
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-limiting surface leaching stabilizes Ru-based catalysts for acidic water oxidation†
Yang
Liu‡
ab,
Xiyu
Li‡
cd,
Haeseong
Jang‡
 ef,
Jianghua
Wu‡
gh,
Min Gyu
Kim
f,
Xiaoke
Xi
a,
Zhanwu
Lei
*a,
Yuchen
Zhang
a,
Yu
Deng
i,
Wensheng
Yan
j,
Jun
Jiang
k,
Shuhong
Jiao
*k,
Jing-Li
Luo
*b and
Ruiguo
Cao
*a
ef,
Jianghua
Wu‡
gh,
Min Gyu
Kim
f,
Xiaoke
Xi
a,
Zhanwu
Lei
*a,
Yuchen
Zhang
a,
Yu
Deng
i,
Wensheng
Yan
j,
Jun
Jiang
k,
Shuhong
Jiao
*k,
Jing-Li
Luo
*b and
Ruiguo
Cao
*a
aHefei National Laboratory for Physical Science at the Microscale, Department of Materials Science and Engineering, University of Science and Technology of China, Hefei, 230026, China. E-mail: zwlei@mail.ustc.edu.cn; rgcao@ustc.edu.cn
bShenzhen Key Laboratory of Energy Electrocatalytic Materials, Guangdong Provincial Key Laboratory of New Energy Materials Service Safety, College of Materials Science and Engineering, Shenzhen University, Shenzhen 518055, Guangdong, China. E-mail: jingli.luo@ualberta.ca
cSchool of Physical Sciences, Great Bay University, Dongguan 523000, China
dSongshan Lake Materials Laboratory, Dongguan, Guangdong 523808, China
eDepartment of Advanced Materials Engineering, Chung-Ang University, Gyeonggi-do, 17546, Republic of Korea
fDepartment of Intelligent Energy and Industry, Chung-Ang University, 84 Heukseok-ro, Dongjak-gu, Seoul 06974, Republic of Korea
gDepartment of Chemistry, Guangdong Provincial Key Laboratory of Catalysis, Southern University of Science and Technology, Shenzhen 518055, China
hSuzhou Institute of Nano-Tech and Nano-Bionics (SINANO), Chinese Academy of Sciences, Suzhou 215123, China
iNational Laboratory of Solid-State Microstructures, Jiangsu Key Laboratory of Artificial Functional Materials, College of Engineering and Applied Sciences and Collaborative Innovation Center of Advanced Microstructures, Nanjing University, Nanjing 210093, China
jNational Synchrotron Radiation Laboratory, University of Science and Technology of China, Hefei, 230029, China
kKey Laboratory of Precision and Intelligent Chemistry, University of Science and Technology of China, Hefei, 230026, China. E-mail: jiaosh@ustc.edu.cn
First published on 25th February 2025
Abstract
Ru-based catalysts are a promising alternative to Ir-based catalysts for the acidic oxygen evolution reaction (OER), but their poor long-term stability remains a significant challenge. Continuous leaching-induced loss of active sites and structural collapse are the primary causes of this instability, severely limiting the practical application of Ru-based catalysts in proton exchange membrane (PEM) electrolyzers. Here, we present a self-limiting surface leaching mechanism that effectively suppresses continuous leaching, thereby significantly prolonging the lifespan of Ru-based catalysts under acidic OER conditions. Specifically, the Ru–Mn solid solution oxide with a hollow shell structure undergoes surface Mn leaching during the initial OER process, resulting in the formation of a Mn-vacancy-rich stable reconstruction layer. This layer effectively inhibits further leaching of both Ru and Mn, thus self-limiting the further degradation of catalysts. As a result, the reconstructed catalyst exhibits an unprecedented durability of up to 2500 h at 10 mA cm−2 in 0.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) M H2SO4. This remarkable stability was also validated in PEM electrolyzers, highlighting its practical applicability. Operando synchrotron characterization combined with theoretical calculations reveals that the formation of Mn vacancies increases the demetallation energy of Ru species, thereby suppressing the continuous leaching and enhancing the long-term stability. This work provides valuable insights for designing highly stable catalysts through a self-limiting leaching mechanism.
M H2SO4. This remarkable stability was also validated in PEM electrolyzers, highlighting its practical applicability. Operando synchrotron characterization combined with theoretical calculations reveals that the formation of Mn vacancies increases the demetallation energy of Ru species, thereby suppressing the continuous leaching and enhancing the long-term stability. This work provides valuable insights for designing highly stable catalysts through a self-limiting leaching mechanism.
Broader contextProton exchange membrane (PEM) water electrolysis is a promising technology for sustainable hydrogen production. However, its large-scale application is hindered by the high cost and extreme scarcity of iridium-based oxide anodes. Recently, ruthenium-based catalysts have emerged as a viable alternative to Ir-based catalysts due to their lower cost and superior activity for the acidic oxygen evolution reaction (OER). A major challenge for Ru-based catalysts is their poor stability, primarily caused by the continuous leaching of active sites under harsh acidic and oxidative conditions. Here, we present a self-limiting surface leaching mechanism that significantly enhances the long-term stability of RuMn solid oxide (RuMnO2) catalysts by suppressing continuous leaching. Characterization reveals that Mn partially dissolves from RuMnO2 during the initial stages of the acidic OER, leading to the formation of a corrosion-resistant, Mn-vacancy-rich surface reconstruction layer. This unique structure effectively inhibits further leaching of Ru and Mn, thereby self-limiting the continuous degradation of the catalysts. As a result, the Mn-vacancy-rich RuMnO2 catalyst exhibits remarkable durability, surpassing 2500 hours at 10 mA cm−2 in 0.5M H2SO4, along with an ultra-low overpotential of ∼166 mV at 10 mA cm−2. In situ characterization and theoretical simulation demonstrate that the Mn vacancies increase the demetallation energy of Ru species and facilitate the formation of the H-stabilized *OO intermediate, significantly improving both long-term stability and catalytic activity.
|
Introduction
Electrocatalytic water splitting powered by renewable energy provides a sustainable pathway for producing green hydrogen.1–3 Among various water electrolysis techniques, PEM water electrolysis stands out for its advantages over alkaline water electrolysis, including higher hydrogen purity, faster charge and ion conductivity, and quicker response times.4–6 However, a major challenge in PEM water electrolysis is the sluggish four-electron transfer kinetics of the anodic acidic OER, which requires the development of efficient catalysts.7–9 Currently, Ir-based catalysts are considered the only practical acidic OER catalysts in PEM electrolyzers due to their robust stability in withstanding the harsh acidic and strong oxidative conditions.10–12 However, the extreme scarcity and expensive price of Ir resources have seriously impeded their large-scale application in practical PEM electrolyzers.13–15 To address these issues, researchers have focused on developing alternative non-Ir-based catalysts to reduce the cost of acidic OER catalysts. Among these, Ru- and Mn-based catalysts have been studied extensively for their advantages in catalytic activity for acidic OERs.4,16–18 Unfortunately, the long-term stability of most Ru- and Mn-based catalysts reported to date remains limited to within tens of hours under high current density conditions in PEM electrolyzers, far short of the requirements for industrial application.19–23The overoxidation and subsequent leaching of metal active sites under harsh acidic OER conditions often lead to crystal structure collapse and catalytic activity loss, which are the primary reasons for the poor stability of acidic OER catalysts.24–26 Ru- and Mn-based catalysts, especially when used as anodes in PEM electrolyzers at high current densities, are particularly susceptible to this degradation mechanism.27–29 Pourbaix diagrams indicate that the thermodynamically stable forms of Ru and Mn under PEM operating conditions are soluble ruthenate (RuO4) and manganate (MnO4−) species, respectively.30,31 Under the operation window in acidic electrolyzers, Ru and Mn ions tend to continuously leach from the catalyst, causing a gradual decline in catalytic activity.32,33 Subsequently, the continuous leaching often leads to the uncontrollable surface atom rearrangement, forming a reconstructed surface layer on the catalyst.34–36 However, this reconstruction layer typically contains a high density of defects that cannot endure strong acidic and oxidative environments, leading to its rapid collapse and a significant loss in performance during acidic OERs.37–39 Therefore, developing structurally stable reconstruction layers to prevent the continuous leaching of metal active sites is essential for achieving highly stable acidic OER catalysts, though significant challenges remain.
Herein, we introduce a self-limiting surface leaching mechanism to develop a corrosion-resistant surface reconstruction layer on a RuMn-based catalyst, which significantly enhances its long-term stability (Fig. 1a). Detailed characterization revealed that Mn partially dissolves from the RuMn solid solution oxide (RuMnO2) during the initial stages of the acidic OER, resulting in the formation of a Mn-vacancy-rich reconstruction layer on the catalyst surface (VMn–RuMnO2−x). Operando synchrotron studies combined with theoretical calculations demonstrated that Mn vacancies stabilize Ru species by increasing the demetallation energy of RuO4, effectively preventing continuous ion leaching and improving long-term catalyst stability. Additionally, the cationic vacancies facilitate the formation of the H-stabilized *OO intermediate and optimize the electronic structure of Ru, lowering the free energy barrier for O–O bond formation and boosting catalytic activity. As a result, the VMn–RuMnO2−x catalyst achieves unprecedented stability of up to 2500 h at 10 mA cm−2 with an ultra-low overpotential of ∼166 mV at 10 mA cm−2 in 0.5M H2SO4. Moreover, a PEMWE device incorporating VMn–RuMnO2−x as the anode catalyst operated stably for over 600 h at 200 mA cm−2, manifesting its practical application potential.
