
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A ruthenium–titania core–shell nanocluster catalyst for efficient and durable alkaline hydrogen evolution†
Hyun Woo
Lim
 a,
Tae Kyung
Lee
bc,
Subin
Park
b,
Dwi Sakti Aldianto
Pratama
d,
Bingyi
Yan
*ae,
Sung Jong
Yoo
*b,
Chan Woo
Lee
*d and
Jin Young
Kim
*af
a,
Tae Kyung
Lee
bc,
Subin
Park
b,
Dwi Sakti Aldianto
Pratama
d,
Bingyi
Yan
*ae,
Sung Jong
Yoo
*b,
Chan Woo
Lee
*d and
Jin Young
Kim
*af
aDepartment of Materials Science and Engineering, Seoul National University, Seoul 08826, Republic of Korea. E-mail: jykim.mse@snu.ac.kr
bHydrogen Fuel Cell Research Center, Korea Institute of Science and Technology (KIST), Seoul 02792, Republic of Korea. E-mail: ysj@kist.re.kr
cDepartment of Chemical and Biological Engineering, Korea University, Seoul 02841, Republic of Korea
dDepartment of Chemistry, Kookmin University, Seoul 02707, Republic of Korea. E-mail: cwlee1@kookmin.ac.kr
eSchool of Chemistry and Chemical Engineering, Yangzhou University, Yangzhou, Jiangsu 225009, P. R. China. E-mail: bingyiyan@yzu.edu.cn
fResearch Institution of Advanced Materials (RIAM) and Institution of Engineering Research, College of Engineering, Seoul National University, Seoul 08826, Republic of Korea
First published on 22nd January 2025
Abstract
Anion-exchange-membrane water electrolysis (AEMWE) is an emerging technology for hydrogen production. While nanoparticles are used as catalysts to enhance catalytic activity, they face durability challenges due to high surface energy and reactivity. Here we present a core–shell nanocluster catalyst featuring a Ru metal core encapsulated in a porous/reduced titania monolayer, incorporating Mo atoms. This core–shell structure not only protects the unstable metal core but also lowers the energy barriers for water dissociation. The synergistic interface formed by the titania heterostructure and Mo doping modulates the electron density distribution of ruthenium active sites, fine-tuning the d-band electronic structure and optimizing the intermediate binding strengths. As a result, exceptionally low overpotentials of just 2 mV at 10 mA cm−2 and 120 mV at 500 mA cm−2 could be achieved. In a practical AEMWE system, the core–shell catalyst shows an outstanding current density of 3.35 A cm−2 under a cell voltage of 2.0 V at 60 °C, preserving its activity over 530 h of long-term electrolysis at 0.5 A cm−2.
Broader contextAnion-exchange-membrane water electrolysis (AEMWE) has emerged as a sustainable technology for hydrogen production, critical for transitioning to a low-carbon economy. However, the sluggish kinetics of the hydrogen evolution reaction (HER) in alkaline conditions, driven by the energy barriers of water dissociation, necessitates high overpotentials, posing a significant challenge for efficient hydrogen production. While nanoparticle catalysts have shown promise in enhancing catalytic activity, their long-term stability remains a concern due to agglomeration and dissolution under electrochemical conditions. To address these issues, core–shell catalysts have gained attention for improving both activity and durability, as they protect the core material while optimizing interfacial electron transfer. This work presents a Ru-based core–shell nanocluster catalyst, featuring a 1-nm Ru metal core encapsulated in a porous/reduced titania monolayer with incorporated Mo atoms. This structure stabilizes the Ru core and enhances catalytic performance by lowering the energy barrier for water dissociation and hydrogen desorption. The catalyst achieves remarkably low overpotentials of 2 mV at 10 mA cm−2 and 120 mV at 500 mA cm−2, demonstrating excellent durability in AEMWE systems with a current density of 3.35 A cm−2 at 2.0 V and 60 °C, while maintaining stability for over 530 h. |
1. Introduction
H2 is an energy carrier that can help replace fossil fuels and facilitate the transition to a low-carbon economy.1–3 H2 can be feasibly produced via water electrolysis powered by intermittent renewable energy.4 Anion-exchange-membrane water electrolysis (AEMWE) is drawing attention as a next-generation hydrogen production technology due to its low-cost components, zero-gap configuration, and applicability for differential pressure operation.5,6 However, the sluggish kinetics of the hydrogen evolution reaction (HER) under alkaline conditions—owing to the energy barrier of water dissociation—necessitates the use of high overpotentials.7 Additionally, at high current densities, the accumulation of hydrogen gas bubbles can block water transport, resulting in significant mass transport overpotential.8 Furthermore, metal-based HER catalysts, including Fe, Co, and Ni, as well as their hydroxide forms, can dissolve in alkaline electrolytes, leading to the degradation of catalytic performance.9,10 The delicate design and development of alkaline HER catalysts are essential to improving the energy efficiency and operational durability of AEMWE.Nanoparticles (NPs) have been utilized as attractive electrocatalyst materials for enhancing catalytic activity, owing to their high surface area, improved electrical conductivity, and remarkable ability to actively promote reactions through interactions with support and reactants.11–13 In the Pt/C catalyst, ∼2 nm-sized Pt NPs are well dispersed onto a conducting carbon black support, resulting in a 24-times-higher mass activity compared to bulk Pt of the same amount of noble metal at −0.05 V vs. RHE.14 The mass activity also highly depends on the Pt size. The Pt/C exhibits 48-times-higher mass activity when particle size is reduced from 1.85 to 1.30 nm.15 Similarly, the turnover frequency (TOF) of Ru NPs also increases from 4.8 × 10−4 to 0.005 s−1 as the NP size decreases from 7.6 to 2.5 nm.16 However, the decrease in particle size to the nanometer level compromises durability.17,18 Smaller NPs have higher surface energy, contributing to instability and promoting agglomeration or Ostwald ripening.19,20 In electrochemical environments, low-coordination sites are more susceptible to dissolution, causing mass loss, and weak nanoparticle-support interactions increase catalyst detachment.21
Core–shell catalysts are gaining attention for addressing these issues, as their ability to protect the core with a thin layer of corrosion-resistant material leads to stable HER activity.22–24 Metal NPs enclosed by a graphene shell maintain their HER overpotentials over a long duration, in contrast to pristine NPs.23,24 Moreover, the core–shell structure offers an interfacial effect that further optimizes hydrogen binding strength and enhances HER activity through electron transfer between the core and shell atoms.25 Dimensional and morphological control of core–shell catalysts is crucial for tuning catalytic performances.26,27 Typically, reducing the shell thickness to a monolayer level maximizes charge polarization at the shell surface, leading to improved catalytic activity.28,29 Furthermore, controlling the shell coverage can improve HER activity by exposing the interfacial active sites.25,28 Nanostructural properties such as core size, core–shell materials, and interfacial compatibility are also vital for catalytic performance. Advanced synthesis and design methods that finely tailor core–shell catalysts can be an effective strategy for achieving both high catalytic activity and durability.
Here we report a core–shell nanocluster (NC) catalyst featuring a 1-nm-sized Ru metal core surrounded by a porous/reduced titania monolayer with dispersed Mo atoms between them. The core–shell structure not only protects the unstable Ru core but also provides synergistic interfaces, thereby enhancing catalytic activity and durability simultaneously (Fig. 1a). For the synthesis (Fig. 1b), RuO2 NCs are hydrothermally deposited onto the Mo-doped titania surface of 3-D Ti electrodes. Through low-temperature post-annealing at 200 °C, atomic diffusion from the Mo-doped titania surface to the RuO2 NC forms a NC comprising a RuO2 core and a Mo-doped titania shell. The thickness of the shell layer can be precisely controlled by adjusting the annealing time. Subsequent electrochemical reduction reconstructs the core–shell NC into a Ru metal core surrounded by a porous and reduced titania monolayer with reduced Mo atoms between them. DFT calculation indicates that the synergistic interface formed by titania heterostructure, and Mo doping induces electron-deficient and electron-accumulated Ru sites near the interface. These unique interfacial sites facilitate both water dissociation and hydrogen desorption, thereby promoting H2 generation under alkaline conditions (Fig. 1c). The final core–shell Ru catalyst shows extremely low overpotentials of 2 mV at 10 mA cm−2 and 120 mV at 500 mA cm−2, outperforming most benchmark catalysts (Fig. 1d).30–32 Furthermore, in AEMWE application, the core–shell Ru catalyst exhibits one of the highest current densities of 3.35 A cm−2 at a cell voltage of 2.0 V at 60 °C, with negligible catalyst degradation over 530 h of long-term electrolysis at 500 mA cm−2. The core–shell NC catalyst addresses critical challenges associated with Ru-based catalysts, including corrosion, difficult hydrogen desorption, high energy barriers for H2O dissociation, and high material costs, thereby demonstrating significant potential for practical applications in the alkaline HER industry.
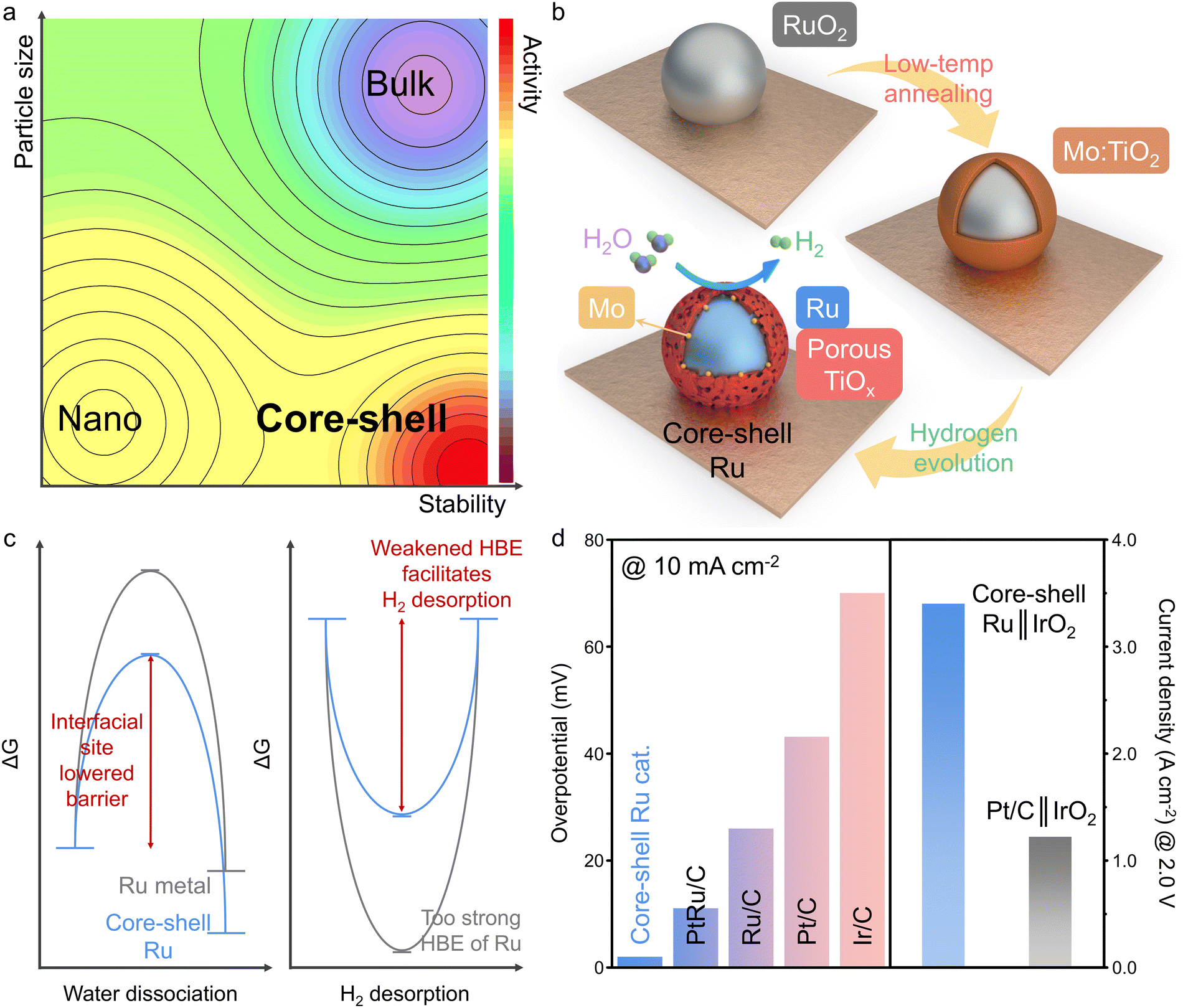 | ||
| Fig. 1 Core–shell Ru catalyst's synthesis strategy, computational results, and catalytic performances. (a) A contour map displaying the activity, stability, and particle size for bulk, nano, and core–shell catalysts. (b) Schematic illustration of the structural transformation of a core–shell Ru catalyst induced by low-temperature post-annealing and electrochemical reduction processes. (c) Schematic illustration of the calculated free energy diagram for water dissociation and hydrogen desorption on a core–shell Ru catalyst. (d) Performance comparison of core–shell Ru and benchmark catalysts in terms of HER overpotential and AEMWE current density. | ||
2. Results and discussion
2.1 Electrocatalytic activity
The HER activities of the core–shell Ru (CS-Ru) catalysts, with varying shell thickness achieved by altering the post-annealing time from 0 to 60 min (denoted as CS-Ru-0, 20, 40, and 60), were evaluated using linear sweep voltammetry (LSV) at a scan rate of 10 mV s−1 in 1.0 M KOH (Fig. 2a and Fig. S1 and S2, ESI†). The formation of the shell results from the diffusion of Ti and Mo from the anatase titania support. A monolayer shell with a thickness of 0.45 ± 0.02 nm is formed after 40 minutes of annealing, while no shell is observed in samples annealed for 0 or 20 minutes (Fig. S3 and Table S1, ESI†). After 60 min of annealing, the shell thickness increased to 1.38 ± 0.07 nm, forming a triple-layer titania structure through controlled diffusion. The LSV scan was performed 50 times for each catalyst, and the final LSV curves are plotted in Fig. 2a. CS-Ru-40 and CS-Ru-60 exhibit superior electrocatalytic activity, with overpotentials of 2 and 14 mV at 10 mA cm−2, respectively, whereas CS-Ru-0 (87 mV) and CS-Ru-20 (22 mV) require higher overpotentials. Online gas chromatography measurements further revealed that CS-Ru-40 achieves a high faradaic efficiency exceeding 96.7% for H2 production at a current density of 10 mA cm−2 and an overpotential of 1.8 mV during chronopotentiometric electrolysis (Fig. S4, ESI†). These results indicate that the controlled diffusion of Ti and Mo through low-temperature annealing for an optimized duration (40 min) achieves the desired shell thickness (a monolayer shell in this study) and significantly improves the alkaline HER activity. The best sample (CS-Ru-40) shows low overpotentials at high current densities, too (51 and 124 mV at 100 and 500 mA cm−2, respectively).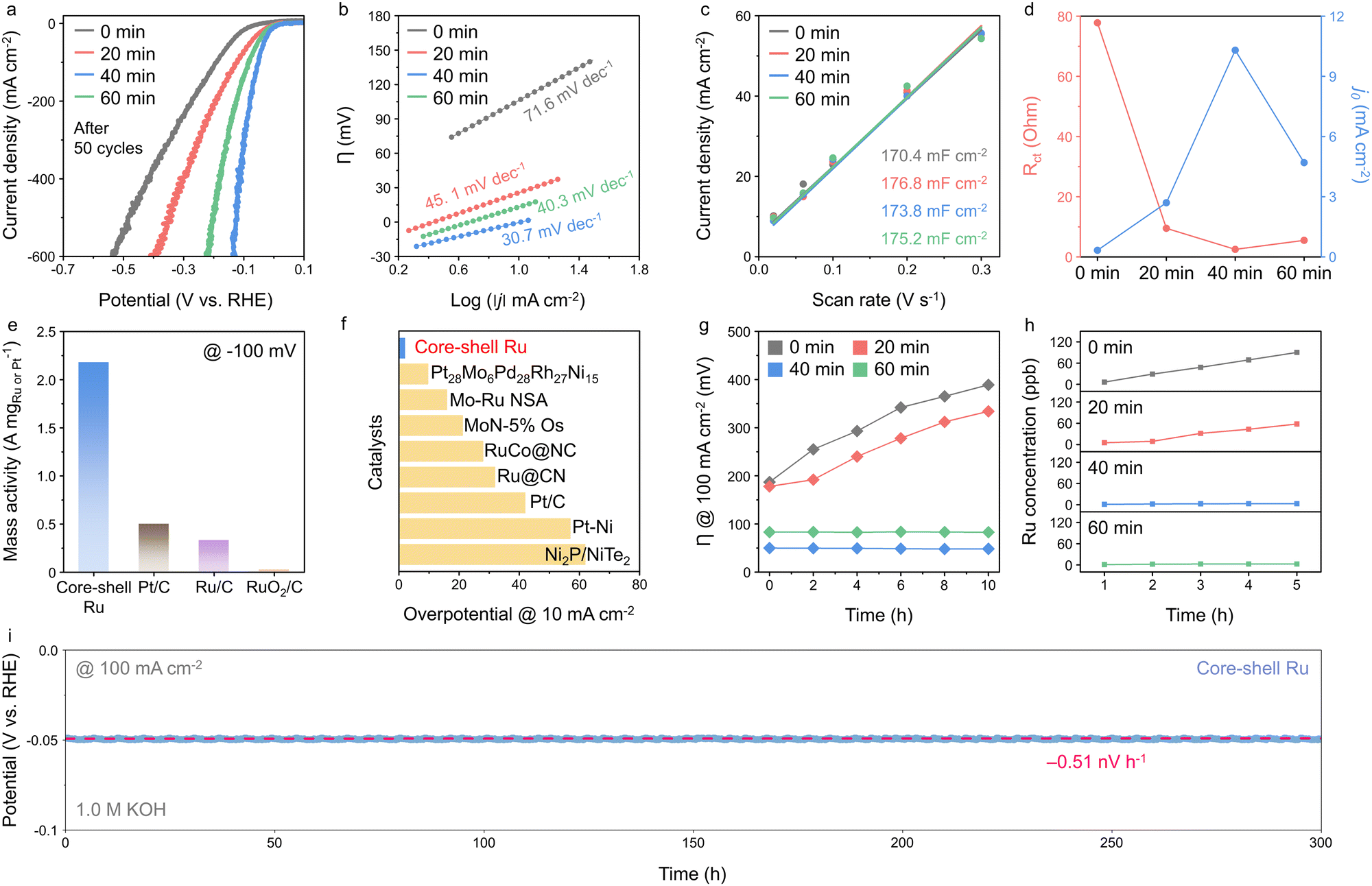 | ||
| Fig. 2 Electrocatalytic activities for HER. (a) LSV curves of core–shell Ru catalysts measured at a scan rate of 10 mV s−1 in 1.0 M KOH electrolyte. (b) Tafel plots, (c) double-layer capacitances, (d) charge transfer resistances, and exchange current densities of core–shell Ru catalysts. (e) Mass activities of core–shell Ru, Pt/C, Ru/C, and RuO2/C calculated at the overpotential of −100 mV. (f) Comparison of HER performance with that of previously reported catalysts. (g) The overpotential values of core–shell Ru catalysts recorded at a current density of 100 mA cm−2 for an initial 10 h. (h) Concentrations of Ru dissolved in electrolyte obtained by ICP-MS during chronopotentiometry at 100 mA cm−2 for 5 h. (i) Long-term stability test for the core–shell Ru at a constant current density of 100 mA cm−2. | ||
Tafel analysis was conducted to clarify the mechanism governing the HER activity of the CS-Ru catalysts (Fig. 2b). CS-Ru-40 exhibits a significantly low Tafel slope (30.7 mV dec−1). The similarity of this Tafel slope to the theoretical value (30 mV dec−1) suggests the occurrence of the Volmer–Tafel mechanism, in which the rate-determining step (RDS) for the HER is the H–H coupling between adsorbed hydrogen intermediates (H*).33 This indicates that CS-Ru-40 effectively ruptures H2O bonds, given that H* is typically produced via the dissociation of H2O bonds under alkaline conditions. In contrast, the Tafel slopes of CS-Ru-0, CS-Ru-20, and CS-Ru-60 (71.6, 45.1, and 40.3 mV dec−1, respectively) suggest that high overpotentials are required to increase the current density by an order of magnitude. These Tafel slope values indicate that the RDS involves water dissociation during the Heyrovsky step,33 as opposed to CS-Ru-40. The double-layer capacitances (Cdl; Fig. 2c and Fig. S5, ESI†) of CS-Ru catalyst electrodes exhibit relatively consistent values (170.4, 176.8, 175.2, and 173.8 mF cm−2). The ECSA of the substrate prepared without nanoclusters was measured at 164.3 mF cm−2, accounting for approximately 94% of the total ECSA (Fig. S5, ESI†). Given the substantial contribution of the substrate to the overall ECSA, the post-annealing time has a negligible impact on the ECSA. The charge-transfer kinetics of CS-Ru catalysts in HER was also investigated through measuring the charge-transfer resistance by electrochemical impedance spectroscopy (EIS).34 Nyquist plots were obtained at 0 V. RHE, which is the equilibrium potential of the HER in 1.0 M KOH at 25 °C (Fig. S6, ESI†). Among the CS-Ru catalysts, CS-Ru-40 exhibits the lowest Rct value (2.5 Ω) and the fastest charge-transfer kinetics, as evidenced by its highest exchange current density (j0 = 10.3 mA cm−2; estimated using the equation Rct = RT/Fj0, where R represents the gas constant and F is the Faraday constant; Fig. 2d).
As a result, the CS-Ru-40 catalyst significantly outperforms the commercial Pt/C (0.2 mgPt cm−2), Ru/C (0.2 mgRu cm−2), and RuO2/C (0.2 mgRu cm−2) catalysts by 41, 24, and 177 mV at 10 mA cm−2, and by 49, 79, and 277 mV at 100 mA cm−2, respectively (Fig. S7, ESI†). Furthermore, the high mass activity of 2.18 A mg−1, calculated based on the small amount of Ru (0.16 mgRu cm−2) obtained by inductively coupled plasma-mass spectroscopy (ICP-MS; Fig. 2e and Table S2, ESI†), is 4.4, 6.6 and 109-times-higher than those of commercial Pt/C (0.5 A mg−1), Ru/C (0.33 A mg−1) and RuO2/C (0.02 A mg−1), respectively (Fig. S8, ESI†). Notably, the HER activity of CS-Ru-40 featured by low overpotentials of 2, 97, and 120 mV at 10, 300, and 500 mA cm−2, respectively, is the best among the reported HER catalysts under alkaline conditions (Fig. 2f and Table S3, ESI†).
Fig. 2g compares the stability of the CS-Ru catalyst under continuous HER operation at 100 mA cm−2. CS-Ru-40 and CS-Ru-60 maintain their overpotentials for an initial 10 h (50 to 48.4 mV and 83.1 to 82.9 mV from 0 to 10 h, respectively), while the activity of CS-Ru-0 and CS-Ru-20 degrades continuously. Moreover, ICP-MS analysis of the electrolyte after chronopotentiometry at 100 mA cm−2 for 5 h shows that the amount of dissolved Ru for the CS-Ru catalysts aligns with the overpotential trend (90.1, 58.1, 2.8, and 2.7 ppb for CS-Ru-0, 20, 40, and 60 after 5 h, respectively; Fig. 2h). This suggests that catalyst deactivation occurs due to the detachment and dissolution of the Ru-based active sites during vigorous HER when the specific shell thickness is not achieved. The long-term stability of CS-Ru-40 is also confirmed by chronopotentiometry at 100 mA cm−2 over a period of 300 h, showing negligible degradation (Fig. 2i). These results indicate that the protective titania shell formed by controlled Ti diffusion around the CS-Ru catalysts alleviates Ru corrosion and enhances catalyst stability.
2.2 Materials characterization
The Ti foam electrode coated with CS-Ru-40 NCs was analyzed before and after the electrochemical reaction (i.e., 50 HER cycles) using high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) in Fig. 3. The results reveal the formation of a 40-nm-thick mixed-metal oxide layer consisting of Ru, Ti, and Mo atoms on the Ti foam electrode (Fig. S9, ESI†). High-resolution HAADF-STEM analysis of the oxide layer shows the distribution of bright core–shell nanoclusters (particle size: 1.9 ± 0.35 nm) on a dark Mo-doped titania support (Fig. 3a and Fig. S10a and S11, ESI†), where bright and dark regions correspond to the (110) plane of rutile RuO2 (JCPDS no. 40-1290) and (101) plane of anatase TiO2 (JCPDS no. 21-1272), respectively. The selected area electron diffraction (SAED) pattern also displays distinct diffraction spots assigned to the rutile and anatase crystal structures (Fig. S10b, ESI†). | ||
| Fig. 3 Characterization of core–shell Ru catalysts. (a) High-resolution HAADF-STEM image of the core–shell Ru NC. (b) Line-scan profile of the core–shell Ru NC. Characterization data in (a) and (b) are measured before the electrochemical reaction. (c) XPS core-level spectra of pre-HER and post-HER samples in Ti 2p and Ru 3p regions. (d) HR-TEM image of the core–shell Ru catalyst measured after 50 LSV scans over a potential range of 0.1 to −1.7 V vs. RHE in 1.0 M KOH (pH = 14). (e) HAADF-STEM image of a post-HER sample. (f) EELS spectrum of the post-HER sample. EELS is measured at NC, indicated by the orange box. Red, blue, and green squares correspond to where the spectra were obtained. (g) Five line-scan profiles obtained from a spherical NC of the post-HER sample to visualize the 3-D elemental distribution. Data in the figures were obtained from the core–shell Ru catalyst post-annealed for 40 min. | ||
Line-scan profile of CS-Ru-40 (Fig. 3b and Fig. S12, ESI†) shows the formation of a core–shell structure, featuring a RuO2 core surrounded by a Mo-doped TiO2 monolayer shell, with Mo atoms predominantly distributed at the interface between the core and the shell. With increasing post-annealing time, the concentration of Ti and Mo in the core increases (Fig. S3 and Fig. S13 and Table S1, ESI†), while the atomic percentages in the anatase titania support remain largely unchanged (Fig. S14, ESI†). X-ray photoelectron spectroscopy (XPS) spectra of the core-level Ti 2p and Ru 3p (Fig. S15, ESI†) show the Ti 2p3/2 peak (Ti4+ at 458.0 eV) and Ru 3p3/2 peak (Ru4+ at 462.7 eV), with an increased Ti4+ peak observed as the post-annealing time increases.35,36 This increased Ti atoms thicken the shell, forming a triple-layer (1.38 nm) titania shell after 60 min as shown in Fig. S3f (ESI†). Overall, these findings confirm the formation of a NC composed of a RuO2 core and a Mo-doped titania shell, facilitated by atomic diffusion from the Mo-doped titania support surface to the RuO2 core.
The XPS analysis reveals that the RuO2 core NCs are significantly reduced to metallic Ru during the electrochemical reduction (Fig. 3c), with the intensity of the Ru0 peak at 461.1 eV increasing after 50 HER cycles. The Ru4+ peak at 462.7 eV of the pre-HER sample decreases at the same time. Interestingly, the Ti4+ peak of the pre-HER sample also decreases and shifts towards lower binding energy after HER, indicating partial dissolution and reduction of Ti4+ ions to form a porous TiOx shell. The reduced core–shell Ru NC catalyst (i.e., CS-Ru-40) was further analyzed by high-resolution transmission electron microscopy (HR-TEM; Fig. 3d), where metallic Ru NCs anchored onto the anatase TiO2 support, as evidenced by the d-spacings assigned to the (100), (101), and (002) planes of hexagonal Ru metal and to the (101) plane of anatase TiO2, were observed. SAED also confirms clear diffraction patterns corresponding to Ru metal and anatase TiO2 (Fig. S16, ESI†). It is noticeable that the Mo 3d XPS profile also shows an increase in the Mo0 peak, implying the reduction of Mo at the surface (Fig. S17, ESI†).37 High-resolution HAADF-STEM analysis is performed to acquire z-contrast images (Fig. 3e and Fig. S18, ESI†). Given that the Ru (z = 44) region is supposed to be brighter than Ti (z = 22), the different brightness can be leveraged to identify the atomic positions of Ru and Ti on the surface of the NCs. Atoms with different brightness are randomly detected from the surface of CS-Ru-40 NC like the single-atom catalysts. These results indicate that the active surface of CS-Ru-40 is atomically heterogeneous, which can be ascribed to the formation of the porous TiOx monolayer shell during HER. This porous feature was examined by electron energy-loss spectroscopy (EELS) spectrum obtained at different locations on a single CS-Ru-40 NC (Fig. 3f). The Ti L-edge signals are partially observed from certain areas of the NC surface, and their intensities are also different depending on the location, corroborating the existence of a porous titania shell.38
The cross-sectional structure of a CS-Ru-40 NC was reconstructed using EDS line-scan profiles of metal elements measured from five different vertical positions (Fig. 3g and Fig. S19, ESI†). It is clearly seen that the core–shell structure consisting of a Ru-based core, Ti-based shell, and interfacial Mo atoms is preserved after HER. However, the intensity ratio of Ti to Ru in the core region decreases after HER, strongly supporting the formation of the porous titania shell, especially considering the same shell thickness before and after HER (0.45 and 0.48 nm, respectively). Furthermore, the presence of the sub-nanometer-sized pores (0.55 nm) was confirmed from the pore-size distribution analysis using the gas adsorption method (Fig. S20, ESI†). The 3-dimensional structure of CS-Ru-40 NC was further investigated using HAADF-STEM imaging and STEM-EDS mapping (Fig. S21, ESI†). The Ru signal is mainly concentrated at the core of the NC (orange rectangle), whereas the Ti and O signals are detected over the entire area of the NC indicating that the Ru core is surrounded by a TiOx shell. The atomic percentages of Ti, Ru, and Mo measured using EDS are 31.5, 64.2, and 4.3 at%, respectively, showing that the Ti-to-Ru ratio decreases from 0.70 to 0.50 after HER. This result supports the partial dissolution of the TiO2 shell and the resulting formation of a porous shell layer, which is consistent with the structural reconstruction.
2.3 Mechanistic understanding
Density functional theory (DFT) calculations were conducted to elucidate the mechanism of the alkaline HER at the catalytically active sites, for which five atomic models of pristine Ru, Ru with a compact TiO2 monolayer (Ru/compact TiO2), Ru with a porous TiO2 monolayer (Ru/porous TiO2), Ru with a porous and reduced TiOx monolayer (Ru/porous TiOx), and Mo-doped Ru with a porous and reduced TiOx monolayer (Mo:Ru/porous TiOx) were constructed (Fig. 4a and Fig. S22, ESI†). Fig. 4b compares the changes in Gibbs free energy during the water dissociation step that is considered as the RDS for HER in alkaline media. The activation energy barriers for the dissociation of H2O into OH* and H* are calculated to be 0.93, 1.02, 0.85, 0.81, and 0.68 eV for pristine Ru, Ru/compact TiO2, Ru/porous TiO2, Ru/porous TiOx, and Mo:Ru/porous TiOx, respectively. The Ru with a compact TiO2 layer shows a higher activation barrier for water dissociation compared with pristine Ru, whereas the Ru surface partially covered with a TiO2 or TiOx monolayer (i.e., porous shell layers) shows lower activation energy values than bare Ru. This suggests that water dissociation is highly facilitated at the open interfacial sites. In addition, the presence of Mo near the active interfacial sites further enhances HER activity by reducing the energy barrier for water dissociation and promoting the supply of hydrogen. The hydrogen binding energy, which is another important activity descriptor for acid HER, was compared in Fig. S23 (ESI†). Pristine Ru metal exhibits the lowest binding energy (−0.95 eV), and thus, the strongest H* adsorption, which is likely to limit HER activity due to the inefficient desorption of H2. The binding energy increases in the presence of a compact TiO2 shell layer (−0.84 eV), and further increases if the shell becomes porous (−0.68 eV) and TiO2 is reduced to TiOx (−0.66 eV). Finally, the binding energy increases to as high as −0.58 eV when a Ru atom near the interface is replaced with a Mo atom, facilitating the turnover of H* to H2 and eventually promoting HER activity. As a result, the synergistic effect, originating from the interface between Ru metal and the reduced titania monolayer, along with the substitution of Ru with Mo, is believed to maximize the performance of alkaline HER.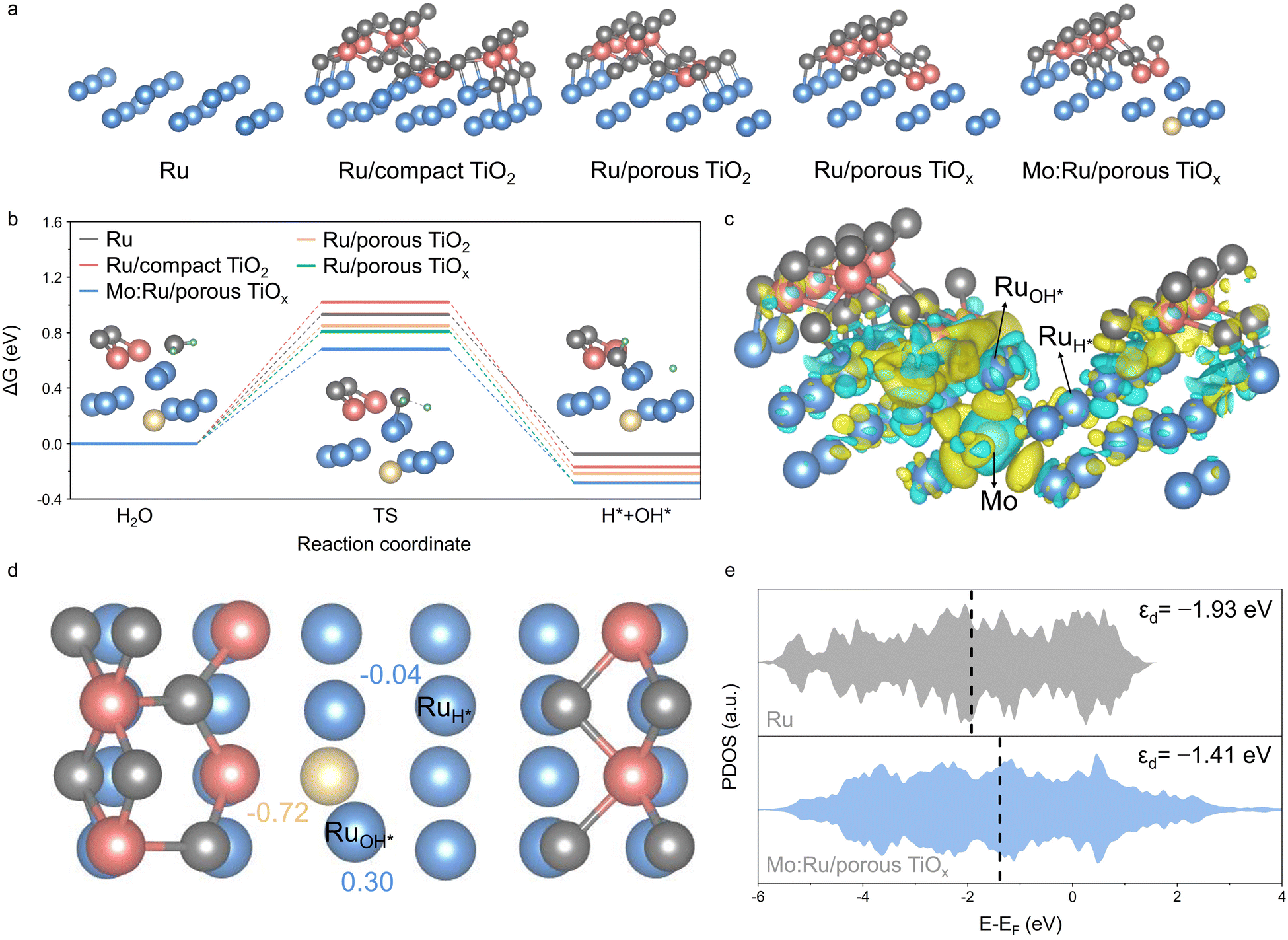 | ||
| Fig. 4 Density functional theory calculations for mechanistic understanding. (a) Five optimized atomic structures used for the DFT calculation. (b) Calculated free energy diagrams of water dissociation on pristine Ru, Ru/compact TiO2, Ru/porous TiO2 or TiOx, and Mo:Ru/porous TiOx surfaces, where the progression of water dissociation in the Mo:Ru/porous TiOx atomic model is illustrated as insets. (c) An atomic model of Mo:Ru/porous TiOx and its charge density distribution. Yellow and cyan regions represent electron-rich and electron-deficient areas, respectively. (d) The calculated Bader charge of Ru and Mo atoms at the interfacial site. The RuOH* and RuH* indicate the atoms where OH* and H* intermediates are attached after water dissociation. (e) Partial density of state (PDOS) and d-band center of Ru atoms interacting with H2O and OH* in pristine Ru and Mo:Ru/porous TiOx. | ||
The localized charge distribution of Mo:Ru/porous TiOx was simulated to comprehensively explore the effects of the interface and elemental substitution (Fig. 4c), and the Bader charge was calculated for the quantitative characterization (Fig. 4d and Fig. S24, ESI†). A relatively electron-deficient Ru atom with a positive Bader charge of 0.3|e| is believed to attract a water molecule, polarize it and weaken its O−H bonds, which facilitates water dissociation and boosts the H* formation rate.39–41 In addition, the electron deficiency causes the d-band center of Ru atoms interacting with OH* in Mo:Ru/porous TiOx to shift closer to the Fermi level compared to that of Ru metal (Fig. 4e). This upward shift enhances adsorption strengths of both H2O and OH*.42,43 The H* escapes from the Ru atom where water dissociation occurs and is attracted to another Ru atom with a partially accumulated electron density and a negative Bader charge of −0.04|e|, where the H2 production step occurs. The accumulated electrons could contribute to a greater filling of the antibonding orbital of Ru−H*, leading to a reduction in the bonding strength between H* and the Ru atom,44 and eventually facilitating the desorption of H* from the Ru atom. The charge redistribution analysis is in good agreement with differences in free energy discussed in association with Fig. 4b, signifying the favorable adsorption/desorption behavior of the intermediates on the Mo:Ru/porous TiOx surface (i.e., CS-Ru-40 sample). These calculation results also align with the observed catalytic activities, indicating that incorporation of Mo significantly reduces the overpotentials—by more than 60-fold—achieving an ideal overpotential of 2 mV at 10 mA cm−2 in the CS-Ru-40 catalyst. This also provides a rational strategy to optimize HER catalysis via controlling the adsorption/desorption behavior of catalysts.
2.4 Anion-exchange-membrane water electrolysis
To test the practical applicability in AEMWE, a membrane electrode assembly (MEA) was constructed using a CS-Ru-40 cathode, IrO2 anode, and a PiperION membrane (Fig. 5a). For preparing the cathode, CS-Ru-40 catalyst was deposited onto a 1.5-mm-thick Ti felt with a porosity of 70%. The current–voltage characteristics was subsequently evaluated in a flowing 1.0 M KOH electrolyte at 60 °C (Fig. 5b). The MEA of CS-Ru-40||IrO2 exhibits a high current density of 3.35 A cm−2 at a cell voltage of 2.0 V, outperforming the benchmark Pt/C||IrO2 system showing a lower current density of 1.22 A cm−2 at the same cell voltage. Notably, CS-Ru-40||IrO2 exhibits record-high performance compared to the previous reports using the catalyst-coated substrate (CCS) method in the AEMWE system (Fig. 5c and Table S4, ESI†), which can be ascribed to the record-high HER activity of the core–shell Ru NC catalyst (Fig. 2f). A voltage breakdown analysis was performed at a current density of 1 A cm−2 to clarify the contribution of each cell component to the overpotential (Fig. 5d).45 The overpotential associated with the anode kinetics exerts the most significant influence on the overpotentials for both MEAs. Regarding cathode kinetics, the CS-Ru-40 cathode demonstrates a substantial reduction in overpotential compared to Pt/C, accounting for the exceptional HER activity of CS-Ru-40. Ohmic resistance during the AEMWE is low enough (0.06 and 0.08 Ω for the CS-Ru-40 and Pt/C cathodes, respectively; Fig. S25, ESI†), so its influence on the AEM performance can be neglected. | ||
| Fig. 5 Performances of core–shell Ru in an AEM electrolyzer. (a) Schematic illustration of an AEM electrolyzer showing key components and their arrangement. (b) Performance evaluation of MEAs utilizing core–shell Ru||IrO2 and Pt/C||IrO2 as cathode||anode in 1.0 M KOH at 60 °C. (c) A comparison of activities of the AEMWE single cells. (d) Voltage breakdown analysis for AEM electrolyzers using core–shell Ru and Pt/C as cathodes. (e) Durability test of an AEM electrolzyer catalyzed by core–shell Ru at 0.5 A cm−2 over 530 h at 60 °C. (f) LSV curves of core–shell Ru measured before and after the AEMWE durability test. Core–shell Ru in this figure corresponds to the catalyst post-annealed for 40 min. | ||
The durability of the AEM electrolyzer with the CS-Ru-40 cathode was examined at a current density of 0.5 A cm−2 and a temperature of 60 °C (Fig. 5e). The initial cell voltage (1.65 V) slightly increases to 1.86 V at the beginning of the operation and remains stable until 430 h with a low degradation rate of 0.18 mV h−1 after being stabilized. The initial increase in the cell voltage can be attributed to the membrane stabilization,46,47 whereas the slow increase over time can be ascribed to the corrosion of IrO2 anode. The image of the IrO2 electrode after durability test shows empty spots and the ICP-MS analysis also confirms that the Ir content decreases from 2 to 0.4 mg cm−2 (Fig. S26, ESI†). The abrupt increase in the cell voltage at approximately 430 h is not associated with the degradation of the HER cathode, but results from the degradation of the membrane, because the cell voltage immediately recovers after replacing the membrane and IrO2 anode. The cell voltage remains stable for an additional 100 h, demonstrating the high stability of the CS-Ru-40 catalyst (over 530 h, at 0.5 A cm−2). Notably, the unique core–shell structure of CS-Ru-40 catalyst was preserved even after the durability test (Fig. S27, ESI†). Furthermore, the HER overpotential of the CS-Ru-40 cathode exhibits almost similar overpotentials before and after the AEMWE durability test over 530 h (before: 2, 65, and 92 mV and after: 2, 69, and 100 mV, at 10, 500, and 1000 mA cm−2, respectively; Fig. 5f), assuring the excellent stability of the core–shell Ru NC catalyst. These findings confirm that CS-Ru-40 as a cathode catalyst of an AEM electrolyzer exhibits high enough activity and stability to be applied to commercial alkaline water electrolysis systems.
3. Conclusions
A core–shell nanocluster catalyst, consisting of a Ru metal core and a porous/reduced titania shell with dispersed Mo atoms in between, was prepared via a hydrothermal process followed by post-annealing and subsequent electrochemical reduction. The best sample featured by a porous and reduced titania monolayer shell with a thickness of ∼0.48 nm (CS-Ru-40) exhibited an almost zero overpotential of 2 mV at 10 mA cm−2 and a low Tafel slope of 30.7 mV dec−1 toward the HER in an alkaline electrolyte, which didn’t increase much even at high current densities (e.g., 51 and 120 mV at 100 and 500 mA cm−2, respectively). These exceptional activities are presumably induced by the unique interfacial sites of the core–shell nanocluster and the electronic modification caused by Mo substitution, leading to the remarkably lowered activation barrier for water dissociation and the energetic barrier for H2 desorption. Additionally, CS-Ru-40 demonstrated high durability, maintaining its low overpotential for 300 h at 100 mA cm−2, owing to the protection imparted by the titania shell. Furthermore, upon being integrated into a practical AEMWE device, CS-Ru-40 delivered one of the highest current densities of 3.35 A cm−2 at 2.0 V (60 °C in 1.0 M KOH). It also showed good stability by maintaining the cell voltage at 0.5 A cm−2 over 530 h at 60 °C with a low degradation rate of 0.22 mV h−1. Overall, the superior activity and durability suggest that the unique core–shell-structured nanocluster is a promising catalyst for practically feasible AEMWE applications.4. Experimental section
4.1 Synthesis of core–shell Ru catalysts
Core–shell Ru catalysts are synthesized through oxidation, hydrothermal method, and low-temperature thermal annealing in air. Initially, the oxidation process occurs by immersing the Ti foam (MTI-KOREA, EQ-TiF-1106, porosity: 40%, thickness: 0.6 mm) into H2O2 solution (Aldrich, 50 wt%) at 70 °C for 30 min. Subsequently, Mo-doping of Ti foam is carried out through a hydrothermal method at 180 °C for 12 h (at a heating rate of 10 °C min−1), utilizing a solution of 0.5 mmol MoCl5 (Aldrich, 99.99%) and 0.5 mmol TiCl4 (Aldrich, >99.0%). After overnight drying in a vacuum oven at 70 °C, the deposition of Ru-based NCs is carried out hydrothermally using a 1 mmol RuCl3·xH2O (Aldrich, 99.98%) solution at 150 °C for 10 h. All solutions are prepared using deionized water generated by Human Corporation (Model Hyman RO 180), and the volume of each solution is fixed at 50 mL. Finally, the synthesized catalysts are annealed at a low temperature of 200 °C, with varying annealing times of 0, 20, 40, and 60 min. Each sample is denoted as CS-Ru-0, 20, 40, and 60. CS-Ru-40, intended for application in practical AEMWE, is synthesized using the same manufacturing process with Ti felt (SINOPRO, porosity: 70%, thickness: 1.5 mm). To prepare the RuO2/C electrode, 1 mg of commercial RuO2, 4 mg of Ketjen black powder, and 40 μL of Nafion solution were mixed in 460 μL of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of water and ethanol. After sonicating for 30 min, 100 μL of the solution was dropped onto a carbon cloth and dried in air overnight. A Ru/C electrode was manufactured using the same process, except for replacing commercial RuO2 with commercial Ru. The Ru mass of both electrodes was measured to be 0.2 mg cm−2.
:1 mixture of water and ethanol. After sonicating for 30 min, 100 μL of the solution was dropped onto a carbon cloth and dried in air overnight. A Ru/C electrode was manufactured using the same process, except for replacing commercial RuO2 with commercial Ru. The Ru mass of both electrodes was measured to be 0.2 mg cm−2.
4.2 Materials characterizations
Morphological analyses (both in wide and magnified views), SAED pattern, elemental mapping, line-scan profiles, and EELS are conducted using a spherical aberration (Cs)-corrected monochromated TEM (Themis Z, Thermo Fisher) at an acceleration voltage of 200 kV. The crystal orientation and lattice distance of the structure were evaluated by X-ray diffraction (XRD) (Model D8 Advance.2020, Bruker) using Cu Kα radiation (λ = 0.1542 nm). The chemical bonding state of the elements within the sample is analyzed using XPS (Versaprobe III, UL-PHI). The binding energy of the XPS spectrum is calibrated to the C 1s peak at 284.6 eV. The quantity of each material in sample and its dissolution in the electrolyte during the reaction are measured using ICP-MS (NexION, PerkinElmer). The pore-size distribution was characterized by adsorption–desorption measurements (ASAP 2020, Micrometritics) using N2 at 77 K.4.3 Electrochemical measurements
Electrochemical measurements are performed using a potentiostat (Model PARSTAT MC, Princeton Applied Research). The H-type cell is utilized to separate the working electrode and counter electrode, with each compartment of the H-type cell separated by a Nafion membrane (NR212, Ion Exchange Materials). The counter electrode and reference electrode are Pt wire (CE-1, Neo Science) and Hg/HgO electrode (RE-61AP, ALS). The HER activity is assessed through a LSV curve in 1.0 M KOH (pH = 14) under a stirring rate of 550 rpm, ranging from 0.1 to −1.7 V vs. Hg/HgO. The LSV is performed at a scan rate of 10 mV s−1. All potentials are initially iR-compensated using eqn (1):| Ecompensated = Emeasured − iRs | (1) |
| ERHE = EHg/HgO + 0.118 V + (RTln10/F) vs. RHE | (2) |
485.3 C mol−1). The overpotential is determined as the difference between the converted potential and 0 V vs. RHE. The Tafel slope is calculated by plotting overpotentials against the logarithm of the current density (log|j|) using the following eqn (3):| η = log(j/j0) | (3) |
4.4 AEM electrolyzer
MEA is assembled using CS-Ru-40 (1 cm2, 0.65 mg of Ru) and Pt/C (1 cm2, 0.65 mg of Pt) as the cathode and IrO2 (1 cm2, 2 mg of Ir) as the anode, with each electrode separated by an PiperION membrane. The Pt/C electrode is manufactured using the CCS method through spray coating on a Ti felt substrate and IrO2 electrode is manufactured using a catalyst coated membrane method. For the cathode, the current collector and gas-diffusion layer (GDL) are made of graphite and Ti felt, while for the anode, they are composed of Au-coated Ti and stainless felt. The electrolyzer is subsequently tested in 1.0 M KOH (pH = 14) electrolyte flowing at a flow rate of 30 mL min−1 at a temperature of 60 °C. The activity is assessed by LSV curve over a potential range of 1.35 to 2.0 V at a scan rate of 10 mV s−1. To evaluate Rs and Rct of CS-Ru-40||IrO2 and Pt/C||IrO2, EIS is measured at a constant current density of 0.1 A cm−2. Overpotential breakdown is performed using Rs obtained through EIS. First, the Ohmic overpotential is calculated based on Ohm's law: ηΩ = iRΩ. The kinetic overpotentials (ηkinetics) of the anode and cathode are then determined by fitting a Tafel plot. Finally, the mass transport overpotential (ηMT) is determined by the residual overpotential calculated through the following equation: ηMT = E − E0 – ηkinetics − ηΩ, where E0 corresponds to the theoretical potential of water electrolysis.Author contributions
H. W. L. designed the experiments, carried out material characterizations and catalytic activity tests. T. K. L and S. P. measured catalytic activity in a practical AEMWE system. D. S. A. P measured hydrogen Faradaic efficiency. B. Y. conducted the DFT calculations. S. J. Y contributed to the evaluation and discussion of catalyst application system. C. W. L. contributed to experimental design, conceptual discussions, and supervision of the research project. J. Y. K. contributed to conceptual discussions and managed and supervised the research project.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) Grant funded by the Korean Government (MSIT) (NRF-2022M3H4A4097525, 2022R1A4A1019296, RS-2023-00210114, 2021M3H4A1A02042948, RS-2023-00273532). This work was supported by National Research and Development Program through the National Research Foundation of Korea funded by Ministry of Science and ICT (No. 2021M3H4A1A03057403).Notes and references
- C. Niether, S. Faure, A. Bordet, J. Deseure, M. Chatenet, J. Carrey, B. Chaudret and A. Rouet, Nat. Energy, 2018, 3, 476–483 CrossRef.
- C. Chen, Y. Kang, Z. Huo, Z. Zhu, W. Huang, H. L. Xin, J. D. Snyder, D. Li, J. A. Herron, M. Mavrikakis, M. Chi, K. L. More, Y. Li, N. M. Markovic, G. A. Somorjai, P. Yang and V. R. Stamenkovic, Science, 2014, 343, 1339–1343 CrossRef PubMed.
- Y. Liu, J. Wu, K. P. Hackenberg, J. Zhang, Y. M. Wang, Y. Yang, K. Keyshar, J. Gu, T. Ogitsu, R. Vajtai, J. Lou, P. M. Ajayan, B. C. Wood and B. I. Yakobson, Nat. Energy, 2017, 2, 1–7 Search PubMed.
- B. Yan, X. Qin, T. Chen, Z. Teng, D. K. Cho, H. W. Lim, H. Hong, Y. Piao, L. Xu and J. Y. Kim, Adv. Funct. Mater., 2024, 34, 1–11 Search PubMed.
- D. Li, A. R. Motz, C. Bae, C. Fujimoto, G. Yang, F. Y. Zhang, K. E. Ayers and Y. S. Kim, Energy Environ. Sci., 2021, 14, 3393–3419 RSC.
- D. Xu, M. B. Stevens, M. R. Cosby, S. Z. Oener, A. M. Smith, L. J. Enman, K. E. Ayers, C. B. Capuano, J. N. Renner, N. Danilovic, Y. Li, H. Wang, Q. Zhang and S. W. Boettcher, ACS Catal., 2019, 9, 7–15 CrossRef CAS.
- M. Bajdich, M. García-Mota, A. Vojvodic, J. K. Nørskov and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 13521–13530 CrossRef CAS.
- A. W. Tricker, J. K. Lee, J. R. Shin, N. Danilovic, A. Z. Weber and X. Peng, J. Power Sources, 2023, 567, 232967 CrossRef CAS.
- J. Staszak-Jirkovský, C. D. Malliakas, P. P. Lopes, N. Danilovic, S. S. Kota, K. C. Chang, B. Genorio, D. Strmcnik, V. R. Stamenkovic, M. G. Kanatzidis and N. M. Markovic, Nat. Mater., 2016, 15, 197–203 CrossRef PubMed.
- Y. Kim, S. M. Jung, K. S. Kim, H. Y. Kim, J. Kwon, J. Lee, H. S. Cho and Y. T. Kim, JACS Au, 2022, 2, 2491–2500 CrossRef CAS PubMed.
- J. Zhu, L. Hu, P. Zhao, L. Y. S. Lee and K. Y. Wong, Chem. Rev., 2020, 120, 851–918 CrossRef CAS.
- D. Astruc, Chem. Rev., 2020, 120, 461–463 CrossRef CAS.
- F. T. Haase, A. Bergmann, T. E. Jones, J. Timoshenko, A. Herzog, H. S. Jeon, C. Rettenmaier and B. R. Cuenya, Nat. Energy, 2022, 7, 765–773 CrossRef CAS.
- W. F. Chen, S. Iyer, S. Iyer, K. Sasaki, C. H. Wang, Y. Zhu, J. T. Muckerman and E. Fujita, Energy Environ. Sci., 2013, 6, 1818–1826 RSC.
- J. Klein, A. K. Engstfeld, S. Brimaud and R. J. Behm, Phys. Chem. Chem. Phys., 2020, 22, 19059–19068 RSC.
- I. L. Simakova, Y. S. Demidova, E. V. Murzina, A. Aho and D. Y. Murzin, Catal. Lett., 2016, 146, 1291–1299 CrossRef CAS.
- R. Borup, J. Meyers, B. Pivovar, Y. S. Kim, R. Mukundan, N. Garland, D. Myers, M. Wilson, F. Garzon, D. Wood, P. Zelenay, K. More, K. Stroh, T. Zawodzinski, J. Boncella, J. E. McGrath, M. Inaba, K. Miyatake, M. Hori, K. Ota, Z. Ogumi, S. Miyata, A. Nishikata, Z. Siroma, Y. Uchimoto, K. Yasuda, K. I. Kimijima and N. Iwashita, Chem. Rev., 2007, 107, 3904–3951 CrossRef CAS.
- Z. Xu, H. Zhang, H. Zhong, Q. Lu, Y. Wang and D. Su, Appl. Catal., B, 2012, 111–112, 264–270 CrossRef CAS.
- L. Xu, H. W. Liang, Y. Yang and S. H. Yu, Chem. Rev., 2018, 118, 3209–3250 CrossRef CAS PubMed.
- H. T. Phan and A. J. Haes, J. Phys. Chem. C, 2019, 123, 16495–16507 CrossRef CAS.
- A. N. Goldstein, C. M. Echer and A. P. Alivisatos, Science, 1992, 256, 1425–1427 CrossRef PubMed.
- X. Zhu, P. Wang, Z. Wang, Y. Liu, Z. Zheng, Q. Zhang, X. Zhang, Y. Dai, M. H. Whangbo and B. Huang, J. Mater. Chem. A, 2018, 6, 12718–12723 RSC.
- J. Su, Y. Yang, G. Xia, J. Chen, P. Jiang and Q. Chen, Nat. Commun., 2017, 8, 1–10 Search PubMed.
- S. Wang, Z. Li, T. Shen and D. Wang, ChemSusChem, 2023, 16, 1–9 Search PubMed.
- H. Liao, C. Wei, J. Wang, A. Fisher, T. Sritharan, Z. Feng and Z. J. Xu, Adv. Energy Mater., 2017, 7, 1–7 Search PubMed.
- X. Zhang, Z. Sun, R. Jin, C. Zhu, C. Zhao, Y. Lin, Q. Guan, L. Cao, H. Wang, S. Li, H. Yu, X. Liu, L. Wang, S. Wei, W. X. Li and J. Lu, Nat. Commun., 2023, 14, 1–13 Search PubMed.
- H. W. Lim, J. H. Park, B. Yan, J. Y. Kim and C. W. Lee, Appl. Catal., B, 2023, 322, 122095 CrossRef.
- Y. Shen, Y. Zhou, D. Wang, X. Wu, J. Li and J. Xi, Adv. Energy Mater., 2018, 8, 1–7 Search PubMed.
- H. W. Lim, D. K. Cho, J. H. Park, S. G. Ji, Y. J. Ahn, J. Y. Kim and C. W. Lee, ACS Catal., 2021, 11, 12423–12432 CrossRef.
- Z. Chen, W. Gong, J. Wang, S. Hou, G. Yang, C. Zhu, X. Fan, Y. Li, R. Gao and Y. Cui, Nat. Commun., 2023, 14, 5363 CrossRef PubMed.
- J. Zhang, L. Zhang, J. Liu, C. Zhong, Y. Tu, P. Li, L. Du, S. Chen and Z. Cui, Nat. Commun., 2022, 13, 1–9 Search PubMed.
- L. Li, S. Liu, C. Zhan, Y. Wen, Z. Sun, J. Han, T. S. Chan, Q. Zhang, Z. Hu and X. Huang, Energy Environ. Sci., 2022, 16, 157–166 RSC.
- T. Shinagawa, A. T. Garcia-Esparza and K. Takanabe, Sci. Rep., 2015, 5, 1–21 Search PubMed.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, Wiley, New York, 2nd edn, 2002, vol. 38 Search PubMed.
- B. Xia, B. He, J. Zhang, L. Li, Y. Zhang, J. Yu, J. Ran and S. Z. Qiao, Adv. Energy Mater., 2022, 12, 2201449 CrossRef.
- J. Zhou, Z. Gao, G. Xiang, T. Zhai, Z. Liu, W. Zhao, X. Liang and L. Wang, Nat. Commun., 2022, 13, 1–10 Search PubMed.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-ray photoelectron spectroscopy: a reference book of standard spectra for identification and interpretation of XPS data, PerkinElmer Corporation, 1992 Search PubMed.
- L. Hong, K. Bhatnagar, R. Droopad, R. F. Klie and S. Öǧüt, Phys. Rev. B, 2017, 96, 035311 CrossRef.
- T. Cheng, L. Wang, B. V. Merinov and W. A. Goddard, J. Am. Chem. Soc., 2018, 140, 7787–7790 CrossRef CAS PubMed.
- N. Yao, P. Li, Z. Zhou, Y. Zhao, G. Cheng, S. Chen and W. Luo, Adv. Energy Mater., 2019, 9, 1–8 Search PubMed.
- J. X. Feng, J. Q. Wu, Y. X. Tong and G. R. Li, J. Am. Chem. Soc., 2018, 140, 610–617 CrossRef CAS.
- Q. Hu, K. Gao, X. Wang, H. Zheng, J. Cao, L. Mi, Q. Huo, H. Yang, J. Liu and C. He, Nat. Commun., 2022, 13, 1–10 Search PubMed.
- Q. Huang, W. Yang, Y. Yan, S. Xie, A. Yu, T. Xu, Y. Zhao, P. Peng and F. F. Li, Adv. Funct. Mater., 2024, 34, 2409406 CrossRef CAS.
- X. Ding, J. Yu, W. Huang, D. Chen, W. Lin and Z. Xie, Chem. Eng. J., 2023, 451, 138550 CrossRef CAS.
- Y. Zheng, A. Serban, H. Zhang, N. Chen, F. Song and X. Hu, ACS Energy Lett., 2023, 8, 5018–5024 CrossRef CAS.
- S. Y. Kang, J. E. Park, G. Y. Jang, O. H. Kim, O. J. Kwon, Y. H. Cho and Y. E. Sung, Int. J. Hydrogen Energy, 2022, 47, 9115–9126 CrossRef CAS.
- R. A. Krivina, G. A. Lindquist, S. R. Beaudoin, T. N. Stovall, W. L. Thompson, L. P. Twight, D. Marsh, J. Grzyb, K. Fabrizio, J. E. Hutchison and S. W. Boettcher, Adv. Mater., 2022, 34, 1–10 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee04867a |
| This journal is © The Royal Society of Chemistry 2025 |