
Stabilizing molecular catalysts on metal oxide surfaces using molecular layer deposition for efficient water oxidation†
Hong
Wang‡
a,
Jian
Li‡
a,
Ke
Liu‡
ad,
Lei
Lei
*ab,
Xun
Chen
*cd and
Degao
Wang
 *abd
*abd
aZhejiang Key Laboratory of Data-Driven High-Safety Energy Materials and Applications, Ningbo Key Laboratory of Special Energy Materials and Chemistry, Laboratory of Advanced Nuclear Materials, Ningbo Institute of Materials Technology and Engineering (NIMTE) of the Chinese Academy of Sciences (CAS), Ningbo, Zhejiang 315201, China. E-mail: leilei0219@nimte.ac.cn
bAdvanced Interdisciplinary Sciences Research (AIR) Center, Ningbo Institute of Materials Technology and Engineering (NIMTE) of the Chinese Academy of Sciences (CAS), Ningbo, Zhejiang 315201, China. E-mail: wangdegao@nimte.ac.cn
cLaboratory of Polymers and Composites, Ningbo Institute of Materials Technology and Engineering (NIMTE) of the Chinese Academy of Sciences (CAS), Ningbo, Zhejiang 315201, China. E-mail: chenxun@nimte.ac.cn
dUniversity of Chinese Academy of Sciences, Beijing, 100049, China
First published on 20th November 2024
Abstract
The stabilization of metal–oxide-bound molecular catalysts is essential for enhancing their lifetime and commercial viability in heterogeneous catalysis. This is particularly relevant in dye-sensitized photoelectrochemical cells (DSPECs), where the surface-bound chromophores and catalysts exhibit instability in aqueous environments, particularly at elevated pH levels. In this work, we have successfully employed molecular layer deposition (MLD) to stabilize ruthenium-based catalysts (RuCP(OH2)2+, denoted as RuCat). The application of polyimide (PI) via MLD onto the porous nanoITO surface significantly improved the stabilization of RuCat molecules for water oxidation. Additionally, time-resolved photoluminescence (TRPL) spectroscopy and femtosecond transient absorption spectroscopy (fs-TAS) results indicated that the MLD-deposited PI effectively preserved the robust redox capacity of the photogenerated electron–hole pairs associated with the catalyst molecules, thereby facilitating more efficient charge transfer. This research presents a novel approach for stabilizing surface-bound small molecules, which may contribute to advancements in heterogeneous catalysis and enhance its commercial viability.
New conceptsWe propose that the surface binding stability of a water oxidation catalyst (RuCP(OH2)2+) over a wide pH range (2–12) can be enhanced by molecular layer deposition of an overlayer of polyimide. (1) The molecular layer deposition of polyimide increased the stability of surface binding, and the reactivity of the bound catalyst. This marks the first instance of using molecular layer deposition of polyimide to stabilize RuCat. (2) This work provides a hybrid approach to heterogeneous catalysis. It combines the advantages of systematic modifications achievable through chemical synthesis with heterogeneous reactivity. |
1. Introduction
Water splitting is a highly promising approach for converting solar energy into chemical bonds.1 In this process, water oxidation is a challenging reaction that requires four electron-hole transfer.2,3 In particular, in pH-neutral media, the slow oxygen evolution reaction (OER) limits the overall efficiency of producing renewable fuels and feedstocks through electricity or photoelectrochemical catalysis. Molecular catalysts provide valuable insights into the water oxidation mechanism. Yet, the stabilization is crucial for their lifetime and commercial feasibility in water oxidation, as surface-bound catalysts tend to be unstable in aqueous environments, particularly at elevated pH levels. Numerous methods have been developed to immobilize coordination complexes on oxide surfaces, including chemical vapor deposition (CVD), atomic layer deposition (ALD), strong metal–support interactions (SMSI), the Stöber method, and non-hydrolytic sol–gel (NHSG) methods. In some instances, a trade-off exists between activity and stability, particularly with CVD.4 SMSI has primarily been employed to create bifunctionality at the interfaces of metal–metal oxide systems, establishing new pathways to enhance catalytic activity; however, this phenomenon is predominantly observed with group 8B metals.5,6 Limitations persist in these processes, especially concerning core–shell structures, Stöber synthesis, and NHSG methods.7,8 The challenges of controlling shell thickness and porosity in these architectures remain significant concerns. The formation of thin layers of inorganic metal oxides via atomical layer deposition (ALD) has been reported to be an effective and reliable approach for stabilizing catalyst molecules on oxides.9 However, the ultra-thin metal oxide layer is only weakly attached to the catalyst. This can add an extra energy barrier to restrict the diffusion and migration of catalyst molecules on the biphasic support surface.10 Furthermore, the necessity for low-temperature processing and the requirement for high vapor pressure precursors at low temperatures have limited the variety of materials applied in ALD to date. Additionally, an ALD-introduced thin layer may change the conduction band potential of the substrate. This leads to a notable reduction in injection yields and an increase in emission, which is detrimental to the photoelectrochemical performance.11,12 The partial deposition of oxide beneath the molecular layer during the ALD process may result in a greater separation distance between the substrate and molecular layer, affecting interfacial charge transfer and the catalysis efficiency.The capability of multistep molecular layer deposition (MLD) to utilize molecular structure and chemistry offers a viable solution to the above issues.13,14 As the sister technique of ALD, MLD was first coined in 1991 by Yoshimura and colleagues.15 MLD is employed to deposit both organic and hybrid (inorganic–organic) films, in contrast to ALD, which is primarily used for the growth of inorganic films. MLD films offer several advantages over their ALD counterparts: (1) due to their high flexibility, MLD films can effectively mitigate the occurrence of defects when subjected to strain, unlike the relatively brittle ALD metal oxide films that are prone to defect formation under similar conditions.16,17 (2) MLD films exhibit a significantly lower density compared to ALD metal oxides. (3) MLD thin films allow for the fine-tuning of the mechanical properties, including density, refractive index, elastic modulus, and hardness, by adjusting the ratios of organic components.18,19 (4) The removal of organic components from MLD films through thermal or chemical processes can yield highly porous metal oxides.20,21 Additionally, MLD is not only low-cost but provides excellent uniform coverage on large planar substrates and achieves highly conformal coatings on complex structures with high aspect ratios.22 In surface engineering, MLD facilitates the adjustment of surface wettability and corrosion resistance.23 MLD is becoming increasingly attractive in energy-related applications.24–28 For example, Zhang et al. prepared a porous structure by annealing the alucone MLD layer. By depositing Pt nanoparticles on this structure, they effectively anchored the Pt catalysts to the surface, resulting in good ORR performance.29 By leveraging the MLD technique, it is possible to produce high-quality thin films of various commercially viable organic polymers as well as inorganic–organic hybrid materials, thereby potentially integrating the advantageous properties of these two distinct chemistries. Yet, currently, there are no studies available that focus on employing MLD to form organic polymer layers to enhance the stability of surface-bound catalysts.
Herein, we took advantage of MLD to prepare polyimide (PI) thin films for systematically stabilizing molecular catalyst RuCP(OH2)2+ (denoted as RuCat) on metal oxide surfaces (denoted as FTO|nanoITO-RuCat|PI). Our approach began with the attachment of a phosphonate-derivatized molecular water oxidation catalyst to the nanoITO particle film-modified electrode. The derivatized electrode was then coated with a conformal nanoscale PI overlayer using MLD. We confirmed the successful synthesis of the MLD-deposited PI. And the electrochemical tests indicated a significant enhancement in catalyst stability for water oxidation, comparable to that achieved with the widely used ALD. Additionally, we also analyzed the kinetic data of the excited states of the samples through time-resolved photoluminescence (TRPL) and transient femtosecond absorption spectroscopy (fs-TAS). The results consistently indicated that the presence of MLD-deposited PI improved charge separation and transfer efficiency in the samples. MLD is considered a vital method for stabilizing heterogeneous catalysis and modifying surfaces in energy production applications.
2. Results and discussion
2.1. Catalysts fabrication and characterizations
The molecular structure of RuCat is shown in Fig. S1 (ESI†), and the detailed synthesis procedures for RuCat, FTO|nanoITO, FTO|nanoITO-RuCat, and FTO|nanoITO-RuCat|PI are available in the Experimental section.30,31Fig. 1a illustrates the schematic representation of PI-stabilized catalyst molecules on an oxide surface (here nanoITO) via the MLD technique. In this study, two cycles of MLD were employed to prepare FTO|nanoITO-RuCat|PI for characterization and performance analysis. The FTO|nanoITO structure features a large porous surface that can adsorb a high percentage of catalyst, facilitating the subsequent electrochemical measurements.32 Additionally, these pores allow precursor molecules to diffuse into the interior of nanoITO, forming conformal PI around the catalyst molecules for efficient stabilization. The deposited PI on the FTO|nanoITO-RuCat surface was initially characterized by Fourier transform infrared (FTIR) spectroscopy (Fig. 1b). The peak at ∼1772 cm−1 was ascribed to the symmetric C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond of the imide, while the peaks at ∼1712 and 1380 cm−1 were associated with the stretching of the asymmetric CC bond and the C–N bond of the imide, respectively.33 Additionally, the peak at ∼728 cm−1 was assigned to the deformation of the imide ring or the carbonyl group.34 These findings indicated that the PI was successfully deposited on the oxide surface.
O bond of the imide, while the peaks at ∼1712 and 1380 cm−1 were associated with the stretching of the asymmetric CC bond and the C–N bond of the imide, respectively.33 Additionally, the peak at ∼728 cm−1 was assigned to the deformation of the imide ring or the carbonyl group.34 These findings indicated that the PI was successfully deposited on the oxide surface.
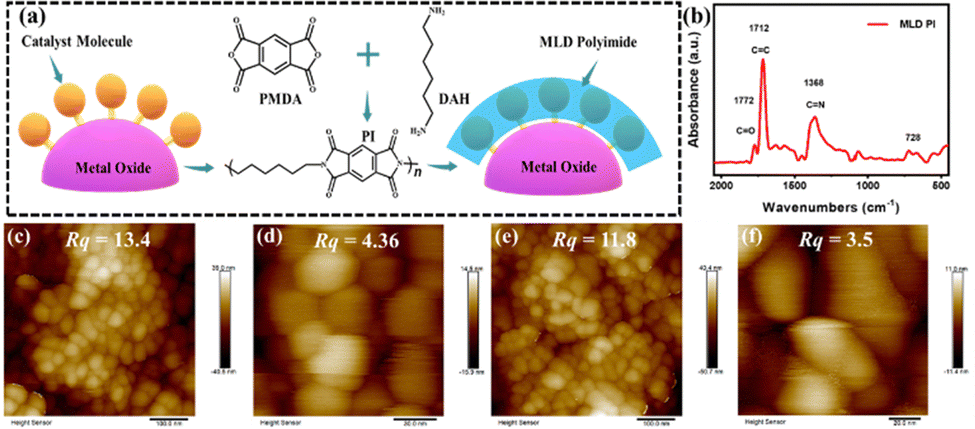 | ||
| Fig. 1 (a) Schematic representation of PI-stabilized catalyst molecules on the electrode via MLD; (b) FTIR absorption spectra of FTO|nanoITO-RuCat|PI; AFM images of (c) and (d) FTO|nanoITO-RuCat, and (e) and (f) FTO|nanoITO-RuCat|PI; Rq denotes roughness. | ||
To study the surface morphology of FTO|nanoITO-RuCat|PI, atomic force microscopy (AFM) was performed. As shown in Fig. 1c and d, the surface roughness of FTO|nanoITO-RuCat was approximately 13.4 nm over a larger area and 4.36 nm over a smaller area; while the surface roughness of FTO|nanoITO-RuCat|PI was around 11.8 nm in a larger area and 3.5 nm in a small area (Fig. 1e and f). The reduction in surface roughness was attributed to the conformal coating of MLD-deposited PI on the ITO nanoparticles. Amorphous PI film planarized the pristine surface, thus further proving the formation of a PI film.35,36 From scanning electron microscopy (SEM) images of FTO|nanoITO (Fig. 2a and b), the porous and rough surface of the nanoITO layer with 5.6 μm thickness was clearly observed. The high-resolution transmission electron microscope (HRTEM) images of FTO|nanoITO-RuCat|PI revealed that the average particle size of the nanoITO was 20 nm, and the lattice stripe space was determined to be 0.22 nm, which is well consistent with the nanoITO's d-value.37 In Fig. 2c and d, an amorphous PI layer was clearly different from the nanoITO lattice observed. The PI layer was evenly coated on the ITO surface with 1–2 nm of thickness through 2 cycles of MLD.
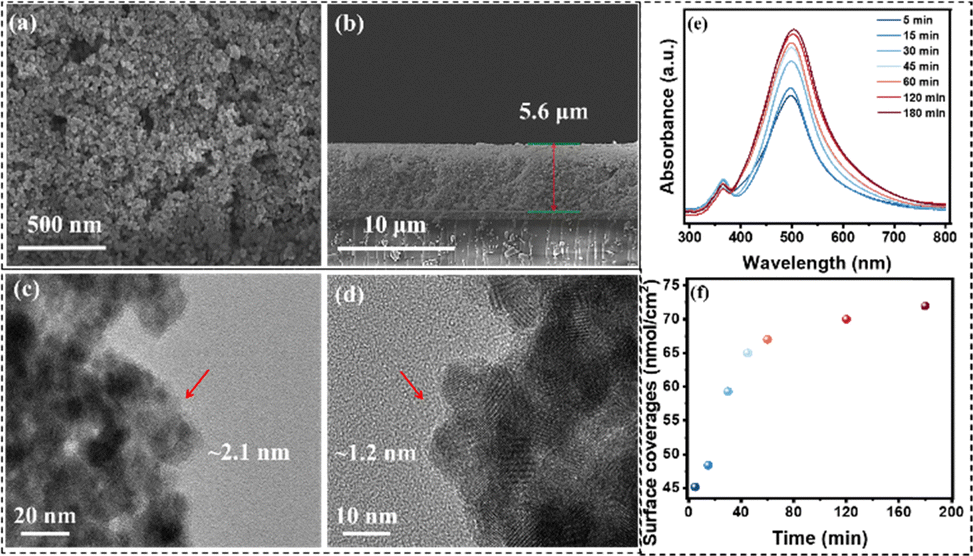 | ||
| Fig. 2 SEM images of FTO|nanoITO from (a) (top view) and (b) cross-sectional view; (c) and (d) HRTEM images of nanoITO-RuCat|PI. (e) UV-vis absorption spectra of FTO|nanoITO electrodes after various soaking times in 150 μM RuCat in methanol; (f) RuCat surface coverages after exposure for different times. | ||
Subsequently, the adsorption capacity of RuCat was investigated by simply adjusting the soaking time of the FTO|nanoITO electrode in the catalyst solution. The surface loading extent (Γ in mol cm−2) of RuCat on FTO|nanoITO was calculated from UV-vis measurements using eqn (1), where A(λ) and ε(λ) are the absorbance and molar absorptivity at wavelength λ. For surface-bound RuCat, λmax = 497 nm, and εmax = 1.5 × 104 M−1 cm−1.38Fig. 2e displays the UV-vis absorption spectra of the FTO|nanoITO electrodes after being soaked for different times in 150 μM RuCat in methanol. The calculations of surface coverage indicated that the absorption intensity increases with extended immersion time (Fig. 2f).
| Γ = A(λ)/[103ε(λ)] | (1) |
2.2. Electrode stability assessment
Repetitive cyclic voltammetry (CV) scans through the RuIII/IICat redox couple were used to evaluate the surface stabilization effect by different cycles of MLD-deposited PI (Fig. S2–S4, ESI†). We quantitatively analyzed the changes in redox peak current for FTO|nanoITO-RuCat and FTO|nanoITO-RuCat|PI over 50 cyclic voltammetry (CV) cycles. As illustrated in Fig. S5 (ESI†), the rate of current decline slows after the deposition of the polymeric interface, indicating the stabilizing effect of PI on the catalyst. In the range of 0 to 0.8 V vs. Ag/AgCl, one set of reversible redox peaks appeared near E1/2 = 0.5 V, corresponding well to the RuIII/IICat redox processes in the Ru metal centers of the catalyst.39 The findings clearly demonstrated that the peak current decreased with increasing CV scans, suggesting the desorption of RuCat from the surface.40,41 However, the presence of PI significantly slowed down the desorption rates of RuCat. To quantitatively analyze the experimental results, surface coverage values (Γ in mol cm−2) were further calculated by integrating the current–potential waveform for the RuIII/IICat redox couple, using eqn (2), where QCV represents the integrated charge from the current–potential waveform, n (=1) is the number of electrons transferred for the redox couple, F is the Faraday constant, and A represents the surface area of the electrode.41| Γ = QCV/nFA | (2) |
Fig. S6 (ESI†) illustrates the relationship between Γ and the number of CV scans for electrodes with different cycles of PI on FTO|nanoITO-RuCat electrodes. In the absence of a PI overlayer, more than 60% of the catalyst is lost from the oxide surface after 50 continuous CV cycles. The electrodes with different catalyst adsorption capacities exhibited a similar trend in surface stability. However, after introducing a PI layer on the oxide surface, the desorption rate of the catalyst significantly decreased. Compared to the pristine FTO|nanoITO-RuCat electrodes, the FTO|nanoITO-RuCat|PI configuration only lost 20% of the surface-bound catalyst after 50 CV cycles. This enhanced stability is attributed to the binding groups that are protected by the added PI. During the MLD process, the molecular precursor diffused into the pores of the ITO nanoparticles, where it adsorbed, reacted, and formed PI molecules within the voids of RuCat, making the phosphate binding group less susceptible to hydrolysis. However, the stability of the FTO|nanoITO-RuCat|PI decreased with further increasing cycles of PI. The catalyst molecules may be completely “buried”, inhibiting electron transfer across the surface and negatively affecting the catalytic reaction.
The stability of the FTO|nanoITO-RuCat|PI electrode under a wide pH range was investigated by electrochemical measurements.42,43 Under acidic conditions (pH = 2), as shown in Fig. 3a, the FTO|nanoITO-RuCat|PI indicated a significant increase in current peaks after 50 CV cycles compared to the FTO|nanoITO-RuCat. Similar boosted stability of the FTO|nanoITO-RuCat|PI is more pronounced during neutral (pH = 7, Fig. 3b) and alkaline (pH = 12, Fig. 3c) electrolytes. The surface molecular loading amount Γ (mol cm−2) after 50 CV cycles at the three different pH levels was calculated. Fig. 3d shows that 79% of the catalysts were preserved with the PI protective layer, while only 51% of the catalyst remained without PI protection at pH = 2. Similar trends were observed in neutral (pH = 7, Fig. 3e) and alkaline (pH = 12, Fig. 3f) electrolytes. Notably, the stability of both electrodes significantly decreased in alkaline media, mainly due to ligand decomposition, consistent with known reactivity in solution,44 and not to surface detachment due to hydrolysis. Additionally, the stabilizing effect of the MLD-PI is comparable to that of ALD-TiO2 protection (Fig. S7, ESI†), further confirming the protective role of the PI overlayer.
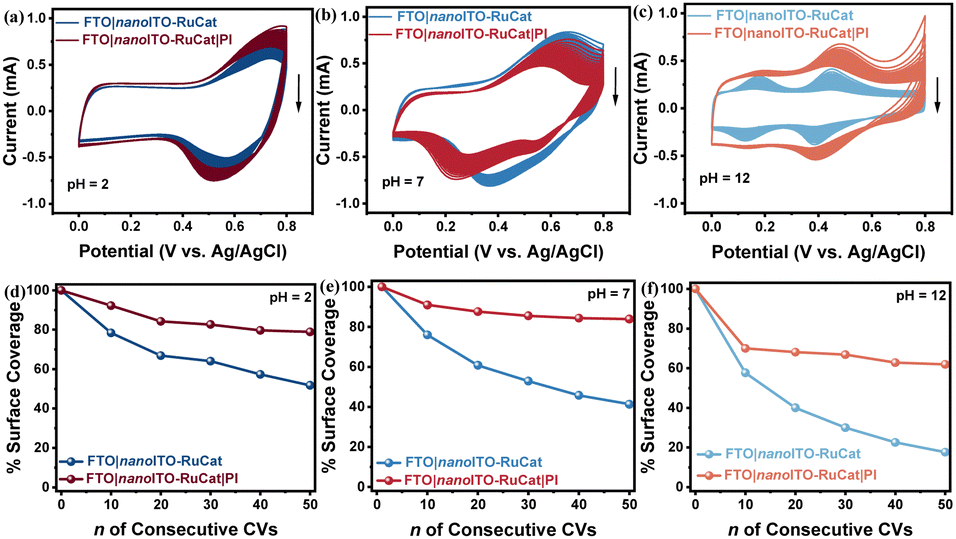 | ||
| Fig. 3 (a)–(c) 50 consecutive CV curve comparisons of FTO|nanoITO-RuCat and FTO|nanoITO-RuCat|PI electrodes at different pH; (d)–(f) variation of Γ/Γo with the number of CV cycles for FTO|nanoITO-RuCat and FTO|nanoITO-RuCat|PI electrodes. | ||
2.3. Catalytic performance and mechanism analysis
To assess the electrode's activity in catalyzing the water oxidation reaction, a quantitative analysis of the oxygen generated is necessary. Gas chromatography was used to analyze the headspace in the electrolysis cell with a controlled potential of ∼1.4 V (vs. Ag/AgCl). As illustrated in Fig. 4a, both electrodes produced oxygen, and Fig. 4b compares the oxygen yields of electrodes with and without the PI layer during water oxidation. The results indicated that the electrode with the PI protective layer achieved a relatively higher oxygen yield, reaching 1281 μmol over 3 hours. This aligns with previous electrochemical performance tests. The MLD-deposited PI layer reduced the desorption of catalyst molecules from the nanoITO surface, thereby enhancing the catalytic efficiency of water oxidation.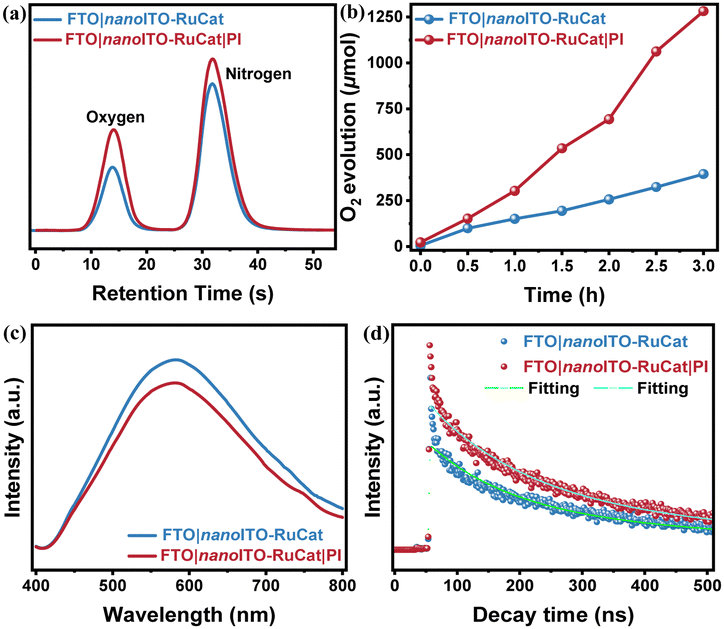 | ||
| Fig. 4 (a) Gas chromatogram following electrolysis of FTO|nanoITO-RuCat and FTO|nanoITO-RuCat|PI; (b) comparison of oxygen production during three hours of electrode catalysis for FTO|nanoITO-RuCat and FTO|nanoITO-RuCat|PI. Eapp = 1.40 V vs. Ag/AgCl, pH = 7. (c) Steady-state fluorescence spectra and (d) transient fluorescence spectra of FTO|nanoITO-RuCat and FTO|nanoITO-RuCat|PI electrodes excited using a 375 nm laser. | ||
Fluorescence spectroscopy was employed to investigate the efficiency of separation for photogenerated carriers and interfacial transfer. As shown in Fig. 4c, the steady-state fluorescence spectra revealed that at an emission wavelength of 580 nm, the FTO|nanoITO-RuCat exhibited a slightly stronger fluorescence response compared to FTO|nanoITO-RuCat|PI. This indicated that the photogenerated electrons and holes were effectively suppressed after PI-coating.45,46 In the transient fluorescence spectra presented in Fig. 4d, the average lifetime of RuCat for FTO|nanoITO-RuCat was 420 ns, whereas for FTO|nanoITO-RuCat|PI, it was longer at 497 ns. The longer lifetimes suggested that the PI layer significantly decreased trap-induced recombination and promoted charge transfer, thereby increasing the chances of holes participating in the water oxidation reaction.47 From Fig. S8(a) (ESI†), the FTO|nanoITO-RuCat|TiO2 composite enhances photocatalytic performance by inhibiting the recombination of photogenerated charge carriers, as evidenced by the decrease in peak intensity. Fig. S8(b) (ESI†) demonstrates that the FTO|nanoITO-RuCat|TiO2 sample has a lifetime of 453 ns, which is significantly longer than that of the FTO|nanoITO-RuCat sample (420 ns). This suggests that the addition of TiO2 considerably slows down trap-mediated recombination. Furthermore, the FTO|nanoITO-RuCat|PI sample exhibited a lifetime of 497 ns across the entire visible wavelength range (400 nm ≤ λ ≤ 800 nm), indicating an even longer lifetime. Therefore, we conclude that the PI provided hole-storing trap states, which extended the lifetime of the trapped holes.
To further validate the role of the introduced PI layer by MLD on photogenerated carrier separation and transport, fs-TAS was performed on FTO|nanoITO-RuCat and FTO|nanoITO-RuCat|PI electrodes. As shown in Fig. 5a and b, after excitation at 430 nm, the FTO|nanoITO-RuCat|PI sample showed an upward trend from 600 nm to 700 nm compared to the FTO|nanoITO-RuCat. This increase is primarily attributed to improved transport of photogenerated electrons, which either eliminated the deeply captured electrons or enhanced the shallowly captured electrons.48,49 We fitted the dynamic decay curves of the photogenerated carriers, as depicted in Fig. 5c and d. The FTO|nanoITO-RuCat sample exhibited a carrier lifetime of 958 ps, while the FTO|nanoITO-RuCat|PI sample demonstrated an exceptionally long carrier lifetime of 2202 ps. Compared to pristine RuCat, the absorption of RuCat|TiO2 decays more slowly over a delay time of 1 to 5000 ps (see Fig. S9(a), ESI†). The absorption-time profiles of RuCat|TiO2 recorded at 515 nm are illustrated in Fig. S9(b) (ESI†). The weighted average lifetime (τave) of the electron–hole pairs is 1590 ps. In contrast, the RuCat|PI electrode exhibits superior electron transport kinetics. In other words, it can be said that the PI acts as a ‘shelter’ for the photoexcited holes to prevent recombination. This implied that the photogenerated electrons and holes can be efficiently separated and transferred to the catalyst surface. The prolonged lifetime of the photogenerated charges facilitates catalytic oxidation performance. These findings aligned well with the results from steady state fluorescence spectroscopy, transient fluorescence spectroscopy, and electrochemical tests mentioned above.