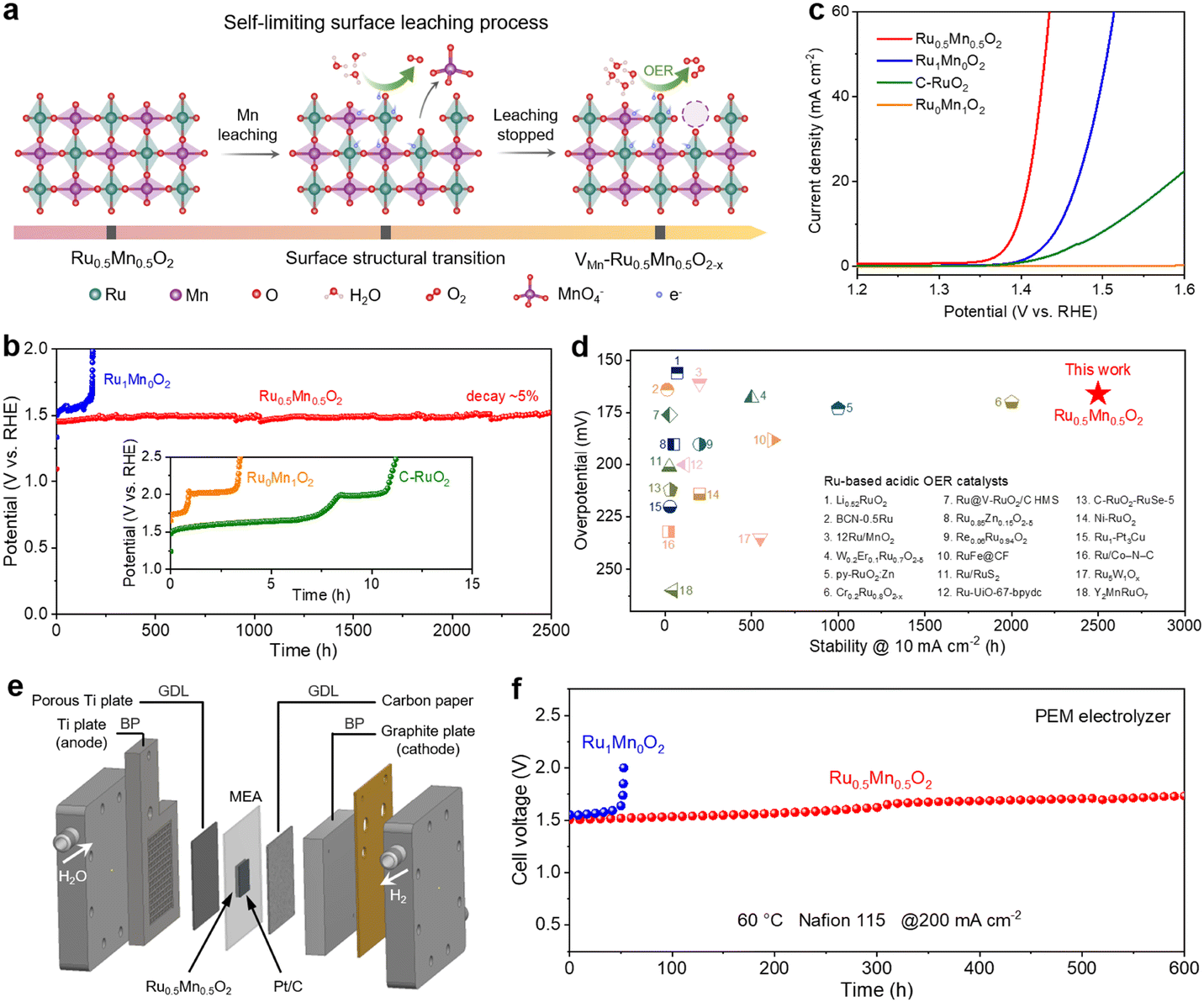 | ||
| Fig. 1 Comparison of activity and stability of various catalysts. (a) Schematic illustration of the self-limiting surface leaching process. (b) Chronopotentiometric curves of Ru0.5Mn0.5O2, Ru1Mn0O2, Ru0Mn1O2, and C–RuO2 at a current density of 10 mA cm−2 in 0.5 M H2SO4 solution. (c) Polarization curves of Ru0.5Mn0.5O2, Ru1Mn0O2, Ru0Mn1O2, and C–RuO2. (d) Comparison of the overpotentials and stabilities for Ru0.5Mn0.5O2 and recently reported Ru-based OER catalysts in acid media. (e) Schematic diagram of the PEM electrolyzer. A typical PEM electrolyzer consists of bipolar plates (BPs), gas diffusion layers (GDLs), and membrane electrode assembly (MEA). (f) Chronopotentiometry testing of PEM electrolyzers using Ru0.5Mn0.5O2 or Ru1Mn0O2 as the anodic catalyst and commercial Pt/C as the cathodic catalyst operated at 200mAcm−2 at 60 °C. | ||
Results and discussion
Self-limiting surface leaching mechanism and electrocatalytic performance of catalysts
To construct a binary metal oxide with a uniform mixture of Ru and Mn, we developed a chelation precipitation method combined with a one-step oxidation process to synthesize a Ru–Mn solid solution oxide with a hollow shell structure (Ru0.5Mn0.5O2) (Fig. S1, ESI†). For comparison, we also synthesized pure Ru oxide (Ru1Mn0O2) and pure Mn oxide (Ru0Mn1O2) using a similar method. As illustrated in Fig. 1a, Ru0.5Mn0.5O2 undergoes a self-limiting surface leaching process under acidic OER conditions. During the initial OER process, Mn atoms may leach from the Ru0.5Mn0.5O2 catalyst with a relatively perfect rutile structure in the form of MnO4−, leading to the formation of a Mn-vacancy-rich surface reconstruction layer (VMn–Ru0.5Mn0.5O2−x). These vacancies can effectively suppress further leaching of both Ru and Mn and inhibit continuous catalyst degradation, thereby enabling the long-term stability of VMn–Ru0.5Mn0.5O2−x during acidic OERs.We examined the long-term OER stability of Ru0.5Mn0.5O2 using chronopotentiometric measurements in 0.5 M H2SO4. Remarkably, Ru0.5Mn0.5O2 retained ∼95% of its initial activity after 2500 hours at 10 mA cm−2 (Fig. 1b). Even at a higher current density of 100 mA cm−2, the potential increased by only 71 mV after 700 hours, highlighting the exceptional durability of Ru0.5Mn0.5O2 (Fig. S2, ESI†). In sharp contrast, C–RuO2 and Ru0Mn1O2 only lasted a few hours of durability testing at 10 mA cm−2 (insert of Fig. 1b). Although Ru1Mn0O2 demonstrated improved long-term stability compared to C–RuO2, it still experienced severe activity loss after 180 h of durability testing (Fig. 1b). We also performed continuous cyclic voltammetry (CV) scanning tests to further evaluate the robustness of Ru0.5Mn0.5O2. As shown in Fig. S3 (ESI†), the overpotential of Ru0.5Mn0.5O2 at 10 mA cm−2 increased by only 7 mV after 1 kth CV scans, significantly lower than the 40 mV increase observed for C–RuO2.
We evaluated the acidic OER performance of Ru0.5Mn0.5O2 using a three-electrode system with a 0.5 M H2SO4 aqueous electrolyte. Linear sweep voltammetry (LSV) results indicate that Ru0.5Mn0.5O2 requires an overpotential of only ∼166 mV to achieve a current density of 10 mA cm−2, outperforming Ru1Mn0O2 (∼218 mV) and C–RuO2 (∼285 mV) (Fig. 1c). Notably, Ru0Mn1O2 shows negligible OER activity due to the absence of Ru active sites. To assess the intrinsic activity of Ru0.5Mn0.5O2, we calculated the electrochemical specific surface area (ECSA) derived from the electrochemical double-layer capacitance (Cdl) (Fig. S4, ESI†). Ru0.5Mn0.5O2 exhibited a larger ECSA than the other catalysts (Fig. S5a, ESI†), indicating more available active sites for the OER. When normalizing the catalytic currents with respect to ECSA, Ru0.5Mn0.5O2 still exhibited the lowest overpotential among these catalysts (Fig. S5b, ESI†), confirming its superior intrinsic activity. In comparison, Ru1Mn0O2 showed a similar overpotential to C–RuO2 based on the ECSA analysis (Fig. S5b, ESI†), indicating that the Ru active sites in both catalysts exhibit comparable intrinsic activity. Furthermore, Ru0.5Mn0.5O2 achieves a high mass activity of 524.6 A gRu−1 (normalized to Ru) at 1.45 V vs. RHE, which is approximately 11.1 times and 38.9 times higher than those of Ru1Mn0O2 (47.2 A gRu−1) and C–RuO2 (13.5 A gRu−1), respectively. These results unambiguously suggest Ru0.5Mn0.5O2 as one of the most active catalysts towards acidic OERs, surpassing many Ru- or Ir-based catalysts reported in the literature (Fig. S6, ESI†). The Tafel slope for Ru0.5Mn0.5O2 was 41.8 mV dec−1, lower than that of Ru1Mn0O2 (60.3 mV dec−1) and C–RuO2 (94.3 mV dec−1), indicating its better OER kinetics (Fig. S7, ESI†). Electrochemical impedance spectroscopy (EIS) further supported these findings, revealing that Ru0.5Mn0.5O2 has the lowest charge transfer resistance (Fig. S8 and Table S1, ESI†), which implies a faster charge transfer rate and OER kinetics. In addition, we investigated how the elemental composition of the solid-solution oxides influences their catalytic activity. By adjusting the ratio of Ru and Mn chlorides, we synthesized a series of RuyMn1−yO2 catalysts (y = 0.9, 0.7, 0.3, and 0.1). Inductively coupled plasma atomic emission spectrometry (ICP-AES) analysis revealed that the Ru/Mn atomic ratios in RuyMn1−yO2 were close to the initial feed ratios (Fig. S9 and Table S2, ESI†). As shown in Fig. S10 (ESI†), Ru0.5Mn0.5O2 demonstrated the highest OER activity among the tested catalysts, suggesting that a 1:1 ratio of Ru to Mn represents the optimal composition. A comparison of the activity and stability of Ru0.5Mn0.5O2 with recent reports in the literature revealed that it outperforms most Ru-based OER catalysts in acidic media (Fig. 1d and Table S3, ESI†).2,36,39–56
Considering the high activity and stability of Ru0.5Mn0.5O2 for acidic OERs, we assembled a PEM electrolyzer using the catalyst as the anode to evaluate its practical application potential. As shown in Fig. 1e and Fig. S11 (ESI†), the PEM electrolyzer was mainly composed of bipolar plates (BPs), gas diffusion layers (GDLs), and membrane electrode assembly (MEA). Among them, MEA with an active area of 4 cm2 was manufactured by sandwiching a proton exchange membrane (Nafion 115) between the anode (Ru0.5Mn0.5O2) and cathode (commercial Pt/C). When the electrolyzer using Ru0.5Mn0.5O2 as the anode was operated at 60 °C, the steady-state polarization curve showed a cell voltage of 1.762 V to achieve a current density of 1 A cm−2 (Fig. S12, ESI†), outperforming the electrolyzer using the Ru1Mn0O2 as the anode (1.947 V@1 A cm−2). We further evaluated the durability of Ru0.5Mn0.5O2 in PEM electrolyzers. Impressively, the PEM electrolyzer with Ru0.5Mn0.5O2 as the anode operated at 200 mA cm−2 for over 600 h without significant performance degradation (Fig. 1f). In sharp contrast, the PEM electrolyzer using Ru1Mn0O2 as the anode exhibited severe decay after just 52 h of durability test at the same current density (Fig. 1f). Furthermore, we increased the operating temperature and current density of the PEM electrolyzer to evaluate the long-term stability of the catalyst under more practical conditions. As shown in Fig. S13 (ESI†), the PEM electrolyzer with Ru0.5Mn0.5O2 as the anode demonstrated stable operation for 330 h under 200 mA cm−2 at 80 °C, and approximately 120 h under 500 mA cm−2 at 60 °C. The elevated temperature and higher current density both adversely impacted the long-term stability of Ru0.5Mn0.5O2, attributed to the harsher operating conditions encountered by the catalyst. These findings demonstrate the excellent activity and stability of Ru0.5Mn0.5O2 for acidic OERs in PEM electrolyzers.
