
Regulating interfacial reactions through electrolyte chemistry enables an anion-rich interphase for wide-temperature zinc metal batteries†
Yimei
Chen
 a,
Kaijie
Zhang
b,
Zhixiao
Xu
a,
Facheng
Gong
b,
Renfei
Feng
c,
Zhehui
Jin
b and
Xiaolei
Wang
*a
a,
Kaijie
Zhang
b,
Zhixiao
Xu
a,
Facheng
Gong
b,
Renfei
Feng
c,
Zhehui
Jin
b and
Xiaolei
Wang
*a
aDepartment of Chemical and Materials Engineering, University of Alberta, 9211-116 Street NW, Edmonton, Alberta T6G 1H9, Canada. E-mail: xiaolei.wang@ualberta.ca
bSchool of Mining and Petroleum Engineering, University of Alberta, 9211-116 Street NW, Edmonton, Alberta T6G 1H9, Canada
cCanadian Light Source Inc., 44 Innovation Blvd., Saskatoon, Saskatchewan S7N 0X4, Canada
First published on 19th November 2024
Abstract
Zinc-ion batteries are challenged by zinc dendrites, notorious side reactions, and poor performance at low temperatures. Here, we present a dual-salt tuned electrolyte exhibiting a wide temperature range (−60 to 25 °C). The Zn(ClO4)2-based electrolyte with high hydrogen bond destruction ability and fast diffusion kinetics is suitable for application at ultralow temperatures. The introduction of Zn(OAc)2 salt enhances cation–anion interaction and facilitates the formation of an anion-rich solvation shell and salt-derived interphase, overcoming issues caused by the strong oxidation of ClO4− in the presence of protons. The selective absorption of OAc− on different zinc crystal planes favors dense zinc deposition towards (101) epitaxial while the as-formed anion-rich SEI layer, featuring 2ZnCO3·3Zn(OH)2 distributed on the surface and ZnCl2 uniformly dispersed throughout, inhibits side reactions of corrosion and hydrogen evolution. Consequently, the batteries employing the designed electrolyte exhibited excellent performances, including a high Coulombic efficiency of 99.5% over 800 cycles at 25 °C; a near-unity Coulombic efficiency (100%) for over 4000 cycles and long cycling stability for over 5 months (16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 cycles) in a Zn//I2 battery with an accumulative capacity of 7300 mA h cm−2 at −40 °C. Even at −60 °C, the solid-state electrolyte demonstrates practical applicability in Zn‖I2/AC and Zn‖VO2 batteries. This dual salt-tuned pure aqueous electrolyte also allows the reversible operation of a pouch cell for over 10000 cycles with an accumulative capacity of 19.0 A h, indicating its promising potential for constructing safe and environmentally friendly zinc-ion batteries with broad working temperatures.
500 cycles) in a Zn//I2 battery with an accumulative capacity of 7300 mA h cm−2 at −40 °C. Even at −60 °C, the solid-state electrolyte demonstrates practical applicability in Zn‖I2/AC and Zn‖VO2 batteries. This dual salt-tuned pure aqueous electrolyte also allows the reversible operation of a pouch cell for over 10000 cycles with an accumulative capacity of 19.0 A h, indicating its promising potential for constructing safe and environmentally friendly zinc-ion batteries with broad working temperatures.
Broader contextThe practical application of aqueous zinc ion batteries is challenged by rampant side reactions of corrosion, hydrogen evolution, and zinc dendrite formation, as well as the fast decay of electrochemical performance at subzero temperatures. To address these issues, instead of introducing organic solvents that compromise the inherent advantages of zinc-ion batteries, this paper proposes a simple dual-salt strategy that demonstrates superior performance across an ultrawide temperature range (−60 °C to 25 °C). In this system, the chaotropic Zn(ClO4)2 serves as the primary salt, reconstructing the hydrogen bonds and disrupting the tetrahedral structure of water, which enables the excellent anti-freezing property of the designed electrolyte. The Zn(OAc)2 salts, possessing a strong coordinate affinity towards Zn2+, buffer the electrolyte pH, enhance cation–anion interaction, and facilitate the formation of an anion-rich solvation shell. This as-formed solvation structure suppresses water decomposition and promotes the formation of an inorganic-rich interphase. Consequently, these designed pure aqueous electrolytes demonstrate high reversibility and dendrite-free deposition across wide temperature ranges. The proposed strategy paves a way toward electrolyte design for constructing wide-temperature Zn metal batteries with high safety, low cost, and fast reaction kinetics. |
1. Introduction
Rechargeable aqueous zinc-ion batteries (AZIBs), that are safe, affordable, and exhibit high theoretical capacity, are finding their niche in large-scale renewable energy storage.1–3 The inherent chemical instability of zinc in water triggers the formation of zinc dendrites and gives rise to side reactions of corrosion and hydrogen evolution reaction (HER), leading to poor electrochemical performance.4,5 The high freezing point of pure aqueous electrolytes results in sluggish reaction kinetics stemming from the sharp decrease in ionic conductivity at subzero temperatures. These issues are closely related to the hydrated Zn(OH2)62+ ion and overabundant water molecules in bulk electrolytes.4,6–10 Engineering electrolyte chemistry has proven effective in simultaneously mitigating the two issues of water crystallization and interfacial reaction.8At ambient temperatures, water is stable with the predominance of a tetrahedral structure that becomes increasingly prevalent as water undergoes cooling.11 The introduction of zinc-containing salts into solutions effectively disrupts the original hydrogen bonds (HBs) and reconstructs HB interactions with water molecules.7 According to the Hofmeister series, ClO4− in Zn(ClO4)2 (ZClO) emerges as the most chaotropic ion among commonly used salts, maintaining high conductivity even in the crystal state.12,13 Consequently, ZClO-based electrolytes are promising choices for constructing AZIBs working at low temperatures attributed to the reconstructed HB network and suppressed water reactivity.7,14,15 However, under ambient conditions, the heightened water hydrolysis enhances the oxidation ability of ClO4−, aggravating anode corrosion and loose zinc deposits, further leading to battery failure. Most reported studies introduced organic solvents to solve these issues of interfacial reactions and water solidification, such as the cosolvent or deep eutectic solvent (DES).16–19 Regrettably, the introduction of organic solvents into aqueous electrolytes often comes with elevated viscosity, reduced conductivity, increased costs, and flammability, compromising the safety and environmental sustainability of aqueous electrolyte systems.20,21 Formulating pure aqueous electrolytes that can handle this significant temperature range remains a huge challenge.22
An effective method to mitigate side reactions and suppress zinc dendrites is to induce an in situ formed solid–electrolyte interphase (SEI) layer. The solvent-derived SEI composed mainly of organic compounds is deemed less favorable due to low ionic conductivity and Young's modulus.23,24 Conversely, the salt-derived inorganic-rich SEI layer demonstrates superior ion conductivity, high electrochemical stability, and high mechanical strength. Enhanced cation–anion coordination facilitates the formation of an anion-derived interphase. Herein, we innovatively design a pure aqueous electrolyte system to regulate competitive coordination between salts and water. Compared to other commonly used aqueous electrolytes for ZIBs, the designed ZClO-based electrolyte with the Zn(OAc)2 salt additive exhibits excellent electrochemical stability across wide temperatures. Both experimental and theoretical evidence shows that the OAc− with strong affinity toward Zn2+ infiltrates the primary solvation shell alongside ClO4−, displacing water molecules and forming a water-poor solvation structure of Zn(OAc)0.32(ClO4)0.89(H2O)4.790.79+, assisting the formation of a 2ZnCO3·3Zn(OH)2(Zn5(CO3)2(OH)6) rich SEI layer. Of note, compared with the only ZnCl2 containing interphase, the as-formed zinc carbonate-rich SEI featuring low solubility and fast desolvation ability effectively suppresses side reactions. Additionally, the absorption of OAc− on the zinc surface guides a compact deposition and homoepitaxial growth of zinc along the (101) plane, effectively impeding zinc dendrite growth and advancing reaction kinetics. The plating behaviors in different electrolytes are shown in Scheme 1. Consequently, batteries employing the designed electrolyte demonstrate excellent electrochemical performance, including high Coulombic efficiencies (CE) of 99.5% over 800 cycles and stable zinc plating/stripping in symmetric cells for 3000 hours at 25 °C, a high CE of 99.7% over 900 cycles at −20 °C, and a near-unity CE (100%) over 4000 cycles at −40 °C. This dual salt-derived SEI-favored pure aqueous electrolyte has low-cost, high-safety, and environmentally friendly features, paving a new path for constructing AZIBs operating under challenging conditions.