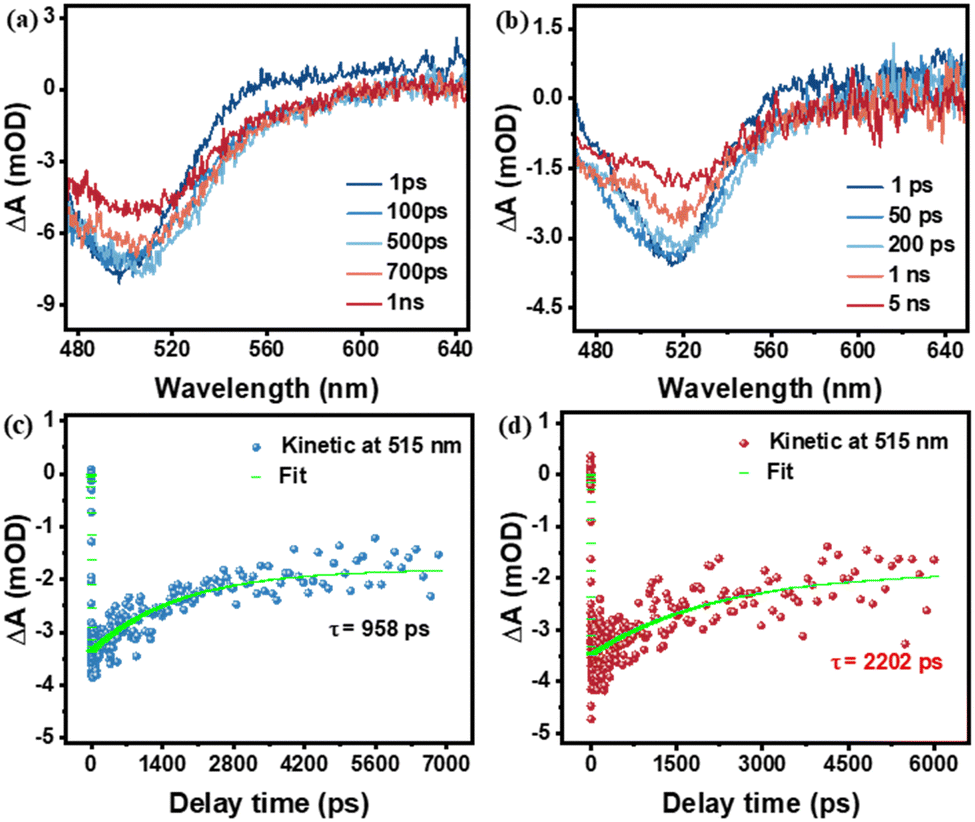 | ||
| Fig. 5 Ultrafast absorption spectra of (a) FTO|nanoITO-RuCat and (b) FTO|nanoITO-RuCat|PI; normalized transient time absorption spectra of (c) FTO|nanoITO-RuCat and (d) FTO|nanoITO-RuCat|PI. | ||
3. Conclusions
In summary, we employed the MLD method to deposit a PI layer on FTO|nanoITO-RuCat, showcasing the excellent stability of MLD-deposited PI on surface-attached molecular catalysts and enhanced catalytic performance for water oxidation. This work provides a novel approach for stabilizing molecular catalysts, revealing significant insights: (1) the surface properties and reactivity were maintained after adding an MLD-deposited PI layer; (2) at elevated pH levels, there is a strong stability in surface binding and water oxidation reactivity; and (3) it lays a foundational approach for creating custom surfaces by integrating molecular and heterogeneous catalytic properties. Our results have explicit implications for employing the MLD-deposited PI protective layer strategy to stabilize molecular catalysts and components on oxide electrodes and semiconductor surfaces, which could be beneficial in the field of photoelectrochemistry.4. Experimental
4.1. Materials
Pyromellitic dianhydride (PMDA), 1,6-diaminohexane (DAH), and hydroxypropyl cellulose (HPC) were purchased from Sigma-Aldrich. Sodium dihydrogen phosphate (NaH2PO4), disodium hydrogen phosphate (Na2HPO4), and sodium phosphate (Na3PO4) were purchased from Macklin. Anhydrous ethanol and methyl alcohol were purchased from Sinopharm Chemical Reagent Co. Indium tin oxide (ITO) nanoparticle dispersion (20%) was purchased from Nangong Longao Trading Co. Fluorine-doped tin oxide (FTO) coated glass (sheet resistance 15 Ω cm−2) was purchased from Wuhan Jingge Solar Technology Co.4.2. Synthesis of RuCP(OH2)2+
In a 100 mL Teflon microwave vessel, [Ru(2,6-bis(1-methyl-1H-benzo[d]imidazol-2-yl) pyridine) (Cl)]2[Cl]2 (41.2 mg, 4.03 × 10−5 mol) and 4,4-(H2O3P–CH2)2-2,2-bipyridine (27.8 mg, 8.08 × 10−5 mol) were suspended in ethanol (20 mL) and water (10 mL). The vessel was briefly subjected to sonication (∼1 minute). The vessel was placed in a microwave reactor where, following a 5-minute ramping period, it was heated at 160 °C for 30 minutes. The pressure of the vessel did not exceed 300 psi. The vessel was allowed to cool to room temperature. The solution was filtered. From the filtrate, the solvent was removed on a rotary evaporator. The residue was dried under vacuum overnight. To the residue, anhydrous methylene chloride (50 mL) was added. The suspension was de-aerated with argon for 15 minutes. With a vent needle in place and under continuous flow of argon (Caution: HCl gas is evolved), trifluoromethanesulfonic acid (1.8 mL) was added slowly. The reaction immediately releases HCl (gas). The reaction was stirred at room temperature overnight (note: the flow of argon was high enough to vent HCl but low enough as to not evaporate methylene chloride over the time of the experiment). Following the reaction, diethyl ether was added to precipitate the solid, which was collected on a glass frit and washed with diethyl ether (62.4 mg, 5.67 × 10−5 mol, 70%).4.3. Preparation of FTO|nanoITO
Take 5 mL of 20% nanoITO dispersion, and 5 mL of anhydrous ethanol, then add 500 mg of HPC, and magnetically stir for 2 hours. The FTO glass was ultrasonically cleaned in ethanol and acetone for 30 min, after which the dispersion was coated with an area of 1 × 1 cm on a clean FTO glass, and then annealed for 2 hours at 500 °C in air.4.4. Preparation of FTO|nanoITO-RuCat
A packed RuCat monolayer was formed on FTO|nanoITO by soaking the slides in 150 μM RuCP(OH2)2+ in methanol solution overnight.4.5. Preparation of FTO|nanoITO-RuCat|PI
PI thin films were deposited in a hot-wall, crossflow F120 ALD reactor (ASM Microchemistry) from GEM Star with an operating pressure of around 10 mbar. Nitrogen (N2, AGA, 99.999%) was used as the carrier and purge gas. Pyromellitic dianhydride (PMDA) and 1,6-diaminohexane (DAH) were evaporated inside the reactor at 190 and 75 °C, respectively. The temperature of the chamber is set to 170 °C. MLD cycles consisted of a 5 s PMDA dose, 40 s hold, 60 s purge, 5 s DAH dose, 40 s hold, and 60 s purge. Holds were accomplished by closing gate valves on both sides of the reaction zone. In this study, two cycles of MLD were employed to prepare FTO|nanoITO-RuCat|PI for characterization and performance analysis.4.6. Film characterization
Bonding information about the as-deposited PI films was obtained from attenuated total reflection Fourier transform infrared (FTIR) spectra (INVENIO). Determination of catalyst adsorption was done by UV-visible spectroscopy (UH-4150). The SEM images were taken by scanning electron microscopy (SEM, S-8230) to characterize surface topography. The TEM images and high-resolution TEM (HRTEM) were taken on a Talos F200X to characterize the PI layer morphology. Surface roughness was characterized with AFM (NT-MDT Spectrum Instruments). Time-resolved photoluminescence (TRPL) spectroscopy (FLS1000) and femtosecond transient absorption spectroscopy (fs-TAS, Helios) were employed to characterize the charge transfer transport kinetics.4.7. Electrochemical measurements
Electrochemical measurements were performed with a model 601D electrochemical workstation from CH Instruments by a three-electrode system. The fabricated sample acted as the working electrode (roughly 1 cm2 area), a Pt wire acted as the counter electrode, and Ag/AgCl was used as the reference electrode. Oxygen yield was monitored using gas chromatography (8890).Author contributions
Degao Wang proposed the project and conceived the idea. Degao Wang supervised the project. Degao Wang and Hong Wang designed the experiments. Degao Wang, Hong Wang, Jian Li and Lei Lei carried out the experiments and analyzed the data. Degao Wang, Hong Wang and Lei Lei organized and wrote the manuscript.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Ningbo Top-talent Team Program (2018A610005), Program for the National Natural Science Foundation of China (22106166), the Yong jiang Innovative Individual Introduction, China, the China Postdoctoral Science Foundation (2022M723253, 2023M733597), and Zhejiang Provincial Natural Science Foundation of China (LQ24B030006).Notes and references
- Y. Du, B. Li, G. Xu and L. Wang, InfoMat, 2022, 5, e12377 CrossRef.
- M. Yang, C. H. Zhang, N. W. Li, D. Luan, L. Yu and X. W. Lou, Adv. Sci., 2022, 9, 2105135 CrossRef CAS.
- J. Plutnar and M. Pumera, Small, 2021, 17, 212088 CrossRef.
- T. N. Phaahlamohlaka, D. O. Kumi, M. W. Dlamini, L. L. Jewell and N. J. Coville, Catal. Today, 2016, 275, 76–83 CrossRef CAS.
- H. Tang, Y. Su, B. Zhang, A. F. Lee, M. A. Isaacs, K. Wilson, L. Li, Y. Ren, J. Huang, M. Haruta, B. Qiao, X. Liu, C. Jin, D. Su, J. Wang and T. Zhang, Sci. Adv., 2017, 3, e1700231 CrossRef PubMed.
- H. Tang, J. Wei, F. Liu, B. Qiao, X. Pan, L. Li, J. Liu, J. Wang and T. Zhang, J. Am. Chem. Soc., 2016, 138, 56–59 CrossRef CAS PubMed.
- H. Wang, J.-f Zhang, Y.-x Bai, W.-f Wang, Y.-s Tan and Y.-z Han, J. Fuel Chem. Technol., 2016, 44, 548–556 CrossRef CAS.
- A. H. Habibi, R. E. Hayes and N. Semagina, Catal. Sci. Technol., 2018, 8, 798–805 RSC.
- K. A. Adegoke and N. W. Maxakato, Coord. Chem. Rev., 2022, 457, 214389 CAS.
- Q. Fu and X. Bao, Chem. Soc. Rev., 2017, 46, 1842–1874 CAS.
- M. Zeng, X. G. Peng, J. J. Liao, G. Z. Wang, Y. F. Li, J. B. Li, Y. Qin, J. S. Wilson, A. M. Song and S. W. Lin, Phys. Chem. Chem. Phys., 2016, 18, 17404–17413 RSC.
- M. J. Kim, J. S. Bae, M. J. Jung, E. Jeon, Y. Park, H. Khan and S. H. Kwon, ACS Appl. Mater. Interfaces, 2023, 15, 45732–45744 CrossRef CAS.
- M. Putkonen, J. Harjuoja, T. Sajavaara and L. Niinistö, J. Mater. Chem., 2007, 17, 664–669 RSC.
- D. C. Cameron and T. V. Ivanova, ECS Trans., 2013, 58, 263–275 CrossRef.
- M. G. Steven, Y. Byunnghoon and A. D. Arrelaine, Acc. Chem. Res., 2009, 42, 498–508 CrossRef.
- T. Yoshimura, S. Tatsuura, W. Sotoyama, A. Matsuura and T. Hayano, Appl. Phys. Lett., 1992, 60, 268–270 CrossRef CAS.
- M. Heikkinen, R. Ghiyasi and M. Karppinen, Adv. Mater. Interfaces, 2024, 2400262 CrossRef.
- B. Yoon, J. L. O'Patchen, D. Seghete, A. S. Cavanagh and S. M. George, Chem. Vap. Deposition, 2009, 15, 112–121 CrossRef CAS.
- D. Choudhury, S. K. Sarkar and N. Mahuli, J. Vac. Sci. Technol., A, 2015, 33, 4900934 CrossRef.
- B. H. Lee, V. R. Anderson and S. M. George, Chem. Vap. Deposition, 2013, 19, 204–212 CrossRef CAS.
- C. X. Zhao, J. N. Liu, B. Q. Li, D. Ren, X. Chen, J. Yu and Q. Zhang, Adv. Funct. Mater., 2020, 30, 2003619 CrossRef CAS.
- X. Meng, J. Mater. Chem. A, 2017, 5, 18326–18378 RSC.
- K. Gregorczyk and M. Knez, Prog. Mater. Sci., 2016, 75, 1–37 CrossRef CAS.
- H. Ablat, I.-K. Oh, N. E. Richey, S. T. Oyakhire, Y. Yang, W. Zhang, W. Huang, Y. Cui and S. F. Bent, ACS Appl. Energy Mater., 2023, 6, 5806–5816 CrossRef CAS.
- D. S. Bergsman, J. G. Baker, R. G. Closser, C. MacIsaac, M. Lillethorup, A. L. Strickler, L. Azarnouche, L. Godet and S. F. Bent, Adv. Funct. Mater., 2019, 29, 1904129 CrossRef CAS.
- S. B. Son, Y. Wang, J. Xu, X. Li, M. Groner, A. Stokes, Y. Yang, Y.-T. Cheng and C. Ban, ACS Appl. Mater. Interfaces, 2017, 9, 40143–40150 CrossRef CAS PubMed.
- J. Heiska, M. Nisula, E. L. Rautama, A. J. Karttunen and M. Karppinen, Dalton Trans., 2020, 49, 1591–1599 RSC.
- J. Lei, S. Sun, Y. Li, P. Xu, C. Liu, S. Chang, G. Yang, S. Chen, W. Fa, D. Wu and A. D. Li, J. Phys. Chem. Lett., 2023, 14, 1389–1394 CrossRef CAS.
- L. Zhang, Y. Zhao, M. N. Banis, K. Adair, Z. Song, L. Yang, M. Markiewicz, J. Li, S. Wang, R. Li, S. Ye and X. Sun, Nano Energy, 2019, 60, 111–118 CrossRef CAS.
- A. M. Lapides, B. D. Sherman, M. K. Brennaman, C. J. Dares, K. R. Skinner, J. L. Templeton and T. J. Meyer, Chem. Sci., 2015, 6, 6398–6406 RSC.
- W. Rabten, M. D. Kärkäs, T. Åkermark, H. Chen, R. Z. Liao, F. Tinnis, J. L. Sun, P. E. M. Siegbahn, P. G. Andersson and B. Åkermark, Inorg. Chem., 2015, 54, 4611–4620 CrossRef CAS PubMed.
- S. Chandrasekaran, N. Kaeffer, L. Cagnon, D. Aldakov, J. Fize, G. Nonglaton, F. Baleras, P. Mailley and V. Artero, Chem. Sci., 2019, 10, 4469–4475 RSC.
- S. Xiong, T. Sheng, L. Kong, Z. Zhong, J. Huang and Y. Wang, Chin. J. Chem. Eng., 2016, 24, 843–849 CrossRef CAS.
- H. Wang, M. Wei, Z. Zhong and Y. Wang, J. Membr. Sci., 2017, 535, 56–62 CrossRef CAS.
- X. Li, M. Vehkamäki, M. Chundak, K. Mizohata, A. Vihervaara, M. Putkonen, M. Leskelä and M. Ritala, Adv. Compos. Hybrid Mater., 2023, 6, 183 CrossRef CAS.
- T. J. Myers, J. A. Throckmorton, R. A. Borrelli, M. O'Sullivan, T. Hatwar and S. M. George, Appl. Surf. Sci., 2021, 569, 150878 CrossRef CAS.
- Z. Ma, Z. Tang, P. Zou, C. Xiao, J. Zhang, H. Wang and L. Jia, Talanta, 2025, 281, 126814 CrossRef CAS.
- Z. F. Chen, J. J. Concepcion, J. W. Jurss and T. J. Meyer, J. Am. Chem. Soc., 2009, 131, 15580–15581 CrossRef CAS.
- H. M. Shahadat, H. A. Younus, N. Ahmad, M. A. Rahaman, Z. A. K. Khattak, S. Zhuiykov and F. Verpoort, Catal. Sci. Technol., 2019, 9, 5651–5659 RSC.
- D. Wang, F. Niu, M. J. Mortelliti, M. V. Sheridan, B. D. Sherman, Y. Zhu, J. R. McBride, J. L. Dempsey, S. Shen, C. J. Dares, F. Li and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 12564–12571 CrossRef CAS PubMed.
- A. K. Vannucci, L. Alibabaei, M. D. Losego, J. J. Concepcion, B. Kalanyan, G. N. Parsons and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 20918–20922 CrossRef CAS.
- W. H. Lee, J. Yi, H. N. Nong, P. Strasser, K. H. Chae, B. K. Min, Y. J. Hwang and H. S. Oh, Nanoscale, 2020, 12, 14903–14910 RSC.
- Y. Liu, D. Yang, Z. Liu and J.-H. Yang, J. Power Sources, 2020, 461, 2281165 Search PubMed.
- K. G. Pushpito, S. B. Bruce, C. Mei, C. Carol and S. Norman, J. Am. Chem. Soc., 1984, 106, 4772–4783 CrossRef.
- A. E. A. Aboubakr, W. M. A. El Rouby, M. D. Khan, A. A. Farghali and N. Revaprasadu, Appl. Surf. Sci., 2020, 508, 145100 CrossRef CAS.
- H. Y. Kim, H. Kong, J. H. Kim, W. G. Yang, H. Lee, S. Ko, H. J. Lee, G. Piao, H. Park, W. S. Chae and J. Yeo, J. Mater. Chem. A, 2023, 11, 4598–4607 RSC.
- K. T. Munson, C. Grieco, E. R. Kennehan, R. J. Stewart and J. B. Asbury, ACS Energy Lett., 2017, 2, 651–658 CrossRef CAS.
- X. Wang, H. Jiang, M. Zhu and X. Shi, Chin. Chem. Lett., 2023, 34, 107683 CrossRef CAS.
- L. Wang, J. Zhang, H. Yu, I. H. Patir, Y. Li, S. Wageh, A. A. Al-Ghamdi and J. Yu, J. Phys. Chem. Lett., 2022, 13, 4695–4700 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4mh01274g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |