
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Simple bifunctional salts for synthesising block copolymers from anhydrides/epoxides and vinyl acetate†
Anna
Lykkeberg
 and
Jennifer A.
Garden
*
and
Jennifer A.
Garden
*
EaStCHEM School of Chemistry, University of Edinburgh, Edinburgh, EH9 3FJ, UK. E-mail: j.garden@ed.ac.uk
First published on 17th December 2024
Abstract
Herein, we report the first synthesis of poly(ester-block-vinyl acetate) via epoxide/anhydride ring-opening copolymerisation and reversible addition–fragmentation chain transfer polymerisation. This was achieved using simple, robust and bifunctional alkali metal carboxylates featuring a xanthate unit.
Block copolymers (BCPs) are a multi-billion-dollar market. Yet the growing drive for sustainability, and the demand for tailored material properties for specific applications, are still driving the need for novel polymer materials. BCPs can harness the distinct and beneficial properties of both parent polymers, with applications including drug delivery, phase compatibilizers and nanolithography.1 Incorporating a polyester block into a BCP can also deliver tuneable biodegradation.2 However, combining two different mechanisms, such as olefin polymerisation and ring-opening (co)polymerisation (RO(CO)P) to produce poly(olefin-block-esters), is synthetically challenging.3
Reversible addition–fragmentation chain transfer (RAFT) polymerisation provides a well-controlled route to an array of functionalised polyolefins,4 whilst epoxide/anhydride ROCOP is a versatile method of generating polyesters because the functional groups can be varied on both monomers (Fig. 1).5,6 However, methods of combining controlled radical polymerisation with ROCOP remain limited.7 Cobalt salen catalysts can elegantly shift between ROCOP and controlled olefin mechanisms upon introducing a gaseous switch reagent such as O2 or CO,8,9 yet these air-sensitive or gaseous chemicals are challenging to handle. Simple and tuneable routes towards poly(olefin-block-esters) are thus an attractive target.
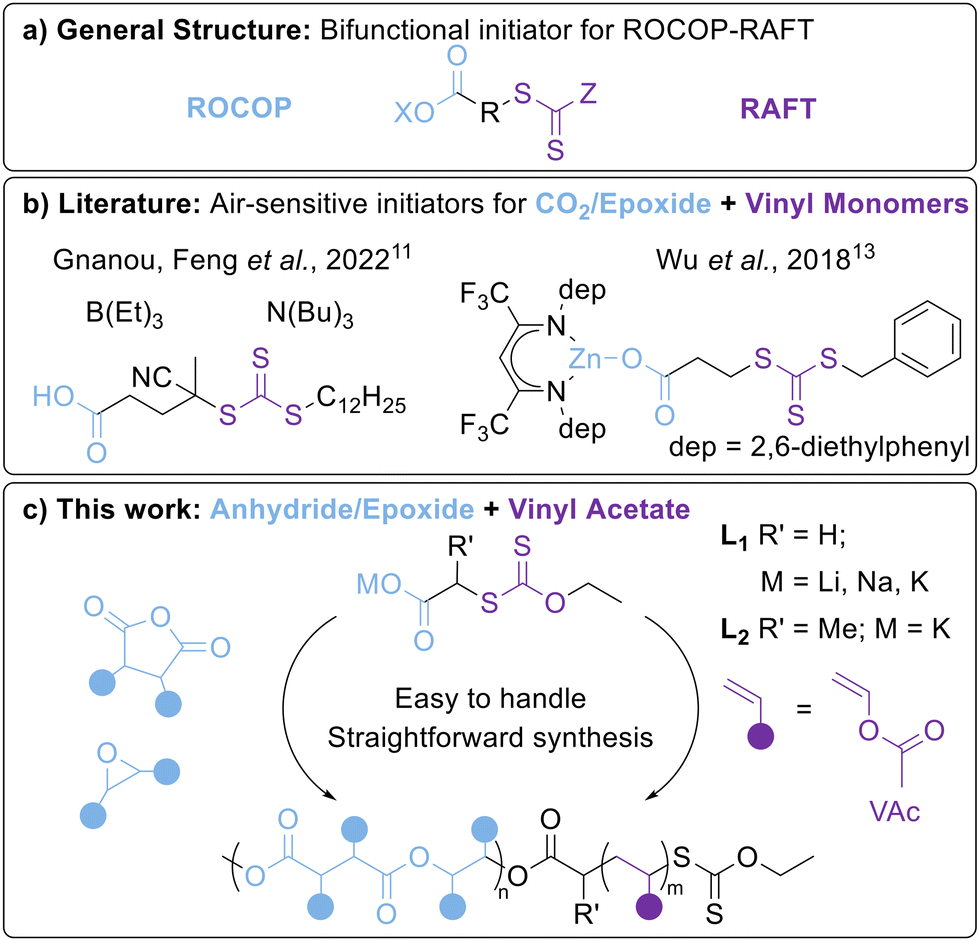 | ||
| Fig. 1 (a) General structure of a bifunctional ROCOP-RAFT initiator, (b) examples of literature systems for preparing ROCOP-RAFT derived BCPs (see Fig. S59 for further details, ESI†) and (c) the carboxylate salt system used in this work. | ||
Bifunctional initiators are emerging as an effective method of combining ROCOP and RAFT polymerisations (Fig. S59, ESI†).10 Here, we use the term “bifunctional initiators” to describe reagents capable of propagating a polyester and a polyolefin chain from different ends of the molecule, although it should be noted that the RAFT-agent is strictly a chain transfer agent rather than an initiator.4 These bifunctional initiators can be RAFT agents with terminal COOH or metal carboxylate units (Fig. 1b). In the former, the COOH unit initiates ROCOP in the presence of a catalyst, such as a Lewis pair boron/amine organocatalyst or a metal complex such as an Al–porphyrin.11,12 As an example of the latter, Wu et al. reported a Zn–β-diiminate complex containing a Zn–carboxylate unit.13 In all cases, the polymerisation requires inert reaction conditions and the use of stringently purified monomers. So far, bifunctional ROCOP-RAFT initiators have mostly focused on CO2/epoxide ROCOP for polycarbonates and the RAFT polymerisation of activated vinyl monomers e.g. styrene and acrylates. Very few studies have combined epoxide/anhydride ROCOP with RAFT polymerisation,14–16 and to the best of our knowledge, none with vinyl acetate (VAc). Notably, VAc is a “less activated monomer” than styrene/acrylates due to a lack of conjugation.
As the air-sensitivity of the ROCOP moiety generally dictates the sensitivity of the bifunctional initiator, we surveyed the literature to identify synthetically simple and robust ROCOP initiators. Wang, Liu et al. reported alkali metal acetates as efficient catalysts for epoxide/anhydride ROCOP. These salts are easy to handle and based on low-toxicity earth abundant metals.17 To translate this concept to RAFT, we designed alkali metal carboxylates featuring xanthate-based RAFT agents (Fig. 1c) for the production of poly(ester-co-vinyl acetate) BCPs under accessible benchtop conditions rarely tested in ROCOP.
Both L1H and L2H (Fig. 1c) were prepared from adapted literature procedures, and were deprotonated by the relevant alkali metal hydride.18,19 As potassium carboxylates have been reported to outperform their Li and Na analogues in epoxide/anhydride ROCOP (K > Na > Li),17 complexes L1K and L2K were generated from both ligands, along with L1Li and L1Na. To the best of our knowledge, L1M (M = Li, Na, K) and L2K have not been previously isolated, and while L1Na has been used as a reaction intermediate,20,21 no characterisation data was reported. The salts were characterised through a combination of multinuclear NMR and FT-IR spectroscopy and elemental analysis (refer to ESI† for details). Alternatively, the L1M carboxylates could be synthesised under benchtop conditions (i.e. in air) by using the corresponding metal hydroxide as the base. In contrast to L1M, L2K was deliquescent and thus could not be isolated under benchtop conditions.
The different initiators were screened for the ROCOP of phthalic anhydride (PA) and cyclohexene oxide (CHO) at 100 °C, with an [initiator]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[PA]:[CHO] ratio of 1:100:400 (Table 1). This screening was performed under inert conditions using purified monomers, to allow direct activity comparisons for the different salts. Overall, L1K and L2K displayed similar activities, and potassium was the best-performing of the alkali metals with activity decreasing in the order of L1K > L1Na > L1Li. For example, after 18 h L1Li gave 45% PA conversion, whereas L1Na achieved 97% conversion. Similarly, L1K significantly outperformed L1Na, giving respective PA conversions of 74% vs. 6% after just 2 h. This activity trend is consistent with the decreasing strength of the anion–cation interaction of the carboxylate and alkali metal cation, enabling improved initiation and propagation. This mirrors reports of the increasing activity of alkali metal acetates for epoxide/anhydride ROCOP upon descending Group 1.17
:[PA]:[CHO] ratio of 1:100:400 (Table 1). This screening was performed under inert conditions using purified monomers, to allow direct activity comparisons for the different salts. Overall, L1K and L2K displayed similar activities, and potassium was the best-performing of the alkali metals with activity decreasing in the order of L1K > L1Na > L1Li. For example, after 18 h L1Li gave 45% PA conversion, whereas L1Na achieved 97% conversion. Similarly, L1K significantly outperformed L1Na, giving respective PA conversions of 74% vs. 6% after just 2 h. This activity trend is consistent with the decreasing strength of the anion–cation interaction of the carboxylate and alkali metal cation, enabling improved initiation and propagation. This mirrors reports of the increasing activity of alkali metal acetates for epoxide/anhydride ROCOP upon descending Group 1.17
| Initiator | Time (h) | Conv.b (%) | PEsb (%) | TOFc (h−1) | M n,calc (kDa) | M n,obs (kDa) | Đ | |
|---|---|---|---|---|---|---|---|---|
|
a [Initiator]:[PA]:[CHO] = 1:100:400, [PA] = 2.5 M, 100 °C, with duplicates run to ensure consistency and a single result reported.
b Conversion and polyester selectivity (PEs) calculated by 1H NMR.
c TOF = (moles of PA consumed)/(moles of catalyst) × time.
d
M
n,calc calculated from monomer conversion: Mn,calc = M0 × ([M]/[I]) × conversion assuming 1 chain per catalyst.
e
M
n,obs and Đ determined by size exclusion chromatography (SEC) calibrated with narrow dispersity polystyrene standards.
f Bimodal traces observed for all samples; average result reported (refer to ESI for details).
g Trimodal traces observed for L1Li sample, averaged bimodal peak reported as for other samples with the additional third peak (at high molecular weight) reported separately.
|
||||||||
| 1 | L1Li | 2 | 3 | 31 | 2 | — | — | — |
| 2 | L1Li | 18 | 45 | 72 | 3 | 11.1 | 4.3g, 28.9g | 1.22, 1.27 |
| 3 | L1Na | 2 | 6 | 48 | 3 | — | — | — |
| 4 | L1Na | 18 | 97 | 84 | 5 | 23.8 | 9.8 | 1.25 |
| 5 | L1K | 0.25 | 11 | 80 | 44 | — | — | — |
| 6 | L1K | 2 | 74 | 90 | 37 | 17.6 | 6.6 | 1.17 |
| 7 | L2K | 0.25 | 11 | 79 | 44 | — | — | — |
| 8 | L2K | 2 | 87 | 93 | 44 | 21.4 | 7.1 | 1.16 |
The perfectly alternating copolymerisation of epoxides/anhydrides generates polyester linkages, whereas sequential epoxide insertion generates polyether linkages.5 In general, the highest polyester selectivity was observed with L1K (90%), which also decreased in the order K > Na > Li (L1Na, 84%; L1Li, 72%). Intriguingly, a higher proportion of polyether linkages were observed at lower monomer conversions, suggesting that polyether formation is favoured in the early stages of the polymerisation. While the exact reasons for this are unclear, a similar phenomenon has been reported for CO2/epoxide ROCOP with an aluminium porphyrin initiator.22
In terms of the different RAFT agents, L1K and L2K demonstrated similar activities and polyester selectivities after 2 h (L1K, 72% conversion, 90% selectivity; L2K, 87% conversion, 93% selectivity). This is perhaps unsurprising, as the propagating metal–Opolymer chain end migrates further away from the initiating unit after each monomer insertion. The RAFT agent thus has a lesser influence on catalyst activity as propagation continues. As L1K and L2K gave the highest activities and selectivities, these initiators were evaluated in a kinetic study for PA/CHO ROCOP (Fig. S42, ESI†). Both showed comparable kobs values, (L1K: 1.0 h−1; L2K: 1.1 h−1). The linear plots of [PA] vs. time show a zero-order dependence on the anhydride, which is consistent with previous literature studies.23
For all initiators, the Mn values increased linearly with conversion and narrow dispersities were obtained (Fig. S43, ESI†). The SEC traces show bimodal molecular weight distributions when using L1Na, L1K and L2K, and a trimodal distribution with L1Li. Bimodal distributions are common in ROCOP, even when stringently purified reagents and air-sensitive conditions are used, and are attributed to trace diols and diacids formed via epoxide or anhydride hydrolysis acting as bifunctional chain transfer agents.14,24,25 As the potassium salts delivered improved performance, L1Li and L1Na were not investigated further. To use the resultant polyesters as macro-RAFT agents, it is essential that the RAFT unit is incorporated into the polymer chain. The presence of RAFT agent end groups was thus established via1H NMR and MALDI-ToF analysis, including diagnostic 1H NMR resonances around 4.25 ppm from the ethoxy CH2 of the L1 RAFT end group. MALDI-ToF analysis shows a series initiated by the desired RAFT agent, as well as a telechelic polymer terminated by hydroxyl end groups (Fig. S45, ESI†); this is common in epoxide/anhydride ROCOP due to initiation from trace diols/diacids.
As L1K displayed similar performance to L2K but was not deliquescent, it was investigated for non-air and moisture-sensitive conditions as well as alternative monomers. Initially, PA/1,2-epoxybutane (EB) ROCOP was evaluated under inert conditions. In contrast to CHO, which features a rigid cyclohexane ring, EB is a flexible aliphatic monomer bearing a linear alkyl side chain. Due to the lower boiling point of EB, the polymerisations were performed at 60 °C, and perfectly alternating poly(PA-alt-EB) was obtained after 48 h (60% conversion, Table 2, entry 1). It is important to note that the polar L1K salt is not completely soluble under these conditions, which may partly contribute to the lower conversions obtained along with the lower reaction temperature.
| Ep | Conv.d (%) | PEsd (%) | M n,calc (kDa) | M n,obs (kDa) | Đ | |
|---|---|---|---|---|---|---|
|
a [L1K]:[PA]:[CHO] = 1:100:400, [PA] = 2.5 M, 100 °C, 2 h; [L1K]:[PA]:[EB] = 1:100:400, [PA] = 2.9 M, 60 °C, 48 h.
b Conducted under air-sensitive conditions, with purified and dried reagents.
c Conducted under benchtop conditions with unpurified reagents.
d Conversion and polyester selectivity (PEs) calculated by 1H NMR.
e
M
n,calc calculated from monomer conversion: Mn,calc = M0 × ([M]/[I]) × conversion assuming 1 chain per catalyst.
f
M
n,obs and Đ determined by SEC calibrated with narrow dispersity polystyrene standards.
g Bimodal traces observed for all samples prepared under air-sensitive conditions, with average result reported.
|
||||||
| 1b | EB | 60 | >99 | 13.3 | 9.1g | 1.15 |
| 2c | EB | >99 | 86 | 22.0 | 1.8 | 1.08 |
| 3b | CHO | 72 | 90 | 17.7 | 6.6g | 1.17 |
| 4c | CHO | 94 | 63 | 23.0 | 1.8 | 1.17 |
Notably, L1K also remained active under “benchtop conditions”. Here, we use this phrase to refer to the polymerisation of unpurified monomers in air, which remains underexplored within ROCOP. This introduces more unknowns and variations in the monomer purity, and so here we evaluated whether L1K would generally tolerate these benchtop conditions. For both PA/EB and PA/CHO, L1K gave higher conversions under benchtop conditions. For example, full conversion of PA/EB was obtained after 48 hours in air, compared with only 60% conversion under air-sensitive conditions (i.e. using purified monomers and an argon atmosphere). This demonstrates the robustness of this system towards oxygen, water and other protic impurities such as phthalic acid, cyclohexane diol and 1,2-butane diol. This is particularly gratifying, as monomer purification and the use of inert conditions can be time and energy intensive. However, the Mn,obs values were significantly lower under benchtop conditions. These observations mirror work by Fieser et al., who reported increased catalyst activity and lower molecular weights when respectively using yttrium salts and deep eutectic solvents for ROCOP in the presence of water and air, although these systems still used purified monomers.26,27 With L1K, the polyester selectivity was higher under inert conditions. For example, perfectly alternating PA/EB polyester was produced under inert conditions, which dropped to 86% polyester selectivity under benchtop conditions (Table 2, entries 1 and 2). Multiple factors have been reported to influence the polyester selectivity, including the catalyst and monomer choice, and the concentration of anhydride.28,29 Here, the difference between benchtop and inert conditions suggests that other factors may also influence the polyester selectivity.
These polyesters were consequently tested as macro-RAFT agents for poly(ester-co-vinyl acetate) BCPs. This was initially studied utilising poly(PA-alt-CHO) prepared under air-sensitive conditions to maximise end group fidelity, and thereby maximise conversion to the target BCP. Copolymerisation with VAc was performed in bulk conditions (RAFT:VAc 1:400, AIBN, 65 °C); 1H NMR spectroscopy showed 90% conversion of VAc to PVAc. SEC analysis showed a new peak of higher molar mass (Fig. 2a), as well as a decrease of the peak attributed to the macro-RAFT agent polyester (refer to ESI,† Fig. S57 for details). Unreacted polyester was also observed, which was attributed to the presence of telechelic hydroxyl-capped polyester (vide supra). Diffusion ordered NMR spectroscopy (DOSY NMR) analysis of the product showed two diffusion co-efficients for the polyester; one corresponds to the polyester precursor and the other is a higher molecular weight species featuring both PVAc and polyester resonances, which provides further support for the formation of BCPs as well as unreacted polyester (Fig. S52, ESI†). In contrast, combining the polyester precursor with PVAc homopolymer gave no change to the diffusion coefficient (Fig. S53, ESI†). The copolymer structure was further probed by collecting separate fractions from the SEC analysis, and analysing these fractions by DOSY NMR (Fig. 2b, see ESI† for details). Fractions collected at low retention times (high Mn) clearly showed the same diffusion coefficient for the 1H NMR signals from both the polyester and PVAc, providing evidence for the new peak in the SEC being poly(PA-alt-CHO-co-vinyl acetate). Taken together, the SEC and DOSY analysis provides good evidence for the formation of BCPs. Thus, the purification of the BCP via stirring and filtering from methanol was investigated (see ESI† for details); SEC analysis showed the successful removal of the vast majority of the unreacted polyester (Fig. 2a).
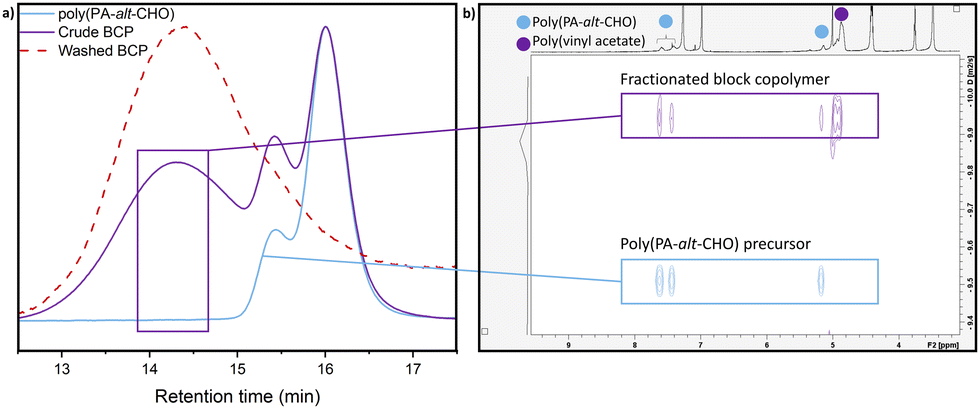 | ||
| Fig. 2 (a) Normalised SEC traces of macro-RAFT agent poly(PA-alt-CHO) (blue), crude poly(PA-alt-CHO)-block-PVAc (purple) and purified poly(PA-alt-CHO)-block-PVAc (red). (b) DOSY NMR of high Mn fraction from SEC fractionation (fraction collected during timeframe shown by purple box) overlaid with poly(PA-alt-CHO) precursor (see also Fig. S54, ESI†). | ||
The robustness of the approach was also trialled utilising the polyester precursors synthesised under benchtop conditions from unpurified monomers (Table 2). Both the benchtop poly(PA-alt-CHO) and poly(PA-alt-EB) were polymerised with VAc (RAFT:VAc 1:100) and gratifyingly, the target BCPs were still observed, as evidenced by new peaks of higher molecular weight in both SEC traces (along with unreacted polyester). DOSY NMR analysis also showed two diffusion coefficients for the polyester signals, one corresponding to the polyester precursor and the other a higher molecular weight BCP species with both polyester and PVAc resonances (refer to ESI,† Fig. S55, S56 and S58).
This work demonstrates proof-of-concept that poly(olefin-co-ester) BCPs, prepared via ROCOP and RAFT polymerisation, can be accessed using simple alkali metal salts. These salts can be used under benchtop conditions with unpurified monomers and under air, and while this understandably reduces the end group fidelity, it demonstrates the potential to use facile conditions and robust initiators to prepare poly(PA-alt-CHO-co-vinyl acetate) and poly(PA-alt-EB-co-vinyl acetate). To the best of our knowledge, this represents the first synthesis of poly(ester-co-vinyl acetate) BCPs via ROCOP and RAFT. The exploration of more accessible reaction conditions and use of unpurified monomers also emphasises the broad potential of this approach as an area that warrants further investigation. This simple synthetic strategy opens up extensive opportunities to exploit different monomer combinations and even extend into multi-BCPs, to prepare a wealth of new materials for broad-ranging applications.
We gratefully acknowledge UKRI Future Leaders Fellowship (J. A. G., MR\T042710\1), Royal Society (J. A. G., RSG\R1\180101), EPSRC SOFI2 Centre for Doctoral Training (A. L. EP/S023631/1) and Croda (A. L.) for funding. We also acknowledge the peak deconvolution template by the Konkolewicz Group provided by the Macromolecular Alliance for Community Resources and Outreach.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- D. J. Walsh, M. G. Hyatt, S. A. Miller and D. Guironnet, ACS Catal., 2019, 9, 11153–11188 CrossRef.
- C. Diaz and P. Mehrkhodavandi, Polym. Chem., 2021, 12, 783–806 RSC.
- Y. You, C. Hong, W. Wang, W. Lu and C. Pan, Macromolecules, 2004, 37, 9761–9767 CrossRef.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef.
- J. M. Longo, M. J. Sanford and G. W. Coates, Chem. Rev., 2016, 116, 15167–15197 CrossRef PubMed.
- S. Paul, Y. Zhu, C. Romain, R. Brooks, P. K. Saini and C. K. Williams, Chem. Commun., 2015, 51, 6459–6479 RSC.
- A. Denk, S. Kernbichl, A. Schaffer, M. Kränzlein, T. Pehl and B. Rieger, ACS Macro Lett., 2020, 9, 571–575 CrossRef PubMed.
- Y. Zhao, Y. Wang, X. Zhou, Z. Xue, X. Wang, X. Xie and R. Poli, Angew. Chem., Int. Ed., 2019, 58, 14311–14318 CrossRef.
- Y. Wang, Y. Zhao, S. Zhu, X. Zhou, J. Xu, X. Xie and R. Poli, Angew. Chem., Int. Ed., 2020, 59, 5988–5994 CrossRef PubMed.
- S. Zhu, Y. Zhao, M. Ni, J. Xu, X. Zhou, Y. Liao, Y. Wang and X. Xie, ACS Macro Lett., 2020, 9, 204–209 CrossRef PubMed.
- N. Patil, Y. Gnanou and X. Feng, Polym. Chem., 2022, 13, 2988–2998 RSC.
- Y. Wang, Y. Zhao, Y. Ye, H. Peng, X. Zhou, X. Xie, X. Wang and F. Wang, Angew. Chem., Int. Ed., 2018, 57, 3593–3597 CrossRef PubMed.
- Y.-Y. Zhang, G.-W. Yang and G.-P. Wu, Macromolecules, 2018, 51, 3640–3646 CrossRef.
- C. A. L. Lidston, B. A. Abel and G. W. Coates, J. Am. Chem. Soc., 2020, 142, 20161–20169 CrossRef PubMed.
- T. M. McGuire, M. Miyajima, M. Uchiyama, A. Buchard and M. Kamigaito, Polym. Chem., 2020, 11, 5844–5850 RSC.
- Y. Zhao, S. Zhu, X. Li, X. Zhao, J. Xu, B. Xiong, Y. Wang, X. Zhou and X. Xie, CCS Chem., 2021, 4, 122–131 CrossRef.
- C.-M. Chen, X. Xu, H.-Y. Ji, B. Wang, L. Pan, Y. Luo and Y.-S. Li, Macromolecules, 2021, 54, 713–724 CrossRef.
- R. Fleet, J. B. McLeary, V. Grumel, W. G. Weber, H. Matahwa and R. D. Sanderson, Macromol. Symp., 2007, 255, 8–19 CrossRef.
- K. Hakobyan, T. Gegenhuber, C. S. P. McErlean and M. Müllner, Angew. Chem., Int. Ed., 2019, 58, 1828–1832 CrossRef.
- M. M. Milosavljević, A. D. Marinković, V. B. Veljković and D. D. Milenković, Montashefte Chem., 2012, 143, 43–49 CrossRef.
- V. N. Yuskovets, E. P. Anan’eva, Yu. A. Trukhanova, N. M. Chernov, I. P. Yakovlev and G. V. Ksenofontova, Russ. J. Gen. Chem., 2022, 92, 1378–1383 CrossRef.
- R. Qu, Z. Wei, H. Suo, Y. Gu, X. Wang, Z. Xin and Y. Qin, J. Polym. Sci., 2023, 61, 777–786 CrossRef.
- B. A. Abel, C. A. L. Lidston and G. W. Coates, J. Am. Chem. Soc., 2019, 141, 12760–12769 CrossRef.
- G.-P. Wu and D. J. Darensbourg, Macromolecules, 2016, 49, 807–814 CrossRef.
- Z. Hošťálek, O. Trhlíková, Z. Walterová, T. Martinez, F. Peruch, H. Cramail and J. Merna, Eur. Polym. J., 2017, 88, 433–447 CrossRef.
- Z. A. Wood, M. K. Assefa and M. E. Fieser, Chem. Sci., 2022, 13, 10437–10447 RSC.
- M. D. C. L. Cheng-Tan, A. N. Nguyen, C. T. Gordon, Z. A. Wood, Y. Manjarrez and M. E. Fieser, ACS Sustainable Chem. Eng., 2024, 12, 7246–7255 CrossRef.
- H. Kyun Ryu, D. Young Bae, H. Lim, E. Lee and K. Son, Polym. Chem., 2020, 11, 3756–3761 RSC.
- W. Thumrongpatanaraks, T. Pongpanit, P. Chumsaeng, T. Jaenjai, S. Yimthachote and K. Phomphrai, ChemistrySelect, 2022, 7, e202104450 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: The data supporting this article have been included as part of the ESI. See DOI: https://doi.org/10.1039/d4cc05501b |
| This journal is © The Royal Society of Chemistry 2025 |