 | ||
| Scheme 1 Schematic diagram of the action mechanism of OAc− in different electrolytes. | ||
2. Results and discussion
2.1 Exploring ideal electrolytes for wide-temperature AZIBs
To assess the anti-freezing property of zinc-based salts with various anions and ascertain the optimal concentration of perchlorate-based electrolytes, the thermodynamic property, HB structure, and diffusion kinetics are investigated. The anti-freezing property was initially observed by storing various electrolytes in the fridge (Fig. S1, ESI†), where 2 M and 3 M ZClO electrolytes demonstrated the best anti-freezing properties. Differential scanning calorimetry (DSC) further revealed the thermal behaviors of various electrolytes (Fig. 1a), where exothermic peaks can be observed for 1 M ZClO, 3 M ZSO, ZnCl2, and Zn(OTF)2 electrolytes, revealing their crystallization temperatures (Tg) of −33, −36, −42, and −57 °C, respectively. No crystallization temperatures were observed for 2.0 and 3.0 M ZClO even down to −90 °C, implying a relatively high tetrahedral entropy of water in these solutions.7 The melting temperature of these electrolytes shows the same trend (Fig. S2, ESI†). The anti-freezing mechanism was reflected by HBs obtained through Raman spectra, where peaks ranging from 2900–3800 cm−1 correspond to the O–H stretching vibration of H2O and can be fitted into three Gaussian components, including strong HB (3300 cm−1), medium HB (3450 cm−1), and weak HB (3600 cm−1) (Fig. S3, ESI†).25,26 The results in Fig. 1b reveal that the 3.0 M ZClO electrolyte possesses the highest weak HBs and the HB disruption ability of four different anions follows the order ZClO > Zn(OTF)2 > ZnCl2 > ZSO, consistent with the Hofmeister series (Fig. S4, ESI†). For ZClO electrolytes with various concentrations, both Raman and Fourier-transform infrared spectroscopy (FTIR) results illustrate that increasing the solute content causes a decrease in the strong HBs and an increase in the weak HBs (Fig. S3 and S5, ESI†).27 Molecular dynamics (MD) simulations (Fig. 1c) reveal that the average number of HBs within water molecules follows the order of 1.0 M ZClO > 3.0 M ZSO> 3.0 M ZnCl2 > 3.0 M Zn(OTF)2 > 2.0 M ZClO > 3.0 M ZClO, demonstrating the establishment of a continuous HB network around the ClO4− anion is challenging due to its inherent “structure-breaking” property.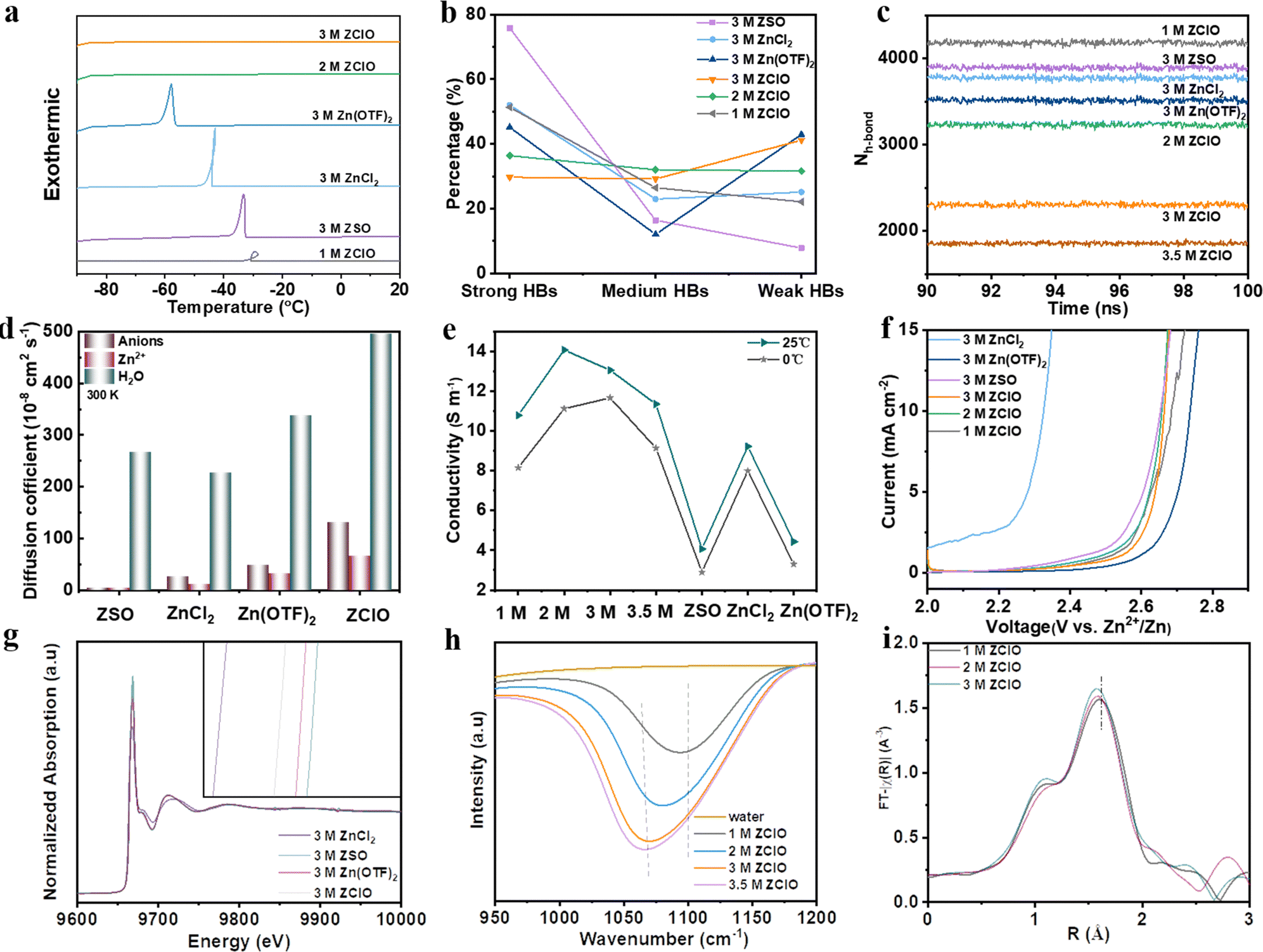 | ||
| Fig. 1 Analysis of the HB structure and dynamic relaxation behavior of water. (a) DSC curve of various electrolytes. (b) Statistics of HBs in different electrolytes derived from Raman results. (c) Average HB number among water clusters based on MD simulations. (d) Self-diffusion coefficients of water molecules in different electrolytes. (e) Ionic conductivity of various electrolytes (1 M, 2 M, 3 M, and 3.5 M represent X M ZClO, and the concentration for ZSO, ZnCl2, and Zn(OTF)2 is 3 M). (f) Anti-oxidation property of various electrolytes. (g) Zn K-edge XANES spectra. (h) FTIR results of various concentrations of ZClO electrolyte. (i) Fourier transform EXAFS curves of various concentrations of ZClO electrolytes. | ||
The diffusion kinetics of electrolyte particles can be analyzed by the mean square displacement and the derived diffusion coefficient. It is obvious that all H2O, Zn2+, and ClO4− particles are transported faster in the 3.0 M ZClO electrolyte than other systems at both 300 and 273 K (Fig. 1d, and Fig. S6, ESI†), which is consistent with their ionic conductivity difference (Fig. 1e). This behavior is attributed to the disrupted tetrahedral structure of water which releases more free water molecules. Meanwhile, ZClO electrolytes also show an enhanced anti-oxidation property superior to ZSO and ZnCl2 electrolytes (Fig. 1f), and comparable to Zn(OTF)2 but with lower cost (Table S1, ESI†). The affinity of these various anions towards the Zn2+ is also studied by the X-ray adsorption fine structure (XAFS) technique. The X-ray adsorption near-edge spectra (XANES) show that SO42− donates higher electrons to Zn, followed by OTF− and ClO4− (Fig. 1g). To explore whether the ClO4− can compete with water and enter the first solvation sheath of Zn2+, we conducted an FTIR test (Fig. 1h), where the representative ClO4− peak shifts from a high wavenumber of 1105 cm−1 to a lower value of 1060 cm−1 as the concentration increases, indicating a transition from free ClO4− ions to a Zn2+–ClO4− ion pair.28 MD simulations further show the transformation of hydrated Zn2+ from Zn(H2O)62+ in 1.0 M ZClO to Zn(H2O)5(ClO4)+ in 3.5 M ZClO (Fig. S7 and Table S2, ESI†), which is consistent with the R-space Fourier-transformed extended X-ray adsorption fine structure (FT-EXAFS) analysis (Fig. 1i) that shows a diminishing Zn–O radical distribution distance with increasing concentration. The participation of ClO4− ions in the solvation shell decreases the number of coordinated water and contributes to the formation of an anion-rich SEI layer, driven by anion decomposition in aqueous electrolytes. The anion-rich SEI layer is desired for regulating electron conduction and ion transport phenomena at the electrode/electrolyte interface. However, the high solubility of ZnCl2 in aqueous electrolyte leads to the dissolution of the Cl-containing SEI during the repeated cycling process.
2.2 Tailoring the solvation structure of electrolytes
The solvation structure of the electrolyte significantly impacts Zn2+ desolvation and interfacial chemistry. Although ZClO electrolyte demonstrates commendable anti-freezing properties, its application in ZIBs with pure formulation yields unsatisfactory CE and lifespan under ambient conditions (Fig. S8, ESI†). This deficiency is attributed to aggravated corrosion and HER reaction as well as the oxidizing property of ClO4− in the presence of abundant protons,29 which complicates the formation of a complete SEI layer. Hence, a triple-functional Zn(OAc)2 salt is introduced as an additive in 3.0 M ZClO electrolyte (Fig. S9, ESI†).30 Anions can directly modify the Zn2+ solvation structures, with their positioning and arrangement within the solvation sheath playing a crucial role in enhancing electrochemical performance. The ClO4− anion exhibits a weak interaction with Zn2+ (−0.7 eV, Fig. 2a), comparable to the Zn2+–H2O interaction. This suggests high mobility for the ClO4− anion around the electrode surface. Consequently, the free-moving ClO4− anions result in low CE and Zn corrosion during the cycling process.31 When Zn(OAc)2 is introduced as a co-salt, the Zn2+–anion interaction is significantly enhanced, and the strong interaction of Zn2+–OAc− (−1.72 eV) causes the Zn2+–OAc− to approach the electrode and inhibit serious side reactions. Besides, the OAc−–Zn2+ interaction weakens the Zn2+–solvent interaction and decreases the population proportion of the solvent molecule in the first solvation, as revealed by molecular dynamics (MD) calculations and a few characteristics. This interfacial situation effectively suppresses H2O reduction. Furthermore, the optimized SEI film inhibits the decomposition of the H2O and provides a favorable environment for the uniform deposition of Zn2+. | ||
| Fig. 2 Investigation of competitive coordination of Zn2+. (a) Binding energy of Zn2+ with different species. (b) and (c) MD results of 3.0 M ZClO electrolyte (b) and 3.0 M ZClO − 0.3 M Zn(OAc)2 additive (c). (d) 1H NMR of different electrolytes. (e) Zn K-edge XANES spectra of different electrolytes. (f) Fourier transform EXAFS curves of ZClO electrolyte with various concentrations of additive. (g) and (h) WT-EXAFS of 3.0 M ZClO (g) and 3.0 M ZClO − 0.3 M Zn(OAc)2 (h). (i) HOMO–LUMO energy levels of various groups. (j) Step-by-step desolvation process of hydrated zinc ions calculated by Gaussian. | ||
This coordination environment of Zn2+ is quantitively elucidated by MD calculations (Fig. 2b, c and Table S2, ESI†), where hydrated zinc ions transform from Zn(H2O)5(ClO4)+ in 3.0 M ZClO to Zn(H2O)4.79(OAc)0.32(ClO4)0.890.79+ in the designed electrolyte with a few species coexisting in the system (Fig. S10, ESI†). The entry of Zn(OAc)2 in the first solvation shell is experimentally confirmed by 1H nuclear magnetic resonance (NMR) and EXAFS results. The signal of 1H originating from the –CH3 group of OAc− shifts downfield in the designed electrolyte compared to that in the pure 0.3 M Zn(OAc)2 solution, indicating fewer electrons around the hydrogen nucleus due to the competition of combined Zn2+ (Fig. 2d). The XAFS results in Fig. 2e show increased energy adsorption edges while Fig. 2f shows a decreased Zn–O bond length with the increase of OAc− additive in the ZClO electrolytes. These phenomena can be attributed to the stronger interactions between OAc− and Zn2+. Simultaneously, the wavelet-transformed (WT) EXAFS analysis reveals a greater Zn–O coordination distance in the 3 M ZClO electrolyte (Fig. 2g) compared to the 3 M ZClO + 0.3 M Zn(OAc)2 solution (Fig. 2h). Both calculation and experimental results demonstrate that the OAc− anions are entering the first solvation sheath, suppressing the water decomposition and side reactions. Furthermore, the introduction of Zn(OAc)2 can act as a pH regulator. With 0.3 M Zn(OAc)2 additive, the pH increases from 2.1 to 2.9, manifesting lowered free H+ and suppressed corrosion and HER reaction (Fig. S11, ESI†).
The effect of the competitive coordination of Zn2+–OAc− and Zn2+–H2O on the electrochemical stability of the electrolytes is studied by the HOMO and LUMO energy levels and the desolvation process. The Zn2+–H2O complex possesses the lowest LUMO energy of −3.46 eV, indicating its preference for gaining electrons producing hydrogen (Fig. 2i). With Zn(OAc)2 inclusion, the enhanced cation–anion contact pairs weaken the Zn2+–H2O interaction and result in a significantly reduced energy barrier for water stripping compared to the Zn(H2O)5(ClO4)− species (Fig. 2j). This is conducive to the desolvation process and the repelling of water from the electrode/electrolyte interface, suppressing the reduction of Zn2+–H2O. Fig. 2j and Fig. S12 (ESI†) also demonstrate that in pure ZClO electrolyte, the stripping of H2O from Zn(H2O)5(ClO4)− is kinetically favored to occur first, due to the lower energy barrier than for ClO4− stripping. This process generates Zn–ClO4−, followed by the reduction of Zn2+ and ClO4−, forming a Cl− rich SEI layer. For the designed system, the H2O is also stripped first, leaving the Zn–OAc− complex around the electrode surface. This brings a large number of OAc− around the electrode surface, facilitating water-poor environment formation and pH regulation benefiting from the OAc− buffer effects.
2.3 Investigating the composition of the salt-derived SEI layer
The X-ray diffraction (XRD) patterns in Fig. 3a show the composition evolution of the SEI layers. The Zn electrodes cycled in the 3 M ZClO electrolyte exhibit apparent peaks of ZnCl2 and Zn4ClO4(OH)7, originating from zinc salt reduction and the HER. Conversely, the zinc electrode subjected to 50 cycles in the tailored electrolyte displays no additional peaks aside from the zinc metal, indicative of the suppressed by-product formation. Upon extending the cycling to 500 cycles, Zn5(CO3)2(OH)6 and a minor peak belonging to ZnCl2 were detected on the zinc surface in the OAc−-containing designed electrolyte, which is also confirmed by the EDX element mapping result (Fig. S13, ESI†), showing the co-existence of Zn, Cl, C, and O. Zn5(CO3)2(OH)6 was reported to be a robust solid–electrolyte interface (SEI) for water-repelling and dendrite suppression.18 Additionally, the designed electrolyte with OAc− facilitates the reversible Zn plating/stripping and the vertical epitaxial growth of Zn, as evidenced by the increasing intensity of the (100) and (101) planes. In particular, the ratio of I(101)/I(002) from 0.83 to 1.37 after 50 cycles, and further to 3.92 after 500 cycles. The preferential electro epitaxial growth of Zn along the (101) direction was further ascertained by 2D synchrotron grazing-incidence X-ray diffraction (GIXRD) analysis. Fig. S14a–c (ESI†) show that the (101) diffraction ring in the designed system is more predominant than the (002) ring, contrasting with stochastic rings in pure ZClO electrolyte (Fig. S14d, ESI†). The intensity ratio of the (101)/(002) plane obtained from the integration towards 2θ directions increases with the prolonged plating of 50, 100, and 500 cycles (Fig. S14d–f, ESI†), consistent with the 1D XRD results. The regulated (101) epitaxial zinc growth with the presence of Zn5(CO3)2(OH)6 in SEI components is also observed in the reported literature.32,33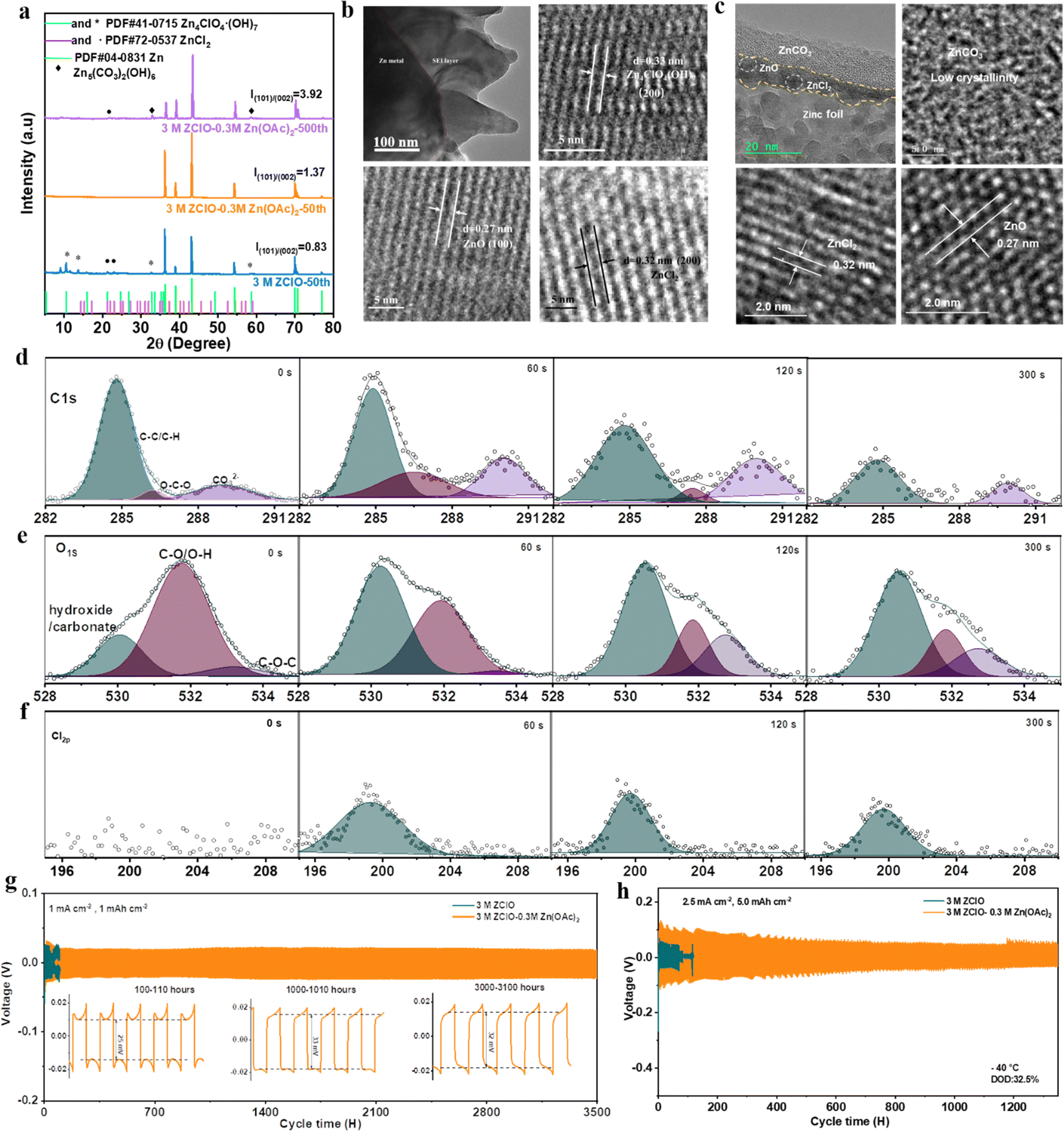 | ||
| Fig. 3 SEI layer characterizations. (a) XRD results of cycled zinc. (b) and (c) TEM results of cycled zinc in 3.0 M ZClO (b) and designed electrolyte (c). (d)–(f) In-depth XPS of C 1s, O 1s, and Cl 2p in the designed electrolyte. (g) Long-term cycling stability of the symmetric cell at 25 °C. (h) Symmetric cell's long cycling performance with DOD of 32.5% at −40 °C. | ||
To obtain detailed information on the SEI layer, the cross-sectional image of the cycled zinc was intuitively observed using a focused ion beam-transmission electron microscope (FIB-TEM). The electrode cycled in 3.0 M ZClO shows an uneven surface with an SEI thickness of around 200 nm, indicating severe electrode corrosion and electrolyte decomposition. The fringes with spacing (d) of 0.27 nm, d = 0.32 nm, and d = 0.33 nm were assigned to the (100) plane of ZnO, (200) plane of ZnCl2, and (200) plane of Zn4ClO4 (OH)7, respectively (Fig. 3b). For the electrode obtained in the designed electrolyte, a dual-SEI layer with a thickness of around 20 nm was observed, featuring an amorphous outer layer and a crystalline inner layer (Fig. 3c). The ZnO and ZnCl2 components were observed in the inner layer. The outer amorphous layer belongs to the ZnCO3-rich component, which is proved later. Additionally, Fig. 3c and the TEM mapping results in Fig. S15 (ESI†) show that the zinc content above SEI and within the SEI is low, demonstrating the zinc is primarily deposited beneath the SEI layer. This behavior is beneficial for suppressing dendrite formation.
The composition of the SEI layer in the designed electrolyte system was further investigated via in-depth X-ray photoelectron spectroscopy (XPS) with Ar ion sputtering. In the C 1s spectrum of the zinc surface obtained in 3.0 M ZClO electrolyte, one apparent peak at 284.8 eV was detected which is assigned to the C–C/C–H groups.34 Meanwhile, a minor peak at 290 eV appears, attributed to CO32− groups originating from the air atmosphere (Fig. S16, ESI†). In comparison, the C 1s signal obtained in the designed electrolyte demonstrates a predominant –CO3 signal, indicative of the formation of 2ZnCO3·3Zn(OH)2. With the increasing sputtering time, the –CO3 intensity decreases but remains noticeable (Fig. 3d). This is consistent with the O 1s spectrum, where hydroxide/carbonate peaks at 530 eV are more pronounced with the OAc− containing electrolyte than in the pure ZClO system (Fig. 3e and Fig. S17, ESI†). The Cl 2p peaks located at around 199 eV and 208 eV belong to the Cl− and ClO4−, respectively. Fig. S18 (ESI†) demonstrates a high Cl-content through various depths of the SEI layer, deriving from the by-products of ZnCl2, and a moderate signal of ClO42−, possibly stemming from the by-products of Zn4ClO4(OH)7 and some electrolyte residues. In contrast, the results for the designed electrolyte display an absence of conspicuous ClO4− peaks and consistently low intensity of Cl− signal across different depths of the SEI layer, indicating an alleviated electrolyte consumption (Fig. 3f). The XPS analysis and XRD results suggest that the designed electrolyte favors the in situ formation of the Zn5(CO3)2(OH)6–ZnCl2-rich interface, where ZnCl2 distributes evenly across the various depths of the SEI layer and basic zinc carbonate displays a decreased gradient distribution from top to bottom, isolating the electrode from H+ attack. Unlike the solvent-derived, inherently unstable, and heterogeneous SEI layers, which cannot bear large volume changes, leading to a continual degradation of active material and rapid dendrite growth, those salt-derived anion-rich SEI layers enhance ionic conductivity and inhibit dendrite growth, as documented in lithium metal batteries.35,36
The possible formation process of the SEI layer in different systems is proposed. For pure ZClO systems, the severe HER reaction and the reduction of perchlorate anions lead to the formation of Zn4ClO4(OH)7 (eqn (1)) and ZnCl2 (eqn (2)) by-products, respectively. The lowest LUMO energy of ClO4− facilitates the continuous decomposition of electrolytes, and considering the high solubility of ZnCl2 in the aqueous electrolyte, this layer is not stable during cycling. For the designed system, the addition of OAc− increases electrolyte pH, promoting the continuous dissolution of CO2 (eqn (3)), which combines with OH− (leftover from the HER) and Zn2+ forming 2ZnCO3·3Zn(OH)2 on the Zn surface (eqn (4)).32,37 Compared with Zn4ClO4(OH)7–ZnCl2 components, the 2ZnCO3·3Zn(OH)2-based SEI with lower solubility and fast Zn2+ conduction, assures the long-lasting protection of the SEI layer and can isolate the electrolyte from reduction. To investigate if the CO32− formation is derived from dissolved CO2, we assembled a battery in a CO2-free atmosphere and tested the in-depth XPS of the cycled zinc (Fig. S19, ESI†). The C 1s peaks at 290 eV are quite the same as that obtained in 3 M ZClO electrolyte (Fig. S16, ESI†). Compared with the C 1s peaks obtained in Fig. 3d, the C 1s signal at 290 eV in a CO2-free atmosphere indicates significantly lowered CO32− content. The remaining CO32− may come from the slow permeation of CO2 from the environment during cycling. After 120 s sputtering, the CO32− peak almost disappears, revealing the significance of CO2 participation in forming 2ZnCO3·3Zn(OH)2.
| 4Zn + ClO4− + 8H2O → Zn4ClO4(OH)7 + 4H2↑ + OH− | (1) |
| Zn + 2ClO4− + 8H2O + 14e− → ZnCl2 + 16OH− | (2) |
| CO2 + OH− ⇌ HCO3− ⇌ CO32− + H+ | (3) |
| 5Zn + 2CO32− + 6OH− → 2ZnCO3 + 3Zn(OH)2 + 10e− | (4) |
Under the synergy effects of OAc− additive, the designed 3.0 M ZClO + 0.3 M Zn(OAc)2 electrolyte system allows for ultralong cycling of symmetric cells for over 3500 hours at 1.0 mA cm−2 and 1.0 mA h cm−2 at room temperature, contrasting to 100 hours in the pure ZClO electrolyte system (Fig. 3g). Besides, the battery assembled in a CO2-free environment failed after 350 hours likely due to the incomplete formation of the 2ZnCO3·3Zn(OH)2 layer in the absence of CO2 (Fig. S20, ESI†), highlighting the benefits of the 2ZnCO3·3Zn(OH)2 layer in protecting the anode. Meanwhile, a significantly lower voltage hysteresis of around 30 mV throughout the process is observed attributed to the favorable wettability of the Zn(OAc)2-based electrolyte, reducing surface energy and promoting Zn2+ diffusion towards the electrode (Fig. S21, ESI†). This stability of the designed system is also observed at both higher and lower current densities (over 2800 hours at 3.0 mA cm−2 in Fig. S22, and over 2100 hours at 0.2 mA cm−2 in Fig. S23, ESI†). The battery with the designed electrolyte also displayed excellent rate capability over a current range of 1.0–5.0 mA cm−2 compared with that cycled in pure ZClO electrolyte (Fig. S24, ESI†). The designed electrolyte system also allows for operating under more challenging conditions. With a Zn foil of 30 μm, long cycling over 550 hours and 300 hours is achieved at a depth of discharge (DOD) of 32.5% (5.0 mA h cm−2, and 5.0 mA h cm−2) and 52.0% (4.0 mA cm−2, and 8.0 mA h cm−2), respectively (Fig. S25 and S26, ESI†), indicating the zinc dendrite growth and side reaction of corrosion and the HER under ambient conditions are greatly suppressed. At −40 °C, stable cycling over 1300 hours is obtained at a DOD of 32.5%, whereas the pure 3.0 M ZClO electrolyte system fails within 100 hours (Fig. 3h). The excellent electrochemical performance strongly supports that the designed electrolyte with OAc− inclusion not only inhibits the water decomposition, alleviating the H+ attack and the subsequent HER but also regulates the zinc deposition, preventing the formation of porous flakes and dendrites.
2.4 Analyzing the interfacial chemistry in the designed electrolyte
The difference in morphology of Zn grown at the electrode surface directly reflects the different diffusion and deposition behaviors in various electrolytes. The electrode surface before cycling shows large scratches and presents no orientation preference (Fig. S27, ESI†). After 100 hours of cycling in 3.0 M ZClO electrolyte, the zinc surface displays a loose and random structure composed of flakes with various sizes (Fig. 4a), mainly from corrosion and subsequent uneven deposition of Zn. By comparison, a highly dense structure with vertically packed nanosheets, resembling the preferred (101) plane, is observed in the designed 3.0 M ZClO + 0.3 M Zn(OAc)2 electrolyte (Fig. 4b). This morphology provides a more intuitive reveal of the zinc deposition on the (101) plane. Compared to facilitating reversible Zn stripping and plating toward the (002) plane, the Zn surface with vertically packed morphology and largely exposed (101) plane enhances mass transfer kinetics and suppresses side reactions by minimizing the grain boundary area (Fig. 4c).38,39 The preferred orientation is the result of the contribution of surface energy and work function of the crystallographic planes. The reduced surface energy and increased nucleation overpotential in the designed electrolyte facilitate the formation of smaller zinc nuclei with higher areal density. The dynamic adsorption of OAc− on the (002) plane and the high nucleation overpotential further directs zinc deposition preferentially in the vertical direction, particularly along the (101) plane by blocking the horizontal growth. Detailed discussion is shown in the ESI,† Discussion S1.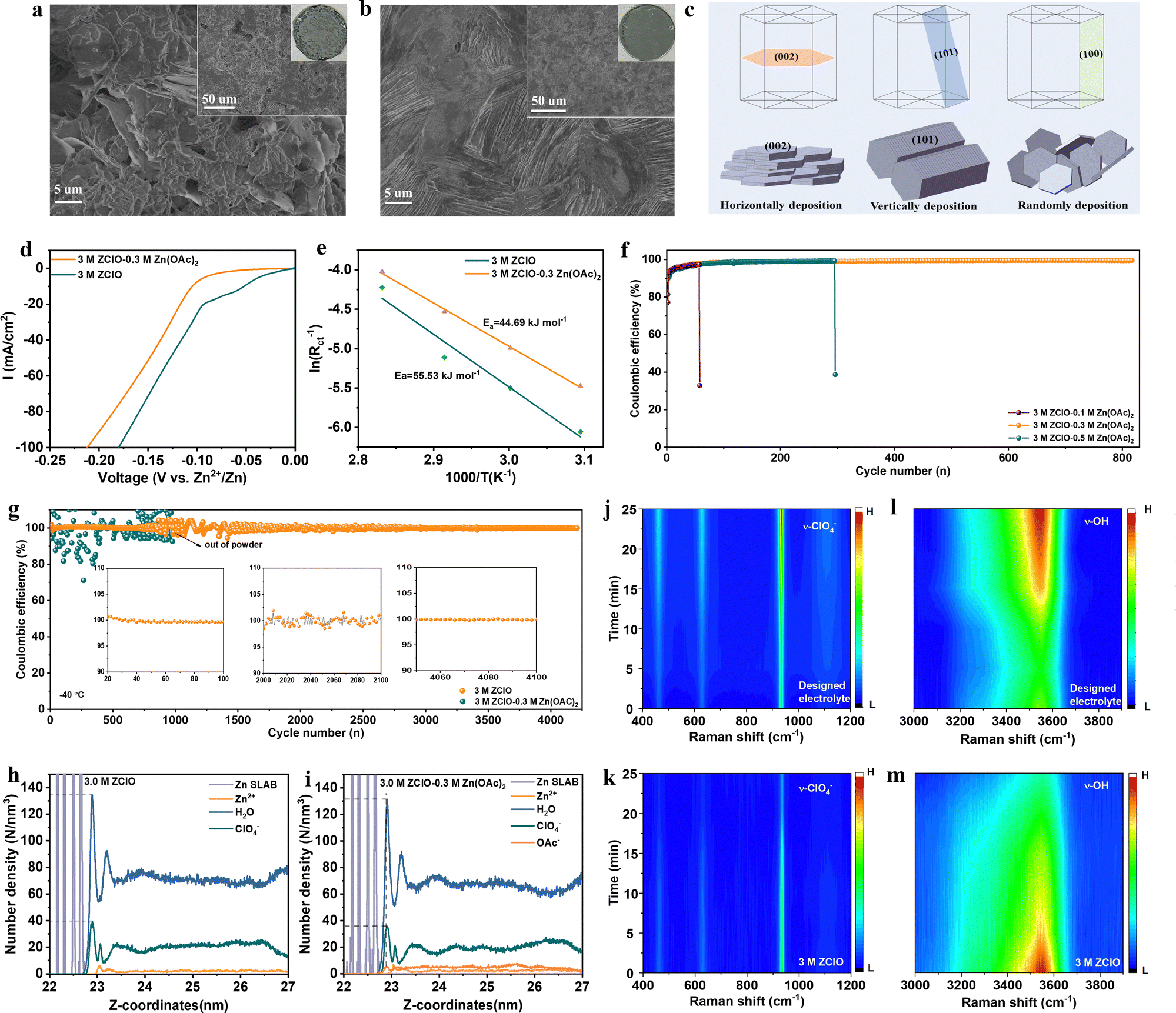 | ||
| Fig. 4 Interface chemistry and deposition morphology. (a) and (b) SEM images obtained in 3.0 M ZClO (a) and OAc-containing electrolyte (b). (c) Schematic depictions of the Zn plating mechanism. (d) LSV curves of different electrolytes. (e) Activation energy of different electrolytes. (f) Coulombic efficiency of Zn‖Cu cells at 1.0 mA cm−2. (g) Coulombic efficiency at 2.0 mA cm−2 at −40 °C. (h) and (i) Surface electrolyte composition simulated by MD. (j) and (l) In situ Raman results of the electrode/electrolyte interface in 3 M ZClO − 0.3 M Zn(OAc)2 electrolyte. (k) and (m) In situ Raman results of the electrode/electrolyte interface in 3 M ZClO electrolyte. | ||
Aside from deposition morphology, the tuned interfacial chemistry is studied with a series of electrochemical tests. Specifically, the inclusion of Zn(OAc)2, buffering electrolyte pH and suppressing water decomposition, lowered the HER potential from 0 to nearly −0.1 V vs. Zn2+/Zn (Fig. 4d). The Tafel plots manifest that the electrolyte containing the Zn(OAc)2 additive shifts the corrosion potential to a more positive value (−0.71 V vs. −0.72 V for 3 M ZClO) along with a diminished corrosion current density (3.32 mA cm−2vs. 5.13 mA cm−2 for 3 M ZClO), illustrating the lowered corrosion rate in the presence of the Zn5(CO3)2(OH)6 protection layer (Fig. S28, ESI†). The CV curves present the reversibility of different electrolyte systems (Fig. S29, ESI†). The cell with 3 M ZClO − 0.3 M Zn(OAc)2 presents overlapped CV curves while the reference one shows great fluctuations. These results suggest that the water-induced corrosion and HER reactions are effectively controlled under the assistance of OAc− additive. The activation energy, which influences the reaction kinetics, is studied by electrochemical impedance spectroscopy (EIS) (Fig. 4e and Fig. S30, ESI†). Within the temperature increase from 50 to 80 °C, the resistance of both electrodes decreases, and at any given temperature, the charge transfer resistance (Rct) of the symmetric cell employing the designed electrolyte is considerably lower than that using pure ZClO electrolyte, highlighting an improved charge transfer capability. The activation energy (Ea) for the designed electrolyte determined through the Arrhenius equation is also lower than that obtained in pure ZClO electrolyte (44.69 kJ mol−1vs. 55.53 kJ mol−1), illustrating that the in situ formed SEI layer promotes the Zn2+ desolvation process.
The mitigated side reactions at the Zn–electrolyte interface in the designed electrolyte are further reflected by the CE in Zn‖Cu cells. The cell using pure ZClO electrolyte failed in a few cycles whereas with an optimal concentration of 3 M ZClO + 0.3 M Zn(OAc)2, a high CE of 98.0% is achieved within 50 cycles and maintained at 99.5% for over 800 cycles, along with a low voltage hysteresis of 35.67 mV (Fig. 4f and Fig. S31, ESI†). In contrast, lower Zn(OAc)2 concentrations (e.g., 0.1 M) fail to achieve effective regulation while higher concentrations (e.g., 0.5 M) adversely impact battery conductivity and the desolvation process (Fig. 4i). The ionic conductivity and viscosity of the electrolyte with 0.3 M Zn(OAc)2 addition are presented in Fig. S32 (ESI†). The gradually increased high CE in the designed electrolyte system is attributed to the progressive formation of a protective SEI layer. The CE was also tested at a low current density of 0.5 mA cm−2, where the HER is more pronounced (Fig. S33, ESI†). The results show the battery with the designed electrolyte shows satisfactory performance, a high average CE of over 98.6% is obtained, along with a long cycling stability of over 1600 cycles, demonstrating the regulated zinc deposition and suppressed side reactions. In contrast, the cell using pure 3.0 M ZClO electrolyte failed in 200 cycles. Under an elevated current of 5.0 mA cm−2, an ultrastable operation is obtained for over 1500 cycles, maintaining an average CE of 99.5% (Fig. S34, ESI†). At a low temperature of −20 °C, the CE rises quickly to 99.5% in 10 cycles and remains at 99.7% for over 900 cycles, in contrast to the poor cycling stability of 150 cycles with a low CE of 97% in pure ZClO solution (Fig. S35, ESI†). More impressively, a near-unity CE (100%) over 4200 cycles is achieved at −40 °C in the designed system with a current density of 2.0 mA cm−2, while the pure ZClO system exhibits a large fluctuance in the CE and is dead within only less than 1000 cycles (Fig. 4j). These results strongly support that the water-induced side reactions of corrosion and the HER reaction are largely suppressed in the designed electrolyte.
To further investigate the interfacial electrolyte configuration in different electrolytes, molecular dynamics (MD) simulations and in situ Raman spectroscopy were conducted. In the 3 M ZClO electrolyte, the zinc surface is adsorbed by a layer of H2O molecules with a peak position at 0.4 nm and a number density of 135 N nm−3, followed by a layer of ClO4− ions at 0.41 nm with a number density of 39 N nm−3 (Fig. 4h). When 0.3 M Zn(OAc)2 is added, a new peak corresponding to OAc− ions appears, with a number density of 6 N nm−3, accompanied by a decrease in the intensity of H2O (131 N nm−3) and ClO4− (36 N nm−3) at the interface (Fig. 4i). Furthermore, compared to the pure ZClO system, the electric double layer in the designed electrolyte is significantly suppressed. The peak positions of both H2O and ClO4− shift closer to the interface, reducing to 0.25 nm, while OAc− ions exhibit an even closer adsorption position at 0.24 nm, indicating a strong affinity toward the zinc surface. Furthermore, in situ Raman spectroscopy was employed to monitor the changes in interfacial substances on the zinc anode during Zn2+ deposition in various electrolytes. The process of Zn2+ plating was studied under a constant-current density of 1 mA cm−2. The designed electrolyte exhibits a peak at 2945 cm−1, corresponding to the symmetric vibrations of the –CH3 group from OAc− (Fig. S36, ESI†). Upon discharging, the intensity of the –CH3 group gradually increases, which can be attributed to the diffusion of Zn2+ towards the cathode, bringing a large amount of OAc− to the electrode/electrolyte interface. The accumulation of OAc− during the Zn2+ plating not only excludes H2O from the surface area but also weakens the Zn–H2O interaction due to the strong binding affinity of OAc− towards Zn2+, thus suppressing the HER. The ClO4− in the designed electrolyte shows a low intensity at the open circuit potential (OCP), and the intensity gradually increases upon discharging due to the co-diffusion with Zn2+ towards the anode (Fig. 4j). Conversely, in the 3 M ZClO electrolyte, the ClO4− intensity decreases with continued discharge (Fig. 4k). This is due to the severe HER in which the H+ competes with Zn2+ gaining electrons, as reflected by the initial low CE in aqueous electrolyte. The leftover OH− and electrostatic repulsions push the ClO4− away from the electrode. The HB evolution at the cathode/electrolyte interface upon discharging was also investigated. The O–H stretching band can be deconvoluted into three types of O–H stretching vibrations: strong HBs at 3250 cm−1 (symmetric H2O), medium HBs (solvated H2O) at 3450 cm−1 and weak HBs at around 3600 cm−1. The O–H stretching band at the OCP in the designed electrolyte is low, attributing to the absorption of OAc− on the zinc surface. Upon discharging, the O–H stretching signal intensified, and the signal at 3550 cm−1 corresponding to the HBs formed between ClO4−/OAc− and H2O increases, ultimately causing the breakage of HBs between water molecules (Fig. 4l). In contrast, the HBs in the 3 M ZClO electrolyte is high at the OCP, consistent with the MD results that in 3 M ZClO electrolyte, the electrode surface is absorbed by a layer of H2O molecules. Upon discharging, the intensity at 3550 cm−1 gradually decreases, attributed to the low ClO4− level and the decomposition of water.
2.5 Electrochemical performance of full cells
To test the compatibility of the designed electrolyte, full cells were assembled using I2/AC as the cathode material due to its high redox potential and superior kinetics. The EIS of the cell utilizing the designed electrolyte displays both lowered charge transfer resistance (R3/Rct) and SEI layer-related resistance (R2/RSEI) than that using pure ZClO electrolyte, indicating faster reaction kinetics and higher ionic conductivity with a Zn5(CO3)2(OH)6-containing interphase (Fig. S37 and Table S3, ESI†). Of note, a higher internal resistance (R1) is also observed with the designed system due to the slightly lowered ionic conductivity with Zn(OAc)2 additive. The CV curves with various scan rates were conducted to study the reaction kinetics of the I2/AC cathode in different electrolyte systems (Fig. 5a and Fig. S38, ESI†). The Zn‖I2/AC cell shows a high working plateau of ∼1.2 V from the redox of I2/I− and an operating window of 0.2–1.7 V5. The b value which reflects the diffusion-controlled process (b = 0.5) and the capacitive-controlled (b = 1) behavior, was determined according to Dunn's method.40 The oxidation (Peak 1) and reduction (Peak 2) peaks show high values of 0.84 and 0.86 for the designed system, and 0.81 and 0.87 for the pure ZClO electrolyte, revealing high capacitive contributions (Fig. 5b and Fig. S39, ESI†). The capacitive contribution for the designed system is 79.45%, 86.65%, 90.13%, 92.53%, and 94.54% at scan rates of 0.2, 0.4, 0.6, 0.8, and 1.0 mV s−1, respectively, higher than that obtained in the pure ZClO system, especially at higher scan rates (Fig. 5c and Fig. S40, ESI†). These higher capacitive contributions may result from the co-adsorption of OAc− anions on the porous carbon and can lead to fast reaction kinetics.41 One main issue that hinders the I2/AC cathode is the dissolution of I2 causing poor cycling performance and severe side reactions.42 Specifically, the solubility of the polyiodide intermediates produced during the reversible I−/I2 redox reaction induces serious shuttle effects, resulting in the irreversible depletion of active mass and zinc corrosion. Apart from controlling the Zn interface chemistry, the effects of OAc− addition on inhibiting the shuttle effect of I3− and alleviating the reduction of I3− by Zn metal were also disclosed by Raman and UV-visible (UV-vis). The Raman results in Fig. S41 (ESI†) suggest that the CH3 peak experiences a redshift with the I3− concentration increasing from 10–30 mM, indicating the interaction between OAc− and I3−. The UV-vis results indicate that compared with pure ZClO electrolyte, the OAc−-containing electrolyte shows a lowered decay rate of the I3− signal, revealing the inhibited reduction of I3− and the derived zinc corrosion (Fig. 5d and e). This conclusion is further supported by the inset images, where a darker brown color of the OAc−-containing electrolyte is observed after 26 hours of resting. | ||
| Fig. 5 Full cell's performance. (a) CV curves of the battery employing the designed electrolyte. (b) Fitted b value in the designed electrolyte. (c) Capacitive contribution in the designed electrolyte. (d) and (e) UV-vis of different electrolytes following the immersion of zinc foils for various durations. (f) Rate performance of the Zn‖I2/AC full cell. (g) Long-term cycling performance at 1.0 A g−1. (h) Long-term cycling performance at an N/P ratio of 4:1 and a lean electrolyte of 11 μL mA−1 h−1. (i) XRD results of the I2/AC cathode following 12000 cycles in the Zn/I2@AC full cell. (j) Comparison of cell performance in ZClO-based electrolytes. | ||
The enhanced reaction kinetics and suppressed side reactions were then studied by electrochemical tests. The cell deploying the designed 3.0 M ZClO + 0.3 M Zn(OAc)2 electrolyte shows higher specific capability than that using pure ZClO electrolyte at various current densities (e.g., 165 vs. 151 mA h g−1 at 0.1 A g−1) (Fig. 5f and Fig. S42, ESI†). Meanwhile, surprisingly long-term stability for over 12000 cycles at 1.0 A g−1 with an extremely low decay rate of 0.0025% per cycle is achieved, contrasting with the lower specific capacity and faster capacity drop (from 120 to 70 mA h g−1) in pure ZClO electrolyte (Fig. 5g). Full cells were also tested under more harsh conditions of a low N/P ratio of 4:1 and a lean electrolyte of only 11 μL mA−1 h−1. A high initial capacity of 120 mA h g−1 is obtained at 2.0 A g−1, with a high-capacity retention of over 82% after 2700 cycles in the designed system (Fig. 5h). Conversely, the cell with pure ZClO electrolyte experiences a substantial capacity decline after only 1200 cycles. This high capacity, long lifetime, and high accumulated capacity obtained with the designed electrolytes are very competitive among reported papers using ZClO as electrolytes6,27,29,43–45 (Fig. 5j). The self-discharge in various electrolytes was also tested, where the designed electrolyte demonstrated higher voltage retention (1.5 V vs. 1.38 V in 3 M ZClO) after 48 hours of resting, illustrating the inhibited shuttle effect of I3− and alleviated reduction of I3− with the introduction of OAc− (Fig. S43, ESI†).
XRD analyses were further performed to study the surface evolution of the cathode (Fig. 5i). The identified peaks in the XRD spectrum correspond to ZnO and 2ZnCO3·3Zn(OH)2. Additionally, a discernible signal associated with ClO4− is observed, likely originating from residual electrolyte remnants on the cathode surface. The observed formation of a composite CEI comprising ZnO and 2ZnCO3·3Zn(OH)2 has significance in mitigating undesirable reactions at the cathode–electrolyte interface, contributing to the enhanced stability of the cathode materials. These results illustrate that the OAc− serves a dual role by not only suppressing dendrite growth and side reactions at the zinc anode but also inhibiting the dissolution and decomposition of cathode materials.
2.6 Full cell performance at low temperatures
Under hydrogen bonding (HB) reconfiguration within ZClO-based electrolytes, the Zn‖I2/AC full cell demonstrates excellent performance at low temperatures. The full cell using the designed electrolyte presents a high initial capacity of 117 mA h g−1 and sustains an ultralong lifespan of 5 months (16500 cycles) at −40 °C, marked by a significantly low decay rate of 0.0009% per cycle (Fig. 6a and Fig. S44a, ESI†). In contrast, the cell utilizing pure 3.0 M ZClO electrolyte displays a low initial capacity of 85 mA h g−1 and suffers a faster capacity decay due to the persistent side reactions (Fig. 6a and Fig. S44b, ESI†). The Zn–I2 full cell with designed electrolyte demonstrates an impressive fast-charging capability. Notably, 9 cycles of charging/discharging can be completed in 2 hours at −40 °C, attributed to the excellent ionic conductivity and fast ion diffusion of the designed system (Fig. 6b). To assess the viability of this electrolyte system under extremely low temperatures, the battery underwent testing at −60 °C (Fig. 6c). Remarkably, despite the complete freezing of the aqueous electrolyte at this temperature (Fig. S1, ESI†), the battery with I2/AC exhibited stable operation for over 3000 cycles. A noteworthy capacity of 94 mA h g−1 at 1.0 A g−1 was attained, showing its exceptional temperature adaptability. Fig. S45 (ESI†) compares the performance of Zn‖I2/AC full cells using 3.0 M Zn(ClO4)2 and 3.0 M Zn(ClO4)2 + 0.3 M Zn(OAc)2 electrolytes across various temperatures, demonstrating that the designed electrolyte operates efficiently over a wide temperature range from −60 °C to 25 °C. The Zn‖VO2 full cell was also assembled to test the applicability of the designed electrolyte. The battery demonstrates a stable long-term cycling performance with a high capacity of 160 mA h g−1 at 1.0 A g−1 at −60 °C and has cycled nearly 1000 cycles (Fig. S46, ESI†). The charge/discharge curves in Fig. S47 (ESI†) present two pairs of redox peaks at 0.56 V/0.72 V and 0.95 V/1.11 V that belong to the multistep reversible Zn2+ insertion/extraction process. These curves exhibit significant overlap, indicating a highly reversible intercalation/deintercalation process.
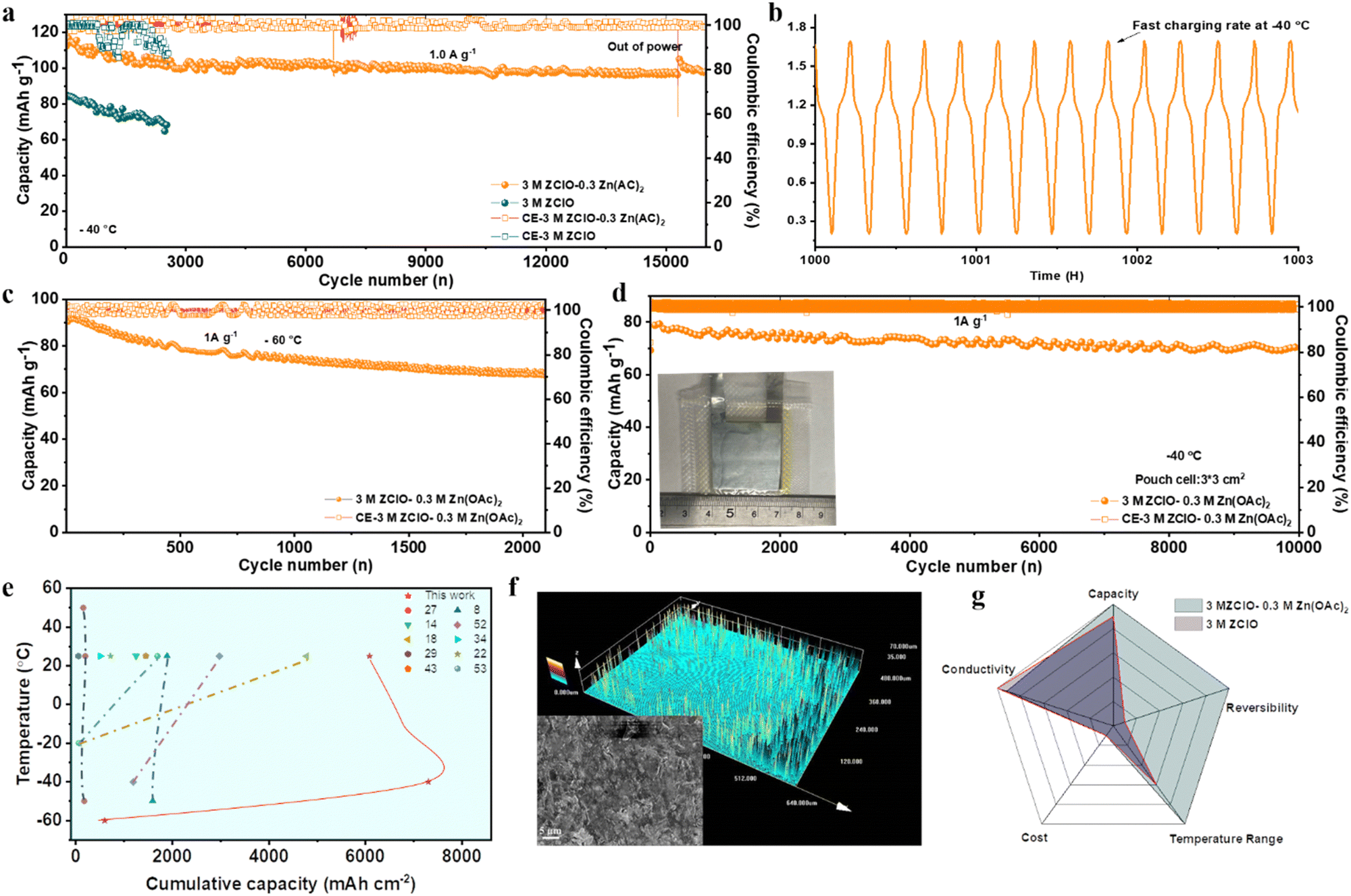 | ||
| Fig. 6 Full cell performance at low temperatures. (a) Long cycling performance of Zn‖I2/AC at 1.0 A g−1 at −40 °C. (b) The voltage-time curves from (a). (c) Long-term cycling performance of Zn‖I2/AC at 1.0 A g−1 at −60 °C. (d) Long-term cycling performance of the Zn‖I2/AC pouch cell at 1.0 A g−1 at −40 °C. (e) Comparing cumulative capacity across diverse temperatures. (f) 3D confocal microscope images and SEM images of zinc after 15000 cycles in the Zn‖I2/AC full cell. (g) Radar plots: promising properties of the designed electrolyte. | ||
Under the favorable SEI formation, the pressure-free pouch cell of I2‖AC with an electrode area of 3 × 3 cm2 also demonstrates promising performance. At a current density of 1.0 A g−1, the cell delivers a high capacity of 80 mA h g−1 at −40 °C, maintaining a high accumulative capacity of 19 A h and a high-capacity retention of 90% over an extended running life of 10000 cycles (Fig. 6d). Compared with coin cells, a bit lower capacity is obtained in pouch cells resulting from the poor contact of electrode and electrolyte. Meanwhile, the charge/discharge curves over different cycles show consistent shapes (Fig. S48, ESI†). The powder density and accumulated energy density of the Zn–I2 full cell, utilizing the designed electrolyte, are highly competitive compared to the reported electrochemical performance achieved with pouch cells17,46–51(Fig. S49, ESI†). These results underscore its considerable potential for practical applications. Even at −60 °C, three pouch cells connected in series can power an LED light (Fig. S50, ESI†). Notably, the Zn‖I2 full cells in the designed system also exhibit a rather competitive cumulative capacity across an extensive temperature range (Fig. 6e), far outperforming those of the reported aqueous ZIBs.8,14,18,22,27,29,34,43,52,53 Subsequent SEM and confocal microscope analyses were conducted on the zinc anode after 16500 cycles (Fig. 6f). The images reveal a consistently flat surface and a dense morphology of zinc deposits, contrasting with the uneven and rough surface obtained in pure ZClO electrolyte (Fig. S51, ESI†). The 2D XRD images in Fig. S52 (ESI†) also prove the (101) epitaxial electrodeposition, indicating well-regulated zinc deposition throughout the prolonged cycling of the full cell. The obtained satisfactory performance with the introduction of Zn(OAc)2 suggests that the SEI-favored electrolyte featuring excellent zinc reversibility, low price, high conductivity, and broad working temperature range, has significant promise for constructing reliable zinc-based batteries (Fig. 6g).
3. Conclusion
In summary, a dual-salt tuned electrolyte is designed by introducing a trifunctional salt of Zn(OAc)2 into 3.0 M zinc perchlorate solutions. Comprehensive theoretical simulations and experimental studies reveal that the perchlorate anions play a crucial role in destroying the original HB network among water clusters and reconstructing the HBs with water, imparting superior anti-freezing properties. The incorporation of triple functional anions of OAc− effectively modulates the low pH value of the designed system and enhances cation–anion interaction, facilitating the formation of an inorganic rich SEI layer of Zn5(CO3)2(OH)6–ZnCl2. Additionally, the OAc− inclusion assists the dense zinc deposition towards the (101) direction, suppressing zinc dendrite growth and advancing reaction kinetics. Benefiting from that, the battery using the designed electrolyte can operate efficiently over a broad temperature range between −60 °C and 25 °C. The Zn‖Cu cells exhibit an exceptional level of Coulombic efficiency, surpassing 99.5% at diverse temperatures. Zn symmetric batteries display notable stability, enduring beyond 1300 hours at −40 °C with a high Zn utilization ratio of 32.5% and over 3000 hours at room temperature. More impressively, the Zn‖I2/AC full cell employing the designed electrolyte presents a high initial capacity of 117 mA h g−1 and sustains an ultralong lifetime exceeding 16500 cycles over 5 months with a remarkably high-capacity retention of 85%. The battery with a low N/P ratio of 4:1 and a lean electrolyte of 11 μL mA−1 h−1 also manifests stable cycling of more than 2700 cycles with high-capacity retention of 82% at ambient temperature. This work provides a feasible route to achieve highly stable ZIBs under extreme conditions from the perspective of electrolyte chemistry engineering.
Data availability
All the data for this article are available in the main text and the ESI,† or upon reasonable request from the corresponding author.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Natural Sciences and Engineering Research Council of Canada (NSERC), through the Discovery Grant Program (RGPIN-2018-06725), the Discovery Accelerator Supplement Grant program (RGPAS-2018-522651), and the Alliance Grants-Mission program (ALLRP 586649-2023), and by the New Frontiers in Research Fund-Exploration program (NFRFE-2019-00488). The authors also acknowledge financial support from the Canada First Research Excellence Fund as part of the University of Alberta's Future Energy Systems research initiative (FES-T06-Q03). Prof. X. Wang acknowledges the support from the Canada Research Chair Program (CRC-2022-00059). All the authors acknowledge the support of nanoFAB in sample preparation and electron microscopy at the University of Alberta. Yimei Chen is supported by the Chinese Scholarship Council (CSC) (Grant No. 202006450027).References
- Z. Xu, et al., An Ultrafast, Durable, and High-Loading Polymer Anode for Aqueous Zinc-Ion Batteries and Supercapacitors, Adv. Mater., 2022, 34, 2200077 CrossRef CAS.
- Y. Chen, et al., Ultrathin Zincophilic Interphase Regulated Electric Double Layer Enabling Highly Stable Aqueous Zinc-Ion Batteries, Nano-Micro Lett., 2024, 16, 96 CrossRef CAS PubMed.
- T. Zhang, et al., Fundamentals and perspectives in developing zinc-ion battery electrolytes: A comprehensive review, Energy Environ. Sci., 2020, 13, 4625–4665, 10.1039/d0ee02620d.
- Z. Hu, et al., Reconstructing Hydrogen Bond Network Enables High Voltage Aqueous Zinc-Ion Supercapacitors, Angew. Chem., Int. Ed., 2023, 135, e202309601 CrossRef.
- Y. Chen, F. Gong, W. Deng, H. Zhang and X. Wang, Dual-function electrolyte additive enabling simultaneous electrode interface and coordination environment regulation for zinc-ion batteries, Energy Storage Mater., 2023, 58, 20–29 CrossRef.
- G. Yang, et al., An aqueous zinc-ion battery working at −50 °C enabled by low-concentration perchlorate-based chaotropic salt electrolyte, EcoMat, 2022, 4, e12165 CrossRef CAS.
- M. Qiu, et al., Tailoring water structure with high-tetrahedral-entropy for antifreezing electrolytes and energy storage at −80 °C, Nat. Commun., 2023, 14, 601 CrossRef CAS PubMed.
- W. Wang, et al., Regulating interfacial reaction through electrolyte chemistry enables gradient interphase for low-temperature zinc metal batteries, Nat. Commun., 2023, 14, 5443 CrossRef CAS PubMed.
- J. Xie, et al., Electrolyte and Interphase Engineering of Aqueous Batteries Beyond “Water-in-Salt” Strategy, Adv. Mater., 2024, 36, 2306508 CrossRef CAS.
- L. Suo, et al., ‘Water-in-Salt’ Electrolyte Enables High-Voltage Aqueous Lithium-Ion Chemistries, Science, 2015, 350, 938–943 CrossRef CAS PubMed.
- P. Kumar, S. V. Buldyrev and H. E. Stanley, A tetrahedral entropy for water, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 22130–22134 CrossRef CAS PubMed.
- Y. Marcus, Effect of ions on the structure of water: Structure making and breaking, Chem. Rev., 2009, 109, 1346–1370, DOI:10.1021/cr8003828.
- Q. Zhang, et al., Chaotropic Anion and Fast-Kinetics Cathode Enabling Low-Temperature Aqueous Zn Batteries, ACS Energy Lett., 2021, 6, 2704–2712 CrossRef CAS.
- T. Sun, S. Zheng, H. Du and Z. Tao, Synergistic Effect of Cation and Anion for Low-Temperature Aqueous Zinc-Ion Battery, Nano-Micro Lett., 2021, 13, 1–10 CrossRef.
- Y. Sun, et al., Salty Ice Electrolyte with Superior Ionic Conductivity Towards Low-Temperature Aqueous Zinc Ion Hybrid Capacitors, Adv. Funct. Mater., 2021, 31, 2101277 CrossRef CAS.
- R. Chen, et al., A hydrated deep eutectic electrolyte with finely-tuned solvation chemistry for high-performance zinc-ion batteries, Energy Environ. Sci., 2023, 16, 2540–2549 RSC.
- Q. Ma, et al., Regulation of Outer Solvation Shell Toward Superior Low-Temperature Aqueous Zinc-Ion Batteries, Adv. Mater., 2022, 34, 2207344 CrossRef CAS.
- W. Xu, et al., Fluoride-Rich, Organic-Inorganic Gradient Interphase Enabled by Sacrificial Solvation Shells for Reversible Zinc Metal Batteries, J. Am. Chem. Soc., 2023, 145, 22456–22465 CrossRef CAS PubMed.
- C. Li, et al., Enabling selective zinc-ion intercalation by a eutectic electrolyte for practical anodeless zinc batteries, Nat. Commun., 2023, 14, 3067 CrossRef CAS.
- F. Ming, et al., Co-Solvent Electrolyte Engineering for Stable Anode-Free Zinc Metal Batteries, J. Am. Chem. Soc., 2022, 144, 7160–7170 CrossRef CAS PubMed.
- Q. Zhang, et al., Modulating electrolyte structure for ultralow temperature aqueous zinc batteries, Nat. Commun., 2020, 11, 4463 CrossRef CAS.
- W. Deng, Z. Deng, Y. Chen, R. Feng and X. Wang, Competitive Coordination Structure Regulation in Deep Eutectic Electrolyte for Stable Zinc Batteries, Angew. Chem., Int. Ed., 2024, 63, e202316499 CrossRef CAS.
- S. Liu, et al., An Inorganic-Rich Solid Electrolyte Interphase for Advanced Lithium-Metal Batteries in Carbonate Electrolytes, Angew. Chem.,– Int. Ed., 2021, 60, 3661–3671 CrossRef CAS.
- J. Wu, et al., Electrostatic Interaction Tailored Anion-Rich Solvation Sheath Stabilizing High-Voltage Lithium Metal Batteries, Nano-Micro Lett., 2022, 14, 147 CrossRef CAS.
- D. Dong, T. Wang, Y. Sun, J. Fan and Y. C. Lu, Hydrotropic solubilization of zinc acetates for sustainable aqueous battery electrolytes, Nat. Sustainability, 2023, 6, 1474–1484 CrossRef.
- Y. Wang, et al., Lean-water hydrogel electrolyte for zinc ion batteries, Nat. Commun., 2023, 14, 3890 CrossRef CAS.
- M. Han, et al., Hydrated Eutectic Electrolyte with Ligand-Oriented Solvation Shell to Boost the Stability of Zinc Battery, Adv. Funct. Mater., 2022, 32, 2110957 CrossRef CAS.
- J. Hao, et al., Studying the Conversion Mechanism to Broaden Cathode Options in Aqueous Zinc-Ion Batteries, Angew. Chem., Int. Ed., 2021, 60, 25114–25121 CrossRef CAS PubMed.
- J. Wan, et al., Hydrated Eutectic Electrolyte Induced Bilayer Interphase for High-Performance Aqueous Zn-Ion Batteries with 100 °C Wide-Temperature Range, Adv. Mater., 2024, 36, 2310623 CrossRef CAS PubMed.
- A. V. Ghule, et al., Simultaneous thermogravimetric analysis and in situ thermo-Raman spectroscopic investigation of thermal decomposition of zinc acetate dihydrate forming zinc oxide nanoparticles, Chem. Phys. Lett., 2003, 381, 262–270 CrossRef.
- L. Zhang and Y. Chen, Electrolyte solvation structure as a stabilization mechanism for electrodes, Energy Mater., 2022, 1, 100004 CrossRef.
- C. Lin, et al., High-Rate, Large Capacity, and Long Life Dendrite-Free Zn Metal Anode Enabled by Trifunctional Electrolyte Additive with a Wide Temperature Range, Adv. Sci., 2022, 9, 2201433 CrossRef CAS PubMed.
- Z. Liu, et al., Construct Robust Epitaxial Growth of (101) Textured Zinc Metal Anode for Long Life and High Capacity in Mild Aqueous Zinc-Ion Batteries, Adv. Mater., 2024, 36, 2305988 CrossRef CAS.
- H. Jiang, et al., Chloride electrolyte enabled practical zinc metal battery with a near-unity Coulombic efficiency, Nat. Sustainability, 2023, 6, 806–815 CrossRef.
- G. Park, K. Lee, D. J. Yoo and J. W. Choi, Strategy for Stable Interface in Lithium Metal Batteries: Free Solvent Derived vs. Anion Derived, ACS Energy Lett., 2022, 7, 4274–4281 CrossRef CAS.
- N. Yao, et al., The Anionic Chemistry in Regulating the Reductive Stability of Electrolytes for Lithium Metal Batteries, Angew. Chem., Int. Ed., 2022, 134, e202210859 CrossRef.
- Z. Hou, et al., A Solid-to-Solid Metallic Conversion Electrochemistry toward 91% Zinc Utilization for Sustainable Aqueous Batteries, Sci. Adv., 2022, 8, eabp8960 CrossRef CAS.
- Z. Liu, et al., Construct Robust Epitaxial Growth of (101) Textured Zinc Metal Anode for Long Life and High Capacity in Mild Aqueous Zinc-Ion Batteries, Adv. Mater., 2024, 36, 2305988 CrossRef CAS.
- W. Zhang, et al., Dynamic Interphase-Mediated Assembly for Deep Cycling Metal Batteries, Sci. Adv., 2021, 7, eabl3752 CrossRef CAS PubMed.
- M. Wang, et al., A Multifunctional Organic Electrolyte Additive for Aqueous Zinc Ion Batteries Based on Polyaniline Cathode, Small, 2023, 19, 2302105 CrossRef CAS PubMed.
- G. Landi, et al., Impact of Acetate-Based Hydrogel Electrolyte on Electrical Performance and Stability of Eco-Friendly Supercapacitors, ChemElectroChem, 2023, 10, e202300443 CrossRef CAS.
- K. Qiu, et al., Highly Compact Zinc Metal Anode and Wide-Temperature Aqueous Electrolyte Enabled by Acetamide Additives for Deep Cycling Zn Batteries, Adv. Funct. Mater., 2024, 34, 2313358 CrossRef CAS.
- W. Yang, et al., Hydrated Eutectic Electrolytes with Ligand-Oriented Solvation Shells for Long-Cycling Zinc-Organic Batteries, Joule, 2020, 4, 1557–1574 CrossRef CAS.
- G. Ma, et al., Non-flammable, dilute, and hydrous organic electrolytes for reversible Zn batteries, Chem. Sci., 2022, 13, 11320–11329 RSC.
- Y. Zhu, et al., Concentrated dual-cation electrolyte strategy for aqueous zinc-ion batteries, Energy Environ. Sci., 2021, 14, 4463–4473 RSC.
- J. Zhou, et al., Establishing Thermal Infusion Method for Stable Zinc Metal Anodes in Aqueous Zinc-Ion Batteries, Adv. Mater., 2022, 34, 2200782 CrossRef CAS.
- W. Ling, et al., Ion Sieve Interface Assisted Zinc Anode with High Zinc Utilization and Ultralong Cycle Life for 61 Wh/kg Mild Aqueous Pouch Battery, ACS Nano, 2024, 18, 5003–5016 CrossRef CAS PubMed.
- H. Zhang, et al., Inducing the Preferential Growth of Zn (002) Plane for Long Cycle Aqueous Zn-Ion Batteries, Adv. Energy Mater., 2023, 13, 2203254 CrossRef CAS.
- H. Ge, et al., An ionically cross-linked composite hydrogel electrolyte based on natural biomacromolecules for sustainable zinc-ion batteries, Nanoscale Horiz., 2024, 9, 1514–1521 RSC.
- M. Fu, et al., Global “Interface-Space” Dual-Modulation by Functional Supramolecules Organic Frameworks on Aqueous Zinc-Ion Batteries, Adv. Funct. Mater., 2024, 34, 2311680 CrossRef CAS.
- Y. Lin, et al., Dendrite-free Zn anode enabled by anionic surfactant-induced horizontal growth for highly-stable aqueous Zn-ion pouch cells, Energy Environ. Sci., 2023, 16, 687–697 RSC.
- S. Huang, S. He, Y. Li, S. Wang and X. Hou, Hydrogen bond acceptor lined hydrogel electrolyte toward Dendrite-Free aqueous Zn ion batteries with low temperature adaptability, Chem. Eng. J., 2023, 464, 142607 CrossRef CAS.
- Y. Wang, et al., Solvent control of water O−H bonds for highly reversible zinc ion batteries, Nat. Commun., 2023, 14, 2720 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee04803b |
| This journal is © The Royal Society of Chemistry 2025 |