
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient pathways to improve electrode performance of P′2 Na2/3MnO2 for sodium batteries†
Yosuke
Ugata‡
 ab,
Tomohiro
Kuriyama‡
a and
Naoaki
Yabuuchi
*ab
ab,
Tomohiro
Kuriyama‡
a and
Naoaki
Yabuuchi
*ab
aDepartment of Chemistry and Life Science, Yokohama National University, 79-5 Tokiwadai, Hodogaya-ku, Yokohama 240-8501, Japan. E-mail: yabuuchi-naoaki-pw@ynu.ac.jp
bAdvanced Chemical Energy Research Center (ACERC), Institute of Advanced Sciences, Yokohama National University, 79-5 Tokiwadai, Hodogaya-ku, Yokohama 240-8501, Japan
First published on 29th November 2024
Abstract
A Mn-based sodium-containing layered oxide, P′2-type Na2/3MnO2, is revisited as a positive electrode material for sodium-ion batteries, and factors affecting its electrochemical performances are examined. The cyclability of Na2/3MnO2 is remarkably improved by increasing the lower cut-off voltage during cycling even though the reversible capacity is sacrificed. Furthermore, the use of highly concentrated electrolytes, in which the presence of free solvent molecules is eliminated, effectively suppresses the dissolution of Mn ions, thus enabling stable cycling with >85% capacity retention for 300 continuous cycles.
In recent years, the rapid growth in the market of electric vehicles has resulted in an increase in the prices for lithium, cobalt, and nickel resources used in lithium-ion batteries. Therefore, great research effort has been conducted to explore new battery systems based on more abundant resources with lower costs. Recently, rechargeable sodium-ion batteries have been attracting much attention for energy storage applications in terms of abundant resources. In the past decade, numerous research efforts have been devoted to developing cost-effective and high-performance electrode materials, especially for positive electrodes, made from earth abundant elements.1–3 Among a variety of potential positive electrode materials, layered sodium transition metal oxides (NaxMeO2, Me = transition metal ions) have been intensively studied.4,5 From the viewpoint of material abundance and energy density, Mn-based layered oxides (NaxMnO2) are considered to be promising positive electrode candidates. The structure of layered NaxMnO2 is divided into two groups with different layered stacking manners, O3-type and P2-type, based on Delmas's notation.6 Here, the letter “O” or “P” denotes the octahedral or prismatic site accommodating Na ions between MnO2 slabs, and the number “2” or “3” represents the number of MnO2 slabs in a cell unit. An in-plane distorted O3-type, denoted as O′3-type, NaMnO2 delivers a relatively large reversible capacity approaching 200 mA h g−1.7 However, the reversible capacity fades upon cycling due to the dissolution of Mn ions into electrolyte solutions and internal stress generated by structural changes on electrochemical cycles.8,9 On the other hand, compared with O′3-type NaMnO2, P′2-type Na2/3MnO2 shows better cyclability without sacrificing a large reversible capacity over 200 mA h g−1, but its cyclability is still unacceptable as an electrode material for practical applications.10,11 In this study, P′2 Na2/3MnO2 is revisited as a positive electrode material for battery applications. The cycling performance of P′2 Na2/3MnO2 is significantly improved by controlling the operating voltage during electrochemical cycling. In addition, the use of highly concentrated electrolytes enables highly reversible and stable cycling of P′2 Na2/3MnO2 for 300 continuous cycles with a large reversible capacity, ∼200 mA h g−1. On the basis of these results, factors affecting the electrode performance of P′2 Na2/3MnO2 and its practical feasibility are discussed.
P′2-type Na2/3MnO2 was synthesized by a solid-state reaction. Starting materials composed of Na2CO3 (FUJIFILM Wako Pure Chemical) and MnCO3·nH2O (FUJIFILM Wako Pure Chemical) were mixed by wet ball milling with ethanol and then dried in air. A mixture of the precursors was pressed into a pellet at 20 MPa, and the obtained pellet was heated at 1050 °C for 12 h in air followed by quenching to room temperature.11Fig. 1a shows a synchrotron X-ray diffraction pattern of the synthesized Na2/3MnO2. Synchrotron XRD measurement was conducted at the beamline BL5S2 in the Aichi Synchrotron Radiation Center (AichiSR) in Japan. The sample powder was sealed in a glass capillary in an Ar atmosphere and measured using the 2D detector (PILATUS 100 K, DECTRIS Ltd). The wavelength of X-rays was 0.775 Å. An XRD pattern of the sample is assigned into a single-phase P′2-type layered structure with a space group of Cmcm. Note that the ratio of b/a values is higher than √3, indicating an in-plane structural distortion induced by Jahn–Teller active Mn3+ ions (Fig. 1b).11 From a morphological observation using a scanning electron microscope (JCM-6000, JEOL), P′2 Na2/3MnO2 consists of mainly large particles with a size of >10 μm and partially small particles (<5 μm) in Fig. 1c. The crystal structure of P′2 Na2/3MnO2 was further characterized by high-angle annular dark field (HAADF) and annular bright field (ABF) scanning transmission electron microscopy (STEM) using a JEOL JEM-ARM200F instrument equipped with a spherical aberration corrector operated at an acceleration voltage of 200 kV (Fig. 1d, e and Fig. S1, ESI†). In a HAADF-STEM image viewed along [100], heavier Mn ions are clearly highlighted as bright spots (Fig. 1d), and their arrangement along the c-axis direction is in good agreement with the P2-type layered stacking sequence. In contrast, for an ABF-STEM image, lighter O and Na ions are also distinctly visualized as dark spots (Fig. 1e).
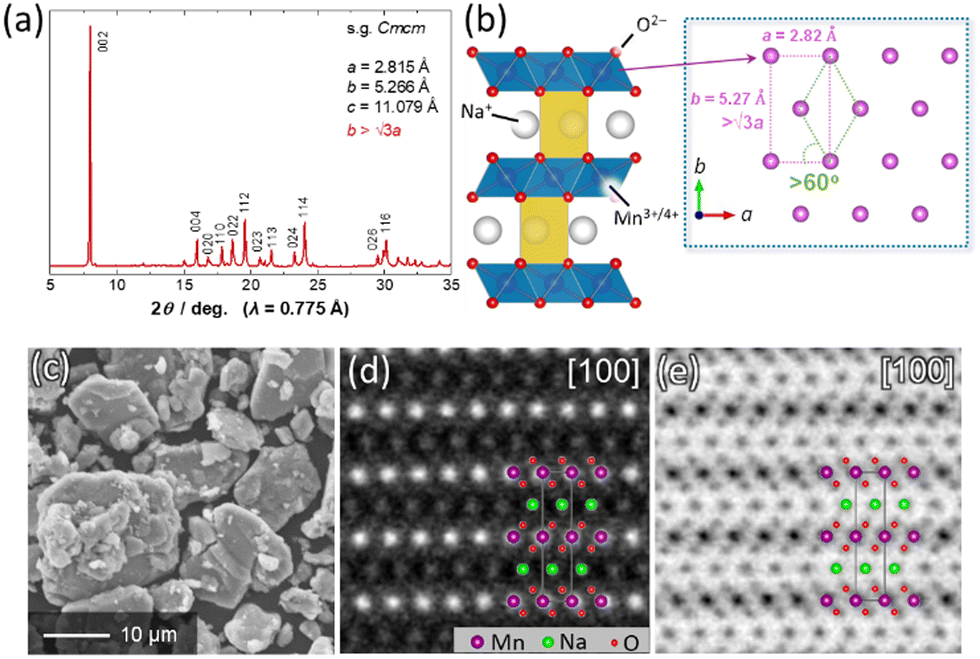 | ||
| Fig. 1 (a) A synchrotron XRD pattern of Na2/3MnO2 and (b) schematic illustration of the P′2-type layered structure, which was drawn using the program, VESTA.12 (c) A SEM image and (d) HAADF- and (e) ABF-STEM images of magnified regions of Fig. S1 (ESI†) along [100] for Na2/3MnO2. | ||
The electrochemical performance of P′2 Na2/3MnO2 was examined in two-electrode electrochemical cells with metallic sodium. The configuration of the two-electrode cell is described in the ESI.† Galvanostatic charge/discharge tests of Na/Na2/3MnO2 cells were performed with two different lower cut-off voltages, 1.5 and 2.4 V, and a fixed upper cut-off voltage of 4.0 V at a current rate of 10 mA g−1. The corresponding charge/discharge curves are shown in Fig. 2a. In the first cycle, the cell discharged to 1.5 V shows a reversible capacity of over 200 mA h g−1, which is consistent with that of a previous report.11 However, in the subsequent cycles, a gradual increase in polarization on the charge/discharge process is observed upon cycling, and 35% of the initial reversible capacity is lost after 30 cycles (Fig. 2b). In contrast, when the lower cut-off voltage is increased to 2.4 V, the increased polarization and capacity fading with cycling are significantly mitigated even though the reversible capacity is limited to approximately 60% of that in the cell with a 1.5 V cut-off. Moreover, coulombic efficiency is also improved, ∼98 and 93% on average for 2.4 and 1.5 V cut-off, respectively, as shown in Fig. 2c. In differential capacity plots shown in Fig. 2d, a stepwise charge/discharge profile is lost after 30 cycles in the voltage range of 1.5–4.0 V, but mostly maintained in the range of 2.4–4.0 V. In general, the dissolution of Mn ions into electrolyte solution is considered as a major degradation mechanism of Mn-based electrode materials.13,14 Moreover, the dissolved Mn ions deposit on the negative electrode surface,15 and deposited Mn causes the electrolyte decomposition and formation of by-products, for instance, alkoxide species.16 When the by-products are soluble species into electrolytes, they are oxidatively decomposed at the surface of the positive electrode, leading to lower coulombic efficiency. Because the same upper cut-off voltage, 4.0 V, is used for both tests, the difference is found with the accumulation of Mn3+ ions for the sample with 1.5 V cut-off. Similarly, the electrode reversibility for a spinel-type oxide, LiMn2O4, is influenced by the average oxidation states for Mn ions, and much better electrode reversibility is achieved for Li1+xMn2−xO4 and LiMgxMn2−xO4 with higher Mn oxidation states.17 Such high reversibility for spinel-type oxides with higher Mn valence is attributed to the smaller amount of Mn dissolution.18 According to the first-principles computational study, the higher Mn oxidation state increases the covalency and decreases the anti-bonding nature for Mn and O bonds, which energetically stabilize Mn–O bonds and reduce Mn dissolution.19 Therefore, increasing the lower cut-off voltage maintains a higher average oxidation state of Mn ions in the electrode (i.e., a larger Mn4+/Mn3+ ratio), thereby mitigating the Mn dissolution into the electrolyte and resulting in superior cyclability for the sample with 2.4 V cut-off. In fact, brown coloration of the glass filter used as a separator associated with Mn dissolution is found after 30 cycles with 1.5 V cut-off but not with 2.4 V (Fig. S2, ESI†). Similarly, for LiMn2O4, the concentration of dissolved Mn ions in the electrolyte decreases with increasing the lower cut-off voltage.20 The difference in average oxidation state of Mn ions in NaxMnO2 at different discharge states is evidenced by hard X-ray absorption spectroscopy (XAS) at the Mn K-edge (Fig. 2e). Hard XAS spectra were collected at beamline BL-12C of the Photon Factory Synchrotron Source in Japan. Composite electrode samples were rinsed with dimethyl carbonate and sealed in a water-resistant polymer film in an Ar-filled glovebox. The Mn K-edge XAS spectra were found in a lower energy region for the sample after discharge to 1.5 V compared to 2.4 V. Furthermore, in situ XRD studies reveal that the structural evolution of NaxMnO2 upon cycling is highly reversible in both voltage ranges of 1.5–4.0 V and 2.4–4.0 V. Although a slightly larger volume change associated with additional Na insertion into Na2/3MnO2 on discharge to 1.5 V is observed, as shown in Fig. S3 (ESI†), clear changes in XRD patterns are observed on charge to >3.7 V for both conditions. The clear shrinkage of the interlayer distance as observed for the 002 diffraction line on charge, ∼4.0 V, is noted. The interlayer distance after charge to 4.0 V is calculated to be 5.2 Å. Note that other peaks almost disappear, indicating that through-plane stacking faults are enriched associated with the glide of the transition metal layer from P2 to O2 phase after desodiation.21 Nevertheless, the interlayer distance is much wider compared with the pure O2 phase (d = 4.4 Å),22 suggesting that P2 domains with wider interlayer distances still coexist with O2 domains. Because the volume changes are pronounced on charge to ∼4.0 V, phase transitions and volume changes on additional sodiation from 2.4 to 1.5 V are not expected to be responsible for the degradation. Hence, the dissolution of Mn ions is considered as a primary reason for the loss of electrode reversibility of P′2 NxMnO2 on reduction to 1.5 V.
 | ||
| Fig. 2 (a) Charge/discharge curves, (b) capacity retention, (c) coulombic efficiency, and (d) differential capacity plots of P′2 Na2/3MnO2 cycled with 1 M NaPF6 in propylene carbonate (PC) electrolyte in the voltage ranges of 1.5–4.0 V and 2.4–4.0 V at a rate of 10 mA g−1. (e) Changes in Mn K-edge XAS spectra for P′2-Na2/3−yMnO2 on electrochemical cycles. | ||
Another key strategy to solve the Mn dissolution issue is the use of sparingly solvating electrolytes. Among these electrolytes, highly concentrated electrolytes (HCEs), in which the presence of free solvent molecules is eliminated, have gained much attention as a new trend in electrolyte design for Li and Na batteries.23–25 Several research groups reported that ether-based HCEs consisting of sodium bis(fluorosulfonyl)amide (NaFSA) and 1,2-dimethoxyethane (DME) exhibit good compatibility with both a 4 V-class positive electrode and Na metal negative electrode and are expected to be promising electrolyte candidates for high energy density Na metal batteries.26,27 However, the insufficient wettability of HCEs to a conventional polyolefin membrane hinders their use for practical applications.28,29 Although a porous glass fiber filter has been used as a substitute for a polyolefin membrane in laboratory-scale studies of HCEs,30 an internal short circuit associated with the penetration of Na dendrites through the separator is inevitable during repeated charge/discharge cycling for the Na cell with the glass fiber filter. To overcome the practical issue of HCEs, the surface coating with a meta-aramid resin has been reported to greatly enhance the wettability of the polyolefin membrane to HCEs with LiFSA.31,32 Similar to LiFSA-based HCEs, as shown in Fig. 3a, the aramid-coated polyolefin membrane (LIELSORT®, Teijin Limited) is successfully applied to the HCE with NaFSA (5 M NaFSA in DME, Kishida Chemical). Therefore, the aramid-coated polyolefin membrane is used as a separator for HCE. To compare the electrode reversibility of P′2 Na2/3MnO2 in conventional 1 M NaPF6 in PC and highly concentrated 5 M NaFSA in DME electrolytes, galvanostatic charge/discharge tests were conducted using Na/Na2/3MnO2 cells. Fig. 3b and Fig. S4 (ESI†) show the charge/discharge curves of Na/Na2/3MnO2 cells with different electrolytes measured at a current rate of 50 mA g−1. In the case of the conventional electrolyte, a severe increase in polarization upon cycling is noted, and the reversible capacity after 100 cycles is decreased to 43 mA h g−1 (Fig. 3c). This degradation can be attributed to the dissolution of Mn ions into the electrolyte. In contrast, the cell with HCE can be cycled without significant polarization increase and capacity loss and achieve a stable cycling with >85% capacity retention and high average coulombic efficiency of 99.9% for 300 continuous cycles (Fig. 3d). This is possibly because of the suppression of Mn dissolution associated with a scarcity of uncoordinated solvent molecules in the HCE with NaFSA.33,34 In addition, although a widely used PF6-based salt reacts with trace amounts of water in the electrolyte to produce hydrofluoric acid, which dissolves the Mn ions in active materials,15,35 the Mn dissolution can be effectively suppressed by using more stable NaFSA as an electrolyte salt. Indeed, no coloration of the glass filter is noted for the cell with HCE even after 200 cycles (Fig. S5, ESI†). Moreover, the use of an aramid-coated polyolefin separator significantly increases the maximum reversible Na deposition/stripping capacity in HCEs compared with a glass fiber filter and enables stable Na deposition/stripping cycling with high coulombic efficiency of ∼98.4% for 100 continuous cycles at a practical areal capacity of 2 mA h cm−2 (Fig. S6, ESI†). Thus, electrolyte engineering is an effective strategy to improve the reversibility of sodium metal batteries.36 Although the ionic conductivity of HCE is one order of magnitude lower than that of a conventional electrolyte at room temperature (Fig. S7, ESI†), the combined use of HCEs and the aramid-coated polyolefin separator would be one of the promising routes to realize stable and long-life operation of high-energy sodium metal batteries with a Mn-based layered oxide under practical conditions.
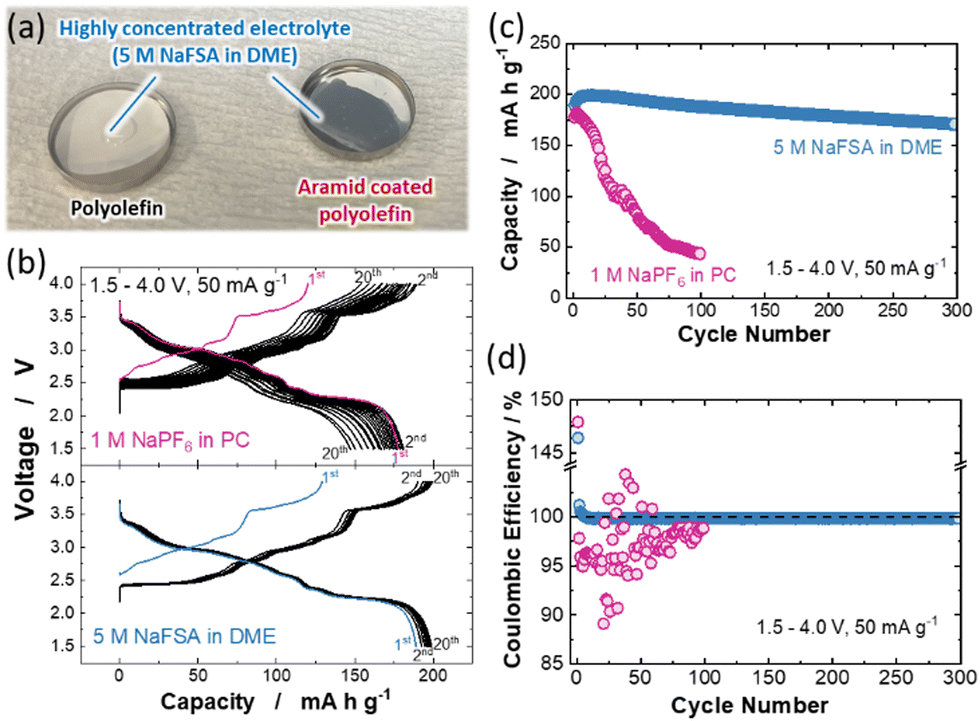 | ||
| Fig. 3 (a) Comparison of wettability of 5.0 M NaFSA in DME electrolyte to polyolefin (left) and aramid-coated polyolefin (right) membranes used as a separator. (b) Charge/discharge curves, (c) capacity retention, and (d) coulombic efficiency of P′2 Na2/3MnO2 cycled with 1 M NaPF6 in PC and 5 M NaFSA in DME electrolytes in the voltage range of 1.5–4.0 V at a rate of 50 mA g−1. | ||
In summary, suppressing the dissolution of Mn ions into the electrolyte is a critical strategy to improve the cycling performance of NaxMnO2. Because Mn3+ ions are enriched after reduction to the lower-cutoff voltage, control of the operating voltage to maintain a high average oxidation state of Mn ions (i.e., a larger Mn4+/Mn3+ ratio) in the electrode during cycling is beneficial to inhibit the dissolution of Mn ions. In addition, the use of HCEs without free solvent molecules is effective in suppressing the Mn dissolution owing to their low solvating ability. These two approaches are helpful to mitigate Mn dissolution and for future research development of cost-effective and long-life rechargeable sodium batteries with abundant Na/Mn ions.
NY acknowledges the partial support from JSPS, Grant-in-Aid for Scientific Research (Grant Numbers 21H04698 and 24H02204). YU also thanks the Grant-in-Aid for Scientific Research (Grant Number 23K13822) from JSPS. This work was partially supported by JST, CREST Grant Number JPMJCR21O6, Japan and by MEXT Program: Data Creation and Utilization-Type Material Research and Development Project, Grant Number JPMXP1122712807. NY acknowledges the partial support by JST as part of Adopting Sustainable Partnerships for Innovative Research Ecosystem (ASPIRE), Grant Number JPMJAP2313. This work was also partially supported by JST, Grant Number JPMJPF2016 and JPMJMS2282-13. We thank Dr Yoshinobu Miyazaki from Sumika Chemical Analysis Service, Ltd for the STEM observation.
Data availability
All relevant experimental and theoretical data within the article will be provided by the corresponding author on reasonable request.Conflicts of interest
There are no conflicts to declare.Notes and references
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636–11682 CrossRef CAS PubMed.
- H. Kim, H. Kim, Z. Ding, M. H. Lee, K. Lim, G. Yoon and K. Kang, Adv. Energy Mater., 2016, 6, 1600943 CrossRef.
- J. Y. Hwang, S. T. Myung and Y. K. Sun, Chem. Soc. Rev., 2017, 46, 3529–3614 RSC.
- K. Kubota, N. Yabuuchi, H. Yoshida, M. Dahbi and S. Komaba, MRS Bull., 2014, 39, 416–422 CrossRef CAS.
- M. H. Han, E. Gonzalo, G. Singh and T. Rojo, Energy Environ. Sci., 2015, 8, 81–102 RSC.
- C. Delmas, C. Fouassier and P. Hagenmuller, Phys. Pol., B, 1980, 99, 81–85 CAS.
- X. Ma, H. Chen and G. Ceder, J. Electrochem. Soc., 2011, 158, A1307–A1312 CrossRef CAS.
- K. Kubota, M. Miyazaki, E. J. Kim, H. Yoshida, P. Barpanda and S. Komaba, J. Mater. Chem. A, 2021, 9, 26810–26819 RSC.
- T. Sato, K. Yoshikawa, W. Zhao, T. Kobayashi, H. B. Rajendra, M. Yonemura and N. Yabuuchi, Energy Mater. Adv., 2021, 2021, 9857563 Search PubMed.
- J. Billaud, G. Singh, A. R. Armstrong, E. Gonzalo, V. Roddatis, M. Armand, T. Rojo and P. G. Bruce, Energy Environ. Sci., 2014, 7, 1387–1391 RSC.
- S. Kumakura, Y. Tahara, K. Kubota, K. Chihara and S. Komaba, Angew. Chem., Int. Ed., 2016, 55, 12760–12763 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- C. Zhan, T. Wu, J. Lu and K. Amine, Energy Environ. Sci., 2018, 11, 243–257 RSC.
- S. K. Sharma, G. Sharma, A. Gaur, A. Arya, F. S. Mirsafi, R. Abolhassani, H.-G. Rubahn, J.-S. Yu and Y. K. Mishra, Energy Adv., 2022, 1, 457–510 RSC.
- A. Blyr, C. Sigala, G. Amatucci, D. Guyornard, Y. Chabre and J.-M. Tarascon, J. Electrochem. Soc., 1998, 145, 194–209 CrossRef CAS.
- S. Komaba, T. Ishikawa, N. Yabuuchi, W. Murata, A. Ito and Y. Ohsawa, ACS Appl. Mater. Interfaces, 2011, 3, 4165–4168 CrossRef CAS PubMed.
- R. J. Gummow, A. D. Kock and M. M. Thackeray, Solid State Ionics, 1994, 69, 59–67 CrossRef CAS.
- W. Choi and A. Manthiram, J. Electrochem. Soc., 2006, 153, A1760–A1764 CrossRef CAS.
- Y. K. Lee, J. Park and W. Lu, J. Electrochem. Soc., 2016, 163, A1359–A1368 CrossRef CAS.
- L.-F. Wang, C.-C. Ou, K. A. Striebel and J.-S. Chen, J. Electrochem. Soc., 2003, 150, A905–A911 CrossRef CAS.
- A. K. Pandey, B. D. L. Campéon, I. Konuma and N. Yabuuchi, Energy Adv., 2023, 2, 98–102 RSC.
- Z. Lua and J. R. Dahn, J. Electrochem. Soc., 2001, 148, A1225–A1229 CrossRef.
- Y. Yamada, J. Wang, S. Ko, E. Watanabe and A. Yamada, Nat. Energy, 2019, 4, 269–280 CrossRef CAS.
- O. Borodin, J. Self, K. A. Persson, C. Wang and K. Xu, Joule, 2020, 4, 69–100 CrossRef CAS.
- Y. Ugata and N. Yabuuchi, Trends Chem., 2023, 5, 672–683 CrossRef CAS.
- R. Cao, K. Mishra, X. Li, J. Qian, M. H. Engelhard, M. E. Bowden, K. S. Han, K. T. Mueller, W. A. Henderson and J.-G. Zhang, Nano Energy, 2016, 30, 825–830 CrossRef CAS.
- J. Lee, Y. Lee, J. Lee, S. M. Lee, J. H. Choi, H. Kim, M. S. Kwon, K. Kang, K. T. Lee and N. S. Choi, ACS Appl. Mater. Interfaces, 2017, 9, 3723–3732 CrossRef CAS.
- J. Zheng, S. Chen, W. Zhao, J. Song, M. H. Engelhard and J.-G. Zhang, ACS Energy Lett., 2018, 3, 315–321 CrossRef CAS.
- Y. Maeyoshi, K. Yoshii and H. Sakaebe, Electrochemistry, 2022, 90, 047001 CrossRef CAS.
- R. Tatara, Y. Okamoto, Y. Ugata, K. Ueno, M. Watanabe and K. Dokko, Electrochemistry, 2021, 89, 590–596 CrossRef CAS.
- Y. Ugata, C. Motoki, S. Nishikawa and N. Yabuuchi, Energy Adv., 2023, 2, 503–507 RSC.
- I. Konuma, N. Ikeda, B. D. L. Campéon, H. Fujimura, J. Kikkawa, H. D. Luong, Y. Tateyama, Y. Ugata, M. Yonemura, T. Ishigaki, T. Aida and N. Yabuuchi, Energy Storage Mater., 2024, 66, 103200 CrossRef.
- Y. Zhang, Y. Ugata, B. L. Campéon and N. Yabuuchi, Adv. Energy Mater., 2024, 14, 2304074 CrossRef CAS.
- Y. Miyaoka, T. Sato, Y. Oguro, S. Kondo, K. Nakano, M. Nakayama, Y. Ugata, D. Goonetilleke, N. Sharma, A. M. Glushenkov, S. Hiroi, K. Ohara, K. Takada, Y. Fujii and N. Yabuuchi, ACS Cent. Sci., 2024, 10, 1718–1732 CrossRef CAS PubMed.
- R. Benedek and M. M. Thackeray, Electrochem. Solid-State Lett., 2006, 9, A265–A267 CrossRef CAS.
- P. Xu, F. Huang, Y. Sun, Y. Lei, X. Cao, S. Liang and G. Fang, Adv. Funct. Mater., 2024, 34, 2406080 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc04719b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |