
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An exploration of the electrocatalytic activity of nickel boride nanocrystals in the oxidation of 5-HMF†
Jennifer
Hong
 ab,
Matteo
Miola
a,
Dominic
Gerlach
b,
Marc C. A.
Stuart
c,
Petra
Rudolf
b,
Dulce M.
Morales
a,
Loredana
Protesescu
b and
Paolo P.
Pescarmona
*a
ab,
Matteo
Miola
a,
Dominic
Gerlach
b,
Marc C. A.
Stuart
c,
Petra
Rudolf
b,
Dulce M.
Morales
a,
Loredana
Protesescu
b and
Paolo P.
Pescarmona
*a
aEngineering and Technology institute Groningen (ENTEG), University of Groningen, 9747AG Groningen, Netherlands. E-mail: p.p.pescarmona@rug.nl
bZernike Institute for Advanced Materials, University of Groningen, 9747AG Groningen, Netherlands
cGroningen Biomolecular Sciences and Biotechnology institute, University of Groningen, 9747AG Groningen, Netherlands
First published on 28th November 2024
Abstract
In this work, we investigated the inherent electrocatalytic activity of nickel borides in an important reaction in the context of electrochemical valorization of biomass as the oxidation of hydroxymethylfurfural (5-HMF) to furan dicarboxylic acid (FDCA). For this purpose, nickel borides (NixB, x = 2 and 3) in the form of phase-pure nanocrystals (NCs) were synthesized through a solid-state synthesis method, supported on carbon paper and then tested as electrocatalysts for the oxidation of hydroxymethylfurfural (pH 12.9 or 13.9, 1.8 V vs. RHE, 3 h) by comparing their activity to that of Ni nanocrystals of similar average particle size (36–39 nm). Ni3B NCs achieved the highest 5-HMF conversion and Faradaic efficiency towards 5-HMF oxidation (Conv.5-HMF = 70%, FE = 94%), which is a markedly better performance compared to Ni2B NCs (Conv.5-HMF = 57%, FE = 72%) and to Ni nanoparticles (Conv.5-HMF = 58%, FE = 65%), thus unequivocally demonstrating for the first time the superior activity brought about by Ni3B. Based on a combination of physicochemical and electrochemical characterization (XPS, SEM, TEM, Cdl analysis), the better performance of the Ni3B-based electrocatalyst is attributed to differences in surface composition compared to the Ni2B-based electrocatalyst and to differences in terms of electrochemical surface area and/or bulk chemical features compared to the Ni-based electrocatalyst. Notably, these results were achieved with a remarkably low electrocatalyst loading (0.05 mg cm−2), leading to significantly higher turnover frequency compared to state-of-the-art nickel boride electrocatalysts for this reaction. A kinetic study showed that NixB NCs catalyze the electrosynthesis of FDCA from 5-HMF both through a direct and indirect mechanism, with the contribution of each changing as a function of the pH of the electrolyte.
Introduction
Although global issues related to non-renewable resources depletion, growing environmental pollution, and the ensuing climate change are generally recognized by the scientific community and, more in general, by society, finding a solution to them remains a complex challenge that requires efforts in different research fields. One of them is the development of efficient strategies for converting renewable resources into useful chemical products. Lignocellulosic biomass is one of the most abundant bio-based feedstock.1–5 Therefore, its conversion into a set of compounds, generally indicated as platform chemicals, has been an important topic of research in recent years. Among these platform chemicals, 5-hydroxymethylfurfural (5-HMF) is considered particularly relevant because it can be obtained from the dehydration of monosaccharides derived from cellulose, and it can be converted into several high-value products: 5-hydroxymethyl-2-furancarboxylic acid (HMFCA); 2,5-diformylfuran (DFF), 5-formyl-2-furancarboxylic acid (FFCA), and 2,5-furandicarboxylic acid (FDCA).3,6 The latter is the most desired product as it is the building block for polyethylene 2,5-furandicarboxylate (PEF), a prospective renewable substitute for the widely used polyethylene terephthalate (PET).The thermochemical oxidation of 5-HMF generally requires the use of a noble metal catalyst (Pt, Au, Pd, Ru) in order to achieve FDCA yield and 5-HMF conversion above 90%.7,8 These reactions also need to be performed at a relatively high temperature (above 100 °C) and high pressure of O2 (10 bar) or air (40 bar). On the other hand, non-noble transition metal catalysts (Fe, Ce, Co, Mn) are reported to be active in promoting the oxidation of 5-HMF (>96%), albeit at harsher reaction conditions compared to the noble-metal catalyzed systems and with lower selectivity towards FDCA.8,9 As an alternative to the conventional thermochemical route, the oxidation of 5-HMF can be also carried out through an electrochemical route (Fig. 1). This route offers some advantages because the electrochemical oxidation proceeds in water at ambient conditions, removing the need for high energy input and organic solvents.10–14 Generally, the electrochemical oxidation of 5-HMF is coupled with the reduction of water generating green hydrogen, although in principle other electrochemical reduction reactions can be chosen to produce other added-value chemicals (e.g. fructose reduction to sorbitol, CO2 reduction to C1–C2 products, levulinic acid reduction to valeric acid, nitrate reduction to ammonia, nitrobenzene reduction to aniline, etc.).14–25 Indeed, the electrochemical conversion of small organic molecules to bulk or specialty chemicals has steadily garnered attention as a possible direction in making the chemical industry more sustainable. The electrochemical oxidation of bio-based organic molecules can generally proceed at a lower potential compared to the oxygen evolution reaction (OER), which is the anodic reaction in conventional water electrolysis, while preventing the competition between the two reactions.4,6,14,26,27 Furthermore, as in the case of the oxidation of 5-HMF, the product(s) generated from electrochemical oxidation have higher industrial value than the O2 produced from OER. As a result, increasing efforts have been placed in investigating and optimizing the electrochemical oxidation of 5-HMF towards the production of FDCA.3,6 In this context, a crucial item is the development of active, selective, stable and affordable electrocatalysts.
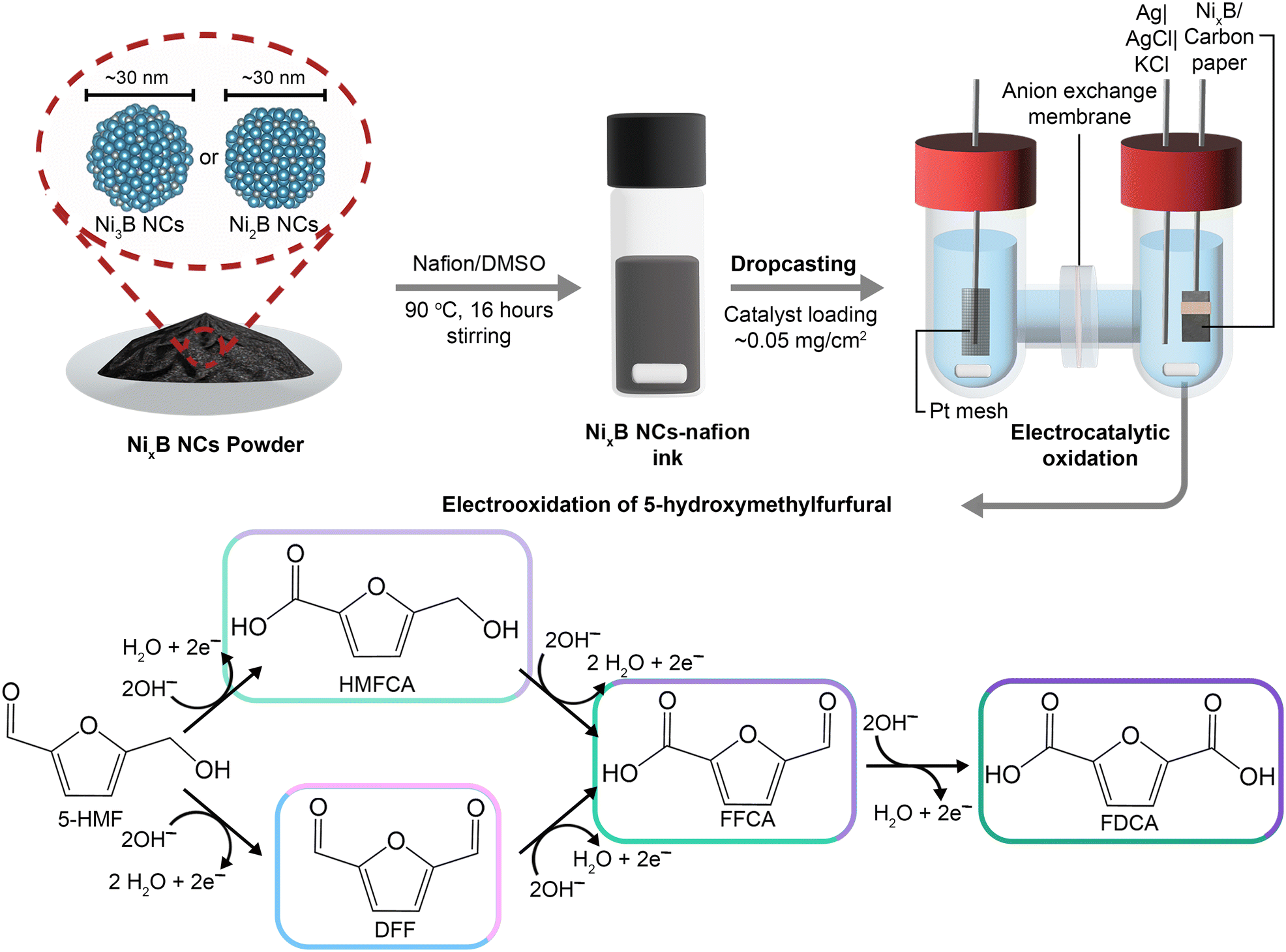 | ||
| Fig. 1 Top: Schematic representation of the preparation and electrocatalytic testing process of NixB NCs. Bottom: Possible 5-HMF oxidation pathways towards FDCA. Note that since the reaction is carried out at high pH (12.9 or 13.9), the carboxyl groups will be mainly in their deprotonated form. | ||
Noble metals (Pt, Ru, Au, Pd, AuPd) have been investigated as active species in electrocatalysts for 5-HMF oxidation. While they show relatively low onset potential for 5-HMF oxidation (∼0.25–0.34 V vs. RHE), selective oxidation of 5-HMF to FDCA was rarely achieved.28–39 Multiple reports on Pt and Ru as electrocatalysts for this reaction showed the main product to be DFF, generally with trace amounts of FDCA.28–33,37–39 The same problem in reaching high FDCA yields was also observed for studies done using Au- and Pd-based electrocatalysts, with the main products obtained being either DFF or HMFCA.34–37 Due to the high cost of noble metals, non-noble metal alternatives have been explored as electrocatalysts for 5-HMF oxidation. In particular, Ni-based electrocatalysts are of interest for the electrochemical oxidation of 5-HMF.7–13,36,40–60 Preceding studies have reported Ni-based electrocatalysts to be very promising for this reaction, reaching complete 5-HMF conversion with FDCA as the only detected product, and >99% Faradaic efficiency (FE) towards 5-HMF oxidation, generally measured in 1 M KOH electrolyte (pHnominal = 13.87) at 25 °C, using 5-HMF concentration of 10 mM, and electrode potentials between 1.3 and 1.7 V vs. RHE. Tables S1 and S2† report the state-of-the-art Ni-based electrocatalysts for oxidation of 5-HMF and key indicators of their performance, including conversion, yield, and FE at various electrolysis conditions.
Among the various types of Ni-based materials (e.g. NixBy, Ni2P, Ni2S3, Ni3N, NiSe, NiO), nickel borides (NixBy), are a class of materials that has slowly garnered attention in recent years as electrocatalysts for water-splitting due to reportedly high activity and stability.61 The use of amorphous NixB supported on Ni foam (NF) as electrocatalyst for the oxidation of 5-HMF has been previously studied in an electrochemical flow-through reactor at pHnominal = 13.87.41 The amorphous NixB was synthesized through the reduction of aqueous NiCl2·6H2O with NaBH4 under argon atmosphere at 0 °C, and subsequently thermally annealed at 300 °C. The resulting NixB was then dispersed in water in the presence of Nafion and dropcast onto 1 × 1 cm2 Ni foam (NixB loading ∼1 mg cm−2).41 With this electrocatalyst, 5-HMF was preferentially oxidized to HMFCA, which was then converted into FDCA with a 98.5% yield and reaching a nearly 100% Faradaic efficiency. Amorphous NixB supported on Ni foam was also studied as electrocatalyst for the paired electrochemical oxidation of 5-HMF and hydrogenation of nitrophenol.56 This NiBx/Ni_foam was prepared through electrodeposition, resulting in a layer of relatively large, amorphous Ni–NiBx particles (![[d with combining macron]](https://www.rsc.org/images/entities/i_char_0064_0304.gif) ≥ 300 nm) on the Ni foam. With this electrocatalyst, full conversion to FDCA (with FE ∼100%) was reached after 100 minutes of electrolysis at a constant potential of 1.4 V vs. RHE. Although both systems proved efficient in achieving high conversion of 5-HMF into FDCA, it is difficult to establish whether the nickel boride phase is actually more active than nickel alone, or the enhanced performance observed in the presence of NixB is just due to the fact that the Ni loading per unit surface and possibly the roughness were increased. The interpretation is made even more complex by the presence of Ni in the Ni foam used as the support, which is also active towards this reaction. Consequently, the role of the nickel boride phase in defining the electrocatalytic activity in the oxidation of 5-HMF has not yet been fully elucidated. Furthermore, both reported systems employ amorphous NixB, whereas the activity of nanocrystalline nickel borides has not been explored so far, possibly because of the harsh reaction conditions generally required to synthesize these materials (T > 600 °C, pressure > 1 GPa).41,56
≥ 300 nm) on the Ni foam. With this electrocatalyst, full conversion to FDCA (with FE ∼100%) was reached after 100 minutes of electrolysis at a constant potential of 1.4 V vs. RHE. Although both systems proved efficient in achieving high conversion of 5-HMF into FDCA, it is difficult to establish whether the nickel boride phase is actually more active than nickel alone, or the enhanced performance observed in the presence of NixB is just due to the fact that the Ni loading per unit surface and possibly the roughness were increased. The interpretation is made even more complex by the presence of Ni in the Ni foam used as the support, which is also active towards this reaction. Consequently, the role of the nickel boride phase in defining the electrocatalytic activity in the oxidation of 5-HMF has not yet been fully elucidated. Furthermore, both reported systems employ amorphous NixB, whereas the activity of nanocrystalline nickel borides has not been explored so far, possibly because of the harsh reaction conditions generally required to synthesize these materials (T > 600 °C, pressure > 1 GPa).41,56
In this work, we investigated for the first time the electrocatalytic performance of crystal-phase-pure nanosized nickel borides NixB, namely Ni3B ( = 39 nm) and Ni2B ( = 38 nm) nanocrystals (NCs) supported on carbon paper, as electrocatalysts for the electrochemical oxidation of 5-HMF under two different pH conditions (Fig. 1). By comparing these crystalline, nanosized NixB systems with a counterpart consisting of Ni particles with similar average size ( = 36 nm), we demonstrated that the Ni3B-based electrocatalyst displays the highest activity and Faradaic efficiency during the electrochemical oxidation of 5-HMF. The tests were carried out with lower Ni loading and higher 5-HMF concentration (100 mM), and thus under more challenging conditions, compared to most literature reports. These conditions were chosen to achieve intermediate conversions of 5-HMF, as this allows to highlight more clearly the differences in activity between the electrocatalysts. Importantly, the fact that the 5-HMF concentration that we used is 10-fold higher than the one conventionally used in the literature (10 mM) can also bring this technology closer to industrial-scale application.
Experimental section
Materials
Anhydrous nickel(II) chloride (NiCl2, 98%), nickel (0) powder (<100 nm, 99%), methanol (MeOH, HPLC grade, ≥99.9%), dimethyl sulfoxide (DMSO, anhydrous, >99.9%), Nafion 117 solution (∼5 wt% solution in a mixture of lower aliphatic alcohols and water), 5-hydroxymethyl furfural (5-HMF, 99%), methyl phenyl sulfone (≥98.0%, GC grade) and potassium hydroxide (KOH, ACS reagent, ≥99.9%, pellets) were purchased from Sigma-Aldrich and used without any further purification. Sodium borohydride (NaBH4, fine granular, >98%) was purchased from Sigma-Aldrich and was ball-milled (2 h, 8000 rpm, zirconia balls) prior to use. Diformyl furan (DFF, >98.0%), 5-(hydroxymethyl)furan-2-carboxylic acid (HMFCA, >98.0%), 5-formyl-2-furancarboxylic acid (FFCA, >98.0%), and 2,5-furandicarboxylic acid (FDCA, >98.0%) were purchased from TCI Europe N.V. and used without any further purification. Carbon paper (CP) electrode was purchased from Fuel Cell Store. Adhesive Teflon tape (TVT acrylic adhesive) was purchased from Metalalloy Italiana. Forblu Selemion AMVN anion-exchange membrane was kindly supplied by AGC Chemical.Solid-state synthesis of Ni3B nanocrystals (Ni3B NCs)
In a standard synthesis of Ni3B NCs,62 NiCl2 (400 mg, 3.08 mmol) and NaBH4 (116.5 mg, 3.08 mmol) were weighed in a 25 mL round-bottomed flask equipped with a glass stirring bar inside a N2-filled glove box. The well-closed flask containing the reaction mixture was quickly transferred and connected to a Schlenk line and placed under alternating cycle of Ar and vacuum for 1 min each. The Ar-vacuum cycle was repeated three times. The reaction mixture was then kept under continuous Ar flow, followed by heating to 400 °C while stirring. The reaction was allowed to proceed at the said temperature for 24 h. Once the specified time was reached, the heating was stopped and the reaction mixture was let to cool down to room temperature. The resulting solid product was washed with MeOH, then twice with deionized water and one final time with MeOH. The washed product was collected as a black fine powder and dried in an oven under vacuum overnight at 60 °C.Solid-state synthesis of Ni2B nanocrystals (Ni2B NCs)
In a typical synthesis,62 commercial Ni0 nanopowder (400 mg, 6.16 mmol) and NaBH4 (257.8 mg, 6.16 mmol) were weighed in a 25 mL round-bottomed flask inside a N2-filled glovebox. The reaction mixture was placed on a Schlenk line under vacuum, then flushed with Ar. The alternating vacuum-Ar cycle (1 min each) was repeated three times. The reaction mixture was then heated to 400 °C and stirred under Ar atmosphere. After 72 h of reaction, the heating was stopped and the reaction mixture was left to cool down naturally to room temperature. The obtained solid product was washed with MeOH, then twice with deionized water and finally again with MeOH. The solid was collected as a black fine powder and dried under vacuum overnight at 60 °C.Solid-state synthesis of Ni nanocrystals (Ni NCs)
In a typical synthesis of Ni NCs,62 NiCl2 (400 mg, 3.08 mmol) and NaBH4 (60.5 mg, 1.03 mmol) were weighed in a round-bottomed flask inside a N2-filled glove box and then placed on a Schlenk line. The reaction mixture was put under vacuum and flushed with Ar alternatingly (1 min each) three times. The mixture was left to react at 400 °C for 24 h under continuous Ar flow. The resulting product (black fine powder) was washed in the same manner as the Ni3B and Ni2B NCs.Working electrode preparation
An aliquot (1, 3 or 15 mg) of the synthesized NCs (Ni, Ni3B or Ni2B) was added to a 0.1 M Nafion/DMSO solution (1 mL), and the mixture was stirred overnight at 90 °C. This resulted in a well-dispersed NCs ink. The NCs ink was sonicated for 45 min in an ultrasonic bath (HBM GL series 2.5 L). A 50 μL aliquot of the electrocatalyst ink was taken with a micropipette and then dropcast onto a 1 × 1 cm2 carbon paper to afford a set of electrodes with different NCs loadings (nominal loadings: 0.05, 0.15, 0.75 mg cm−2).B2O3 ink was prepared by dissolving 20 mg of B2O3 in 0.1 M Nafion/ethanol solution (1 mL). A 50 μL aliquot of the solution was taken with a micropipette and then dropcast onto a 1 × 1 cm2 carbon paper to give B2O3@CP with B2O3 loading of 1 mg cm−2.
Electrochemical measurements
All the electrochemical measurements were performed at room temperature (without temperature control) in a two-compartment H-type cell with a three-electrode configuration at pHnominal = 12.87 (0.1 M KOH electrolyte) or pHnominal = 13.87 (1.0 M KOH electrolyte) using a Gamry Interface 1000 E electrochemical workstation (setup shown in Fig. S1†). The KOH electrolyte solutions were prepared using Milli-Q water. It should be noted that pH 12.87 and 13.87 are the nominal pH values of 0.1 M KOH and 1.0 M KOH electrolyte solution, respectively. The pH of the these electrolytes was measured using Mettler Toledo pH Meters and was found to be 12.9 for 0.1 M KOH and 13.9 for 1.0 M KOH and, therefore, will be referred to as such in the rest of the text. The two compartments were separated by an anion-exchange membrane and a cellulose-based foam to prevent crossover of organic compounds between compartments. A Pt-mesh was used as the counter electrode (CE), Ag|AgCl|KCl (3 M) as the reference electrode (RE), and the carbon paper loaded with the NCs as the working electrode (WE). The anode and cathode compartments were filled with 10 mL of electrolyte.The uncompensated resistance (RU) was determined through electrochemical impedance spectroscopy (EIS) measurement from 10 to 100 kHz. Cyclic voltammograms (CVs) (5 cycles) were measured at a scan rate of 10 mV s−1 from 0 to 1.5 V vs. Ag|AgCl|KCl, first with no 5-HMF in the anodic compartment. Afterwards, 5-HMF (126.1 mg, corresponding to a 100 mM concentration in the electrolyte solution) was added only into the anodic compartment. We measured the pH of the electrolytes for both the 0.1 M KOH and 1.0 M KOH systems after the addition of 5-HMF and found that this did not alter the pH (12.9 and 13.9, respectively). A sample was collected from this anodic electrolyte solution to determine the initial concentration of 5-HMF, followed by CVs measurement (5 cycles) at a scan rate of 10 mV s−1 from 0 to 1.5 V vs. Ag|AgCl|KCl. Then, a constant-potential electrolysis (chronoamperometry) measurement was performed at 0.85 V vs. Ag|AgCl|KCl in 0.1 M KOH and at 0.8 V vs. Ag|AgCl|KCl in 1.0 M KOH for 3 h, unless indicated otherwise. Generally, the studies were repeated three times to test the reproducibility of the results (Fig. S2 and S3†). All the potentials were converted to the reversible hydrogen electrode (RHE) scale through the Nernst equation below:
| E vs. RHE = Eapplied vs. Ag|AgCl|KCl + E0(Ag|AgCl|KCl) + 0.059 pH |
E 0(Ag|AgCl|KCl) = 0.2 V is the standard potential of the Ag|AgCl reference electrode at 25 °C.63
The pH of the electrolyte was measured throughout the reaction (t = 30, 60, 90, 120, 180 min) and found to decrease slightly and gradually. At the end of the reaction, the pH of the electrolyte for the 0.1 M KOH and 1.0 M KOH systems were 12.6 and 13.6, respectively.
To monitor the substrate conversion, 100 μl of the anodic electrolyte were sampled at t = 0, 30, 60, 90, 120, and 180 min for product analysis (details in next section).
The double-layer capacitance (CDL) of the elecrocatalysts was determined by recording CVs with different scan rates (500, 1000, 2000, 3333 mV s−1) within the region where redox activity is absent (0.9 V vs. RHE to 1.1 V vs. RHE). The difference in the measured current density between the cathodic and anodic scans (Δj/2) was plotted as a function of the scan rate (ν), and the slopes of these plots provided the CDL values.
Electrode recycling tests were conducted using the same setup described above (two-compartment H-type cell at pH 13.9 (1.0 M KOH electrolyte), Pt mesh as counter electrode and Ag|AgCl|KCl as reference electrode). After the chronoamperometric test at 1.8 V vs. RHE for 3 h, the electrode was washed with Milli-Q water. The H-cell was cleaned thoroughly with Milli-Q water and fresh electrolyte solution was added to the clean setup. The previously-tested electrode was used as the working electrode once again and underwent the same testing procedures (CV with and without 5-HMF and chronoamperometry at 1.8 V vs. RHE for 3 h). The procedure was repeated for a total of five tests.
High-performance liquid chromatography (HPLC) measurements
Quantitative analysis of the organic compounds present in the electrolyte samples from the 5-HMF oxidation was performed by HPLC. The electrolyte samples (100 μl) taken during the chronoamperometric experiments were diluted in 1 mL of Milli-Q water and neutralized to pH 7 by dropwise addition of 0.5 M H2SO4. 100 μl of methyl phenyl sulfone solution (5.6 mg mL−1) was added to the sample as an internal standard. The HPLC measurements were performed on an Agilent Technologies 1200 series chromatographer equipped with a Bio-Rad Aminex HPX-87H 300 × 7.8 mm column at T = 60 °C with 0.5 mM aqueous H2SO4 as the eluent (flow rate: 0.55 mL min−1) and a refractive index detector. The calibration curve for 5-HMF and the expected oxidation products (HMFCA, DFF, FFCA, FDCA) were made using solutions of the corresponding standard of known concentrations in the presence of methyl phenyl sulfone as the internal standard.The conversion of 5-HMF, oxidation products yield, and the Faradaic efficiency (FE) were calculated as follows:
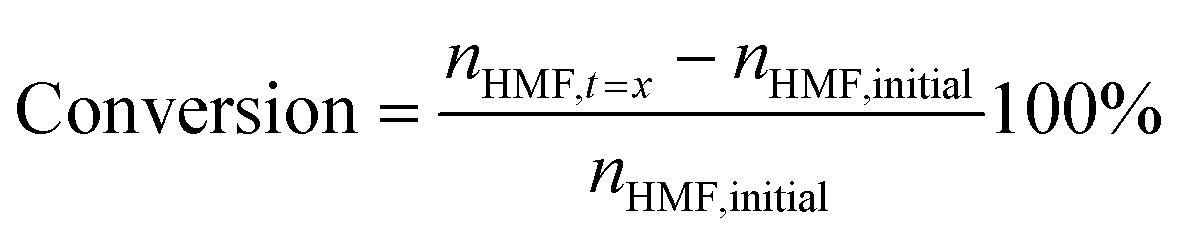
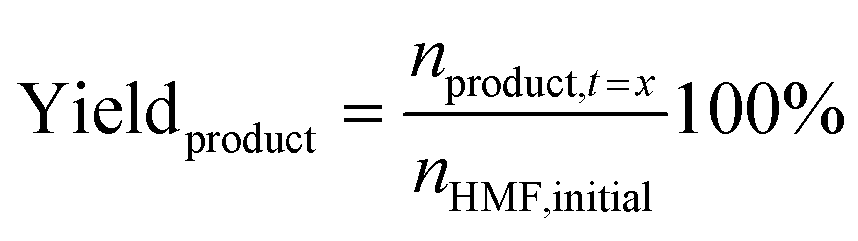
where n is the number of moles of each specific compound measured at the start (t = 0) or during electrolysis (t = x), F is the Faraday constant (96
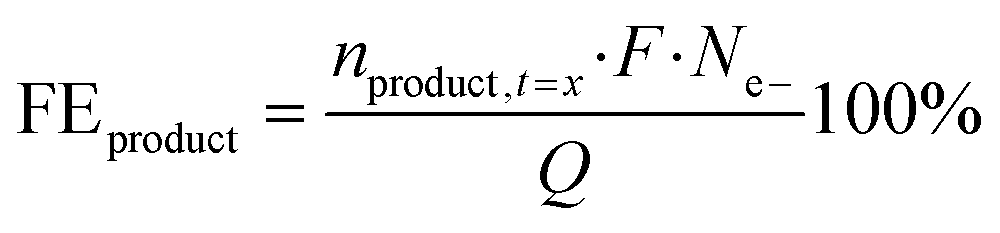
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1), Ne- is the number of electrons required for the oxidation of 5-HMF towards a certain product, and Q is the charge passing through the system.
485 C mol−1), Ne- is the number of electrons required for the oxidation of 5-HMF towards a certain product, and Q is the charge passing through the system.
It is worth noting that we chose to take into account the degradation into humins in our calculations of the conversion of 5-HMF, as this provides a more complete evaluation of the process, though in the literature on the electrochemical oxidation of 5-HMF this aspect if often omitted.
Characterization methods
X-ray diffraction (XRD) patterns were recorded on a Bruker D8 Advance diffractometer equipped with θ-θ Bragg-Bretano geometry and a Cu X-ray tube operated at 40 kV and 40 mA.Transmission Electron Microscopy (TEM) images of the nanocrystals mixed with Nafion in DMSO (dropcasted on Cu grid) were taken on a Philips CM120 electron microscope operating at 120 keV. The particle size average and distribution were measured by analyzing 50 randomly selected particles from multiple TEM images of each sample, using the software ImageJ.
Scanning Electron Microscopy (SEM) images of the nanocrystals cast on carbon paper were taken using a Fei NovaNanoSEM 650 operated at 1 kV, with a spot size of 3.5 nm, and high voltage of 15 kV.
X-ray Photoelectron Spectroscopy (XPS) was carried out on a Surface Science Instruments SSX-100 ESCA equipment with a monochromatic Al Kα X-ray source (hν = 1486.6 eV). The pressure in the measurement chamber was kept below 8 × 10−9 mbar during data acquisition. The electron take-off angle was 37° relatively to the surface normal. The diameter of the analyzed area was 1000 μm. The energy resolution was 1.26 eV (or 1.67 eV for broad survey scans). The XPS data were analyzed using the least-squares curve fitting program Winspec (developed at the LISE, University of Namur, Belgium) and included a Shirley baseline subtraction and a peak deconvolution performed utilizing a linear combination of Gaussian and Lorentzian functions, taking into account the experimental resolution. The spectra were fitted with a minimum number of peaks. Binding energies of isolated peaks typically have an uncertainty of ±0.05 eV. When more than one component was needed to reproduce the raw data, the uncertainty in the peak position increased to ±0.1 eV. Binding energies were referenced to the C 1s photoemission peak at a binding energy of 284.8 eV, which originates from adventitious carbon (C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C).64
C).64
Inductively Coupled Plasma Mass Spectrometry (ICP-MS) measurements were performed with ICP-MS/MS iCAP TG Thermo Scientific. All samples were prepared through acid digestion. A small amount of sample was weighed in a poly propylene cup then 100 μL of 32.5% or 65% HNO3 was added. Following sample dissolution, 400 μL of water was added.
Results and discussion
In this work, we investigated the electrocatalytic activity of two different crystalline phases of NixB NCs, i.e. Ni3B and Ni2B, in the electrochemical oxidation of 5-HMF (Fig. 1). We aimed to establish the electrocatalytic activity of Ni borides and to determine whether these compounds outperform Ni (in the form of nanoparticles with similar size distribution to that of the NixB). To prepare the phase-pure nanocrystalline Ni3B and Ni2B NCs (where phase-pure refers to the bulk of the nanoparticles containing a single crystalline NixB phase), we employed a novel method that was recently developed by our groups.62 We synthesized the NixB NCs through solid-state reaction of nickel precursors with sodium borohydride (NaBH4) at a relatively low temperature (400 °C) under atmospheric pressure, i.e. under significantly less harsh conditions compared to what is typically required to achieve crystallinity in Ni borides. The Ni NCs were prepared through a similar solid-state reaction, but with lower NaBH4:Ni ratio. The XRD patterns of the resulting Ni, Ni3B, and Ni2B powders match the references, with no other crystalline phase being observed in any of the three samples (Fig. 2a–c). This indicates that only a single crystalline phase is present in each material. The particle size estimated using the Scherrer equation was ca. 30 nm for all the three materials.
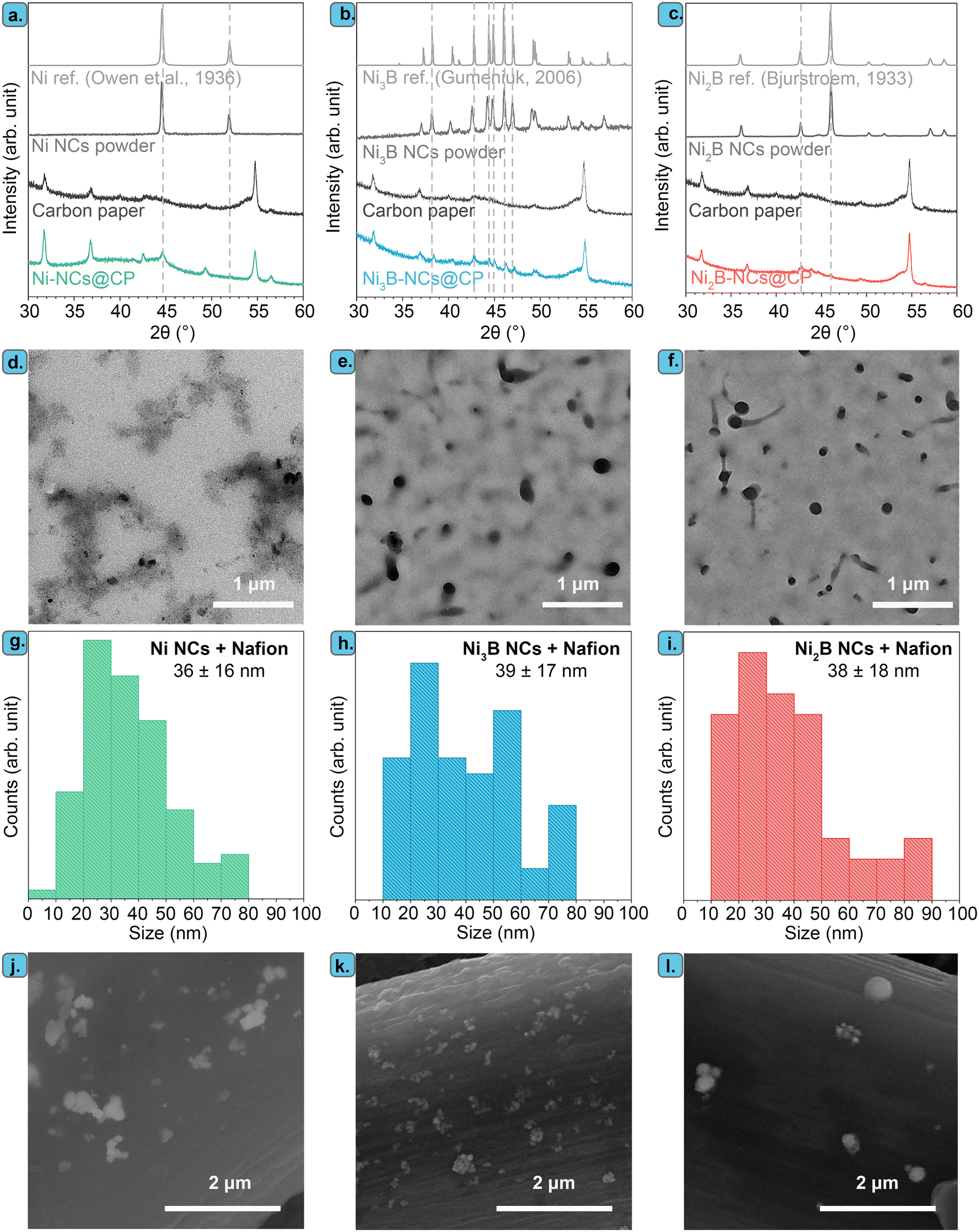 | ||
| Fig. 2 XRD patterns of (a) Ni NCs powder and Ni-NCs@CP, (b) Ni3B NCs powder and Ni3B-NCs@CP, and (c) Ni2B NCs powder and Ni2B-NCs@CP prepared with a catalyst loading of 0.05 mg cm−2. The XRD pattern of carbon paper, Ni, Ni3B, and Ni2B are provided as references. TEM images of (d) Ni NCs mixed with Nafion, (e) Ni3B NCs mixed with Nafion, and (f) Ni2B NCs mixed with Nafion. Particle size distribution based on TEM images of (g) Ni NCs mixed with Nafion, (h) Ni3B NCs mixed with Nafion, and (i) Ni2B NCs mixed with Nafion. SEM images of (j) Ni-NCs@CP, (k) Ni3B-NCs@CP, and (l) Ni2B-NCs@CP prepared with a catalyst loading of 0.05 mg cm−2. | ||
TEM images of the Ni, Ni3B, and Ni2B stabilized by Nafion (i.e. the components of the inks used to prepare the electrocatalysts) show in all cases discrete particles along with aggregated particles (Fig. 2d–f and S4†). For all the materials, the average size of the discrete nanoparticles is similar (36 nm for Ni, 39 nm for Ni3B and 38 nm for Ni2B), with the particle size distribution being rather broad in all cases (Fig. 2g–i). These values are consistent with, and yet slightly higher, than those obtained with the Scherrer equation. The larger particles consist of aggregates of the primary particles (Fig. S4†) and have irregular shapes with sizes > 100 nm (Fig. 2d–f and S4†). Selected area electron diffraction (SAED) and Fast Fourier Transform (FFT) analysis coupled to the TEM measurements (Fig. S4†) confirmed the crystal structures that were observed by XRD in the three materials.
In order to be used as electrocatalysts, these powders need to be loaded onto a conductive support. In this study, we chose to use carbon paper (CP) instead of the commonly employed Ni foam,41,56 to exclude the likely electrocatalytic contribution of the Ni support. The use of a non-Ni based support allowed us to study the electrocatalytic properties stemming from the nickel borides and compare them to those of nickel. Additionally, we aimed at minimizing the loading of the Ni-based electrocatalyst as nickel has recently been added to the list of EU critical raw materials.65 Therefore, we explored the effect of the catalyst loading (0.05, 0.15, 0.75 mg cm−2) by varying the concentration of NixB NCs in the catalyst ink (1 to 15 mg of NCs in 1 mL of 0.1 M Nafion/DMSO solution) used for preparing the electrode. These relatively low catalyst loadings were selected to exploit the nanoscale size of the NixB and to minimize particle aggregation. Indeed, SEM analysis evidenced that the obtained Ni3B-NCs@CP electrocatalysts with catalyst loading 0.05 mg cm−2 consist of well-dispersed nickel boride NCs supported on the carbon paper fiber (Fig. 2k), with the nickel borides being present as discrete (∼30–70 nm) or mildly aggregated structures (>100 nm), in agreement with the TEM analysis. With higher catalyst loading of 0.15 and 0.75 mg cm−2, a higher degree of particle aggregation can be observed in both samples as shown by the SEM images (Fig. S5 and S6†). In addition, Ni NCs (Ni-NCs@CP) and Ni2B NCs (Ni2B-NCs@CP) were also supported onto carbon paper through the same preparation method described in details above. SEM images of Ni-NC@CP (Fig. 2j) and Ni2B-NCs@CP (Fig. 2l) showed that also these materials consist of discrete particles and some aggregates (similar to those observed by TEM) supported on the carbon paper. The XRD patterns of the supported Ni NCs, Ni3B NCs and Ni2B NCs display the characteristic peaks of each material alongside the carbon paper diffraction peaks (Fig. 2a–c).
All the electrocatalytic tests were performed in batch mode in a two-compartment H-type electrochemical cell with a three-electrode configuration at pH 12.9 (0.1 M KOH) or pH 13.9 (1.0 M KOH). At the highest KOH concentration, 5-HMF has a tendency to rapidly degrade into undesired by-products and eventually into insoluble humins.39,66 These non-electrochemical side reactions occur in competition with the electrochemical oxidation of 5-HMF. Since our tests were carried with relatively high 5-HMF concentration (100 mM), at which we anticipate the electrochemical oxidation to be incomplete, the formation of humins is expected to be noticeable at pH 13.9. In order to mitigate the occurrence of these unwanted side reactions, we performed the 5-HMF electrochemical oxidation experiments at the lower pH of 12.9. An even lower pH than 12.9 would not be suitable for the reaction due to too low OH− concentration (OH− ions are involved in the half-reactions as shown in the scheme in Fig. 1).
5-HMF electrochemical oxidation in 0.1 M KOH (pH 12.9)
Our investigation of the electrocatalytic performance of Ni borides started by studying the electrochemical behaviour of Ni3B-NCs@CP in the absence of 5-HMF. Cyclic voltammetry (CV) indicated the presence of Ni2+ at the surface of Ni3B-NCs@CP, as evidenced by the Ni2+/Ni3+ redox peaks observed in the potential region between 1.25 and 1.5 V vs. RHE (Fig. 3a). With increasing applied potential (E ≥ 1.5 V vs. RHE), the oxygen evolution reaction (OER) was observed. The presence of Ni2+ species at the surface of Ni3B-NCs@CP is in agreement with previously reported characterization by XPS and iDPC STEM, which showed that an amorphous layer containing Ni(OH)2 and B2O3 is present at the surface of Ni3B NCs.62 | ||
| Fig. 3 Electrochemical oxidation of 5-HMF (100 mM) in 0.1 M KOH (pH 12.9) catalyzed by Ni3B-NCs@CP. (a) Cyclic voltammetry of Ni3B-NCs@CP in the presence and absence of 5-HMF with a scan rate of 10 mV s−1. (b) Cyclic voltammetry of Ni3B-NCs@CP with different electrocatalyst loadings in the presence of 100 mM 5-HMF recorded with a scan rate of 10 mV s−1. (c) Chronoamperometry measured at 1.8 V vs. RHE for 3 h using Ni3B-NC@CP with different electrocatalyst loadings with an initial concentration of 100 mM of 5-HMF. The plot also shows the sum of the concentrations of 5-HMF and of the reaction intermediates as a function of time obtained with an electrocatalyst loading of 0.05 mg cm−2. | ||
Next, we investigated the effect of the presence of 5-HMF by means of cyclic voltammetry with Ni3B-NCs@CP as the electrode with a Ni3B NCs loading of 0.05 mg cm−2 (Fig. 3a). Following the addition of 5-HMF, a few notable changes can be observed in the CV of Ni3B-NCs@CP. Firstly, the voltammogram displays a much more intense oxidation peak. The large oxidation peak corresponding to the oxidation of 5-HMF overlaps with the potential region at which Ni2+ is oxidized to Ni3+, thereby covering the Ni2+/Ni3+ oxidation peak, and extending towards higher potential. At 1.8 V vs. RHE, a higher current density (11.5 mA cm−2) is observed in the presence of 5-HMF than in its absence (7.8 mA cm−2). This strongly suggests that the oxidation of 5-HMF is taking place over the Ni3B NCs-based electrocatalyst. It is also worth noting that in the absence of 5-HMF a current density higher than 20 mA cm−2 (attributed to OER and possibly to oxidation of the carbon paper support – see Fig. S7†) is reached in the higher potential region (E > 2.2 V vs. RHE) compared to the measurements done in the presence of 5-HMF, in which case a current density of less than 10 mA cm−2 is observed (Fig. 3a). The hampering of the OER is possibly due to a preferential adsorption of 5-HMF or of its oxidation products on the active sites compared to that of OH−.
The effect of catalyst loading on the electrochemical oxidation of 5-HMF was investigated using Ni3B-NCs@CP electrodes with different Ni3B NCs loadings: 0.05, 0.15, and 0.75 mg cm−2 (Fig. 3b and S7† for the CVs in the presence and in the absence of 5-HMF, respectively). The comparison of the CV of carbon paper alone with that of Ni3B-NCs@CP in the presence of 5-HMF (Fig. 3b) clearly demonstrates the contribution of Ni3B NCs as electrocatalyst, allowing the 5-HMF oxidation to start at a much lower potential (>1.4 V vs. RHE) than on carbon paper (>1.8 V vs. RHE). However, the effect of the catalyst loading on the oxidation of 5-HMF is almost negligible as the CV of the three Ni3B-NCs@CP samples with increasing catalyst loading are nearly identical (Fig. 3b). The similarity in performance is further exemplified by the chronoamperometry (Fig. 3c) conducted at 1.8 V vs. RHE. The 5-HMF conversion, yields, and FE are also found to be almost the same for the different loadings tested (Fig. S8†). To gain better understanding on the similar activity observed despite the increased catalyst loading, we measured the double-layer capacitance (CDL) of the Ni3B-NCs@CP 0.05, 0.15, and 0.75 mg cm−2 electrocatalysts (Fig. S9†). The CDL values of the three samples are very similar, with that of Ni3B-NCs@CP 0.75 mg cm−2 (0.0494 mF cm−2) being only slightly higher than those of Ni3B-NCs@CP 0.15 mg cm−2 (0.0486 mF cm−2) and Ni3B-NCs@CP 0.05 mg cm−2 (0.0478 mF cm−2). This result is in agreement with the higher degree of aggregation of the Ni3B NCs observed at higher loading (vide supra). Therefore, we attribute the similar activity at different loadings to the decreased accessibility of the Ni3B particles at higher loading caused by their aggregation, and reflected by their similar CDL values. Based on the results of this study, we concluded that using the lower Ni3B NCs loading of 0.05 mg cm−2 for the electrochemical oxidation of 5-HMF at pH 12.9 leads to a similar performance compared to the higher loadings and, hence, this loading was used for all the subsequent studies.
It should be noted that the gradually decreasing current density observed in the chronoamperometric test (Fig. 3c) is mainly attributed to the decreasing concentration of reactants (5-HMF and intermediates), due to their conversion during the experiment, rather than being caused by a deactivation of the electrocatalyst. This was inferred based on the fact a plot of the sum of the concentration profiles of 5-HMF and of the intermediates (DFF and FFCA) as a function time follows a similar trend to that of the current density (Fig. 3c). The similarity between the sum of the concentration profiles of the reactants (5-HMF and intermediates) and the current density as a function of time was also observed for the Ni3B-NCs@CP 0.15 mg cm−2 and 0.75 mg cm−2 electrodes (Fig. S10 and S11†). Furthermore, when chronoamperometry at 1.8 V vs. RHE was measured for 3 h without 5-HMF with Ni3B-NCs@CP, after an initial, small current density drop (∼1.5 mA cm−2) in the first 15 min, the current density remains nearly constant at 4.5 mA cm−2 throughout the rest of the measurement (Fig. S12†). It is also worth noting that although 5-HMF was added to the anodic compartment to have an initial concentration of 100 mM, at the start of the CA measurement (i.e. after 5 CV cycles), the sum of the 5-HMF, DFF, and FFCA concentration was below 100 mM (∼95 mM). This can be attributed to the well-known fast degradation of 5-HMF into humins in basic medium.39,66
The fact that the chronoamperometry of the carbon paper alone shows a much lower, constant current density (Fig. 3c) and that virtually no electrochemical oxidation of 5-HMF was observed in this test (Fig. S13†) confirms that Ni3B NCs is the electrocatalytically active species and supports our hypothesis that the change in current density in the tests Ni3B-NCs@CP is related to the decrease in the concentration of reactants (5-HMF and intermediates) as the test proceeds. While there is a 14% decrease in the 5-HMF concentration throughout the test with carbon paper, no 5-HMF oxidation product was detected, implying that 5-HMF was not electrochemically oxidized but had instead non-electrochemically degraded.
Furthermore, characterization of the most active among the tested electrocatalysts (Ni3B-NCs@CP) after the chronoamperometric test showed that no pronounced structural or morphological changes occurred during the electrocatalytic test, as shown by XRD and SEM (Fig. 4). On the other hand, the XPS of the Ni3B-NCs@CP showed that the Ni and B surface atoms are mostly in oxidized state after the electrochemical test (Fig. 5). XPS analysis of the Ni3B NCs powder Ni 2p core level region showed the presence of a mixture of Ni0 and Ni2+ on the surface of the nanocrystals (see Fig. 5a, grey line).62 After being dropcast onto a carbon paper support and tested for the electrochemical oxidation of 5-HMF, a higher degree of surface oxidation is observed as the XPS spectrum of Ni3B-NCs@CP shows mainly a contribution from Ni2+, while the contribution from Ni0 expected for Ni3B is barely visible (Fig. 5a). This indicates that oxidation occurs deeper into the nanocrystal layers during the electrocatalytic test. B 1s XPS analysis shows two distinct peaks: at 188 eV, which corresponds to the characteristic B0 peaks in metal borides, and at 192 eV, which can be attributed to oxidized B species (Fig. 5b).62 For the Ni3B NCs powder, the main boron species is B0, as evidenced by the higher intensity of the signal at 188 eV compared to the one at 192 eV (Fig. 5b, grey). On the other hand, the B 1s XPS of Ni3B-NCs@CP after the electrocatalytic test (Fig. 5b), shows that the spectral intensity due to B0 is lower than that of oxidized B, indicating that the applied oxidizing potential leads to surface oxidation of the Ni3B NCs. In addition, the elemental ratio of Ni:B based on the XPS analysis was found to have increased from 7.2 in the Ni3B NCs powder to 8.6 in the Ni3B-NCs@CP after the electrocatalytic test, suggesting loss of surface B atoms which are possibly displaced by oxygen. In all the samples, XPS also highlighted the presence of Cl (Fig. 5b), which originates from residual Cl− coordinated to Ni2+ on the surface of the Ni3B.62 The C 1s and O 1s XPS signals of the used electrocatalyst were not particularly informative and are thus provided in the ESI† (Fig. S14).
 | ||
| Fig. 4 Characterization of the Ni3B-NCs@CP electrocatalyst before and after chronoamperometric test (electrochemical oxidation of 5-HMF at 1.8 V vs. RHE for 3 h, 100 mM 5-HMF). (a) XRD pattern of Ni3B-NCs@CP as prepared and Ni3B-NCs@CP after the electrocatalytic test at pH 12.9. (b) SEM image of Ni3B-NCs@CP as prepared. (c) SEM image of Ni3B-NCs@CP after the electrocatalytic test at pH 12.9. | ||
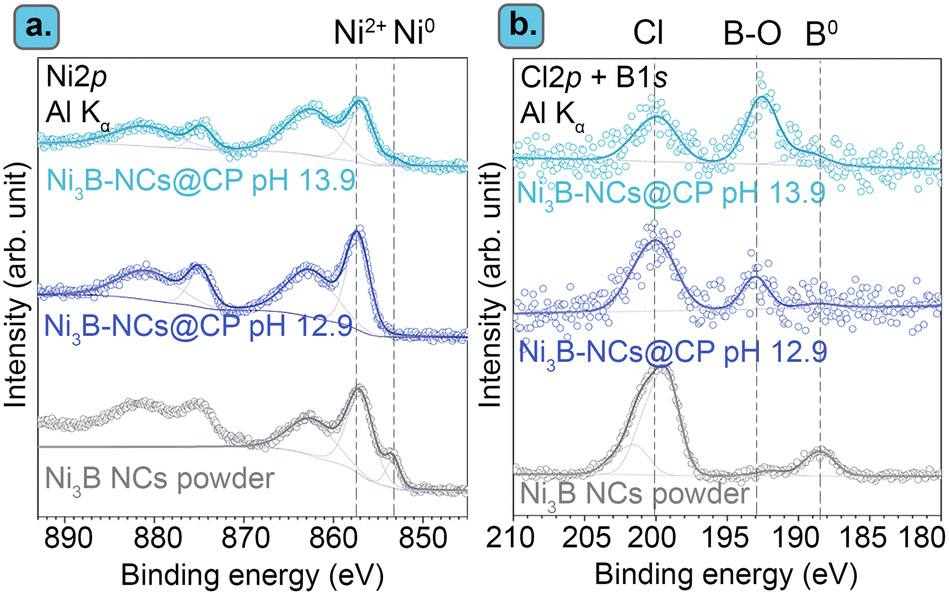 | ||
| Fig. 5 XPS spectra of the (a) Ni 2p and (b) Cl 2p/B 1s core level regions of Ni3B NCs powder (grey), Ni3B-NCs@CP after the electrocatalytic test at pH 12.9 (purple), and at pH 13.9 (blue). Data of Ni3B NCs powder was reproduced with permission under a Creative Commons CC-BY 4.0 from Hong et al.62 Copyright 2023 Americal Chemical Society. | ||
Combining the XPS data with the XRD analysis before and after the electrocatalytic tests suggests that, while the surface of Ni3B NCs underwent oxidation during electrolysis in the presence of 5-HMF, the crystalline Ni3B core was preserved.
Effect of electrolyte concentration and pH on the oxidation mechanism
A higher OH− concentration, and thereby a more basic environment, increases the conductivity of the electrolyte and is thus generally beneficial for the reaction rate. Therefore, the effect of pH on the Ni3B-NCs@CP-catalyzed electrochemical oxidation of 5-HMF was investigated by performing the studies in 1.0 M KOH (pH 13.9) and comparing the results with this higher pH to those obtained in 0.1 M KOH (pH 12.9). The studies were performed with our best-performing electrocatalyst, Ni3B-NCs@CP (0.05 mg cm−2 catalyst loading), as the working electrode in the presence of 100 mM 5-HMF at constant potential of 1.8 V vs. RHE for 3 h (Fig. 6 and Fig. S15–S17†). Comparison of the cyclic voltammograms of Ni3B-NCs@CP at the two aforementioned pH values (Fig. 6a) reveals that the Ni3B-NCs@CP electrocatalyst is able to reach a higher current density at the same potential at pH 13.9 compared to pH 12.9. The difference in activity is also reflected in the 5-HMF conversion: at pH 12.9, a 5-HMF conversion of 40% was reached in 3 h, while at pH 13.9, 70% of 5-HMF was converted in the same time interval. On the other hand, a lower carbon balance was found at pH 13.9 (73%, see Table S4†) than what observed at pH 12.9 (95%, see Table S3†). This difference in carbon balance is attributed to a higher degree of formation of undesired degradation products (e.g. humins) at pH 13.9, which were observed visually but that cannot be quantified by HPLC.39,66 Furthermore, at pH 12.9 a FE > 99% towards the oxidation of 5-HMF was reached, whereas at pH 13.9 the FE was lower but still high (94%), with the remaining 6% likely corresponding to the oxygen evolution reaction (though further oxidation of the organics to volatile C1–C2 compounds cannot be excluded). The hypothesized increased competition with OER can be explained considering that at higher pH the ratio between [OH−] and [5-HMF] increases because the amount of 5-HMF in solution is the same in all experiments. As a consequence, the ratio between adsorbed OH− and adsorbed 5-HMF on the electrode increases at higher pH, which could favor the OER (which requires only OH−) compared to the oxidation of 5-HMF (which involves both this reactant and OH−).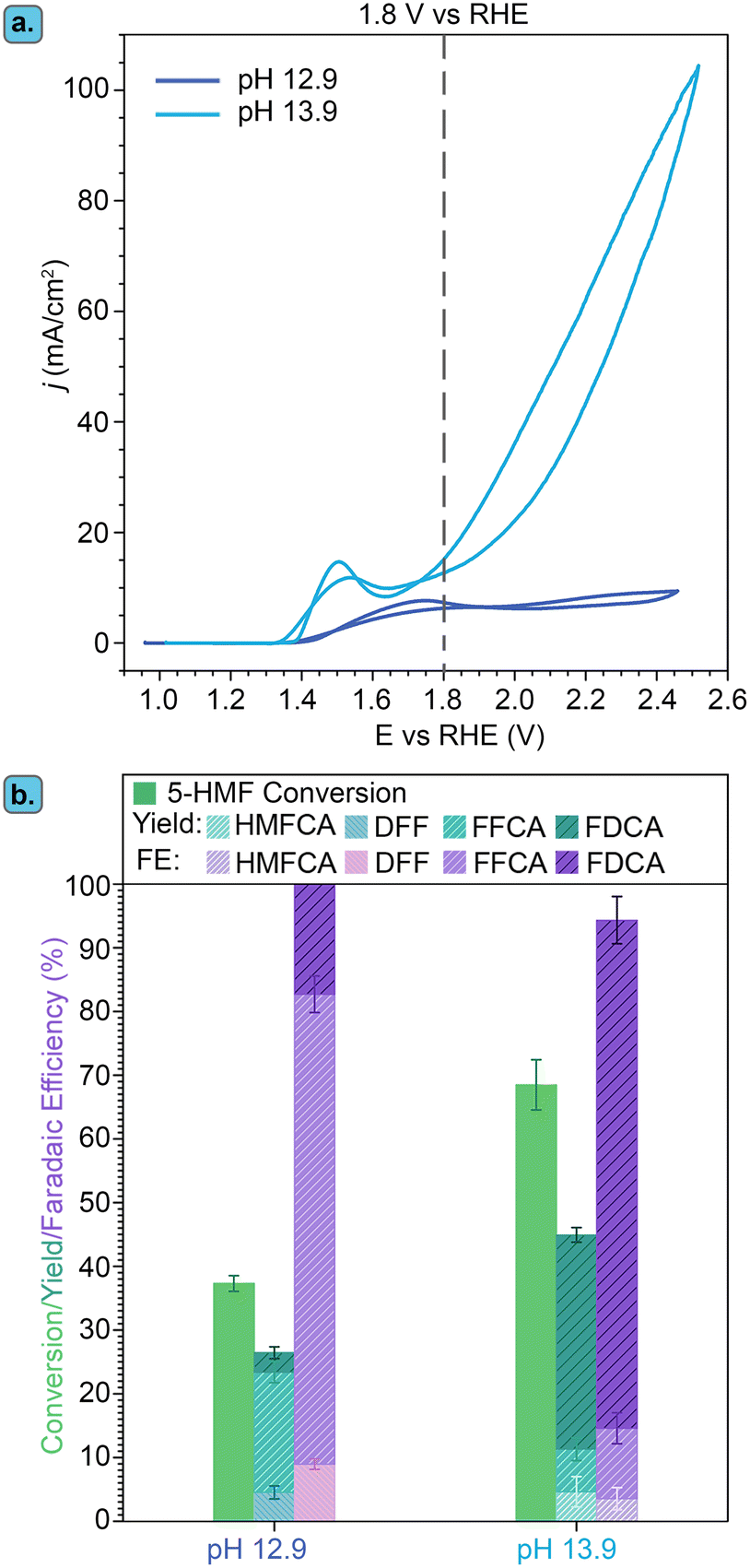 | ||
| Fig. 6 Effect of pH (12.9 and 13.9) on the electrocatalytic performance of Ni3B-NCs@CP. (a) Cyclic voltammetry of Ni3B-NCs@CP at pH 12.9 (dark blue) and pH 13.9 (light blue) in the presence of 5-HMF. (b) Conversion, product yields and Faradaic efficiency (FE) obtained after 3 h of electrochemical oxidation of 5-HMF (initial concentration: 100 mM) catalyzed by Ni3B-NCs@CP at a constant potential of 1.8 V vs. RHE. | ||
Another relevant difference between the tests carried out at pH 12.9 and 13.9 is the products distribution (Fig. 6b). Both formyl furan carboxylic acid (FFCA) and furan dicarboxylic acid (FDCA) were observed as oxidation products in the studies performed at both pHs. However, at pH 12.9 diformyl furan (DFF) was also observed as a minor product with low concentration throughout the reaction (Fig. S18†). Conversely, at pH 13.9 DFF was not observed whereas 5-(hydroxymethyl)furan-2-carboxylic acid (HMFCA) was detected (Fig. S19†). HMFCA and DFF are the two possible products of the initial 2-e− oxidation of 5-HMF, with HMFCA being formed through the oxidation of the aldehydic carbonyl of 5-HMF to a carboxyl group and with DFF being generated through the oxidation of the alcohol group of 5-HMF to a carbonyl group (Fig. 1). The fact that different amounts of these two compounds were observed at the two pH values is an indication of a difference in the mechanism(s) followed in the two cases. Preceding studies on the electrochemical oxidation of 5-HMF proposed two distinct mechanisms for this half-reaction depending on the applied potential: direct and indirect oxidation.6 In the former case, the applied potential is directly driving the oxidation of the substrate, thereby the oxidation state of the electrocatalyst remains unaffected during the reaction (Fig. 7, left box).38,67,68 Conversely, in the case of indirect oxidation, the oxidation state of the electrocatalyst changes throughout the reaction as it functions as a redox mediator, i.e. the metal in a high oxidation state undergoes a non-electrochemical redox reaction with the reactant, thereby getting reduced, after which it is electrochemically reoxidized to its original high oxidation state (Fig. 7, right box).55,56,69–71 By monitoring the electrochemical oxidation of 5-HMF with in situ UV-vis spectroscopy and surface-enhanced Raman spectroscopy, the group of Sun concluded that Ni-based electrocatalysts follow the indirect oxidation pathway in which Ni2+ is initially oxidized to Ni3+, which then acts as the oxidant of the organic compounds. The resulting Ni2+ is then reoxidized to Ni3+ under the applied potential.56 This reaction mechanism corresponds well to the CV at pH 13.9 in Fig. 6a, where we can see the oxidation peak in the 5-HMF-containing electrolyte. The formed Ni3+ species then oxidize one of the functional groups (–OH or –CHO) of 5-HMF. The hydrogen from the functional group is chemically transferred to the Ni3+ (NiOOH to Ni(OH)2) hence reducing it back to Ni2+. The reduction of Ni3+ occurs without contribution from the applied potential resulting in the lack of reduction peak in the CV at pH 13.9 (Fig. 6a).
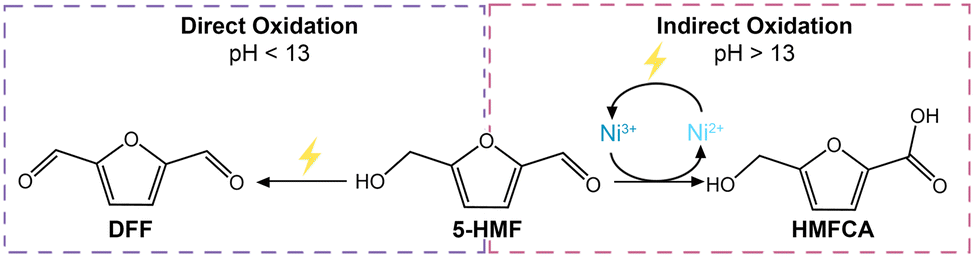 | ||
| Fig. 7 Schematic of the two possible reaction mechanisms in the electrochemical oxidation of 5-HMF: direct oxidation (left) and indirect oxidation (right). | ||
To gain better understanding of the role of the potential and of the OH− concentration on the oxidation pathway and, consequently, on the product selectivity, we performed a study on 5-HMF oxidation by Ni3+ in the form of γ-NiOOH at the two said pH values, without applying a potential. γ-NiOOH was synthesized following the procedure reported by Menezes et al.72 The Ni3+-containing powder was mixed in equimolar amount with 5-HMF in 0.1 M KOH and 1.0 M KOH and then stirred for 24 h. Within 1 h, the black NiOOH powder gradually turned green, suggesting its conversion to Ni(OH)2 (see pictures in Fig. S20†), and thus the reduction of Ni3+ to Ni2+. HPLC analysis of the reaction mixture showed the gradual conversion of 5-HMF and formation of HMFCA in both samples (Fig. S21 and S22†). The formation of HMFCA and the change in oxidation state of nickel indicate that the oxidation of 5-HMF can occur via indirect oxidation through a potential-independent mechanism. This experiment also showed that this indirect oxidation mechanism can occur both at pH 12.9 and pH 13.9, though the reaction rate is significantly higher at higher pH (compare Fig. S21 with S22†). Interestingly, DFF was not detected in the sample at pH 12.9. This contrasts with the experiment carried out under an applied potential at pH 12.9, in which DFF was observed in relevant yields (Fig. 6b). This suggests that the formation of DFF occurs through a direct oxidation mechanism, which requires application of potential to take place, in line with what suggested by previous reports.70 Combining these experimental evidences, it can be concluded that in the electrochemical test at pH 12.9 both indirect oxidation and direct oxidation can occur simultaneously and are competing with each another. On the other hand, in the electrochemical test at pH 13.9 the reaction is dominated by the indirect mechanism. This is attributed to the observed higher rate of the non-electrochemical oxidation of 5-HMF by Ni3+ species at this pH and to the fact that the concentration of Ni3+ is 5 times higher at pH 13.9 than it is at pH 12.9 (based on integration of the Ni2+/Ni3+ oxidation peaks in Fig. S15†).
Characterization by XPS of the Ni3B-NCs@CP electrocatalysts after the chronoamperometric test at 13.9 showed that the Ni and B atoms at the surface of the material are mostly in the oxidized state after the electrochemical test, which is a similar behaviour to the one observed after the test at 12.9 (compare top and bottom signals in Fig. 5). There are some observable differences however between the samples tested at the two pH. At pH 12.9, the Ni3B-NCs@CP displays a more intense Cl peak than both peaks corresponding to B–O and B. However, at pH 13.9, the signal of B–O is more prominent than those of Cl and B. This implies that at pH 13.9, a higher fraction of surface B in the Ni3B NCs is oxidized and the Cl− on the Ni3B surface is displaced by oxygen species, thereby resulting in lower B0 and Cl signals. Secondly, the Ni/B ratio significantly changes for the Ni3B-NCs@CP tested at pH 13.9 (Ni/B = 2.4) when compared to the Ni3B NCs powder (Ni/B = 7.2) and Ni3B-NCs@CP tested at pH 12.9 (Ni/B = 8.6), which suggests that at pH 13.9 surface Ni preferentially leached out throughout the electrocatalytic test.62 It is worth noting that in a previous report on amorphous nickel boride measured under similar conditions (pHnominal = 13.87), XPS did not show any signal in the B 1s core level region, thereby leading to the conclusion that the surface boron species leached out during the reaction.41
Effect of the applied potential on the oxidation rate and selectivity
The effect of the applied potential on the kinetics of the electrochemical oxidation of 5-HMF was studied more in depth at pH 13.9, which gave the most promising results in terms of 5-HMF conversion (vide supra). For this purpose, chronoamperometric experiments were conducted for 3 h at different potentials (1.5, 1.6, 1.7, 1.8 V vs. RHE) with Ni3B-NCs@CP (0.05 mg cm−2) as the electrocatalyst. Chronoamperometry performed at 1.5 and 1.8 V vs. RHE showed a gradual decrease in current density over time that is coupled to the change in reactants concentration (Fig. 8a and b) with a similar trend to that observed at pH 12.9 (vide supra). As discussed previously, this suggests that the drop in current density is caused by the decrease in the concentration of reactants (5-HMF and the reaction intermediates, HMFCA and FFCA) and not to deactivation of the electrocatalyst. In addition, it is also worth noting that in Fig. 8a and b, the sum of the concentrations of 5-HMF and reaction intermediates is <100 mM. As mentioned in the previous section, this is because in between the addition of 5-HMF (nHMF,initial) and the start of the chronoamperometry (nHMF, t=0), there was a partial degradation of 5-HMF into humins. The hypothesis that the drop in current density is caused by the decrease in 5-HMF and reaction intermediates concentration is further supported by the increase in current density observed by varying the initial 5-HMF concentration from 10 to 200 mM (Fig. S23 and S24†). Applying the potential from 1.5 to 1.8 V vs. RHE did not lead to marked differences in terms of 5-HMF conversion, product yields and Faradaic efficiency towards monomeric oxidation products (HMFCA, FFCA, FDCA) – see Fig. S19 and S25–S27.† Although it might seem surprising that the performance in the electrochemical conversion of 5-HMF conversion is similar at 1.5 and 1.8 V vs. RHE, this is actually in line with the analogous current density observed at these two potentials by cyclic voltammetry (Fig. 6a) and at the start of the chronoamperometric tests (Fig. 8a and b). However, the profiles of the current density and of the sum of the concentrations of the reactants (5-HMF and the intermediates) as a function of time are different at the two potentials, with a higher initial rate of conversion at 1.8 V vs. RHE (Fig. 8a and b). This difference combined with the fact that the current density profile measured by cyclic voltammetry shows a minimum in the region between 1.5 and 1.8 V vs. RHE might indicate that the reaction mechanism is not the same at the two potentials. We can tentatively propose that at 1.5 V vs. RHE, which corresponds to the potential where Ni2+ is oxidized to Ni3+, the indirect mechanism discussed above is predominant, whereas by increasing the potential the contribution of a direct mechanism, in which 5-HMF and its derivatives are electrochemically oxidized, becomes more relevant.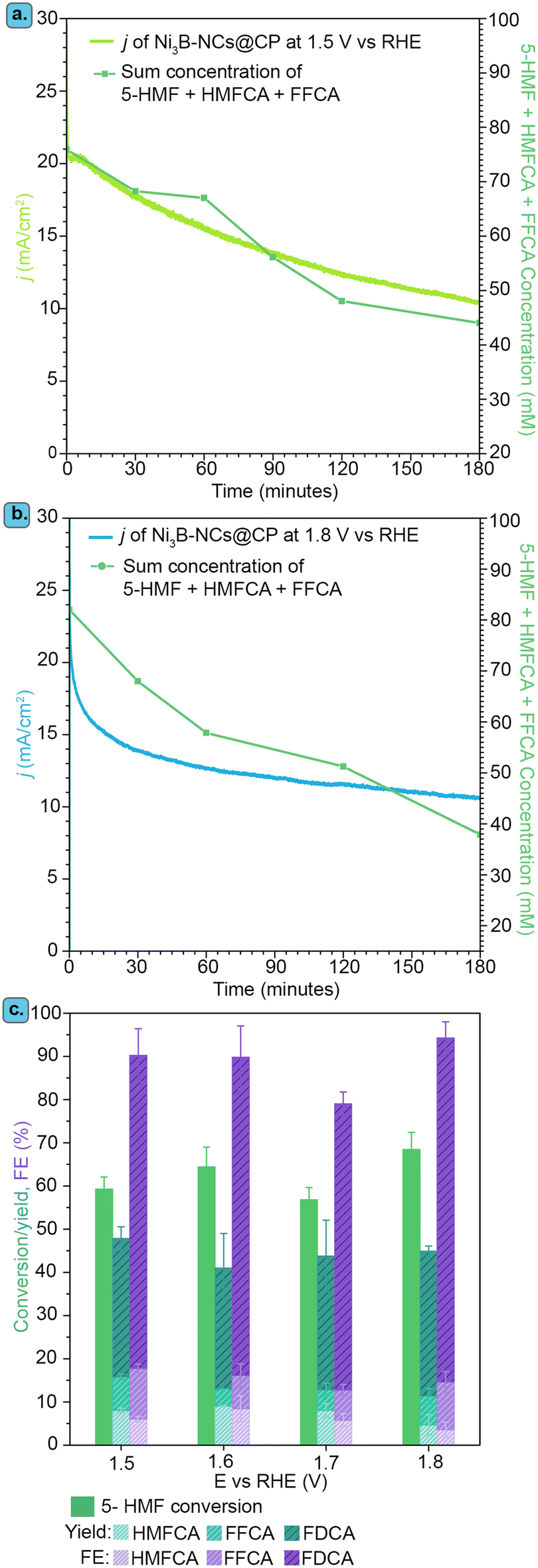 | ||
| Fig. 8 Electrolysis of 5-HMF at different potentials in pH 13.9. Current density and sum of the concentrations of 5-HMF and reaction intermediates (green), measured during chronoamperometric tests of Ni3B-NCs@CP at a constant potential of (a) 1.5 V vs. RHE (light green line) and (b) 1.8 V vs. RHE (blue line). (c) Conversion, product yields and FE in the electrochemical oxidation of 5-HMF catalyzed by Ni3B-NCs@CP after 3 h at different constant applied potentials. | ||
Comparing the performances of Ni NCs with NixB NCs
We compared the performance of our Ni3B-NCs@CP electrocatalyst in the oxidation of 5-HMF to that of the Ni2B-NCs@CP and Ni-NC@CP electrocatalysts under the same testing conditions. Importantly, the dinickel boride (Ni2B NCs), which was prepared using commercial Ni nanopowder as precursor, and the nickel nanocrystals (Ni NCs) were obtained with a similar average particle size to the Ni3B NCs (see above for the TEM analysis). The similarity in average size is a useful feature, as it allows comparing the differences in electrocatalytic activity between the three nickel-based materials. These studies were carried out in all three cases with an electrocatalyst loading of 0.05 mg cm−2, initial 5-HMF concentration of 100 mM HMF, and both at pH 12.9 and 13.9. For the tests carried out at pH 12.9, cyclic voltammetry of Ni-NCs@CP, Ni3B-NCs@CP and Ni2B-NCs@CP in the presence of 5-HMF (Fig. 9a) showed no significant differences between Ni- and NixB-based electrodes. The similar activity of the three electrocatalysts at this pH is further demonstrated during the chronoamperometric tests, which were performed at 1.8 V vs. RHE for 3 h. The three materials displayed a similar initial current density (∼11–13 mA cm−2), which gradually decreased along with the 5-HMF depletion (Fig. 9b). Accordingly, the three materials also show similar 5-HMF conversion (∼35–39%) and total product yields (∼25%), see Fig. 9c. Ni and Ni2B showed slightly lower FE (97%) compared to Ni3B, which achieved complete FE (>99%) towards 5-HMF oxidation. However, these differences are likely to fall within the experimental error margin.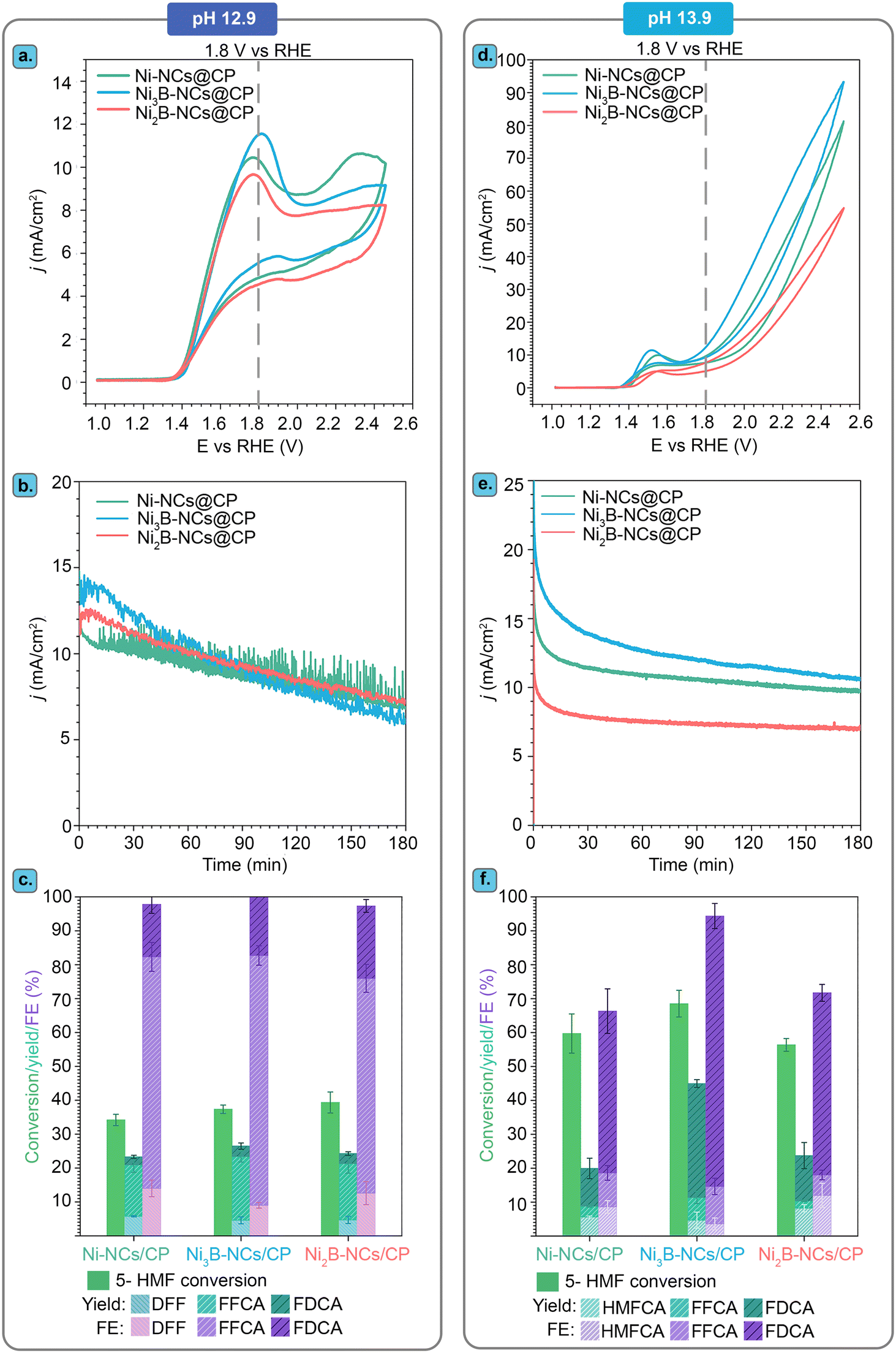 | ||
| Fig. 9 Performance of Ni-NCs@CP (green), Ni3B-NCs@CP (blue), and Ni2B-NCs@CP (red) in electrocatalyzing the oxidation of 5-HMF (100 mM) at pH 12.9 and 13.9. (a) Cyclic voltammetry in the presence of 100 mM 5-HMF at (a) pH 12.9 and (d) pH 13.9. Chronoamperometry at constant potential 1.8 V vs. RHE for 3 h at (b) pH 12.9 and (e) pH 13.9. Conversion, product yields and FE in the electrochemical oxidation of 5-HMF catalyzed by Ni-NCs@CP, Ni3B-NCs@CP, and Ni2B-NCs@CP at (c) pH 12.9 and (f) pH 13.9. | ||
On the other hand, when comparing the performance of the electrocatalysts at pH 13.9, a more substantial difference in the activity of the three electrocatalysts was observed. Under these conditions, Ni3B-NCs@CP is the most active electrocatalyst (Fig. 9d), reaching the highest current density (14 mA cm−2) at the applied potential of 1.8 V vs. RHE, followed by Ni-NCs@CP (11 mA cm−2) and finally Ni2B-NCs@CP (8 mA cm−2). The same trend in activity can be observed in the chronoamperometric test (1.8 V vs. RHE, 3 h, Fig. 9e), in which Ni3B displayed the highest initial current density, which gradually dropped as 5-HMF was converted. It is worth noting that while these differences in current density are not major, the ranking in activity of these electrocatalysts was confirmed by repeating each of these tests 3 times, showing consistently the same trend (Fig. S28–S30†). In agreement with the CV and the chronoamperometry, Ni3B-NCs@CP was able to achieve the highest 5-HMF conversion of 70%, with a total yield of 45% of oxidation products and FE towards 5-HMF oxidation of 94% (Fig. 9f). Compared to the Ni3B-based material, the Ni-based electrocatalyst achieved a lower 5-HMF conversion (60%), total product yield (20%), and FE (65%). Another important difference is that significantly higher FDCA yield was obtained with Ni3B-NCs@CP (∼34%) compared to that achieved with Ni-NCs@CP (13%). Since the test conditions and the preparation of the electrodes were the same, and the size of the Ni-containing active species were very similar in these experiments, we can conclude that the higher activity of the Ni3B-based electrocatalyst stems from the intrinsic features of this material.
The fact that the activity of the Ni3B NCs is higher than both a material that is richer in boron (Ni2B NCs) and a material that does not contain boron (Ni NCs) suggests that not only the presence but also the configuration of boron in the crystal structure (which differs significantly between Ni3B and Ni2B) might play a role in determining the electrocatalytic activity. However, these electrocatalysts differ also in terms of the species present at the surface of the material. While the surface of Ni3B NCs mainly consists of Ni2+ species (e.g. Ni(OH)2) that can participate to the oxidation of 5-HMF (either through a direct or indirect mechanism), the surface of the Ni2B NCs is dominated by oxidized B species, which are not expected to contribute significantly as electrocatalytically active sites.62,73–75 This hypothesis is supported by a control test in which equimolar B2O3 and 5-HMF were mixed in 0.1 M KOH (pH 12.9) and 1.0 M KOH (pH 13.9), and the 5-HMF conversion was followed by HPLC (Fig. S31 and S32†). After 24 h, no 5-HMF oxidation products were detected at any of the two pH values. On the other hand, degradation of 5-HMF to insoluble, brown side products undetectable by HPLC, most likely humins, was observed, as expected at the highly basic condition used in this study. Furthermore, we supported B2O3 on carbon paper (B2O3 loading of ~1 mg cm−2) and tested the obtained electrode by cyclic voltammetry in 1.0 M KOH with 100 mM 5-HMF (Fig. S33†). The performance of the B2O3/carbon paper electrode is very similar to that of an electrode consisting of carbon paper alone, with both samples showing 5-HMF oxidation/OER only at >2 V vs. RHE. This indicates that B2O3 does not contribute significantly to the electrocatalytic activity. Based on these considerations, it can be inferred that the difference in activity between the two nickel borides stems from their different surface compositions. With the purpose of getting further insight on the electrocatalytic behaviour of the tested materials, we measured their double layer capacitance (CDL, see Fig. 10). Notably, the trend observed in the electrocatalytic activity based on the current density (tests at pH = 13.9) is the same one found for the CDL values: Ni3B-NCs@CP (0.0478 mF cm−2) > Ni-NCs@CP (0.0368 mF cm−2) > Ni2B-NCs@CP (0.0322 mF cm−2). The bare carbon paper, which is nearly inactive in the electrochemical oxidation of 5-HMF (vide supra), has lower CDL (0.0222 mF cm−2) compared to the Ni-containing electrocatalysts. The double layer capacitance is related to the electrochemical surface area (ECSA = CDL/CS, where CS is the specific capacitance of the electrode), and this could be considered as an indication that the differences in activity between the Ni-containing electrocatalysts are due to differences in electrochemical surface area. However, this would be the case only if the CS value does not differ significantly between the three materials, which is not necessarily the case as evidenced by the different chemical features of these materials. Additionally, TEM and SEM analysis of the materials (Fig. 2) did not show significant differences in terms of particle size at the nanoscale or in dispersion on the carbon paper, which suggests that the exposed surface area of the three materials should be similar. Based on these considerations, while we observe a correlation between double layer capacitance and electrocatalytic activity, this is probably not (only) correlated to differences in electrochemical surface area but (also) to the chemical features of the materials such as the differences in the crystal structure.
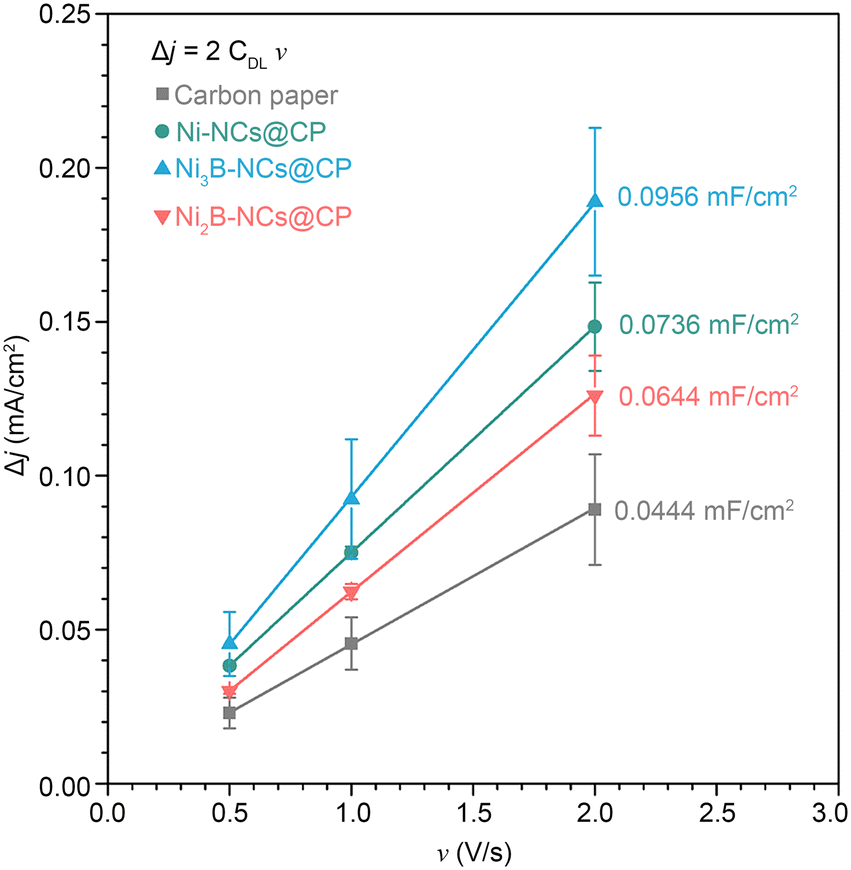 | ||
| Fig. 10 Difference of anodic to cathodic current density plotted against the scan rate. From the value of slope of the plots (2CDL), the double layer capacitance (CDL) can be calculated. | ||
When compared to the state-of-the-art nickel boride electrocatalysts for 5-HMF oxidation (Table S1†), our best performing electrocatalyst (Ni3B-NCs@CP) displayed much higher turnover frequency (expressed based on the moles of C6-products per mass of nickel boride per hour - see SI for the formula). This is related to the fact that this electrocatalyst proved to work optimally with very low loading and high 5-HMF concentration. On the other hand, these conditions implied that the conversion of 5-HMF and product yields that we obtained were lower compared to those over the best nickel boride electrocatalyst in the literature. To improve this feature, we carried out a longer reaction time chronoamperometry (1.8 V vs. RHE, 8 h, 1.0 M KOH, 100 mM 5-HMF, see Fig. 11). This allowed reaching nearly complete conversion of 5-HMF (∼97%), with the following product yields: FDCA 54%, HMFCA 4%, FFCA 8%. These results show an increase in the selectivity towards FDCA among the oxidation products, in line with the higher degree of oxidation of this compound compared to HMFCA and FFCA. Despite the higher conversion and yield, there are a few drawbacks in the longer electrochemical reaction. Firstly, the carbon balance decreases from 73% in the 3 h reaction to 58% in the 8 h reaction, indicating that the degree of degradation of 5-HMF to undesired by-products increases with the reaction time. Additionally, the FE efficiency towards 5-HMF oxidation is lower at this longer reaction time, dropping to 47% (FDCA 42%, HMFCA 1%, FFCA 4%) compared to the 94% after 3 h. This is attributed to the increased competition with the OER caused by the lower concentration of 5-HMF as a consequence of its conversion.
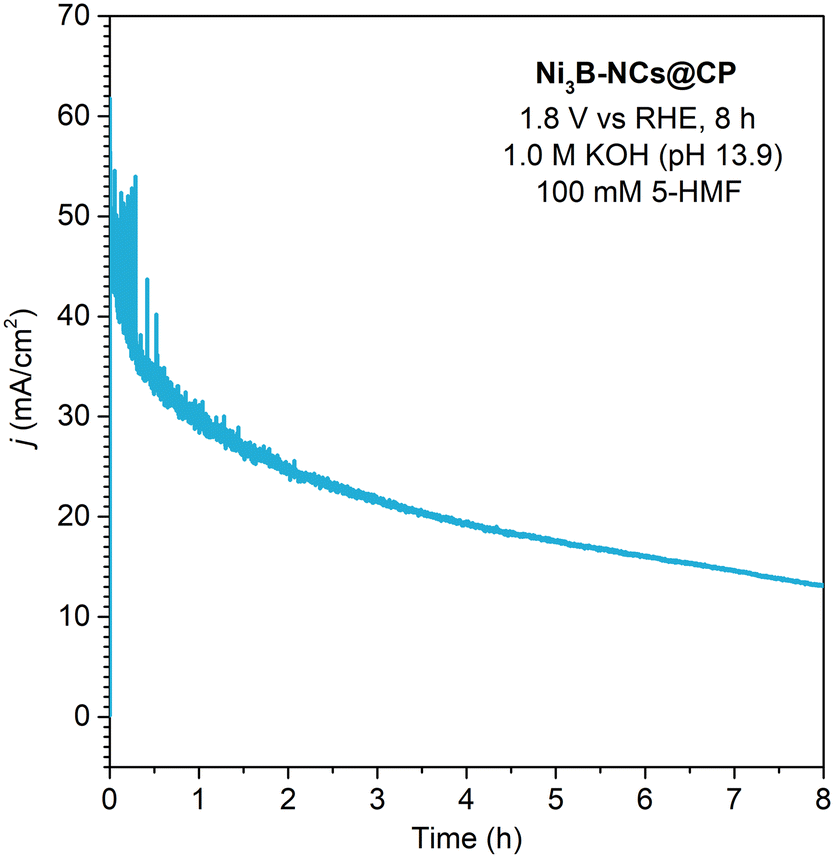 | ||
| Fig. 11 Chronoamperometry of Ni3B-NCs@CP (0.05 mg cm−2) conducted at constant potential of 1.8 V vs. RHE for 8 h in the presence of 5-HMF (100 mM) at pH 13.9 (1.0 M KOH). | ||
Stability of Ni3B NCs as electrocatalyst
Besides showing promising electrocatalytic activity, metal borides have been proposed to display high electrochemical stability under operating conditions. In this context, we studied the stability of the best-performing electrocatalyst identified in this work, Ni3B-NCs@CP, in 5 consecutive chronoamperometric runs of 5-HMF electrochemical oxidation at 1.8 V vs. RHE for 3 h at pH 13.9 (Fig. 12). After each chronoamperometric run, the Ni3B-NCs@CP electrode was rinsed with Milli-Q water and a fresh new electrolyte was added to the cell. The Ni3B-NCs@CP was then tested for another run of electrochemical oxidation of 5-HMF, which includes CVs and CA measurement.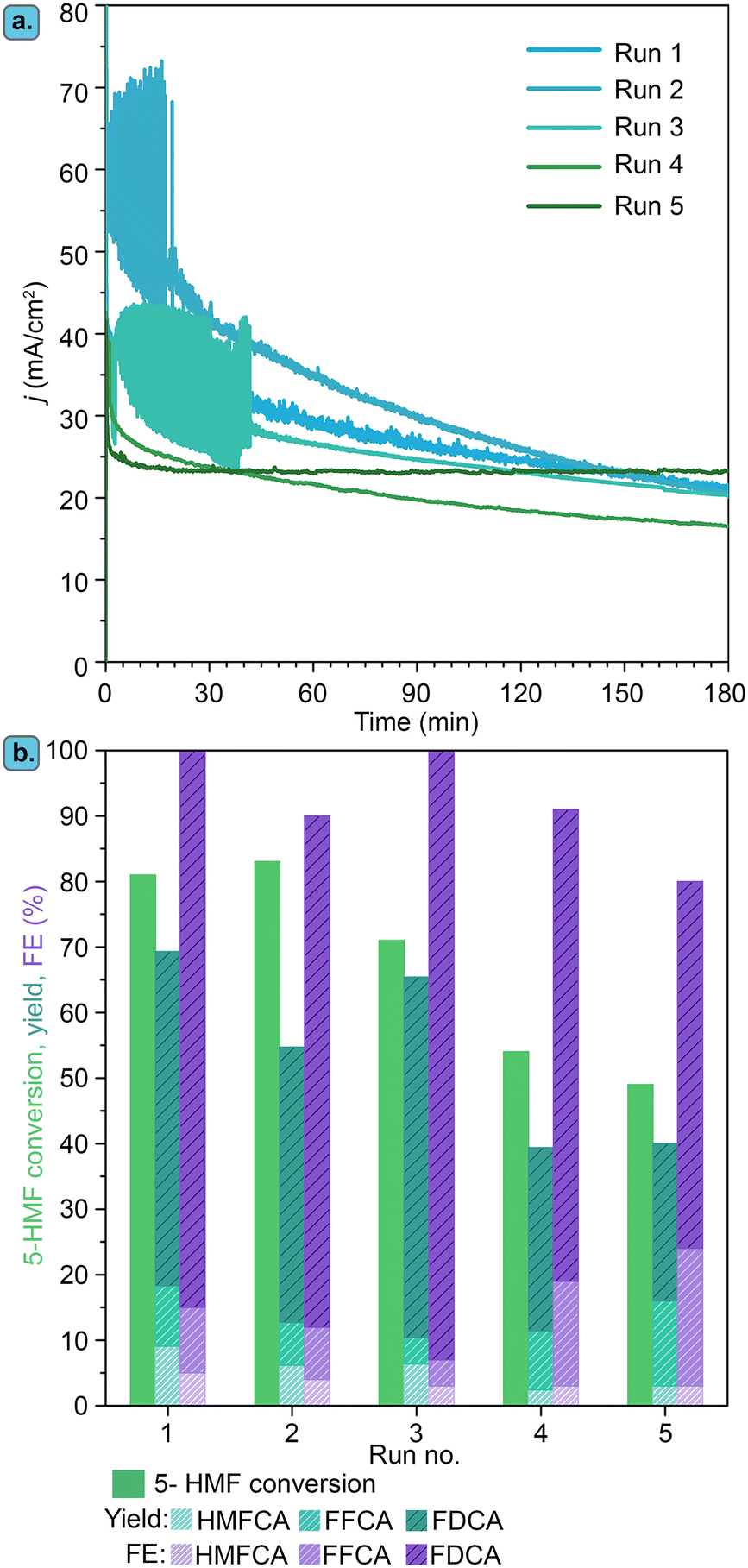 | ||
| Fig. 12 (a) Chronoamperometry of Ni3B-NCs@CP (∼0.05 mg cm−2) in 5 consecutive runs of electrochemical oxidation of 5-HMF (100 mM initial concentration) at constant potential of 1.8 V vs. RHE. (b) 5-HMF conversion, product yields and FE obtained at the end of each run of 5-HMF electrochemical oxidation with Ni3B-NCs@CP as the electrocatalyst. | ||
As can be observed in Fig. 12a, chronoamperometry conducted at 1.8 V vs. RHE for 3 h at pH 13.9 with the Ni3B-NCs@CP electrocatalyst maintained a similar profile during the first three runs, with some fluctuations (the current density in run 2 seems higher), most likely related to the high level of noise observed in some of these tests. The preserved electrochemical activity in the first three runs is reflected in the similar 5-HMF conversion for each run (∼70–80%) and FE (>90%), see Fig. 12b and S34–S36.† From the fourth electrochemical oxidation run, the activity profile becomes different, with a lower initial current density and a lower slope as a function of time, which led to lower degree of conversion of 5-HMF (Fig. S37–S38†). This drop in activity might be attributed to gradual degradation of the carbon paper support, reorganization of the Ni3B surface, catalyst detachment and/or gradual leaching of Ni from the electrode to the electrolyte. ICP-MS analysis of the electrolyte after each test showed a minor degree of leaching of nickel species (ca. 0.2 to 0.6% of the initial amount of Ni was lost in each step), suggesting that leaching is not the main cause of the observed deactivation. On the other hand, the degradation of the carbon paper support was clearly observed by visual analysis: after the fifth run, the carbon paper tended to crumble upon manual handling, making further recycling tests impossible.
XRD analysis (Fig. S39†) of the Ni3B-NC@carbon paper at different stages of the recycling tests showed that after five runs, the Ni3B characteristic peaks observed in the fresh Ni3B-NCs@CP are still present, indicating that the bulk crystal structure is retained. After the electrocatalytic test runs, XRD also showed new peaks that do not correspond to Ni3B. EDS elemental mapping (Fig. 13) showed that besides the elements originally present in the material (the sulfur originating from the Nafion), a large amount of K and minor amounts of Mg and Fe were also present. Most likely, all these elements stem from the KOH in the electrolyte solution, which deposited on the carbon paper either as such or as other K-based species (e.g. KCl, K2CO3, K2C2O6, K2SO3 and K2SO4). Indeed, some of the diffraction peaks of these compounds correspond to those observed in the XRD pattern after the third run, but the complexity of the diffractogram indicates that a mixtures of different compounds is present (Fig. S40†). These K-based compounds can be clearly observed by SEM (Fig. 13) on the Ni3B-NCs@carbon paper already after the third run in the form of flakes occupying the space between the carbon paper fibers. The fact that the activity is largely preserved upon reuse, indicates that these K-based residues either desorb from the electrode surface during the chronoamperometry or, if they remain adsorbed, they are not influencing significantly the electrocatalytic behaviour of the Ni3B phase and can thus be considered as spectator species.
 | ||
| Fig. 13 SEM and EDS analysis of Ni3B-NCs@CP (initial loading: 0.05 mg cm−2) after 3 runs of 5-HMF electrochemical oxidation at 1.8 V vs. RHE for 3 h. | ||
Overall, the Ni3B NCs showed good stability, high crystalline integrity, and retention of most of their activity upon reuse with negligible leaching of active species. The main limitation in terms of long-term stability of the studied electrocatalytic system is thus not related to the nickel boride electrocatalyst but to the carbon support, which can undergo electrochemical (oxidative) and/or mechanical degradation. Therefore, future research might focus on finding an alternative support with increased stability.
Conclusions
The activity of crystal-phase-pure Ni3B and Ni2B NCs as electrocatalysts for the electrosynthesis of FDCA through the oxidation of 5-HMF was investigated and compared to that of Ni NCs with similar average particle size and under the same testing conditions. We demonstrated that varying the Ni3B NCs loading on the carbon paper electrodes has no significant impact on the performance towards the electrochemical oxidation of 5-HMF at pH 12.9 (0.1 M KOH). Therefore, we were able to significantly decrease the electrocatalyst loading, achieving high conversion of 5-HMF with 20 times lower loading (0.05 mg cm−2) and higher concentration of 5-HMF (100 mM) than in previous reports (corresponding to a 200 times lower catalyst loading relative to HMF, see Tables S1 & S2†). This resulted in a one order of magnitude higher turnover frequency compared to the state-of-the-art nickel boride electrocatalysts for this reaction (Table S1†). By studying this organic electrosynthesis at two different pH, namely pH 12.9 and 13.9, we observed two possible oxidation pathways: at pH 12.9, the oxidation of 5-HMF to FDCA proceeds through a DFF intermediate, while at pH 13.9 no DFF was observed. Combining these results with control tests in which γ-NiOOH was used for the non-electrochemical oxidation of 5-HMF, we concluded that both direct and indirect mechanisms operate at pH 12.9, whereas the indirect oxidation mechanism dominates at pH 13.9. When the electrocatalytic performance of Ni-NCs@CP, Ni3B-NCs@CP, and Ni2B-NCs@CP were compared under the optimized electrolysis conditions (at 1.8 V vs. RHE and pH 13.9), Ni3B-NCs@CP showed the highest 5-HMF conversion (70%), FE towards 5-HMF oxidation (94%) and FDCA yield (34%). Since the particle size and dispersion on the carbon support of the three materials are similar, these results demonstrate for the first time that the Ni3B-NCs-based electrocatalysts display enhanced activity for HMF oxidation compared to the Ni NCs and Ni2B NCs counterparts. The higher activity of the Ni3B-NCs-based electrocatalyst compared to the Ni2B NCs counterpart is attributed to the different surface composition evidenced by XPS analysis, with Ni3B NCs mainly presenting oxidized Ni species at the surface and Ni2B NCs displaying mainly oxidized B species at the surface. Furthermore, we observed a correlation between the trend of activity and the trend of double-layer capacitance, although this does not necessarily imply that the electrochemical surface area follows the same trend. Importantly, Ni3B-NCs@CP maintained good activity over 5 consecutive runs of electrochemical oxidation of 5-HMF. Future work should aim at finding an alternative, more robust support compared to the carbon paper used in this work, which should allow the same good dispersion of Ni3B NCs while remaining stable over multiple runs of electrocatalytic testing.Data availability
Data for this article is available either in the Supplementary Information or, in the case of the raw data, at figshare (https://doi.org/10.6084/m9.figshare.27145611).Author contributions
Jennifer Hong performed the experiments and analysis with assistance from Matteo Miola under the supervision of Paolo P. Pescarmona and Loredana Protesescu. Dominic Gerlach performed the XPS measurements and analysis under the supervision of Petra Rudolf. Dulce M. Morales assisted in the data analysis. Jennifer Hong and Paolo P. Pescarmona wrote the manuscript with contributions from all authors, who approved the final version of the manuscript.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
University of Groningen funded Jennifer Hong research through a PhD Scholarship. The authors gratefully acknowledge technical support from Jacob Baas, Léon Rohrbach, Gert-Jan Boer, Jan Nijhoff, and Dr. Gert ten Brink.References
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS.
- L. T. Mika, E. Cséfalvay and Á. Németh, Chem. Rev., 2018, 118, 505–613 CrossRef CAS.
- R.-J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS.
- J. Theerthagiri, K. Karuppasamy, J. Park, N. Rahamathulla, M. L. A. Kumari, M. K. R. Souza, E. S. F. Cardoso, A. P. Murthy, G. Maia, H.-S. Kim and M. Y. Choi, Environ. Chem. Lett., 2023, 21, 1555–1583 CrossRef CAS.
- L. Zhang, T. U. Rao, J. Wang, D. Ren, S. Sirisommboonchai, C. Choi, H. Machida, Z. Huo and K. Norinaga, Fuel Process. Technol., 2022, 226, 107097 CrossRef CAS.
- Y. Yang and T. Mu, Green Chem., 2021, 23, 4228–4254 RSC.
- Z. Zhang and K. Deng, ACS Catal., 2015, 5, 6529–6544 CrossRef CAS.
- M. Sajid, X. Zhao and D. Liu, Green Chem., 2018, 20, 5427–5453 RSC.
- C. Chen, L. Wang, B. Zhu, Z. Zhou, S. I. El-Hout, J. Yang and J. Zhang, J. Energy Chem., 2021, 54, 528–554 CrossRef CAS.
- D. A. Giannakoudakis, J. C. Colmenares, D. Tsiplakides and K. S. Triantafyllidis, ACS Sustain. Chem. Eng., 2021, 9, 1970–1993 CrossRef CAS.
- S. Rezaee and S. Shahrokhian, Appl. Catal., B., 2019, 244, 802–813 CrossRef CAS.
- C. Ma, P. Fang and T.-S. Mei, ACS Catal., 2018, 8, 7179–7189 CrossRef CAS.
- Y. Kwon, K. J. P. Schouten, J. C. van der Waal, E. de Jong and M. T. M. Koper, ACS Catal., 2016, 6, 6704–6717 CrossRef CAS.
- Z. Lin, X. Chen, L. Lu, X. Yao, C. Zhai and H. Tao, Nanotechnol. Rev., 2023, 12, 20220518 CrossRef CAS.
- J. Creus, M. Miola and P. P. Pescarmona, Green Chem., 2023, 25, 1658–1671 RSC.
- K. C. Poon, W. Y. Wan, H. Su and H. Sato, RSC Adv., 2022, 12, 22703–22721 RSC.
- D. Gao, R. M. Arán-Ais, H. S. Jeon and B. Roldan Cuenya, Nat. Catal., 2019, 2, 198–210 CrossRef CAS.
- J. Liu and S. Tao, Appl. Surf. Sci., 2023, 616, 156464 CrossRef CAS.
- G.-F. Chen, Y. Yuan, H. Jiang, S.-Y. Ren, L.-X. Ding, L. Ma, T. Wu, J. Lu and H. Wang, Nat. Energy, 2020, 5, 605–613 CrossRef CAS.
- D. Reyter, D. Bélanger and L. Roué, Electrochim. Acta, 2008, 53, 5977–5984 CrossRef CAS.
- A. D. Stergiou, D. H. Broadhurst and M. D. Symes, STAR Protocols, 2022, 3, 101817 CrossRef CAS PubMed.
- N. Daems, F. Risplendi, K. Baert, A. Hubin, I. F. J. Vankelecom, G. Cicero and P. P. Pescarmona, J. Mater. Chem., 2018, 6, 13397–13411 RSC.
- N. Daems, J. Wouters, C. Van Goethem, K. Baert, C. Poleunis, A. Delcorte, A. Hubin, I. F. J. Vankelecom and P. P. Pescarmona, Appl. Catal., B., 2018, 226, 509–522 CrossRef CAS.
- M. Miola, B. C. A. de Jong and P. P. Pescarmona, Chem. Commun., 2020, 56, 14992–14995 RSC.
- Z. Lin, L. Wang, T. Jia, X. Wang, C. Li, H. Wang, L. Li, Y. Zhou, C. Zhai, H. Tao and S. Li, Chem. Eng. J., 2024, 481, 148429 CrossRef CAS.
- F. W. S. Lucas, R. G. Grim, S. A. Tacey, C. A. Downes, J. Hasse, A. M. Roman, C. A. Farberow, J. A. Schaidle and A. Holewinski, ACS Energy Lett., 2021, 6, 1205–1270 CrossRef CAS.
- E. C. Morales, M. A. Kazakova, A. G. Selyutin, G. V. Golubtsov, D. M. Morales and A. M. Robledo, Surf. Interfac., 2024, 46, 104026 CrossRef CAS.
- K. B. Kokoh and E. M. Belgsir, Tetrahedron Lett., 2002, 43, 229–231 CrossRef CAS.
- S. R. Kubota and K.-S. Choi, ChemSusChem, 2018, 11, 2138–2145 CrossRef CAS.
- T. Cao Tao, ChemSusChem, 2017, 10, 4851–4854 CrossRef PubMed.
- D. J. Chadderdon, L. Xin, J. Qi, Y. Qiu, P. Krishna, K. L. More and W. Li, Green Chem., 2014, 16, 3778–3786 RSC.
- G.-R. Xu, M. Batmunkh, S. Donne, H. Jin, J.-X. Jiang, Y. Chen and T. Ma, J. Mater. Chem. A, 2019, 7, 25433–25440 RSC.
- M. Park Minju, ACS Nano, 2020, 14, 6812–6822 CrossRef PubMed.
- N. Heidary and N. Kornienko, Chem. Commun., 2019, 55, 11996–11999 RSC.
- H. G. Cha and K.-S. Choi, Nat. Chem., 2015, 7, 328–333 CrossRef CAS PubMed.
- G. Liu, T. Nie, Z. Song, X. Sun, T. Shen, S. Bai, L. Zheng and Y.-F. Song, Angew. Chem., 2023, 135, e202311696 CrossRef.
- R. Latsuzbaia, R. Bisselink, A. Anastasopol, H. van der Meer, R. van Heck, M. S. Yagüe, M. Zijlstra, M. Roelands, M. Crockatt, E. Goetheer and E. Giling, J. Appl. Electrochem., 2018, 48, 611–626 CrossRef CAS.
- C. Zhou, W. Shi, X. Wan, Y. Meng, Y. Yao, Z. Guo, Y. Dai, C. Wang and Y. Yang, Catal. Today, 2019, 330, 92–100 CrossRef CAS.
- K. R. Vuyyuru and P. Strasser, Catal. Today, 2012, 195, 144–154 CrossRef CAS.
- X. Hao, T. Cai, J. Ma, G. She, H. Zhang, W. Wang, J. Yu, L. Mu and W. Shi, ChemPlusChem, 2023, 88, e202300285 CrossRef CAS.
- S. Barwe, J. Weidner, S. Cychy, D. M. Morales, S. Dieckhöfer, D. Hiltrop, J. Masa, M. Muhler and W. Schuhmann, Angew. Chem., Int. Ed., 2018, 57, 11460–11464 CrossRef CAS.
- F.-B. Liu, N. Lin, L.-C. Cong, X.-X. Li, F.-Y. Han, D.-Y. Xin and H.-B. Lin, ChemCatChem, 2023, 15, e202300765 CrossRef CAS.
- D. Xiao, X. Bao, D. Dai, Y. Gao, S. Si, Z. Wang, Y. Liu, P. Wang, Z. Zheng, H. Cheng, Y. Dai and B. Huang, Adv. Mater., 2023, 35, 2304133 CrossRef CAS.
- S. Li, S. Wang, Y. Wang, J. He, K. Li, Y. Xu, M. Wang, S. Zhao, X. Li, X. Zhong and J. Wang, Adv. Funct. Mater., 2023, 33, 2214488 CrossRef CAS.
- G. Grabowski, J. Lewkowski and R. Skowroński, Electrochim. Acta, 1991, 36, 1995 CrossRef CAS.
- B. You, X. Liu, N. Jiang and Y. Sun, J. Am. Chem. Soc., 2016, 138, 13639–13646 CrossRef CAS PubMed.
- B. You, N. Jiang, X. Liu and Y. Sun, Angew. Chem., Int. Ed., 2016, 55, 9913–9917 CrossRef CAS.
- B. You, X. Liu, X. Liu and Y. Sun, ACS Catal., 2017, 7, 4564–4570 CrossRef CAS.
- M. Li, L. Chen, S. Ye, G. Fan, L. Yang, X. Zhang and F. Li, J. Mater. Chem. A, 2019, 7, 13695–13704 RSC.
- A. R. Poerwoprajitno, L. Gloag, J. Watt, S. Cychy, S. Cheong, P. V. Kumar, T. M. Benedetti, C. Deng, K.-H. Wu, C. E. Marjo, D. L. Huber, M. Muhler, J. J. Gooding, W. Schuhmann, D.-W. Wang and R. D. Tilley, Angew. Chem., Int. Ed., 2020, 59, 15487–15491 CrossRef CAS PubMed.
- Y. Lu, C.-L. Dong, Y.-C. Huang, Y. Zou, Y. Liu, Y. Li, N. Zhang, W. Chen, L. Zhou, H. Lin and S. Wang, Sci. China Chem., 2020, 63, 980–986 CrossRef CAS.
- X. Chen, X. Zhong, B. Yuan, S. Li, Y. Gu, Q. Zhang, G. Zhuang, X. Li, S. Deng and J.-G. Wang, Green Chem., 2019, 21, 578–588 RSC.
- X. Deng, M. Li, Y. Fan, L. Wang, X.-Z. Fu and J.-L. Luo, Appl. Catal., B., 2020, 278, 119339 CrossRef CAS.
- S. Choi, M. Balamurugan, K.-G. Lee, K. H. Cho, S. Park, H. Seo and K. T. Nam, J. Phys. Chem. Lett., 2020, 11, 2941–2948 CrossRef CAS.
- N. Heidary and N. Kornienko, Chem. Sci., 2020, 11, 1798–1806 RSC.
- P. Zhang, X. Sheng, X. Chen, Z. Fang, J. Jiang, M. Wang, F. Li, L. Fan, Y. Ren, B. Zhang, B. J. J. Timmer, M. S. G. Ahlquist and L. Sun, Angew. Chem., Int. Ed., 2019, 58, 9155–9159 CrossRef CAS.
- M. Cai, Y. Zhang, Y. Zhao, Q. Liu, Y. Li and G. Li, J. Mater. Chem. A, 2020, 8, 20386–20392 RSC.
- N. Zhang, Y. Zou, L. Tao, W. Chen, L. Zhou, Z. Liu, B. Zhou, G. Huang, H. Lin and S. Wang, Angew. Chem., Int. Ed., 2019, 58, 15895–15903 CrossRef CAS.
- Z. Yang, S. Wang, C. Wei, L. Chen, Z. Xue and T. Mu, Energy Environ. Sci., 2024, 17, 1603–1611 RSC.
- Z. Yang, B. Zhang, C. Yan, Z. Xue and T. Mu, Appl. Catal., B., 2023, 330, 122590 CrossRef CAS.
- S. Gupta, M. K. Patel, A. Miotello and N. Patel, Adv. Funct. Mater., 2020, 30, 1906481 CrossRef CAS.
- J. Hong, S. Mutalik, M. Miola, D. Gerlach, R. Mehrabi K, M. Ahmadi, B. J. Kooi, G. Portale, P. Rudolf, P. P. Pescarmona and L. Protesescu, Chem. Mater., 2023, 35, 1710–1722 CrossRef CAS.
- L. Meites, Handbook of analytical chemistry, McGraw-Hill, New York, 1st edn, 1963 Search PubMed.
- J. F. Moulder, J. Chastain and R. C. King, Handbook of X-ray Photoelectron Spectroscopy: A Reference Book of Standard Spectra for Identification and Interpretation of XPS Data, Physical Electronics Division, Perkin-Elmer Corporation, Eden Prairie, Minnesota, 1995 Search PubMed.
- E. Commision, Critical raw materials.
- M. L. Krebs, A. Bodach, C. Wang and F. Schüth, Green Chem., 2023, 25, 1797–1802 RSC.
- S. E. Davis, B. N. Zope and R. J. Davis, Green Chem., 2012, 14, 143–147 RSC.
- M. Zhang, Y. Liu, B. Liu, Z. Chen, H. Xu and K. Yan, ACS Catal., 2020, 10, 5179–5189 CrossRef CAS.
- B. J. Taitt, D.-H. Nam and K.-S. Choi, ACS Catal., 2019, 9, 660–670 CrossRef CAS.
- M. T. Bender, Y. C. Lam, S. Hammes-Schiffer and K.-S. Choi, J. Am. Chem. Soc., 2020, 142, 21538–21547 CrossRef CAS.
- W. Chen, C. Xie, Y. Wang, Y. Zou, C.-L. Dong, Y.-C. Huang, Z. Xiao, Z. Wei, S. Du, C. Chen, B. Zhou, J. Ma and S. Wang, Chem, 2020, 6, 2974–2993 CAS.
- P. W. Menezes, S. Yao, R. Beltrán-Suito, J. N. Hausmann, P. V. Menezes and M. Driess, Angew. Chem., Int. Ed., 2021, 60, 4640–4647 CrossRef CAS.
- J. Masa, P. Weide, D. Peeters, I. Sinev, W. Xia, Z. Sun, C. Somsen, M. Muhler and W. Schuhmann, Adv. Energy Mater., 2016, 6, 1502313 CrossRef.
- J. Hong, S. Mutalik, P. P. Pescarmona and L. Protesescu, Chem. Mater., 2024, 36, 2147–2164 CrossRef CAS.
- J. Masa and W. Schuhmann, ChemCatChem, 2019, 11, 5842–5854 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The ESI consists of tables listing prior works on electrochemical oxidation of 5-HMF with Ni-based electrodes and figures with cyclic voltammetry, chronoamperometry, SEM images, setup images, HPLC and XPS data for the Ni-NCs@CP, Ni3B-NCs@CP, and Ni2B-NCs@CP. See DOI: https://doi.org/10.1039/d4cy01220h |
| This journal is © The Royal Society of Chemistry 2025 |