Res., 2019, 12, 2774–2780 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ya00295d |
| ‡ Y. N. Pan and Q. Yang contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |