
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unravelling the potential role of polyethyleneimine (PEI)-based nanosystems in skin cancer therapy
Ajeet
Kumar†
 a,
Sabya
Sachi Das†
*b,
Srushti
Tambe
c,
Babita
Kaundal
d,
Sunny Kumar
Sarraf
b and
Kavindra Kumar
Kesari
*ef
a,
Sabya
Sachi Das†
*b,
Srushti
Tambe
c,
Babita
Kaundal
d,
Sunny Kumar
Sarraf
b and
Kavindra Kumar
Kesari
*ef
aDepartment of Chemistry, University of Florida, Gainesville, FL 32611, USA
bSchool of Pharmaceutical and Population Health Informatics, DIT University, Dehradun, Uttarakhand 248009, India. E-mail: ss.das@dituniversity.edu.in
cDepartment of Pharmaceutical Science and Technology, Institute of Chemical Technology, Mumbai, Maharashtra 400019, India
dDepartment of Epigenetics and Molecular Carcinogenesis, The University of Texas MD Anderson Cancer Center, Houston, TX 301429, USA
eDepartment of Applied Physics, School of Science, Aalto University, Espoo, 00076, Finland. E-mail: kavindra.kesari@aalto.fi
fUniversity Center for Research and Development, Chandigarh University, Mohali, Punjab, India
First published on 11th November 2024
Abstract
Skin cancer is one of the most common cancer types affecting a major portion of the world's population, particularly in fair-skinned populations. Broadly, skin cancer is categorized into two major forms, carcinoma and melanoma, based on their physiological conditions. Skin carcinoma, but more particularly melanoma, remains a significant global health concern, with increasing incidence rates observed across various demographics. While traditional approaches such as surgery, chemotherapy, and radiation therapy remain cornerstones of treatment, latest developments in skin cancer treatment encompass novel therapeutic modalities, targeted drug delivery systems, and personalized approaches to patient care. Polyethyleneimine (PEI)-based nanosystems have emerged as a promising avenue for personalized cancer immunotherapy and also as a potential targeted therapeutic approach to combat skin cancer. PEI is a highly cationic polymer that has garnered significant interest in the field of nanomedicine for its potential in delivering therapeutic agents, including nucleic acids and small molecules, specifically to cancer cells. In this review, we discuss and summarize the challenges associated with PEI and strategies for its modification, PEI as a potential therapeutic carrier, skin cancer types and pathogenesis, and the potential role PEI-based nanosystems play in effective skin cancer management.
1. Introduction
Polyethyleneimine (PEI) is a positively charged polymer consisting of repeating units of amino groups and ethylene (CH2CH2). It is derived from the ring opening of aziridine and is also referred to as polyfunctional aziridine (polyaziridine).1–5 PEI has two main structural variants: linear PEI ((CH2CH2NH)n; L-PEI) and branched PEI (H(NHCH2CH2CH2)nNH2; B-PEI).1,3,4 B-PEI may consist of all kinds of primary (1°), secondary (2°), and tertiary (3°) amino groups whereas L-PEI mainly comprises 1° and 2° amino groups (Fig. 1). L-PEI exists as a solid at room temperature (it has a melting point of about 73–75 °C) whereas B-PEI exists as a liquid, regardless of the molecular weight. L-PEI is soluble in hot water at low pH, and in various organic solvents including chloroform, ethanol, and methanol. PEI is widely utilized as a synthetic polycation due to its chemical configuration and functionality, characterized by the existence of cationic 1° (25%), 2° (50%), and 3° amines (25%).6 The polymer's high positive charge potential, attributed to protonable amino nitrogen chains occurring every third atom, enables efficient complex formation with nucleic acids and protects cells against nuclease-mediated degradation. Additionally, the abundance of protonable amino nitrogen allows PEI to act as a proton sponge, which buffers the endosomal pH and induces osmotic swelling and rupture of the endosomal membrane. This helps in the release of polymer–nucleic acid complexes (polyplexes) into the cytoplasm.4,7 PEI first forms complexes with DNA through counter-condensation, which helps in reducing intramolecular repulsions and releasing chloride and salt ions.8 Its unique structure and properties enable stabilization or modification of various inorganic hybrid nanoparticles (NPs).9–12 Also it can bind to anionic remains within the DNA templates and polymerase through electrostatic interaction which significantly enhances the effectiveness of transfection.13 PEI has a high buffering capacity, which makes it beneficial for the endosomal escape of gene payloads during transfection.14 Compared to other polycations, the polycation PEI has high intrinsic endosomal activity and is effective at condensing DNA.15,16 To improve the effectiveness of targeted medications and gene therapy, it is frequently utilized as a transfection reagent and nanocarrier for drug delivery systems.4,12 The high transfection efficiency of its polyplexes has led to its status as the gold standard for polymer-based gene carriers.17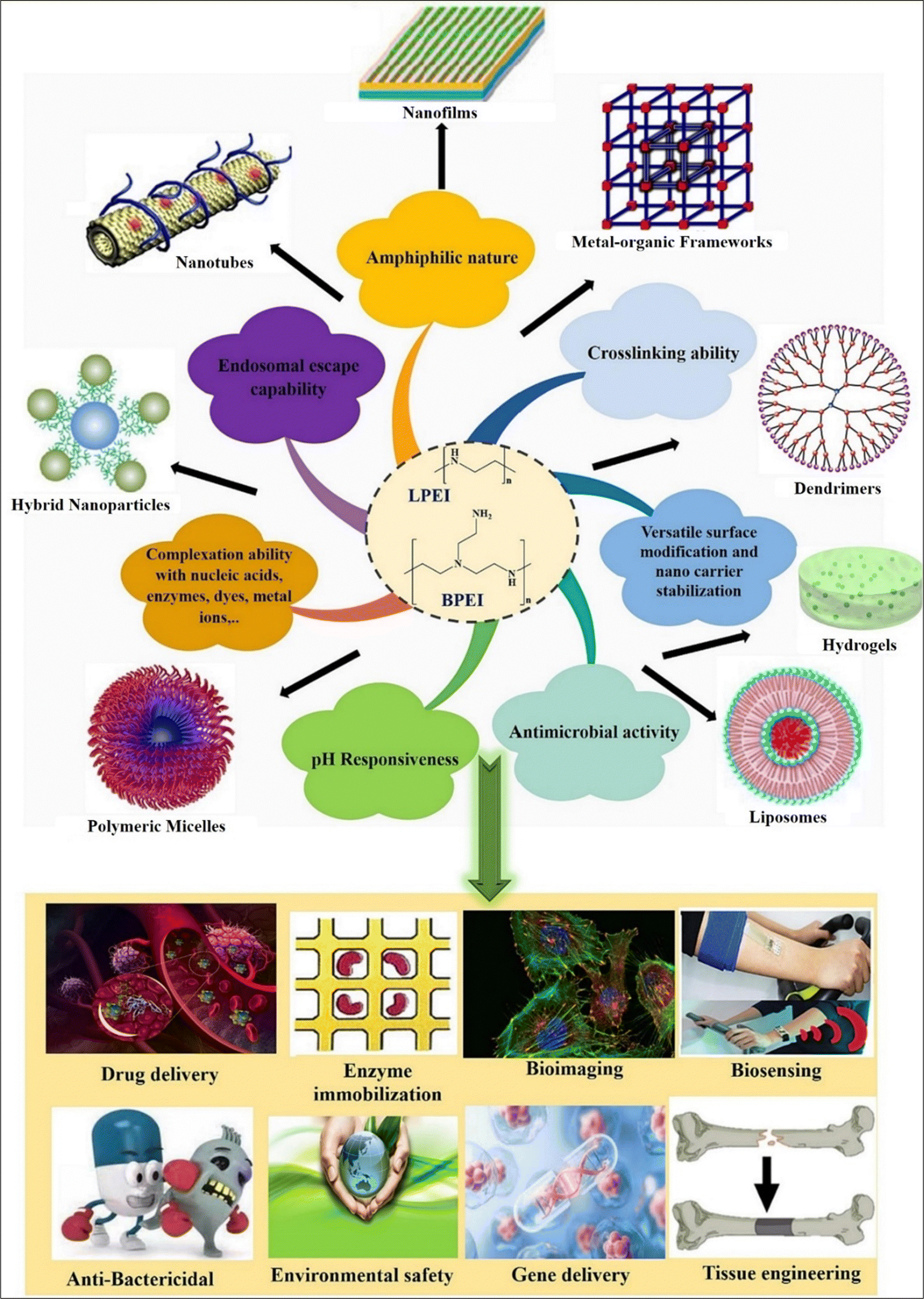 | ||
| Fig. 1 Schematic diagram showing various properties and applications of PEI-based nanosystems. Reproduced from ref. 18 with permission from Elsevier, copyright 2024. | ||
Beyond biomedical use, PEI's unique structure and abundance of amino groups find applications in various industries (Fig. 1). For example, PEI serves as a flocculant in oil removal from synthetic water and as a wet-strength agent in paper and shampoo production.13,17,19 PEI is frequently employed in biomedicine to immobilize enzymes,13 immobilize viruses on cellulose,20 promote cell adhesion,13,21 transfect genes,22 and synthesize NPs to increase their stability as well as anticancer effectiveness.13,23 To explore a potential mechanism for the activation of apoptosis, Kafil V. et al. (2010) evaluated the effects of linear and branched PEI (L-PEI and B-PEI) over cytogenomic changes in A431 lung cancer cells. A431 cells were treated with PEI at the prescribed dose for 4 hours when they were 40–50% confluent. Additionally, analyses using flow cytometry showed that the B-PEI caused more internalization when compared to linear PEI, leading to more cytotoxicity. B-PEI's early and late apoptosis was validated via the annexin V assay, imposing part of the DNA damage seen in the comet experiment. Akt-kinase, a biomolecule that may be impacted by PEI was identified using western blot assay. These findings demonstrate that B-PEI can induce apoptosis in target cells even when Akt-kinase is activated.24
According to Wightman et al.16 under salt conditions, linear PEI22/DNA complexes generally had a higher transfection effectiveness in vitro than branched PEI/DNA complexes.16 Also, they claimed that kinetic instability that exists naturally may be the cause of linear PEI's higher transfection effectiveness.4 Studies have revealed that the most reliable indicator of the effectiveness of gene transfection and cytotoxicity is the molecular weight of PEI.25–27 While cytotoxicity seems to increase with increasing polymer size, it was noticed that PEI gene transfer actions augmented with an increase in molecular weight. For instance, low molecular weight (LMW) PEI (2 kDa) was safe but had very poor transfection ability and could not efficiently condense DNA. High molecular weight (HMW) PEI (25 kDa) on the other hand demonstrated good transfection efficiency but also notable cytotoxicity. Moreover, HMW-PEI's long-term safety is troublesome because it is nonbiodegradable and likely would result in the development of cytotoxicity in vivo.28 Variations in its properties may impact PEI's capacity for drug delivery. It has been reported that polymer-based nanocarriers are used as co-delivery systems for gene-targeted therapy and anticancer medications.29,30 PEI spontaneously attaches and condenses nucleic acids to produce toroidal multiplexes that are easily endocytosed by the cells, although few studies are using PEI for co-delivery in cancer. At practically every physiological pH, PEI can maintain a considerable buffering capacity, preserving nucleic acids and preventing lysosomal nuclease degradation.3
1.1. Challenges with PEI and strategies for modification
The safety of PEI, however, remains a constant worry for its clinical application.4 The safety of PEIs in clinical applications can vary based on factors such as molecular weight, chemical structure, and concentration. High MW PEIs can adversely affect various cell types, including neurons. High-MW PEIs have been observed to alter the plasma membrane, leading to changes in a manner resembling necrosis, followed by the activation of mitochondrial-mediated apoptosis across different cell types.3,31,32 Low-MW PEIs, in contrast, showed significantly reduced toxicity but generally lack transfection efficiency. These PEIs have been reported to induce necrotic cell death and apoptosis both in vitro and in vivo, leading to toxicity, which limits their clinical application.4 In the interim the buffering capacity of these protonable polymers causes extra proton accretion within endosomes resulting in counterion and water gathering which causes osmolysis. PEI toxicity seems to be associated with the interruption of the endosome–lysosome complexes, the same mechanism accountable for their transfection efficiency. The solubility, biodegradability, and chemical homogeneity of high-MW PEIs have all been altered in an effort to increase their biocompatibility.3PEI, being a cationic polymer with multiple amino groups, exhibits inherent cytotoxicity. By attaching to negatively charged transmembrane heparan proteoglycans, cationic PEI enters the cellular environment and can damage or impair the cellular components or whole cell by disrupting the membrane.5,33 Moreover, PEI induces apoptosis by creating pores in the mitochondrial membrane, leading to cell death.34,35 PEI exhibits poor biodegradability in living organisms, and its cytotoxicity is directly influenced by the molecular weight and degree of branching.36 A higher molecular weight, branched PEI has a higher cytotoxicity. Simple changes that shield PEI's surface amines greatly increase its biocompatibility.5,37 However, the cytotoxicity of PEI is reduced and the biocompatibility is increased through nullifying its surface potential using various physical or chemical modifications. It is important to note that these surfaces allow it to acquire additional capabilities, like biomarker and targeting.5
To overcome several issues associated with PEIs as previously discussed, researchers have investigated several physical and chemical modifications of PEI (Fig. 2). Zintchenko et al. have developed various derivatives of branched PEI through methods such as ethyl acrylate modification of amines, acetylation of 1° amines, or coupling of negatively charged groups such as propionic acid or succinic acid. These modifications led to enhanced gene silencing efficiency in siRNA-mediated gene knockdown experiments. Furthermore, they found that succinylation of branched PEI significantly reduced the toxicity of polymer compared to unmodified PEI. This reduced cytotoxicity was presumably due to the incorporation of biodegradable linkages in the PEI structure which facilitate polymer decomposition.38 In 2008, Xu et al. reported the synthesis of a biodegradable PEI biscarbamate conjugate (PEIC) by combining LMW-PEI (MW = 800) with 1,4-butanediol bis(chloroformate). The resulting PEIC had a molecular weight of 2800 and a number-average molecular weight (Mn) of 910. When compared to PEIs with molecular weights of 2 kDa or 25 kDa, this modified PEIC demonstrated reduced cytotoxicity. A similar biodegradable carbamate linkage was utilized in subsequent works to synthesize a variety of polymers.39 Wen et al. improved the biocompatibility of PEI with various chemical modifications such as carboxylation, acetylation, hydroxylation, and PEGylation of the polymer.37 The cytotoxicity of the PEI was significantly decreased or masked by these techniques. For biomedical applications, functional groups including folic acid (FA), polyethylene glycol (PEG), fluorescent tags, hyaluronic acid (HA), and protein can be altered with PEI.5,40–42
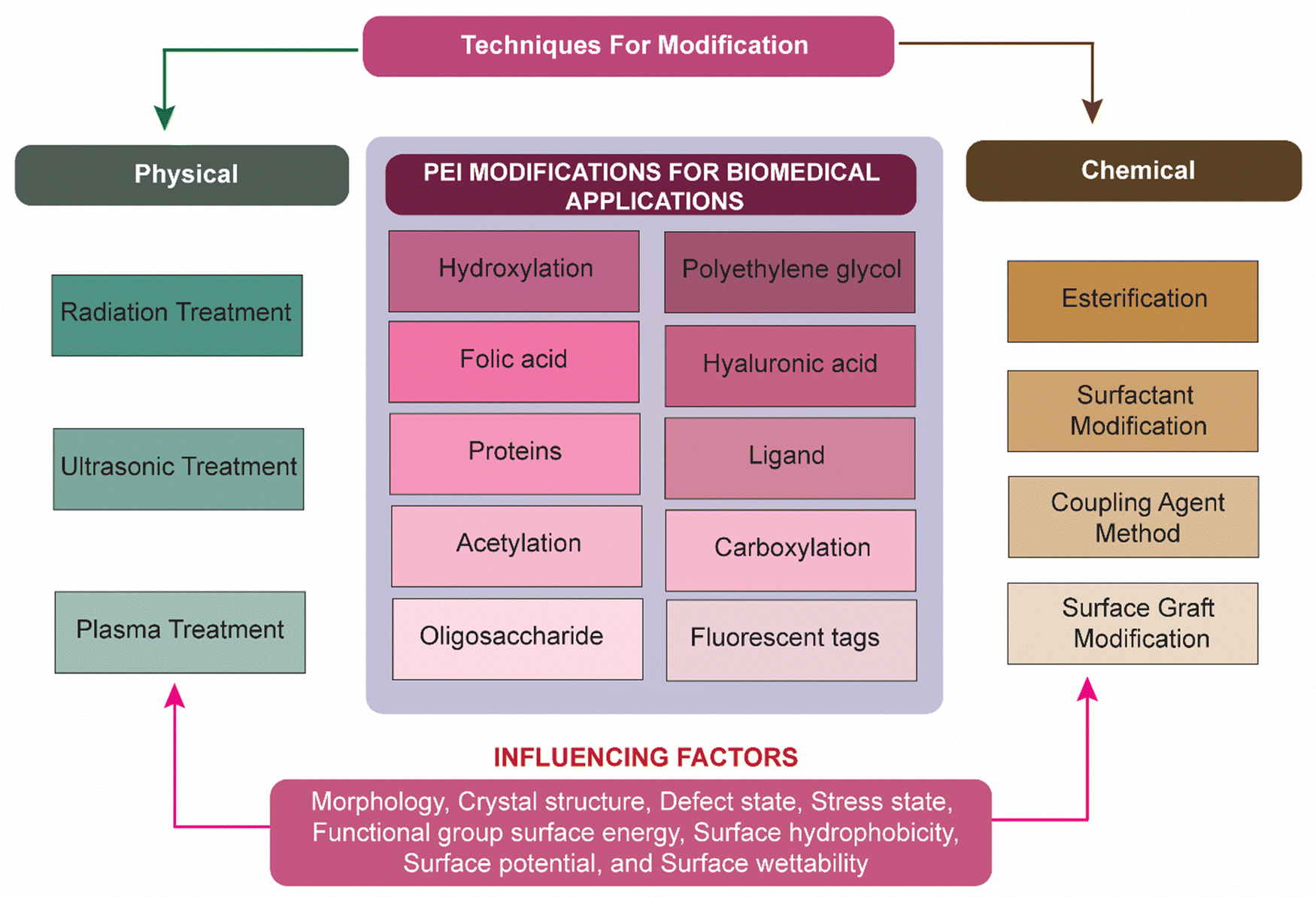 | ||
| Fig. 2 Illustration showing various techniques used for the modification of PEI for biomedical applications. The figure also represents various influencing factors for PEI stability and those considered during PEI modification using various approaches. | ||
Degradable cross-linking reagents, including diacrylate,26 glutaraldehyde,43 dithiobis(succinimidyl propionate), oligo-(L-lactic acid-co-succinic acid),44 and dimethyl-3,3′-dithiobispropionimidate·2HCl45 have also been utilized for modifications. The results presented that the transfection efficacy of these crosslinked-PEIs was comparatively more than PEI (MW 25k or even more), and also exhibited comparatively less cytotoxicity. Another method involved connecting LMW-PEIs using linear chains. Wong et al. produced chitosan-graft-PEI (PEI-g-CH) through a cationic polymerization method using aziridine and oligochitosan. PEI-g-chitosan exhibited greater transfection effectiveness than PEI (MW 25k), confirmed through both in vitro and in vivo experiments.46 Tang et al. crosslinked LMW-branching PEI (MW 600) with cyclodextrins to produce HMW-cationic polymers with an average molecular weight of 61 kDa. These polymers exhibited higher gene transfection efficiency and lower cytotoxicity in vitro.47 Jiang et al., synthesized a PEI-g-CH copolymer through imine reactions amongst periodate (IO4−)-oxidized CH and LMW-PEI (MW 1800 Da). Their research demonstrated that PEI-g-CH exhibited superior cell transfection capabilities compared to PEI 25k, along with reduced cytotoxicity.48 Stearic acid blocks added to PEI demonstrated increased transfection in antigen-presenting cells (APCs) and decreased toxicity, resulting in improved immune responses against the HIV-1 gag protein with high antigen-specific antibody secretion and pro-inflammatory cytokine production.49
2. Skin cancer: types, pathogenesis and causing factors
Broadly, skin cancer is split into two major forms, carcinoma and melanoma. Skin carcinoma encompasses a spectrum of malignancies, including basal cell carcinoma (BCC), squamous cell carcinoma (SCC), and melanoma, each presenting unique challenges in diagnosis and treatment. Skin cancer progresses through three main stages. Initially, it is confined to the epidermis, the outermost skin layer (Fig. 3). In this early stage, BCC appears as pearly bumps, while SCC shows as rough, scaly patches, and melanoma is identified by the ABCDE rule (asymmetry, border, color, diameter, evolving).50 As the cancer advances, it penetrates the dermis, the deeper skin layer, increasing the risk of rapid growth and local damage. In the final stage, the cancer spreads to the lymph nodes and other organs, significantly increasing mortality risks.51,52 Key risk factors include genetics, age, UV radiation, nevi, and skin pigmentation.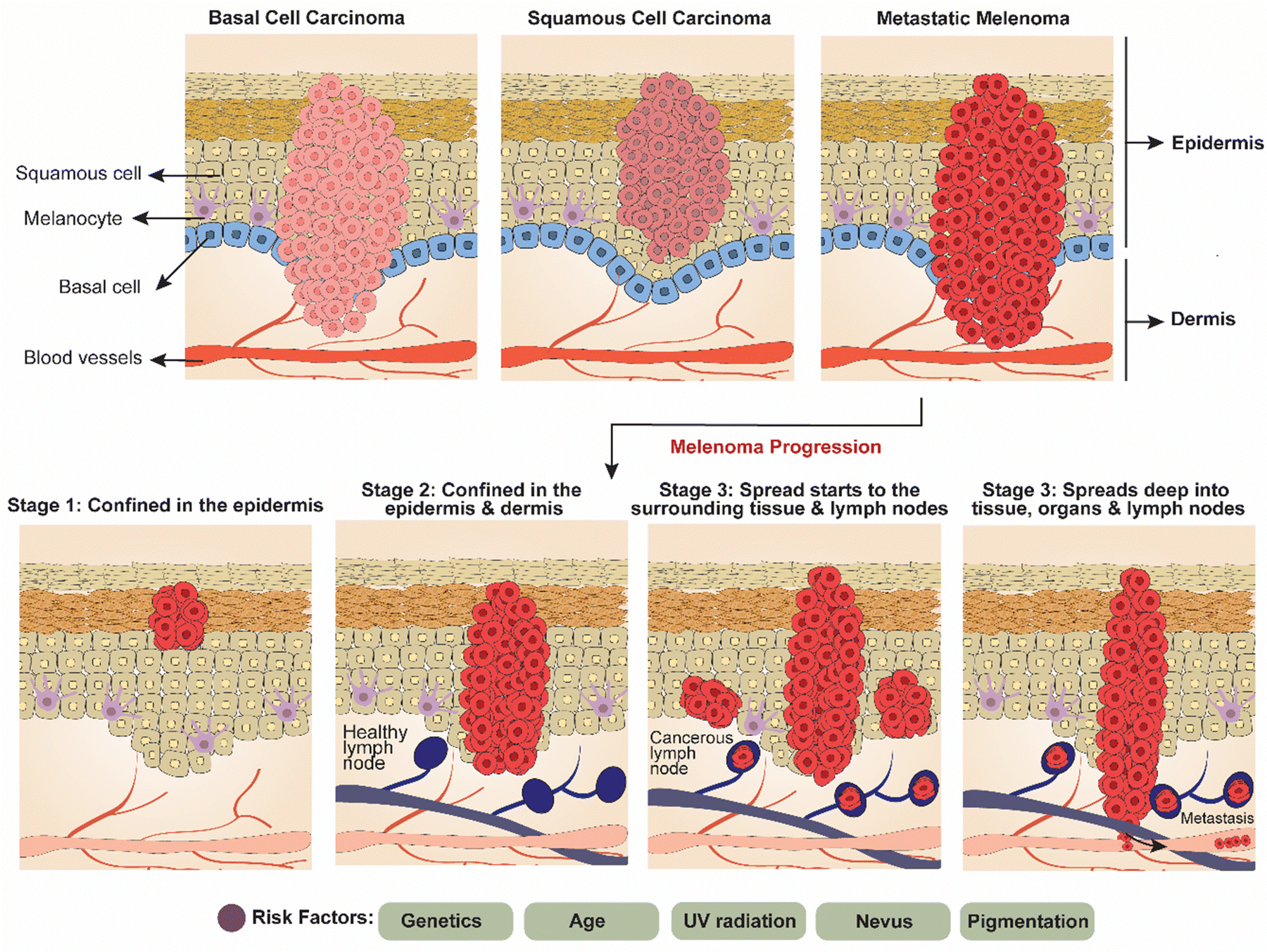 | ||
| Fig. 3 Diagrammatic representation of types of skin carcinoma (basal cell carcinoma, squamous cell carcinoma, and metastatic melanoma) and melanoma progression stages. | ||
Melanoma is a type of cancer that originates in melanocytes, the cells responsible for pigment production.53,54 This cancer develops when normal melanocytes acquire somatic mutations or inherit genetic flaws, transforming into malignant melanoma.55 Melanocytes, derived from the neural crest, are pigment-producing cells found in various parts of the body including the skin, hair follicles, uvea of the eye, inner ear, heart, and mucosal tissues.56–58 Melanocytes produce melanin within specialized structures called melanosomes through complex biochemical processes. Skin pigmentation, both across different ethnicities and within them, is determined by the amount of melanin produced by melanocytes and the size of melanosomes, rather than the number of melanocytes present.56,59,60 Due to higher concentrations of melanin in the epidermis, individuals with darker skin tones are less prone to developing melanoma compared to those with lighter skin tones, who typically have lower epidermis melanin contents.61–64 Melanoma must be recognized as a heterogeneous cluster of diseases rather than as a singular disease, with faults affecting critical physiological functions involving cell cycle regulation, cell signaling pathways, cell adhesion, cell differentiation, and cell death.65,66 The requirement for individualized melanoma diagnosis, prognosis, and treatment is highlighted by the variability of molecular defects.67 Also, early detection of melanoma is crucial because it is “curable” at this stage.56 Cytological features such as an uneven and thick nuclear membrane, along with prominent nucleoli are characteristics of malignant melanoma. The risk of developing melanoma increases significantly with intermittent sun exposure due to the intensely acute and complex link between ultraviolet (UV) light exposure and the disease.68,69 Melanoma can be categorized into two main types based on site specificity: cutaneous and non-cutaneous.
Cutaneous melanoma arises from a complex interplay of phenotypic and constitutional variables. Along with the incidence of melanoma, this interaction also affects the clinical traits and oncogenic pathways utilized by the tumor for growth.70 Typically, cutaneous melanoma starts in the epidermis, the outermost layer of skin, and can progress to become invasive. It can further be divided into four subtypes:
(a) Superficial spreading melanoma (70%): the most common type of melanoma. It experiences lateral (radial) growth first and then vertical (invasive) growth arises.
(b) Nodular melanoma (15–30%): rapidly expanding raised or polypoid abrasions that are frequently bluish or blackish in appearance and display an initial vertical growth stage.
(c) Lentigo maligna melanoma (4–10%): occurs more commonly in elderly patients with frequently sun-exposed skin. It characteristically commences as a minor freckle-like macula and later grows, becomes darker and irregular shaped, and displays a vertical growth stage.
(d) Acral lentiginous (<5%): lesions occur most frequently on soles, palms, or subungual, and infrequently over mucosal surfaces.
Any non-cutaneous sites with melanocytes, such as the ophthalmic, genitourinary,53 nasopharyngeal, gastrointestinal and vaginal regions, can also develop melanoma.71 According to an analysis of the National Cancer Data Base, which has information on 84![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 836 people with cutaneous and non-cutaneous melanoma, these are far less common than cutaneous melanoma. 91.2% of the diagnosed melanomas were cutaneous melanomas.53 Ocular melanoma accounts for 5.2% of cases, 1.3% of which are primary melanoma of the mucosa, and 2.2% of which have an undetermined primary location. Melanoma prognosis is based on the thickness of the lesion, with thicker lesions correlating to a higher fatality rate. Therefore, to stop metastasis, melanoma lesions must be found early and removed.53
836 people with cutaneous and non-cutaneous melanoma, these are far less common than cutaneous melanoma. 91.2% of the diagnosed melanomas were cutaneous melanomas.53 Ocular melanoma accounts for 5.2% of cases, 1.3% of which are primary melanoma of the mucosa, and 2.2% of which have an undetermined primary location. Melanoma prognosis is based on the thickness of the lesion, with thicker lesions correlating to a higher fatality rate. Therefore, to stop metastasis, melanoma lesions must be found early and removed.53
3. PEI-based nanosystems in skin cancer: molecular mechanism and drug targeting
Skin cancer, including both carcinoma and melanoma, remains a significant global health concern, with increasing incidence rates observed across various demographics. However, recent years have witnessed remarkable advancements in treatment strategies, revolutionizing the management of this disease.51,52 The latest developments in skin cancer treatment, encompass novel therapeutic modalities, targeted drug delivery systems, and personalized approaches to patient care. Skin cancer encompasses a spectrum of malignancies, including melanoma, BCC, and SCC, each presenting unique challenges in diagnosis and treatment.51,52,72 While traditional approaches like surgery, chemotherapy, and radiation therapy remain cornerstones of treatment, recent breakthroughs have expanded the therapeutic landscape, offering new hope for patients. PEI-based nanomaterials facilitate the delivery of therapeutic agents into cancer cells primarily through endocytosis. Once inside the cell, the nanomaterials escape the endosomes via the “proton sponge effect,” which is characteristic of PEI's high buffering capacity. This effect leads to osmotic swelling and ruptures the endosomes, releasing the therapeutic agents into the cytoplasm.73 Due to the higher permeability and retention (EPR) effect in tumors, PEI-based nanomaterials can accumulate more in the tumor tissue than in normal tissues. This selective accumulation allows for higher local concentrations of the therapeutic agent, improving its efficacy and reducing systemic side effects.743.1. Cellular uptake and intracellular trafficking
PEI-based NPs exhibit a remarkable capacity to facilitate the internalization of therapeutic agents into cancer cells via endocytosis. Once inside the cell, PEI's proton sponge effect triggers endosomal disruption, enabling the release of therapeutic cargo into the cytoplasm.73 This mechanism ensures efficient delivery of therapeutic payloads, enhancing their effectiveness in combating skin cancer.3.2. Gene silencing and immunomodulation
PEI-based nanomaterials play a pivotal role in gene silencing strategies aimed at targeting specific oncogenes implicated in skin cancer progression. By delivering nucleic acid-based therapeutics, such as siRNA or miRNA, PEI NPs can selectively silence key genes involved in tumor proliferation and survival.75 Additionally, PEI-based nanomaterials can modulate the immune response within the tumor microenvironment, promoting anti-tumor immunity and enhancing the efficacy of cancer immunotherapy.763.3. Immune activation and antigen presentation
Immunotherapy has emerged as a game-changer in the treatment of advanced melanoma, leveraging the immune system to combat cancerous cells. Checkpoint inhibitors, including anti-PD-1 and anti-CTLA-4 antibodies, have established remarkable efficacy in prolonging survival and inducing durable responses in patients with metastatic melanoma. Additionally, targeted therapies targeting specific genetic mutations, such as BRAF and MEK inhibitors, have shown promise in patients harboring BRAF-mutant melanomas, offering personalized treatment options.77 PEI-based NPs can be designed to promote immune activation and antigen presentation within the tumor microenvironment, thereby enhancing the efficacy of cancer immunotherapy. By delivering immunomodulatory agents and tumor antigens directly to antigen-presenting cells, these NPs facilitate the priming and activation of anti-tumor immune responses.76 PEI-based nanomaterials, for instance, offer a versatile platform for targeted drug delivery, facilitating the efficient delivery of therapeutic agents into cancer cells while minimizing systemic toxicity. These nanomaterials can be tailored to respond to the unique microenvironment of skin tumors, enabling precise release of therapeutic payloads and enhancing treatment efficacy.76Nanoparticles target cancer cells in skin cancer through both passive and active targeting mechanisms (Fig. 4). Passive targeting exploits the EPR effect, where the leaky vasculature and meagre lymphatic draining of tumor tissues allow NPs to accumulate more in the tumor site, thereby concentrating the therapeutic agents where needed while reducing exposure to healthy tissues.78–80 Active targeting, on the other hand, involves modifying the NPs’ surfaces with specific ligands, such as antibodies or peptides, which bind to receptors overexpressed on cancer cells. This binding ensures that the NPs are more precisely delivered to the cancer cells, enhancing the therapeutic effect, and minimizing side effects by sparing normal cells.81
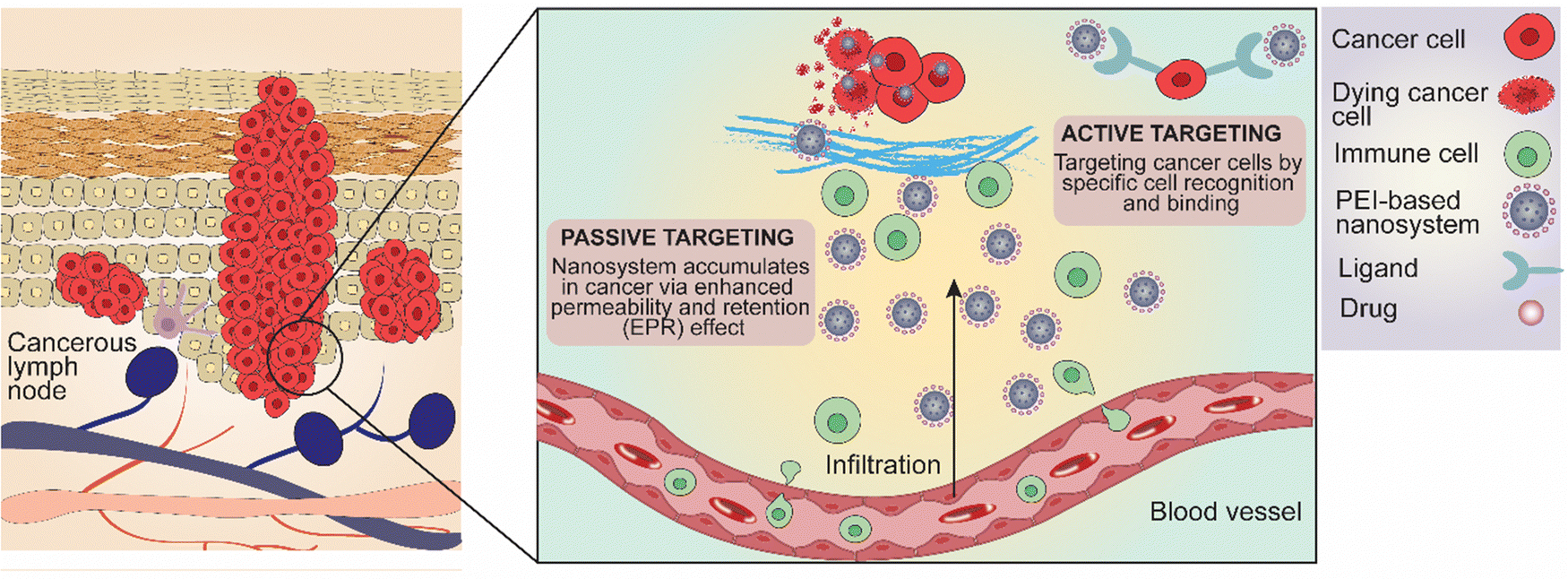 | ||
| Fig. 4 Major molecular mechanism and drug release strategies of PEI-based and PEI-functionalized nanosystems targeting skin carcinoma and melanoma. | ||
3.4. Drug release strategies
Several strategies have been employed to achieve controlled release of drugs from PEI-based nanocarriers, thereby optimizing their therapeutic efficacy and minimizing systemic toxicity.5,25 These include stimulus-responsive drug release systems, such as pH-responsive or enzyme-responsive NPs, which exploit the acidic tumor microenvironment or specific enzymatic activities within cancer cells to trigger drug release.82,83 Other strategies involve the use of stimuli such as light, heat, or ultrasound to remotely trigger drug release from nanocarriers at the site of the tumor. Furthermore, the design of biodegradable PEI derivatives or hybrid nanomaterials composed of PEI and other biocompatible polymers can facilitate sustained drug release kinetics and reduce the risk of long-term toxicity.5,25Tumor microenvironments typically have a lower pH compared to normal tissues. PEI-based nanomaterials can be designed to release their cargo in response to the acidic conditions found in tumor tissues. This pH-sensitive drug release ensures that the therapeutic agents are released primarily within the tumor, enhancing their therapeutic effect while minimizing damage to healthy cells.84 Another innovative strategy involves the use of photo-responsive PEI-based nanomaterials. Upon exposure to specific wavelengths of light, these nanomaterials can release their therapeutic cargo. This approach allows for precise spatial and temporal control over drug release, particularly useful in treating accessible skin cancers.85 Skin cancer cells often overexpress certain enzymes, such as matrix metalloproteinases (MMPs). PEI-Ns can be engineered to degrade in the presence of these enzymes, triggering the release of their therapeutic payload. This strategy leverages the unique enzyme profile of the tumor to achieve targeted drug delivery.86
4. Various PEI-based nanosystems in skin cancer therapy
PEI is a highly cationic polymer that has garnered significant interest in the field of nanomedicine for its potential in delivering therapeutic agents, including nucleic acids and small molecules, specifically to cancer cells. PEI-Ns (lipid, polymeric and inorganic NPs) can be engineered using various physical, chemical, and biological strategies for cancer theranostic applications (Fig. 5). In the context of skin cancer, PEI-Ns offer a promising approach due to their ability to enhance cellular uptake, facilitate endosomal escape, and enable targeted delivery.17,87 The initial step in the therapeutic action of PEI-Ns is their uptake by cancer cells. This process primarily occurs through endocytosis, a cellular mechanism where the cell membrane engulfs the nanomaterial to form an endosome. PEI's high positive charge promotes its interaction with the negatively charged cell membrane, enhancing endocytosis efficiency. Studies have shown that PEI-based NPs are predominantly internalized via clathrin-mediated endocytosis, although caveolae-mediated endocytosis and macropinocytosis may also play roles.88,89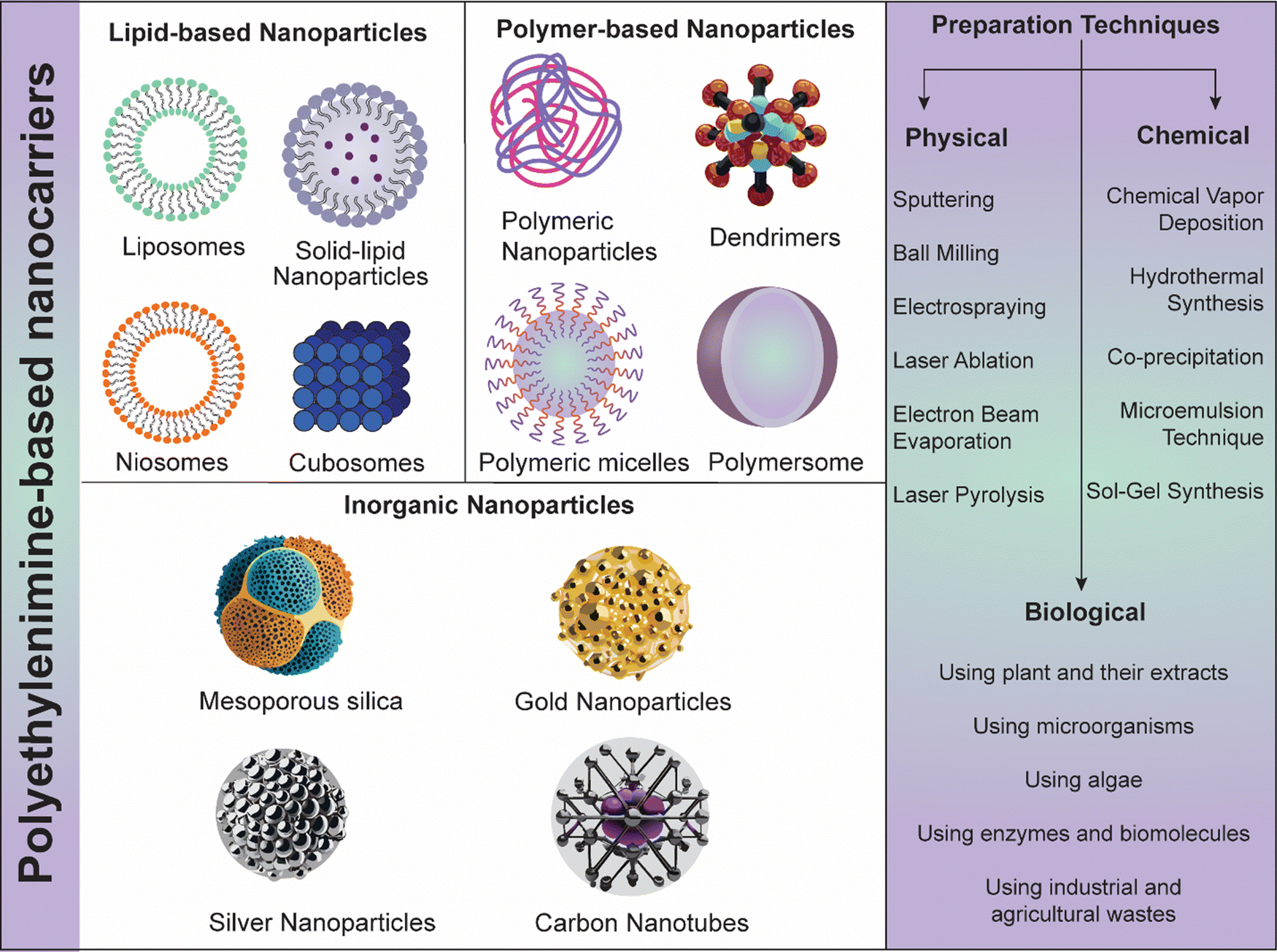 | ||
| Fig. 5 Schematic representation of the utilization of nanosystems in skin cancer therapy. | ||
