
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNear-IR absorbing tetraene-linked π-conjugated porous polymers for energy storage and electrical conductivity†
Vinutha K.
Venkatareddy
a,
Hamidreza
Parsimehr
b,
Anna
Ignaszak
*b and
Rajeswara Rao
M
 *a
*a
aDepartment of Chemistry, Indian Institute of Technology, Dharwad, Karnataka, India. E-mail: rajesh@iitdh.ac.in
bDepartment of Chemistry, University of New Brunswick, Fredericton, Canada
First published on 21st November 2024
Abstract
Tetraene-linked diketopyrrolopyrrole (DPP)-based CMPs were developed via Knoevenagel condensation of ditopic active hydrogen containing DPP with tritopic aryl aldehydes. The “tetra-ene” π-arrangement in the molecular framework promotes uninterrupted π-delocalization, resulting in near-infrared (NIR) absorption (∼red edge of 1200 nm), high electrical conductivity in the pristine (10−3 S m−1) and doped states (0.2 S m−1), and moderate energy storage (70 F g−1).
Conjugated microporous polymers (CMPs; 2D polymers) are an important subclass of porous materials that integrate extended π-conjugated frameworks with permanent nanopores.1,2 The CMPs possess unique features such as high surface area, flexible structural design, and intriguing optical properties, which make them suitable for gas/energy storage,3 catalysis, and semiconducting applications.4–10 In pursuit of promoting π-delocalization within the CMP network, several linkers, including vinylene (–C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–),11–14 cyano vinylene [–(CN)CC–],15–17 imine (–CN–),18–20 and keto-enamine (–CO–CC–NH–),21,22 hydrazine, azine,23,24etc., have been introduced. Strong π-bond polarization in these linkers, except for vinylene, restricts the facile electron delocalization. Thus, the vinylene linkers have gained prominence due to their efficient π-electron delocalization and the associated high chemical stability, leading to the development of several CMPs.
C–),11–14 cyano vinylene [–(CN)CC–],15–17 imine (–CN–),18–20 and keto-enamine (–CO–CC–NH–),21,22 hydrazine, azine,23,24etc., have been introduced. Strong π-bond polarization in these linkers, except for vinylene, restricts the facile electron delocalization. Thus, the vinylene linkers have gained prominence due to their efficient π-electron delocalization and the associated high chemical stability, leading to the development of several CMPs.
Among the several strategies known for constructing vinylene linkages, Knoevenagel condensation is found to be the most versatile and broadly advantageous for 2D polymer synthesis due to its simple reaction conditions, high yields, building block scope and reversible covalent bonds to form ordered structures.13,25 A typical reaction involves the addition of an active hydrogen compound to an aromatic aldehyde. Several building blocks containing active hydrogen compounds (Chart 1) have been designed and incorporated into a 2D-polymer network. These polymers have demonstrated high π-delocalization in the backbone, leading to superior properties and functions. However, most of these building blocks are either tritopic, resulting in cross-conjugation,26 or have polarizable π-bonds, significantly minimizing π-delocalization. Thus, more sophisticated building blocks are needed to benefit from the advantage of vinylene linkages. In this line, we identified that C2 topic dimethyl diketopyrrolopyrrole (1) would serve as an ideal building unit, which can render a unique “tetra-ene” linkage upon integration into the polymeric network and promote π-electron delocalization seamlessly. Colleen N. Scott and co-workers27 reported its synthesis and explored its versatility for developing small molecules, while our group later introduced redox-active groups and tested them for electrochromism.28 Besides these two examples, no other tetra-ene linked DPP containing systems have been developed.
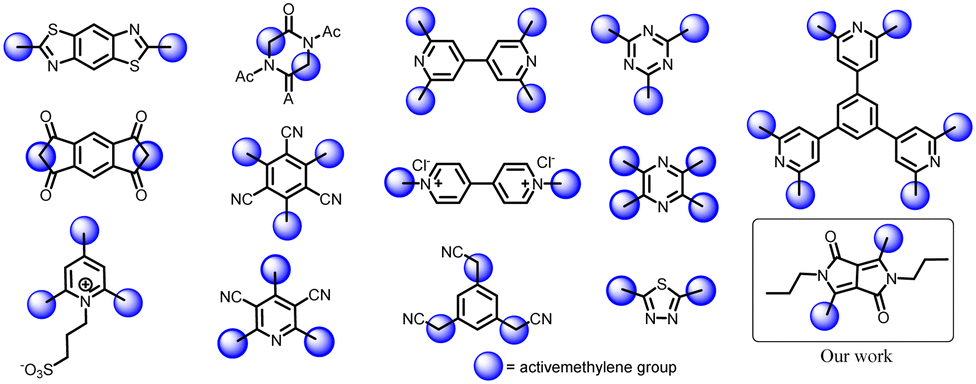 | ||
| Chart 1 Active methylene building blocks used in synthesizing vinylene-linked 2D-polymers. | ||
The diketopyrrolopyrrole (DPP) is a strong electron-accepting chromophore known for its intriguing properties, such as strong light absorption, excellent photostability, strong fluorescence, and high charge carrier mobility.29 It has been widely used in organic field effect transistors, fluorescent probes, ion sensing, etc.30 Due to DPP's extraordinary properties and functions, it has been widely incorporated into various molecular frameworks ranging from small molecules to 2D polymers, although the number of 2D polymers is extremely small. Sabrina et al. developed a DPP containing a 2D-covalent organic framework (COF) employing a boronate ester-linker.31 At the same time, Xu et al. utilized an imine linker to construct a DPP-integrated COF. The former COF (carbon nanotube incorporated) exhibited electrical conductivity of 1.8 × 10−6 S m−1, while the latter could only show 5.8 × 10−10 S m−1 and a battery capacity of 8.7 mA h cm−2.32 Liang Luo et al. recently synthesized another imine-linked DPP-grafted COF exhibiting ambipolar conductivity of 1.2 S m−1 and supercapacitance of 384 F g−1 due to high porosity with the hexagonal framework enhancing charge mobility.33 Shiming Bi et al. synthesized a fluorescent 2D-polymer via Sonogashira coupling for fluoride ion sensing.34 The linkers utilized here are weak π-communicating; thus, the bandgap of the polymers lies around 1.8–2.1 eV, which also dramatically moderates the conducting properties. This can be attributed to the lower charge carrier mobilities arising from poor π–π interactions and charge carrier mobilities.
Here, we report vinylene-linked DPP integrated 2D CMPs through Knoevenagel condensation. The resulting CMPs possess tetra-ene linkage, resulting in a unique π-electron delocalization pathway. The polymer showed NIR absorbance with the red edge extending up to ∼1200 nm and a narrow band gap as low as ∼1 eV. The facile π-electron delocalization in DT-P led to high electrical conductivity in its pristine form (3 × 10−3 S m−1) and doped form (0.2 S m−1). Moreover, the polymer also shows a moderate supercapacitor efficiency of 70 F g−1.
The 2D polymers, DP-P and DT-P, were synthesized by knitting dimethyl DPP (1) with benzene-1,3,5-tricarbaldehyde (2) or 4,4′,4′′-nitrilotribenzaldehyde (3) through Knoevenagel condensation. In a typical reaction procedure, the building units are reacted in the presence of diisopropylamine and L-proline (catalyst) in ethanol solvent at 90 °C for five days (Scheme 1). At the end of five days, the resultant deeply coloured insoluble solids were filtered and purified by washing with various solvents (dichloromethane, THF, methanol, and DMF) to eliminate oligomers and subsequently dried under vacuum to achieve the polymers (DP-P and DT-P) in 85–90% yields. To evaluate the properties of the polymers, soluble model compounds (DP-M and DT-M) have also been synthesized by reacting 1 with p-tolualdehyde and 4-(diphenylamino)benzaldehyde, respectively, in ∼90% yields by following the same synthetic procedure.
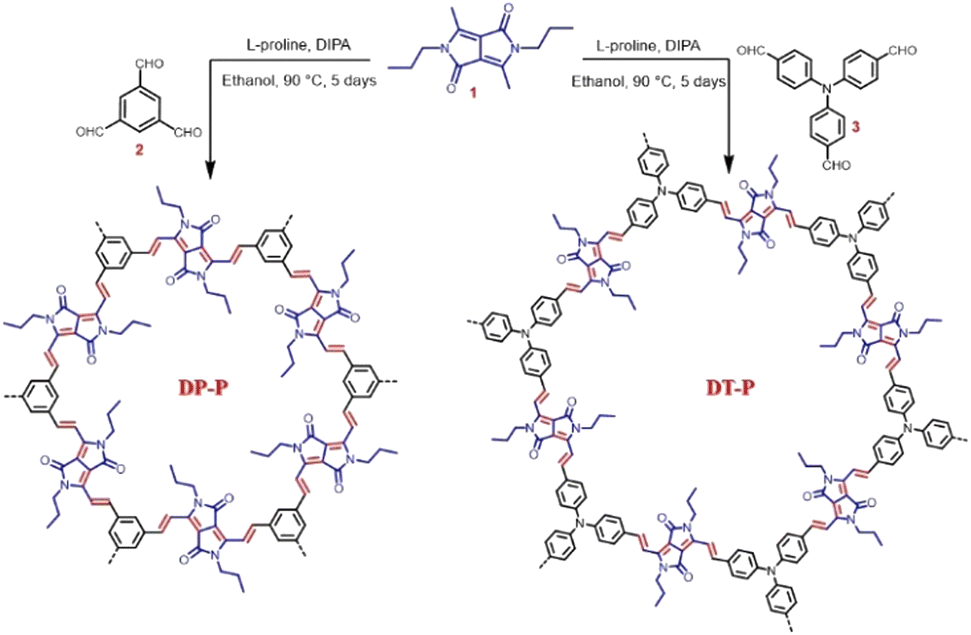 | ||
| Scheme 1 Synthesis of DP-P and DT-P CMPs via Knoevenagel condensation. | ||
The formation and molecular connectivity of the polymers have been confirmed using FT-IR and CP-MAS 13C NMR. The CP-MAS 13C NMR of the polymers shows a characteristic signal of carbonyl carbon at 165 ppm and vinylene carbons at 110 & 105 ppm, supporting the presence of DPP and the vinylene linkers. The other DPP and aryl signals appear at 150 ppm and 130–140 ppm, respectively. The signals corresponding to N-alkyl appear in the up-field region between 10 and 50 ppm (Fig. 1a). FT-IR of the polymers shows bands at 1580 cm−1 and 1625 cm−1 (Fig. 1b) corresponding to ν(CO) and ν(CC), respectively. It is important to note that the CO stretching experiences a blue shift of 70 cm−1 from 1650 cm−1 compared to 1 (Fig. S1, ESI†) due to extended π-delocalization caused by the vinylene linkages. The 13C NMR and FT-IR spectra of the polymers match closely with the corresponding soluble model compounds, providing additional proof of the formation of the polymers. The structural and electronic properties of DT-P and DP-P and their model compounds DT-M and DP-M are studied by DFT using the B3LYP-631G(d) basis set level. In the case of DT-P and DP-P, the HOMO resides primarily on the TPA and phenyl moieties with minimal delocalization on the DPP unit. On the contrary, the LUMO is predominantly confined to the DPP, with negligible contributions from the TPA and aryl units. The energies of the HOCO and LUCO of DT-P and DP-P were found to be −4.0 eV, −4.5 eV and −3.2 eV, −3.4 eV with band gaps 0.8 eV and 1.1 eV, respectively (Fig. S9–S11, ESI†). A similar observation has also been noted for their model compounds (DT-M and DP-M); however, with significant differences in the HOMO and LUMO energy levels, which are −4.8 eV, −4.5 eV and −2.6 eV, −2.5 eV with the band gap of 2.2 eV and 2.0 eV, respectively (Fig. S10, ESI†). A facile π-electron delocalization in the two-dimensional plane is envisaged for narrow band gaps. DT-P possessing strong D–A interactions between the polymers further tunes the energy levels and displays the lowest bandgap compared to DP-P.
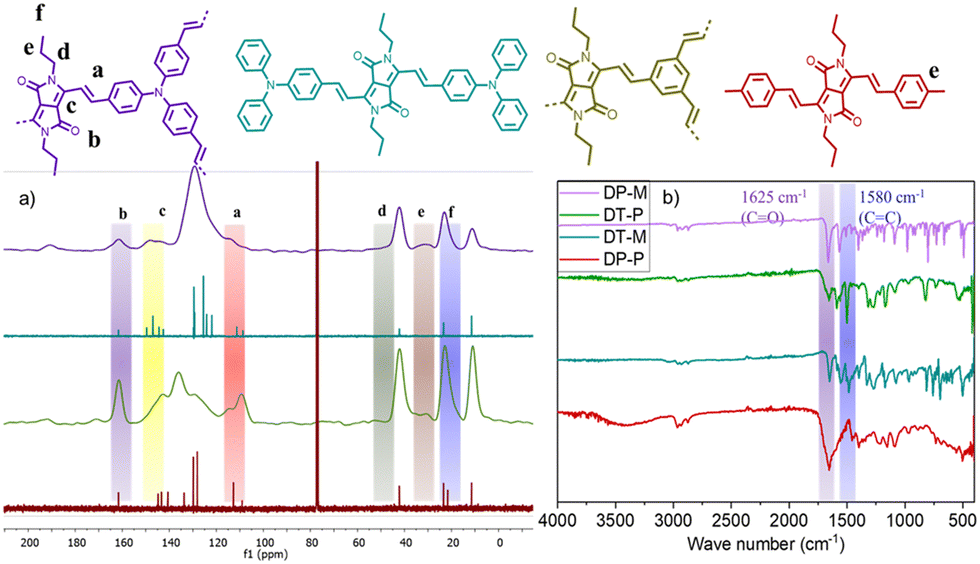 | ||
| Fig. 1 (a) 13C CPMAS spectra (b) FTIR spectra of DP-P and DT-P along with their model compounds (DP-M, DT-M). | ||
Thermogravimetric analysis was done to understand the thermal stability of the polymers. The synthesized powders of DT-P and DP-P were found to have thermal stability up to 330 °C without losing weight (Fig. S4, ESI†). The polymers are also chemically stable even in harsh conditions, such as 1 M KOH and 0.5 M HCl solutions for five days. The acid or base-treated samples displayed no sign of decomposition, as confirmed by FT-IR (Fig. S2, ESI†). The morphology of the CMPs investigated by field emission scanning electron microscopy (FE-SEM) revealed distinct morphology. Spherical sheets for DT-P and fibre-like structures for DP-P at the magnitude of 500 nm (Fig. S3, ESI†) have been observed. The variation in the building units and the porous network could be the reason for such differences. It is important to note that fibrillar morphology is quite rare.31,35 In powder X-ray diffraction (XRD) analysis, both polymers exhibited broad peaks, indicating the amorphous nature of the polymers (Fig. S5, ESI†). The polymers have also been characterized by XPS, which confirmed the chemical state of the polymers (Fig. S6 and S7, ESI†).
The porous properties of the polymers were studied by nitrogen adsorption/desorption experiments at 77 K. The Brunauer–Emmett–Teller (BET) surface area of DT-P and DP-P was 8.7 m2 g−1 and 8.1 m2 g−1 with pore volume and diameter of 0.026 cc g−1, 0.054 cc g−1 and 2.5 nm, 3.7 nm, respectively. The increased uptake of N2 at the lower relative pressure (p/p0) indicates the microporous nature of the polymers, along with the presence of the mesopores due to the hysteresis loop at the higher p/p0. Both the polymers exhibited type-IV adsorption isotherms (Fig. S8, ESI†). The low surface area of the polymers could be attributed to the interference of alkyl chains and their amorphous nature. The amorphosity of polymers will often lead to the disordered arrangement of the molecular units, resulting in the blocking of the pores.36
The diffuse reflectance absorption spectra of DT-P and DP-P exhibited broad absorption spanning across the visible to NIR region, with the red edge appearing at 1130 nm (band gap – 1.19 eV) and 784 nm (band gap – 1.60 eV), respectively (Fig. 2a). Such low energy absorption is a manifestation of facile π-electron delocalization enabled by the tetraene bridge within the polymeric network. The model compounds display a red edge around 888 nm (band gap – 1.43 eV) (DT-M) and 784 nm (bandgap – 1.63 eV) (DP-M). It is important to note that among all the DPP-integrated CMPs/2D-polymers reported so far, DT-P and DP-P display the lowest optical band gap (Table S2, ESI†), indicating the importance of tetra-ene based π-delocalization.
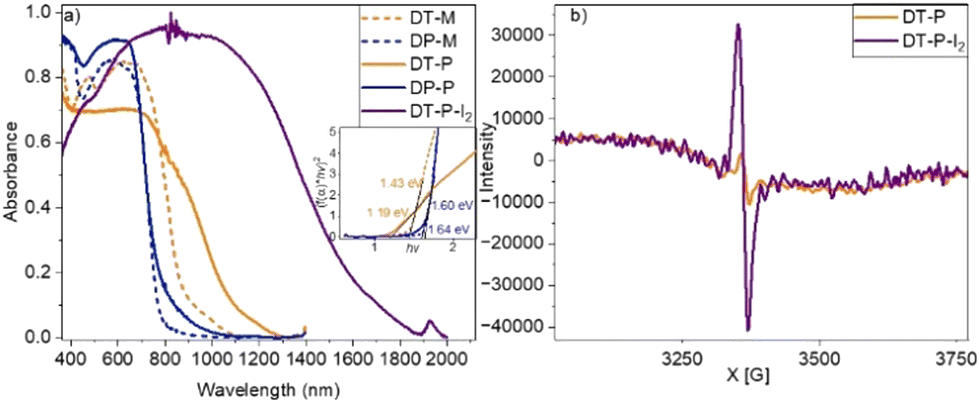 | ||
| Fig. 2 Absorption spectra of model compounds DT-M, DP-M and polymers DT-P, DP-P & and the iodine doped DT-P (DT-P-I2) (a), Inset: Kubelka Munk plot of DT-P and DP-P. (b) EPR spectra of DT-P in its pristine and doped states. | ||
Inspired by the low band gap and facile π-electron delocalization of the polymers, we explored the electrical conductivity of DT-P. Two-probe electrical conductivity measurements with a cylindrical pellet (0.15 × 0.4 cm) of the pristine polymer revealed a conductivity of 3.4 × 10−3 S m−1 (Fig. 3a). The conductivity in the pristine material is significantly high and, in fact, among the best compared to the reported CMPs. Generally, organic polymers possess poor electrical conductivity (insulating behaviour) in their virgin form due to a deficiency of electronic pathways and a strong covalent nature, leading to insufficient charge transfer.37 So far, only a few examples have been reported with appreciable conductivity in their pristine form (Table S3, ESI†). Our polymer is superior to the reported ones. However, introducing charge carriers into the polymeric network boosts the conductivity manifold. Following the same trend, doping DT-P by iodine via oxidizing TPA into TPA˙+ increased the conductivity of the polymer by two orders of magnitude to 0.2 S m−1. This value is comparable to the best conducting 2D polymers reported (Table S3, ESI†). Most importantly, the doped DT-P showed good stability without any sign of dropping the electrical conductivity (0.1 S m−1) for 150 h (Fig. 3b). The high stability of the doped polymer can be assigned to various resonance structures supported by vinylene linkages and the DPP core (Fig. 3d). The optimal time for doping to achieve the best conductivity is 14 hours (Fig. 3c), assigned to forming the highest charge carriers. The successful doping and the formation of the charge carriers (radical cations) in DT-P have been confirmed by the electron paramagnetic resonance (EPR) and UV-Vis-NIR absorption spectroscopy, which showed a high intense signal with a g value of around 2.0043 (Fig. 2b) and a broad absorption band with a red edge of 1700 nm (Fig. 2a), respectively. Such features indicate the presence of charge carriers and their facile delocalization along the polymeric backbone. Interestingly, the pristine polymer (DT-P) was also EPR-active and showed a signal; however, it had a low intensity. This indicates that the pristine polymer also possesses free radicals due to high electron delocalization.
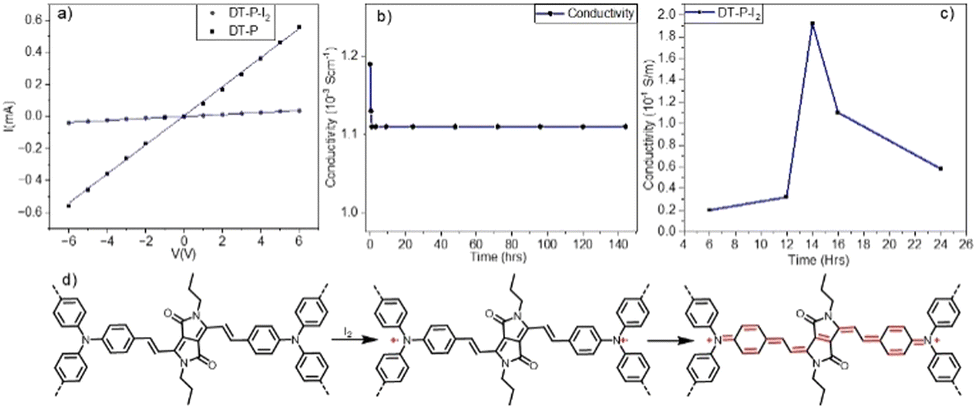 | ||
| Fig. 3 The I–V plot of the DT-P polymer (a); stability test of the doped DT-P polymer (b); conductivity of the DT-P polymer at different doping times (c) and the proposed mechanism for the I2 doped DT-P polymer (d). | ||
Considering the electroactive unit (TPA) in the DT-P polymer, we studied the polymer's ability for energy storage and its potential use as pseudo/supercapacitor. As detailed in the ESI,† we have attempted several electrochemical tests in 1 M KOH in three-electrode systems. Fig. S15 (ESI†) displays the cyclic voltammetry (CV) curves of DT-P (and Fig. S16, ESI†DP-P) in its pristine form (a); their combination with the carbon black (b) added to observe the effect of increased electrical conductivity on the pseudo-capacitance and with the addition of graphene oxide to seek the possible synergistic effect between redox-active materials (DT-P) and other capacitive components (c). A distinctive quasi-reversible redox event is present for the polymer and its combination with different additives. DT-P showed the highest redox peak currents for the electrode made of bare polymer. The redox signals are attributed to the redox reactions of the electrochemically active group in a quasi-reversible manner. We considered electrochemical oxidation to be centred on motifs involved in chemical doping, as presented in Fig. 3d. With this in mind, we hypothesized that the electrochemical activity is associated with oxidation steps that can be compared to chemical oxidation by I2 (Fig. 3d). CV scans recorded at 5 mV s−1 in an aqueous electrolyte were analyzed to calculate gravimetric specific capacitance (Cs, F g−1) according to eqn (S1) (ESI†). In summary, comparing the bare polymer and their mixture with conducting carbon black, we have learned that DT-P is conducting enough to support the electron transfer effectively and does not need additives to improve the electrode performance. This can be seen by comparing the values of Cs presented in Table S4 (ESI†). This confirms that a pristine polymer has sufficient conductivity and can be directly used as an electrode component. This agrees with the above-presented conductivity study. Concerning the value of Cs, we compare our performance to work demonstrated by researchers working on similar polymers. Their Cs values of DPP-TBB-COF and DPP-TPP-COF are 227.1 and 49.8 F g−1 at the scan rate of 2 mV s−1, respectively, and gradually decreased to 54.9 and 24.8 F g−1 as the scan rate increased.38 We could speculate that higher Cs could be related to the higher surface area of their materials.
It is well-established that GO can be considered capacitive at approximately 154 F g−1 in 6 M KOH.39 For this reason, we introduced a small quantity to the electrode formulation and repeated the same electrochemical tests. In summary, as shown in Table S4 (ESI†), Cs has improved due to the presence of the second capacitive component. Electronic interaction between the carbon and the polymer can create this capacitance improvement.
Furthermore, we quantified a diffusion coefficient (D) of ion transport for charge compensation due to the polymer redox activity. The analysis was carried out using CVs recorded at various potential scan rates (eqn (S2) and Fig. S10–S13, ESI†). D for DT-P is lower than this parameter reported, for example, for polypyrrole pseudo capacitors (10−6–10−9 cm2 s−1). However, this is a reasonable value because polymers presented in this work are giant molecules compared to polypyrrole.40
In conclusion, we introduced a novel DPP-based building block for developing π-conjugated vinylene-linked diketopyrrolopyrrole (DPP)-based CMPs. The backbone of the polymers consists of a “tetra-ene” π-arrangement, which is unique in terms of structural design and contributes to near-infrared (NIR) absorption (∼1200 nm) and narrow band gap (1.19 eV). The uninterrupted π-delocalization enabled high electrical conductivity for the TPA-containing polymer (DT-P) in its pristine (10−3 S m−1) and doped state (0.2 S m−1). In addition, the polymer (in combination with graphene oxide) can also behave as an electrode and exhibit moderate energy storage of 70 F g−1. The strategy reported here is novel and will offer valuable insights and impetus to develop new polymers with improved π-delocalization, resulting in superior properties and applications.
Rajeswara Rao thanks SERB, India, and IIT Dharwad for partially supporting this research through a Core Research Grant (CRG/2023/002129) and an Energizing MoU grant, respectively. Vinutha thanks DST for the inspire fellowship (IF190711). The authors are grateful to the sophisticated central instrumentation facility (SCIF), IIT Dharwad, and all its staff members for letting us use the facilities and assisting us with the material characterizations. We also thank Ms Saraswati and Prof. Ruma Ghosh, Department of Electrical Engineering, for providing GO.
Data availability
The supporting data of this article have been included in the ESI.†Conflicts of interest
There are no conflicts to declare.References
- J.-S. M. Lee and A. I. Cooper, Chem. Rev., 2020, 120, 2171–2214 CrossRef CAS
.
- H. V. Babu,
et al., ACS Appl. Mater. Interfaces, 2019, 11, 11029–11060 CrossRef CAS
- M. G. Mohamed,
et al., ACS Appl. Energy Mater., 2024, 7, 5582–5593 CrossRef CAS
- J. Xie, P. Gu and Q. Zhang, ACS Energy Lett., 2017, 2, 1985–1996 CrossRef CAS
- Z. P. Yu,
et al., ACS Appl. Polym. Mater., 2021, 3, 60–92 CrossRef CAS
- Y. Xu,
et al., Chem. Soc. Rev., 2013, 42, 8012 RSC
- U. Karatayeva,
et al., Adv. Sci., 2024, 11, 2308228 CrossRef CAS
- S. Luo,
et al., J. Mater. Chem. A, 2020, 8, 6434–6470 RSC
- S. Wang,
et al., Nat. Commun., 2023, 14, 6891 CrossRef CAS PubMed
- Y. Su,
et al., J. Am. Chem. Soc., 2024, 146, 15479–15487 CrossRef CAS PubMed
- Z. Zhang,
et al., J. Am. Chem. Soc., 2023, 145, 16704–16710 CrossRef CAS
- K. Aswani Raj and M. Rajeswara Rao, J. Mater. Chem. C, 2023, 12, 110–117 Search PubMed
- T. Jadhav,
et al., Angew. Chem., Int. Ed., 2019, 58, 13753–13757 CrossRef CAS PubMed
- A. Acharjya,
et al., Angew. Chem., Int. Ed., 2019, 58, 14865–14870 CrossRef CAS PubMed
- R. Bu,
et al., ACS Appl. Mater. Interfaces, 2021, 13, 26431–26440 CrossRef CAS
- Y. Yang,
et al., ACS Catal., 2022, 12, 10718–10726 CrossRef CAS
- Y. Su,
et al., J. Am. Chem. Soc., 2022, 144, 18218–18222 CrossRef CAS PubMed
- M. Afshari,
et al., ACS Appl. Mater. Interfaces, 2022, 14, 22398–22406 CrossRef CAS PubMed
- F. Niu,
et al., J. Mater. Chem. C, 2021, 9, 8562–8569 RSC
- F. Huang,
et al., J. Mater. Chem. A, 2024, 12, 7036–7046 RSC
- C. R. DeBlase,
et al., J. Am. Chem. Soc., 2013, 135, 16821–16824 CrossRef CAS PubMed
- M. Rajeswara Rao,
et al., J. Am. Chem. Soc., 2017, 139, 2421–2427 CrossRef
- V. S. Vyas,
et al., Nat. Commun., 2015, 6, 8508 CrossRef CAS PubMed
- S. Dalapati,
et al., J. Am. Chem. Soc., 2013, 135, 17310–17313 CrossRef CAS PubMed
- S. Xu, M. Richter and X. Feng, Acc. Mater. Res., 2021, 2, 252–265 CrossRef CAS
- S. Bi,
et al., J. Am. Chem. Soc., 2020, 142, 11893–11900 CrossRef CAS
- D. Feng,
et al., Org. Lett., 2019, 21, 1973–1978 CrossRef CAS PubMed
- V. K. Venkatareddy and M. Rajeswara Rao, RSC Adv., 2024, 14, 10017–10023 RSC
- N. Luo,
et al., Org. Chem. Front., 2021, 8, 4560–4581 RSC
- D. Molina,
et al., J. Mater. Chem. C, 2021, 9, 16078–16109 RSC
- S. Rager,
et al., Chem. Mater., 2019, 31, 2707–2712 CrossRef CAS PubMed
- J. Xu,
et al., ACS Energy Lett., 2021, 6, 3053–3062 CrossRef CAS
- L. Luo,
et al., Small, 2024, 20, 2402993 CrossRef CAS PubMed
- S. Bi,
et al., J. Mater. Chem. C, 2018, 6, 3961–3967 RSC
- C. Yang,
et al., Adv. Sci., 2024, 11, 2400626 CrossRef CAS
- M. C. Senarathna,
et al., Angew. Chem., Int. Ed., 2023, 62, e202312617 CrossRef CAS
- S. Bommakanti,
et al., J. Phys. Chem. C, 2023, 127, 8352–8361 CrossRef CAS
- L. Luo,
et al., Small, 2024, 20, 2402993 CrossRef CAS PubMed
- L. W. Le Fevre,
et al., ChemistryOpen, 2019, 8, 418–428 CrossRef CAS PubMed
- N. Patterson,
et al., RSC Adv., 2020, 10, 20162–20172 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc05074f |
| This journal is © The Royal Society of Chemistry 2025 |