
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective separation of Am(III)/Eu(III) using heterocyclic bistriazolyl phosphonate grafted zirconia and titania solid phase extractants†
O.-M.
Hiltunen
 a,
T.
Suominen
a,
J.
Aho
a,
M.
Otaki
a,
A.
Zupanc
ab,
S.
Hietala
a,
G.
Silvennoinen
a and
R.
Koivula
a
a,
T.
Suominen
a,
J.
Aho
a,
M.
Otaki
a,
A.
Zupanc
ab,
S.
Hietala
a,
G.
Silvennoinen
a and
R.
Koivula
a
aDepartment of Chemistry – Radiochemistry Unit, FI-00014 University of Helsinki, A. I. Virtasen Aukio 1, P.O. Box 55, Helsinki, Finland. E-mail: otto-matti.hiltunen@helsinki.fi
bFaculty of Chemistry and Chemical Technology, University of Ljubljana, Večna pot 113, 1000 Ljubljana, Slovenia
First published on 21st November 2024
Abstract
Surface functionalization of metal oxides with phosphonic acid monolayers by covalent bonding enables the generation of robust hybrid materials with enhanced separation properties. Mesoporous crystalline zirconia and titania serve as applicable inorganic supports with high thermal stability and resistance to oxidation, acidity and radiolysis. We have fabricated selective solid phase extractants that efficiently separate americium and europium from each other, via straightforward grafting of the zirconia and titania surfaces with N- and S-donor complexing agents, namely 2,6-bis-triazolyl-pyridine derivatives. Separation factors (Am/Eu) up to 13 were obtained in binary solution at pH 2 and preference for Am over Eu was observed even in Eu excess solution. These stable hybrid materials can be utilized for separation purposes without substantial degradation, providing advantageous reusability and a greener option in comparison to commonly used solvent extraction methods.
Introduction
Spent nuclear fuel (SNF) treatment and recycling has emerged as one of the most significant barriers in the development of sustainable nuclear energy. A remarkable amount of high-level liquid waste (HLLW) is composed during the common plutonium uranium extraction separation (PUREX) process, still containing minor actinides (MA) with long half-lives, which need to be separated and transmutated before final disposal in ideal cases.1–3 The neutron capture properties of lanthanides will hamper the possible transmutation process making the separation of actinides (An) and lanthanides (Ln) necessary prior to optimal treatment of the SNF.4,5 To date, this separation has remained challenging due to their parallel oxidation state, electrostatic properties and ionic radii.6,7 One of the most significant chemical differences between these two groups is the greater availability of valence electrons within actinides in comparison to lanthanides. This allows higher covalent contribution to metal–ligand bonding compared to lanthanides.8–10 Both lanthanides and actinides are classified as hard acids, according to Pearson's HSAB principle,11,12 but lanthanides with smaller frontier 4f orbitals are considered slightly harder. Therefore they are preferentially coordinated to hard oxygen donor ligands, such as commonly reported malonamide and diglycolamide (DGA) moieties, that represent the Lewis basic character.13,14 In comparison, the utilization of soft, large and polarizable nitrogen or sulfur donors is preferable for actinide extraction, and it has been shown that ligands including these N-or S-donor atoms can exploit this small but significant difference between the An/Ln chemistry.15,16 N-heterocyclic pyridyl ligands are a class of extractants that have recently attracted increased interest for their potential to carry out efficient An/Ln separation by solvent extraction methods.6,13,17–19 However, solvent extraction strategies have encountered challenges, such as the radiolytic and hydrolytic stability of the ligands and generation of notable amounts of secondary liquid waste with expensive disposal demands during multiple cycles of extraction.20,21A wide range of applicable solid-phase extraction (SPE) materials have been developed to overcome the difficulties with solvent extraction methods. These solid sorbents provide an alternative approach for separation purposes in a greener fashion due to minimized waste production and higher reusability.
Solid supports, such as silica, polymers or activated carbon-based materials, can be exploited for their high surface area and active pore surfaces which can be functionalized by organic extractants.21,22 Moreover, the properties of the well-designed frameworks can be tailored, i.e. pore size and volume. Most of the reported surface modification techniques, in which the coordinating ligands are anchored onto solid supports, entail impregnation, coating or post-synthetic grafting via covalent bonding.22–25 The preorganization of ligands fixed on a solid support can promote the extraction of trivalent actinides and lanthanides and selective enrichment due to aggregate formation between the ligands and the metal cations.26 Additionally, when applying the SPE materials for dynamic pressurized flow-through separation columns, the sorbents should be able to show compatible pore structure and tunable pore volume, as well as adjustable pore connectivity and surface properties.21,27
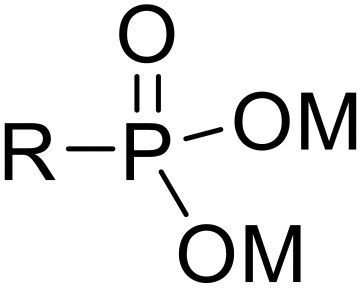
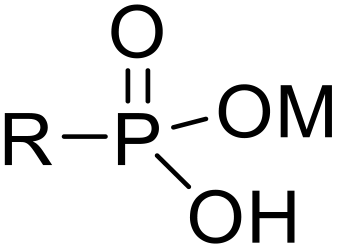
Recently, surface phosphonation has attracted great research interest in the field of interfacial chemistry.28 Metal oxides provide a robust framework which can be grafted with phosphonic acid monolayers to produce phosphonate oxide hybrid materials. The covalent nature of P–O–M bonds within these solid sorbents has remarkable hydrolytic durability, and inorganic supports, such as zirconia and titania, are known to be resistant against heat, oxidation and radiation.28,29
Heterocyclic triazinyl-based N-donor ligands such as terdentate bis-triazinyl-pyridines (BTPs) (Fig. 1a), bis-triazinyl-bipyridines (BTBPs) and bis-triazinyl-phenanthrolines (BTPhens) have emerged as very promising classes of ligands for selective separation of minor actinides from lanthanides in nitric acid media over a wide pH range among solvent extraction studies.1,7,23,30 The strong binding capability of these ligands is mainly related to the σ-donation of the nitrogen into the metal orbitals together with the π-accepting character of the heteroring. Both central pyridyl nitrogen and triazinyl nitrogen atoms participate in the formation of this tridentate binding cavity.17,18 Triazinyl pyridine ligands are also a promising class of extractants due to their convenient “click” chemistry synthesis pathway and the resulting functionalization possibilities.31 Most of the reported SPE materials utilized for selective MA–lanthanide separation entail silica, titania beads and polymer resin frameworks functionalized with BTP derivative ligands with post-synthetic grafting methods.22,23,25,32
 | ||
| Fig. 1 The structure of triazinyl (a) and triazolyl (b) pyridine ligands recently used for MA–lanthanide separation and triazolyl phosphonic acid ligands used in this study (c–e). | ||
Besides the aforementioned N-donor ligands, 2,6-bis(1,2,3-triazol-4-yl)pyridine (commonly PyTri) and its lipophilic derivatives with a relatively similar chelating unit (Fig. 1b) have proved as efficient candidates for selective actinide extraction under SANEX conditions with fast kinetics and good radiochemical stability.33,34 Along with symmetric bistriazinyl ligands, N-heterocyclic pyridyl ligands bearing the five membered pyrazole ring (Pypz and PypzH) have shown promising Am/Eu separation properties via bidentate coordination, with a strong effect of alkyl substituents of the pyrazole on the selectivity.35,36 Furthermore, S-donor candidates, such as dithiophosphinic acids, dithiophosphinates and dithiocarbamates, have been shown to preferentially extract actinides over lanthanides.37,38 Thioethers, in turn, are suspected to be more stable than thiophosphinates since the C–S bond is stronger than the S–P bond. Thioethers with a thiophene center unit have exhibited high selectivity and complex formation toward actinides in recent DFT studies.39
Herein, we report novel hybrid separation materials that employ phosphonates as compatible linkages for anchoring the desired heterocyclic triazolyl derivates into a zirconia or titania support (Fig. 1c–e). The ligands can be further hydrolyzed to the phosphonic acid form prior to coordination to the metal oxide framework via condensation between surface hydroxyl groups of the metal oxide and P–OH groups of phosphonic acid. This enables straightforward grafting of the ligands onto the inorganic support via covalent P–O–M bonding. The post-synthetic grafting method was adopted for the modification of the surface of porous zirconia and titania since the macroscopic structures of the porous metal oxides are predicted to remain unchanged during the grafting process while the surface and interface properties can be controlled. In addition, the phosphonate ligands are durable under acidic conditions and tend to form robust and uniform phosphonate layers.28 The advantageous stability of the inorganic matrix and the selective nature of the extracting ligands are combined in these inorganic–organic hybrid sorbents.
Materials and methods
Chemicals
All organic reactants were purchased from Sigma Aldrich and they were of reagent grade or higher. Reagents were used as supplied without further purification. Zirconium dioxide pellets (monoclinic) were purchased from Saint-Gobain NorPro. Crystalline titanium dioxide was prepared according to the literature procedure with modifications (details in ESI,† chapter II). 2-Azidoethylphosphonate was synthesized following the procedure reported by Ma et al.40 2,6-Bis(1,2,3-triazol-1-diethylethanephosphonate)pyridine (PyTri) and ethynylpyridine-1,2,3-triazol-1-diethylethanephosphonate (EPTEP) ligands were synthesized according to the previously reported synthesis pathway by Brunet et al. with modifications (details in ESI,† chapter I).41 The 2,5-diethynylthiophene precursor was prepared according to the previously reported literature protocol by Roy et al.42 2,5-Bis(1,2,3-triazol-1-diethylethanephosphonate)thiophene (BTEPT) was synthesized from 2,5-diethynylthiophene and 2-azidoethylphosphonate utilizing a similar synthesis route to that used also with PyTri and EPTEP ligand synthesis (details in ESI,† chapter I).41 Carrier-free Am-241 was obtained from Eckert&Ziegler Nuclitec GmBH and carrier-free Eu-152 tracer was obtained from Isotope Product Laboratories, respectively.Synthesis of Zr and Ti surface grafted hybrid materials
Hybrid SPE materials were prepared by a straightforward reaction between the phosphonate ligand solutions and the porous metal oxide supports. The metal oxides were ground into small particles and then sieved to a particle size between 74 and 149 μm. ZrPyTri1, ZrPyTri2 and TiPyTri materials were prepared by first dissolving the PyTri ligand in 1 M HCl to afford approximately 10 mM (ZrPyTri2 and TiPyTri) or 5 mM (ZrPyTri1) solution (details in ESI† chapter II). The solution was then mixed with zirconium dioxide (300 mg, 2.44 mmol) or titanium dioxide (200 mg, 2.50 mmol in a sealed vessel. The resulting suspension was shaken vigorously and mixed in a rotary mixer at a constant speed of 50 rpm for 24 h at room temperature. The mixture was centrifuged at 2773 rcf for 10 minutes and the supernatant was discarded. The remaining solid material was washed with 10 mL of distilled water using the centrifuge for phase separation. The water phase was removed, and the material was treated in the same way five times. Finally, the material was transferred to a crucible and dried in oven at 70 °C for 24 h. EPTEP and BTEPT ligands were used in a similar manner to obtain ZrEPTEP and ZrBTEPT hybrid materials (details in ESI,† chapter II).Instrumentation for characterization of hybrid materials
X-ray diffraction (XRD)
The crystal phases of the products were identified by their X-ray diffraction patterns. XRD-measurements were carried out on a Malvern PANalytical X'Pert monochromatic powder diffractometer. Copper Kα1 X-rays of wavelength 1.54056 Å were used operating at 40 kV and 40 mA. Unit cell parameters were calculated using UnitCell software.Infrared (IR) spectroscopy
Infrared transmittances were determined over wavenumbers from 4000 to 400 cm−1 with a Bruker Alpha FTIR spectrometer fitted with a single reflection attenuated total reflection (ATR) sampling accessory and Spectrum software.Scanning electron microscopy (SEM)
The morphology of the zirconia and titania hybrids was investigated with a Hitachi S-4800 field emission scanning electron microscope (FESEM). The samples were coated with a 10.9 nm carbon layer in order to prevent the surface charge effect.Compositional analysis
The contents of carbon, nitrogen and sulfur in the hybrid materials were analyzed by combustion methods with the use of a Thermo Scientific Flash 2000 elemental analyzer.Nitrogen adsorption–desorption isotherms
For the nitrogen adsorption–desorption isotherms, the samples were measured at 77 K and the isotherms were obtained using a Quantachrome Instruments Autosorb iQ by degassing at 180 °C for 7 h. The surface areas were calculated according to the Brunauer–Emmett–Teller (BET) method. The pore volumes and size distributions were obtained by using the internal density functional theory (DFT) method of equipment.Liquid state NMR
The 1H, 13C and 31P NMR spectra of the ligands synthesized were obtained using a 400 MHz Bruker Avance Neo 400 spectrometer with the CDCl3 central line at 7.26 ppm, D2O at 4.79 and (CD3)2SO at 2.50 as a reference.Solid-state nuclear magnetic resonance (NMR)
The spectral data of solid-state 31P magic angle spinning (MAS) NMR of the hybrid phosphonic acid materials was recorded on a Bruker Avance III 500 MHz spectrometer equipped with a 4 mm H/X/Y MAS probe. The solid samples were filled into a 4 mm zirconia rotor and spun at a rate of 10 kHz in order to separate the spinning side bands with the main resonance. 31P NMR spectra were acquired with a 90° pulse (77 kHz RF), a 100 s recycle delay, and 64 scans. The 31P chemical shift reference was external 85% H3PO4 at 0 ppm. The peak fitting iteration algorithm with Gaussian distribution of peaks was performed for peak deconvolution using the OriginLab Origin 2020 program.Thermogravimetric analysis (TGA)
The thermogravimetric analysis (TGA) was conducted using a Netzsch STA 449F3 Jupiter, and the heating rate was set to 10 °C min−1 under an air gas and nitrogen flow of 20 mL min−1, with a temperature range of 25 to 1000 °C.Sorption methodology
Elemental sorption studies for synthesized materials were performed at pH 1–3 in nitric acid media representing PUREX raffinate conditions via a batch method. 10 ± 0.2 mg of the material was placed in a 20 mL polyethylene vial with 5 mL of nitric acid solution. The mixture was spiked with gamma-emitting carrier-free 241Am3+ or 152Eu3+ tracers separately to result in ∼30 Bq mL−1 solution. The pH was adjusted by adding small amounts of HNO3 or NaOH aqueous solution. Binary solution experiments with competitive sorption were performed with mixed 241Am/152Eu solutions. In order to obtain an equimolar solution, inactive Eu(III) nitrate was added to obtain a 9.8 × 10−10 mol L−1 solution of 241Am and 9.8 × 10−10 mol L−1 of 152Eu/Eu. Mixed 241Am and 152Eu solution with a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 Am:Eu, containing 9.8 × 10−10 mol L−1 241Am and 39.2 × 10−9 mol L−1 Eu, was similarly prepared by using inactive Eu(III) nitrate, to render the approximate MA:Ln ratio in SNF.4 Batch experiments with varying amounts of added NaNO3 were performed in equimolar solution of 241Am/152Eu and the samples were treated in a similar manner but adding the desired amount of NaNO3 to the sample. All the batch sorption experiments were carried out with duplicate samples for both the samples and the standards. The batch factor V/m in eqn (1) for the samples was 500 mL g−1. The samples were equilibrated in a constant rotary mixer (50 rpm) for 24 h followed by phase separation using a Thermo Fisher Scientific Heraeus Megafuge 1.0R with 2773 × g for 10 minutes. The solutions were then filtered through 0.2 μm PVDF membrane syringe filters (Supor, Pall Laboratory Corporation). The 241Am and 152Eu gamma activities in the solutions were measured before (A0) and after (A) contact with the sorbents by GEM-25195 and GC4519 gamma counting detectors (GEM Series HPGe coaxial detector system). The equilibrium pH of each sample after sorption was measured from the filtrate with a Thermo Scientific Orion 3 Star pH Benchtop Meter. Errors reported for Kd values were calculated as standard deviation of the gamma counting results.
:40 Am:Eu, containing 9.8 × 10−10 mol L−1 241Am and 39.2 × 10−9 mol L−1 Eu, was similarly prepared by using inactive Eu(III) nitrate, to render the approximate MA:Ln ratio in SNF.4 Batch experiments with varying amounts of added NaNO3 were performed in equimolar solution of 241Am/152Eu and the samples were treated in a similar manner but adding the desired amount of NaNO3 to the sample. All the batch sorption experiments were carried out with duplicate samples for both the samples and the standards. The batch factor V/m in eqn (1) for the samples was 500 mL g−1. The samples were equilibrated in a constant rotary mixer (50 rpm) for 24 h followed by phase separation using a Thermo Fisher Scientific Heraeus Megafuge 1.0R with 2773 × g for 10 minutes. The solutions were then filtered through 0.2 μm PVDF membrane syringe filters (Supor, Pall Laboratory Corporation). The 241Am and 152Eu gamma activities in the solutions were measured before (A0) and after (A) contact with the sorbents by GEM-25195 and GC4519 gamma counting detectors (GEM Series HPGe coaxial detector system). The equilibrium pH of each sample after sorption was measured from the filtrate with a Thermo Scientific Orion 3 Star pH Benchtop Meter. Errors reported for Kd values were calculated as standard deviation of the gamma counting results.
Determination of the distribution coefficient
The distribution coefficient (Kd, mL g−1) describes the distribution of Am3+ and Eu3+ between the solution and the solid sorbent. Values of Kd were determined for 152Eu and 241Am individually using eqn (1): | (1) |
 denotes the activity concentration of the metal in the sorbent after contact. Correspondingly, [M3+]i and [M3+]t represent the activity or concentration of the metal in the initial solution and the liquid phase after contact with the sorbent. V stands for the volume of the solution and m is the mass of the sorbent. The separation factor (SFAm/Eu) between Am3+ and Eu3+ cations can be determined based on their Kd values (eqn (2)):
denotes the activity concentration of the metal in the sorbent after contact. Correspondingly, [M3+]i and [M3+]t represent the activity or concentration of the metal in the initial solution and the liquid phase after contact with the sorbent. V stands for the volume of the solution and m is the mass of the sorbent. The separation factor (SFAm/Eu) between Am3+ and Eu3+ cations can be determined based on their Kd values (eqn (2)): | (2) |
Results and discussion
Characterization of the hybrid materials
The reason for using high hydrochloric acid concentration in the hybrid synthesis is to enhance the esterification between the phosphonic acid ligand and the hydroxyl groups on the metal oxide surface.43 Functionalization of zirconia and titania matrices was confirmed with FTIR spectra, elemental combustion (CHNS), solid state NMR and thermogravimetric analysis. In addition, X-ray diffraction, nitrogen porosimetry and scanning electron microscopy were used to evidence the effect of the hybridization on the pore structures and the substantial impact on the morphology of the materials.Infrared spectroscopy
FTIR-ATR spectroscopy gave insights into the structure and coordination of the grafted zirconia and titania materials. The spectra for all the grafted hybrid materials, as well as bare zirconia and titania are shown in Fig. 2. IR transmittance of the grafted products showed strong absorption peaks around 900–1200 cm−1, which were assigned as vibrations of the orthophosphate group (P–O stretching),44 with the P–OH vibration located at the lower end of the shoulder around 900–970 cm−1. In this region, the obtained products showed a slight difference in their absorbance and wavenumber shifts. The absorbance is stronger for ZrPyTri in comparison to ZrEPTEP, most probably due to the greater size of the phosphonate ligand involved. It is also evident from the spectra that TiPyTri contains more protonated free P–OH moieties in comparison to the grafted zirconia materials, which can be seen from the P–O stretch region being shifted to lower wavenumber. The smaller bands detected between 1200 and 1300 cm−1 in the spectra represent the vibration of the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond.45 The obtained phosphorous-originating bands were not detected for bare zirconia or titania, confirming that the phosphonate moieties were present in all the synthesized hybrid materials and the surface functionalization was successful on the metal oxide surfaces. Due to coordination to metal, the P–O absorbance bands have shifted to higher wavenumbers at around 1100–1150 cm−1 (IR data for bare ligands shown in ESI,† Fig. S2), since P–OH groups have replaced PO32− moieties upon functionalization to a metal center (M–O–P).46 The stronger shifts to higher wavenumber with ZrPyTri and ZrEPTEP in comparison to TiPyTri indicate that these two zirconia hybrids have relatively fewer free phosphonate groups and more P–OZr groups present. Exceptionally, ZrBTEPT has notably weaker P–O stretch in comparison to the other materials, most probably due to lower ligand loading.
O bond.45 The obtained phosphorous-originating bands were not detected for bare zirconia or titania, confirming that the phosphonate moieties were present in all the synthesized hybrid materials and the surface functionalization was successful on the metal oxide surfaces. Due to coordination to metal, the P–O absorbance bands have shifted to higher wavenumbers at around 1100–1150 cm−1 (IR data for bare ligands shown in ESI,† Fig. S2), since P–OH groups have replaced PO32− moieties upon functionalization to a metal center (M–O–P).46 The stronger shifts to higher wavenumber with ZrPyTri and ZrEPTEP in comparison to TiPyTri indicate that these two zirconia hybrids have relatively fewer free phosphonate groups and more P–OZr groups present. Exceptionally, ZrBTEPT has notably weaker P–O stretch in comparison to the other materials, most probably due to lower ligand loading.
 | ||
| Fig. 2 FTIR spectra of bare porous zirconia and titania and all the synthesized hybrid materials. | ||
Solid state NMR results
Solid-state 31P MAS NMR is particularly a demonstrative method for phosphorus speciation analysis and the observed 31P chemical shifts can be related to corresponding coordination states of the phosphonate groups. The quantified phosphonate group speciation for the materials is given in Table 1 (for the spectra see ESI,† Fig. S3). Fitting of the spectral envelopes in all the materials revealed the presence of four signals with the same approximate chemical shifts of 0–3, 6–8, 17–20 and 27–28 ppm. The 31P chemical shift of the hydrolyzed PyTri ligand was found at 23.1 ppm (ESI†) and due to the coordination to zirconium, an upfield shift of approx. 4 ppm is generated by interaction between phosphonates and the metal atoms.47,48 The signals obtained between 17 and 20 ppm can be associated with single coordination of phosphonate to the metal atom, with alternative bond lengths and angles which have an effect on the obtained shifts. More precisely, Zr or Ti bridges two oxygen atoms of the phosphonic acid group P(O)(OMO).49,50 Phosphonate moieties with increased coordination to two metal atoms appear to cause shifts around 27 to 29 ppm. The downfield shift effect might arise from an increased coordination degree.47 The peaks at 0–4 ppm are attributed to bidentate double coordinated phosphonate species on zirconia or titania.49 Monodentate species with uncoordinated OH− groups involved P(O)(OM)(OH) at 6–8 ppm are primarily composed of esterification of the OH-groups on the metal oxide surface. These species are typically detected at the upfield region around 5–15 ppm from free acid, which is in agreement with the obtained shifts of these peaks.49,51 The prevailing coordination mode for all the materials was found to be bidentate with notable monodentate coordination as well, which can be seen from the data in Table 1. The difference between quantified species of the predominant coordination state at 17–20 ppm was relatively small between the materials. Comparing the spectra of ZrPyTri and TiPyTri, it was notable that ZrPyTri had larger signals at bidentate metal coordination species. Consequently, ZrPyTri had smaller signals associated with free phosphonates. For ZrEPTEP it was obvious that the bridged coordination mode was prevalent. ZrBTEPT, in turn, indicated a higher amount of bidentate coordination species with two zirconia atoms but similarly lower coordination with three zirconium atoms. The obtained results indicated, together with the FTIR data, that a higher degree of P–OM coordination species within grafted zirconia hybrids is present and that a larger amount of free phosphonate groups is present with TiPyTri correspondingly. This may suggest enhanced selectivity for zirconia hybrids in comparison to titania.| Chemical shift (ppm) | 0 to 4 | 6 to 8 | 17 to 20 | 27 to 29 |
|---|---|---|---|---|
| P(O)(OM)2 | P(O)(OM)(OH) | P(O)(OMO) | P(O)(OM)(OM2) | |
|
|
|
|
|
|
| ZrPyTri1 | 4.3 | 9.9 | 76.3 | 9.4 |
| ZrPyTri2 | 5.4 | 10.3 | 69.7 | 14.6 |
| TiPyTri | 1.8 | 14.8 | 72.1 | 11.3 |
| ZrEPTEP | 2.4 | 8.7 | 79.7 | 9.3 |
| ZrBTEPT | 15.4 | 11.9 | 70.2 | 2.5 |


Nitrogen porosimetry
All the grafted zirconia materials showed similar nitrogen adsorption isotherms, characteristically type IV of the IUPAC classification due to the presence of a hysteresis loop in the isotherm, suggesting the capillary condensation of mesopores.52 Similar isotherm characteristics could be seen also for titania and therefore it was concluded that both metal oxides contain mesopores. The representative isotherms of ZrPyTri2 and bare zirconia as well as TiPyTri and bare titania are illustrated in Fig. 3a (for other Zr materials see ESI,† Fig. S4). More precisely, a H3 type hysteresis loop evolving at P/P0 ∼ 0.95 is typically observed with agglomerates that are composed of rigidly joined plate-like particles leading to the formation of slit-shaped pores.53 The mesopores are advantageous in sorbent materials since the active surface area is maximized allowing high adsorption capability as well as mass transfer kinetics within the pore system.54,55 The BET surface area was calculated to be approximately 100 m2 g−1 for bare zirconia and all the grafted zirconia hybrids with limited differences, as shown in the data in Table 2. The pore volumes for zirconia hybrids varied in the range of 0.21–0.26 cm3 g−1 and the mode pore widths were obtained as 6.79 nm with a narrow pore size distribution centered around 7–10 nm. For grafted titania the surface area was notably larger, 149 m2 g−1 with also a greater pore volume of 0.35 cm3 g−1 and a pore width of 7.03 nm. There was no significant difference in the surface areas between bare metal oxides and the ligand grafted hybrids as shown in Table 2. The pore width was however decreased after functionalization as illustrated in Fig. 3b. The relatively large pore size of the materials enables high amounts of ligands to be attached to the pores, and subsequently the ligands can efficiently trap M3+ cations into the pores, providing high uptake. The obtained results evidence that the surface grafting takes place only in a thin layer on the metal oxide surfaces while the macroscopic structures remain unchanged. Therefore, the mesoporous structure with large pore sizes is preserved throughout the functionalization, and these functionalized pores will be further accessible to the trivalent Am and Eu cations allowing enhanced adsorption performance.27 | ||
| Fig. 3 a) Nitrogen adsorption–desorption isotherms calculated using the DFT method for bare zirconia and titania, as well as for ZrPyTri2 and TiPyTri being demonstrative hybrids, and b) pore size distributions of the same materials. | ||
| Material | Surface area (m2 g−1) | Pore volume (cm3 g−1) | Mode pore width (nm) |
|---|---|---|---|
| ZrO2 | 102 | 0.252 | 8.46 |
| ZrEPTEP | 108 | 0.207 | 6.79 |
| ZrPyTri1 | 104 | 0.213 | 7.03 |
| ZrPyTri2 | 111 | 0.203 | 6.79 |
| ZrBTEPT | 116 | 0.257 | 6.79 |
| TiO2 | 168 | 0.415 | 9.77 |
| TiPyTri | 149 | 0.346 | 7.03 |
Thermogravimetric analysis
TG analysis results shown in Fig. 4 imply that the obtained mass loss for ZrPyTri was approximately 7% and the mass had a continuous decrease up to 600 °C. TiPyTri, in turn, showed a slightly higher mass drop around 9% with a similar trend. Both materials established a notable mass drop at approximately 350 °C, indicating the decomposition of the PyTri ligand involved. It can be clearly seen from the curves of different hybrids that the mass drop related to the ligand decomposition is detected at different temperatures with different slopes; for instance, ZrEPTEP seems to decompose at slightly higher temperature at ∼400 °C while ZrBTEPT accomplishes a smoother mass drop around 250 °C. In addition, the mass drop for ZrBTEPT is clearly smaller which is most probably related to the lower ligand loading. When comparing the curves of the bare metal oxides and grafted materials it is obvious that the mass loss observed below 200 °C is mainly related to moisture and hydrated water. The fast mass drop of bare zirconia and titania at even lower temperature is assigned to the loss of loosely bound surface water molecules and this drop for titania was larger indicating a higher amount of surface water molecules present. Consequently, the surfaces of the bare zirconia and titania are considered more hydrophilic in comparison to the grafted materials due to the hybridization of the surface. | ||
| Fig. 4 Thermogravimetric analysis of all the grafted hybrid materials and bare metal oxides. | ||
X-ray diffraction
According to the XRD data shown in Fig. 5, the synthesized hybrid zirconia materials were shown to be monoclinic (ZrPyTri2 for example), while grafted titania was mostly anatase (TiPyTri for example, for the rest of zirconia hybrids, see ESI,† Fig. S6). The obtained diffraction peaks also confirmed that the grafted zirconia and titania had a similar crystal structure, in comparison to their corresponding metal oxides (Fig. 5). The results are consistent with nitrogen adsorption isotherm results, confirming that the ligand grafting does not alter the morphology of the material. The ordered and crystalline structure of the material is desirable because it will provide chemical stability,28 and the possible size exclusion effect which would cause enhanced selectivity to the material. According to the Scherrer equation, a mean ordered size of the crystallite was calculated to be approximately 14.9 nm for ZrPyTri and 14.0 nm for TiPyTri (details in ESI,† chapter III). The obtained crystal sizes are comparable or higher to other reported porous hybrid sorbent materials.32,56 | ||
| Fig. 5 Powder XRD patterns presented for a) bare zirconia and ZrPyTri2, and b) bare titania and TiPyTri. | ||
Scanning electron microscopy
SEM images show that all the porous zirconia materials are composed of rough and irregular particles in shape, and the size range is between approx. 50 and 150 μm. The grafting did not affect the surface structure of zirconia (Fig. 6a and b, S7 ESI†). In comparison, titania materials showed more spherical shape, but approximately the same particle size (Fig. 6c and d). The SEM images indicate that zirconia materials could be considered superior in column experiments due to their sharper edges, and hence they would be less exposed to clogging. These angular shapes can be thought to be more tolerable for dynamic pressurized conditions. Generally, the morphology of the porous zirconia and titania allows the material to be closely packed and easily handled which is desirable for chromatographic column separation. | ||
| Fig. 6 SEM images of porous ZrPyTri2 (a and b) and TiPyTri (c and d). | ||
Elemental analysis
The level of functionalization of zirconia and titania hybrids was quantitatively determined by CHNS microanalysis (Table 3). Consequently, the total organic contents of the functionalized materials were calculated as the weight percentage according to the elemental analysis results. The molecular formulae of the organic functionalized molecules and the measured percentages of CHNS were used to calculate n(C)/n(N) ratios for each material. These ratios with PyTri grafted materials were all close to a theoretical value of 1.85. The deviation from the theoretical value originates mostly from the measurement uncertainty and perhaps from the incomplete hydrolysis step which may promote the carbon contents of the final hybrid material. The estimation of the functionalization level on the metal oxide surfaces can be done utilizing the obtained nitrogen contents, assuming that the structure of the ligands was preserved (see Table 3). The level of the ligand loading was then normalized relative to the surface area of the zirconia or titania matrix. PyTri2 with the larger P/Zr ratio showed a higher loading level of 0.96 molecules per nm2 compared to ZrPyTri1 with 0.67 molecules per nm2 which is consistent with the difference in ligand loading. The corresponding loading for TiPyTri was significantly lower, only 0.59 molecules per nm2 which may be a result of the higher surface area of the titania matrix. ZrEPTEP, in turn, has a loading of 1.69 ligands per nm2 which is remarkably higher in comparison to the PyTri hybrids. The most probable explanation is the smaller size and less complex structure of the EPTEP ligand that has only one phosphonic acid linkage incorporated to the zirconia surface. The BTEPT ligand loading on zirconia was clearly smaller (0.54 molecules per nm2) in comparison to the other zirconia materials.| Material | N wt% | H wt% | C wt% | S wt% | Total% | n(C)/n(N) | Ligand loading/nm2 |
|---|---|---|---|---|---|---|---|
| ZrPyTri2 | 1.56 | 0.46 | 2.70 | 4.72 | 2.02 | 0.96 | |
| ZrPyTri1 | 1.10 | 0.43 | 2.02 | 3.55 | 2.14 | 0.67 | |
| TiPyTri | 1.52 | 0.63 | 2.85 | 5.00 | 2.18 | 0.59 | |
| ZrEPTEP | 1.57 | 0.52 | 3.77 | 5.86 | 2.80 | 1.69 | |
| ZrBTEPT | 0.75 | 0.52 | 1.62 | 0.18 | 3.07 | 2.50 | 0.54 |
Overall, high phosphonate ligand loading was achieved for all the functionalized materials, namely 4.72% for ZrPyTri2 and 5.00% for TiPyTri (Table 3). Previously reported BTP functionalized titania nanoparticles with post-synthetic grafting for instance had a CHN content of only 1.7%.32 The ligand size and its complexity have an impact on the loading level and the results are in agreement with previously reported surface grafted zirconia materials.57 Since the organic compositions of ligands within grafted zirconia and titania are nearly the same it is possible to compare their sorption properties. Thus, the possible effects of the surface area, pore size, ligand density on the surface and phosphonate speciation on the sorption properties and Am/Eu selectivity of the materials can be discussed.
Sorption behaviour
The batch sorption experiments showed that ZrPyTri1 clearly preferred Am over Eu but the selectivity as well as the separation efficiency remained relatively low since the obtained SF was 4 at pH 2.5 (Table 4). The low sorption of Am at relatively high pH indicated that the level of surface grafting could be increased, since previously reported surface grafted zirconia sorbents showed significant uptake for trivalent lanthanides.57 Moreover, solvent extraction studies with similar tridentate N-donor ligand units have recently provided remarkably high SFAm/Eu values.34 The sorption efficiency and selectivity of the PyTri functionalized zirconia could be increased accordingly, still preserving the benefits of SPE materials.| pH | Material | K d (mL g−1) | K d (mL g−1) | SF(Am/Eu) |
|---|---|---|---|---|
| Am-241 | Eu-152 | |||
| 2.5 ± 0.2 | ZrPyTri1 | 800 ± 29 | 200 ± 8 | 4.0 |
| 1.5 ± 0.2 | ZrPyTri2 | 190 ± 20 | 11 ± 2 | 17.3 |
| 2.1 ± 0.2 | 11700 ± 270 |
1700 ± 66 | 6.9 | |
| 2.5 ± 0.2 | 13100 ± 1650 |
1500 ± 32 | 9.0 | |
| 2.1 ± 0.2 | ZrEPTEP | 80 ± 16 | 90 ± 9 | 0.9 |
| 2.1 ± 0.2 | ZrBTEPT | 273 ± 26 | 108 ± 7 | 2.5 |
| 2.1 ± 0.2 | TiPyTri | 16200 ± 700 |
6600 ± 200 | 2.5 |
| 2.5 ± 0.2 | ZrO2 | 54 ± 10 | 53 ± 11 | 1.0 |
| 2.5 ± 0.2 | TiO2 | 80 ± 30 | 110 ± 12 | 0.7 |
Doubling the PyTri ligand loading (ZrPyTri2) resulted in more significant uptake for Am and greatly enhanced the selectivity with SFAm/Eu ∼ 7 at pH 2. The acid concentration was varied to investigate the most optimal conditions for the separation still providing high uptake and selectivity. The highest SF value of 17.3 was observed at pH 1.5 but there the uptake was low for both nuclides. It can be clearly seen from the results that the uptake increases gradually along with increasing solution pH, thereby confirming that the acid concentration has a strong effect on the sorption properties of the materials because of the competition with the acidic H3O+ ions (Table 4). The most efficient separation was achieved at pH 2.5 with the highest uptake for Am while the corresponding Eu uptake was left notably lower, resulting in the increased SFAm/Eu of 9. It is noteworthy that the level of ligand loading plays an important role in terms of sorption and selectivity, since the total organic content was only increased from 3.55% to 4.72% from ZrPyTri1 to ZrPyTri2 (Table 3) but the sorption efficiency and SFAm/Eu values were increased drastically (Table 4).
The effect of symmetry within the coordinating ligand was examined with an alternative monotriazinyl pyridine ligand, denoted as EPTEP that contains only one phosphonic acid group and has a chemical structure relatively close to previously reported pyridylpyrazole (Cn-PypzH) extractants that have been used for efficient extraction of Am. (Fig. 1).35 This ligand was similarly grafted to the zirconia surface, and it was proved to have the same macroscopic properties as ZrPyTri materials as shown in the previous section. Since the ligand loading for ZrEPTEP was also comparable with ZrPyTri2, it was reasonable to compare the sorption and selectivity of these two materials. The sorption efficiency of ZrEPTEP was low for both Am and Eu in comparison to ZrPyTri materials and no significant selectivity was detected (Table 4). It is suggested that the steric hindrance of the branched chain and alkyl substituents within pyridyl-pyrazole ligands used in SX methods must play a key role on its selectivity as reported.37 ZrEPTEP has a lack of these alkyl chains. The center pyridyl-triazinyl moiety within EPTEP is considered as a good bidentate coordinating unit as well when functionalized on the zirconia support but the electron withdrawing effect of the alkyne substituent may result in an inferior donating effect of the electrons by the pyridyl group. The coordination kinetics may be slower in comparison to liquid–liquid interactions as well.
Zirconia grafted with thiophene-centered bistriazinyl phosphonate (ZrBTEPT) was also tested for Am/Eu separation and the results showed that the sorption efficiency and selectivity were decreased when compared to ZrPytri2. It can be seen from Table 3 that the ligand loading and ligand density on the surface are lower for this material which may be caused by incomplete hydrolysis of the BTEPT ligand as discussed earlier. However, this hybrid showed small but notable selectivity towards Am with SFAm/Eu = 2.5 (Table 4). Moreover, the soft and polarizable S-donating thiophene core, together with triazinyl coordination moieties, is a promising candidate for MA–lanthanide separation. The previously reported 2,5-thiophenedicarboxylic acid ligand bearing a similar thiophene center was suggested to show higher selectivity towards An than Ln due to higher formation constant values. The computed bond lengths, as well as EXAFS studies, indicated that the An–S bond is shorter than the corresponding Ln–S bond, allowing S to interact more strongly with the actinides.39 However, the rigid nature of the thiophene unit may limit the complexation properties of the BTEPT ligand and commonly N-donor extractants have provided better chemical stability.58
TiPyTri, in turn, was synthesized in the same manner as ZrPyTri2, and the elemental analysis indicates that the ligand loading of these hybrids was approximately the same. TiPyTri showed higher uptakes for both nuclides at the same pH 2.5, resulting in a lower SF value (Table 4). The increased sorption is possibly related to the larger pore size of the titania matrix (Table 2). Due to the higher surface area of the titania support, the ligand density on the surface was decreased, probably leading to lower selectivity (Table 3). On the other hand, the phosphonate speciation upon grafting can explain the difference in selectivity. The most likely rationale is that the coordinated phosphonate groups increase the selectivity while the free phosphonates are considered less selective species. The decreased selectivity of TiPyTri can thereby be predicted to be originating from the higher amount of uncoordinated free phosphonate species involved. This can be seen by 31P MAS-NMR (Table 1), which confirms that a relatively higher number of free P–O species were present for TiPyTri in comparison to ZrPyTri2. The NMR results also showed that ZrPyTri2 had more coordinated P–O groups at 0–3 ppm and 27–28 ppm. These P–O–M groups can be thought to be more hindered to bind cations than uncoordinated P–O groups, providing enhanced selectivity of the phosphonate species.
Finally, the uptake of bare zirconia and titania was studied. The ion exchange of Am and Eu on the bare porous zirconia and titania matrices was rather low when the solution pH is below 3, as expected. Neither the ZrO2 nor TiO2 support is selective for extraction as such, which can be seen from the results given in Table 4.
Competitive uptake experiments
Competitive sorption experiments were also performed since ZrPyTri2 demonstrated considerable Am/Eu selectivity and relatively high sorption in separate solutions. The binary solutions included Am and Eu in equimolar ratios and the obtained results indicated that the selectivity was only marginally reduced. The preference for the extraction of Am over Eu was preserved in all tested acid concentrations with SFAm/Eu varying between 4 and 11 (Fig. 7). However, the Am uptake was decreased due to increased competitive sorption caused by the higher amount of Eu ions present in equimolar solution. It is noticeable that at pH 3 the dramatic increase in both Am and Eu uptake results in a reduced separation factor. This is probably related to the participation of the zirconia matrix in the sorption, and the consequent increase in the ion exchange effect of the material. The results indicate that the separation factor seem to be decreasing along with the increasing uptake and thus, the most optimal conditions in terms of sorption efficiency and separation factor were achieved within the pH range 2–2.5. However, it should be noted that the separation factor values may have considerable variation depending on the composition of the liquid phase involved.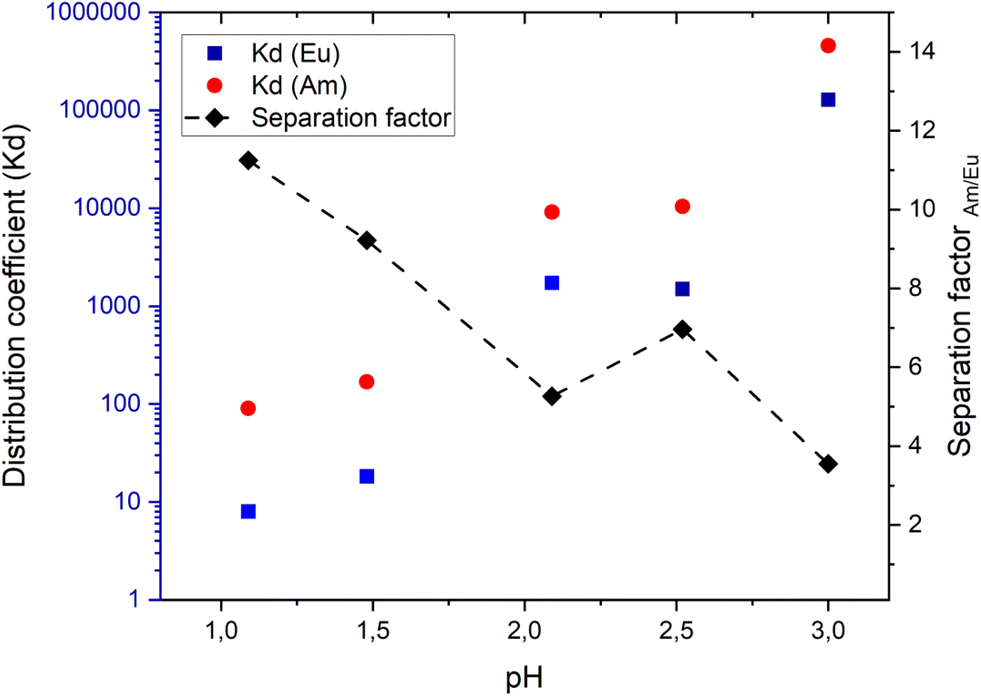 | ||
| Fig. 7 Am-241 and Eu-152 sorption in binary competitive solution with an equimolar ratio by ZrPyTri2 (pH 1–3, contact time: 24 h). Vertical error bars represent counting errors that are too small to be visible on the scale of this figure. | ||
Furthermore, competitive sorption experiments were undertaken in equimolar Am/Eu solution with TiPyTri, in the pH range 1.5–3.0. The results showed that the SF values remained almost invariant, while the uptake increased drastically along with the pH (Table 5). Overall, TiPyTri showed significantly higher Am and Eu uptakes but lower SF values also in binary solution in comparison to ZrPyTri2.
| pH | K d (mL g−1) | K d (mL g−1) | SF(Am/Eu) |
|---|---|---|---|
| Am-241 | Eu-152 | ||
| 1.5 ± 0.2 | 200 ± 8 | 60 ± 4 | 3.3 |
| 2.1 ± 0.2 | 17100 ± 400 |
7300 ± 400 | 2.4 |
| 2.5 ± 0.2 | 21500 ± 500 |
9300 ± 340 | 2.3 |
| 3.1 ± 0.2 | 1376000 ± 143000 |
416000 ± 30300 |
3.3 |
Since the obtained SF values were high in binary equimolar solution, especially for ZrPyTri2, competitive sorption was studied for this hybrid also from the 1:40 Am:Eu solution in order to mimic the MA:Ln ratio under SNF conditions. Despite the decreased uptake, the achieved extraction for Am under these conditions corresponds to more than 92% with a remarkable separation factor of 6, indicating that the material could be used to selectively extract Am from excess Ln (Table 6). To our knowledge, only a few examples of selective Am sorbents in the presence of excess europium in SPE materials have been reported previously.59 According to the obtained results, the total uptake of ZrPyTri2 was approximately 3.85 μg (Am + Eu) per gram in 0.01 M HNO3 solution. In comparison to previously reported surface grafted SPE materials, for instance, the silica coated nanoparticles functionalized with BTPhen derivatives achieved an extraction of approx. 1 μg (Am, Eu) g−1 under similar conditions, while BTP grafted titania beads showed approx. 1 μg (Am) g−1 extraction from 0.01 M nitric acid.1,32
:Eu molar ratio 1:40 by ZrPyTri2 (pH 2.1, contact time: 24 h)
| pH | Solution | K d (mL g−1) | K d (mL g−1) | SF(Am/Eu) |
|---|---|---|---|---|
| Am-241 | Eu-152 | |||
| 2.1 ± 0.2 |
1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 Am/Eu :1 Am/Eu
|
9200 ± 320 | 1700 ± 100 | 5.4 |
| 2.1 ± 0.2 |
1:40 Am/Eu
|
6400 ± 140 | 1050 ± 30 | 6.0 |
Effect of nitrates on the sorption properties
The difference in speciation of trivalent cations plays an important role in terms of selective separation and therefore, the effect of NaNO3 in the separation solution was investigated. The equilibrium sorption of Am and Eu was studied in three different NaNO3 concentrations at pH 2 for ZrPyTri2 and TiPyTri. The recent DFT studies suggest that the most indicative form of the trivalent metal cations in the solution would be a tetraaquatrinitrito (M(NO3)3(H2O)4) complex.60,61 J. Burk et al. reported DFT calculation studies for a triazinyl-based BTPhen ligand from SANEX with a structure close to the PyTri ligand. BTPhen was complexed with Am and Eu for energetic calculations which were made for naked M3+ ions and (NO3)3(H2O)4 correspondingly, and the results predicted that the most stable complexes were the ones containing nitrate anions due to the highest coordination, while also having the lowest charge. The calculations showed that Am(NO3)3(H2O)4 species formed a more stable complex with the ligand in all of the studied complexation forms.61 These findings could be used to explain the increased Am/Eu selectivity for both ZrPyTri and TiPyTri in added NaNO3 solutions demonstrated in Table 7. The increase in uptake would be related to higher nitrate concentration in the case of neutral extractant ligands but according to our experimental results the uptake decreases along with higher NaNO3 concentration. Potentially PyTri ligands can bind also to charged species such as M3+ or M(NO3)2+ and thereby establish an ion exchange character. The determination of the sorption mechanism would however require further study.| Material | NaNO3 conc. (M) | K d (mL g−1) | K d (mL g−1) | SF(Am/Eu) |
|---|---|---|---|---|
| Am-241 | Eu-152 | |||
| ZrPyTri2 | 1 | 6300 ± 200 | 700 ± 20 | 9.0 |
| 0.1 | 11700 ± 850 |
1370 ± 30 | 8.5 | |
| 0.01 | 14600 ± 900 |
1100 ± 60 | 13.3 | |
| — | 9200 ± 320 | 1700 ± 100 | 5.4 | |
| TiPyTri | 1 | 7000 ± 550 | 1900 ± 50 | 3.7 |
| 0.1 | 17000 ± 1200 |
4350 ± 250 | 3.9 | |
| 0.01 | 18100 ± 400 |
4570 ± 150 | 4.0 | |
| — | 17100 ± 400 |
7300 ± 400 | 2.4 |
Conclusions
This study provides an approach for the design of new organophosphorus-based hybrid SPE materials by utilization of M–O–P linkage bonds. These materials were prepared by introducing complexing PyTri ligand derivatives onto porous crystalline zirconia and titania frameworks and they were used for selective Am/Eu separations in nitric acid media. The phosphonate coupling molecules were covalently bound via a straightforward post-synthetic grafting method to provide sorbents with enhanced separation properties. The chemical and hydrolytic stability of these hybrids is advantageous in comparison to polymer- or silica-based materials which have been functionalized via impregnation methods, since impregnated materials are prone to leakage of functional organic ligands from the hybrid.22,62 In addition, mesoporous structure is preferable because the active surface area of the material will be maximized which promotes the sorption capacity. The present hybrid separation materials can be considered highly applicable for MA–lanthanide separation from PUREX raffinates and they could be also advantageous over common solvent extraction strategies used for this purpose, such as SANEX.Batch experiments proved that the functionalized zirconia and titania materials showed high sorption efficiency with remarkable preference for Am both in separate and binary solutions. The material with the highest selectivity presented in this study turned out to be ZrPyTri2 with SFAm/Eu 7–17 in various nitric acid concentrations. The acid concentration strongly affected the sorption efficiency and the selectivity of the sorbents. The selectivity was shown to decrease along with higher pH due to the increased sorption for both Am and Eu. The most optimal separation conditions were found at pH 2–2.5 in terms of high Am sorption performance of the hybrid but also significant SFAm/Eu. Moreover, ZrPyTri2 was proved to selectively extract Am in competitive equimolar solution and even in the presence of excess Eu concentration. TiPyTri indicated lower selectivity but better sorption efficiency and higher capacity, compared to ZrPyTri2. The selectivity is suggested to originate from the higher phosphonate coordination to metal centers by ZrPyTri2 and greater ligand density on the surface. The presence of added sodium nitrates in the equimolar Am/Eu solutions apparently promoted the separation efficiency of Am, resulting in increased SFAm/Eu from 5.4 up to 13.3 at pH 2, which may be due to the more stable complex of Am(NO3)2+ and the PyTri ligand. Similarly, the SFAm/Eu values were increased for TiPyTri under the same conditions. In conclusion, PyTri phosphonate derivatives were successfully applied as selective coordinating ligands, and they showed high potential for efficient MA–Ln separation when incorporated into inorganic supports. Meanwhile, the EPTEP functionalized zirconia lacked this selectivity probably due to its asymmetric coordination structure. The novel S-donating thiophene-centered bistriazinyl bisphosphonate (BTEPT) ligand was introduced to the zirconia surface as well, and the resulting hybrid showed lower Am/Eu selectivity, but it may be a promising candidate for further MA–Ln separation studies. The sorption of trivalent Am and Eu by the grafted hybrid materials originates from the ligand–metal interactions in acidic media; the phosphonic acid groups are covalently bound to the hydroxyl groups of the zirconia or titania surface, allowing the N- or S-donating heterocyclic units to form a coordination sphere with the cation. Therefore, the surface complexation process is the most probable sorption mechanism. Comparing the Am/Eu selectivity of the hybrid materials presented in this study to other reported surface grafted solid sorbents, e.g. BTP grafted titania beads32 have provided an SFAm/Eu of 64 and BTPhen coated silica nanoparticles an SFAm/Eu up to 18 (ref. 1) under the same acidic sorption conditions.
Separation factor values derived from batch experiments are however indicative results and therefore the separation efficiency that could be accomplished via column separation studies may be different due to variance in solid/liquid ratios and kinetics between these two experimental approaches. Consequently, the future work will focus on eluting agents and the use of the presented separation materials in column experiments since the elution processes are key attributes of a selective separation and recovery.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research is partly funded by State Nuclear Waste Management Fund, on the basis of proposals by the Ministry of Employment and Education of Finland. A. Z. acknowledges the Magnus Ehrnrooth Foundation and Slovenian Research and Innovation Agency (Research Core Funding Grant P1-0230) for financial support.References
- A. Afsar, L. M. Harwood, M. J. Hudson, P. Distler and J. John, Chem. Commun., 2014, 50, 15082–15085 RSC.
- S. A. Darda, H. A. Gabbar, V. Damideh, M. Aboughaly and I. Hassen, J. Radioanal. Nucl. Chem., 2021, 329, 15–31 CrossRef CAS.
- S. A. Ansari, P. Pathak, P. K. Mohapatra and V. K. Manchanda, Sep. Purif. Rev., 2011, 40, 43 CrossRef CAS.
- Actinide and fission product partitioning and transmutation status and assessment report, OECD/NEA, Paris, 1999 Search PubMed.
- M. Salvatores, Nucl. Eng. Des., 2005, 235, 805 CrossRef CAS.
- P. J. Panak and A. Geist, Chem. Rev., 2013, 113(2), 1199–1236 CrossRef CAS PubMed.
- H. H. Dam, D. N. Reinhoudt and W. Verboom, Chem. Soc. Rev., 2007, 36, 367 RSC.
- M. P. Jensen and A. H. Bond, J. Am. Chem. Soc., 2002, 124, 9870–9877 CrossRef CAS PubMed.
- S. A. Kozimor, P. Yang, E. R. Batista, K. S. Boland, C. J. Burns, D. L. Clark, S. D. Conradson, R. L. Martin, M. P. Wilkerson and L. E. Wolfsberg, J. Am. Chem. Soc., 2009, 131, 12125–12136 CrossRef CAS PubMed.
- G. R. Choppin and K. L. Nash, Radiochim. Acta, 1995, 70/71, 225–236 CrossRef CAS.
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef CAS.
- K. L. Nash, Solvent Extr. Ion Exch., 1993, 11, 729–768 CrossRef CAS.
- A. Leoncini, J. Huskens and W. Verboom, Chem. Soc. Rev., 2017, 46, 7229–7273 RSC.
- M. Wehbie, G. Arrachart, C. Arrambide Cruz, I. Karamé, L. Ghannam and S. PelletRostaing, Sep. Purif. Technol., 2017, 187, 311–318 CrossRef CAS.
- L. Harwood, F. Lewis and M. Hudson, Synlett, 2011, 2609–2632 CrossRef.
- M. P. Kelley, N. P. Bessen, J. Su, M. Urban, S. I. Sinkov, G. J. Lumetta, E. R. Batista, P. Yang and J. C. Shafer, Chem. Commun., 2018, 54, 10578–10581 RSC.
- A. Geist, U. Müllich, D. Magnusson, P. Kaden, G. Modolo, A. Wilden and T. Zevaco, Solvent Extr. Ion Exch., 2012, 30, 433–444 CrossRef CAS.
- A. F. Henwood, I. N. Hegarty, E. P. McCarney, J. I. Lovitt, S. Donohoe and T. Gunnlaugsson, Coord. Chem. Rev., 2021, 449, 214206 CrossRef CAS.
- F. W. Lewis, L. M. Harwood, M. J. Hudson, M. G. Drew, J. F. Desreux, G. Vidick, N. Bouslimani, G. Modolo, A. Wilden, M. Sypula, T.-H. Vu and J.-P. Simonin, J. Am. Chem. Soc., 2011, 133, 13093–13102 CrossRef CAS PubMed.
- B. C. Ekberg, A. Fermvik, T. Retegan, G. Skarnemark, M. R. S. Foreman, M. J. Hudson, S. Englund and M. Nilsson, Radiochim. Acta, 2018, 96, 225–233 CrossRef.
- Y. Hu, J. Florek, D. Larivière, F. Fontaine and F. Kleitz, Chem. Rec., 2018, 18, 1261–1276 CrossRef PubMed.
- Y.-Z. Wei, H. Hoshi, M. Kumagai, T. Asakura and Y. Morita, J. Alloys Compd., 2004, 374, 447–450 CrossRef CAS.
- P. Deepika, K. N. Sabharwal, T. G. Srinivasan and P. R. Vasudeva Rao, Sep. Sci. Technol., 2014, 48, 2020 CrossRef.
- T. Chen and D. Zhao, Coord. Chem. Rev., 2023, 491, 215259 CrossRef CAS.
- Y. L. Xu, S. Y. Kim, T. Ito, K. Hitomi, E. Kuraoka, S. Usuda and K. Ishii, J. Radioanal. Nucl. Chem., 2014, 299, 149 CrossRef CAS.
- L.-Y. Yuan, L. Zhu, C.-L. Xiao, Q.-Y. Wu, N. Zhang, J.-P. Yu, Z.-F. Chai and W.-Q. Shi, ACS Appl. Mater. Interfaces, 2017, 9, 3774–3784 CrossRef CAS PubMed.
- Z. Bayram-Hahn, B. A. Grimes, A. M. Lind, R. Skudas, K. K. Unger, A. Galarneau, J. Iapichella and F. Fajula, J. Sep. Sci., 2007, 30, 3089–3103 CrossRef CAS PubMed.
- Y.-P. Zhu, T.-Y. Ma, Y.-L. Liu, T.-Z. Ren and Z.-Y. Yuan, Inorg. Chem. Front., 2014, 1, 360–383 RSC.
- C. S. Griffith, M. D. Reyes, N. Scales, J. V. Hanna and V. Luca, ACS Appl. Mater. Interfaces, 2010, 2, 3436–3446 CrossRef CAS PubMed.
- A. Ossola, E. Mossini, E. Macerata, W. Panzeri, A. Mele and M. Mariani, Ind. Eng. Chem. Res., 2022, 61, 4436–4444 CrossRef CAS PubMed.
- B. Schulze and U. S. Schubert, Chem. Soc. Rev., 2014, 43, 2522 RSC.
- J. Veliscek-Carolan, K. A. Jolliffe and T. L. Hanley, Chem. Commun., 2015, 51, 11726 RSC.
- E. Mossini, E. Macerata, N. Boubals, C. Berthon, M.-C. Charbonnel and M. Mariani, Ind. Eng. Chem. Res., 2021, 60, 11768–11777 CrossRef CAS.
- A. Ossola, E. Macerata, E. Mossini, M. Giola, M. C. Gullo, A. Arduini, A. Casnati and M. Mariani, J. Radioanal. Nucl. Chem., 2018, 318, 2013–2022 CrossRef CAS.
- D. Su, Y. Liu, S. Li, S. Ding, Y. Jin, Z. Wang, X. Hu and L. Zhang, Eur. J. Inorg. Chem., 2017, 2017, 651–658 CrossRef CAS.
- X.-H. Kong, Q.-Y. Wu, C.-Z. Wang, J.-H. Lan, Z.-F. Chai, C.-M. Nie and W.-Q. Shi, J. Phys. Chem. A, 2018, 122, 4499–4507 CrossRef CAS PubMed.
- N. P. Bessen, J. A. Jackson, M. P. Jensen and J. C. Shafer, Coord. Chem. Rev., 2020, 421, 213446 CrossRef CAS.
- J. R. Klaehn, D. R. Peterman, M. K. Harrup, R. D. Tillotson, T. A. Luther, J. D. Law and L. M. Daniels, Inorg. Chim. Acta, 2008, 361, 2522–2532 CrossRef CAS.
- N. P. Bessen, I. A. Popov, C. R. Heathman, T. S. Grimes, P. R. Zalupski, L. M. Moreau, K. F. Smith, C. H. Booth, R. J. Abergel, E. R. Batista, P. Yang and J. C. Shafer, Inorg. Chem., 2021, 60, 6125–6134 CrossRef CAS PubMed.
- C. Ma, T. Bian, S. Yang, C. Liu, T. Zhang, J. Yang, Y. Li, J. Li, R. Yang and W. Tan, Anal. Chem., 2014, 86, 6508 CrossRef CAS PubMed.
- E. Brunet, O. Juanes, L. Jimenez and J. C. Rodrıguez-Ubis, Tetrahedron Lett., 2009, 50, 5361 CrossRef CAS.
- S. Roy, S. Chowdhury, S. Mishra and S. Patra, Chem. – Asian J., 2020, 15, 3304–3313 CrossRef CAS PubMed.
- W. A. Schafer, P. W. Carr, E. F. Funkenbusch and K. A. Parson, J. Chromatogr. A, 1991, 587, 137–147 CrossRef CAS.
- J. Veliscek-Carolan, A. Rawal, D. T. Oldfield, G. J. Thorogood and N. M. Bedford, ACS Appl. Nano Mater., 2020, 3, 3717–3729 CrossRef CAS.
- F. Askari, E. Ghasemi, B. Ramezanzadeh and M. Mahdavian, Dyes Pigm., 2016, 124, 18–26 CrossRef CAS.
- M. C. Zenobi, C. V. Luengo, M. J. Avena and E. H. Rueda, Spectrochim. Acta, Part A, 2010, 75(4), 1283–1288 CrossRef PubMed.
- M. M. Gómez-Alcántara, A. Cabeza, P. Olivera-Pastor, F. Fernandez-Moreno, I. Sobrados, J. Sanz, R. E. Morris, A. Clearfield and M. A. G. Aranda, Dalton Trans., 2007, 2394–2404 RSC.
- G. Grossmann, K. A. Burkov, G. Hägele, L. A. Myund, S. Hermens, C. Verwey and S. M. Arat-ool, Inorg. Chim. Acta, 2004, 357, 797–808 CrossRef CAS.
- W. Gao, L. Dickinson, C. Grozinger, F. G. Morin and L. Reven, Langmuir, 1996, 12(26), 6429–6435 CrossRef CAS.
- D. Massiot, S. Drumel, P. Janvier, M. Bujoli-Doeuff and B. Bujoli, Chem. Mater., 1997, 9(1), 6–7 CrossRef CAS.
- X.-Z. Lin and Z.-Y. Yuan, Eur. J. Inorg. Chem., 2012, 2661–2664 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603 CrossRef CAS.
- D. Gu and F. Schuth, Chem. Soc. Rev., 2014, 43, 313 RSC.
- J. G. Wang, Z. F. Bian, J. Zhu and H. X. Li, J. Mater. Chem. A, 2013, 1, 1296 RSC.
- X.-J. Zhang, T.-Y. Ma and Z. Y. Yuan, J. Mater. Chem., 2008, 18, 2003–2010 RSC.
- M. Otaki, T. Suominen, V. Suorsa, S. Hietala and R. Koivula, Mater. Adv., 2023, 4, 551–560 RSC.
- Z. Kolarik, U. Müllich and F. Gassner, Solvent Extr. Ion Exch., 1999, 17, 23–32 CrossRef CAS.
- J. Veliscek-Carolan and A. Rawal, Chem. Commun., 2019, 55, 1168 RSC.
- C. Xiao, Q. Wu, C. Wang, Y. Zhao, Z. Chai and W. Shi, Sci. China:Chem., 2014, 57, 1439–1448 CrossRef CAS.
- J. Burk, L. Sikk, K. Tämm and P. Burk, Comput. Theor. Chem., 2022, 1214, 113786 CrossRef CAS.
- Y. L. Xu, S. Y. Kim, T. Ito, K. Hitomi, E. Kuraoka, S. Usuda and K. Ishii, J. Radioanal. Nucl. Chem., 2014, 299, 149–155 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lf00277f |
| This journal is © The Royal Society of Chemistry 2025 |