4.1. PEI-based nanosystems for gene delivery and immunotherapy
PEI-based nanomaterials can deliver siRNA or miRNA to target specific oncogenes in skin cancer cells, leading to gene silencing. This process can downregulate the expression of proteins critical for cancer cell survival and proliferation, thereby inducing apoptosis. For example, siRNA targeting BRAF mutations common in melanoma have shown potential in reducing tumor growth.75PEI-Ns have gained considerable consideration in the field of cancer therapy due to their ability to efficiently deliver therapeutic agents, including small interfering RNA (siRNA), to target cells. In the context of skin cancer, such as melanoma, the progress of effective delivery systems is vital for enhancing the therapeutic effectiveness of chemotherapeutics with reduced or negligible systemic toxicity. PEI-based NPs complexed with plasmid DNA encoding tumor suppressor genes have shown promise in treating melanoma. These complexes can efficiently transfect skin cancer cells, restoring the function of tumor suppressor genes and inhibiting tumor growth.87 Doxorubicin, a chemotherapeutic agent, can be conjugated to PEI to improve its delivery to melanoma cells. This conjugation enhances the drug's solubility and stability, allowing for more effective targeting of cancer cells while reducing systemic toxicity.24 PEI-Ns have been used to deliver siRNA targeting specific mutations in melanoma cells. These systems can effectively silence oncogenes such as BRAF, leading to reduced tumor cell proliferation and increased apoptosis.73 A recent study highlighted the use of poly(D,L-lactic-co-glycolic acid) (PLGA)-PEI NPs covered with poly(I) for personalized cancer immunotherapy. These NPs leverage the immune-stimulating properties of poly(I), a synthetic analog of double-stranded RNA, to enhance anti-tumor immune responses. This strategy has shown potential in boosting the effectiveness of immunotherapy in skin cancer by facilitating the delivery of immune-modulating agents directly to the tumor site.84
Once internalized, PEI-Ns are trapped within endosomes. Efficient endosomal escape is crucial for the release of the therapeutic payload into the cytoplasm, where it can exert its intended effect. PEI facilitates endosomal escape through the “proton sponge effect.” The polymer's high buffering capacity causes an invasion of protons and chloride ions into the endosome, causing osmotic swelling and subsequently disrupts the endosomal membrane. This process releases the encapsulated drug or genetic material into the cytoplasm.15,90 After endosomal escape, the nanomaterials must navigate the intracellular environment to reach their specific target, such as the nucleus or cytoplasmic components. PEI can be modified with various targeting ligands, such as peptides, antibodies, or small molecules, to enhance its specificity for cancer cells. For example, folic acid and RGD peptides have been used to target folate receptors and integrins, respectively, which are overexpressed in many types of cancer, including skin cancer.91,92 For gene therapy applications, the therapeutic DNA or RNA delivered by PEI-Ns must enter the nucleus. PEI facilitates this process through its interaction with the nuclear pore complex. Additionally, the polymer's positive charge helps condense the genetic material into compact structures that are more easily transported into the nucleus.25,93
Several factors influence the efficiency of PEI-Ns in delivering therapeutic agents to skin cancer cells: The structure of PEI (branched vs. linear) affects its cellular uptake and toxicity. Branched PEI is often more effective in gene delivery due to its higher buffering capacity and ability to condense DNA, but it also tends to be more toxic compared to linear PEI.94 The molecular weight of PEI influences its transfection efficiency and cytotoxicity. Higher molecular weight PEI typically shows higher transfection efficiency but also higher toxicity. Therefore, an optimal balance between molecular weight and safety must be achieved.95 Modifying the surface of PEI-Ns with PEG or other biocompatible polymers can reduce their cytotoxicity and improve their stability in biological environments. PEGylation also enhances the circulation time of the nanosystems in the bloodstream, promoting their accumulation in tumors through the EPR effect.96 The incorporation of targeting ligands enhances the specificity of PEI-Ns for cancer cells, reducing off-target effects and improving therapeutic outcomes. For instance, targeting ligands such as folic acid and RGD peptides have been shown to significantly enhance the uptake of PEI-based NPs by skin cancer cells.97,98
PEI-Ns have garnered substantial consideration in the field of cancer immunotherapy due to their capability to improve the immune responses against tumors (Fig. 6). One promising approach involves the use of PLGA-PEI NPs covered with poly (IC) for personalized cancer immunotherapy.76 When incorporated into PLGA-PEI NPs, poly (IC) acts as a potent immunostimulant, promoting the maturation and activation of dendritic cells and enhancing the presentation of tumor antigens to T cells. PLGA-PEI NPs can be engineered to target specific tumor antigens, facilitating their uptake by antigen-presenting cells (APCs) like dendritic cells. Once internalized, the NPs release poly (IC), which activates APCs and promotes the priming of tumor-specific T cell responses. Poly (IC) promotes the production of pro-inflammatory cytokines and chemokines, which recruit and activate cytotoxic T lymphocytes (CTLs) to the tumor site. By enhancing the CTL response, PLGA-PEI NPs covered with poly (IC) can effectively target and kill cancer cells, leading to tumor regression.76
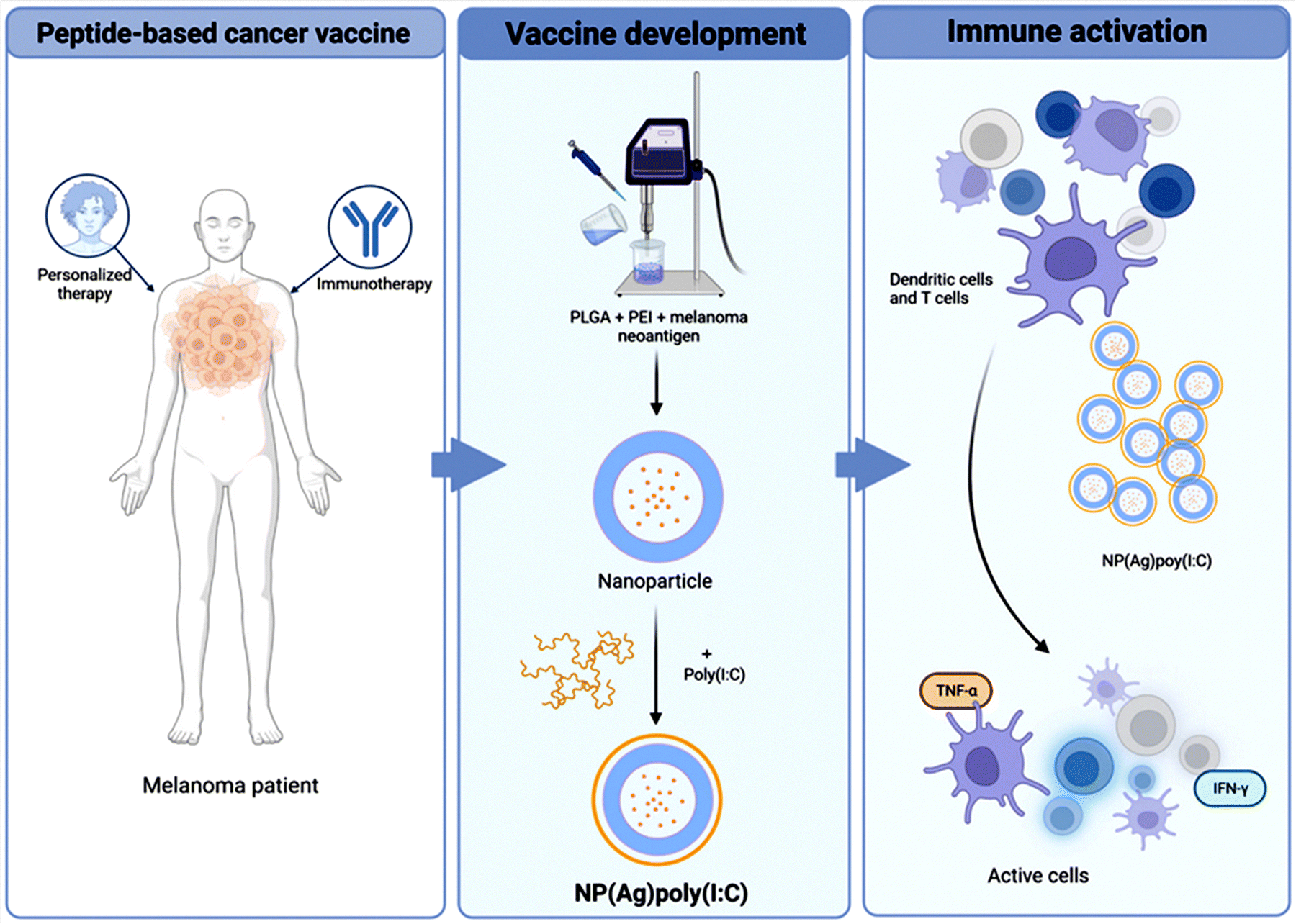 | ||
| Fig. 6 A schematic illustration highlighting the potential immune response generated using PLGA-PEI based NPs conjugated with immunotherapy for the specific elimination of melanoma tumor cells. Reproduced from ref. 76 with permission from Springer Nature, copyright 2024. | ||
Ragelle et al. fabricated chitosan-based NPs tailored for intravenous delivery of small interfering RNA (siRNA) which demonstrated remarkable stability in biological environments such as blood, along with a strong gene silencing capability with minimal cytotoxicity.99 Importantly, they observed that the formulations had a significant level of gene silencing when amine-rich PEI polymer was added. Furthermore, the activity of gene silencing and its cytotoxicity were assessed in luciferase-expressing B16 melanoma cells. The study highlighted the critical importance of nanocarrier stability in achieving the desired therapeutic effects. Notably, the inclusion of PEG and utilization of high-MW chitosan contributed to the structural integrity of the NPs, leading to high levels of in vitro gene silencing.99 Kurosaki et al. developed a new vector by electrostatically coating cationic PEI/pDNA complexes with folic acid (FA). The coating significantly reduced their cytotoxicity towards the melanoma cell line B16-F10, which expresses the folate receptor (FR).100 Moreover, the anionic FA60/PEI/pDNA complexes demonstrated excellent transgene efficiency in B16-F10 cells via the FR-mediated pathway. Importantly, these complexes did not exhibit any erythrocyte agglutination. Several organs (liver, kidney, spleen, and lung) with FR showed better transgenic effectiveness than PEI/pDNA complexes following intravenous injection of FA60/PEI/pDNA complexes into mice. Pre-administration of FA substantially reduced the gene expression of FA60/PEI/pDNA complexes. Overall, the FA60/PEI/pDNA complexes demonstrated promise for improving the efficacy of gene therapy, particularly in FR-expressing cells and tissues.100 In 2017, Lojk et al. investigated the stress responses of polyacrylic acid (PAA) and PEI coated NPs against the primary human myoblasts (MYO) and the B16 mouse melanoma cell lines.101 Even at high concentrations (100 g mL−1), negatively charged PAA did not activate the transcription factor NF-κB, cause cell toxicity, or produce reactive oxygen species (ROS). In contrast, positively charged PEI NPs caused necrosis and an increase in ROS after 24 h of incubation, even at lower concentrations (>4 g mL−1). Furthermore, 30 min after incubation, PEI NPs caused NF-κB activation in MYO cells, most likely by activating the TLR4 receptor. Surprisingly, B16 cells did not exhibit an NF-κB response.101
In 2014, Pyshnaya et al. synthesized linear PEI-modified gold nanorods (PEI-GNRs) and compared their physical, chemical, and biological characteristics to those of GNRs modified with bovine serum albumin (BSA) and spherical gold NPs (sGNPs) modified with the same substances (Fig. 7A(a and b)). BHK-21 and HeLa cells were unaffected by PEI-GNRs and GNPs (MTT test). Using TEM ultrathin sections, the diffusion of GNPs within the melanoma (B16), BHK-21, and HeLa cells was evaluated post-incubation after 30 min, 3 h, and 24 h (Fig. 7A(c)). Through caveolin-dependent and lipid raft-mediated endocytosis, PEI-GNRs and PEI-sGNPs showed rapid and active cell penetration and accumulated in endosomes and lysosomes. GNPs that had been altered with BSA exhibited protracted floating and a sizable delay in cell penetration. The findings demonstrate that penetration into cells is determined by the initial charge of NPs. As a result, the created PEI-GNRs were safe, stable in cell culture media, and capable of effectively penetrating cells.102 Jiang and team investigated the impact of modifying hyaluronic acid (HA) on receptor-associated endocytosis by tagging HA-derivatives with quantum dots (QDots) (Fig. 7B(a–c)). HA-QDot conjugates (<25 mol% degree of modifications) were more effectively taken up via HA receptor-mediated endocytosis compared to QDots alone.103 In B16-F1 cells expressing HA receptors, the siRNA/PEI-HA combination demonstrated superior gene silencing efficacy compared to the siRNA/PEI complex. Specifically, the anti-PGL3-Luc siRNA/PEI-HA complex achieved gene silencing levels in the range of 50% to 85%, depending on serum levels up to 50%. siRNA/PEI-HA combination primarily accumulated in tissues rich in HA receptors, including the kidney, liver, and tumors. Intratumoral injection of the anti-VEGF siRNA/PEI-HA complex in C57BL/6 mice demonstrated effective tumor growth inhibition through HA receptor-mediated endocytosis of tumor cells (Fig. 7B(d and e)).103
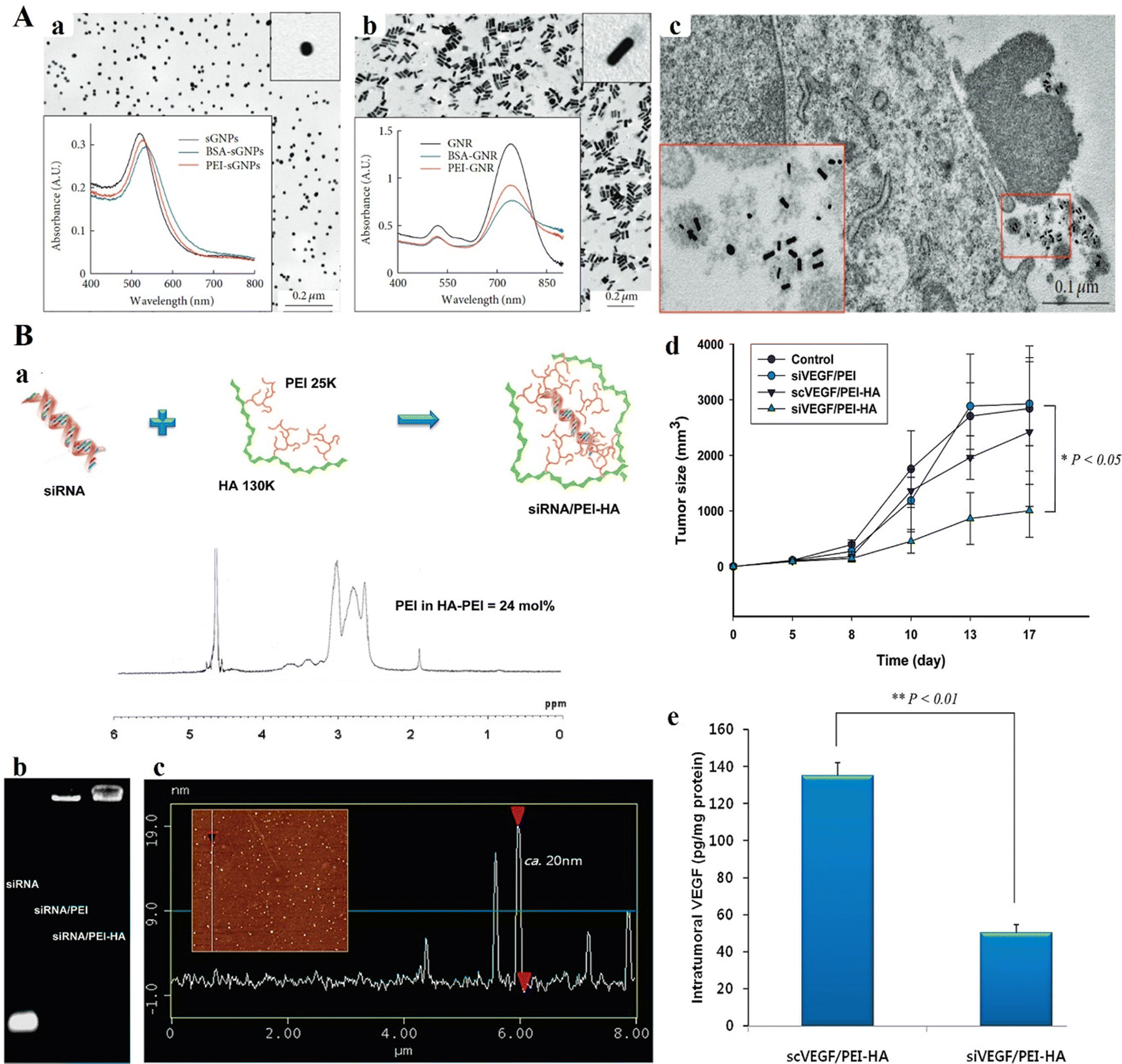 | ||
| Fig. 7 (A) TEM images and absorption spectra of (a) citrate-sGNP and (b) CTAB-GNR suspensions in aqueous medium. (c) Characteristic images of BSA-GNRs related with cell detritus (enlarged in the box) nearby the surface of B16 cells after 3 h of incubation. TEM of ultrathin sections. Reproduced from ref. 102 with permission from Wiley, copyright 2014. (B) (a) Schematic representation of the siRNA/PEI–hyaluronic acid (HA) complex. 1H NMR analysis established the effective development of PEI–HA conjugates with ca. 24 mol% PEI amount. (b) Agarose gel electrophoresis of siRNA, siRNA/PEI complex and siRNA/PEI–HA complex. (c) AFM particle size analysis of the siRNA/PEI–HA complex (20 nm with a little negative surface charge). (d) Tumor volume change in tumor-bearing C57BL/6 mice with increasing time after intertumoral injection of a control (5% glucose solution), siVEGF/PEI complex, scrambled siVEGF (scVEGF)/PEI–HA complex, and siVEGF/PEI–HA complex. The results were represented as mean ± SD (n = 3). *P < 0.05 versus control. (e) VEGF levels in tumor tissues at 17 days post-treatment with the scVEGF/PEI–HA complex and siVEGF/PEI–HA complex. The excised tumors were homogenized in PBS with protease inhibitor. After centrifugation, the amount of VEGF in each supernatant was measured by ELISA. **P < 0.01 versus scVEGF/PEI–HA. Reproduced from ref. 103 with permission from Springer Nature, copyright 2024. | ||
Chen and colleagues developed and reported a chitosan-linked PEI (CP) as a nonviral vector for dendritic cell (DC) gene delivery. They demonstrated that plasmid DNA can form positive nanoparticle complexes with CP. The CP/DNA complexes showed superior transfection efficiency in DCs compared to Lipofectamine 2000 (Lipo), a commercial transfection reagent. The study revealed that antigen plasmid-engineered DCs using CP have the potential to mediate an antitumor immune response. The study revealed that antigen plasmid-engineered DCs using CP have the potential to mediate an antitumor immune response. Physicochemical analysis confirmed the formation of cationic NPs by CP/DNA complexes. Transfection of DCs with CP/DNA complexes resulted in higher transfection efficiency and lower cytotoxicity compared to Lipofectamine 2000. Furthermore, DCs transfected with CP/DNA expressing gp100 exhibited increased resistance to B16BL6 melanoma challenges after vaccination.104 In a study by Yao and team, a nanopolymer for delivering Interleukin-2 (IL-2) was discussed. The nanopolymer, termed H1, consists of low MW PEI (600 Da) linked by β-cyclodextrin and conjugated with folate. H1 was utilized to form polyplexes with IL-2 plasmid (H1/pIL-2) that resulted in polyplexes with a diameter of approximately 100 nm. These polyplexes were injected into the tumors of C57/BL6 mice carrying B16-F1 melanoma grafts, which inhibited the growth of the tumors and increased overall survival. They also discovered that H1/pIL-2 boosted the infiltration of CD4 T and CD8 cells as well as natural killer cells into the tumor environment, as well as the activation and proliferation of these cells in peripheral blood. To summarize, the outcomes demonstrate that H1/pIL-2 is a safe and efficient melanoma treatment, with efficacy comparable to rAdv-IL-2. This innovative approach represents an alternative method of gene therapy for melanoma.85 Cheng et al. developed and reported reduction-sensitive gene carriers using diselenide bonds cross-linked to oligoethylenimine 800 Da (OEI800). The findings demonstrated that OEI800-SeSex, containing diselenide bonds, had the same reduction sensitivity as OEI800-SSx (cross-linked with disulfide bonds), effectively binding plasmid DNA to form nanosized particles. Compared to the nondegradable PEI25k control, in vitro tests demonstrated that the reducible OEI800-SeSex and OEI800-SSx exhibited significantly lower cytotoxicity and higher transfection activity. Specifically, OEI800-SeSex outperformed OEI800-SSx in terms of transfection efficiency in B16F10 cells at a C/P ratio of 10, while in HeLa cells, OEI800-SeSex outperformed OEI800-SSx across all C/P ratios tested. Interestingly, the reduction sensitivity of diselenide bonds was found to be cell-dependent.105
In a study by Alshamsan et al., siRNA polyplexes of PEI or its stearic acid derivative (PEI-StA) were investigated for their ability to induce B16 cell death both in vitro and in vivo through signal transducer and activator of transcription 3 (STAT3) knockdown in B16 murine melanoma. The study focused on the physical encapsulation of siRNA/PEI and PEI-StA polyplexes within PLGA NPs for STAT3 knockdown in dendritic cells (DCs). The average diameter and zeta potential of PLGA NPs comprising siRNA polyplexes of PEI and PEI-StA ranged from 350 to 390 nm and −13 to −19 mV, respectively. Additionally, the encapsulation efficiencies for siRNA in PLGA-P and PLGA-PS were 26% and 43%, respectively. SiRNA release from both types of NPs exhibited a triphasic pattern, with PLGA-PS showing a faster release rate. Their fluorescence microscopy uptake research verified both the NP types' endosomal localization and uptake by DC. DCs displayed high STAT3 and low CD86 expression after being exposed to B16-F10 conditioned media, indicating decreased function. DC maturation and functionality were restored by STAT3 siRNA when it was silenced by PLGA-P and PLGA-PS, as shown by the elevation in CD86 expression, strong TNF-R production, and considerable allogenic T cell proliferation. Moreover, encapsulation in PLGA NPs dramatically decreased the toxicity of PEI on DCs.106 In 2013, Kurosaki and team reported a unique and secure gene delivery vector coated with polyglutamic acid (PGA), offering efficient transfection capabilities.107 The PGA-coated NPs were precisely engineered into spherical shapes, enhancing their stability and performance. In mouse models, intravenously administered plasmid DNA/PEI complexes (non-coated) demonstrated notable transgenic efficacy in the spleen and lung but led to severe liver damage and mortality. In contrast, the PGA-coated complexes selectively demonstrated excellent transgenic efficacy in the spleen without causing toxicity. The PGA-coated complexes showed significant accumulation and high levels of gene expression in the spleen's marginal zone. These findings highlight the potential of PGA-coated complexes for delivering DNA vaccines effectively. They also utilized a melanoma DNA vaccine, pUb-M, with the PGA-coated complex. This formulation notably inhibited the growth and metastasis of the B16-F10 melanoma cell line, underscoring the therapeutic promise of the pUb-M-containing PGA-coated complex.107 In a recent study by Zhang et al. a precise targeting delivery system of cRGD-R9-cholesterol-PEI-PEG (RRCPP) NPs, was developed by incorporating cholesterol, PEG, and the cell-penetrating peptide conjugate cRGD (R8-cRGD) into a low-MW PEI.108 The R8-cRGD alteration helped the RRCPP delivery system's improved siRNA absorption efficiency The study focused on Wee1, an oncogenic nuclear kinase that controls the G2/M checkpoint in the cell cycle and is often overexpressed in melanoma and indicates a poor prognosis. Wee1 siRNA was delivered using RRCPP NPs, forming an RRCPP/siWee1 complex. This complex strongly suppressed the expression of the Wee1 gene (>60% suppression), and it also incited death in B16 tumor cells by inhibiting the G2M checkpoint and DNA damage in vitro. Moreover, the complex effectively inhibited lung metastasis (almost 66% inhibition rate) and subcutaneous xenograft model B16 tumor growth (nearly 85% inhibition rate) (Fig. 8).108
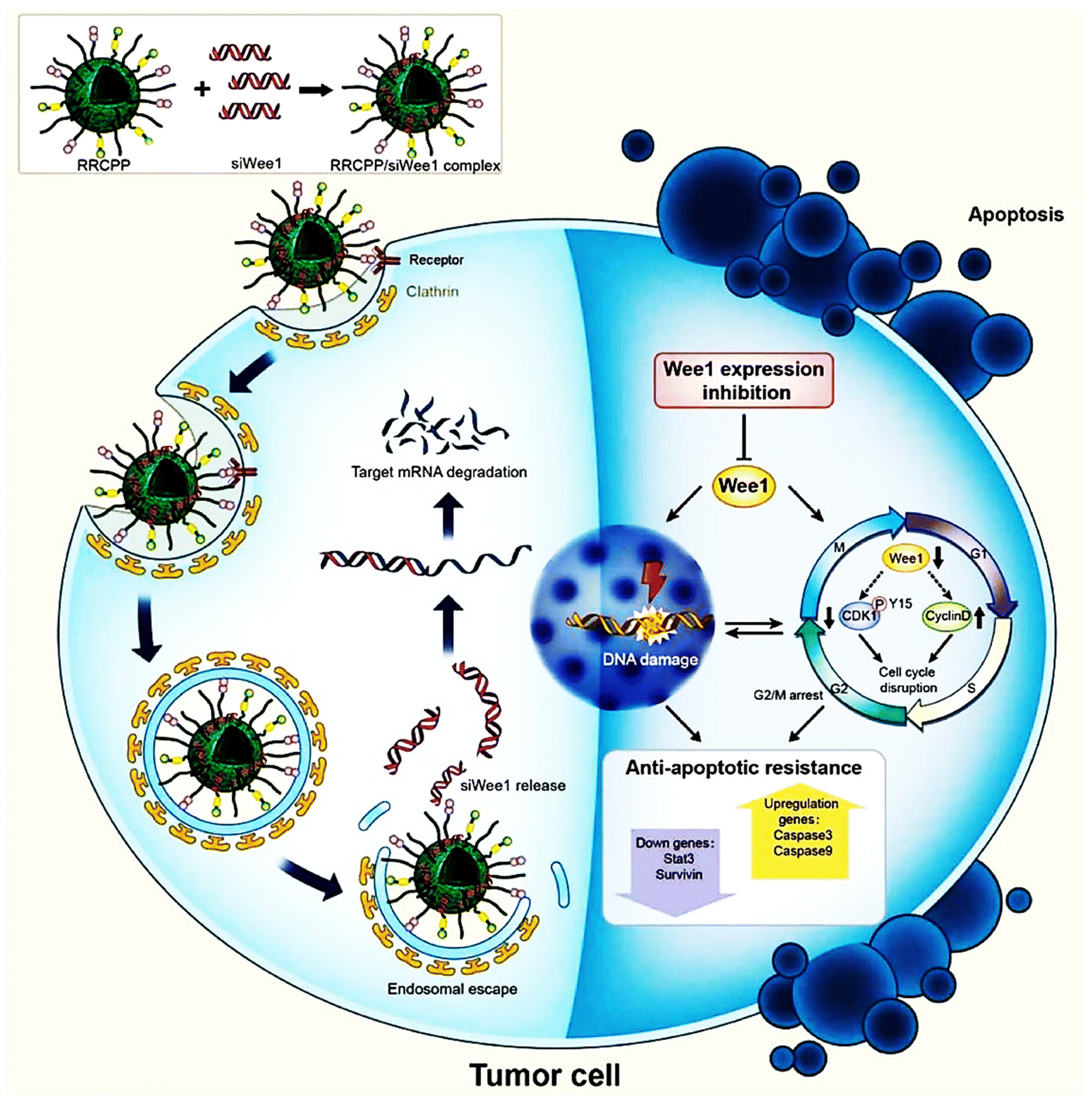 | ||
| Fig. 8 Scheme representing that the cRGD-R9-cholesterol-PEI-PEG (RRCPP) NPs/siWee1 (Wee1-targeting siRNAs) complex capably induced B16 cell apoptosis by activating cell-cycle disorder and DNA damage. Reproduced from ref. 108 with permission from American Chemical Society, copyright 2024. | ||
In 2011, Zhou et al. reported a novel cationic nanogel called heparin-PEI (HPEI) for efficient gene delivery. In their study, they constructed a highly efficient interleukin-15 (IL-15) plasmid and investigated the impact of HPEI-pIL-15 complexes on lung distribution levels and their anticancer effects against lung metastases of CT26 colon cancer and B16-F10 melanoma.109 Their work demonstrated that animals treated with HPEI-pIL-15 exhibited a reduced tumor metastasis index compared to other treatments. Intravenous injection of the HPEI-pIL-15 complex resulted in the maximum plasmid circulation levels in the lungs, as compared to PEI2K-pIL-15 and PEI25K-pIL-15 complexes. Additionally, levels of interferon-γ and tumor necrosis factor-α in the serum increased, along with an increment in the number of natural killer cells infiltrating the tumor tissues in pIL-15-treated mice. In addition, the HPEI-pIL15 group showed activation of apoptosis and suppression of cell proliferation in lung tumor foci.109 Liu et al. have established a nonviral gene vector termed PEI-P123-R13 by crosslinking pluronic P123 (P123) and LMW PEI, and subsequently conjugating a bifunctional peptide R13 (arginine–glycine–aspartate–cysteine, RGDC) to the polymer for tumor targeting and enhanced cellular absorption.110
In comparative studies using two different cell lines, PEI-P123-R13 exhibited significantly low cytotoxicity and high gene transfection efficacy compared with PEI 25 kDa (HeLa and B16). This novel polymer has promise as a low-cytotoxicity and highly effective gene delivery agent. However, it is important to determine if PEI-P123-R13 could demonstrate the same exceptional qualities, including stability over multiple cycles, tumor targeting specificity, non-cytotoxicity, and efficient in vivo transfection.110 Following a single subcutaneous dose, the NP vaccination significantly increased the frequency of neoantigen-specific CD8+ T cells in systemic circulation, reaching up to 23 ± 7%. This was attributed to the vaccine's ability to enhance dendritic cell activation and antigen cross-presentation. Despite the potent immune response, the initial anti-tumor efficacy was modest, as the activated CD8+ T cells in circulation showed restricted tumor infiltration. However, they achieved high anti-tumor efficacy by promoting tumor infiltration of vaccine-primed CD8+ T cells through local delivery of a STING agonist. In animal models of MC-38 colon cancer and B16F10 melanoma, the NP vaccine and STING agonist therapy eradicated the tumors and created long-lasting immune memory. For individualized cancer immunotherapy, this method offers a fresh treatment approach based on combination nano-immunotherapy.111
4.2. PEI-based drug co-delivery nanosystems
PEI-based nanosystems, for instance, offer a versatile platform for targeted drug delivery, facilitating the efficient delivery of therapeutic agents into cancer cells while minimizing systemic toxicity. Several strategies have been employed to achieve controlled release of drugs from PEI-based nanosystems, thereby optimizing their therapeutic efficacy and minimizing systemic toxicity.5,25 These include stimulus-responsive drug release systems, such as pH-responsive or enzyme-responsive NPs, which exploit the acidic tumor microenvironment or specific enzymatic activities within cancer cells to trigger drug release.82,83 Kodama et al. reported a novel gene delivery vector utilizing methotrexate (MTX)-coated plasmid DNA–PEI (pDNA–PEI) complexes through electrostatic binding. In the B16-F10 mouse melanoma cell line, these pDNA-PEI-MTX demonstrated gene expression efficiency comparable to cationic pDNA–PEI complexes. The MTX complexes were absorbed by the folate receptor through the cell-specific uptake pathways. Notably, the MTX120 complexes exhibited no signs of blood aggregation. After being administered intravenously, the MTX120 complexes had a higher transgene efficiency in the liver and spleen than the PEI complexes. The MTX complexes are therefore anticipated to be potential gene vectors in the future.112Xu et al. fabricated PEI-CA-DOX conjugates, through the conjugation of doxorubicin (DOX) to PEI through cis-aconitic anhydride (CA; a pH-sensitive linker). They subsequently utilized these conjugates to form PEI-CA-DOX/siRNA complex NPs through electrostatic interaction between anionic siRNA and cationic PEI-CA-DOX. The drug release experiments revealed that DOX was released more quickly at acidic pH levels compared to neutral pH (7.4). Also, when applied to B16F10 cells, PEI-CA-DOX/Bcl2 siRNA combination NPs exhibit greater cytotoxicity and apoptosis induction than DOX or Bcl2 siRNA alone (Fig. 9A). The efficacy of PEI-CA-DOX/Bcl2 siRNA complex NPs was further demonstrated in vivo, when administered directly into the lungs of B16-F10 melanoma-bearing mice. This localized codelivery approach led to enhanced anticancer activity with minimal adverse effects on healthy lung tissues (Fig. 9B). A significant portion of these drugs and siRNA accumulate in lung tumor tissues, but very infrequently in normal lung tissues. This pulmonary delivery strategy shows promise for effective cancer therapy while reducing systemic toxicity.113
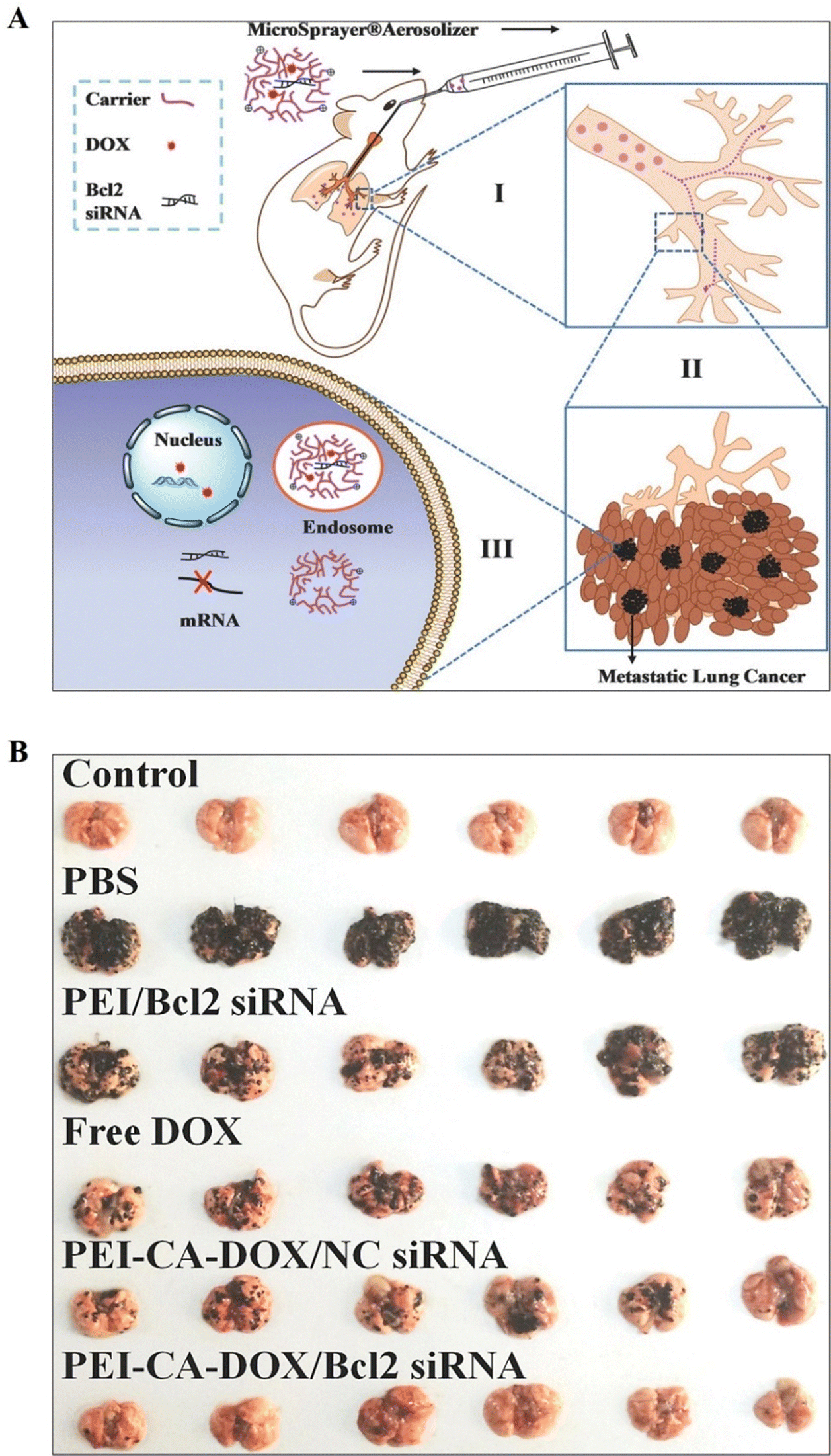 | ||
| Fig. 9 (A) Representation of pulmonic co-delivery of DOX and siRNA to the lungs: (I, II) DOX and siRNA pass within the trachea, bronchi, and alveolus; (III) DOX and siRNA were co-delivered to cancerous cells (metastatic). (B) Images of lungs attained from C57BL/6 mice. Healthy lungs of mice without B16F10 cell implantation were considered as the control group (n = 6) and other groups represent the lung status of mice with B16F10 cell implantations via pulmonary administration of PBS, PEI/Bcl2 siRNA nanoparticles, free DOX, PEI-CA-DOX/Nc siRNA NPs, and PEI-CA-DOX/Bcl2 siRNA NPs, respectively (n = 6). Reproduced from ref. 103 with permission from American Chemical Society, copyright 2024. | ||
In 2008, Wang et al. developed Thioketal-crosslinked PEI (TK-PEI) to condense the p53 gene into nanocomplexes (NCs), which were then coated with hyaluronic acid (HA) modified with pheophytin a (Pha) to enhance their colloidal stability and enable cancer cell targeting.114 In their studies they observed that endosomal membranes were disrupted and the p53 gene was more expressed for anti-cancer gene therapy using short-term (8-minute) light irradiation, which in turn induced controlled levels of ROS. Subsequently, long-term (30-minute) light irradiation during the post-transfection stage produced fatal levels of ROS, synergistically inducing cancer cell death, and supporting the efficacy of p53 gene therapy. These results demonstrate a novel approach utilizing light-triggered ROS generation to enhance targeted cancer therapy via TK-PEI/p53-HA-Pha nanocomplexes.114 In 2021, Park et al. reported a personalized immunization platform based on PEI for the coordinated delivery of neoantigen peptides and CpG adjuvants encapsulated in small NPs.111
Huang et al. reported a novel strategy comprising a combinatorial chemo-photodynamic approach for targeting melanoma, utilizing targeted drug delivery and the charge-reversal phenomenon to enhance cellular uptake (Fig. 10). They synthesized an amphiphilic Pt(IV)-PEI-chlorin e6 (Pt(IV)-PEI-Ce6) polymer, which self-assembled into NPs termed PPC in an aqueous solution.115 These NPs were further layered with HA to form negatively charged PPC@HA. The negatively charged PPC@HA decreased the monocyte-phagocyte system (MPS) clearance throughout system circulation and improved its targeted delivery to melanoma cells that overexpress CD44. Hyaluronidase overexpression in the tumor caused HA breakdown upon accumulation to release the positively charged PPC, which led to an increase in PPC internalization into tumor cells.115
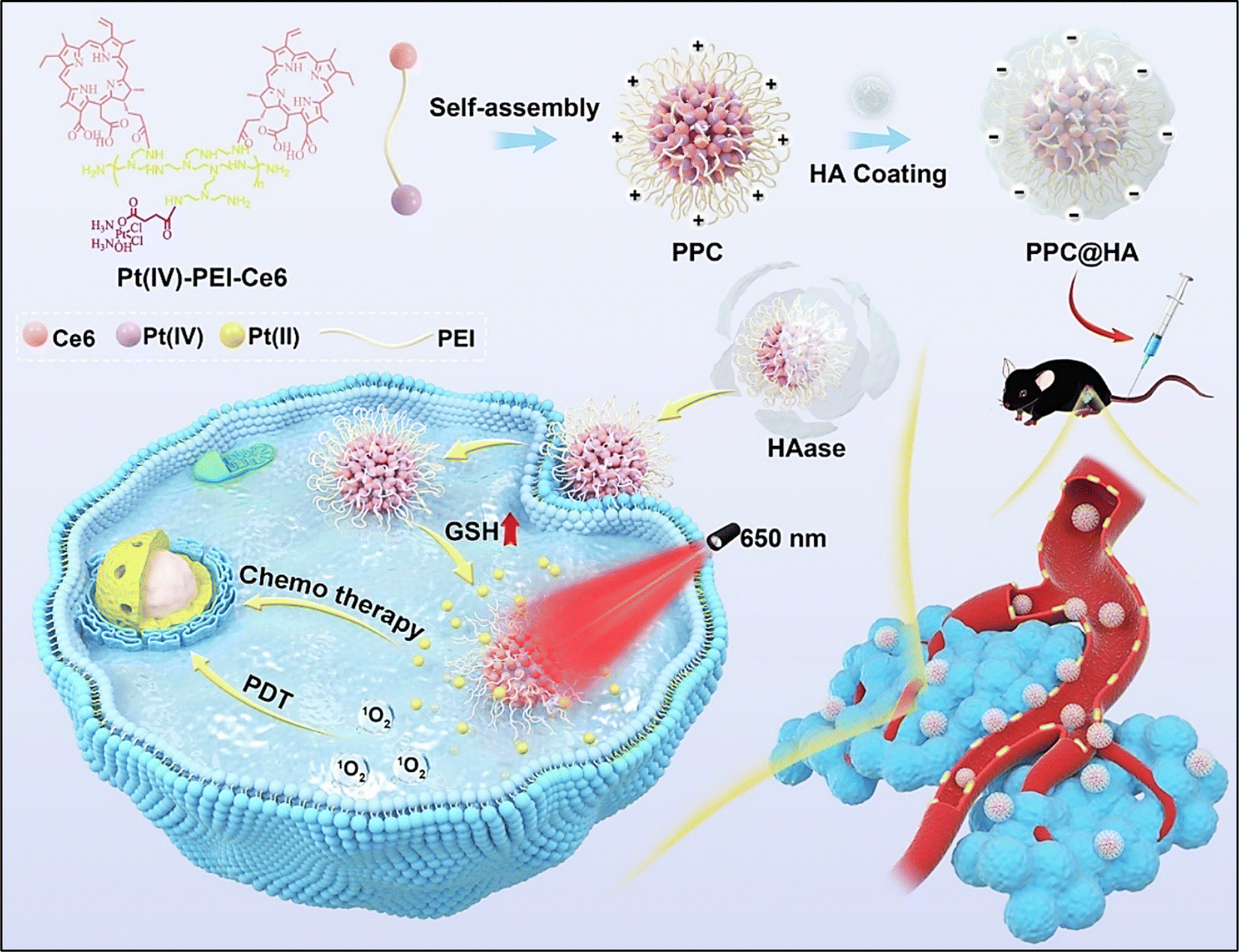 | ||
| Fig. 10 Schematic representation of the synthesis of charge-reversal micelles (PPC@HA), targeted drug delivery and improved cellular uptake of the fabricated chemo-photodynamic nanomedicine for melanoma therapy. Reproduced from ref. 115 with permission from Elsevier, copyright 2024. | ||
In 2023, researchers developed a novel gene delivery system called GEMNS-PEI/pDNA, designed to transport genes into B16F10 cells using green fluorescent protein (GFP) expression in pDNA and graphene-encapsulated magnetic nanoparticles (GEMNS) coated with branched PEI. The transfection efficacy of this system was assessed using PCR and confocal microscopy. The results highlighted that the complex effectively facilitated gene transfection in melanoma cells when used as intended. This approach represents a promising non-viral vector for delivering naked nucleic acids into eukaryotic cells for targeted gene delivery in melanoma therapy.116 Lei et al.117 fabricated HA-mediated microneedle biomineralized melanin NPs (derived from cuttlefish ink (CFI) which possess antioxidative and photothermal properties), and further entrapped inside an amorphous silica shell (a resource for bioactivity towards stimulating skin tissue regeneration). PEI and silica were coated over these materials for improving the zeta potential, leading to superior penetrability and release of entrapped compounds through microneedles systems. Furthermore, this hybrid platform showed superior activity towards scavenging ROS leading to controlled anti-inflammatory activity and regulating angiogenic gene expressions. Also, due to their superior penetrability through skin, they exhibited excellent photothermal annihilation of the residual subcutaneous tumor cells, and therefore avoided the reappearance and inhibition of Staphylococcus aureus infections in the wound areas.117
5. Conclusions and future perspectives
Significant advancements in polymer science, materials science, chemistry, and nanotechnology have facilitated the research and development of a diverse array of PEI-based nanosystems. These systems are designed with the conjugation of various biological and therapeutic molecules, making them highly versatile for numerous biomedical applications, especially for skin cancer. Due to the distinct structural properties of PEI, it is possible to physically encapsulate drugs, genes, and proteins within PEI-Nsor to covalently attach these molecules onto the nanosystems. This enables targeted delivery of these therapeutic agents into specific cells or tissues. The versatility of PEI allows for the development of diverse nanosystems tailored to address various challenges associated with skin cancer treatment, including drug resistance, targeted delivery, and minimizing systemic toxicity.118 Despite PEI's potential as a leading second-generation non-viral vector, several critical issues must be discussed before it can be clinically translated for cancer theranostics. Although PEI exhibits notable cytotoxicity, various surface modification techniques can enhance its biocompatibility. However, extensive research studies are warranted to determine the most suitable surface modification methods for specific research objectives. Moreover, the PEI's type and molecular weight significantly influence the drug loading efficiency, yet the connection among these factors remains indistinct. It is also important to note that although varied targeting agents have been established, their drug delivery efficacy remains low, characteristically under 5%. Thus, the delivery effectiveness of PEI-based systems must be enhanced for effective cancer theranostics. Nonetheless, while instant toxicity at in vivo levels seems negligible with suitable surface modifications, the enduring biodegradability and biosafety of PEI-based systems require thorough investigation.Efforts should be made to design PEI-based drug delivery systems that are either biodegradable or sufficiently small to fall within the renal filtration threshold for rapid renal clearance.5 Lastly, regulatory hurdles and manufacturing challenges must be overcome. The production of PEI-based nanosystems at a scale suitable for clinical use requires robust and reproducible methods that meet stringent quality control standards. Furthermore, gaining regulatory approval demands comprehensive preclinical data demonstrating safety and efficacy, followed by well-designed clinical trials. Although substantial developments have been made in PEI-based nanomaterials for skin cancer treatment, continued research and collaboration across disciplines are essential to overcome the existing challenges. With sustained effort, these innovative nanosystems have the potential to revolutionize skin cancer therapy, offering more effective, targeted, and safer treatment options for patients.
Author contributions
AK, SSD, SKS and KKK: conceptualization of the work. AK, SSD, SKS, ST, and BK: data collection and drafting. SSD and ST: made illustrations. AK, KKK and SSD: finalized and reviewed the manuscript.Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Conflicts of interest
The authors declare that they have no known competing financial interests.Acknowledgements
This research received no external funding.References
- A. I. Fahira, R. Amalia, M. I. Barliana, V. A. Gatera and R. Abdulah, Breast Cancer, 2022, 14, 71–83 Search PubMed.
- T. Patel, J. Zhou, J. M. Piepmeier and W. M. Saltzman, Adv. Drug Delivery Rev., 2012, 64, 701–705 CrossRef CAS.
- I. Posadas, S. Monteagudo and V. Cena, Nanomedicine, 2016, 11, 833–849 CrossRef CAS PubMed.
- W. Yuan and H. Li, Nanostruct. Drug Delivery, 2017, 445–460, DOI:10.1016/b978-0-323-46143-6.00014-2.
- C. Zhao and B. Zhou, J. Funct. Biomater., 2023, 14, 12, DOI:10.3390/jfb14010012.
- J. J. Virgen-Ortiz, J. C. S. Dos Santos, A. Berenguer-Murcia, O. Barbosa, R. C. Rodrigues and R. Fernandez-Lafuente, J. Mater. Chem. B, 2017, 5, 7461–7490 RSC.
- Y. Zhang, Z. Wang and R. A. Gemeinhart, J. Controlled Release, 2013, 172, 962–974 CrossRef CAS PubMed.
- B. Demeneix, J. Behr, O. Boussif, M. A. Zanta, B. Abdallah and J. Remy, Adv. Drug Delivery Rev., 1998, 30, 85–95 CrossRef.
- N. A. Peppas, P. Bures, W. Leobandung and H. Ichikawa, Eur. J. Pharm. Biopharm., 2000, 50, 27–46 CrossRef CAS.
- E. Tsuchida and K. Abe, Interactions Between Macromolecules in Solution and Intermacromolecular Complexes, 1982, ch. 1, pp. 1–119 DOI:10.1007/BFb0017549.
- S. S. Das, D. Sharma, B. V. K. Rao, M. K. Arora, J. Ruokolainen, M. Dhanka, H. Singh and K. K. Kesari, Mater. Adv., 2023, 4, 6064–6091 RSC.
- S. S. Das, A. K. Dubey, P. R. P. Verma, S. K. Singh and S. K. Singh, Nanotherapeutics in Cancer Vaccination and Challenges, Academic Press, 2022, pp. 427–445 Search PubMed.
- S. V. Vinogradov, T. K. Bronich and A. V. Kabanov, Adv. Drug Delivery Rev., 2002, 54, 135–147 CrossRef CAS.
- B. Schwarz and O. M. Merkel, Mater. Matters, 2017, 12, 2 Search PubMed.
- O. Boussif, F. Lezoualc'h, M. A. Zanta, M. D. Mergny, D. Scherman, B. Demeneix and J. P. Behr, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 7297–7301 CrossRef CAS.
- L. Wightman, R. Kircheis, V. Rossler, S. Carotta, R. Ruzicka, M. Kursa and E. Wagner, J. Gene Med., 2001, 3, 362–372 CrossRef CAS PubMed.
- P. Vicennati, A. Giuliano, G. Ortaggi and A. Masotti, Curr. Med. Chem., 2008, 15, 2826–2839 CrossRef CAS.
- N. Fattahi, L. Gorgannezhad, S. F. Masoule, N. Babanejad, A. Ramazani, M. Raoufi, E. Sharifikolouei, A. Foroumadi and M. Khoobi, Adv. Colloid Interface Sci., 2024, 325, 103119 CrossRef CAS.
- K. Kratz, T. Hellweg and W. Eimer, Colloids Surf., A, 2000, 170, 137–149 CrossRef CAS.
- J. W. Goodwin, J. Hearn, C. C. Ho and R. H. Ottewill, Br. Polym. J., 2007, 5, 347–362 Search PubMed.
- A. Fernández-Nieves, A. Fernández-Barbero, B. Vincent and F. J. de las Nieves, Macromolecules, 2000, 33, 2114–2118 CrossRef.
- A. Loxley and B. Vincent, Colloid Polym. Sci., 1997, 275, 1108–1114 CrossRef CAS.
- G. M. Eichenbaum, P. F. Kiser, A. V. Dobrynin, S. A. Simon and D. Needham, Macromolecules, 2001, 34, 6526 CrossRef CAS.
- V. Kafil and Y. Omidi, Bioimpacts, 2011, 1, 23–30 CAS.
- W. T. Godbey, K. K. Wu and A. G. Mikos, J. Biomed. Mater. Res., 1999, 45, 268–275 CrossRef CAS.
- Q. Peng, Z. Zhong and R. Zhuo, Bioconjugate Chem., 2008, 19, 499–506 CrossRef CAS.
- Z. Zhong, J. Feijen, M. C. Lok, W. E. Hennink, L. V. Christensen, J. W. Yockman, Y. H. Kim and S. W. Kim, Biomacromolecules, 2005, 6, 3440–3448 CAS.
- Y. Wen, S. Pan, X. Luo, X. Zhang, W. Zhang and M. Feng, Bioconjugate Chem., 2009, 20, 322–332 CrossRef CAS PubMed.
- J. H. Ryu, H. Y. Yoon, I. C. Sun, I. C. Kwon and K. Kim, Adv. Mater., 2020, 32, e2002197 Search PubMed.
- H. Sun, I. Yarovoy, M. Capeling and C. Cheng, Top. Curr. Chem., 2017, 375, 24 Search PubMed.
- C. H. Lee, D. Kasala, Y. Na, M. S. Lee, S. W. Kim, J. H. Jeong and C. O. Yun, Biomaterials, 2014, 35, 5505–5516 CrossRef CAS.
- D. Zhong, Y. Jiao, Y. Zhang, W. Zhang, N. Li, Q. Zuo, Q. Wang, W. Xue and Z. Liu, Biomaterials, 2013, 34, 294–305 CrossRef CAS.
- E. Wagner and J. Kloeckner, Polymer Therapeutics I, 2006, ch. 23, pp. 135–173 Search PubMed.
- S. M. Moghimi, P. Symonds, J. C. Murray, A. C. Hunter, G. Debska and A. Szewczyk, Mol. Ther., 2005, 11, 990–995 CrossRef CAS.
- P. Xu, G. K. Quick and Y. Yeo, Biomaterials, 2009, 30, 5834–5843 CrossRef CAS PubMed.
- A. C. Hunter, Adv. Drug Delivery Rev., 2006, 58, 1523–1531 CrossRef CAS PubMed.
- S. Wen, F. Zheng, M. Shen and X. Shi, J. Appl. Polym. Sci., 2012, 128, 3807–3813 CrossRef.
- A. Zintchenko, A. Philipp, A. Dehshahri and E. Wagner, Bioconjugate Chem., 2008, 19, 1448–1455 CrossRef CAS PubMed.
- S. Xu, M. Chen, Y. Yao, Z. Zhang, T. Jin, Y. Huang and H. Zhu, J. Controlled Release, 2008, 130, 64–68 CrossRef CAS PubMed.
- J. Li, Y. Hu, J. Yang, W. Sun, H. Cai, P. Wei, Y. Sun, G. Zhang, X. Shi and M. Shen, J. Mater. Chem. B, 2015, 3, 5720–5730 RSC.
- Y. Wang, Z. Xiong, Y. He, B. Zhou, J. Qu, M. Shen, X. Shi and J. Xia, Mater. Sci. Eng., C, 2018, 83, 9–16 CrossRef CAS PubMed.
- B. Zhou, L. Zheng, C. Peng, D. Li, J. Li, S. Wen, M. Shen, G. Zhang and X. Shi, ACS Appl. Mater. Interfaces, 2014, 6, 17190–17199 CrossRef CAS.
- Y. H. Kim, J. H. Park, M. Lee, Y. H. Kim, T. G. Park and S. W. Kim, J. Controlled Release, 2005, 103, 209–219 CrossRef CAS.
- H. Petersen, T. Merdan, K. Kunath, D. Fischer and T. Kissel, Bioconjugate Chem., 2002, 13, 812–821 CrossRef CAS.
- M. A. Gosselin, W. Guo and R. J. Lee, Bioconjugate Chem., 2001, 12, 989–994 CrossRef CAS.
- K. Wong, G. Sun, X. Zhang, H. Dai, Y. Liu, C. He and K. W. Leong, Bioconjugate Chem., 2006, 17, 152–158 CrossRef CAS.
- G. P. Tang, H. Y. Guo, F. Alexis, X. Wang, S. Zeng, T. M. Lim, J. Ding, Y. Y. Yang and S. Wang, J. Gene Med., 2006, 8, 736–744 CrossRef CAS PubMed.
- H. L. Jiang, Y. K. Kim, R. Arote, J. W. Nah, M. H. Cho, Y. J. Choi, T. Akaike and C. S. Cho, J. Controlled Release, 2007, 117, 273–280 CrossRef CAS PubMed.
- M. Zhao, M. Li, Z. Zhang, T. Gong and X. Sun, Drug Delivery, 2016, 23, 2596–2607 CrossRef CAS PubMed.
- E. H. Tracey and A. Vij, Dermatol. Clin., 2019, 37, 73–82 CrossRef CAS PubMed.
- D. Schadendorf, A. C. J. van Akkooi, C. Berking, K. G. Griewank, R. Gutzmer, A. Hauschild, A. Stang, A. Roesch and S. Ugurel, Lancet, 2018, 392, 971–984 CrossRef.
- A. Slominski, J. Wortsman, A. J. Carlson, L. Y. Matsuoka, C. M. Balch and M. C. Mihm, Arch. Pathol. Lab. Med., 2001, 125, 1295–1306 CrossRef CAS PubMed.
- S. Carr, C. Smith and J. Wernberg, Surg. Clin. North Am., 2020, 100, 1–12 CrossRef PubMed.
- R. I. Hartman and J. Y. Lin, Hematol. Oncol. Clin. North Am., 2019, 33, 25–38 CrossRef PubMed.
- Q. Liu, M. Das, Y. Liu and L. Huang, Adv. Drug Delivery Rev., 2018, 127, 208–221 CrossRef CAS.
- K. Eddy and S. Chen, Int. J. Mol. Sci., 2020, 21, 8984 CrossRef CAS PubMed.
- R. L. Mort, I. J. Jackson and E. E. Patton, Development, 2015, 142, 620–632 CrossRef CAS.
- A. H. Shain and B. C. Bastian, Nat. Rev. Cancer, 2016, 16, 345–358 CrossRef CAS PubMed.
- N. G. Crawford, D. E. Kelly, M. E. B. Hansen, M. H. Beltrame, S. Fan, S. L. Bowman, E. Jewett, A. Ranciaro, S. Thompson, Y. Lo, S. P. Pfeifer, J. D. Jensen, M. C. Campbell, W. Beggs, F. Hormozdiari, S. W. Mpoloka, G. G. Mokone, T. Nyambo, D. W. Meskel, G. Belay, J. Haut, N. C. S. Program, H. Rothschild, L. Zon, Y. Zhou, M. A. Kovacs, M. Xu, T. Zhang, K. Bishop, J. Sinclair, C. Rivas, E. Elliot, J. Choi, S. A. Li, B. Hicks, S. Burgess, C. Abnet, D. E. Watkins-Chow, E. Oceana, Y. S. Song, E. Eskin, K. M. Brown, M. S. Marks, S. K. Loftus, W. J. Pavan, M. Yeager, S. Chanock and S. A. Tishkoff, Science, 2017, 358, 6365 CrossRef.
- R. A. Sturm, Hum. Mol. Genet., 2009, 18, R9–R17 CrossRef CAS.
- S. Alaluf, D. Atkins, K. Barrett, M. Blount, N. Carter and A. Heath, Pigm. Cell Res., 2002, 15, 112–118 CrossRef CAS PubMed.
- A. Hennessy, C. Oh, B. Diffey, K. Wakamatsu, S. Ito and J. Rees, Pigm. Cell Res., 2005, 18, 220–223 CrossRef CAS PubMed.
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-Cancer J. Clin., 2018, 68, 7–30 CrossRef PubMed.
- T. Tadokoro, N. Kobayashi, B. Z. Zmudzka, S. Ito, K. Wakamatsu, Y. Yamaguchi, K. S. Korossy, S. A. Miller, J. Z. Beer and V. J. Hearing, FASEB J., 2003, 17, 1177–1179 CrossRef CAS PubMed.
- J. Lomas, P. Martin-Duque, M. Pons and M. Quintanilla, Front. Biosci., 2008, 13, 5071–5093 CrossRef CAS.
- P. Bharadwaj, S. S. Das, S. Beg and M. Rahman, Nanoformulation Strategies Cancer Treat., 2021, 277–289, DOI:10.1016/b978-0-12-821095-6.00020-3.
- A. Naspi, V. Panasiti, F. Abbate, V. Roberti, V. Devirgiliis, M. Curzio, M. Borghi, F. Lozupone, S. Carotti, S. Morini, E. Gaudio, S. Calvieri and P. Londei, PLoS One, 2014, 9, e98641 CrossRef.
- B. Ahmed, M. I. Qadir and S. Ghafoor, Crit. Rev. Eukaryotic Gene Expression, 2020, 30, 291–297 CrossRef.
- C. Berking, R. Takemoto, K. Satyamoorthy, T. Shirakawa, M. Eskandarpour, J. Hansson, P. A. VanBelle, D. E. Elder and M. Herlyn, Cancer Res., 2004, 64, 807–811 CrossRef CAS.
- V. Nikolaou and A. J. Stratigos, Br. J. Dermatol., 2014, 170, 11–19 CrossRef CAS PubMed.
- H. S. Kim, E. K. Kim, H. J. Jun, S. Y. Oh, K. W. Park, D. H. Lim, S. I. Lee, J. H. Kim, K. M. Kim, D. H. Lee and J. Lee, BMC Cancer, 2010, 10, 167 CrossRef PubMed.
- A. Sood, S. S. Das, A. Dev, D. Bhardwaj, A. Kumar, G. Agrawal and S. S. Han, Eur. Polym. J., 2023, 196 Search PubMed.
- X. Guo and L. Huang, Acc. Chem. Res., 2020, 45, 971–979 CrossRef.
- H. Maeda, Adv. Drug Delivery Rev., 2015, 91, 3–6 CrossRef CAS.
- M. S. Al-Dosari and X. Gao, AAPS J., 2009, 11, 671–681 CrossRef CAS PubMed.
- L. Gonzalez-Melero, E. Santos-Vizcaino, R. Varela-Calvino, I. Gomez-Tourino, A. Asumendi, M. D. Boyano, M. Igartua and R. M. Hernandez, Drug Delivery Transl. Res., 2024, 14(10), 2788–2803 CrossRef CAS.
- R. Goyal, S. Husain, K. Wilson, H. Chopra, R. Pahwa, M. Loganathan and R. Sharma, Explor. Med., 2023, 782–812 CAS.
- M. A. Subhan, S. S. K. Yalamarty, N. Filipczak, F. Parveen and V. P. Torchilin, J. Pers. Med., 2021, 11, 571 CrossRef PubMed.
- S. S. Das, S. Alkahtani, P. Bharadwaj, M. T. Ansari, A. L. MDF, Z. Pang, M. S. Hasnain, A. K. Nayak and T. M. Aminabhavi, Int. J. Pharm., 2020, 585, 119556 CrossRef CAS PubMed.
- N. K. Jha, S. Arfin, S. K. Jha, R. Kar, A. Dey, R. Gundamaraju, G. M. Ashraf, P. K. Gupta, S. Dhanasekaran, M. M. Abomughaid, S. S. Das, S. K. Singh, K. Dua, S. Roychoudhury, D. Kumar, J. Ruokolainen, S. Ojha and K. K. Kesari, Semin. Cancer Biol., 2022, 86, 1086–1104 CrossRef CAS.
- Y. Yao, Y. Zhou, L. Liu, Y. Xu, Q. Chen, Y. Wang, S. Wu, Y. Deng, J. Zhang and A. Shao, Front. Mol. Biosci., 2020, 7, 193 CrossRef CAS PubMed.
- H. A. Barkat, S. S. Das, M. A. Barkat, S. Beg and H. A. Hadi, Future Oncol., 2020, 16, 2959–2979 CrossRef CAS PubMed.
- S. S. Das, P. Bharadwaj, M. Bilal, M. Barani, A. Rahdar, P. Taboada, S. Bungau and G. Z. Kyzas, Polymers, 2020, 12 Search PubMed.
- M. Zare, M. Norouzi, F. Yazdian and M. Azizi, Cancer Nanotechnol., 2020, 11, 15–29 CrossRef.
- H. Yao, S. S. Ng, L. F. Huo, B. K. Chow, Z. Shen, M. Yang, J. Sze, O. Ko, M. Li, A. Yue, L. W. Lu, X. W. Bian, H. F. Kung and M. C. Lin, Mol. Cancer Ther., 2011, 10, 1082–1092 CrossRef CAS.
- M. Li, G. Zhao, W. K. Su and Q. Shuai, Front. Chem., 2020, 8, 647 CrossRef CAS.
- A. Zakeri, M. A. J. Kouhbanani, N. Beheshtkhoo, V. Beigi, S. M. Mousavi, S. A. R. Hashemi, A. Karimi Zade, A. M. Amani, A. Savardashtaki, E. Mirzaei, S. Jahandideh and A. Movahedpour, Nano Rev. Exp., 2018, 9, 1488497 CrossRef.
- H. Lv, S. Zhang, B. Wang, S. Cui and J. Yan, J. Controlled Release, 2006, 114, 100–109 CrossRef CAS.
- I. S. Zuhorn, R. Kalicharan and D. Hoekstra, Mol. Ther., 2002, 5, 488–494 Search PubMed.
- N. D. Sonawane, F. C. Szoka, Jr. and A. S. Verkman, J. Biol. Chem., 2003, 278, 44826–44831 CrossRef CAS.
- T. Merdan, J. Kopecek and T. Kissel, Adv. Drug Delivery Rev., 2002, 54, 715–758 CrossRef CAS PubMed.
- S. Wang, P. Huang and X. Chen, Adv. Mater., 2016, 28, 7340–7364 CrossRef CAS PubMed.
- W. T. Godbey, K. K. Wu and A. G. Mikos, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 5177–5181 CrossRef CAS PubMed.
- M. Thomas, Q. Ge, J. J. Lu, J. Chen and A. M. Klibanov, Pharm. Res., 2005, 22, 373–380 CrossRef CAS.
- D. Fischer, T. Bieber, Y. Li, H. P. Elsasser and T. Kissel, Pharm. Res., 1999, 16, 1273–1279 CrossRef CAS.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288–6308 CrossRef CAS PubMed.
- T. Xia, M. Kovochich and A. Nel, Clin. Occup. Environ. Med., 2006, 5, 817–836 Search PubMed.
- H. Zhu, Y. Li, R. Qiu, L. Shi, W. Wu and S. Zhou, Biomaterials, 2012, 33, 3058–3069 CrossRef CAS.
- H. Ragelle, R. Riva, G. Vandermeulen, B. Naeye, V. Pourcelle, C. S. Le Duff, C. D'Haese, B. Nysten, K. Braeckmans, S. C. De Smedt, C. Jerome and V. Preat, J. Controlled Release, 2014, 176, 54–63 CrossRef CAS.
- T. Kurosaki, T. Morishita, Y. Kodama, K. Sato, H. Nakagawa, N. Higuchi, T. Nakamura, T. Hamamoto, H. Sasaki and T. Kitahara, Mol. Pharmaceutics, 2011, 8, 913–919 CrossRef CAS.
- J. Lojk, K. Strojan, K. Mis, B. V. Bregar, I. Hafner Bratkovic, M. Bizjak, S. Pirkmajer and M. Pavlin, Toxicol. Lett., 2017, 270, 108–118 CrossRef CAS PubMed.
- I. A. Pyshnaya, K. V. Razum, J. E. Poletaeva, D. V. Pyshnyi, M. A. Zenkova and E. I. Ryabchikova, BioMed Res. Int., 2014, 2014, 908175 Search PubMed.
- G. Jiang, K. Park, J. Kim, K. S. Kim and S. K. Hahn, Mol. Pharmaceutics, 2009, 6, 727–737 CrossRef CAS PubMed.
- Y. Z. Chen, X. L. Yao, G. X. Ruan, Q. Q. Zhao, G. P. Tang, Y. Tabata and J. Q. Gao, Biotechnol. Appl. Biochem., 2012, 59, 346–352 CrossRef CAS.
- G. Cheng, Y. He, L. Xie, Y. Nie, B. He, Z. Zhang and Z. Gu, Int. J. Nanomed., 2012, 7, 3991–4006 CAS.
- A. Alshamsan, A. Haddadi, S. Hamdy, J. Samuel, A. O. El-Kadi, H. Uludag and A. Lavasanifar, Mol. Pharmaceutics, 2010, 7, 1643–1654 CrossRef CAS PubMed.
- T. Kurosaki, Y. Kodama, T. Muro, N. Higuchi, T. Nakamura, T. Kitahara, M. Miyakoda, K. Yui and H. Sasaki, Biol. Pharm. Bull., 2013, 36, 1800–1806 CrossRef CAS.
- X. Zhang, A. Cai, Y. Gao, Y. Zhang, X. Duan and K. Men, Mol. Pharmaceutics, 2021, 18, 3387–3400 CrossRef CAS.
- X. Zhou, X. Li, M. Gou, J. Qiu, J. Li, C. Yu, Y. Zhang, N. Zhang, X. Teng, Z. Chen, C. Luo, Z. Wang, X. Liu, G. Shen, L. Yang, Z. Qian, Y. Wei and J. Li, Cancer Sci., 2011, 102, 1403–1409 CrossRef CAS.
- K. Liu, X. Wang, W. Fan, Q. Zhu, J. Yang, J. Gao and S. Gao, Int. J. Nanomed., 2012, 7, 1149–1162 CAS.
- K. S. Park, J. Nam, S. Son and J. J. Moon, Biomaterials, 2021, 274, 120844 CrossRef CAS PubMed.
- Y. Kodama, R. Noda, K. Sato, H. Harasawa, T. Kurosaki, H. Nakagawa, T. Nakamura, T. Kitahara, T. Muro and H. Sasaki, Biol. Pharm. Bull., 2018, 41, 1537–1542 CrossRef CAS.
- C. Xu, P. Wang, J. Zhang, H. Tian, K. Park and X. Chen, Small, 2015, 11, 4321–4333 CrossRef CAS.
- J. Wang, H. He, X. Xu, X. Wang, Y. Chen and L. Yin, Biomaterials, 2018, 171, 72–82 CrossRef CAS.
- X. Huang, N. Mu, Y. Ding, H. W. Lam, L. Yue, C. Gao, T. Chen, Z. Yuan and R. Wang, Acta Biomater., 2022, 147, 356–365 CrossRef CAS PubMed.
- M. Bamburowicz-Klimkowska, M. Malecki, M. Bystrzejewski, A. Kasprzak and I. P. Grudzinski, Biochem. Biophys. Res. Commun., 2023, 652, 84–87 CrossRef CAS PubMed.
- Q. Lei, D. He, L. Ding, F. Kong, P. He, J. Huang, J. Guo, C. J. Brinker, G. Luo, W. Zhu and Y. Yu, Adv. Funct. Mater., 2022, 32, 2113269 CrossRef CAS.
- J. Li, X. Yu, X. Shi and M. Shen, Prog. Mater. Sci., 2022, 124, 100871 CrossRef CAS.
Footnote |
| † These authors have contributed equally to the manuscript. |
| This journal is © The Royal Society of Chemistry 2025 |