Self-limiting surface structural evolution of catalysts during acidic OERs
We comprehensively characterized the morphology and structure of the catalysts before and after the OER process to elucidate the structural evolution of Ru0.5Mn0.5O2. First, we carried out scanning electron microscopy (SEM) and X-ray diffraction (XRD) to investigate potential changes in the morphology and crystalline structure of the catalysts. SEM images reveal that the prepared Ru0.5Mn0.5O2 catalysts displayed a hollow branch-like structure with abundant accessible channels (Fig. S14a, ESI†), which is beneficial for enhancing the electrochemical performance by boosting mass transfer of the reactant and product. Notably, the catalyst maintained its original hollow branch-like structure even after a 50 h OER test (Fig. S14b–d, ESI†), as confirmed by low-magnification TEM images (Fig. S15, ESI†). The XRD patterns of the prepared Ru0.5Mn0.5O2 matched the rutile phase solid solution of RuO2 and MnO2 (P42/mnm space group), confirming the formation of a solid-solution structure (Fig. S16, ESI†). Notably, the diffraction peaks of Ru0.5Mn0.5O2 shift to higher angles compared to Ru1Mn0O2, due to the substitution of Ru by Mn with a smaller ionic radius, leading to a reduction in interplanar spacing and an increase in the diffraction angle (θ), as described by the Bragg equation. After the OER testing, no significant changes were observed in the XRD patterns, indicating that post-OER Ru0.5Mn0.5O2 preserved its initial solid-solution phase (Fig. S17, ESI†).Subsequently, we utilized high-resolution TEM (HRTEM) to investigate the evolution of the surface structure on the catalyst during the acidic OER process. HRTEM images of the prepared Ru0.5Mn0.5O2 display clear lattice fringes with lattice spacings of 0.325 nm and 0.259 nm, corresponding to the (110) and (101) planes of rutile RuO2 (JCPDS: 43-1027), respectively (Fig. S18a, ESI†). The selected area electron diffraction (SAED) pattern, with distinct diffraction rings, confirmed its polycrystalline nature (Fig. S18b, ESI†). The average grain size is approximately 4.7 nm (Fig. S19, ESI†). Energy-dispersive X-ray spectroscopy (EDX) mapping demonstrated a homogenous distribution of Ru, Mn, and O elements, further confirming the successful formation of RuMn solid-solution oxides (Fig. S20, ESI†). As a comparison, we also characterized the surface crystal structure of post-OER Ru0.5Mn0.5O2 using HRTEM. As shown in Fig. 2a and b, the surface crystal structure of the post-OER Ru0.5Mn0.5O2 was well-preserved, with interplanar spacings of 0.316 corresponding to the (110) planes of rutile RuO2. Additionally, no noticeable amorphous reconstruction layers were observed on the catalyst surface. Elemental mapping (Fig. 2c) and EDX line-scan analysis (Fig. S21, ESI†) of the post-OER Ru0.5Mn0.5O2 showed a uniform distribution of Ru, Mn, and O without significant segregation. These results collectively demonstrate that the Ru0.5Mn0.5O2 catalyst retains both its morphology and crystalline structure during long-term acidic OER testing, highlighting its stability.
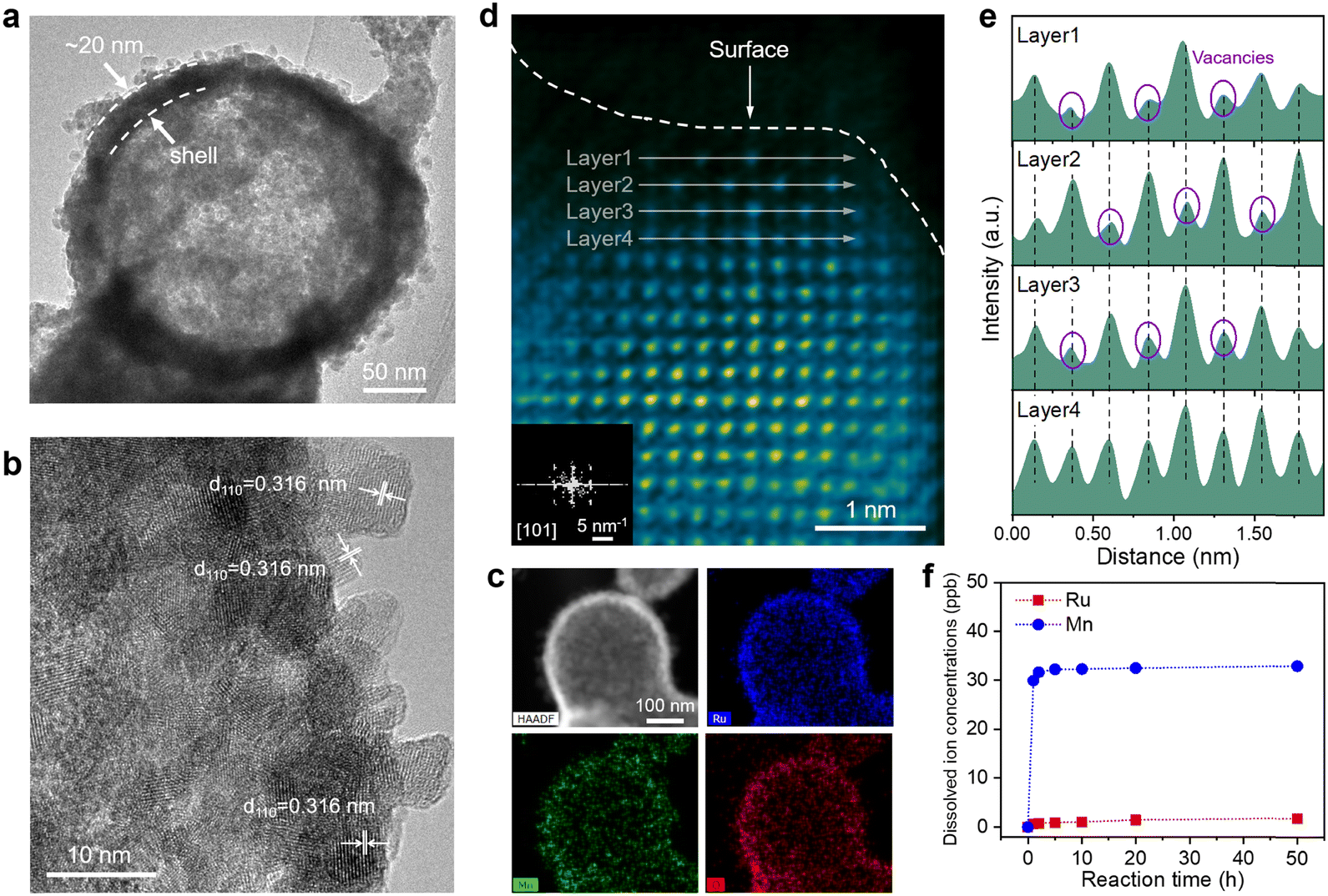 | ||
| Fig. 2 Structural evolution of Ru0.5Mn0.5O2 during the acidic OER process. (a) Low-magnification TEM image of Ru0.5Mn0.5O2 after a 50 h OER test. (b) HRTEM image of Ru0.5Mn0.5O2 after a 50 h OER test. (c) HAADF image of Ru0.5Mn0.5O2 after a 50 h OER test and the corresponding EDS elemental mappings of Ru, Mn, and O. (d) Atomic-resolution HAADF-STEM image of Ru0.5Mn0.5O2 after a 50 h OER test from the [101] zone axis (inset: corresponding FFT pattern). (e) Corresponding intensity profiles over the selected atomic columns (layer 1 to layer 4 in (d)). (f) Dissolved Ru and Mn ion concentrations measured for Ru0.5Mn0.5O2 in electrolyte for different reaction times. | ||
To further probe the atomic structure evolution of catalysts during the acidic OER process, we employed an aberration-corrected high-angle annular dark-field scanning transmission electron microscope (HAADF-STEM) on both the prepared and post-OER Ru0.5Mn0.5O2. HAADF-STEM images of the prepared Ru0.5Mn0.5O2 showed well-crystallized nanocrystals without significant defects (Fig. S22, ESI†). The corresponding Fast Fourier transform (FFT) pattern confirmed that the crystal structure aligns with the tetragonal crystal system of rutile RuO2 in the [111] projection (inset of Fig. S22a, ESI†). In contrast, the cation vacancies marked by purple circles are found in the post-OER Ru0.5Mn0.5O2 due to their lower intensity in the HAADF-STEM images (Fig. 2d and e).57 Despite the presence of these vacancies, the surface atoms remained orderly arranged, and the crystal structure continued to align with the tetragonal crystal system of rutile RuO2 in the [101] projection (inset of Fig. 2d). Notably, these cation vacancies were predominantly located on the surface of the nanoparticles (Fig. 2d), suggesting their formation is closely linked to irreversible ion dissolution occurring during the acidic OER process. This detailed analysis highlights the formation of surface cation vacancies as a key feature in the structural evolution of the catalyst during the OER, which may play a significant role in influencing its performance and stability.
Additionally, we assessed the concentrations of Ru and Mn ions leached into the electrolyte during the acidic OER process from Ru0.5Mn0.5O2 using inductively coupled plasma-mass spectrometry (ICP-MS). As shown in Fig. 2f, the dissolved Ru ion concentration was consistently below 2 ppm throughout the 50 h OER test. In contrast, the dissolved Mn ion concentration rapidly increased to 31.69 ppb within the first 2 h and reached 32.33 ppb after 10 h, then plateaued. This uneven metal ion dissolution was further corroborated by EDS (Fig. S23 and Table S4, ESI†) and X-ray photoelectron spectroscopy (XPS) (Fig. S24 and Table S5, ESI†) analysis. Specifically, the atomic ratio of Ru/(Ru + Mn) increased rapidly during the first 10 hours and then stabilized (Fig. S23e and S24b, ESI†), reflecting the more pronounced leaching of Mn ions in the early stages of the OER process. Given that the leaching amount of Mn from the Ru0.5Mn0.5O2 is much greater than that of Ru, the cation vacancies observed in the post-OER Ru0.5Mn0.5O2 is mainly caused by the leaching of Mn ions. To further confirm the presence of Mn vacancies on the post-OER Ru0.5Mn0.5O2, we employed electron paramagnetic resonance (EPR) spectroscopy. As shown in Fig. S25 (ESI†), the post-OER Ru0.5Mn0.5O2 exhibits a stronger EPR peak at a g value of 2.003, which is attributed to unpaired electrons associated with Mn vacancies, compared to the pristine Ru0.5Mn0.5O2. These Mn vacancies likely alter the local electronic structure surrounding the Ru active sites, thereby modulating the catalytic activity and stability of Ru0.5Mn0.5O2 with surface Mn vacancies (VMn–Ru0.5Mn0.5O2−x).
Understanding the self-limiting surface leaching mechanism
To investigate the effect of cation vacancies on the electronic structure of catalytic active sites, we performed XAFS measurements to analyse the valence state and coordination environment of the active sites. As shown in Fig. 3a and Fig. S26 (ESI†), the valence state of Ru in Ru0.5Mn0.5O2 remained stable and below +4 during the 50 h OER test, which was also demonstrated by Ru M-edge soft XAS (Fig. S27, ESI†), preventing the overoxidation and subsequent dissolution of high-valence Ru species. In contrast, the valence state of Mn increased during the 50 h OER test according to Mn K-edge XANES spectra (Fig. S28, ESI†) and Mn L-edge soft XAS (Fig. S29, ESI†). To investigate the reasons behind the valence state changes of Ru and Mn during surface reconstruction, we first investigated the impact of Mn doping on the Ru valence state in Ru0.5Mn0.5O2. As shown in Fig. S30 (ESI†), Ru K-edge XANES spectra combined with absorption energy (E0) analysis reveal that the average Ru valence state in Ru0.5Mn0.5O2 (+3.60) is higher than that in Ru1Mn0O2 (+3.42), suggesting electron transfer from Ru to Mn via bridging oxygen atoms in Ru0.5Mn0.5O2. This electron transfer is further supported by XPS and soft XAS data (Fig. S31, ESI†), where the peak positions for Ru in Ru0.5Mn0.5O2 shift to higher energies compared to those in Ru1Mn0O2. Next, we analyzed the effect of Mn dissolution on the valence states of Ru and Mn. When Mn dissolves and forms cation vacancies in the catalyst, the oxidation states of both Ru and Mn in VMn–Ru0.5Mn0.5O2−x would be expected to increase to maintain charge neutrality. However, experimental results show that only the oxidation state of Mn increases after Mn dissolution (Fig. S28, ESI†), while the oxidation state of Ru remains nearly unchanged (Fig. 3a). This is because, after Mn dissolution and the formation of cation vacancies in VMn–Ru0.5Mn0.5O2−x, the Ru atoms surrounding the cation vacancies no longer transfer electrons to the Mn atoms that have been replaced by vacancies. Consequently, the electron density of Ru increases, leading to a reduction in its oxidation state. In summary, the increase in oxidation state of Ru induced by charge neutrality and the decrease in its oxidation state due to weakened electron transfer ultimately stabilize the average oxidation state of Ru during the surface reconstruction process (Fig. 3a).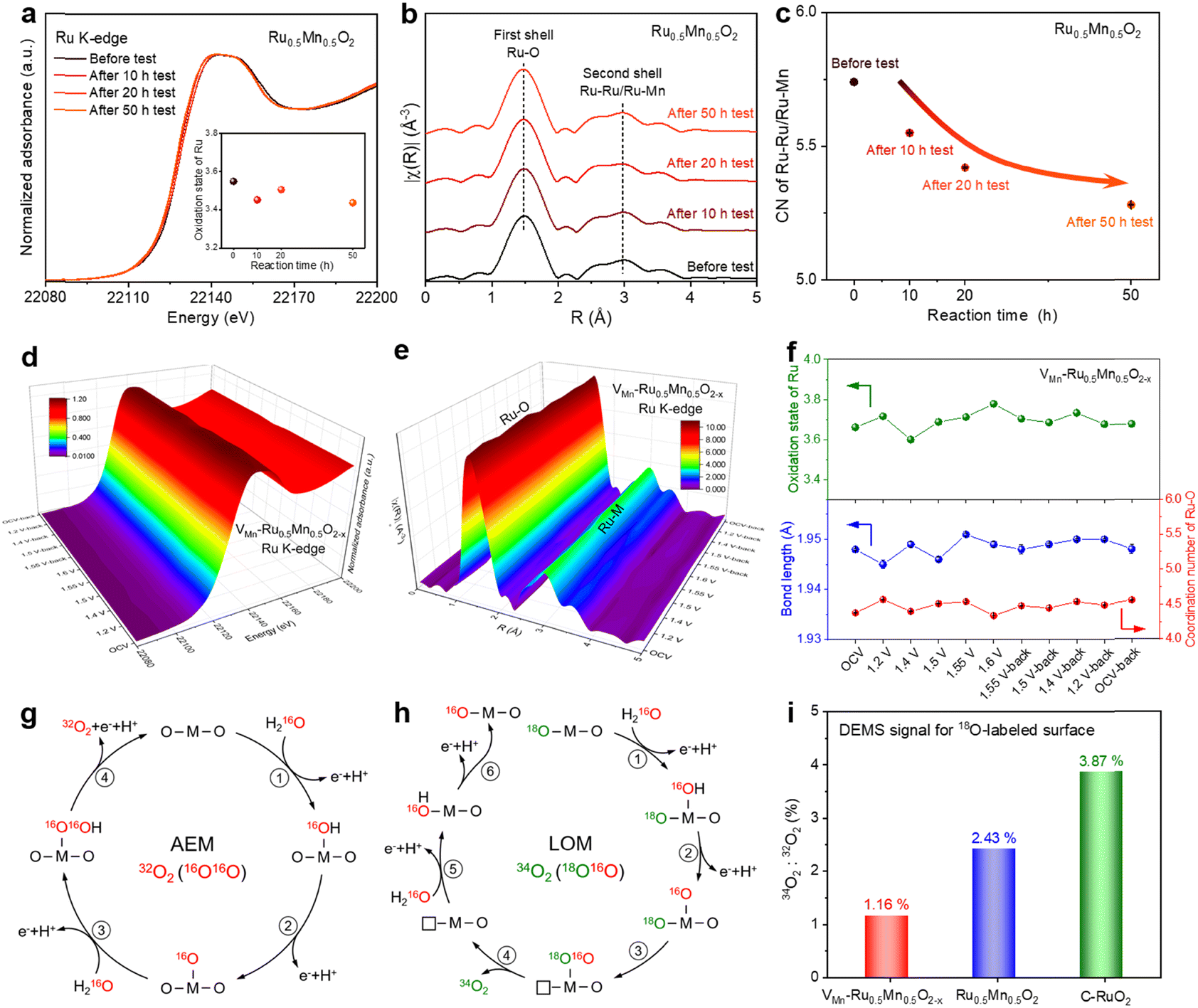 | ||
| Fig. 3
Operando XAFS measurements and mechanism investigation. (a) Ru K-edge XANES spectra of Ru0.5Mn0.5O2 before and after 10 h, 20 h, and 50 h OER tests (inset: the oxidation state of Ru at different reaction times). (b) Fourier-transformed Ru K-edge EXAFS spectra of Ru0.5Mn0.5O2 before and after 10 h, 20 h, and 50 h OER tests. (c) The coordination number of Ru–Ru/Ru–Mn for Ru0.5Mn0.5O2 before and after 10 h, 20 h, and 50 h OER tests. (d) 3D plot of operando Ru K-edge XANES spectra for VMn–Ru0.5Mn0.5O2−x. (e) 3D plot of operando Ru K-edge EXAFS spectra for VMn–Ru0.5Mn0.5O2−x. (f) The top image: the oxidation state of Ru under different applied potentials for VMn–Ru0.5Mn0.5O2−x. The bottom image: the bond length and CN of Ru–O under different applied potentials according to the EXAFS fitting results of VMn–Ru0.5Mn0.5O2−x. Reaction paths and different isotope-labeled O2 products of AEM (g) and LOM (h) in the electrolyte using H216O as the solvent. (i) The ratio of 34O2:32O2 for 18O-surface labeled VMn–Ru0.5Mn0.5O2−x, 18O-surface labeled Ru0.5Mn0.5O2, and 18O-surface labeled C–RuO2 in the electrolyte using H216O as the solvent. | ||
We further examined the coordination environment changes of Ru and Mn by fitting the EXAFS spectra of Ru0.5Mn0.5O2 over different reaction times. As shown in Fig. 3b, the Ru K-edge FT-EXAFS profiles of the Ru0.5Mn0.5O2 exhibited a prominent peak at 1.47 Å, corresponding to the first Ru–O coordination shell, and another peak at 2.97 Å, associated with Ru–Ru/Ru–Mn coordination in the second shell. EXAFS fitting analysis for the first-shell coordination revealed that both the bond length and coordination number (CN) of the Ru–O bond remained largely unchanged throughout the 50 h OER process (Fig. S32, S33, and Table S6, ESI†). Notably, the second-shell EXAFS fitting results showed a decrease in the coordination number of Ru–Ru/Ru–Mn with increasing reaction time (Fig. 3c), which can be attributed to the gradual rise in Mn vacancy concentration in the VMn–Ru0.5Mn0.5O2−x. We also analysed the change in the Mn coordination environment during the OER process according to the Mn K-edge EXAFS spectra (Fig. S34, S35, and Table S7, ESI†). Similarly, the bond length and coordination number of Mn–O remain stable during the OER (Fig. S36, ESI†). Moreover, the coordination number of Mn–Ru/Mn–Mn generally shows a decreasing trend with increasing reaction time, as revealed by the second-shell Mn K-edge EXAFS fitting (Fig. S37 and Table S7, ESI†), further confirming the formation of Mn vacancies in the Ru0.5Mn0.5O2 catalyst during the OER process. These results confirm that the crystal structure framework of the solid solution remains intact throughout the acidic OER process, accompanied by the formation of surface cation vacancies.
To gain an in-depth understanding of the structural evolution of the catalyst under acidic OER conditions, we conducted operando XAFS measurements. Before conducting operando XAFS tests, we activated the Ru0.5Mn0.5O2 electrode under a constant current density to obtain the VMn–Ru0.5Mn0.5O2−x. The resulting VMn–Ru0.5Mn0.5O2−x catalyst, which had undergone the self-limiting surface leaching process and contains Mn vacancies, was used for the operando XAFS testing. During the operando XAFS experiment, the voltage applied to VMn–Ru0.5Mn0.5O2−x was first increased from 1.2 V to 1.6 V vs. RHE, and then reversed back from 1.6 V to 1.2 V vs. RHE (Fig. S38, ESI†). The Ru K-edge XANES spectra recorded at different potentials showed slight changes in absorption energy (Fig. 3d and Fig. S39, ESI†). Specifically, the oxidation state of Ru first increased from +3.59 at 1.4 V to +3.78 at 1.6 V and then returned to its original oxidation state at 1.2 V-back (Fig. 3f, top), indicating that neighboring Ru atoms transferred charge to the intermediates involved in the OER. However, the valence state of Mn remained unchanged throughout the OER process, as evidenced by Mn K-edge XANES spectra (Fig. S40, ESI†). These observations indicate that the main active element and site of VMn–Ru0.5Mn0.5O2−x are Ru and the Ru–O bond. Notably, the valence state of Ru in VMn–Ru0.5Mn0.5O2−x remained below +4 under OER conditions (Fig. 3f, top), preventing overoxidation and dissolution of Ru species. To further examine potential changes in the Ru coordination environment, we conducted Ru K-edge EXAFS fitting analysis for the first-shell coordination (Fig. 3e and Fig. S41, S42, ESI†), with results summarized in Table S8 (ESI†). Changes were observed in the average Ru–O bond length or coordination number (CN) of Ru–O (Fig. 3f, bottom). In addition, after reactions were completed, the coordination number and bond length of Ru–O returned to their original states. Similarly, the Mn–O bond length and coordination number returned to their original state once the applied voltage was removed, as demonstrated by the EXAFS results for Mn K-edge (Fig. S43–S45 and Table S9, ESI†). Based on the operando XAS results, a comprehensive analysis of the changes in both oxidation number and local structure during the reaction reveals that Ru, which exhibits alterations in both its oxidation state and local structure, serves as the primary active site. In contrast, Mn, which undergoes changes solely in its local structure without any variation in its oxidation state, functions as the supporting site. It is important to note that the Mn vacancies in the VMn–Ru0.5Mn0.5O2−x effectively stabilize the structure of catalyst, thereby maintaining the stability of the oxidation state of Mn during the short-term operando XAFS testing (3.47 h) (Fig. S38, ESI†). Overall, the VMn–Ru0.5Mn0.5O2−x catalyst maintained structural stability under OER conditions, confirming its durability for acidic OER applications.
Our study has elucidated the self-limiting surface leaching process of the Ru0.5Mn0.5O2 catalyst by a comprehensive series of characterization tests. Next, we examine the OER mechanisms associated with Ru0.5Mn0.5O2. Two well-known OER mechanisms include the adsorbate evolution mechanism (AEM) and the lattice oxygen mechanism (LOM). Catalysts that follow the LOM often suffer from stability issues due to the bulk oxygen diffusion and structural reconstruction caused by the continuous formation of oxygen vacancies and dissolution of cations during lattice oxygen redox. To determine which mechanism governs the Ru0.5Mn0.5O2 catalyst, we conducted operando DEMS measurements via the isotope 18O-labelling method. We loaded VMn–Ru0.5Mn0.5O2−x, prepared Ru0.5Mn0.5O2, and C–RuO2 onto porous gas-permeable carbon paper electrodes and subjected them to cyclic voltammograms (CV) cycles in the H218O electrolyte. If the catalyst operates via the LOM, 18O would be incorporated into the catalyst surface through lattice oxygen exchange (Fig. S46a, ESI†). Conversely, catalysts following the AEM would not be labeled with 18O on their surface (Fig. S46b, ESI†). Subsequently, we thoroughly washed the catalysts with abundant H216O to remove any surface-adsorbed H218O, followed by three CV cycles in an H216O electrolyte. During the CV cycle process, we measured the isotope signal of evolved O2. As shown in Fig. S47 (ESI†), all three catalysts predominantly released 32O2, with a smaller amount of 34O2. The 32O2 signal corresponds to the AEM (Fig. 3g), while the 34O2 signal is related to the LOM (Fig. 3h). This suggests that the three catalysts primarily follow the AEM rather than the LOM. Notably, VMn–Ru0.5Mn0.5O2−x exhibited the lowest ratio of 34O2 to 32O2 (1.16% for VMn–Ru0.5Mn0.5O2−x, 2.43% for Ru0.5Mn0.5O2, and 3.87% for C–RuO2) (Fig. 3i), indicating minimal involvement of the LOM. These findings suggest that Mn doping and cation vacancies play a crucial role in inhibiting lattice oxygen participation, thereby stabilizing the catalyst surface and facilitating high catalyst stability.
In light of the above findings, we have summarized the self-limiting surface leaching mechanism of the Ru0.5Mn0.5O2 catalyst, as illustrated in Fig. 1a. Initially, Mn is uniformly doped into the RuO2, creating a Ru–Mn solid solution oxide with a relatively perfect rutile structure (Ru0.5Mn0.5O2). Subsequently, Mn on the catalyst surface dissolves through the LOM pathway during the initial acidic OER process, resulting in a defective Ru0.5Mn0.5O2 structure with abundant surface Mn vacancies (VMn–Ru0.5Mn0.5O2−x). Lastly, the newly formed surface primarily follows the AEM pathway, with the LOM pathway being suppressed, thus inhibiting further leaching of Ru/Mn and achieving a stable acidic OER.
Theoretical calculations
Density functional theory (DFT) calculations were performed to investigate the effects of self-limiting surface leaching on catalytic activity and stability for the OER. The fully oxidized (110) surface was chosen to be used in our DFT calculations because the RuO2(110) is the most stable surface in the C–RuO2 catalyst. As shown in Fig. 4a and Fig. S48 (ESI†), there are two types of Ru sites in the terminated layer of the RuO2(110) surface: the coordinatively unsaturated site (CUS) with five coordinated O atoms and the fully coordinated bridge site (BRI) with six coordinated O atoms. Our DFT results suggest that, for a Mn dopant, it is energetically favorable to replace the BRI Ru site not the CUS site (Fig. 4b). Nevertheless, the experimental results show that the Mn demetallation occurs during the OER process, leading to the formation of Mn vacancies. In our simulations, the Mn vacancy at the BRI site and the formation of the Mn vacancy are shown in Fig. 4c and Fig. S49 (ESI†), using as the model the VMn–RuMnO2−x(110) surface. By operando DEMS measurements, we have demonstrated that the OER process mainly follows the AEM pathway on the C–RuO2, RuMnO2, and VMn–RuMnO2−x. As depicted schematically in Fig. 4d and Fig. S50 (ESI†), at the CUS Ru site, the 4e− OER on VMn–RuMnO2−x(110) can be triggered from one H2O molecule adsorption. After two sequential deprotonation steps, it forms the surface-adsorbed O (*O), which is followed by water nucleophilic attack to form an *OOH intermediate and then the deprotonation of *OOH to produce an O2 molecule. Based on the free energy profile of RuO2(110), the calculated overpotential, as the descriptor of catalytic activity, is about 0.81V (Fig. 4e and Fig. S51, Table S10, ESI†). It is contributed by the step of *OOH formation, indicating that the *OOH formation step is the rate-determining step, consistent with previous results.44,45,58 Nevertheless, on the RuMnO2(110) surface, the *OOH intermediate donates a proton to a neighboring oxygen, forming an H-stabilized *OO species (Fig. S52, ESI†).48 This process lowers the free energy barrier for O–O bond formation during the water nucleophilic attack step. As a result, the overpotential of RuMnO2(110) is 0.68 V (Fig. 4e and Table S10, ESI†), meaning that the strategy of Mn doping improves the catalytic activity of RuO2. Meanwhile, the H-stabilized *OO species were also formed on the surface of VMn–RuMnO2−x(110) (Fig. 4d and Fig. S50d, ESI†), promoting the formation of the key *OOH intermediate. More importantly, the Mn vacancy in VMn–RuMnO2−x(110) further lowers the free energy barrier of the step of H-stabilized *OO species formation, thereby regulating the rate-limiting step to obtain the lowest overpotential of 0.50 V among these three different surfaces (Fig. 4e and Table S10, ESI†).
 | ||
| Fig. 4 Theoretical calculations. Atomistic structures of the surfaces of RuO2(110) (a), RuMnO2(110) (b), and VMn–RuMnO2−x(110) (c). (d) Schematic illustration of AEM for the OER on the VMn–RuMnO2−x(110) surface. (e) Computed free energy evolution of the OER via AEM on surfaces of RuO2(110), RuMnO2(110) and VMn–RuMnO2−x(110) under an electrode potential of 0V vs. RHE. (f) The projected densities of states (PDOS) of Mn and Ru atom in the top-atom layer of RuMnO2(110). (g) Calculated Ru demetallation energies for structural degradation of the surfaces of RuO2(110), RuMnO2(110), and VMn–RuMnO2−x(110). | ||
To reveal the formation of Mn vacancies, the PDOS of the Mn and Ru atoms in the top layer of the RuMnO2(110) surface is plotted in Fig. 4f, showing that the density of states below the Fermi level for Mn is lower than that for Ru (such as, ranging from −2 eV to 0 eV). This suggests that the Mn atom is more likely to be oxidized to a higher oxidation state than the Ru atom, which could lead to the easier demetallation of Mn (MnO4−) compared to Ru (RuO4), thereby resulting in the formation of Mn vacancies rather than Ru vacancies in the Ru0.5Mn0.5O2 catalyst. As one the descriptors of structural stability, the Ru demetallation energy can be calculated based on the proposed mechanism for Ru demetallation (Fig. S53–S55, ESI†). Compared to the demetallation energy of the RuO4 for the RuO2(110) surface (1.54 eV), Mn doping increases the demetallation energy to be 2.18 eV (Fig. 4g), indicating that Mn doping stabilizes the CUS Ru atom on the RuMnO2(110) surface. Importantly, the demetallation energy of RuO4 further increases to 2.36 eV in the VMn–RuMnO2−x(110) surface (Fig. 4g), suggesting that the formation of Mn vacancies also improves the stability of the CUS Ru site. Therefore, these calculations shows that the Mn doping and Mn vacancies in VMn–RuMnO2−x not only enhance the catalytic activity of Ru sites, but also improve the stability of the catalyst.
Actually, the Mn dissolution results in the formation of a Mn vacancy (VMn), which changes the coordination of the neighboring CUS Ru site, but slightly affects the other CUS Ru atoms that are further away from the VMn site. As a result, the VMn leads to a higher density of states below the Fermi level of the CUS Ru atom than that of other Ru atoms further away from the VMn site (such as, ranging from −2 to 0 eV, Fig. S56, ESI†). This indicates that the Ru atom neighboring the VMn site has a greater ability to resist changes in its valence state compared to other Ru atoms further away from the VMn site. In other words, VMn stabilizes the neighboring Ru atoms, but exerts a weaker stabilizing effect on those further from the VMn site. To further support this conclusion, we calculated the demetallation energies of Ru atoms at various distances from the VMn site. As shown in Fig. S57 (ESI†), demetallation energies of CUS Ru atoms show a dependency on the distance between the Ru site and the VMn site, and it decreases as the distance increases. Therefore, the VMn tends to inhibit the demetallation of the neighboring CUS Ru atom, but slightly affects the Ru atoms that are further away from the VMn site. Meanwhile, the VMn also affects the later Mn dissolution, as confirmed by calculations of the electronic energies of models containing two Mn vacancies at varying distances (VMn–VMn distance). As shown in Fig. S58a and b (ESI†), the electronic energy depends on the VMn–VMn distance, increasing as the VMn–VMn distance decreases. This indicates that the later VMn formation is more likely to be contributed by the Mn site farther from the VMn site, rather than the Mn site closer to the VMn site. Because the VMn changes the coordination of neighboring Mn atom, it has a slight effect on the other Mn atoms further away from this VMn site. It leads to a lower density of states of the Mn atom close to the VMn site compared to those farther away (such as, ranging from −2 eV to 0 eV, Fig. S58c, ESI†). This suggests that the Mn atom further away from the VMn site is likely to be oxidized to a higher oxidation state, leading to Mn demetallation (MnO4−). As a result, these VMn sites tend to be distributed in a scattered manner on the surface.
Conclusions
In summary, we have clarified the self-limiting surface leaching mechanism in Ru–Mn solid solution oxides, demonstrating its effectiveness in enhancing the long-term stability of the catalyst during acidic OERs. Specifically, unstable RuMnO2 undergoes a transformation into a corrosion-resistant VMn–RuMnO2−x with abundant surface Mn vacancies through this self-limiting surface leaching process during the initial OER phase. Operando synchrotron characterization combined with theoretical calculations reveals that cation vacancies and Mn dopants stabilize Ru species by increasing the demetallation energy of Ru active sites, thereby improving the long-term stability of VMn–RuMnO2−x. Furthermore, the synergy between Mn dopants and cationic vacancies facilitates the formation of the H-stabilized *OO intermediate and regulates the rate-limiting step, effectively lowering the activation free energy and enhancing OER activity. Leveraging these advantages, the VMn–RuMnO2−x catalyst achieves unprecedented durability alongside exceptional catalytic activity for the acidic OER, as verified in practical PEM water electrolyzers. These insights into the self-limiting surface leaching mechanism provide valuable guidance for developing efficient and stable acidic OER catalysts.Methods
Synthesis of Ru0.5Mn0.5O2
Firstly, RuCl3·3H2O (0.5 mmol, 130.71 mg) and MnCl2·4H2O (0.5 mmol, 98.96 mg) were dissolved in ultra-pure water with vigorous stirring. Ethylenediaminetetraacetic acid disodium salt dihydrate (EDTA-2Na) (0.5 mmol, 186.12 mg) was then added to the mixture, which was stirred for 1 h to form a homogeneous solution. Subsequently, an excess amount of anhydrous ethanol was added to the solution under stirring, and the precipitated products were collected by centrifugation and washed multiple times with anhydrous ethanol. The obtained sample, referred to as Ru0.5Mn0.5-EDTA precursors, was heated to 400 °C and held for 2 h at a heating rate of 5 °C min−1 in a muffle furnace. The resulting black solid powder was washed several times with ultra-pure water to remove NaCl impurities and was denoted as Ru0.5Mn0.5O2.Synthesis of RuyMn1−yO2 (y = 0, 0.1, 0.3, 0.7, 0.9, and 1)
The preparation of the RuyMn1−yO2 (y = 0, 0.1, 0.3, 0.7, 0.9, and 1) followed the same procedure as for Ru0.5Mn0.5O2, except for the adjustment in the amounts of RuCl3·3H2O and MnCl2·4H2O. Specifically, the total amount of Ru and Mn was kept constant at 0.1 M, with the molar ratio of Ru to Mn set as y:(1 − y).Materials characterization
Field-emission scanning electron microscopy (FESEM) characterization was conducted using a SU-8220 instrument. Low-magnification TEM images were obtained on a Hitachi-7650. XRD patterns were recorded using an Ultima IV using CuKα radiation (λ = 1.5418 Å). ICP-AES was performed on an Optima 7300 DV, and ICP-MS analysis was conducted on a PlasmaQuad 3. XPS data were collected using a Thermo ESCALAB 250Xi with AlKα radiation. Room-temperature EPR spectra of the sample were obtained using a JEOL JES-FA200 ESR spectrometer. Soft XAS spectra were acquired at the BL12B-a (MCD) beamline of the NSRL in Hefei, China. HAADF-STEM images and energy-dispersive X-ray spectra (EDX) were captured using a FEI Titan3 G2 60-300 equipped with double spherical aberration correctors. Operando DEMS measurements were conducted with a Hiden HPR-40 DEMS system to record the mass signals of 32O2 and 34O2. X-ray absorption fine structure (XAFS) data were collected at the Wide Energy XAFS beamline (10C beam line) of the Pohang Light Source-II (PLS-II), operated in top-up mode with a ring current of 250mA at 3.0GeV.
Electrochemical measurement
Electrochemical measurements were carried out using a CHI 760E electrochemical workstation at ambient temperature. Catalyst ink was prepared by dispersing 10 mg of catalyst in 280 μL of ethanol, followed by the addition of 15 μL Nafion solution, and the mixture was stirred vigorously for 2 hours. The OER activity of the catalysts was evaluated using a three-electrode configuration, comprising a carbon rod as the counter electrode, Ag/AgCl (saturated KCl) as the reference electrode, and a glassy carbon electrode (GCE) as the working electrode. For the preparation of the catalyst-coated GCE, 2 μL catalyst ink was dropped onto the GCE (0.19625 cm2), yielding a catalyst loading of 0.35 mg cm−2. To assess catalytic activity, linear sweep voltammetry (LSV) measurements were performed within the designated potential ranges at a scan rate of 1 mV s−1, incorporating iR compensation. The Nyquist plots of electrochemical impedance spectroscopy (EIS) were acquired over the frequency range of 10 kHz to 0.1 Hz with an applied amplitude of 5 mV. Subsequently, OER stability tests were also conducted using a three-electrode system, where the counter electrode was a carbon rod, the reference electrode remained Ag/AgCl (saturated KCl), and carbon paper served as the working electrode. Catalyst-loaded carbon paper electrodes were prepared by applying 40 μL of catalyst ink to carbon paper (0.5 × 1.2 cm2), with a catalyst loading of 2.26 mg cm−2. Stability assessments were made through chronopotentiometry tests at a constant current density of either 10 mA cm−2 or 100 mA cm−2 in a 0.5 M H2SO4 solution without iR compensation. Additionally, cyclic voltammetry (CV) was performed between 1.2 and 1.5 V vs. RHE at a scan rate of 100 mV s−1.To determine the double-layer capacitance (Cdl), cyclic voltammetry (CV) measurements were performed within a non-faradaic potential window of 0.896 to 0.996 V vs. RHE, using scan rates ranging from 10 to 200 mV s−1. A linear plot was generated by correlating the current density (Δj/2 at 0.946 V vs. RHE) with the scan rate. The slope of this linear fit corresponds to the Cdl. The ECSA was then derived from the Cdl using the following equation:
| ECSA = Cdl·S/Cs |
The mass activity values (A gRu−1) of the catalysts were calculated using the following equation:

In this work, all measured potentials were referenced to the RHE using the equation: E(RHE) = E(Ag/AgCl) + 0.198 V. The saturated Ag/AgCl reference electrode was calibrated in a high-purity hydrogen-saturated acidic electrolyte (0.5 M H2SO4), with platinum wire as the working electrode, a carbon rod as the counter electrode, and saturated Ag/AgCl as the reference electrode. Cyclic voltammetry (CV) was performed at a scan rate of 1.0 mV s−1. The thermodynamic potential for the hydrogen electrode reaction was determined by averaging the two potentials at which the current crossed zero (Fig. S59, ESI†).
Operando XAFS measurements
Ru and Mn K-edge X-ray absorption fine structure (XAFS) data, including X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS), were acquired at the wide energy XAFS beamline (10C beamline) of the Pohang Light Source-II (PLS-II) in top-up mode, operating at a ring current of 250 mA at 3.0 GeV. The incident beam from the multipole wiggler source was monochromatized using a Si(111) double-crystal monochromator (Bruker ASC). For operando XAFS analysis, a home-made operando three-electrode cell with polyimide film windows was utilized, consisting of a platinum counter electrode, an Ag/AgCl reference electrode, and a working electrode loaded with electrocatalysts on carbon paper (loading area: 1.5 cm2, catalyst loading: 0.5 mg cm−2). Prior to the operando XAFS measurements, the working electrode was activated by running it at a current density of 50 mA cm−2 in a 0.5 M H2SO4 electrolyte for 10 h. To obtain more surface information, the incidence angle between the incident beam and the sample were set to below 20 degrees. The operando XAFS measurements were performed during chronoamperometry (CA) experiments at selected potentials in 0.5 M H2SO4 electrolyte. After stabilization of the current response, XAFS spectra were recorded in fluorescence mode, with a measurement time of about 20 minutes per spectrum. The intensity of the incident X-ray beam was reduced by around 30% to eliminate higher-order harmonic contributions. Energy calibration was conducted simultaneously using reference metal foils for each measurement. The recorded spectra were converted into normalized XANES and Fourier-transformed radial distribution functions (RDFs) using Athena and Artemis software based on standard XAFS procedures. All EXAFS fitting procedures were performed using the Artemis program.MEA fabrication and PEM water electrolyzer cell test
The catalyst-coated membrane (CCM) method was employed to construct the membrane electrode assembly (MEA). Ru0.5Mn0.5O2 or Ru1Mn0O2 served as the anode catalyst, while commercial Pt/C (75 wt%) was utilized as the cathode catalyst, with a Nafion 115 proton exchange membrane acting as the electrolyte. For the preparation of anode and cathode inks, the catalysts were dispersed in a mixture of isopropanol, distilled water, and 5 wt% Nafion® solution. After ultrasonicating in an ice water bath for at least 30 minutes, a homogeneous catalyst ink was achieved. The anode and cathode catalysts were then air-sprayed directly onto both sides of the Nafion 115 membrane (4 cm2 geometric area) using an ultrasonic spray coating system. Catalyst loadings were controlled at 4 mgcat cm−2 for the anode and 0.5 mgPt cm−2 for the cathode. Finally, the catalyst-coated membranes were hot-pressed at 500 kPa for 3 minutes at 110 °C.The PEM electrolyzer was constructed with a titanium plate (bipolar plate) at the anode and a graphite plate (bipolar plate) with serpentine flow channels at the cathode. The MEA was sandwiched between a sintered porous titanium plate gas diffusion layer (GDL) on the anode side and a carbon paper GDL on the cathode side. The assembly was tightened to a pressure of 4 N m. During testing, both the anode and cathode plates were heated to 60 °C, while deionized water preheated to 60 °C was pumped into the anode at a flow rate of 100 mL min−1. The performance evaluation of the PEM electrolyzer was carried out using an ITECH IT-M3110 device. Polarization curves of the PEM electrolyzers were recorded over a current density range of 0.01 to 1.1 A cm−2. The stability of the electrolyzer was assessed by conducting chronopotentiometry measurements at a constant current density of 200 mA cm−2.
Computational details
We used Vienna ab initio simulation package (VASP),59,60 with generalized gradient approximation (GGA) in Perdew, Burke, and Ernzerhof (PBE) functional,61 and projector augmented wave (PAW) method, to perform all density functional theory (DFT) calculations.62 The plane-waved energy cutoff is set to be 500 eV. The convergence of force between atoms for optimization is set to 0.02 eV Å−1, and the convergence of total energy for wave function self-consistent are set to 10−5 eV. To simulate the surface of RuO2, two bottom atom layers are fixed, while the other are fully relaxed. For simulating the surface of RuMnO2, all four Mn atoms are located at BRI sites on the top atomic layer, as this configuration results in a lower electronic energy. Since these four doped Mn atoms are equivalent, we created a Mn vacancy by removing one BRI Mn atom and one bonded O atom from the surface model to simulate the surface of VMn–RuMnO2−x. The vacuum size is chosen as 18 Å to avoid interaction between two slabs for all structures.The model we used in DFT calculations cannot exactly present the Mn-doping rate, the Mn-doping sites, the proportion of Mn vacancies, as well as the distribution of all VMn sites in the experiment. Nevertheless, our calculations are effective to reveal the effect of VMn on the catalytic activity and stability. This method of DFT investigations has been successfully used to study the doping effect on RuO2 in other experiments.2,48
Author contributions
R. G. Cao, J. L. Luo, and S. H. Jiao conceived the experiments and supervised this project. Y. Liu performed the preparation, characterization and performance measurement of the catalyst. X. Y. Li carried out the theoretical calculation. H. Jiang and M. G. Kim performed the XAFS test and analysed the data. J. H. Wu and Y. Deng carried out the HAADF-STEM characterization. X. K. Xi, Z. W. Lei and Y. C. Zhang revised the manuscript. W. S. Yan assisted in conducting the soft XAS experiments. Y. Liu and R. G. Cao wrote the manuscript. All the authors discussed the results and commented on the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the support from the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDB0450302), the Shenzhen Science and Technology Program (KQTD20190929173914967, ZDSYS20220527171401003), the National Key Research and Development Program of China (Grant No. 2022YFA1504102), the National Natural Science Foundation of China (Grant No. 52225105, 22279127, 52072358, U21A2082, and 22209162), and the Fundamental Research Funds for the Central Universities (Grant No. YD2060002043). The authors appreciate the support from BL10B and BL12B-a beamlines at the National Synchrotron Radiation Laboratory (NSRL), Hefei, China. This work was partially carried out at the Instruments Center for Physical Science, University of Science and Technology of China (USTC). The authors thank the USTC Supercomputing Center for providing computational resources for this project. The authors also acknowledge the financial support from Gotion High-tech Co., Ltd.References
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- Z.-Y. Wu, F.-Y. Chen, B. Li, S.-W. Yu, Y. Z. Finfrock, D. M. Meira, Q.-Q. Yan, P. Zhu, M.-X. Chen, T.-W. Song, Z. Yin, H.-W. Liang, S. Zhang, G. Wang and H. Wang, Nat. Mater., 2022, 22, 100–108 CrossRef PubMed.
- I. Slobodkin, E. Davydova, M. Sananis, A. Breytus and A. Rothschild, Nat. Mater., 2024, 23, 398–405 CrossRef CAS PubMed.
- L. An, C. Wei, M. Lu, H. Liu, Y. Chen, G. G. Scherer, A. C. Fisher, P. Xi, Z. J. Xu and C. H. Yan, Adv. Mater., 2021, 33, 2006328 Search PubMed.
- Z. Shi, J. Li, Y. Wang, S. Liu, J. Zhu, J. Yang, X. Wang, J. Ni, Z. Jiang, L. Zhang, Y. Wang, C. Liu, W. Xing and J. Ge, Nat. Commun., 2023, 14, 843 Search PubMed.
- M. F. Lagadec and A. Grimaud, Nat. Mater., 2020, 19, 1140–1150 Search PubMed.
- J. Kibsgaard and I. Chorkendorff, Nat. Energy, 2019, 4, 430–433 CrossRef.
- Z. Chen, L. Guo, L. Pan, T. Yan, Z. He, Y. Li, C. Shi, Z. F. Huang, X. Zhang and J. J. Zou, Adv. Energy Mater., 2022, 12, 2103670 CrossRef CAS.
- Q. Shi, C. Zhu, D. Du and Y. Lin, Chem. Soc. Rev., 2019, 48, 3181–3192 Search PubMed.
- F. Liao, K. Yin, Y. Ji, W. Zhu, Z. Fan, Y. Li, J. Zhong, M. Shao, Z. Kang and Q. Shao, Nat. Commun., 2023, 14, 1248 CrossRef CAS PubMed.
- L. Zu, X. Qian, S. Zhao, Q. Liang, Y. E. Chen, M. Liu, B.-J. Su, K.-H. Wu, L. Qu, L. Duan, H. Zhan, J.-Y. Zhang, C. Li, W. Li, J. Y. Juang, J. Zhu, D. Li, A. Yu and D. Zhao, J. Am. Chem. Soc., 2022, 144, 2208–2217 CrossRef CAS PubMed.
- L. C. Seitz, C. F. Dickens, K. Nishio, Y. Hikita, J. Montoya, A. Doyle, C. Kirk, A. Vojvodic, H. Y. Hwang, J. K. Norskov and T. F. Jaramillo, Science, 2016, 353, 1011–1014 CrossRef CAS PubMed.
- S. Hao, H. Sheng, M. Liu, J. Huang, G. Zheng, F. Zhang, X. Liu, Z. Su, J. Hu, Y. Qian, L. Zhou, Y. He, B. Song, L. Lei, X. Zhang and S. Jin, Nat. Nanotechnol., 2021, 16, 1371–1377 CrossRef CAS PubMed.
- Y.-R. Zheng, J. Vernieres, Z. Wang, K. Zhang, D. Hochfilzer, K. Krempl, T.-W. Liao, F. Presel, T. Altantzis, J. Fatermans, S. B. Scott, N. M. Secher, C. Moon, P. Liu, S. Bals, S. Van Aert, A. Cao, M. Anand, J. K. Nørskov, J. Kibsgaard and I. Chorkendorff, Nat. Energy, 2021, 7, 55–64 Search PubMed.
- A. Mayyas, M. Ruth, B. Pivovar, G. Bender and K. Wipke, Manufacturing Cost Analysis for Proton Exchange Membrane Water Electrolyzers, 2019, https://www.nrel.gov/docs/fy19osti/72740.pdf Search PubMed.
- J. Gao, H. Tao and B. Liu, Adv. Mater., 2021, 33, 2003786 CrossRef CAS PubMed.
- J. Ni, Z. Shi, Y. Wang, J. Yang, H. Wu, P. Wang, M. Xiao, C. Liu and W. Xing, eScience, 2025, 5, 100295 CrossRef.
- L. Hou, X. Gu, X. Cui, J. Tang, Z. Li, X. Liu and J. Cho, EES Catal., 2023, 1, 619–644 RSC.
- K. A. Stoerzinger, R. R. Rao, X. R. Wang, W. T. Hong, C. M. Rouleau and Y. Shao-Horn, Chem, 2017, 2, 668–675 CAS.
- C. Roy, R. R. Rao, K. A. Stoerzinger, J. Hwang, J. Rossmeisl, I. Chorkendorff, Y. Shao-Horn and I. E. L. Stephens, ACS Energy Lett., 2018, 3, 2045–2051 CrossRef CAS.
- C. Rong, K. Dastafkan, Y. Wang and C. Zhao, Adv. Mater., 2023, 35, 2211884 CrossRef CAS PubMed.
- H. Li, Y. Lin, J. Duan, Q. Wen, Y. Liu and T. Zhai, Chem. Soc. Rev., 2024, 53, 10709–10740 RSC.
- Q. Wang, Y. Cheng, H. B. Tao, Y. Liu, X. Ma, D. S. Li, H. B. Yang and B. Liu, Angew. Chem., Int. Ed., 2023, 62, e202216645 CrossRef CAS PubMed.
- S. Cherevko, A. R. Zeradjanin, A. A. Topalov, N. Kulyk, I. Katsounaros and K. J. J. Mayrhofer, ChemCatChem, 2014, 6, 2219–2223 CrossRef CAS.
- A. Grimaud, O. Diaz-Morales, B. Han, W. T. Hong, Y.-L. Lee, L. Giordano, K. A. Stoerzinger, M. T. M. Koper and Y. Shao-Horn, Nat. Chem., 2017, 9, 457–465 CrossRef CAS PubMed.
- K. Klyukin, A. Zagalskaya and V. Alexandrov, J. Phys. Chem. C, 2019, 123, 22151–22157 CrossRef CAS.
- A. Li, H. Ooka, N. Bonnet, T. Hayashi, Y. Sun, Q. Jiang, C. Li, H. Han and R. Nakamura, Angew. Chem., 2019, 131, 5108–5112 CrossRef.
- F.-Y. Chen, Z.-Y. Wu, Z. Adler and H. Wang, Joule, 2021, 5, 1704–1731 CrossRef CAS.
- W. Zhao, Y. Liu, X. Fu and W. Wang, Renewables, 2023, 1, 638–667 CrossRef.
- Z. Wang, X. Guo, J. Montoya and J. K. Nørskov, npj Comput. Mater., 2020, 6, 160 CrossRef.
- L. Chong, G. Gao, J. Wen, H. Li, H. Xu, Z. Green, J. D. Sugar, A. J. Kropf, W. Xu, X. M. Lin, H. Xu, L. W. Wang and D. J. Liu, Science, 2023, 380, 609–616 CrossRef CAS PubMed.
- S. Cherevko, S. Geiger, O. Kasian, N. Kulyk, J.-P. Grote, A. Savan, B. R. Shrestha, S. Merzlikin, B. Breitbach, A. Ludwig and K. J. J. Mayrhofer, Catal. Today, 2016, 262, 170–180 CrossRef CAS.
- M. Guo, R. Deng, C. Wang and Q. Zhang, J. Energy Chem., 2023, 78, 537–553 CrossRef CAS.
- G. Zhao, W. Guo, M. Shan, Y. Fang, G. Wang, M. Gao, Y. Liu, H. Pan and W. Sun, Adv. Mater., 2024, 36, e2404213 CrossRef PubMed.
- W. Liu, Z. Duan and W. Wang, J. Phys. Chem. C, 2023, 127, 5334–5342 CrossRef CAS.
- J. Chen, Y. Ma, T. Huang, T. Jiang, S. Park, J. Xu, X. Wang, Q. Peng, S. Liu, G. Wang and W. Chen, Adv. Mater., 2024, 36, 2312369 CrossRef CAS PubMed.
- L. An, F. Yang, C. Fu, X. Cai, S. Shen, G. Xia, J. Li, Y. Du, L. Luo and J. Zhang, Adv. Funct. Mater., 2022, 32, 2200131 CrossRef CAS.
- C. F. Dickens and J. K. Nørskov, J. Phys. Chem. C, 2017, 121, 18516–18524 CrossRef CAS.
- D. Galyamin, J. Torrero, I. Rodríguez, M. J. Kolb, P. Ferrer, L. Pascual, M. A. Salam, D. Gianolio, V. Celorrio, M. Mokhtar, D. Garcia Sanchez, A. S. Gago, K. A. Friedrich, M. A. Peña, J. A. Alonso, F. Calle-Vallejo, M. Retuerto and S. Rojas, Nat. Commun., 2023, 14, 2010 CrossRef CAS PubMed.
- Y. Qin, T. Yu, S. Deng, X.-Y. Zhou, D. Lin, Q. Zhang, Z. Jin, D. Zhang, Y.-B. He, H.-J. Qiu, L. He, F. Kang, K. Li and T.-Y. Zhang, Nat. Commun., 2022, 13, 3784 CrossRef CAS PubMed.
- X. Bai, X. Zhang, Y. Sun, M. Huang, J. Fan, S. Xu and H. Li, Angew. Chem., Int. Ed., 2023, 62, e202308704 CrossRef CAS PubMed.
- C. Lin, J.-L. Li, X. Li, S. Yang, W. Luo, Y. Zhang, S.-H. Kim, D.-H. Kim, S. S. Shinde, Y.-F. Li, Z.-P. Liu, Z. Jiang and J.-H. Lee, Nat. Catal., 2021, 4, 1012–1023 Search PubMed.
- S. Hao, M. Liu, J. Pan, X. Liu, X. Tan, N. Xu, Y. He, L. Lei and X. Zhang, Nat. Commun., 2020, 11, 5368 CrossRef CAS PubMed.
- D. Zhang, M. Li, X. Yong, H. Song, G. I. N. Waterhouse, Y. Yi, B. Xue, D. Zhang, B. Liu and S. Lu, Nat. Commun., 2023, 14, 2517 CrossRef CAS PubMed.
- Y. Shen, X.-L. Zhang, M.-R. Qu, J. Ma, S. Zhu, Y.-L. Min, M.-R. Gao and S.-H. Yu, Nat. Commun., 2024, 15, 7861 CrossRef CAS PubMed.
- Y. Li, W. Wang, M. Cheng, Y. Feng, X. Han, Q. Qian, Y. Zhu and G. Zhang, Adv. Mater., 2023, 35, 2206351 CrossRef CAS PubMed.
- L. Hou, Z. Li, H. Jang, Y. wang, X. Cui, X. Gu, M. G. Kim, L. Feng, S. Liu and X. Liu, Adv. Energy Mater., 2023, 13, 2300177 CrossRef CAS.
- H. Jin, X. Liu, P. An, C. Tang, H. Yu, Q. Zhang, H.-J. Peng, L. Gu, Y. Zheng, T. Song, K. Davey, U. Paik, J. Dong and S.-Z. Qiao, Nat. Commun., 2023, 14, 354 CrossRef CAS PubMed.
- J. Zhu, Y. Guo, F. Liu, H. Xu, L. Gong, W. Shi, D. Chen, P. Wang, Y. Yang, C. Zhang, J. Wu, J. Luo and S. Mu, Angew. Chem., Int. Ed., 2021, 60, 12328–12334 CrossRef CAS PubMed.
- N. Yao, H. Jia, J. Zhu, Z. Shi, H. Cong, J. Ge and W. Luo, Chem, 2023, 9, 1882–1896 CAS.
- J. Wang, C. Cheng, Q. Yuan, H. Yang, F. Meng, Q. Zhang, L. Gu, J. Cao, L. Li, S.-C. Haw, Q. Shao, L. Zhang, T. Cheng, F. Jiao and X. Huang, Chem, 2022, 8, 1673–1687 CAS.
- Y. Yao, S. Hu, W. Chen, Z.-Q. Huang, W. Wei, T. Yao, R. Liu, K. Zang, X. Wang, G. Wu, W. Yuan, T. Yuan, B. Zhu, W. Liu, Z. Li, D. He, Z. Xue, Y. Wang, X. Zheng, J. Dong, C.-R. Chang, Y. Chen, X. Hong, J. Luo, S. Wei, W.-X. Li, P. Strasser, Y. Wu and Y. Li, Nat. Catal., 2019, 2, 304–313 CrossRef CAS.
- C. Rong, X. Shen, Y. Wang, L. Thomsen, T. Zhao, Y. Li, X. Lu, R. Amal and C. Zhao, Adv. Mater., 2022, 34, 2110103 CrossRef CAS PubMed.
- Y. Wen, C. Liu, R. Huang, H. Zhang, X. Li, F. P. García de Arquer, Z. Liu, Y. Li and B. Zhang, Nat. Commun., 2022, 13, 4871 CrossRef CAS PubMed.
- J. Liu, T. Wang, Z. Lin, M. Liao, S. Liu, S. Wang, Z. Cai, H. Sun, Y. Shen, Y. Huang and Q. Li, Energy Environ. Sci., 2024, 17, 3088–3098 RSC.
- K. Wang, Y. Wang, B. Yang, Z. Li, X. Qin, Q. Zhang, L. Lei, M. Qiu, G. Wu and Y. Hou, Energy Environ. Sci., 2022, 15, 2356–2365 RSC.
- Z. L. Zhao, Q. Wang, X. Huang, Q. Feng, S. Gu, Z. Zhang, H. Xu, L. Zeng, M. Gu and H. Li, Energy Environ. Sci., 2020, 13, 5143–5151 RSC.
- Y. Wang, X. Lei, B. Zhang, B. Bai, P. Das, T. Azam, J. Xiao and Z. S. Wu, Angew. Chem., Int. Ed., 2023, 63, e202316903 CrossRef PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary figures and tables. See DOI: https://doi.org/10.1039/d4ee05220j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |