
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Heterogeneous catalysis strategies for polyolefin plastic upcycling: co-reactant-assisted and direct transformation under mild conditions
Haokun Wang†
,
Sijie Huang†
and
Shik Chi Edman Tsang
 *
*
Wolfson Catalysis Centre, Department of Chemistry, University of Oxford, Oxford OX1 3QR, UK. E-mail: edman.tsang@chem.ox.ac.uk
First published on 18th December 2024
Abstract
The large-scale production and inadequate disposal of polyolefin (PO) plastics pose significant environmental challenges. Traditional recycling methods are energy-intensive and often ineffective, prompting a need for more sustainable approaches. In recent years, catalytic upcycling under mild conditions has emerged as a promising strategy to transform PO plastics into valuable products. Co-reactants such as hydrogen, short-chain alkanes or alkenes, oxygen, and CO2 play a crucial role in driving these transformations, influencing reaction mechanisms and broadening the range of possible products. This review categorizes recent advancements in PO plastic upcycling based on the type of co-reactant employed and compares these with direct, co-reactant-free processes. Despite these advances, challenges remain in improving catalytic stability, product selectivity, and overcoming diffusion limitations in viscous plastic feedstocks. This review underscores the catalytic chemistry underpinning the development of efficient PO plastic upcycling processes with co-reactants, offering insights into future directions for sustainable plastic chemical management.
 Haokun Wang | Haokun Wang received his BSc degree in Chemical Engineering from Beijing University of Chemical Technology and University of Birmingham, followed by an MSc degree in Advanced Chemical Engineering from Imperial College London. In 2020, he joined Professor Edman Tsang's research group as a PhD student in the Department of Inorganic Chemistry at the University of Oxford. His research focuses on heterogeneous catalysis, plastic upcycling, and reaction engineering. |
 Sijie Huang | Sijie Huang obtained her BSc in Materials Science and Engineering from Beijing University of Chemical Technology. In 2023, she joined Prof. Edman Tsang's research group as a DPhil student in Inorganic Chemistry at the University of Oxford (UK). Her research focuses on the design, synthesis, and mechanistic study of zeolites as active catalysts. |
 Shik Chi Edman Tsang | Edman Tsang is a Professor of Chemistry and Head of the Wolfson Catalysis Centre at the University of Oxford, UK. His research focuses on nanomaterials and heterogeneous catalysis with applications in energy and environmental sustainability. His work includes the development of catalytic, photocatalytic, and electrocatalytic technologies for green chemistry, fine chemical synthesis, energy storage, and ammonia and hydrogen technologies, including fuel cells. He has particular expertise in the design and architecture of nanocatalysts, coupled with in situ diffraction and spectroscopic techniques, to elucidate catalytic mechanisms. |
1. Introduction
Plastics have become deeply integrated into modern life due to their large-scale production and advantageous properties, including low-cost, high strength-to-weight ratio, thermal and electrical insulation, and durability.1–3 These characteristics have led to their widespread use in daily life, spanning diverse sectors such as packaging, textiles, building and construction, and more.4,5 However, despite their pivotal role in modern society, the massive volume of plastic production, coupled with inadequate waste management, has resulted in severe environmental and public health concerns.6–8 It has been estimated that global plastic production reached approximately 400 million metric tons (Mt) in 2022, yet only about 9% of this total has been recycled, while 12% has been incinerated, and the remaining 79% has been accumulated in landfills or the natural environment.5,9 Furthermore, the vast majority of plastics are derived from fossil fuels, with around 7% of global crude oil and natural gas resources being consumed for their production.10 As a result, the low recyclability of plastic waste not only exacerbates environmental issues but also contributes to significant material losses. Addressing the plastic waste crisis from both environmental and resource sustainability perspectives has therefore become a pressing global challenge.Traditional methods of plastic waste disposal, such as landfills and incineration, are cost-effective but present considerable environmental risks. For instance, landfills can lead to soil and water contamination, while incineration produces harmful gases that degrade air quality and contribute to climate change.3,11–13 In contrast, recycling approaches including mechanical recycling and chemical recycling methods such as pyrolysis and gasification are more environmentally friendly and capable of producing valuable products. However, these processes are energy- and labor-intensive, with high reaction temperature requirement for pyrolysis and gasification and significant labor demands for the thorough sorting and collection of plastic waste in mechanical recycling.3,14–16 Catalytic cracking has emerged as a promising alternative for plastic upcycling, offering potential to convert plastic waste into higher-value products at relatively lower temperature (around 400 °C) and with reduced environmental impact. This method has gained significant attention in recent years, but challenges remain, particularly concerning catalyst deactivation due to carbon deposition, poisoning by contaminants, and pore blockage. These issues necessitate additional treatment for both spent catalysts and feedstock, thereby increasing operating costs, complicating the process, and potentially degrading product quality.3 Given these challenges, there is an urgent need for the development of more sustainable and environmentally friendly chemical upcycling strategies that operate under milder conditions.
Among the various categories of plastics, polyolefin (PO) plastics, including low-density polyethylene (LDPE), linear low-density polyethylene (LLDPE), high-density polyethylene (HDPE), polypropylene (PP), and polystyrene (PS), collectively account for over 60% of global plastic consumptions.5,17 As major contributors to plastic waste, PO plastics present distinct challenges for recycling due to their unique physical and chemical characteristics, which differentiate them from more readily recyclable plastics such as polyethylene terephthalate (PET).18 One of the major challenges stems from their high molecular weight, which hampers heat and mass transfer during melting and inhibits the accessibility of catalytic active sites.19 Thus, there is a critical need to prioritize the development of efficient PO upcycling methods under mild conditions.
In recent years, significant progress has been made in the field of plastic upcycling, particularly with regard to the transformation of PO plastics under mild reaction conditions (typically below 300 °C). Numerous reviews have been published that summarize these advancements, focusing on various aspects such as catalyst development, reaction mechanism, process and reaction engineering, and diverse products through thermal catalysis, electrocatalysis, photocatalysis, and even emerging techniques like microwave-catalysis.1,3,20–27 However, many of these plastic conversion processes under mild conditions are challenging to achieve without the use of effective co-reactants, such as hydrogen, short-chain alkanes and alkenes, oxygen, and CO2. Furthermore, the introduction of different co-reactants can lead to changes in thermodynamics and distinct reaction pathways, profoundly influencing both process design and catalyst development. Selecting the appropriate co-reactants can also open avenues to broaden the product spectrum by incorporating heteroatoms, such as oxygen and nitrogen, thus driving PO upcycling towards a wider array of valuable products.
Although co-reactants play a crucial role in dictating reaction mechanisms and outcomes in plastic upcycling, there remains a significant gap in the literature regarding the systematic classification and discussion of their impact. In this review, we aim to bridge this gap by categorizing recent advances in PO upcycling under mild conditions according to the specific co-reactants employed. The associated experimental results and reaction conditions are comprehensively summarized in Table 1, with the overarching motivation depicted in Fig. 1. Notably, we emphasize the catalytic chemistry that underpins their transformations. Each section provides a detailed examination of the underlying reaction mechanisms, conditions, catalysts, and products associated with particular co-reactants, followed by a comparative analysis of processes conducted without co-reactants under mild conditions. Finally, we highlight key opportunities and challenges that the field of PO upcycling must address in the pursuit of more sustainable and efficient plastic upcycling technologies.
| Catalyst system | Plastic(s) | Co-reactant (s) | Plastic/catalyst mass ratio | Temperature (°C) | Pressure (bar) | Time (h) | Major product | Ref. |
|---|---|---|---|---|---|---|---|---|
| Pt/USY | PE, PP | H2 | 20 | 280 | 30 | 3 | C5–12 | 28 |
| Pt/WZr | LDPE | H2 | 10 | 250 | 30 | 2 | C7–12 | 29 |
| PtSnCe/SiAl | HDPE | H2 | 10 | 270 | 30 | 2 | C5–12 | 30 |
| Ru/ZrO2 | LDPE | H2 | 34 | 400 | 35 | 2 | C5–21 | 31 |
| NiCoAlx | LDPE | H2 | 20 | 280 | 20 | 4 | Liquid-range hydrocarbons | 32 |
| Ru/SBA | LDPE | H2 | 30 | 230 | 20 | 5 | Liquid fuels | 33 |
| Ru/TiO2 | PP | H2 | 5 | 240 | 20 | 4 | C1–5 | 34 |
| Ru/TiO2-A-SG | PP | H2 | 7 | 260 | 30 | 18 | Liquid-range hydrocarbons | 35 |
| WOx/SiO2 and Pt/Al2O3 | LDPE | n-Decane | 0.16 | 300 | 30 | 3 | Short-chain alkane | 36 |
| SnPt/γ-Al2O3 and Re2O7/γ-Al2O3 | PE | n-Pentane | n/a | 200 | 40 | 15 | Liquid alkanes | 37 |
| MTO/Cl–Al2O3 | PE | Ethylene | 1.67 | 100 | 1 | 5 | Propylene | 38 |
| Na/Al2O3 and WO3/SiO2 | PE | Ethylene | 1.25 | 320 | 15 | 1.5 | Propylene and isobutylene | 39 |
| Ru/TiO2 | PE | O2 (in air) | 2 | 160 | 15 | 24 | Dicarboxylic acids | 40 |
| NiO/TiO2 | PS | O2 (in air) | 2 | 200 | 10 | 18 | Benzoic acid | 41 |
| Co/MCM-41 | PE | O2 | 10 | 125 | 10 | 12 | Dicarboxylic (C10–20) acids | 42 |
| Cu–Fe3O4 and Zn/ZSM-5 | PE | CO2 | 5 | 360 | 30 | 1 | Aromatics | 43 |
| CuZnZrOx and ZSM-5 | PE | CO2 | 15 | 380 | 5 | 3.3 | BTX | 44 |
| Ga/ZSM-5-meso | LDPE | CO2 | 1.33 | 550 | n.a. | n.a. | BTEX | 45 |
| Pt/MnOx and ZSM-5 | LDPE | CO2 | 2.5 | 280 | 10 | 3 | BTX | 46 |
| Ru/SiO2 | PS | Methanol | 10 | 280 | 40 | 6 | Alkylbenzenes | 47 |
| MnO2 | PS | CO(NH2)2, O2 | 60 | 200 | 15 | 36 | Benzonitrile, benzamide | 48 |
| Pt/γ-Al2O3 | PE | None | 0.59 | 280 | n.a. | 24 | Long-chain alkylaromatics and alkylnaphthalenes | 49 |
| Pt/F-Al2O3 | PE | None | 0.6 | 280 | n.a. | 8 | Alkylaromatics | 50 |
| Ru/HZSM-5(300) | HDPE | None | 10 | 280 | 20 | 24 | C1–15 | 51 |
| Layered self-pillared zeolite | PE | None | 5 | 240 | 0.1 | 4 | C4–12 isoalkanes | 52 |
| ZSM-5 nanosheet | LDPE | None | 5 | 280 | 0.1 | 7 | C3–6 olefins | 53 |
 | ||
| Fig. 1 Progress in plastic upcycling according to their co-reactants. | ||
2. Polyolefin upcycling with co-reactants
2.1 Hydrogen
Although the use of appropriate catalysts significantly reduces the reaction temperatures during catalytic cracking, the process still necessitates relatively high thermal conditions, typically at around 400 °C, which, while lower than those required for pyrolysis (commonly above 500 °C), are still substantial.3 The need for these elevated temperatures arises from the inherent thermodynamic nature of both pyrolysis and catalytic cracking, where endothermic reactions necessitates high temperatures to cleave the robust C–C bond in PO. To overcome this temperature barrier and facilitate PO plastic upcycling under milder conditions, the introduction of high-pressure hydrogen has been explored. This strategic shift directs the reaction mechanism toward hydrocracking or hydrogenolysis pathways, where the overall process becomes exothermic. As a result, these hydrogen-assisted routes enable PO upcycling to occur at lower temperatures, typically below 300 °C, offering a more energy-efficient alternative for PO plastic waste upcycling.27Hydrocracking conventionally utilizes bifunctional catalysts, with metal sites facilitating dehydrogenation and hydrogenation, while acid sites drive the cleavage of C–C bonds. In some cases, hydrocracking can also be initiated using monofunctional acid catalysts, resembling catalytic cracking. Different from catalytic cracking, under high pressure, Brønsted acid sites (BASs) become significantly more active, altering both product selectivity and yield. Additionally, hydrogen serves a critical role in suppressing coke formation on the catalyst surface, thereby enhancing the catalysts' durability and extending their operational life. Hydrogenolysis, in contrast to hydrocracking, follows metal-catalyzed pathways involving alkylidyne intermediates, diverging from carbonium ion-driven mechanisms observed in hydrocracking. This mechanistic variation results in a distinct product profile, with hydrogenolysis often leading to elevated methane production due to terminal C–C bond cleavage and the removal of methyl branching reactions that are thermodynamically disfavored in hydrocracking.27,54,55 Despite these mechanistic disparities, both processes yield analogous value-added products, such as fuel-range hydrocarbons (e.g., naphtha, gasoline, jet fuel, and diesel), alongside waxes and lubricants.3,27
Recent innovations in hydrogen-assisted upcycling of PO plastics have concentrated on two major fronts: the development of more efficient and stable catalytic systems, and a deeper understanding of structure–performance relationships. In hydrocracking, the focus has been refining metal–acid proximity, fine-turning the metal–acid balance (MAB), and intensifying the synergistic interplay between metal and acid sites. Conversely, hydrogenolysis improvements have been achieved through precise control of the geometric and electronic properties of active metals, and the employment of confinement effects to augment the catalytic performance.
A notable example comes from Han et al., who achieved significant improvements in hydrocracking performance by meticulously controlling the nanoscale proximity between Pt nanoparticles and USY zeolites.28 Their study revealed that positioning Pt nanoparticles on the surface of USY led to a remarkable 450% increase in catalytic activity compared to configurations where Pt nanoparticles were embedded within the USY channels or physically mixed with zeolite. While Pt nanoparticles within the USY pores exhibited superior activity for smaller n-alkane substrates (C6 and C8), their catalytic activity dropped significantly for larger substrates, such as larger n-alkanes or PE. This reduction in activity was attributed to diffusion constraints, as the bulk molecules faced difficulties accessing the Pt nanoparticles embedded deep within the narrow USY channels, impeding the formation of alkene intermediates as illustrated in Fig. 2(a). Conversely, when the proximity was increased from the nanoscale to the microscale (i.e., physical separation of Pt and USY), conversion also decreased, as the extended distance hindered the diffusion of alkene intermediates from Pt sites to acid sites. Liu et al. similarly underscored the critical role of proximity in bifunctional catalytic systems.56 Their findings demonstrated that in Pt/WZr + HY systems, the spatial separation of Pt nanoparticles from the zeolite acid sites facilitated more precise control over reaction intermediates, resulting in improved selectivity towards fuel-range hydrocarbons. By contrast, Pt nanoparticles in close proximity to zeolite acid sites induced excessive cracking, thereby shifting product selectivity towards lighter hydrocarbons (C5–C7). These studies collectively highlight the pivotal role of the metal–acid proximity in modulating the accessibility of active sites, which directly influences the overall efficiency, selectivity, and stability of the hydrocracking process.
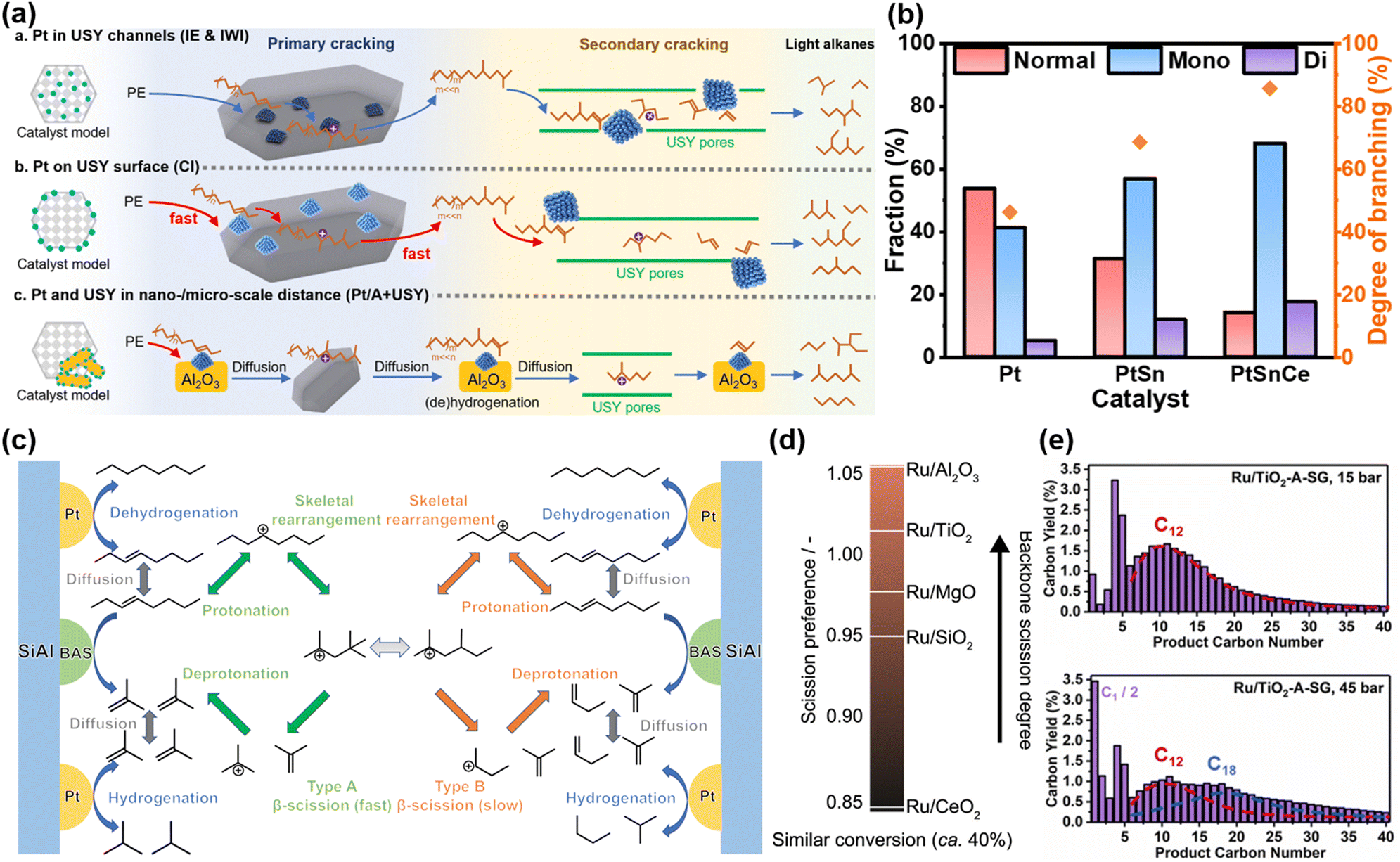 | ||
| Fig. 2 Recent advances in PO plastic upcycling via hydrogen-assisted processes. (a) Proposed mechanism of hydrocracking of PE on Pt/USY catalysts with varying metal–acid proximities. Reproduced with permission from the Royal Society of Chemistry, copyright 2024.28 (b) and (c) Investigation of the hydrocracking reaction mechanism. (b) Catalytic tests using n-octadecane as the substrate. (c) Reaction mechanism analysis using n-octane as the reactant model. Reproduced with permission from American Chemical Society, copyright 2023.30 (d) Scission preferences in PP hydrogenolysis on supports with different basicities. Reproduced with permission from Elsevier, copyright 2023.34 (e) Product distribution at varying hydrogen pressures. Reproduced with permission from Elsevier, copyright 2023.35 | ||
Aside from the importance of metal–acid proximity, the role of MAB has also been investigated for its impact on hydrocracking performance. Vance et al. reported that an increase in the MAB drives product selectivity (C1–C35) towards heavier hydrocarbons, while simultaneously promoting greater branching in the residual polymer.29 However, unlike the hydrocarbon of smaller alkanes, where a higher MAB typically enhances the selectivity for branched products, the overall fraction of branched isomers in the liquid phase remains largely independent of MAB in polymer hydrocracking. This deviation can be attributed to the preferential adsorption of the larger polymer chains onto the catalyst's active sites, which limits the accessibility of smaller liquid products to these sites, thus resulting in a uniform distribution of branched products across varying MAB.
Regarding the synergetic interactions between metal and acid sites, our recent work presented an optimized approach to amplify them, significantly elevating the hydrocracking performance.30 In the initial Pt/SiAl system, a high solid yield and low selectivity for fuel-range products exhibited significant limitations. To address this, the introduction of Sn as a promoter enhanced the dehydrogenation and hydrogenation activities, effectively lowering the solid yield while increasing selectivity towards gasoline-range products. Deeping this exploration, the subsequent incorporation of a finely tuned amount of Ce promoter further augmented the catalytic performance, achieving gasoline yields surpassing 77 wt%. This substantial improvement can be ascribed to a combination of the electronic modulation of metallic Pt and the enhanced strength and concentration of BASs. Mechanistic investigation revealed a distinct evolution in the reaction pathways across different catalytic systems. In the Pt/SiAl catalyst, the strong adsorption of HDPE and its fragments led to more extensive cracking, followed by a type B β-scission mechanism, which produced higher gaseous yields. The introduction of Sn in the PtSn/SiAl system mitigated this extensive adsorption by reducing the Pt particle size and altering its electronic properties, promoting a balance between type A and B β-scission pathways. The further addition of Ce in the PtSnCe/SiAl catalyst amplified the synergistic effect between Pt and BASs, favoring type A β-scission as the dominant reaction pathway. This shift is significant as type A β-scission generally produces fewer gaseous by-products and leads to a more selective conversion into valuable gasoline-range hydrocarbons. These catalyst-induced modulations of reaction pathways highlight the critical role of rational catalyst design in precisely tuning the reaction kinetics and controlling the product selectivity. The progressive transition in reaction mechanisms was evaluated by measuring the isomerization degree analysis conducted with n-octadecane as a model substrate, as illustrated in Fig. 2(b) and (c). Furthermore, the strong interaction between Pt, Sn, and Ce played a crucial role in preventing sintering and suppressing coke formation, thereby contributing to enhanced stability.
In the realm of hydrogenolysis, significant advancements have been made through the deliberate modulation of the geometric and electronic properties of active metal sites to optimize catalytic performance. A prime example is the work by Tamura et al., who systematically investigated the impact of the Ru particle size on hydrogenolysis efficiency.31 Their findings demonstrated a volcano-shaped correlation between the Ru particle size and catalytic conversion, with peak activity observed at an optimal particle size of around 2.5 nm. The turnover frequency (TOF) per surface Ru atom increased with the particle size but exhibited a sharp decline for smaller Ru particle sizes. This trend is attributed to the prevalence of plane sites of Ru particles as the primary active centers, where multipoint interactions between the Ru surface and PE molecules facilitated improved adsorption and catalytic activity. However, as the Ru particle size increases, metal dispersion diminishes, reducing the number of accessible active sites and thereby resulting in the characteristic volcano-shaped conversion profile.
Beyond geometric optimization, strategic turning of the electronic properties of active sites has also emerged as a pivotal factor in enhancing the hydrogenolysis efficiency. A notable contribution in this area comes from Chu et al., who introduced a Ni promoter into a CoAl-LDH framework to engineer the electronic structure of Co centers.32 This modification induced electronic deficiency at the Co sites through electron redistribution between Co and Ni. Such electronic tuning not only suppressed the solid yield but also reduced the number of C–C cleavages, attributed to the disruption of the continuous Co–Co site network by the introduction of Ni, which inherently exhibits weaker C–C bond cleavage activity. Furthermore, the incorporation of Ni improved the hydrogenation capability of catalysts, effectively mitigating over-hydrogenolysis to methane and selectively driving the formation of liquid fuel products.
In addition to tuning metal active sites, the confinement effect has emerged as a pivotal strategy for optimizing the hydrogenolysis performance. Kang et al. demonstrated that the precise encapsulation of Ru nanoparticles uniformly within the mesoporous channels of SBA-15 induced a pronounced ‘‘entropy confinement effect’’ on polymer chains.33 This confinement substantially reduced the entropy of chains and facilitated nearly a twofold increase in solid conversion compared to conventional Ru/SBA where Ru particles were more diffusely distributed both inside and outside the channels. By restricting the conformational freedom of macromolecular chains, this confinement effect stabilizes transition states, enhancing the adsorption process and significantly boosting the catalytic efficiency.
On top of advancements in the development of more efficient and stable catalytic systems, substantial progress has been made in elucidating clear structure–performance relationships. A prominent example is the work by Jaydev et al., who introduced the novel metric of “scission preference” to quantify the ratio of backbone scission to demethylation events during PP hydrogenolysis.34 This metric provides a crucial tool for disentangling the competing scission pathways and identifying catalysts that selectively favor backbone cleavage over demethylation. Leveraging this metric, the study established a robust correlation between the surface basicity of metal oxide supports and scission preference, demonstrating that lower surface basicity consistently favors backbone scission as depicted in Fig. 2(d). The breakthrough in catalyst evaluation offers valuable guidance for catalyst design and performance in PP hydrogenolysis.
While hydrocracking and hydrogenolysis are traditionally viewed as mechanistically distinct, Chen et al. revealed that both pathways can occur concurrently over a Ru/TiO2-A-SG catalyst during PP hydroconversion.35 Their investigation into the effect of reaction conditions showed that hydrocracking dominates at low hydrogen pressure, whereas hydrogenolysis becomes increasingly significant at high hydrogen pressure, as illustrated by the product distribution in Fig. 2(e). Furthermore, they demonstrated that a higher Ru loading dramatically enhances the contribution of hydrogenolysis, evidenced by an increase in methane production. This research provides profound insights into the mechanistic overlap of these two pathways and underscores the potential for tuning reaction routes through the manipulation of reaction parameters.
2.2 Short-chain hydrocarbons (alkanes and alkenes)
Short-chain hydrocarbons, particularly alkanes and alkenes, have gained prominence as effective co-reactants in facilitating PO plastic upcycling under mild conditions. These co-reactants primarily drive metathesis reaction pathways, where C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds, either inherently present in the polymer structure or formed through catalytic dehydrogenation, undergo cross-metathesis with additional olefins, such as solvents or ethylene. Unlike the exothermic nature of hydrogen-assisted hydrocracking and hydrogenolysis pathways, metathesis-driven chain disproportionation is generally thermoneutral, allowing the process to proceed at moderate temperatures (typically below 300 °C). In the absence of hydrogen, the use of excess light alkanes or alkenes is necessary to shift the equilibrium towards the production of intermediate-length hydrocarbons by reacting with macromolecular polymers. Recent progress in this field has focused on utilizing solvents to produce fuel-rang products and ethylene to generate valuable intermediates like propylene and butene.
C bonds, either inherently present in the polymer structure or formed through catalytic dehydrogenation, undergo cross-metathesis with additional olefins, such as solvents or ethylene. Unlike the exothermic nature of hydrogen-assisted hydrocracking and hydrogenolysis pathways, metathesis-driven chain disproportionation is generally thermoneutral, allowing the process to proceed at moderate temperatures (typically below 300 °C). In the absence of hydrogen, the use of excess light alkanes or alkenes is necessary to shift the equilibrium towards the production of intermediate-length hydrocarbons by reacting with macromolecular polymers. Recent progress in this field has focused on utilizing solvents to produce fuel-rang products and ethylene to generate valuable intermediates like propylene and butene.
Ellis et al. introduced the first fully heterogenous catalytic system for PE decomposition, employing a tandem dehydrogenation and olefin cross-metathesis pathway.37 In this innovative system, PtSn/Al2O3 catalyzed dehydrogenation and hydrogenation reactions, while Re2O7/Al2O3 facilitated olefin metathesis, with n-pentane as the co-reactant. This ‘‘olefin-intermediate’’ mechanism effectively activated the aliphatic PE backbone via dehydrogenation, enabling subsequent selective cleavage of the resulting olefin intermediate, as illustrated in Fig. 3(a). This approach yielded a remarkable 73% reduction in the molecular weight of the PE substrate at 200 °C after 15 hours. Informed by these insights, Kim et al. refined the system by employing WOx/SiO2 as a metathesis catalyst in conjunction with Pt/Al2O3 for dehydrogenation.36 To further enhance the catalytic stability, zeolite 4A was introduced into the reaction environment, serving to protect metathesis active sites from in situ-generated oxygenates. This strategic adjustment substantially improved the catalyst's durability. Furthermore, the study demonstrated that increasing the pretreatment temperature significantly boosted both conversion and product yields. The incorporation of longer-chain alkane co-reactants also enhanced their catalytic reactivity, shifting the molecular weight distribution towards heavier hydrocarbons post-reaction, as illustrated in Fig. 3(b).
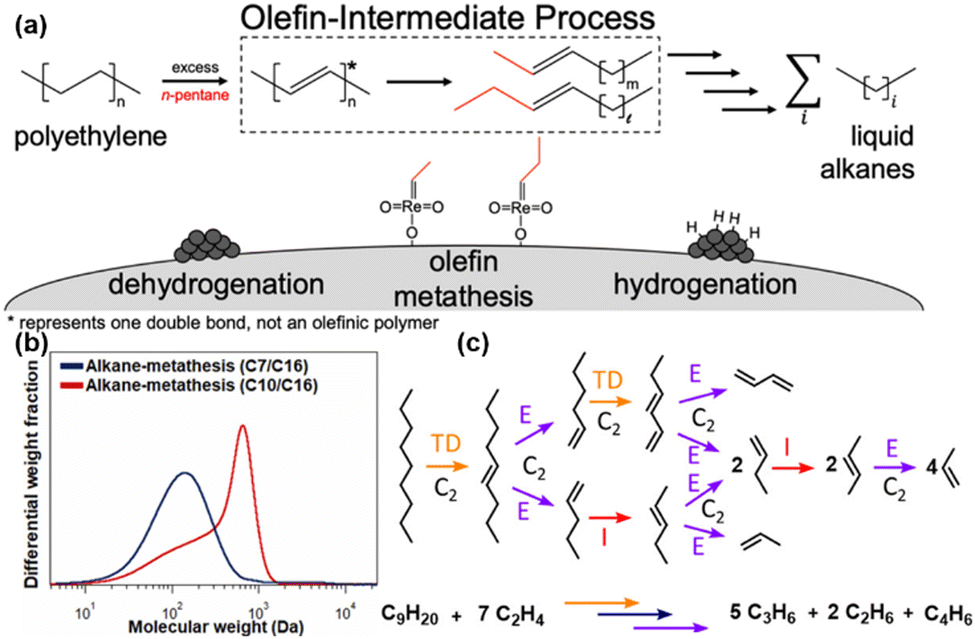 | ||
| Fig. 3 Recent advances in PO plastic upcycling via short-chain hydrocarbon-assisted processes. (a) Schematic representation of the ‘‘olefin-intermediate’’ process. Reproduced with permission from American Chemical Society, copyright 2021.37 (b) Molecular weight distribution after the alkane metathesis reaction. Reproduced with permission from Elsevier, copyright 2022.36 (c) Example of the complete conversion of n-nonane into five molecules of propylene, two molecules of ethane, and one molecule of butadiene via two transfer dehydrogenation (TD) steps, two isomerization (I) steps, and eight ethenolysis (E) steps. Reproduced with permission from American Chemical Society, copyright 2022.38 | ||
Wang et al. pioneered a novel metathesis pathway for the efficient transformation of PE into propylene through co-feeding excess ethylene.38 This strategy employed a tandem catalytic process, integrating rapid olefin metathesis (ethenolysis) with a rate-limiting isomerization step, resulting in selective propylene production as the major product, as illustrated in Fig. 3(c). For fully saturated PE, an initial dehydrogenation step was necessary to introduce unsaturation, enabling subsequent ethenolysis and isomerization. Their approach, validated using both homogeneous and heterogeneous catalysts, achieved exceptional propylene selectivity (≥94%) in a semi-continuous operation, offering a transformative solution to advancing circular economy initiatives for PO upcycling. Expanding upon these findings, Conk et al. optimized this methodology by employing a dual-catalyst system composed of Na/Al2O3 and WO3/SiO2, enabling the direct conversion of PE and PP into valuable light olefins such as propylene and isobutylene, without the prerequisite of pre-dehydrogenation.39 The catalytic approach synergistically harnessed the ability of Na/Al2O3 to cleave polymer chains to form olefins, while the robust olefin metathesis activity of WO3/SiO2 drove further conversion to light olefins. The optimized process exhibited enhanced yields of target olefins, effectively addressing key limitations such as low product selectivity, high greenhouse gas emissions, and reliance on expensive single-use catalytic systems.
2.3 Oxygen
Oxygen, as a highly versatile co-reactant, plays a pivotal role in the oxidative upcycling of PO, enabling the incorporation of oxygenated functional groups and thereby broadening the range of upcycled products. Unlike co-reactants such as hydrogen or short-chain alkanes and alkenes, oxygen-facilitated upcycling can occur under significantly milder conditions, often at temperatures below 200 °C. The ability to use ambient air as an oxygen source further enhances the environmental and economic sustainability of the process by eliminating the need for purified gases or other costly inputs. Notably, PO plastic can degrade in the presence of oxygen even in the absence of a catalyst under mild conditions, something not possible with hydrogen or alkanes and alkenes as co-reactants. However, while the exothermic nature of the oxidation reaction permits lower operating temperatures and non-catalytic degradation, uncontrolled oxidation can lead to over-oxidation, yielding low-value products like CO2. Therefore, the development of selective catalysts and fine-tuning of reaction conditions are crucial to enhancing both conversion and product selectivity towards high-value compounds.Regarding the reaction mechanism underlying oxygen-assisted PO upcycling, radical intermediates play a pivotal role. Two primary radicals are involved: alkyl radicals, formed via the abstraction of H atoms from the C–H bond, and peroxyl radicals, generated from the reaction with molecular O2. These radical intermediates drive the oxidation transformation of polymer chains, often involving intramolecular hydrogen abstraction, condensation, and decomposition reactions, leading to primary products like fatty acids with varying carbon chain lengths (Fig. 4(a)). In this aspect, Smak et al. conducted a seminal study on non-catalytic oxidative upcycling of PE, providing critical mechanistic insights through advanced analytical techniques, including 2D nuclear magnetic resonance (2D-NMR) and in situ transmission infrared (IR) spectroscopy.57 Their findings revealed that the thermal initiation of PE leads to the formation of aliphatic radicals, which readily incorporate oxygen at radical sites. This incorporation triggers subsequent intramolecular hydrogen transfers, leading to the generation of hydroperoxyl or terminal carbonyl groups. As the reaction progresses, these intermediates undergo further decomposition, yielding alcohols, ketones, and acids, as well as higher-order oxidation products.
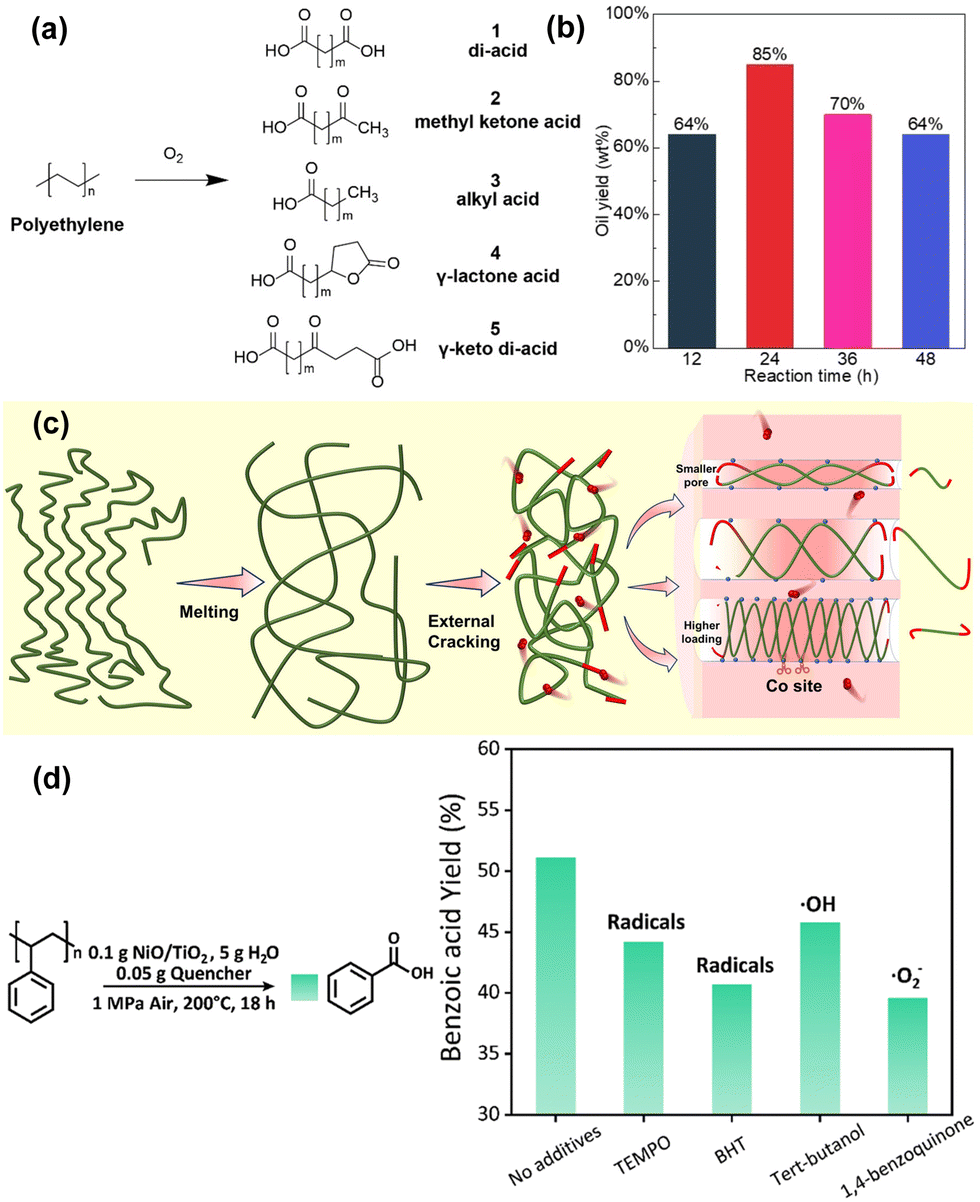 | ||
| Fig. 4 Recent advances in PO plastic upcycling via oxygen-assisted processes. (a) Common products of PE oxidative upcycling. Reproduced with permission from Wiley-VCH, copyright 2023.57 (b) Analysis of the oil products by oxi-upcycling LDPE pellets in terms of Mw and the proton integration ratio. Reproduced with permission from Wiley-VCH, copyright 2023.40 (c) Schematic degradation mechanism of PE on Co/MCM-41. Reproduced with permission from Wiley-VCH, copyright 2024.42 (d) Radical inhibition experiments to scavenge the intermediate radicals (TEMPO: 2,2,6,6-tetramethyl-1-piperidinyloxyl; BHT: 2,6-di-tert-butyl-4-methylphenol). Reproduced with permission from the Royal Society of Chemistry, copyright 2024.41 | ||
In the context of catalytic oxidative PO upcycling, the role of active metal sites becomes crucial. These metal centres, particularly noble metals, facilitated oxygen adsorption, activation, and subsequent formation of reactive radical species. For instance, Ru-based catalysts have been extensively studied due to their ability to efficiently activate oxygen and promote the oxidative degradation of polymers.58,59 Wang et al. demonstrated the remarkable catalytic performance of Ru/TiO2, achieving 95% conversion of PE within 24 hours under 1.5 MPa of air at 160 °C.40 Their analysis showed a gradual increase in the ratio of α-carbonyl-H to total H, as depicted in Fig. 4(b), indicating progressive oxidation and a shift towards selective formation of aliphatic dicarboxylic acids.
Non-noble metals, such as Co, Mn, and Ni, have garnered significant attention as viable alternatives to nobler metals for facilitating oxygen adsorption and activation in the oxidative plastic upcycling process.60,61 Zhang et al. demonstrated that Co-doped MCM-41 molecular sieves exhibited remarkable catalytic efficiency, achieving 85.9% carbon conversion, while 58.9% of the products comprising valuable long-chain dicarboxylic acids under relatively mild reaction conditions (1 MPa O2 at 125 °C).42 The proposed mechanism for PE degradation, illustrated in Fig. 4(c), was corroborated by density functional theory (DFT) calculations using pentane as a model compound. In this system, PE undergoes thermal melting at the set temperature, enhancing its contact with a catalytic surface. The primary cracking of polymer is initiated on Co sites located on the external surface of the catalyst, while oxygen, adsorbed on Co(II) sites, plays a crucial role in facilitating further oxidative degradation. This stepwise process, driven by the smaller oxygen species, leads to the generation of oxygen radicals that actively participate in the breakdown of polymer chains. The presence and role of reactive oxygen species (ROS), especially ˙O2− radicals, in oxidative polymer degradation were further substantiated by Sun et al., who employed NiO/TiO2 catalysts to convert PS into benzoic acid.41 The mechanistic role of ˙O2− as the dominant oxidative species was experimentally verified through the use of radical scavengers, which significantly reduced the yield of benzoic acid, as depicted in Fig. 4(d). The introduction of 1,4-benzoquinone, in particular, resulted in the most substantial reduction (∼12%) in product yield, providing strong evidence that ˙O2− radicals are the primary species driving the depolymerization of PS. This study highlights the pivotal role that ROSs play in facilitating oxidative polymer degradation, particularly in non-noble metal-catalyzed systems, and underscores the potential of tailoring such species to achieve targeted upcycling products.
While the aforementioned processes demonstrated significant potential as single-step transformations with minimal need for extensive pre-treatment or post-treatment, the integration of additional steps can broaden the range of products and enhance the overall economic viability of plastic upcycling techniques. Xu et al. provided a compelling example of this approach with their two-step process for converting PO-derived waxes into high-value fatty acids.62 In the first step, temperature-gradient thermolysis selectively breaks down PE and PP into waxes, which are subsequently subjected to oxidation using oxygen and a manganese stearate catalyst. This oxidative step yields a wide distribution of fatty acids with varying carbon chain lengths, which can be further processed through saponification to produce surfactants. The resultant surfactants not only hold a significantly higher economic value but also have a larger market demand compared to traditional petrochemical-derived products, such as fuels or alkylaromatics. This multi-step approach offers a more comprehensive and economically attractive pathway for valorizing PO plastic waste and underscores the potential for innovation in the design of PO plastic upcycling processes. The advancement of novel oxidative upcycling using oxygen, leveraging noble and non-noble metal catalysts, represents a significant frontier in the quest to develop environmentally benign and economically sustainable methods for addressing the global plastic waste challenge. Despite recent progress, in-depth mechanistic studies especially targeting PO feedstocks still remain limited, and the development of highly efficient and selective catalytic systems is still in its early stages. This presents significant opportunities for advancing both the fundamental understanding and practical applications of oxygen-assisted upcycling technologies targeting high conversion and high selectivity products.
2.4 Carbon dioxide as a co-reactant
Cracking plastics using CO2 as a co-reactant in recent years presents an innovative, dual-purpose strategy to mitigate both plastic and CO2 problems simultaneously. However, given the inherent chemical inertness of CO2, the reaction conditions are usually more demanding relative to other co-reactants. Typically, temperatures exceeding 300 °C and pressures near 3 MPa are necessary to overcome the thermodynamic barriers. Additionally, these reactions often rely on a bifunctional or tandem catalytic system, where the plastic decomposition and CO2 activation processes are spatially and functionally separated. This is generally achieved by coupling zeolites, which drive plastic decomposition, with metal oxides that activate CO2. The reactions are interconnected by hydrogen spillover between the two catalyst phases. Such metal oxide/zeolite catalyst pairings are already well-established in the CO2 hydrogenation domain, where metal oxides convert CO2 into intermediates like methanol, which are subsequently transformed into hydrocarbons via BASs in the zeolite.63–66 In these systems, CO2 plays a dual role, not only assisting in hydrocarbon production but also in generating reactive oxygen species to mitigate coke deposition, thereby improving the long-term stability of the catalyst.67 Moreover, CO2 has been shown to be incorporated into aromatic products when coupled with n-butane through the formation of methyl-substituted lactones, and it acts as a hydrogen acceptor in ethane-to-ethene transformations, preventing excessive C–C bond cleavage.68,69In the context of plastics, the introduction of CO2 during co-conversion stabilizes the hydrogen generated during plastic aromatization, facilitating a forward-driving reaction that yields valuable unsaturated products such as benzene, toluene, and xylene (BTX) (Fig. 5(a)). For example, Chen et al. exploited CO2 as a mild oxidant and hydrogen acceptor in a Cu–Fe3O4 and Zn/ZSM-5 catalyst system, enabling efficient PE aromatization.43 This clever coupling exceeded the theoretical selectivity threshold of 50%, achieving over 64% aromatic selectivity under 3 MPa of CO2 at 390 °C. Notably, as CO2 conversion increased from approximately 5% to 15%, the yield of aromatic compounds rose proportionally, reaching around 12% at 300 °C. Isotope-labelling studies confirmed that CO2 was not incorporated into the aromatic rings or alkyl chains of the final products, but was instead converted into CO via reverse water–gas shift (RWGS) reaction. Similarly, Liu et al. found no evidence of carbon from CO2 being integrated into aromatic species using a CuZnZrOx and ZSM-5 system.44
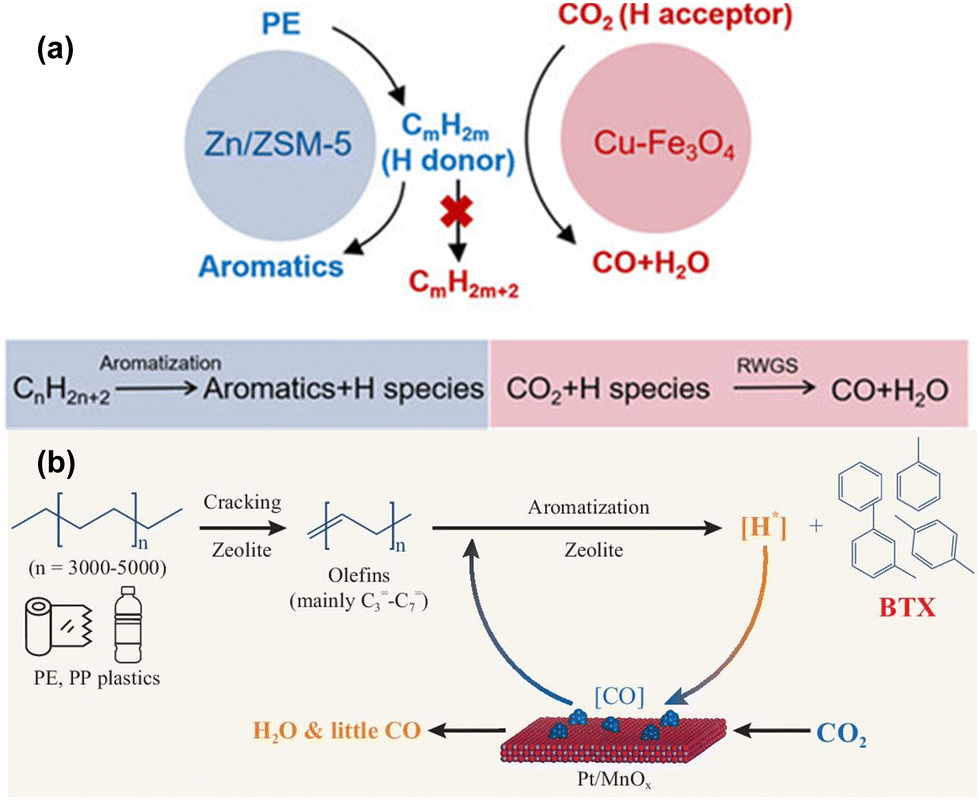 | ||
| Fig. 5 Recent advancements in the design of multifunctional and tandem catalysts for PO plastic upcycling using carbon dioxide as a co-reactant. (a) Proposed mechanism for the coupling reaction of CO2 with PE. Reproduced with permission from Chinese Chemical Society, copyright 2023.43 (b) Proposed reaction pathway for CO2-facilitated upcycling of PO with CO2 incorporated into BTX products. Reproduced with permission from Oxford University Press, copyright 2023.46 | ||
Advancing beyond these initial findings, Zhang et al. integrated the catalytic roles typically performed by separate catalysts into single, multifunctional mesoporous Ga/ZSM-5 catalysts, successfully achieving an aromatics yield of 63.3%.45 The products are mainly benzene, toluene, ethylbenzene, and xylene (BTEX). Although CO2 incorporation into product carbon chains was not observed, these studies laid critical groundwork for future breakthroughs. A significant leap was made by Ding et al., who demonstrated that up to 90% of the consumed CO2 could be incorporated into aromatic products using a bifunctional Pt/MnOx-ZSM-5 catalyst system (Fig. 5(b)).46 Direct isotope-labelling provided unequivocal evidence of 13C from 13CO2 being present in both benzene and toluene molecules, which marked a significant development in CO2 valorization during plastic upcycling. Interestingly, while the reaction mechanism involving CO2 and plastic conversion mirrored those seen in the earlier studies, this system uniquely facilitated the further transformation of CO into aromatics. The isotope experiments conclusively demonstrated that CO2 hydrogenation intermediates like methanol were not involved in the reaction pathways, confirming that Pt/MnOx catalyst promotes RWGS reaction by scavenging hydrogen and activating CO2. This catalytic system achieved an impressive 60% yield of aromatics at a notably lower temperature of 300 °C, making it a promising approach for scalable plastic upcycling and CO2 utilization.
These findings underscore the transformative potential of CO2 as a co-reactant in plastic upcycling, not only enabling simultaneous waste mitigation but also expanding the scope of products that can be derived from plastic upcycling. The ability to efficiently utilize CO2 in this context—whether as a hydrogen acceptor, a reactant for further chemical transformations, or a stabilizer in aromatization reactions—opens new avenues for circular carbon economies. However, further research is needed to refine these catalytic systems, particularly in optimizing the integration of CO2 into the carbon backbone of final products, and to explore how CO2 can be employed to selectively tailor the product distribution towards high-value chemicals.
2.5 Other co-reactants
Beyond conventional co-reactants like H2, short-chain alkanes or alkenes, O2, and CO2, emerging strategies for PO upcycling now include alternative co-reactants, multi-step processes, and synergistic application of multiple co-reactants. These approaches build upon earlier foundational research, pushing the boundaries of catalytic plastic upcycling by exploring more complex reaction pathways and leveraging novel co-reactant combinations.In Section 2.1, we discussed the direct use of hydrogen as a co-reactant. However, other hydrogen-donating compounds, such as methanol, can serve as alternative hydrogen sources during plastic decomposition. For instance, Zeng et al. demonstrated the efficiency of methanol-assisted upcycling of PS using a Ru/SiO2 catalyst.47 The coupling of methanol decomposition and PS hydrogenolysis not only enabled a high liquid yield of liquid products, predominantly monocyclic aromatics but also rendered the process thermodynamically favorable at a moderate temperature of 280 °C. This approach notably shifted product selectivity towards alkylbenzenes and diphenyl alkanes, which accounted for 84.3% of the liquid products. In contrast, traditional PS hydrogenolysis, when using hydrogen alone, tends to generate more methane products and saturate benzene rings, thereby lowering the degree of PS depolymerization and increasing the formation of undesirable products. The mechanistic pathway, as illustrated in Fig. 6(a), involved the decomposition of methanol into CO and active hydrogen species. These hydrogen species facilitated the selective cleavage of C–C bonds in PS, driving the hydrogenolysis process to completion. Isotopic labeling studies with 13C-labeled methanol confirmed that all CO, CO2, and over 90% of CH4 were derived from methanol, not PS. Kinetic analysis using deuterated methanol (CH3OD and CD3OD) revealed that the rate-limiting step was the cleavage of O–D bonds, underscoring the critical role of methanol decomposition in enabling the plastic upcycling reaction.
 | ||
| Fig. 6 Recent advancements in the design of multifunctional and tandem catalysts for PO plastic upcycling using other co-reactants. (a) Reaction route of methanol-assisted PS hydrogenolysis. Reproduced with permission from Wiley-VCH, copyright 2024.47 (b) PP upcycling using H2O2 as a co-reactant. Reproduced with permission from the Royal Society of Chemistry, copyright 2023.70 (c) Schematic degradation of HDPE. Step 1, dehydrogenation using 1 wt% Pt/Al2O3 in dry toluene for 36–72 hours at 250 °C under N2. Step 2, microwave-assisted oxidation in dilute nitrate solution (0.10 g mL−1) for 1 hour at 180 °C under 60 bar N2. Reproduced with permission from the Royal Society of Chemistry, copyright 2024.71 (d) Upcycling of PO to nitrogenated compounds. Reproduced with permission from American Chemical Society, copyright 2024.48 | ||
Similarly, hydrogen peroxide (H2O2), a reactive oxygen species, has been employed as an oxidative co-reactant for the upcycling of PO plastics. In a novel approach, Wei et al. integrated exothermic oxidative cracking with endothermic thermal cracking to facilitate a self-sustaining exothermic PP degradation reaction, as shown in Fig. 6(b).70 The reaction system required only 5 minutes of external heating at 80°, after which the reaction progressed autonomously without further energy input. This represents a significant advancement in terms of energy efficiency and process intensification, highlighting the potential of hybrid thermal-oxidative systems to minimize energy consumption while delivering effective plastic upcycling.
Building on the concept of multi-step processes, Klingler et al. introduced a two-stage strategy to enhance the upcycling of PO.71 Initially, the polymer undergoes a dehydrogenation pre-treatment over Pt/Al2O3, producing approximately 10% CC double bonds in a toluene solution. This pre-treatment step enhances the homogeneity of the feedstock while the introduction of unsaturated imperfections via CC double bonds significantly improves subsequent reactivity. The second stage involves oxidation in nitric acid solution under microwave irradiation at 180 °C for 1 hour, as depicted in Fig. 6(c). Following this oxidative step, long-chain dicarboxylic acids (primarily C9 and C13) are generated with an overall carbon conversion of approximately 58%. These dicarboxylic acids are subsequently employed in a molten catalytic condensation process to synthesize high-value polyester, offering a novel and economically viable route for the valorization of PO plastic waste.
In addition to incorporating oxygen-based co-reactants, nitrogen-containing compounds have also emerged as promising co-reactants in the upcycling of PO plastic. Zhao et al. reported a groundbreaking approach wherein urea, an inexpensive and readily available nitrogen source, was used to convert PO plastic into nitrogen-containing chemicals, such as benzonitrile and benzamide, at temperatures below 200 °C in an O2 atmosphere (Fig. 6(d)).48 This method marked the first decomposition of nitrogen incorporation into plastic upcycling and was successfully applied to a wide range of real-world plastic waste, including PS, acrylonitrile butadiene styrene (ABS), PP, and rubber waste. This reaction proceeds via the oxidative degradation of polymers to form benzaldehyde as an oxygenated intermediate. Subsequently, in the presence of O2 and ammonia derived from urea, benzaldehyde undergoes two potential reaction pathways to form nitrile products. In the first pathway, benzaldehyde reacts with ammonia through nucleophilic attack, followed by the oxidation of phenylmethanimine. Alternatively, benzaldehyde is further oxidized to benzoic acid, which then reacts with ammonia to form benzamide. Benzamide undergoes dehydration, forming benzonitrile as the final product. The ability to transform waste plastics into high-value nitriles and amides under mild conditions represents a significant leap forward in diversifying the chemical products derived from plastic waste. Moreover, the introduction of nitrogen functional groups into upcycled products opens new avenues for producing value-added chemicals, which are in high demand across various industries.
Collectively, these innovative methodologies underscore the transformative potential of leveraging diverse co-reactants and multi-step processes to expand the product spectrum in plastic upcycling. By strategically integrating different reaction pathways, it is possible to not only enhance process efficiency but also produce a wider array of high-value chemical products. As research in this field progresses, the continued exploration of novel co-reactants and process integration will be essential for overcoming the remaining challenges in plastic waste management.
3. Polyolefin upcycling without co-reactants
Despite significant efforts to incorporate co-reactants for facilitating PO plastic upcycling under mild conditions, as discussed in the previous sections, traditional upcycling methods without such assistance typically rely on catalytic cracking mechanisms that demand elevated reaction temperatures (around 400 °C). To overcome this thermodynamic limitation and achieve efficient upcycling at more moderate conditions, recent research has pivoted towards innovative strategies. These approaches primarily emphasize either tandem reactions that couple thermodynamically distinct processes to drive conversion at lower temperatures, or the design of tandem or multifunctional catalysts capable of circumventing conventional energy barriers.A groundbreaking contribution by Zhang et al. introduced the concept of tandem hydrogenolysis and aromatization for the selective transformation of PO plastics into long-chain alkylaromatics.49 This method employed a Pt/Al2O3 catalyst to synergize hydrogenolysis and aromatization processes at a relatively mild temperature of 280 °C, yielding high-value C30 alkylaromatics and alkylnaphthenes. Remarkably, no external hydrogen source was required, as PE chains themselves served as internal hydrogen donors via reactions such as dehydrocyclization, providing the necessary hydrogen for the hydrogenolysis step. The coupling of exothermic hydrogenolysis with endothermic aromatization rendered this transformation thermodynamically feasible under mild conditions, establishing a novel pathway for PO plastic upcycling without co-reactants.
Building on this innovative work, Sun et al. further enhanced the catalytic system by incorporating halogen modifiers, such as Cl and F, to strengthen the BASs over the Pt/Al2O3 catalyst.50 This modification resulted in a five-fold increase in the rate of C–C bond cleavage, while also doubling the molar yield of alkylaromatics products. Detailed mechanistic studies revealed that, while Pt-catalyzed dehydrogenation and hydrogenation steps approached quasi-equilibrium, the acid-catalyzed C–C bond cleavage and skeletal arrangements were the rate-limiting steps. This underscores the critical role of enhanced acidity in the catalyst support, which was instrumental in driving higher product yields and improving the overall catalytic performance. In a similar vein, Du et al. employed Ru/ZSM-5 in tandem reactions into separable cyclic hydrocarbons, achieving an 18-folder increase in catalytic activity compared to Pt/Al2O3.51 Mechanistic analysis elucidated the synergistic interaction between Ru and the acid sites of ZSM-5, which promoted the dehydrogenation of polymer chains to generate CC bonds. These unsaturated bonds, in turn, facilitated the formation of carbonium ions on the acid sites, driving the cyclization process. Notably, this process necessitates the co-presence of a CC bond and a carbonium ion within a molecular chain at an optimal distance. Furthermore, the confined pore structure of ZSM-5 effectively inhibited the formation and diffusion of polyaromatic species, thereby significantly enhancing the catalyst's stability during HDPE upcycling.
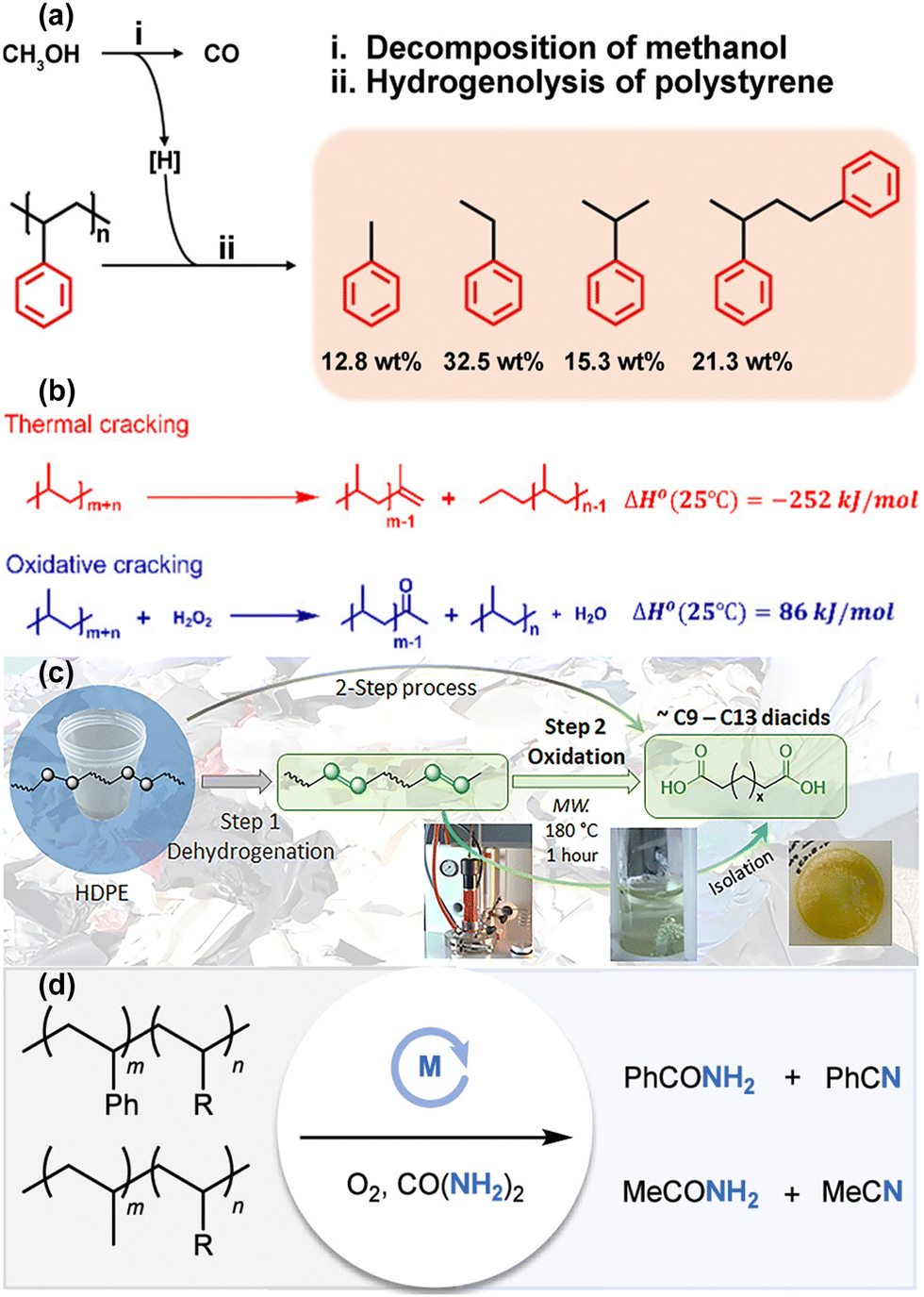
Beyond the strategy of reaction coupling to overcome thermodynamic limitations in PO plastic upcycling under mild conditions, recent innovations in catalyst design have been proven highly effective in addressing these challenges. For example, Cen et al. developed a novel layered self-pillared zeolite that successfully converted PE into high-quality gasoline with remarkable selectivity (99%) and yields exceeding 80% at just 240 °C.52 This low-temperature conversion was attributed to the catalyst's unique structure, which features an open framework with tri-coordinated Al sites capable of activating the inert C–H bonds in PE. The catalysts operate through a self-supplied hydrogen (SSH) mechanism, where the hydrogen required for the reaction is generated internally. Additionally, BASs within the zeolite structure drive β-scission and isomerization processes, facilitating the conversion of PE into a branched alkane, as shown in Fig. 7(a). Further advancing this area, Feng et al. engineered a hierarchical tandem porous catalyst, inspired by clefts found in natural protease, achieving high yields of light hydrocarbons at 280 °C.53 The catalyst's architecture, spanning from macropores to mesopores to micropores, plays a critical role in reducing the decomposition temperature of PO plastics. The outer hydrophobic macropore arrays facilitate the rapid transport of bulky PO into the mesopores, where weak acid sites initiate pre-cracking into smaller intermediates. These intermediates subsequently migrate into inner microporous ZSM-5 nanosheets, where strong BASs complete the cracking process, producing light hydrocarbons and olefin products. This process, as depicted in Fig. 7(b), benefits from a synergistic gradient porous structure (from macropores to mesopores to micropores), combined with a gradient of acidity (from weak to strong), enabling efficient PO cracking, suppressing coke formation, and facilitating the desorption of the desired products.
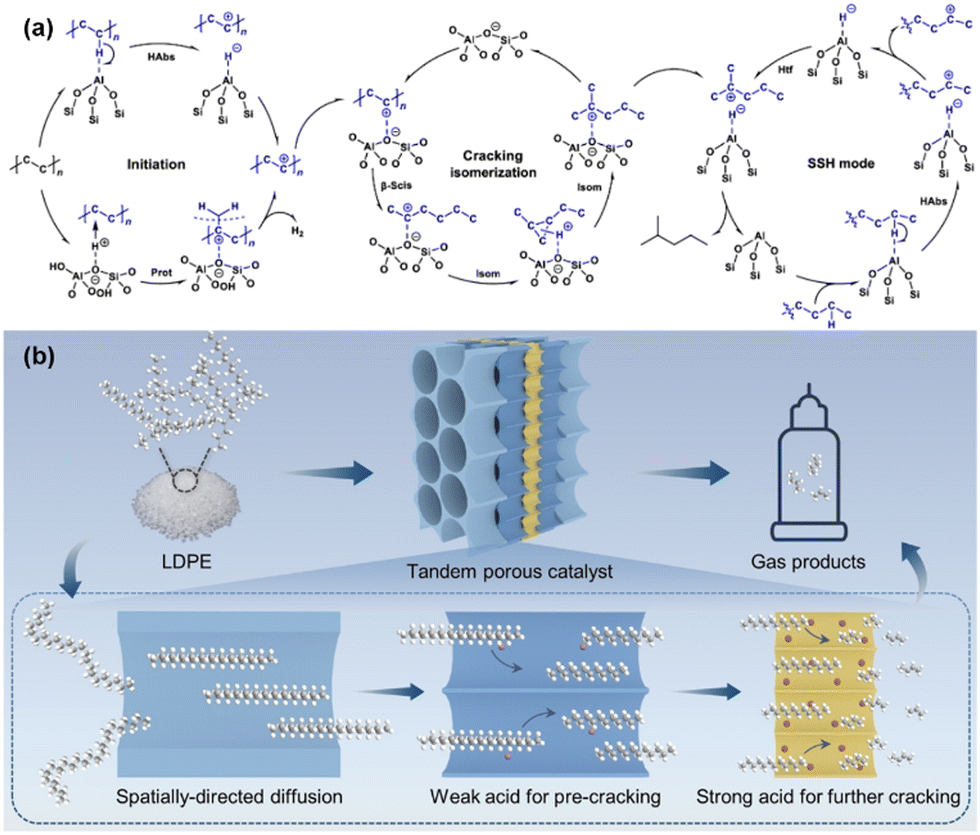 | ||
| Fig. 7 Recent advancements in the design of multifunctional and tandem catalysts for PO plastic upcycling without the use of co-reactants. (a) Proposed primary reaction pathways for the conversion of PE to iso-alkanes over LSP-Z100, involving key steps such as initiation, cracking/isomerization, and hydride transfer. Habs, hydride abstraction; Prot, protonation; β-scis, β-scission; Isom, isomerization; and Htf, hydrogen transfer. Reproduced with permission from Springer Nature, copyright 2024.52 (b) Schematic representation of the cascade cracking of LDPE over tandem porous catalysts. Reproduced with permission from Wiley-VCH, copyright 2024.53 | ||
4. Conclusions and future perspectives
Significant progress has been made in recent years in the upcycling of PO plastics through heterogeneous catalytic strategies under mild conditions. This review has systematically categorized key advancements, distinguishing between co-reactant-assisted pathways and direct transformations without co-reactants. Despite these breakthroughs, several critical challenges remain to be addressed to fully realize the potential of PO plastic upcycling.Firstly, although considerable strides have been made in enabling PO plastic upcycling at lower temperatures, catalytic performances, particularly with respect to selectivity, continue to present substantial limitations.72 Current approaches often result in a wide distribution of liquid-range hydrocarbons; with few cases achieving precise control over specific chemicals, only propylene and methane were achieved as main products up to now. Except for their use as fuels, the broad range of products may have to introduce expensive separation facilities for chemical production from plastic upcycling. Thus, there remains a pressing need for innovative catalytic strategies that can selectively drive the formation of targeted, high-value products. Furthermore, catalytic stability is often underreported, with most studies only demonstrating short-term stability over limited reaction cycles (typically fewer than three) in academic laboratories. Most reported works may not be practically implemented especially the high carbon to oxidant (H, O) ratios could be subject to carbon deposition on the catalyst. For example, aromatic or high olefin by-products may also be prone to carbon deposition more favorably. For real-world applications, long-term durability, particularly when processing mixed plastic waste with impurities (i.e., organic dyes and additives), of feedstocks is essential. A deeper investigation into the catalyst deactivation mechanism will provide crucial insights for the rational design of more robust and resilient catalysts with kinetic barriers for deactivation reactions.
In addition, while substantial research has elucidated reaction mechanisms for PO plastic upcycling, much of this understanding has been derived from ex situ techniques, which do not fully capture the dynamics of the reaction under operating conditions. In situ characterization methods under realistic reaction conditions are imperative for revealing the intricate reaction pathways and kinetics. Such mechanistic insights are vital for the next generation of catalyst design, facilitating more efficient and selective processes.
Another fundamental challenge stems from the inherent nature of plastics as macromolecules. Plastics exhibit distinctive chemical and physical properties, particularly high viscosity upon melting, which imposes significant diffusion limitations and constrains reaction kinetics. The slow kinetics between liquefied plastics/fragments with solid catalysts forming gaseous products from trickle bed reactor systems with complex kinetics would seriously affect the techno-economic evaluation of a new process. Recent studies have highlighted strategies to mitigate these diffusion barriers and improve catalytic efficiency.73,74 However, further advances in reactor designs and process engineering are urgently needed to optimize heat and mass transfer, particularly in the processing of highly viscous plastic streams, to achieve more effective upcycling.
In addition, recent studies indicate that finely dispersed microplastics in contaminated water could pose health issues to living organisms on earth. For example, it has been reported that microplastics in mice body could reduce sperm count and caused abnormalities and hormone disruption.75 Furthermore, microplastics have been recently found in human semen sperm.76 The catalytic science and technology for the plastic upgrading processes could be somehow related to the development of potential new catalytic removal of microplastics from water with different challenges.
In conclusion, the upcycling of PO plastics is a deeply emerging new interdisciplinary field, requiring seamless expertise from heterogeneous catalysis, reaction engineering, environmental science, and industrial implementation. Addressing the critical challenges within this domain, ranging from enhancing catalytic performance and stability to improving process scalability, will be essential for advancing sustainable strategies for plastic management. By fostering collaborations across these diverse disciplines, we can unlock innovative solutions that have potential to significantly reduce plastic waste, mitigate environmental impacts, and contribute to the global transition towards a more circular economy.
Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the EPSRC, UK (EP/K040375/1), for supporting catalytic research at the Wolfson Catalysis Centre, University of Oxford, and the Siam Cement Group (SCG), Thailand, for kindly providing the PO samples.References
- K. Zheng, Y. Wu, Z. Hu, S. Wang, X. Jiao, J. Zhu, Y. Sun and Y. Xie, Chem. Soc. Rev., 2023, 52, 8–29 RSC.
- D. J. Walsh, M. G. Hyatt, S. A. Miller and D. Guironnet, ACS Catal., 2019, 9, 11153–11188 CrossRef.
- H. Wang and S. C. Edman Tsang, Cell Rep. Phys. Sci., 2024, 5, 102075 CrossRef.
- W. Zhang, S. Kim, L. Wahl, R. Khare, L. Hale, J. Hu, D. M. Camaioni, O. Y. Gutiérrez, Y. Liu and J. A. Lercher, Science, 2023, 379, 807–811 CrossRef PubMed.
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- P. G. C. Nayanathara Thathsarani Pilapitiya and A. S. Ratnayake, Cleaner Mater., 2024, 11, 100220 CrossRef.
- R. Geyer, in Plastic Waste and Recycling, ed. T. M. Letcher, Academic Press, 2020, pp. 13–32 Search PubMed.
- H. Chen, K. Wan, Y. Zhang and Y. Wang, ChemSusChem, 2021, 14, 4123–4136 CrossRef PubMed.
- Europe, Plastics – the fast Facts 2023, Plastics Europe, 2023.
- G. Celik, R. M. Kennedy, R. A. Hackler, M. Ferrandon, A. Tennakoon, S. Patnaik, A. M. LaPointe, S. C. Ammal, A. Heyden, F. A. Perras, M. Pruski, S. L. Scott, K. R. Poeppelmeier, A. D. Sadow and M. Delferro, ACS Cent. Sci., 2019, 5, 1795–1803 CrossRef PubMed.
- I. M. Steensgaard, K. Syberg, S. Rist, N. B. Hartmann, A. Boldrin and S. F. Hansen, Environ. Pollut., 2017, 224, 289–299 CrossRef PubMed.
- H.-G. Ni, S.-Y. Lu, T. Mo and H. Zeng, Environ. Pollut., 2016, 214, 70–76 CrossRef PubMed.
- D. Pan, F. Su, C. Liu and Z. Guo, Adv. Compos. Hybrid Mater., 2020, 3, 443–461 CrossRef CAS.
- K. Ragaert, L. Delva and K. Van Geem, Waste Manage., 2017, 69, 24–58 CrossRef.
- J. Hopewell, R. Dvorak and E. Kosior, Philos. Trans. R. Soc., B, 2009, 364, 2115–2126 CrossRef PubMed.
- A. Angyal, N. Miskolczi and L. Bartha, J. Anal. Appl. Pyrolysis, 2007, 79, 409–414 CrossRef.
- J. E. Rorrer, G. T. Beckham and Y. Román-Leshkov, JACS Au, 2021, 1, 8–12 CrossRef PubMed.
- I. Vollmer, M. J. F. Jenks, M. C. P. Roelands, R. J. White, T. van Harmelen, P. de Wild, G. P. van der Laan, F. Meirer, J. T. F. Keurentjes and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2020, 59, 15402–15423 CrossRef PubMed.
- A. Bin Jumah, V. Anbumuthu, A. A. Tedstone and A. A. Garforth, Ind. Eng. Chem. Res., 2019, 58, 20601–20609 CrossRef.
- D. Ma, Y. Wang, M. L. Sarazen, C. Boyer, T. Saito, H. Duan and W. Huang, Cell Rep. Phys. Sci., 2024, 5(10), 102209 CrossRef.
- M. Wang, M. Wang and D. Ma, J. Phys. Chem. C, 2024, 128, 16302–16307 CrossRef.
- H. Zhou, Z. Li, L. Ma and H. Duan, Chem. Commun., 2022, 58, 897–907 RSC.
- X. Lou, F. Liu, Q. Li, M. Chu, G. Wang, J. Chen and M. Cao, Chem. Commun., 2024, 60, 2828–2838 RSC.
- S. Sun and W. Huang, JACS Au, 2024, 4, 2081–2098 CrossRef PubMed.
- S. Xu, J. Tang and L. Fu, Langmuir, 2024, 40, 3984–4000 CrossRef.
- M. Gardette, A. Perthue, J.-L. Gardette, T. Janecska, E. Földes, B. Pukánszky and S. Therias, Polym. Degrad. Stab., 2013, 98, 2383–2390 CrossRef.
- J. Sun, J. Dong, L. Gao, Y.-Q. Zhao, H. Moon and S. L. Scott, Chem. Rev., 2024, 124, 9457–9579 CrossRef.
- X. Han, X. Zhou, T. Ji, F. Zeng, W. Deng, Z. Tang and R. Chen, EES Catal., 2024, 2, 300–310 RSC.
- B. C. Vance, P. A. Kots, C. Wang, Z. R. Hinton, C. M. Quinn, T. H. Epps, L. T. J. Korley and D. G. Vlachos, Appl. Catal., B, 2021, 299, 120483 CrossRef.
- H. Wang, T. Yoskamtorn, J. Zheng, P.-L. Ho, B. Ng and S. C. E. Tsang, ACS Catal., 2023, 13, 15886–15898 CrossRef.
- M. Tamura, S. Miyaoka, Y. Nakaji, M. Tanji, S. Kumagai, Y. Nakagawa, T. Yoshioka and K. Tomishige, Appl. Catal., B, 2022, 318, 121870 CrossRef.
- M. Chu, X. Wang, X. Wang, P. Xu, L. Zhang, S. Li, K. Feng, J. Zhong, L. Wang, Y. Li, L. He, M. Cao, Q. Zhang, L. Chi and J. Chen, J. Am. Chem. Soc., 2024, 146, 10655–10665 CrossRef.
- Q. Kang, M. Chu, P. Xu, X. Wang, S. Wang, M. Cao, O. Ivasenko, T.-K. Sham, Q. Zhang, Q. Sun and J. Chen, Angew. Chem., Int. Ed., 2023, 62, e202313174 CrossRef.
- S. D. Jaydev, M.-E. Usteri, A. J. Martín and J. Pérez-Ramírez, Chem. Catal., 2023, 100564 CrossRef.
- L. Chen, J. B. Moreira, L. C. Meyer and J. Szanyi, Appl. Catal., B, 2023, 335, 122897 CrossRef.
- D. Kim, Z. R. Hinton, P. Bai, L. T. J. Korley, T. H. Epps and R. F. Lobo, Appl. Catal., B, 2022, 318, 121873 CrossRef.
- L. D. Ellis, S. V. Orski, G. A. Kenlaw, A. G. Norman, K. L. Beers, Y. Román-Leshkov and G. T. Beckham, ACS Sustainable Chem. Eng., 2021, 9, 623–628 CrossRef.
- N. M. Wang, G. Strong, V. DaSilva, L. Gao, R. Huacuja, I. A. Konstantinov, M. S. Rosen, A. J. Nett, S. Ewart, R. Geyer, S. L. Scott and D. Guironnet, J. Am. Chem. Soc., 2022, 144, 18526–18531 CrossRef.
- R. J. Conk, J. F. Stahler, J. X. Shi, J. Yang, N. G. Lefton, J. N. Brunn, A. T. Bell and J. F. Hartwig, Science, 2024, 385, 1322–1327 CrossRef PubMed.
- K. Wang, R. Jia, P. Cheng, L. Shi, X. Wang and L. Huang, Angew. Chem., Int. Ed., 2023, 62, e202301340 CrossRef PubMed.
- C. Sun, Y. Guo, X. Liu and Y. Wang, Catal. Sci. Technol., 2024, 14, 6584–6591 RSC.
- Q. Zhang, J. He, X. Wei, C. Shen, P. Ye, W. An, X. Liu, H. Li, S. Xu, Z. Su and Y.-Z. Wang, Angew. Chem., Int. Ed., 2024, 63, e202407510 CrossRef.
- W. Chen, Y. Jiao, Y. Liu, M. Wang, F. Zhang and D. Ma, CCS Chem., 2024, 6, 1422–1429 CrossRef.
- Y. Liu, B. Ma, J. Tian and C. Zhao, Sci. Adv., 2024, 10, eadn0252 CrossRef PubMed.
- J. Zhang, D. Lai, Z. Chen, X. Wang, Q. Xiong, J. Li, X. Zhang, B. O. Oboirien and G. Xu, ACS Sustainable Chem. Eng., 2024, 12, 13137–13148 CrossRef.
- Y. Ding, S. Zhang, C. Liu, Y. Shao, X. Pan and X. Bao, Natl. Sci. Rev., 2024, 11, nwae097 CrossRef PubMed.
- L. Zeng, T. Yan, J. Du, C. Liu, B. Dong, B. Qian, Z. Xiao, G. Su, T. Zhou, Z. Peng, Z. Wang, H. Li and J. Zeng, Angew. Chem., Int. Ed., 2024, 63, e202404952 CrossRef PubMed.
- B. Zhao, Z. Hu, Y. Sun, R. Hajiayi, T. Wang and N. Jiao, J. Am. Chem. Soc., 2024, 146(42), 28605–28611 CrossRef PubMed.
- F. Zhang, M. Zeng, R. D. Yappert, J. Sun, Y.-H. Lee, A. M. LaPointe, B. Peters, M. M. Abu-Omar and S. L. Scott, Science, 2020, 370, 437–441 CrossRef PubMed.
- J. Sun, Y.-H. Lee, R. D. Yappert, A. M. LaPointe, G. W. Coates, B. Peters, M. M. Abu-Omar and S. L. Scott, Chem, 2023, 9(8), 2318–2336 Search PubMed.
- J. Du, L. Zeng, T. Yan, C. Wang, M. Wang, L. Luo, W. Wu, Z. Peng, H. Li and J. Zeng, Nat. Nanotechnol., 2023, 18, 772–779 CrossRef PubMed.
- Z. Cen, X. Han, L. Lin, S. Yang, W. Han, W. Wen, W. Yuan, M. Dong, Z. Ma, F. Li, Y. Ke, J. Dong, J. Zhang, S. Liu, J. Li, Q. Li, N. Wu, J. Xiang, H. Wu, L. Cai, Y. Hou, Y. Cheng, L. L. Daemen, A. J. Ramirez-Cuesta, P. Ferrer, D. C. Grinter, G. Held, Y. Liu and B. Han, Nat. Chem., 2024, 16, 871–880 Search PubMed.
- J. Feng, J. Duan, C.-T. Hung, Z. Zhang, K. Li, Y. Ai, C. Yang, Y. Zhao, Z. Yu, Y. Zhang, L. Wang, D. Zhao and W. Li, Angew. Chem., Int. Ed., 2024, 63, e202405252 CrossRef CAS PubMed.
- J. Weitkamp, ChemCatChem, 2012, 4, 292–306 Search PubMed.
- W.-T. Lee, A. van Muyden, F. D. Bobbink, M. D. Mensi, J. R. Carullo and P. J. Dyson, Nat. Commun., 2022, 13, 4850 Search PubMed.
- S. Liu, P. A. Kots, B. C. Vance, A. Danielson and D. G. Vlachos, Sci. Adv., 2021, 7, eabf8283 Search PubMed.
- T. J. Smak, P. de Peinder, J. C. van der Waals, R. Altink, I. Vollmer and B. M. Weckhuysen, ChemSusChem, 2024, 17, e202301198 CrossRef CAS PubMed.
- T. Reier, M. Oezaslan and P. Strasser, ACS Catal., 2012, 2, 1765–1772 Search PubMed.
- J. Ying, J.-B. Chen, Y.-X. Xiao, S. I. Cordoba de Torresi, K. I. Ozoemena and X.-Y. Yang, J. Mater. Chem. A, 2023, 11, 1634–1650 RSC.
- B. Zhao, H. Tan, J. Yang, X. Zhang, Z. Yu, H. Sun, J. Wei, X. Zhao, Y. Zhang, L. Chen, D. Yang, J. Deng, Y. Fu, Z. Huang and N. Jiao, Innovation, 2024, 5, 100586 CAS.
- Z.-P. Wu, X. F. Lu, S.-Q. Zang and X. W. Lou, Adv. Funct. Mater., 2020, 30, 1910274 Search PubMed.
- Z. Xu, N. E. Munyaneza, Q. Zhang, M. Sun, C. Posada, P. Venturo, N. A. Rorrer, J. Miscall, B. G. Sumpter and G. Liu, Science, 2023, 381, 666–671 Search PubMed.
- X. Liu, M. Wang, H. Yin, J. Hu, K. Cheng, J. Kang, Q. Zhang and Y. Wang, ACS Catal., 2020, 10, 8303–8314 CrossRef CAS.
- Z. Li, J. Wang, Y. Qu, H. Liu, C. Tang, S. Miao, Z. Feng, H. An and C. Li, ACS Catal., 2017, 7, 8544–8548 Search PubMed.
- M. Tong, E. Hondo, L. Gapu Chizema, C. Du, Q. Ma, S. Mo, C. Lu, P. Lu and N. Tsubaki, New J. Chem., 2020, 44, 9328–9336 RSC.
- A. Ramirez, P. Ticali, D. Salusso, T. Cordero-Lanzac, S. Ould-Chikh, C. Ahoba-Sam, A. L. Bugaev, E. Borfecchia, S. Morandi, M. Signorile, S. Bordiga, J. Gascon and U. Olsbye, JACS Au, 2021, 1, 1719–1732 CrossRef PubMed.
- K. Ma, S. Zhao, M. Dou, X. Ma and C. Dai, ACS Catal., 2024, 14, 594–607 CrossRef.
- C. Wei, W. Zhang, K. Yang, X. Bai, S. Xu, J. Li and Z. Liu, Chin. J. Catal., 2023, 47, 138–149 CrossRef.
- J. Yang, L. Wang, J. Wan, F. El Gabaly, A. L. Fernandes Cauduro, B. E. Mills, J.-L. Chen, L.-C. Hsu, D. Lee, X. Zhao, H. Zheng, M. Salmeron, C. Wang, Z. Dong, H. Lin, G. A. Somorjai, F. Rosner, H. Breunig, D. Prendergast, D.-E. Jiang, S. Singh and J. Su, Nat. Commun., 2024, 15, 911 CrossRef.
- X. Wei, Q. Zhang, C. Shen, X. Zhao, F. Zhang, X. Liu, G. Wu, S. Xu and Y. Z. Wang, Mater. Horiz., 2023, 10, 3694–3701 RSC.
- W. Wu Klingler, L. Perret, P. Rupper, S. Lehner, X. Zhou, H. Eliasson, R. Muff, M. Heuberger and S. Gaan, Green Chem., 2024, 26, 10422–10433 RSC.
- L. Li, M. Leutzsch, P. Hesse, C. Wang, B. Wang and F. Schüth, Angew. Chem., Int. Ed., 2024, e202413132 Search PubMed.
- S. Rejman, I. Vollmer, M. J. Werny, E. T. C. Vogt, F. Meirer and B. M. Weckhuysen, Chem. Sci., 2023, 14, 10068–10080 RSC.
- S. D. Jaydev, A. J. Martín, D. Garcia, K. Chikri and J. Pérez-Ramírez, Nat. Chem. Eng., 2024, 1, 565–575 CrossRef PubMed.
- H. Jin, T. Ma, X. Sha, Z. Liu, Y. Zhou, X. Meng, Y. Chen, X. Han and J. Ding, J. Hazard. Mater., 2021, 401, 123430 CrossRef PubMed.
- L. Montano, E. Giorgini, V. Notarstefano, T. Notari, M. Ricciardi, M. Piscopo and O. Motta, Sci. Total Environ., 2023, 901, 165922 CrossRef.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |