
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrochemical aldehyde hydrogenation: probing the inner-sphere strategy with nickel-bipyridine complexes†
Gabriel Durin‡
 ab,
Mijung Lee‡a,
Martina A. Pogany‡a,
Christian Kahla,
Thomas Weyhermüllera,
Walter Leitnerac and
Nicolas Kaeffer*a
ab,
Mijung Lee‡a,
Martina A. Pogany‡a,
Christian Kahla,
Thomas Weyhermüllera,
Walter Leitnerac and
Nicolas Kaeffer*a
aMax Planck Institute for Chemical Energy Conversion, Stiftstrasse 34-36, 45470 Mülheim an der Ruhr, Germany. E-mail: nicolas.kaeffer@cec.mpg.de
bUniversité Grenoble Alpes, DCM, CNRS, 38000 Grenoble, France
cInstitut für Technische und Makromolekulare Chemie, RWTH Aachen University, Worringerweg 2, 52074 Aachen, Germany
First published on 22nd November 2024
Abstract
Developing electrohydrogenation routes for organics is crucial in synthesis electrification. Herein, we examine the electrocatalytic hydrogenation of aldehydes through an inner-sphere mechanism at a nickel-bipyridine complex. An (electro)reduction triggers the coordination of the aldehyde into a key nickeloxirane species, which affords hydrogenation products by stoichiometric protonations. Turnover yet remains challenging with acids suitable for electrocatalytic conditions due to sluggish proton transfers, which we probed by combined reactivity and computational studies.
The electrification of synthetic processes is empowering the use of renewable and sustainable resources in chemistry.1–5 This virtuous approach is often propelled by electrocatalysis to incorporate energy efficiency and selectivity. A key to the rational development of these electrocatalytic systems is the decoding of the mechanisms involved. The well-defined nature of molecular complexes facilitates access to mechanistic information. This strategy has led to molecular electrocatalytic systems for the hydrogenation of organic multiple bonds.6–17 In contrast to CO2 reduction, the electrocatalytic hydrogenation (ECH) of organic carbonyls with molecular complexes remains largely under-explored.18
Siewert and co-workers demonstrated the repurposing of {Mn(bpy)}-type hydrogenation electrocatalysts from CO2 to organic carbonyls, including aliphatic aldehydes,15,16 while Waymouth and co-workers addressed the challenging ECH of benzaldehyde using a Mo Shvo-type complex14 (Scheme 1). These systems perform hydrogenation through the evolution of a metal hydride that is transferred to the electrophilic C–O carbon.
 | ||
| Scheme 1 Electrocatalytic hydrogenation of C–O and C–C multiple bonds by molecular complexes, including nickel-bipyridine complexes. | ||
In our exploration of electrocatalyzed synthesis, we recently employed the coordination ability of nickel-bipyridine complexes to achieve alkyne ECH through successive electron and proton transfers, bypassing metal hydride intermediates.19,20 This approach exploits hydride-free selective pathways that are at odds with catalysis in thermal hydrogenation and reported ECHs. Herein, we investigate the challenges in transposing the hydride-free strategy from the hydrogenation of C–C multiple bonds to that of C–O bonds (Scheme 1).
Knowing that [Ni(bpy)(BzO)2] (1) is a hydrogenation electrocatalyst for alkyne substrates,20 we aimed at probing this complex for carbonyl substrates.
However, electrolysis of benzaldehyde (S1) in the presence of 1 and benzoic acid (BzOH) as a proton source leads to marginal conversion and no detected hydrogenation product S1H2, once approximately 2 electrons were passed per S1 (Scheme 2A). Varying the solvent (e.g., DMSO, THF) or even the substrate (p-MeO-PhCHO) was not productive. Moreover, greater cathodic applied potential (see ESI,† Section S3.4) or higher acidity (e.g., [DMFH]OTf)21 trigger the direct reduction of S1 at the electrode, and hence are not compatible with this system.
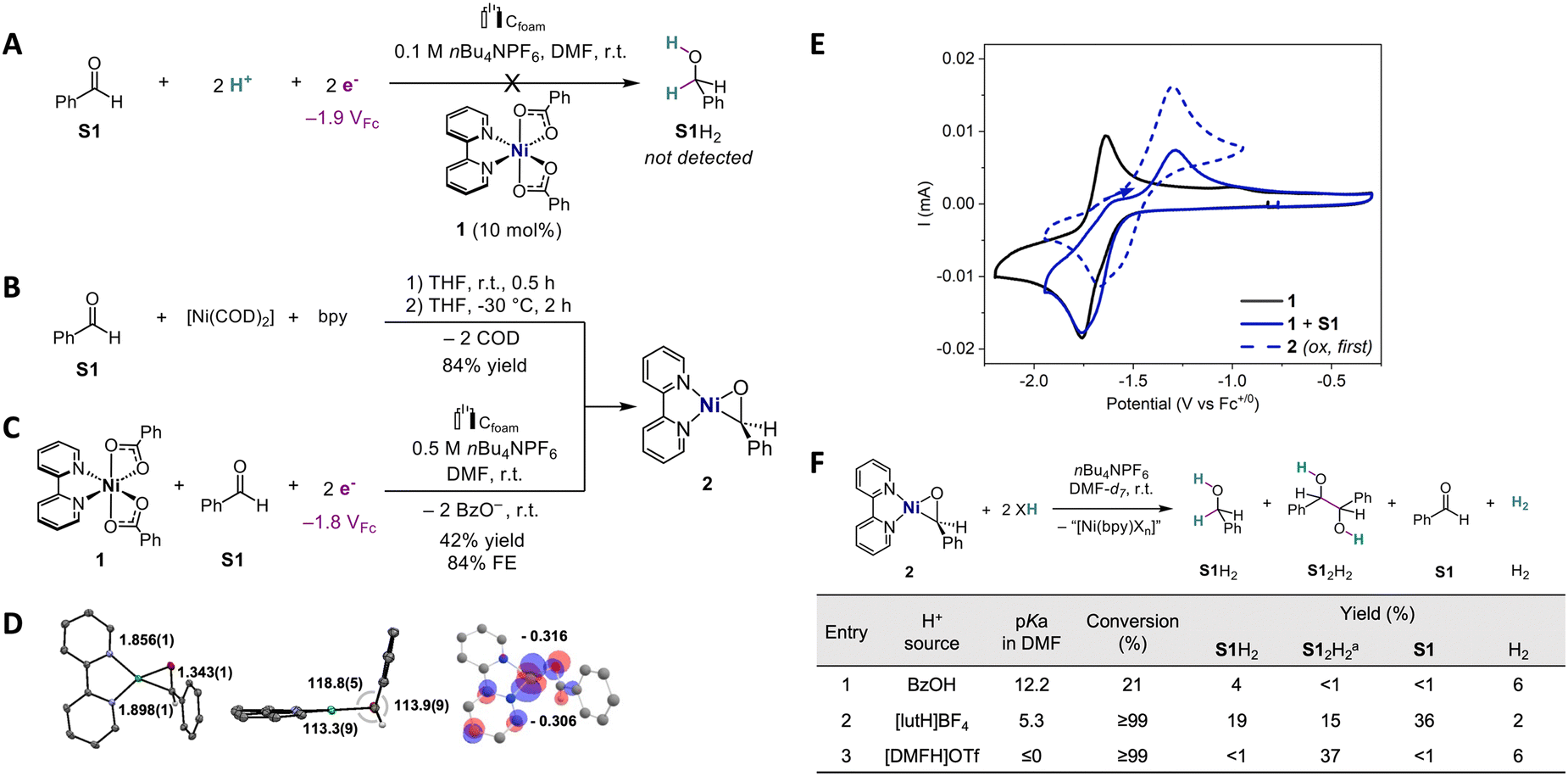 | ||
Scheme 2 (A) Electrocatalytic attempt using 1 (1 mM), S1 (10 mM), and BzOH (22 mM) after passing 8.5C (approximately 2e−/S1). (B) Synthesis of 2 from [Ni(COD)2]. (C) Electrosynthesis of 2 from 1 at Eapp = −1.80 VFc (charge = 4.56C; approximately 1e−/1). (D) Molecular structure of 2 obtained by XRD (ORTEP; 50% probability; color code: gray: C; purple: N; green: Ni; red: O; white: H) and computed electron density of the HOMO of 2 with Mulliken charges at aldehydic C and O (H atoms omitted for clarity, except aldehydic ones). (E) CVs of 1, 1 and S1, and 2 (1 mM each; 0.1 V s−1 scan rate). F Protonation reactions starting from 2 (at 30 min). Unless otherwise stated, the supporting electrolyte is DMF 0.1 M nBu4NPF6. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Maximum theoretical yield 50%. Maximum theoretical yield 50%. | ||
In the absence of catalysis, it is thus questionable whether a pathway involving a nickelacyclic intermediate, in analogy to the nickelacyclopropene reported for alkyne semi-hydrogenation, is viable in the case of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond hydrogenation. We examined the formation of such species in the case of C–O unsaturations by reacting a 1:1 mixture of [Ni(COD)2] (COD = 1,4-cyclooctadiene) and 2,2′-bipyridine with benzaldehyde (S1) in THF, inspired by previous studies and our precedents.20,22,23 The reaction quickly evolves a green precipitate identified after the workup as nickeloxirane [Ni(bpy)(PhCHO)]22–24 (2), obtained in 84% yield (Scheme 2B and ESI,† Section S2.1).
O bond hydrogenation. We examined the formation of such species in the case of C–O unsaturations by reacting a 1:1 mixture of [Ni(COD)2] (COD = 1,4-cyclooctadiene) and 2,2′-bipyridine with benzaldehyde (S1) in THF, inspired by previous studies and our precedents.20,22,23 The reaction quickly evolves a green precipitate identified after the workup as nickeloxirane [Ni(bpy)(PhCHO)]22–24 (2), obtained in 84% yield (Scheme 2B and ESI,† Section S2.1).
The molecular structure was elucidated by single crystal X-ray diffraction (XRD), and indicates a planar geometry for nickeloxirane 2 (Scheme 2D; ESI,† Section S2.4) with a C–O bond length of 1.343 Å, which was quite elongated relative to S1 (1.210 Å).25 Moreover, the 1H NMR signature (DMSO-d6) attributed to the aldehydic hydrogen of 2 is manifested by a broad singlet at 5.05 ppm, in a region typical of benzylic resonances (vs. 10.02 ppm in free S1). Additionally, the C–O stretching frequency at 1360 cm−1 in 2 undergoes a substantial redshift compared to free S1 (ν(CO) = 1693 cm−1; see ESI†, Section 2.3). This spectroscopic evidence further reinforces the C–O single-bond characteristic in 2.
Investigating the voltammetric behavior of 1 shows that the addition of aldehyde S1 (1 equiv.) triggers the reduction wave at Ep,c = −1.76 VFc20 (VFc stands for V vs. Fc+/0) to evolve a shoulder (E ≈ −1.70 VFc) and lose its reversibility (Scheme 2E). Most importantly, these changes are accompanied by the buildup of an anodic wave at Ep,a = −1.29 VFc, matching that observed for the oxidation of native 2. These points suggest that 2 can also be formed by reductive electrogeneration from 1 and S1. Bulk reductive electrolysis of a mixture of 1 and S1 at Eapp = −1.80 VFc (1/S1 1:1 ratio, approximately 1e−/1 at saturation) (Scheme 2C) further confirms the electrogeneration of 2 in 42% yield and 84% faradaic efficiency, as revealed by the characteristic voltammetric and 1H NMR signatures (ESI,† Section S4.4).
While these results demonstrate the binding and activation of S1 in nickelacyclic species 2, including under electroreductive conditions, protonation is required to release the hydrogenated product and induce turnover. After 30 min, addition of 2 equivalents of BzOH (pKa 12.2 in DMF26) to 2 dissolved in DMF-d7 and nBu4NPF6 20 mM (r.t.) affords 21% conversion of 2 and a 4% yield in S1H2 with H2 as a byproduct (6% yield) (Scheme 2F; entry 1). Extending the reaction time to 24 h leads to full conversion and S1H2 in 17% yield (see ESI,† Section S4.1 for additional details). These results indicate the slow reactivity of BzOH with 2, leading to hydrogenation into S1H2 and the hydrogen evolution reaction (HER). The latter reactivity suggests the formation of nickel hydride species, while the former can also proceed by protonations of the metallacycle as observed with alkynes.20
To determine if stronger acids would accelerate the reaction or promote one reaction pattern over the other, we tested 2,6-lutidinium (lutH+ pKa 5.3 in DMF;27,28 Scheme 2F; entry 2). After 30 min, 2 is fully converted and affords S1H2 in 19% yield and low hydrogen evolution (2%). It is worth noting two additional points. First, the pinacol coupling product hydrobenzoin (S12H2) is detected in substantial yields (15%). Second, significant amounts of unreacted S1 are released (36%), which indicates the displacement of that substrate from the Ni center.
We surmise that this expulsion is induced by the coordination of the proton source at Ni, potentially leading to [Ni(bpy)(lut)(H)]+, a hydride species. That hypothesis is reinforced by the observation of a 1H NMR signal at −21.74 ppm (Fig. S14, ESI†), which was attributed to such Ni–H hydride compounds by comparison with literature data.29,30 Evidence consisting of the concomitant observation of the Ni–H species and free S1 indicates that this hydride is not highly reactive towards the aldehyde. In the case of the more acidic DMFH+ (pKa ≤0 in DMF26,28), the selectivity in S1-derived products is now fully switched to S12H2 (Scheme 2F; entry 3), while Ni–H is also detected (Fig. S14, ESI†). The increased selectivity for the product S12H2 when Ni–H is detected suggests 1e−/1H+ reactivity31 of these hydrides.
We further explored the conditions for hydrogenation (see ESI,† Sections S4.1–S4.3). With 2, the use of alternative Brønsted acids does not increase yields in hydrogenated products, and Lewis acids do not afford hydrogenation. Regarding ligands, the more electron-poor 4,4-bis(trifluoromethyl)-2,2′-bipyridine fails to generate an aldehyde adduct, but a nickeloxirane forms when electron-donating 4,4-bis(methoxy)-2,2′-bipyridine is used, which in turn leads to similar protonation results. The combination of 2 and lutH+ provides the highest yield of S1H2.
These results indicate that the (electro)reductive generation of the nickelacyclic species 2 from S1 is feasible, but subsequent reactivity is undermined by competing hydride formation or slow protonation. While lutH+ or DMFH+ afford fast conversion of 2 into the hydrogenation products of S1, these proton sources are too acidic for the investigated electrocatalytic potentials. In contrast, the less acidic BzOH appropriate to ECH of alkyne leads to sluggish reactivity.
To further uncover the mechanistic limitations and the underlying distinctions of the operating alkyne ECH case, we employed DFT calculations (Scheme 3). We considered an initiation from 1 by 2-electron reduction associated with the release of a benzoate ligand, leading to I1 as previously described,20 and used this as the reference entry point. The binding of S1 to I1 by associative displacement of BzO− is kinetically facile (TSI1-2 at +2.8 kcal mol−1) and highly favored, leading to nickeloxirane 2 as the most stable computed intermediate (ΔG = −23.8 kcal mol−1). We note that, aside from being a relevant species by electrochemical access, 2 is also the starting material in our stoichiometric experiments. We then investigated protonation at the substrate sites in 2, namely at the C or the O atom in the nickelacycle.
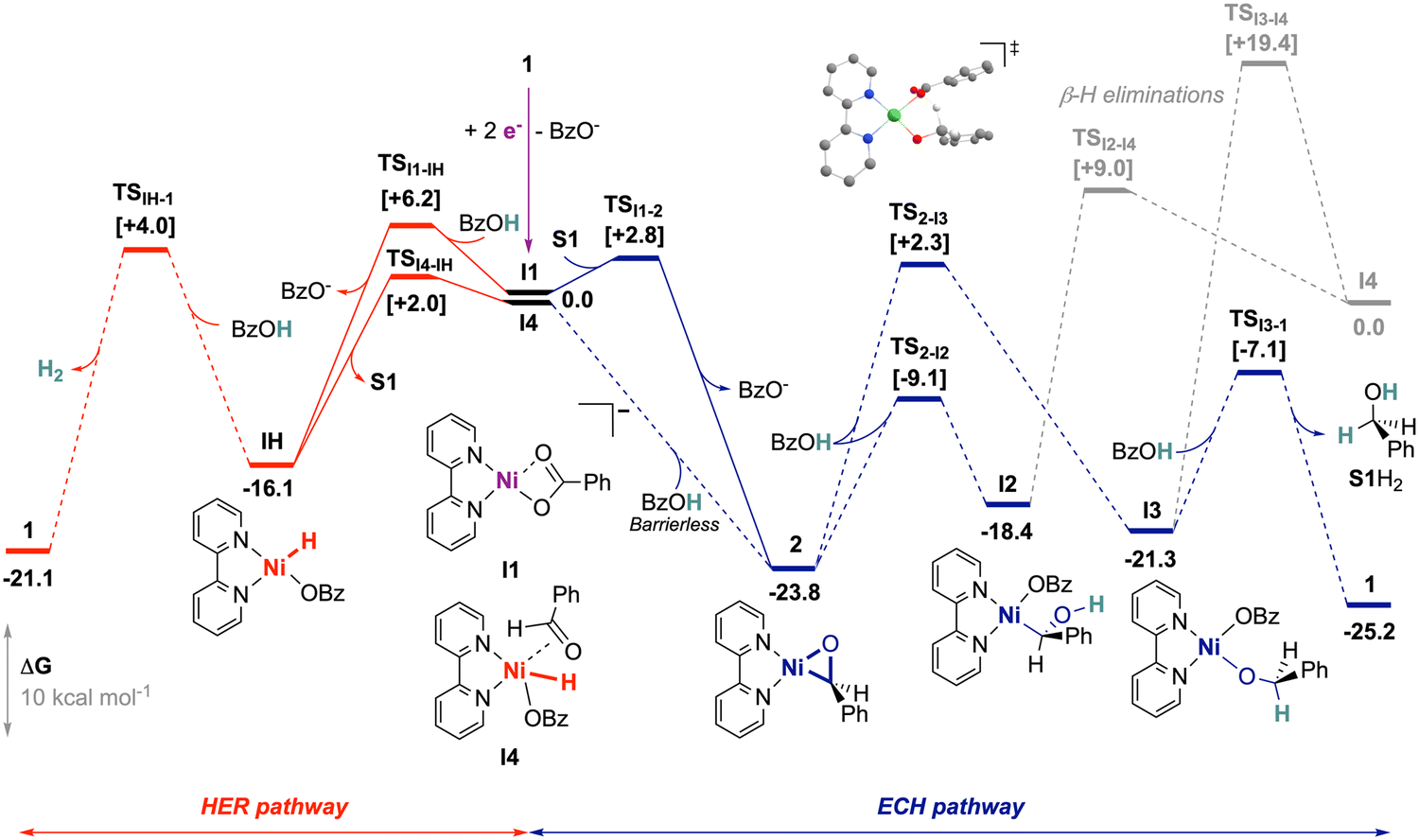 | ||
| Scheme 3 Computed HER (red)20 and ECH (blue) pathways at the PBE-D3/6-311+G(d,p) level of theory using the CPCM model to account for the solvent effect of DMF. | ||
There is a reachable transition state (TS) for the calculated O-protonation (TS2-I2 at ΔΔG‡ = +14.7 kcal mol−1), but the formation of the nickel-alkyl species I2 is endergonic (ΔΔG = +5.4 kcal mol−1). Furthermore, this intermediate appears unproductive because neither protonation nor isomerization seems plausible (see ESI,† Section S5.2). C-protonation is computed to afford a relatively stable benzolate complex I3 (ΔG = −21.3 kcal mol−1), from which a second O-protonation is feasible to release the desired product S1H2. However, the initial C-protonation is hindered by high energy cost at TS2-I3 (ΔΔG‡ = +26.1 kcal mol−1). It was noted that β-H elimination, which would have contributed to the hydride pathway (HER) from I2 and I3, is unlikely, with TSs >27 kcal mol−1. Consequently, the computational results support the difficulty of protonations of 2 by BzOH and, within computational uncertainty, are close to feasibility limits at RT (>23 kcal mol−1), further corroborating the slow and understoichiometric evolution of S1H2 observed in chemical conditions.
We next turned to the formation of hydride species to trace the HER with our system. The electrogeneration of I1 from precatalyst 1 can lead to a slow HER via the hydride complex IH, as previously described.20 However, I1 is not relevant in stoichiometric experiments with 2, yet it produces H2. In that case, we found that the oxidative addition of BzOH at 2 leads to hydride I4 without barriers, thus restoring the occurrence of the HER. The span of that pathway is relatively high (+25.8 kcal mol−1), but it cannot be fully discarded within the uncertainty of the calculations, and hence can possibly account for H2 produced.
For the formation of the hydrobenzoin S12H2, a radical pathway resulting from the homolytic cleavage of the Ni–C bond of I2 (BDE = 34.6 kcal mol−1) seems reasonable. Another pathway involving ring expansion by addition of S1 to 2 proved to be too high in energy (>29 kcal mol−1; see ESI,† Section S5.2).
S1 binding into 2 and IH hydride generation are expected to rapidly occur from I1, with respective computed TSs of 2.8 and 6.2 kcal mol−1, but 2 is strongly stabilized versus IH by 7.7 kcal mol−1. The accumulation of the nickelaoxirane is therefore plausible in the hypothesis of slow subsequent kinetics supported by the elevated spans. In this regard, in the voltammetry of 1 recorded with excess BzOH, the addition of 1 equivalent of S1 restores the electrochemical signature of 2 (Fig. S9, ESI†). This displacement of the electrochemical systems towards 2 even in the presence of BzOH corroborates the thermodynamic preference of the nickelaoxirane versus the hydride species and further shows that S1 inhibits the HER. We note that this relative stability with respect to IH is lower for nickeloxirane as compared to nickelacyclopropene20 (ΔG(metallacycle → IH) = 7.7 vs. 13.9 kcal mol−1), which supports a more accessible nickel hydride in the case of aldehyde as a substrate.
In summary, in an approach to transpose a hydride-free electrocatalytic hydrogenation from C–C to C–O π-bonds, we demonstrated the successful (electro)generation of a key nickeloxirane species, leading to hydrogenation products by stoichiometric protonation. However, identifying efficient electrocatalytic conditions remains challenging. We determined that inner-sphere substrate binding is preferred over hydride formation, but subsequent protonations of the nickelacyclic species are obstructive. These findings will guide further molecular catalysis for carbonyl electrohydrogenation.
The authors gratefully acknowledge basic support from the Max Planck Society and the RWTH Aachen University. They thank Annika Gurowski, Alina Jakubowski and Justus Werkmeister for assistance with analytical measurements. Open Access funding provided by the Max Planck Society.
Data availability
The data supporting this article are included as part of the ESI.† The crystallographic data for 2 have been deposited at the CCDC under number 2374778.Conflicts of interest
There are no conflicts to declare.Notes and references
- A. Wiebe, T. Gieshoff, S. Mohle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 CrossRef CAS PubMed.
- N. Kaeffer and W. Leitner, JACS Au, 2022, 2, 1266–1289 CrossRef CAS PubMed.
- N. von Wolff, O. Rivada-Wheelaghan and D. Tocqueville, ChemElectroChem, 2021, 8, 4019–4027 CrossRef CAS.
- G. Centi, S. Perathoner, C. Genovese and R. Arrigo, Chem. Commun., 2023, 59, 3005–3023 RSC.
- Z. Liu, L. Zhang, Z. Ren and J. Zhang, Chem. – Eur. J., 2023, 29, e202202979 CrossRef CAS PubMed.
- J. Derosa, P. Garrido-Barros and J. C. Peters, J. Am. Chem. Soc., 2021, 143, 9303–9307 CrossRef CAS PubMed.
- J. Derosa, P. Garrido-Barros, M. Li and J. C. Peters, J. Am. Chem. Soc., 2022, 144, 20118–20125 CrossRef CAS PubMed.
- S. Gnaim, A. Bauer, H.-J. Zhang, L. Chen, C. Gannett, C. A. Malapit, D. E. Hill, D. Vogt, T. Tang, R. A. Daley, W. Hao, R. Zeng, M. Quertenmont, W. D. Beck, E. Kandahari, J. C. Vantourout, P.-G. Echeverria, H. D. Abruna, D. G. Blackmond, S. D. Minteer, S. E. Reisman, M. S. Sigman and P. S. Baran, Nature, 2022, 605, 687–695 CrossRef CAS PubMed.
- X. Wu, C. N. Gannett, J. Liu, R. Zeng, L. F. T. Novaes, H. Wang, H. D. Abruña and S. Lin, J. Am. Chem. Soc., 2022, 144, 17783–17791 CrossRef CAS PubMed.
- I. M. F. De Oliveira and J.-C. Moutet, J. Mol. Catal., 1993, 81, L19–L24 CrossRef CAS.
- J.-C. Moutet, L. Yao Cho, C. Duboc-Toia, S. Ménage, E. C. Riesgo and R. P. Thummel, New J. Chem., 1999, 23, 939–944 RSC.
- H. Shimakoshi, Z. Luo, K. Tomita and Y. Hisaeda, J. Organomet. Chem., 2017, 839, 71–77 CrossRef CAS.
- M. H. Rønne, D. Cho, M. R. Madsen, J. B. Jakobsen, S. Eom, É. Escoudé, H. C. D. Hammershøj, D. U. Nielsen, S. U. Pedersen, M.-H. Baik, T. Skrydstrup and K. Daasbjerg, J. Am. Chem. Soc., 2020, 142, 4265–4275 CrossRef PubMed.
- K. C. Armstrong and R. M. Waymouth, Organometallics, 2020, 39, 4415–4419 CrossRef CAS.
- I. Fokin and I. Siewert, Chem. – Eur. J., 2020, 26, 14137–14143 CrossRef CAS PubMed.
- I. Fokin, K.-T. Kuessner and I. Siewert, ACS Catal., 2022, 12, 8632–8640 CrossRef CAS.
- D. P. Marron, C. M. Galvin, J. M. Dressel and R. M. Waymouth, J. Am. Chem. Soc., 2024, 146, 17075–17083 CrossRef CAS PubMed.
- G. Durin, N. Kaeffer and W. Leitner, Curr. Opin. Electrochem., 2023, 41, 101371 CrossRef CAS.
- M. Y. Lee, C. Kahl, N. Kaeffer and W. Leitner, JACS Au, 2022, 2, 573–578 CrossRef CAS PubMed.
- G. Durin, M. Y. Lee, M. A. Pogany, T. Weyhermuller, N. Kaeffer and W. Leitner, J. Am. Chem. Soc., 2023, 145, 17103–17111 CrossRef CAS PubMed.
- M. Chandrasekaran, M. Noel and V. Krishnan, J. Electroanal. Chem., 1991, 303, 185–197 CrossRef CAS.
- E. Dinjus, I. Gorski, H. Matschiner, E. Uhlig and D. Walther, Z. Anorg. Allg. Chem., 1977, 436, 39–46 CrossRef CAS.
- E. Dinjus, H. Langbein and D. Walther, J. Organomet. Chem., 1978, 152, 229–237 CrossRef CAS.
- We note that the synthesis of 2 had been previously reported in ref. 23, but with partial characterization (elemental analysis, infrared and ultraviolet-visible spectroscopies).
- K. B. Borisenko, C. W. Bock and I. Hargittai, J. Phys. Chem., 1996, 100, 7426–7434 CrossRef CAS.
- V. Fourmond, P. A. Jacques, M. Fontecave and V. Artero, Inorg. Chem., 2010, 49, 10338–10347 CrossRef CAS PubMed.
- A. Kütt, S. Tshepelevitsh, J. Saame, M. Lõkov, I. Kaljurand, S. Selberg and I. Leito, Eur. J. Org. Chem., 2021, 1407–1419 CrossRef.
- L. Sooväli, I. Kaljurand, A. Kütt and I. Leito, Anal. Chim. Acta, 2006, 566, 290–303 CrossRef.
- J. Breitenfeld, O. Vechorkin, C. Corminboeuf, R. Scopelliti and X. Hu, Organometallics, 2010, 29, 3686–3689 CrossRef CAS.
- A. K. Ravn, M. B. Johansen and T. Skrydstrup, Angew. Chem., Int. Ed., 2022, 61, e202112390 CrossRef CAS PubMed.
- M. J. Chalkley, P. Garrido-Barros and J. C. Peters, Science, 2020, 369, 850–854 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2374778. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc04050c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |