
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Harnessing mesoporous g-C3N4-based photocatalytic materials for sustainable fuel production via solar energy conversion: a review
Pratikshya Dash
a,
Ashish Kumar Kar
 b,
Rajendra Srivastava
*b and
Kulamani Parida
*a
b,
Rajendra Srivastava
*b and
Kulamani Parida
*a
aCentre for Nano Science and Nano Technology, Institute of Technical Education and Research, Siksha ‘O’ Anusandhan University, Bhubaneswar-751030, India. E-mail: kulamaniparida@soa.ac.in; paridakulamani@yahoo.com; Fax: +91 674 2350642; Tel: +91 674 2351777
bCatalysis Research Laboratory, Department of Chemistry, Indian Institute of Technology Ropar, Rupnagar-140001, India. E-mail: rajendra@iitrpr.ac.in; Tel: +91-1881-232064
First published on 8th May 2025
Abstract
The current energy production technology is associated with incompetent and unsustainable global conditions like climate change, the greenhouse effect, etc. Therefore, the call for sustainable and renewable energy practices is essential to address future energy crises, preserve ecological balance and combat climate change. Harnessing solar energy conversion via artificial photosynthesis over an efficient semiconductor is a key strategy to maintain the circular energy cycle and achieve zero-emission energy missions. g-C3N4 is one of the most exclusively studied semiconductor-photocatalysts; however, its bulk structure suffers from several significant limitations. Rational modifications of morphology and porosity led to the development of a mesoporous-C3N4 (mp-g-CN) framework, which has excellent photoresponsive features. mp-g-CN enriched with superior physicochemical properties, improved optoelectronic features, and well-dispersed active site distribution can be synthesized by either template-assisted or template-free synthesis methods. The template-free synthesis approach is more appealing than the template-assisted synthesis method, which can reduce the multiple synthesis steps and avoid the use of hazardous chemicals. Further, the strategic functional modifications deliver more efficient mp-g-CN structures, which can be considered as a reference photoactive material for producing H2, H2O2, NH3, carbonated fuel and biofuels from renewable precursors. Finally, some unresolved hitches in advancing mp-g-CN photocatalysts to achieve high efficiency in artificial photosynthesis have been encountered as current challenges and future prospects.
 Pratikshya Dash | Pratikshya Dash completed her MSc degree in Chemistry from Sambalpur University, Sambalpur, Odisha, India. She qualified GATE 2021. She is currently pursuing her PhD under the supervision of Prof. Kulamani Parida at the Centre for Nano Science and Nano Technology (CNSNT), Institute of Technical Education and Research (ITER), Siksha ‘O’ Anusandhan (Deemed to be University). Her current research interests are focused on the development of mesoporous graphitic nitride-based nanomaterials for solar energy conversion applications. |
 Ashish Kumar Kar | Dr Ashish Kumar Kar completed his Master of Science (MSc) in Organic Chemistry at Maharaja Sriram Chandra Bhanja Deo University, Odisha, India. He received his PhD from the Department of Chemistry at IIT Ropar in 2023 and then joined ESPCI, Paris, as a postdoctoral researcher. His current research areas are focused on the development of carbon-based materials, including MOFs, mesoporous carbon, metal/metal oxide-embedded N-doped carbon, etc., for sustainable catalytic and photocatalytic processes. |
 Rajendra Srivastava | Dr Rajendra Srivastava is a Professor at the Department of Chemistry, IIT Ropar. He acquired a PhD from CSIR-NCL Pune. He worked as a postdoctoral fellow at KAIST, South Korea, and as a JSPS fellow at Hokkaido University, Japan. He received the Best Thesis award, the NASI-SCOPUS Young Scientist, the Young Scientist award from the Catalysis Society of India, and the Research awards from IIT Ropar and Punjab University. He is the editorial board member of ChemCatChem. His research interest includes the synthesis of nanostructured functional materials for fuel and chemical production. He has published more than 200 peer-reviewed research articles in reputed international journals and holds nine patents. |
 Kulamani Parida | Prof. Kulamani Parida is a globally distinguished scientist in the areas of Materials Science, Catalysis and Nanotechnology. He superannuated as a Chief Scientist and a Professor of Academy of Scientific and Innovative Research (AcSIR) from CSIR-IMMT, Bhubaneswar, in 2014. His adoration for chemistry and passion for research couldn't let Prof. Parida to sit idle after retirement. With the same devotion and dedication, Prof. Parida (Director) started the ‘Centre for Nano Science and Nano Technology’ a research unit of Siksha ‘O’ Anusandhan (Deemed to be University), Bhubaneswar. His research interests include the design and development of advanced materials encompassing fine chemical, energy generation/storage, and environmental applications. He has published more than 550 research articles and review articles in renowned international journals. He bags a citation of 35040 to his credit with an h-index of 103 and an i10-index of 452. He has featured in the global 2% scientist list for the last five consecutive years. For more details: https://kmparidaimmt.weebly.com/. |
Wider impactIn light of the growing energy demand and potential future energy crises, transitioning from fossil fuels to sustainable energy sources is crucial. One promising approach is solar-to-energy conversion through artificial photosynthesis over a cost-effective and robust photocatalyst by utilizing naturally abundant precursors. g-C3N4 is a well-researched semiconductor photocatalyst recognized for its affordability, absence of metals, durability, and non-toxic characteristics. Further, the rational modifications of the morphology and porosity of g-C3N4 lead to the development of mesoporous g-C3N4-based materials. Overall, we offer a comprehensive evaluation of the emergence of mp-g-CN-based photocatalytic materials and the current landscape of their synthesis approaches, emphasizing structural and functional modifications to achieve effective solutions for semiconductor-based photocatalysis aimed at solar energy production. Furthermore, we also propose the associated research challenges and future prospects in this direction. The promising prospects of this advanced photocatalytic material emphasize the potential for interdisciplinary collaboration across science and engineering, which could foster innovative solutions and advancements in semiconductor photocatalysis, ultimately contributing to sustainable energy technologies. We believe this review intends to benefit the research communities, who are interested in semiconductor photocatalysis for sustainable energy production. We also believe this review could be a guiding tool for fabricating efficient g-C3N4-based photocatalysts. |
1. Introduction
The continuous increase in energy demand over time is due to the industrial revolution, population growth, and advancements in daily life such as electricity, heating, and transportation. Additionally, global energy consumption is projected to double by 2050 due to rapid population growth.1–3 Currently, fossil fuels such as oil, coal, and natural gas collectively meet approximately 80% of the current global energy demand and are being depleted continuously. Hence, these conventional energy sources need to be replaced by renewable precursors to meet the ever-increasing demands of energy.4,5 Also, the over-exploitation of these fossil fuels is leading to their rapid depletion and rendering them inadequate to satisfy the rising energy demands in the near future.6 Moreover, the combustion of fossil fuels is associated with the emission of an enormous amount of CO2, unwanted hydrocarbons, sulphur (S), and nitrogen (N) into the atmosphere. Excess CO2 emission causes the greenhouse effect and global warming.7 Similarly, the unwanted hydrocarbon deposition disturbs the carbon neutrality cycle. In contrast, S and N can oxidize to various acids, causing acid rain, hence posing a threat to the ecological balance.8 Therefore, it is absolutely essential to establish a sustainable alternative to fossil fuels by using renewable sources for energy production. This is a fundamental challenge to counter future energy crisis and ensure a cleaner environment.9,10 Renewable sources such as solar energy stand out as one of the major sources of vital energy, and can potentially become an epitome of high-input energy producer. As per the estimation, the earth receives a substantial amount of energy (1.9 × 108 TW h per year) from the Sun, which is almost 1000 times greater than the global energy consumption (1.3 × 105 TW h per year).11 Hence, it is absolute to utilize this primary energy resource through several opportunistic approaches in order to develop sustainable energy grids such as green electricity using solar cells.12 Distinctly, the use of solar energy for sustainable fuel production by harnessing the natural abundant renewable resources such as water, CO2, lignocellulosic biomass, and the earth's abundant natural gases can be a significant move towards the construction of a sustainable world (Fig. 1a). | ||
| Fig. 1 (a) Schematic outline of semiconductor-mediated solar fuel production by utilizing abundant renewable precursors and (b) photocatalytic processes involved in semiconductor photocatalysis. | ||
A recent report from March 2023 statistics of the U.S. National Oceanic and Meteorological Administration revealed that the CO2 emission already has surpassed 414.49 ppm, demanding the immediate implementation of necessary measures to take care of the surplus CO2 content.13 Providentially, the utilization of this excess anthropogenic CO2 gas for the production of various high-density gaseous and liquid fuels by harnessing solar energy can be an effective solution for the energy catastrophe as well as to mitigate the global warming impact by maintaining carbon neutrality.14 In CO2, ‘C’ exists in its highest oxidation state and can be reduced to form various value-added chemical products having one or more carbon atoms, named C1 and C2+ compounds such as CO, HCHO, HCOOH, CH3OH, CH3CHO, CH3CH2CHO, etc. These photoreduced products have great potential as modern-day fuel, currently gaining huge research interest.14,15 Similarly, the thirst for sustainable development of carbon-free fuels such as H2, H2O2, NH3, etc., is an emerging topic in recent times to prevent carbon emissions into the environment.16,17 Indeed, hydrogen (H2) fuel is a truly sustainable and cleanest mode of energy with zero pollution. H2 has a very high specific energy density (142 MJ kg−1), typically greater than that of the currently used fuels. However, the current production of H2, primarily through the steam reforming process, is not ideal, as this process involves high CO2 emanation.18,19 Therefore, it is crucial to develop a sustainable route for H2 production instead of the fossil fuel steam reformation process. There is significant research activity focused on sustainable H2 production by considering using water as a renewable precursor via photocatalytic water-splitting.20,21 The fate of H2 production via the photocatalytic pathway depends upon several critical factors, which have been extensively elaborated later in this review. However, the major concern associated with the use of this high-energy-density H2 fuel is its successful storage. The storage and transportation of H2 fuel is highly challenging and requires delicate measures due to the explosive nature of H2.22 Therefore, chemicals such as formic acid and NH3 suitably serve as the source of H2 carrier, and hence, their sustainable production is concurrently crucial, like sustainable H2 production. For instance, NH3 is a reasonably higher volumetric energy density fuel than H2, and its storage and transportation is simpler than H2. NH3 (in liquid state) possesses optimum gravimetric and volumetric energies (18.8 MJ kg−1 and 11.5 MJ L−1, respectively), suggesting its reasonable comparability to those of CH3OH.23,24 The worldwide requirement of NH3 crosses 200 million tons per annum, suggesting its huge demand because of its numerous applications. Besides, NH3 can be stored as a liquid under milder and lower pressure; it can be utilized as both a hydrogen precursor and an energy vector in applications like fuel cells and ammonia turbines.23–25 Currently, industrial NH3 production is carried out through the well-known Haber–Bosch process, where H2 and N2 undergo a reaction at high temperature and pressure over Fe-based heterogeneous catalysts. As reported several times, this is an energy-consuming process in which a large quantity of fossil fuel is utilized to produce H2, resulting in significant CO2 emissions.26,27 Hence, developing a sustainable and greener route for NH3 production using N2 fixation is crucial yet challenging. Although biological N2 fixation and electrochemical N2 reduction seem greener, these processes are far from practical implementation due to techno-economic barriers.23,24 However, the photochemical pathway for NH3 production via N2 reduction reaction (NRR) by harnessing solar energy is an attractive example of solar energy production from renewable precursors. Similarly, the photocatalytic production of hydrogen peroxide (H2O2) is an excellent example of solar energy production using molecular O2 as a precursor. H2O2 is widely known as a green oxidant and acts as a bleaching agent in the pulp, paper, and textile industry.28 Moreover, H2O2 has attracted significant interest in recent times as a promising energy carrier compared to H2 due to its high energy density, complete solubility in aqueous medium, and easy storage and transportation as a liquid fuel, making it a viable option to partially substitute fossil fuels.28,29 It is also used in the construction of hydrogen peroxide fuel cells to generate electricity with +1.09 V vs. NHE, which is comparable to that of the hydrogen fuel cell (+1.23 V) and methanol fuel cell (+1.21 V). Due to these several characteristics, the annual global demand keeps increasing every year. In 2021, the global production of CH3OH crossed 107 million tons and is expected to reach up to 500 million tons by 2050.30 However, the current industrial method for H2O2 production is the anthraquinone method, which is energy-intensive and produces waste.28–30 Therefore, the economical and sustainable H2O2 production, by harnessing renewable solar energy, can significantly reduce fossil fuel consumption and eliminate environmental impact to achieve the energy security goal.
Biofuels are another class of sustainable energy alternatives to fossil fuels that can be derived from the earth-abundant lignocellulose biomass. They are regarded as green, sustainable and modern-day fuels that can meet future energy demands.31,32 Approximately 170 billion metric tons per year of biomass is produced by nature via photosynthesis, making it the fourth largest naturally occurring energy provider after oil, coal and natural gas.31 The lignocellulosic biomass entities, mainly comprised of cellulose and hemicellulose, can be hydrolyzed to sugars, which can be derivatized further to produce second-generation bioethanol. Lignocellulosic biomass is known to produce several aromatic moieties, sustainable chemicals, diesel-like biofuels and various platform chemical commodities such as furfural, 5-hydroxymethylfurfural, 2,5-dimethylfuran, 2-methylfuran, and vanillin.31–33 Long carbon chain alkanes, which are found in bio-diesel and jet fuels, can be mainly synthesized from non-edible oils and vegetable oils extracted from biomass.34 Often, biomass transformations and several biorefineries are primarily operated by thermal catalytic methods, but harsh reaction conditions and very high energy consumption obviate their practical applications.35 However, the solar energy-driven photocatalytic approach for converting lignocellulosic biomass to several value-added chemicals and fuels can be regarded as a green, sustainable and eco-friendly energy-saving process for future biofuel production, which has recently drawn huge attention.35,36 Hitherto, solar fuel production using lignocellulosic biomass is still an underdeveloped process and requires a huge advanced research focus to embark on some eye-catching progress.
Semiconductor-based photocatalysis is a progressive green technology that utilizes everlasting solar energy as a primary energy provider using an appropriate semiconductor to execute the photochemical process (Fig. 1b). Semiconductor photocatalysis can offer promising pathways toward clean energy production, as highlighted by the incessantly increasing numbers of publications each year.37,38 Despite many reported inorganic and organic semiconductor photocatalytic materials, researchers have never stopped exploring g-C3N4 with multiple arenas in photocatalysis due to its exceptional structural features (Fig. 2).
 | ||
| Fig. 2 Schematic evolution of g-C3N4 from various precursors embedded with surface active sites and structural features. | ||
g-C3N4 possesses a unique graphene-like structural arrangement with a two-dimensional (2D) conjugated structure, formed by triazine rings or tri-s-triazine rings as basic structural units by the thermal condensation of either melamine, dicyanamide, urea or thiourea (Fig. 2). Such structural arrangements make g-C3N4 a potential photocatalytic material embedded with multifunctional active sites (Fig. 2).17,39–41 Moreover, these active sites and structural features not only exclusively produce the photogenerated charge carriers but also allow the fast charge separation to smoothly execute the surface redox reaction. Furthermore, the presence of the C–N robust covalent framework in the triazine unit provides outstanding thermal and chemical stability to g-C3N4. The bandgap energy (Eg) of g-C3N4 is estimated around 2.7 eV, and the associated energy bands, i.e., conduction band (CB) and valence band (VB), are located at −1.4 eV and 1.3 eV vs. normal hydrogen electrode (NHE), respectively. This band alignment provides numerous opportunities to target various photocatalytic applications and allows the possibility of further band engineering to increase efficiency.39–43 Hence, g-C3N4 has been vastly explored and reported to exhibit excellent performance in photocatalytic H2 production, H2O2 generation, CO2 reduction, organic pollutant degradation, water remediation and other artificial photosynthesis applications.17,41–51 Further, g-C3N4-based materials, signifying as visible light active metal-free photocatalysts, have currently gained tremendous research interest for sustainable chemical production by photocatalytic biomass transformation.52–54 In contrast, pristine g-C3N4 is also associated with some major drawbacks, which lead to suboptimal performance.39,40 Consequently, the rational modifications of the morphology and porosity of g-C3N4 have been reported to lead to excellent photoresponsive properties. Porosity in nanomaterials is generally classified into three types based on pore size: micropores (less than 2 nm), mesopores (2–50 nm), and macropores (greater than 50 nm). Materials with smaller pore sizes, such as micropores and mesopores, generally exhibit higher specific surface areas and more accessible active sites, making them highly advantageous for catalytic applications.55 In contrast, macropores are more effective in adsorbing organic macromolecules.56 Conversely, mesopores are more advantageous in the transportation and diffusion of reactants, products and solvent molecules because of the smaller void space between pores and a large void fraction within the framework.55,57,58 This offers multiple light reflections/scattering phenomena to enhance light harvesting efficacy. Therefore, mesoporous materials have been immensely explored in catalysis and photocatalysis over the past few decades.57–59 In addition, mesoporous materials can be a suitable support matrix for various nanoscopic substances, such as metal nanoparticles (MNps) in catalysis and photocatalysis. Specifically, active components embedded in mesoporous materials demonstrate improved catalytic/photocatalytic activity and stability compared to their unsupported counterparts.57–60
In recent years, several reports have emphasized the remarkable progress in various structurally modified g-C3N4-based photocatalysts, elucidating various synthesis methods and structural enhancements such as narrow and tunable pore size distributions, customizable morphologies, optimized optoelectronic properties, pore surface engineering, etc.61–66 These modifications aim to enhance photocatalytic performance by improving light absorption, charge separation, and surface reactivity. For instance, Wang et al. and Hasnan et al. have reported in their review that the strategic structural modifications can significantly improve the photoresponsive features.61,62 Khan et al. and Liu et al. have also demonstrated that deliberate elemental doping and significant defect engineering can exclusively modulate the band structure, making the g-C3N4-based photocatalysts more promising for efficient charge separation dynamics.65,66 All these review articles acknowledge the importance of surface area and porosity in enhancing light absorption. However, they often overlook systematic methodologies for fabricating porous g-C3N4 with tailored mesopore volume and high surface area. Only a handful of reviews have reported on the development of mp-g-CN-based photocatalysts over the past years. In 2017, Lakhi et al. and Sun et al. primarily reported the synthesis of mp-g-CN-based photocatalysts mainly dominated by a templated approach.67,68 They have also discussed that the subsequent structural modifications along with the creation of a mesoporous structure significantly boosted the photocatalytic activity. Since then, significant research progress has focused on template-free strategies, offering advantages such as atom economy and reduced reliance on silica precursors and corrosive chemicals. Further, the multifunctionality induced in the mp-g-CN-based photocatalyst's framework holds substantial potential in addressing various environmental concerns, like sustainable energy production via artificial photosynthesis. Therefore, it is absolutely imperative to cover the research gap in the context of the development of mp-g-CN-based photocatalysts and their prevailing prospects in solar energy conversion, which is yet to be explored. This review comprehensively outlines recent developments and advancements in mp-g-CN photocatalysts along with their potential applications in solar energy conversion (Fig. 3). Besides the description of various synthesis routes with particular emphasis on the emerging template-free approaches for mp-g-CN synthesis, this review provides a detailed testimony from fundamental principles to mechanistic pathways underlying mp-g-CN framework-driven solar energy conversion for sustainable fuel production. In addition, we critically evaluate the advances of mp-g-CN-based photocatalyst mediated biofuel production. Lastly, we also propose the associated research challenges and future prospects in this rapidly evolving field. We believe this review can resonate with a broad range of readers, from academia to industry, to create a reference value for developing highly efficient g-C3N4-based materials for artificial photosynthesis.
 | ||
| Fig. 3 Schematic outline of general synthesis routes, associated structural modifications and potential applications of mp-g-CN discussed in this review. | ||
2. mp-g-CN-based photocatalysts
g-C3N4 produced by the traditional synthesis method has a crammed and close-packed structure, commonly referred to as bulk g-C3N4, which hinders its photoresponsive performance. The strategic mesoporosity modifications in the g-C3N4 structure can ameliorate the optoelectronic response and photoactivity. In contrast to bulk g-C3N4, mp-g-CN-based photocatalysts thereby attracted considerable research interest in the field of photocatalysis owing to their distinctive and appealing structural features, as illustrated in Fig. 4a.67–70 However, the strategic structural modulation followed by doping, vacancy, or defect engineering in mp-g-CN can further modify the optoelectronic state to optimize light absorption, reducing band gap energy, charge carrier dynamics, and surface reactivity. Moreover, by successfully creating mesopores in the g-C3N4 framework, both surface and bulk functionalities can be achieved.70 The mesoporous channel in mp-g-CN serves as a potential scaffold that incorporates both organic and inorganic nanostructures within the mesopores by various surface engineering and nano functionalization phenomena. This creates closed contacts between mp-g-CN and the adopted semiconductor nanostructures in the resultant composite structure.67–71 These closed contacts, in terms of the grain boundary overlapping between the semiconductors, are responsible for the better charge separation and migration of the photoexcited charge pairs.71 Therefore, despite the neat mp-g-CN, the assessment of mesoporous creation has also been extended to a wide range of functionalized mp-g-CN hybrid composite semiconductors (mp-g-CN/metal oxide, mp-g-CN/metal sulfide, etc.).70–72 | ||
| Fig. 4 (a) Schematic of the comparison of bulk g-C3N4 and mp-g-CN, (b) various synthesis approaches for mp-g-CN. | ||
The synthesis of mp-g-CN-based photocatalytic systems is generally classified into two categories, i.e., templated-assisted method and template-free method, as shown in Fig. 4b. It is noted that the template-assisted mp-g-CN-based photocatalysts exhibit a more ordered mesoporous channel and high surface area compared to those synthesized via the template-free method.73 The general synthesis approach for the production of mp-g-CN-based photocatalytic systems is briefly discussed below.
2.1. Synthesis of mp-g-CN-based photocatalysts using the template-assisted method
The templates are considered an indispensable tool for the creation of ordered mesoporous structures in materials. Template-assisted synthesis methods are based on the use of inorganic or organic frameworks as templates and are considered the most effective approaches for the synthesis of target frameworks having an absolute size, tunable morphology, as well as a well-defined porous structure.73 Mesoporous channels with well-defined porosity provide cross-plane electron diffusion channels, which results in the diminution of the surface recombination sites for the photogenerated excitons.74 It is worth mentioning that, the deliberate creation of such ordered and dimensionally controlled mesoporous channels in g-C3N4-based photocatalysts using the template-assisted strategy can lead to several beneficial aspects such as (i) significant improvement in photogenerated charge separation by providing active sites for self-trapping of the charges, (ii) exposure of catalytic active sites, dispersed on the surface and in the bulk, to resonate with the charge transportation phenomena, and (iii) creation of a suitable platform for hosting the other semiconductors or co-catalysts in order to advance the overall photocatalytic activity. Hence, these beneficial aspects of mp-g-CN-based photocatalytic systems demand further exploration to construct highly efficient photocatalytic materials, which largely depends on the synthesis approach and the choice of a suitable template. In the literature, mp-g-CN materials have generally been synthesized and fabricated by using two types of templates: hard template and soft template.73 The morphology and the pore structure of g-C3N4 could be regulated by adjusting the type of template involved in template-assisted synthesis. The details of both types of template-assisted synthesis strategies are discussed below. | ||
| Fig. 5 (a) Schematic representation of various hard templates, (b) stepwise synthesis procedures for the hard template-assisted mp-g-CN photocatalyst. | ||
Initially, the appropriate g-C3N4 precursor in solution form is infused into the ordered porous channels of the template, followed by evaporation to create a solid template-precursor mixture. The fixed and rigid nature of the porous channels triggers the shaping of g-C3N4 within the fixed meso channels. Finally, removing the hard template through chemical etching results in the formation of regular and interconnected ordered mp-g-CN.73 The earliest work on mp-g-CN synthesis was reported by Vinu et al., using ethylenediamine and carbon tetrachloride as CN precursors in the presence of SBA-15.76 Similarly, Groenewolt et al. reported another silica monolith mediated mp-g-CN using cyanamide as the CN precursor.77 Following this work, in another attempt, cyanamide as the CN precursor was mixed with silica Nps with an average particle size of 12 nm.78 After thermal condensation at 550 °C and removal of silica using ammonium hydrogen difluoride (NH4HF2), the resultant mp-g-CN was achieved. The specific surface area was improved from 86 to 327 m2 g−1 by varying the silica-to-cyanamide ratio from 0.5 to 1.6. Afterwards, a series of silica template-mediated mp-g-CN-based photocatalytic systems were prepared. Of note, although the use of a hard template is associated with a multiple-step process and requires fluoride-based reagents for the removal of the silica-based template, the hard template-mediated mp-g-CN-based materials continue to attract significant research interest due to the superior quality of the mesoporous framework with the improved physicochemical properties. For example, Liu et al. reported an ordered mp-g-CN (mpg-C3N4) photocatalyst using SBA-15 as a silica-based hard template via an impregnation method.79 At first, SBA-15 was prepared using the Pluronic P123 symmetric triblock copolymer as a soft template to create ordered porous channels in SBA-15 and tetraethyl orthosilicate (TEOS) as the silica source. After the successful synthesis of SBA-15, it was gradually impregnated with an aqueous solution of cyanamide and subsequently calcined at three different temperatures and etched with NH4HF2 to obtain the ompg-C3N4 photocatalyst (Fig. 6a). The resultant synthesized CN materials were designated as ompg-C3N4-450, ompg-C3N4-500, and ompg-C3N4-550, corresponding to the calcination temperature 450 °C, 500 °C and 550 °C, respectively. The developed mesoporosity was confirmed using several characterization techniques, and the material obtained at 550 °C calcination temperature was claimed to be the finest photocatalytic CN material in comparison to other synthesized materials. The surface and nanostructure morphology of ompg-C3N4-550 indicated that after the removal of SBA-15, the successful rod-shaped morphology of ompg-C3N4-550 was achieved due to the templating effect of SBA-15, whereas bulk g-C3N4 exhibited an irregular agglomerated morphology (Fig. 6b and c). Additionally, the mesoporosity was confirmed by small-angle X-ray scattering (SAXS) analysis. The SAXS measurement analysis indicated that the existence of (100) and (110) reflections corroborated with the ordered 2D hexagonal mesoporous structure having p6mm space group. The mesoporous channel creation was further investigated by N2 adsorption–desorption (BET analysis). All the CN samples exhibited a type IV isotherm and a H1 hysteresis loop, suggesting the existence of an ordered mesoporous framework having cylindrical porous channels (Fig. 6d). The surface area was improved from 3.3 m2 g−1 for bulk g-C3N4 to 86.2 m2 g−1 for ompg-C3N4-550.
 | ||
| Fig. 6 (a) Diagrammatic sketch of the fabrication of ompg-C3N4-550, TEM images of (b) bulk g-C3N4 and (c) ompg-C3N4-550, and (d) BET isotherms of all the CN materials and bulk g-C3N4. Reproduced with permission.79 Copyright 2016, Elsevier (for interpretation of the references to color in this figure legend, the reader is referred to the web version of this article). | ||
In another work, Chen et al. reported a 3D mp-g-CN photocatalyst using SiO2 spheres as the hard template and melamine as the CN precursor, as schematically illustrated in Fig. 7A-a.80 In a typical synthesis method, at first, melamine and SiO2 spheres with different particle sizes (15 nm, 30 nm, 100 nm and 200 nm) were vigorously mixed through a ball milling process and then subjected to calcination to obtain g-C3N4@SiO2-x (where x stands for the particle size of SiO2 spheres, i.e., 15, 30, 100 and 200 nm) (Fig. 7A-a). The resultant g-C3N4@SiO2-x materials (g-C3N4@SiO2-15, g-C3N4@SiO2-30, g-C3N4@SiO2-100 and g-C3N4@SiO2-200) were further treated with 10% HF aqueous solution for silica etching to obtain g-C3N4-15, g-C3N4-30, g-C3N4-100, and g-C3N4-200, respectively. The textual analysis study suggested that the CN materials obtained using 15 nm and 30 nm SiO2 hard templates exhibited superior textual properties among all the synthesized g-C3N4-x (Fig. 7A-(b and c)). Both g-C3N4-15 and g-C3N4-30 displayed a type IV isotherm with an average pore diameter of 12.4 nm and 17.5 nm, respectively, estimated using the BJH model. However, the pore diameter for g-C3N4-100 and g-C3N4-200 using the BJH model could not be determined, which might be due to the existence of larger pores or macropores. Additionally, a decreasing trend of the surface area was observed when the particle size of the SiO2 sphere increased (Fig. 7A-b). The well-ordered porous architecture present in all CN materials was further visualized using SEM and TEM investigation (Fig. 7A-(d–l)). SEM analysis demonstrated a 3D cotton wadding structure having a distinguished porous channel present in g-C3N4-15 and g-C3N4-30. g-C3N4-100 and g-C3N4-200 exhibited an ordered honeycomb-like framework with macroporous channels (Fig. 7A-(e–h)). This structural variation was due to the thermal polymerization and formation of the g-C3N4 framework, which was confined to the crevices between the regime of SiO2 particles. Furthermore, the TEM investigation revealed the pore diameter of all the synthesized CN materials (Fig. 7A-(i–l)). The pore diameters were found to be 14 nm, 20 nm, 100 nm and 180 nm, respectively, for g-C3N4-15, g-C3N4-30, g-C3N4-100, and g-C3N4-200 consistent with the SiO2 particle size adopted during the synthesis.
 | ||
| Fig. 7 (A) (a) Schematic depiction of the synthesis of 3D porous g-C3N4, (b) BET isotherms and (c) BJH pore size distribution of all the synthesized materials, (d) SEM image of bulk g-C3N4, SEM and TEM images of (e) and (i) g-C3N4-15, (f) and (j) g-C3N4-30, (g) and (k) g-C3N4-100, and (h) and (l) g-C3N4-200. Reproduced with permission.80 Copyright 2020, Elsevier. (B) (a) Diagrammatic representation of halloysite@nano-g-C3N4, SEM and TEM images of (b) and (e) BCN, (c) and (f) MCN and (d) and (g) halloysite@nano-g-C3N4, (h) and (i) BET isotherms and BJH pore size distribution of the modified halloysite template, halloysite@nano-g-C3N4, MCN and BCN. Reproduced with permission.81 Copyright 2018, Elsevier. | ||
Wang et al. reported an mp-g-CN nanotube (MCN) synthesized using a modified halloysite as a hard template and melamine as a CN source via the chemical vapor deposition (CVD) method.81 At first, the activated halloysite, by calcination and acidification, was placed on top of a cylinder inside a covered crucible containing melamine for the vapor deposition, as shown in Fig. 7B-a. In this process, melamine was thermally polymerized and wrapped over the halloysite surface in the vapor form during thermal treatment. The deposition of the g-C3N4 shell on halloysite formed a khaki powder designated as ‘halloysite@nano-g-C3N4’ (Fig. 7B-a). The resultant halloysite@nano-g-C3N4 was treated with 10% HF solution to form MCN. The successful synthesis of MCN using halloysite was confirmed using SEM and TEM investigation. Compared to the irregular granule-like morphology of bulk g-C3N4 (BCN), MCN exhibited nanotubular morphology (Fig. 7B-(b–g)). Moreover, due to the halloysite-mediated synthesis, the textural properties of MCN were very high compared to that of BCN (Fig. 7B-(h and i)). MCN displayed a specific surface area of 85.5 m2 g−1 with a hierarchical porous structure having small nanopores (<5 nm) and larger mesopores between 10 and 50 nm. In contrast, BCN exhibited an impoverished surface area of only 6 m2 g−1 and low undefined porosity.
In another attempt, a sandwich-like 3D mesoporous graphite-like g-C3N4-based catalyst abbreviated as meso-g-C3N4/WP/meso-g-C3N4 was prepared using solid-phase in situ reduction and calcination.82 Initially, tungsten phosphide (WP) was synthesized and used as a mediator for the development of meso-g-C3N4/WP/meso-g-C3N4 using cyanamide as a CN precursor and colloidal silica as a hard template (Fig. 8A-a). meso-g-C3N4/WP/meso-g-C3N4 after the WP incorporation exhibited sandwich-like laminated heterojunction nanosheets as visualized by TEM investigation (Fig. 8A-(b–e)). A neat mp-g-CN (meso-g-C3N4) was fabricated by using colloidal silica for the comparison study. Based on the TEM analysis conducted for meso-g-C3N4 and WP, a completely different morphology was obtained for meso-g-C3N4/WP/meso-g-C3N4, which exhibited a successful sandwich-like nanosheet morphology. The N2 adsorption–desorption analysis was further conducted to estimate the extent of mesoporosity present in all synthesized materials. The BET surface area was found to be 11, 117, 6, and 82 m2 g−1 for bulk g-C3N4, meso-g-C3N4, WP and meso-g-C3N4/WP/meso-g-C3N4, respectively (Fig. 8A-(f and g)). Among all the materials, only meso-g-C3N4 and meso-g-C3N4/WP/meso-g-C3N4 exhibited a type IV isotherm, which corroborated with the formation of ordered mesoporosity in their framework. Hence, these two materials displayed a much higher surface area and porosity than bulk g-C3N4. Similarly, another hollow mp-g-CN-based composite catalyst was fabricated by Ren et al.83 They reported a Z-scheme g-C3N4/Ag2CrO4 composite by using SiO2 as a hard template. The SiO2 hard template was initially synthesized by using TEOS and C18TMOS as silica sources. Then, the hollow mp-g-CN designated as ‘HCN’ was prepared using cyanamide and the synthesized SiO2 hard template, followed by thermal polymerization and, subsequently, the silica etching process. Afterwards, the as-synthesized hollow HCN acted as a support matrix for the synthesis of the g-C3N4/Ag2WO4 mesoporous composite, designated as the HCN/ACO composite. The hollow sphere morphology and the surface deposition of Ag2CrO4 were investigated using TEM, HR-TEM and EDS elemental mapping analysis (Fig. 8B-(a–e)). The mesoporous architecture was further confirmed using N2 adsorption–desorption analysis and was well corroborated by the TEM investigation. The type IV isotherm, exhibiting a higher surface area than bulk g-C3N4 and a large pore diameter, suggested that the use of a SiO2 hard template successfully created the hollow mesoporous framework in the HCN/ACO composite (Fig. 8B-f).
 | ||
| Fig. 8 (A) (a) Schematic of meso-g-C3N4/WP/meso-g-C3N4 formation, TEM images of (b) meso-g-C3N4, (c) WP, (d) and (e) meso-g-C3N4/WP/meso-g-C3N4, (f) BET isotherms, and (g) BJH pore size distribution plot of all the synthesized materials. Reproduced with permission.82 Copyright 2020, Elsevier. (B) (a–c) TEM images, (d) HR-TEM image and (e) EDS mappings of the HCN/ACO composite, (f) N2 adsorption–desorption isotherms and BJH pore size distribution plot (inset) of CN, HCN, ACO and the HCN/ACO composite. Reproduced with permission.83 Copyright 2021, Elsevier. | ||
So far, the focus has been on the development of mp-g-CN-based materials using the hard template synthesis approach. Similar to hard template-assisted synthesis, the following section provides a brief discussion and highlights the achievements of the development of soft-template-assisted mp-g-CN-based materials.
 | ||
| Fig. 9 Stepwise synthesis of a mesoporous g-C3N4-based photocatalyst using the soft template approach. | ||
A mp-g-CN photocatalytic material having a worm-like porous architecture with narrow pore size distribution was prepared by using Pluronic P123 as a soft template and melamine as the CN precursor.85 The resultant mp-g-CN material exhibited layered and plate-like morphology with worm-like porous channels. The N2 adsorption–desorption analysis suggested the surface area of bulk g-C3N4 to be 9 m2 g−1, which was improved to 90 m2 g−1 after the synthesis using P123 as a soft template and also the resultant mp-g-CN material exhibited a uniform and narrow pore size distribution. Similarly, Wang et al. reported 3D aggregated hollow bubbles of the mp-g-CN photocatalytic system using a combination of supramolecular precursor derived from dicyandiamide (DCDA) and P123 as a soft template.87 Initially, a supramolecular precursor of DCDA was synthesized, which acted as a true CN precursor for the synthesis of mp-g-CN hollow bubbles in the presence of P123. The obtained CN materials were designated as BCN (when synthesized using DCDA directly), HCN (when synthesized using the supramolecular precursor), CN-0% (absence of P123) and CN-2% (when P123 content was 2%). The structural and morphological modulation was achieved by using P123, which can be understood from the SEM and TEM investigation. Both SEM and TEM investigation suggested that an aggregated morphology was obtained for BCN (Fig. 10a and e), while SEM analysis showed flake-like surface morphology for the HCN sample, which consisted of porous nanosheets, as confirmed from the TEM analysis (Fig. 10b and f). It was observed that CN-0% exhibited a porous sheet with larger size and thickness, as confirmed by SEM and TEM analysis (Fig. 10c and g). However, CN-2% exhibited a 3D aggregated hollow bubble morphology (Fig. 10d and h), which is due to the use of the P123 template. The difference in the structural feature was further demonstrated by PXRD observation. All the CN samples, i.e., BCN, HCN, CN-0% and CN-2%, exhibited the standard diffraction peak at 2θ 12.8° and 27.5° attributed to the 100 and 002 plane of pristine g-C3N4. However, their peak intensities differed from each other; the peak intensity followed the order BCN > HCN > CN-0% > CN-2% (Fig. 10i). This could be due to the difference in the morphology, as confirmed by the TEM investigation. The authors also reported that the (002) plane of HCN was slightly shifted to the right due to the decrement in the interlayer stacking distance. However, the (002) plane was observed to be slightly shifted towards the left side in the case of CN-0% and CN-2% compared to HCN, which was hypothesized to be due to the increment in the thickness of the shell wall of the hollow bubble structure. The existence of the type IV isotherm and H3 hysteresis loop from N2 adsorption–desorption analysis suggested the mesoporous framework (Fig. 10j). The BET surface area of CN-2% was higher than that of the other CN materials, signifying the influential role of P123 in the improvement of the microstructure.
 | ||
| Fig. 10 SEM and TEM images of (a) and (e) BCN, (b) and (f) HCN, (c) and (g) CN-0% and (d) and (h) CN-2%, respectively, (i) and (j) PXRD and BET isotherms of BCN, HCN, CN-0% and CN-2%. Reproduced with permission.87 Copyright 2018, Elsevier. | ||
In several reports, the use of triblock polymers as soft templates has been shown to lead to several hitches, such as the uncontrolled growth of size and morphology of the pores.67,73,84 The improper operation of the soft template also led to interlayer collapse or over-stacking during the molecular polymerization, which subsequently impaired the homogeneity in mesoporous channel development. Moreover, since the calcination process is limited (around 550 °C) for the synthesis of g-C3N4 and the soft template removal also occurs in the range of 500–550 °C, the residual carbon from the soft template could remain in the final g-C3N4 material even after the calcination process. Therefore, other than the triblock polymer, several other soft templates, such as ionic liquids and bubble formation agents, i.e., ammonium salts, have recently been utilized to develop efficient mp-g-CN-based photocatalytic materials, which are summarized below.
The ionic liquid-assisted synthesis strategies provide several beneficial aspects for producing mp-g-CN-based photocatalytic materials. In this synthesis approach, the ionic liquid interacts with the CN precursors and forms a supramolecular precursor having an appropriate and definite well-defined molecular arrangement, which leads to the formation of a precise morphology in the final CN material. Moreover, the ionic liquid decomposes during the calcination process to generate in situ pore-forming gases as well as various dopants, which are responsible for the formation of functional and porous CN material.88–91 Wang et al. reported an ionic liquid-assisted boron (B) and phosphorus (P) double-doped mesoporous g-CN photocatalyst using cyanuric acid and melamine as the CN precursors.89 They reported that [Emim]BF4 and [Bmim]PF6 ionic liquids were used as B and P sources as well as mediators for the mesoporous generation. The ionic liquid formed a supramolecular assembly through hydrogen bonding with the imidazole moieties and the –NH2 and –OH groups of melamine and cyanuric acid (Fig. 11A-a). They also mentioned that the ionic liquid started to decompose around 400 °C, and produced several gaseous intermediates such as water vapor, NH3, CO2, etc., which served as in situ intermediates for etching the carbon nitride surface and were responsible for the creation of mesopores. They compared the structural evolution of the resultant B and P doped mp-g-CN (designated as CN-45B-45P, where 45 represents the dosage of both ionic liquids) with bulk g-C3N4 prepared using only melamine (designated as CN) and a mixture of melamine and cyanuric acid (designated as CNN). The surface and nanostructure morphology of the resultant CN, CNN and CN-45B-45P were suggested; CN exhibited a block structure, CNN exhibited a lamellar structure, while CN-45B-45P exhibited a porous structure with thin thickness (Fig. 11A-(b–g)). Upon decomposition of the ionic liquid at elevated temperatures, B and P were successfully doped into the material (Fig. 11A-(h–l)). N2 adsorption–desorption analysis unveiled a type IV BET isotherm for all the synthesized materials, signifying the existence of mesoporous channels (Fig. 11A-m). It was reported that CN-45B-45P exhibited a high surface area and large pore volume of 137.49 m2 g−1 and 0. 8279 cm3 g−1, respectively (Fig. 11A-n). Similar to ionic liquids, other bubble templates have also been used and have recently drawn significant attention for the fabrication of mp-g-CN-based photocatalysts.92 In the bubble template-assisted synthesis method, various gases are released during the thermal decomposition of bubble templates at elevated temperatures, which are responsible for creating abundant porosity and pore modifications, and ultimately improving the microstructure of the final mp-g-CN material. For example, Kadam et al. reported S-doped mp-g-CN nanosheets (p–SCN–x, x = 0.5, 1, 2 and 3, representing the mass ratio of NH4Cl to thiourea) using thiourea as the sulfur and CN precursor and NH4Cl as a gas bubble template (Fig. 11B-a).93 The degree of crystallinity decreased with the increase in NH4Cl content due to the layer separation and formation of a porous structure, as shown by the PXRD observation. The ‘S’ doping was evidenced from the XPS analysis, confirming that ‘S’ was covalently bonded to ‘C’ and ‘N’. A progressive increase of porosity with the increase in NH4Cl content was observed from SEM analysis (Fig. 11B-b). On increasing the NH4Cl content, a larger amount of in situ gas bubble components (NH3, HCl, and H2S) formed, which reduced the nanosheet thickness and led to the collapse of the long-range ordered porous structure. The TEM investigation of p-SCN-2 suggested a very thin nanosheet structure with a surface characterized by numerous ordered holes, which could serve as the active centre (Fig. 11B-(c and d)). All the p-SCN materials exhibited a type IV isotherm and H3 hysteresis, signifying the signature feature of the mesoporous structure. p-SCN-2 exhibited a higher surface area and pore volume than p-SCN-1 and p-SCN-3 (Fig. 11B-(e and f)). This suggested that an optimum content of NH4Cl was required to gain the ideal mesoporous structure. In contrast, minimum dosing failed to create abundant mesoporosity, and excess dosing led to the collapse of the mesoporous framework.
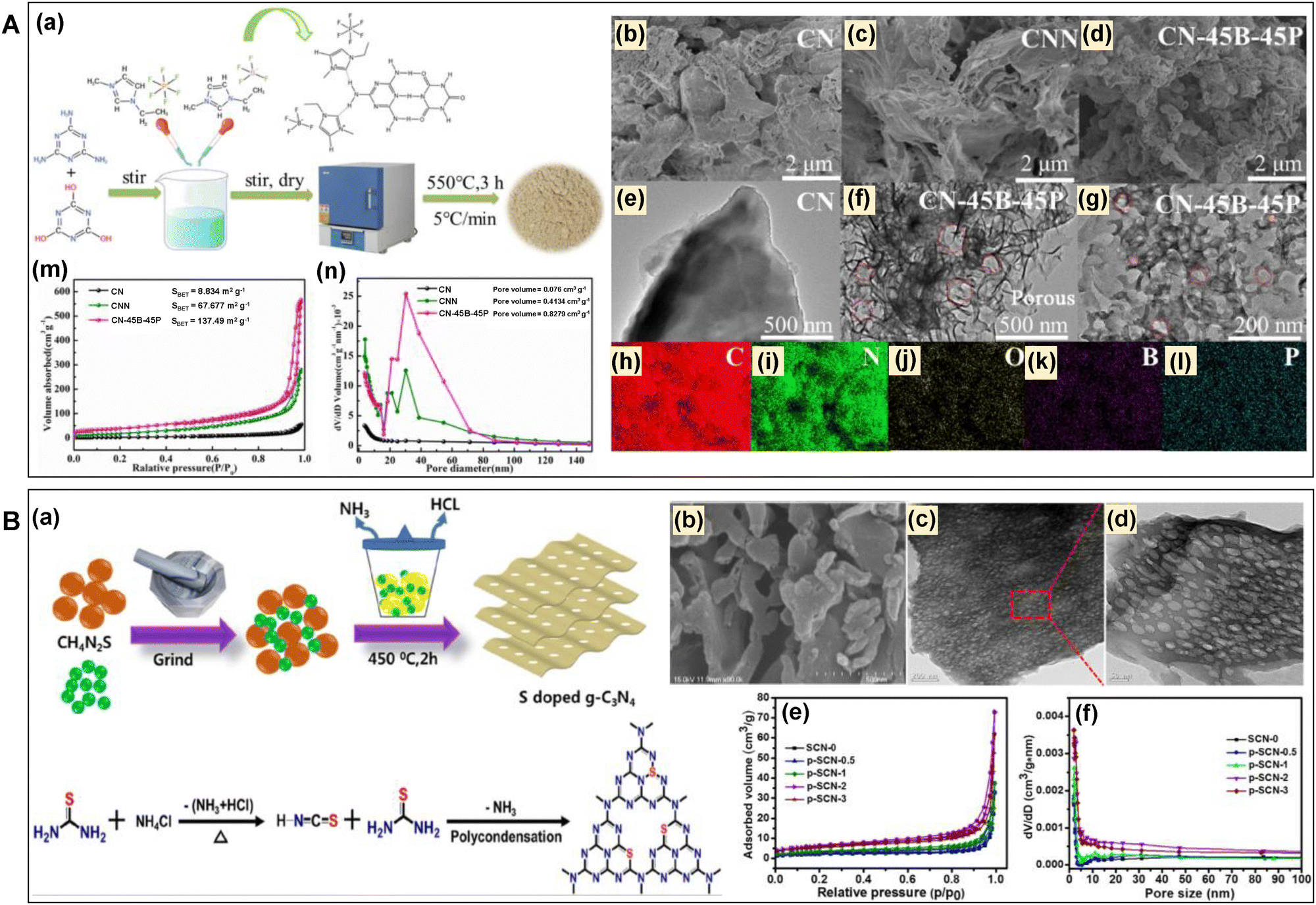 | ||
| Fig. 11 (A) (a) Schematic of the synthesis of B and P doped mp-g-CN (CN-45B-45P), (b) and (e) SEM and TEM images of CN, (c) SEM of CNN, (d), (f) and (g) SEM and TEM images of CN-45B-45P, (h)–(l) elemental mapping of CN-45B-45P, (m) and (n) BET isotherms and pore size distributions of CN, CNN, and CN-45B-45P. Reproduced with permission.89 Copyright 2023, Elsevier. (B) (a) Diagrammatic representation of S doped mp-g-CN (p–SCN–x), (b) SEM image of p–SCN–2, (c) and (d) TEM images of p–SCN–2, (e) and (f) BET isotherms and pore size distribution of all the synthesized materials (here, SCN-0 represents S-doped bulk g-C3N4 synthesized without NH4Cl). Reproduced with permission.93 Copyright 2020, Elsevier. | ||
The primary advantage of using soft templates is their easy removal, less time consumption, and relatively straightforward process compared to hard template-mediated synthesis for the formation of the mp-g-CN matrix. In some instances, the soft templates, such as ionic liquids or bubble templates, can also induce heteroatom doping, such as P, B, S, Cl, I, etc., to avoid the multiple reaction steps.
2.2. Synthesis of mp-g-CN-based photocatalysts using the template-free method
It is noted that being a 2D nanomaterial, pristine g-C3N4 consists of a stacked layered structure, which hinders it from achieving well-ordered porosity and adequate surface area.39,40 The distribution of active sites is masked due to the layered structure, and the inappropriate light adsorption leads to insufficient light harvesting probabilities. Moreover, such bottlenecks were systematically encountered by adapting the structural improvements via the template-assisted method, as briefed in the above section. However, the protocols mentioned above for the synthesis of mp-g-CN-based nanomaterials (using the hard and soft template methods) are somewhat cumbersome and suffer from various concerns such as atom economy, reliance on hazardous chemicals or additives required for the template synthesis, time-consuming processes, etc. Unlike template-assisted strategies, the template-free synthesis approach offers significant advantages due to its innovative features, including sustainable and straightforward synthesis methods, economic viability, and potential for large-scale production.68,94–99 Various strategic synthesis methodologies have been adopted to achieve template-free synthesis of mp-g-CN-based photocatalysts (Fig. 12). | ||
| Fig. 12 Schematic outline of various template-free synthesis approaches of mp-g-CN. | ||
The synthesis of mp-g-CN by supramolecular assembly of various precursors is an ideal example of a template-free approach. In the template-free synthesis method, suitable and appropriate precursors are taken and incubated for the molecular assembly to form an intermediate precursor, and this process truly controls the mesoporosity and provides unique features to the resultant mp-g-CN nanomaterials.68,98,100–103 In this method, the precursors with appropriate composition combine to form a supramolecular assembly through hydrogen bonding. This non-covalent interaction forms a stable aggregate of the supramolecular structure of various precursors, which ultimately controls the morphology and mesostructure of the final g-C3N4 material.100–103 The template-free synthesis method provides the opportunity to tailor the composition and structure of supramolecular aggregates, thereby enabling the production of various structurally modified g-C3N4-based nanomaterials. Further, the synthesis and advancement of mp-g-CN-based photocatalysts using various template-free synthesis strategies have been demonstrated below to realize the popularity and acceptability of these synthesis routes in the development of highly photocatalytic systems.
The supramolecular assembly of melamine–cyanuric acid is capable of forming various morphologically controlled g-C3N4-based nanomaterials. For example, Sun et al. synthesized 3D porous nanovesicles (PCNNVs) using a template-free strategy, involving the supramolecular assembly of melamine and cyanuric acid in DMSO.104 They demonstrated that different size variations of PCNNVs could be achieved by controlling the mass ratio of melamine to cyanuric acid, and the resultant synthesized PCNNV materials were designated as PCNNVs-L, PCNNVs-M and PCNNVs-S (where L, M and S represent large, middle and small sizes, respectively). Fig. 13A-a illustrates the synthesis route for these PCNNV materials. It was noted that, after the dissolution process, cyanuric acid and melamine interacted via H-bonding between the hydroxyl group of cyanuric acid and the amino group of melamine in the DMSO solution. Varying the mass ratio of melamine to cyanuric acid led to the formation of voids due to the collapse of supramolecular aggregates and the in situ generated gas (NH3 and CO2) being soaked during thermal treatment. The generated nanovesicle 3D porous structure was visualized by SEM and TEM investigation (Fig. 13A-(b–g)). It can be seen that PCNNVs exhibited a 3D cavity-like porous vesicle architecture, and the size of PCNNVs-L, PCNNVs-M and PCNNVs-S was found to be 360, 280 and 170 nm, respectively, as obtained from TEM investigation (Fig. 13A-(e–g)). The PXRD pattern suggested the peak intensities corresponding to (002) and (100) in all PCNNVs to be significantly lower than that of BCN, which suggested disruption of the in-plane structure due to the creation of a 3D porous architecture and the resultant porous cavities had smaller shell thickness. Furthermore, the N2 adsorption–desorption analysis was conducted to evaluate the presence of porous architecture in all PCNNV materials. All the PCNNVs showed a type IV isotherm and a H3 hysteresis loop, suggesting the presence of mesoporous channels in these materials (Fig. 13A-(h and i)). Expectedly, BCN exhibited a very poor surface area and pore volume in comparison to all PCNNVs.
 | ||
| Fig. 13 (A) (a) Graphical representation of the synthesis of various 3D PCNNV structures, SEM images of (b) PCNNVs-L, (c) PCNNVs-M, (d) PCNNVs-S, TEM image of (e) PCNNVs-L, (f) PCNNVs-M, (g) PCNNVs-S, (h) BET isotherms, (i) pore size distribution of BCN, PCNNVs-L, PCNNVs-M, and PCNNVs-S. Reproduced with permission.104 Copyright 2022, Elsevier. (B) (a) PXRD of b-CN and p-CN, (b) SEM of p-CN. Reproduced with permission.105 Copyright 2014, Elsevier. | ||
Shen et al. reported a template-free synthesis method for the preparation of a novel g-C3N4 having a 3D hierarchical porous structure.105 In this study, melamine and cyanuric acid were assembled in ethanol to form a supramolecular precursor by stirring at 6 °C for 24 h. Afterwards, the resultant supramolecular complex was heated at 520 °C for 4 h under Ar flow to obtain a 3D porous structure of g-C3N4, designated as p-CN. Cyanuric acid acted as a shaping agent, disrupting the regular polymerization of melamine into a bulk structure. Therefore, PXRD data indicated a notable shift and broadening of the (002) plane in p-CN compared to bulk g-C3N4 (b-CN), attributed to an increase in the interplanar stacking distance or a decrease in the interlayer stacking density and reduced long-range interplanar stacking caused by surface defects during annealing (Fig. 13B-a). Moreover, the morphology and microstructure analysis suggested that b-CN had a stacked lamellar structure, whereas p-CN exhibited a hierarchical 3D porous structure decorated with several hollow-like microstructures (Fig. 13B-b). This led to an enhancement in the textural properties of p-CN relative to b-CN, with the surface area exhibiting a 3.5-fold increase relative to that of b-CN.
Similarly, in another study, Chen et al. reported the fabrication of an ultra-small size Fe-cluster embedded 3D porous g-C3N4-based catalyst, designated as FeNx/CCN, using a template-free approach.106 In this study, melamine and cyanuric acid formed a supramolecular complex and were mixed with the FeNx precursor, which was created by reacting the Fe source with 2-methyl imidazole in the presence of polyvinylpyrrolidone. The resultant melamine–cyanuric acid supramolecular complex was then annealed to form FeNx/CCN. The stepwise synthesis procedure is illustrated in Fig. 14a. The TEM investigation suggested the presence of a hierarchical porous network in FeNx/CCN (Fig. 14b and c). The porous nature of both FeNx/CCN and 3D CN (synthesized without adding the FeNx precursor) was evaluated using N2 sorption analysis. The type IV isotherm with H3 hysteresis loop confirmed the presence of mesoporosity in both the synthesized samples (Fig. 14d). However, both surface area and pore volume were reduced in FeNx/CCN compared to 3DCN, which was believed to be due to the blocking of micro and mesoporous channels due to the deposition of the Fe cluster.
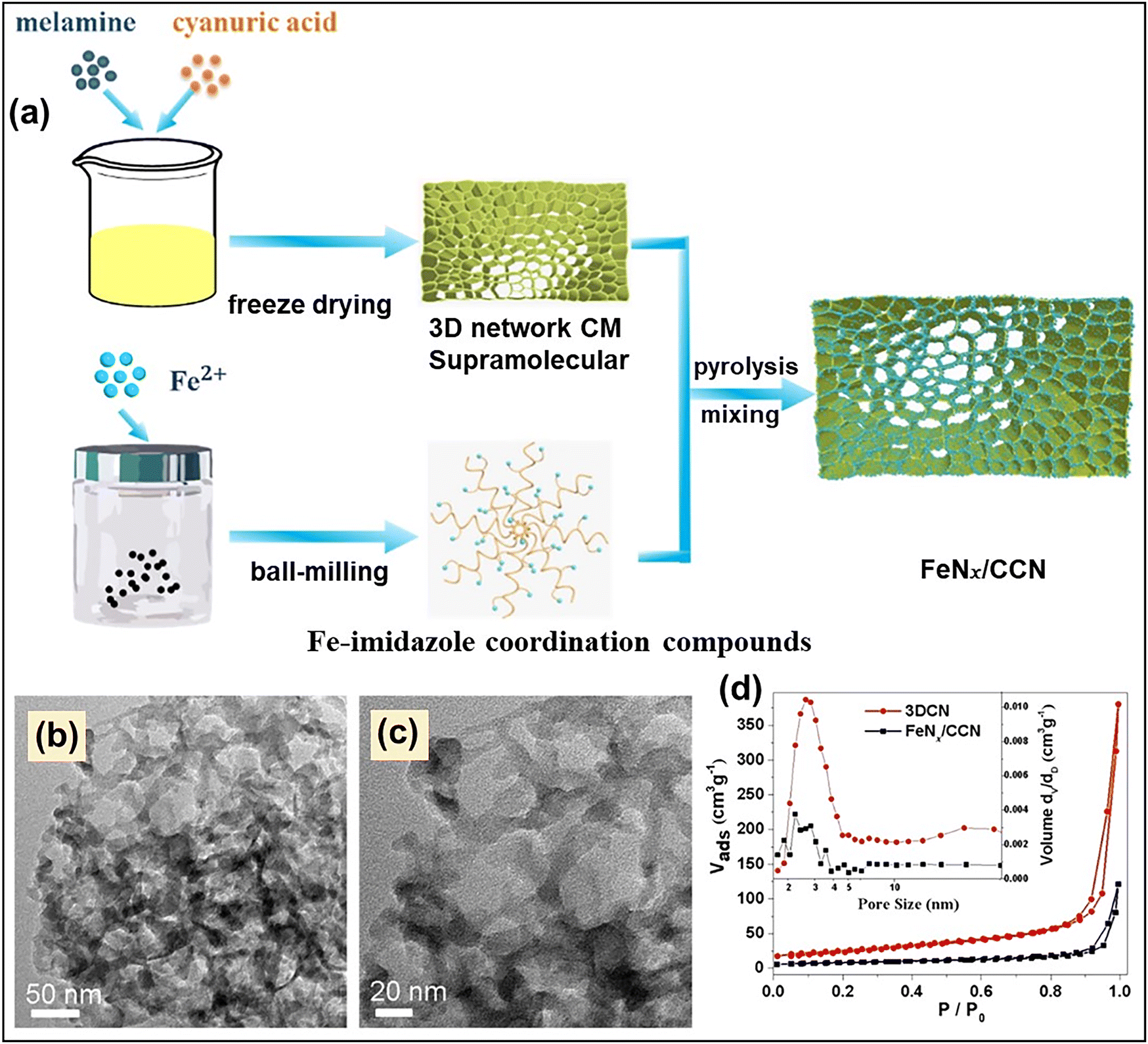 | ||
| Fig. 14 (a) Schematic illustration of the step-wise synthesis of FeNx/CCN, (b) and (c) TEM images of FeNx/CCN, (d) N2 adsorption–desorption isotherm and pore size distribution (inset) of 3DCN and FeNx/CCN. Reproduced with permission.106 Copyright 2023, American Chemical Society. | ||
The synthesis of various mp-g-CN-based materials (including both template-assisted and template-free methods) has been discussed so far, following the bottom-up approach in which appropriate N-containing precursors are used. However, top-down synthesis strategies have also been employed to synthesize mp-g-CN-based materials.107 These top-down synthesis strategies are classified under one of the prominent template-free synthesis methods for synthesizing mp-g-CN-based materials. In this method, mp-g-CN-based materials can be synthesized or derived from bulk g-C3N4 by delamination of its stacked-layer structure. Generally, the top-down method involves an exfoliation or etching process. The weak van der Waals interaction between the stacked layers of bulk g-C3N4 can be exploited to separate it into a few layers using the exfoliation approach.107,108
The improved textural properties and structural modulation can be achieved during the exfoliation process. The ultrathin sheets and pore modulation provide a larger surface area compared to that of bulk g-C3N4 after the exfoliation process. The magnitude of mesoporosity creation also depends on the exfoliation process, as this process opens up the porous channels within the framework.108–110 Mainly, three primary techniques have been employed to exfoliate bulk g-C3N4: (i) ultra-sonication-assisted exfoliation, (ii) thermal exfoliation, and (iii) chemical exfoliation. In the ultra-sonication-assisted exfoliation method, the ultrasonic wave drives the delamination process of the stacked layers in bulk g-C3N4. Generally, water and alcohols are chosen as the solvent medium during the ultra-sonication-assisted exfoliation method.107–110 Kumar and co-workers reported a mp-g-CN photocatalyst using ultra-sonication-assisted exfoliation of as-synthesized bulk g-C3N4.111 Bulk g-C3N4 was exfoliated by the probe sonication method in a water–ethanol mixture to obtain mp-g-CN with an interconnected porous nanostructure. mp-g-CN exhibited a type IV isotherm and a H2 hysteresis loop as evidenced by the N2 sorption analysis (Fig. 15A). This suggested that the exfoliation process significantly improved the textural properties of mp-g-CN compared to bulk g-C3N4 (Fig. 15A).
 | ||
| Fig. 15 (A) BET isotherm and the corresponding textural properties of mesoporous mp-g-CN and bulk g-C3N4. Reproduced with permission.111 Copyright 2014, The Royal Society of Chemistry. (B) (a) Schematic representation of the formation of CNNs, (b) PXRD, SEM and TEM images of (c) and (e) CNB, (d) and (f) CNNs respectively, (g) BET isotherms and pore size distribution (inset) of CNB and CNNs. Reproduced with permission.112 Copyright 2023, American Chemical Society (for interpretation of the references to color in this figure legend, the reader is referred to the web version of this article). | ||
In the thermal exfoliation method, post-heat treatment of synthesized bulk g-C3N4 induces layer separation and creates porosity. For example, thin-layer porous g-C3N4 nanosheets (CNNs) were reported using the thermal exfoliation strategy (Fig. 15B-a).112 After the thermal exfoliation process, the volume of the material increased, and the color of CNNs turned pale yellow, whereas bulk g-C3N4 (CNB) appeared dark yellow (Fig. 15B-a). The intensity of the PXRD patterns at 2θ values of 13.1° and 27.6°, corresponding to the (100) and (002) planes of CNB, respectively, decreased after the exfoliation process (Fig. 15B-b), indicating a successful reduction in the stacked layers. Moreover, the (002) plane of CNNs was slightly shifted to the left, which was attributed to a reduction of the interlayer distance, resulting in a more densely packed g-C3N4 structure after thermal exfoliation. The TEM and SEM investigation suggested that the bulk agglomerated structure of CNB changed to porous nanosheets after the thermal exfoliation process (Fig. 15B-(c–f)). The type IV isotherm and the characteristic H3 hysteresis loop suggested the creation of mesoporous channels after thermal exfoliation (Fig. 15B-g).
The chemical exfoliation process includes delamination of the stacked layered structure of bulk g-C3N4 using suitable etching agents such as acids or bases. Most notably, these etching agents also contribute to the additional functionalization of the resultant g-C3N4 structure.113,114 Liu et al. reported the preparation of mp-g-CN nanosheets (CNS) by the chemical exfoliation of bulk g-C3N4 (BCN).115 SEM and TEM analysis suggested that after the exfoliation and delamination of BCN in the presence of sulphuric acid, the stacked layers were modified to thin layer nanosheets (Fig. 16A-(a–d)). The type IV isotherm with an H3 hysteresis loop confirmed the presence of mesoporous channels in the synthesized materials (Fig. 16A-f). The textural properties were enhanced, with the surface area and the mesoporous volume of CNS being 55.4 m2 g−1 and 0.216 cm3 g−1, respectively, while BCN exhibited a shallow surface area (only 7.61 m2 g−1) and a poor mesoporous volume (0.055 cm3 g−1), signifying the improvement of textural properties after the chemical exfoliation process (Fig. 16A-(e and f)).
 | ||
| Fig. 16 (A) SEM and TEM images of (a) and (c) BCN and (b) and (d) CNS, and BET isotherm and pore size distribution (inset) of (e) BCN and (f) CNS. Reproduced with permission.115 Copyright 2021, Elsevier. (B) (a) Synthesis scheme of 10KCN, (b) and (c) TEM images of 10 KCN, (d) BET isotherms, (e) surface area and pore volume of all the synthesized materials. Reproduced with permission.116 Copyright 2020, American Chemical Society. (C) (a) Schematic illustration of the synthesis of Sg-C3N4/Bi2S3, (b) and (c) SEM of Sg-C3N4 and Sg-C3N4/Bi2S3 respectively, (d) TEM of Sg-C3N4/Bi2S3. Reproduced with permission.117 Copyright 2021, Elsevier. (D) (a) and (b) TEM images of TSCN10, (c) N2 adsorption–desorption isotherm and pore size distribution of TS, CN and TSCN10. Reproduced with permission.118 Copyright 2017, Elsevier. | ||
The presence of additives such as alkali or alkaline earth metal hydroxide during g-C3N4 synthesis can induce mesoporosity and layer separation in the resultant structure. In this process, the additives etch the g-C3N4 framework during the course of calcination to form abundant pores and structural defects.116,119 For example, a defect-modified thin layer porous g-C3N4 (10KCN) was prepared via KOH-assisted thermal polymerization of urea and melamine (Fig. 16B-a).116 During the calcination, KOH enhanced the thermal oxidation process, accelerating the release of CO2, H2O, and NO2 to induce mesopores, decrease layer thickness and introduce defects in the resultant structure. The TEM images revealed the ultra-thin sheets with distinct porous pockets in the 10KCN structure (Fig. 16B-(b and c)). This material exhibited a large surface area of 199.4 m2 g−1 with a pore volume of 1.3 cm3 g−1 compared to all the synthesized materials, suggesting the facile mass transfer phenomena (Fig. 16B-(d and e)).
On the other hand, when synthesizing mp-g-CN-based composites by hybridizing two or more semiconductor materials, mesoporosity with a large surface area can also be induced without the addition of any extra templates. These employed semiconductors other than g-C3N4 can trigger the creation of mesoporous channels in g-C3N4 without the need for any external templates or pore-forming agents during the synthesis process.117,118 In this context, a sulfur-doped porous g-C3N4/Bi2S3 nanocomposite (Sg-C3N4/Bi2S3) was fabricated using a facile one-step template-free approach.117 Fig. 16C-a provides the schematic of the synthesis procedure of Sg-C3N4/Bi2S3. Bi(NO3)3·5H2O and thiourea were used as precursors, and they gradually decomposed and reacted to form Bi2S3. Thiourea acted as a precursor for Sg-C3N4 and as a sulfur source for in situ formation of Bi2S3. Compared to the closed aggregated sheet-like structure of pristine Sg-C3N4, Sg-C3N4/Bi2S3 exhibited a more hierarchical porous morphology as confirmed by the FE-SEM and HR-TEM analysis (Fig. 16C-(b-d)). In another report, a novel mesoporous titanosilicate/g-C3N4 nanocomposite (TSCN10) was fabricated.118 The TEM analysis suggested that titanosilicate (TS) was grown on g-C3N4 (CN) in the nanostructure of TSCN10 (Fig. 16D-(a and b)). N2 adsorption–desorption analysis indicated that TSCN10 exhibited a type IV isotherm with better textural properties, owing to the pronounced mesoporous channel compared to CN due to the presence of TS (Fig. 16D-c).
This section provides an overview of the synthesis of mp-g-CN photocatalysts using template-assisted (hard/soft) and template-free strategies. Table 1 summarizes a list of mp-g-CN-based photocatalysts synthesized using a wide range of precursors and templates, which led to superior textural properties. Various hard templates such as silica spheres, SBA-15, and MCM-41 can utilize their rigid inorganic frameworks to create ordered mesoporous channels in mp-g-CN. The mp-g-CN precursors are first infiltrated into the template's voids via the nanocasting approach. The subsequent calcination and template removal yields precise mesoporous structures with high surface area. However, the complex reaction steps of hard template synthesis, hazardous etching agents, and scalability issues are the key challenges of the hard template assisted mp-g-CN synthesis. In contrast, the soft templates such as surfactants, CTAB, and P123 rely on organic–organic assembly with mp-g-CN precursors, which undergo micelle formation through LCT via the EISA approach to generate mesoporosity. These templates decompose during the thermal polymerization process, leaving behind mesopores, and offer advantages like simplified removal, heteroatom doping, and reduced synthesis time of mp-g-CN. Despite this, the mesopore ordering is comparatively less controlled compared to hard template methods. Further, the ionic liquid and gas bubble soft templates are currently gaining huge attention in the synthesis of mp-g-CN. Ionic liquid-assisted synthesis involves the interaction with the CN precursors, forming a supramolecular assembly, which upon thermal annealing produces mp-g-CN. In some instances, the ionic liquid-assisted synthesis is also responsible for heteroatom doping in mp-g-CN. Several gas-forming agents, such as ammonium halide, glucose, sodium bicarbonate, etc., can directly mix with the CN precursors to form mp-g-CN. During the thermal annealing process, the gas-forming agents break down to gas bubbles, which act as a template to create a porous channel in mp-g-CN. Presently, template-free synthesis methods have emerged as a trending approach for fabricating mp-g-CN-based photocatalysts, which can avoid exogenous templates by leveraging supramolecular precursor self-assembly (e.g., hydrogen-bonded aggregates) or top-down exfoliation techniques (ultrasonication, thermal/chemical etching) to delaminate bulk g-C3N4 into ultrathin, porous layers. The template-free approaches prioritize sustainability and scalability, and avoid reliance on hazardous chemicals but often result in lack of structural precision in mp-g-CN. Although template-assisted strategies yield well-ordered porous and high surface area mp-g-CN, their reliance on hazardous chemicals and multi-step processes has spurred interest in template-free alternatives in recent times. Overall, the choice of the synthesis route dictates the balance between pore ordering, scalability, and microenvironmental impact on optimizing the optoelectronic features and photocatalytic efficiency.
| Photocatalyst | Precursor | Type of template | BET surface area (m2 g−1) | Pore volume (cm3 g−1) | Ref. |
|---|---|---|---|---|---|
| MCN-1 | Carbon tetrachloride, ethylenediamine | Hard template (SBA-15) | 505 | 0.55 | 76 |
| mpg-C3N4/1.6 | Cyanamide | Hard template (Ludox HS40) | 439 | — | 78 |
| ompg-C3N4 | Cyanamide | Hard template (SBA-15) | 86.2 | 0.172 | 79 |
| g-C3N4 | Melamine | Soft template (P123) | 90 | — | 85 |
| CN-2% | Dicyandiamide | Soft template (P123) | 100 | 0.46 | 87 |
| p-SCN-2 | Thiourea | Gaseous bubble template (NH4Cl) | 21.8 | 0.11 | 93 |
| PHCN3 | Melamine, urea | Template-free (supramolecular self-assembly) | 79.29 | 0.28 | 98 |
| CM-K12 | Melamine, cyanuric acid, potassium hydrogen phthalate | Template-free (supramolecular self-assembly) | 127 | 0.88 | 102 |
| PCNNVs | Melamine, cyanuric acid | Template-free (supramolecular self-assembly) | 150.74 | — | 104 |
| FeNx/CCN | Melamine, cyanuric acid, Fe-imidazole | Template-free (supramolecular self-assembly) | 27.3 | 0.186 | 106 |
| CNNs | Melamine | Template-free (thermal exfoliation) | 212 | 1.07 | 112 |
| Meso CN spheres | Carbon tetrachloride, ethylenediamine | Cellular silica foams | ∼550 | 0.90 | 120 |
| MCN-4-130 | Amino guanidine | Hard template (SBA-15) | 321 | 0.44 | 121 |
| MCN-6-150 | Carbon tetrachloride, ethylenediamine | Hard template (KIT-6) | 637 | 0.9 | 122 |
| MCN-ATN | 3-Amino-1,2,4-triazine | Hard template (KIT-6) | 472–635 | 0.71–0.99 | 123 |
| meso-CN | Cyanamide | Hard template (KIT-6) | 171 | 0.84 | 124 |
| M3AT | 3-Amino-1H-1,2,4-triazole | Hard template (SBA-15) | 382 | 0.789 | 125 |
| mpg-CN–0.3C60 | Melamine, fullerene C60 | Hard template (Ludox HS30) | ∼200 | 0.367 | 126 |
| MCN-TMA | 3-Amino 1,2,4 triazole, trimesic acid | Hard template (Ludox HS40) | 69 | 0.19 | 127 |
| CNBF-0.5 | Dicyandiamide | Soft template | 444 | 0.32 | 128 |
| Ionic liquid (BmimBF4) | |||||
| CN-T-800 | Melamine, glutaraldehyde | Soft template (Triton X-100) | 221 | 0.21 | 129 |
| CN-P123-6 | Melamine | Soft template (P123) | 73.29 | 0.27 | 130 |
| Zn-mpg-C3N4 | Melamine, ZnCl2·2H2O | Soft template (PEG 1500) | 9.8 | 0.029 | 131 |
| g-C3N4-ALS 2% | Melamine | Gaseous bubble template (ammonium lauryl sulfate (ALS)) | 40 | 0.21 | 132 |
| HP-CN | Melamine | Template-free (chemical exfoliation) | 108.3 | 0.74 | 133 |
| PCNS | Melamine | Template-free (thermal exfoliation) | 190.1 | 0.61 | 134 |
| PDCN | Melamine, urea | Template-free (supramolecular self-assembly) | 34.5 | 0.19 | 135 |
| HACN | Cyanuric acid, melem powder | Template-free (supramolecular self-assembly) | 54.3 | — | 136 |
3. Structural modification of the mp-g-CN-based nanomaterial
Structural diversification arises in the synthesized mp-g-CN nanomaterials when a specific synthesis methodology (either the template-assisted or template-free method) is adapted. The appropriate synthesis route in the production of mp-g-CN-based nanomaterials, as discussed in the above section, not only improves the textural properties but also provides several structural modifications compared to pristine g-C3N4. The common structural modifications observed are morphological modification, caused by both template-assisted and template-free synthesis approaches, and functional modification, due to doping and defect engineering followed by vacancy creation, which leads to deviation in the lattice arrangements from the pristine structure (Fig. 17). Moreover, functional modifications can also be experienced due to the development of mp-g-CN hybrid nanocomposites with appropriate semiconductors, co-catalysts or sensitizers. These structural modifications bring irregularities in the nanostructure to improve the electronic properties and have been proven to be beneficial in photocatalytic activity.63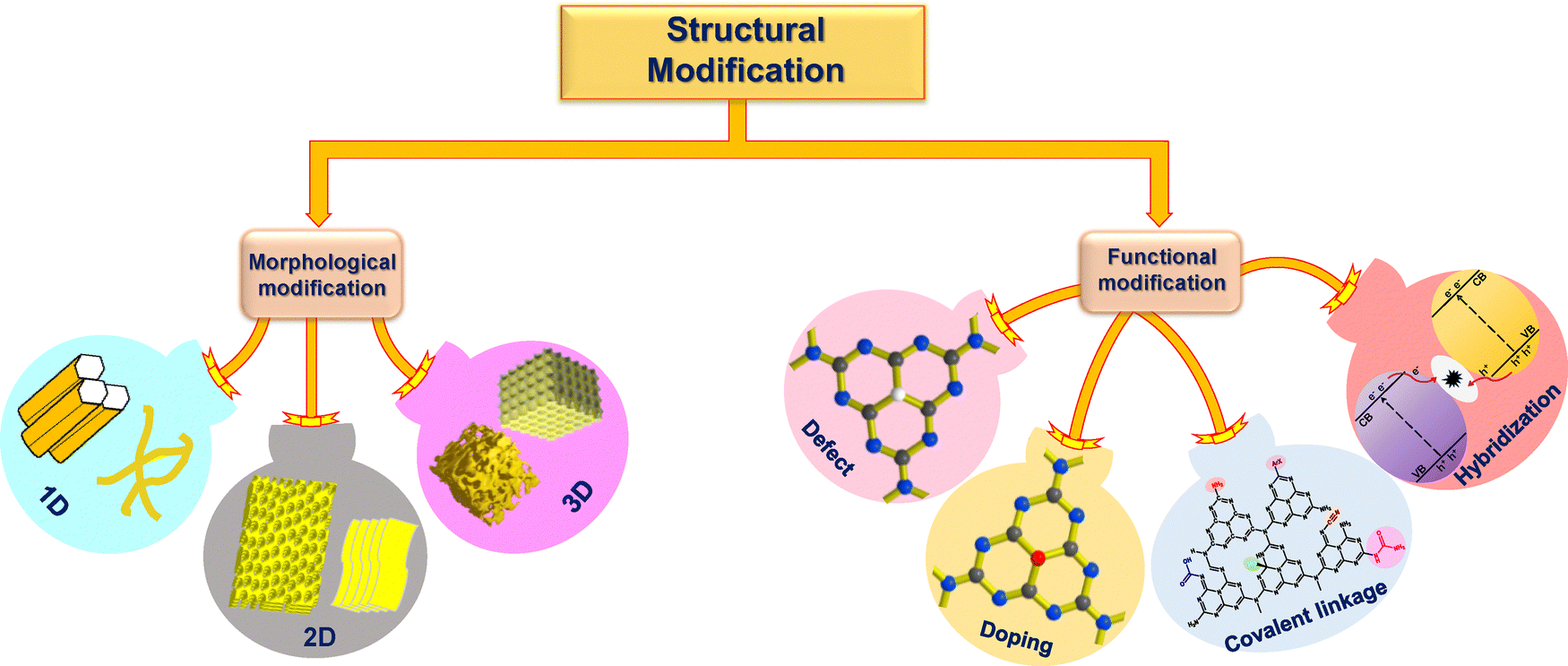 | ||
| Fig. 17 Schematic illustration of structural modifications in mp-g-CN. | ||
3.1. Morphological modifications
During the synthesis of mesoporous g-C3N4 nanostructures using the aforementioned synthesis strategies, it is obvious that the resultant mp-g-CN exhibits pronounced morphological modifications in comparison to the pristine g-C3N4 structure. Moreover, the morphological modulation is also responsible for the superior physiochemical properties, thus endorsing the photocatalytic activity. The morphological modulation also controls the structure–activity relationship in a larger context by altering the photogenerated charge pair separation and migration. The morphological modulation results in different dimensions, which essentially depend on the synthesis procedures. Based on dimensional classification, the g-C3N4 nanomaterial can exhibit (i) 0 D, (ii) 1D, (iii) 2D, and (iv) 3D structures with variable morphologies. The possible morphologies for g-C3N4 materials are quantum dots for 0D, nanorods/nanotubes for 1D, nanosheets for 2D, and nanocubes/nanospheres, etc. for 3D (Fig. 17).110,137 Generally, g-C3N4 materials widely attain 2D nanosheet morphology under regular synthesis strategies. However, altering the synthesis protocols and precursors can lead to morphologies other than the usual nanosheet morphology. It is also noted that the production of mp-g-CN nanomaterials completely depends on the appropriate synthesis conditions as discussed in the above section; hence, it is obvious that the resultant mp-g-CN materials would exhibit various morphologies, which are purely controlled by the choice of synthesis conditions. Notably, 0D g-C3N4 quantum dots do not possess pores; hence, mp-g-CN can only have 1D, 2D, and 3D electronic structures with variable controlled morphologies. The observed morphologies obtained for the mp-g-CN materials are discussed below.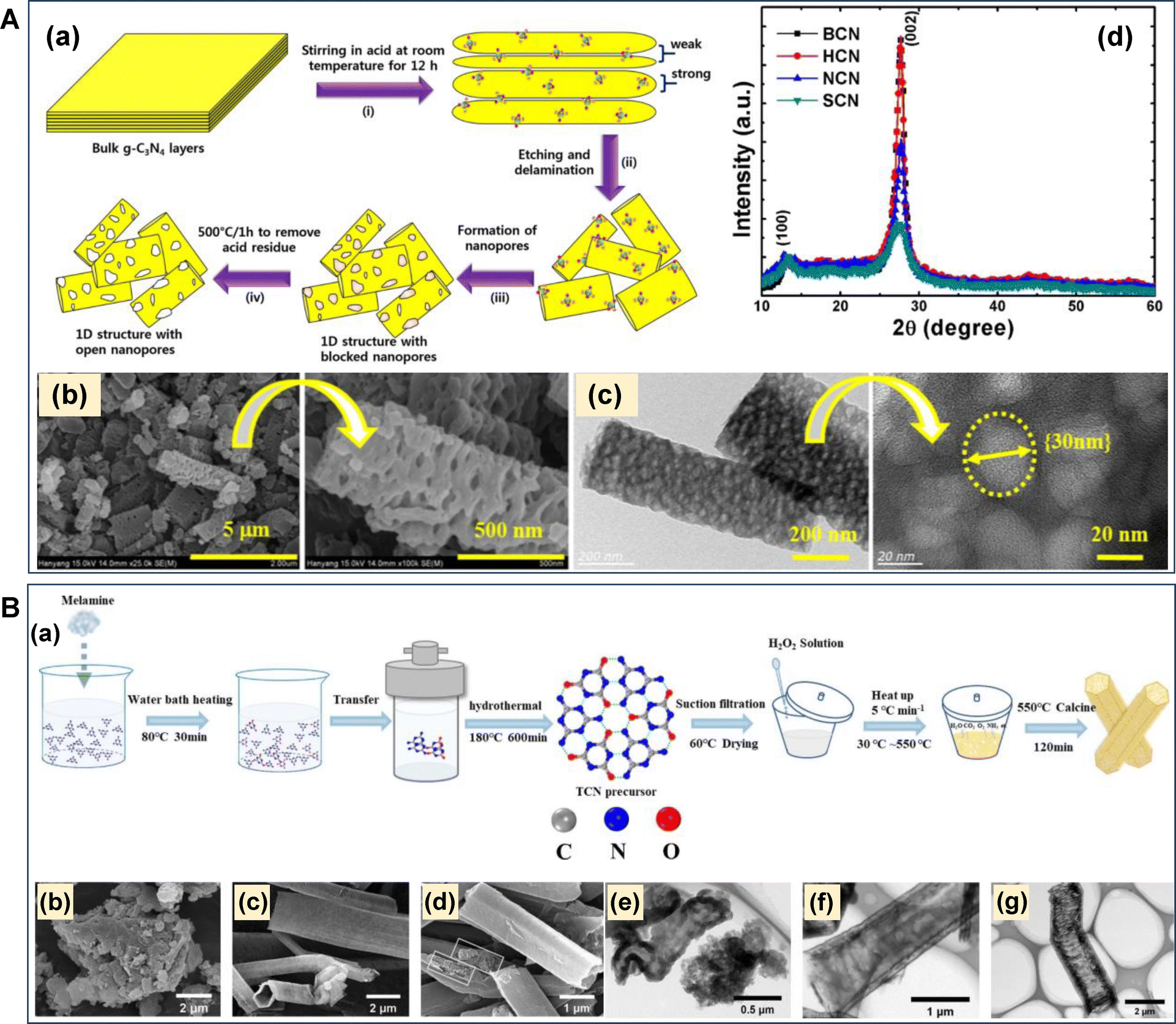 | ||
| Fig. 18 (A) (a) Schematic depiction of the formation of 1D mp-g-CN nanorods via the top-down approach, (b) and (c) SEM and TEM images of SCN, (d) PXRD of all the synthesized materials. Reproduced with permission.139 Copyright 2016, Nature Communication. (B) (a) Scheme for the step-wise synthesis of AN-TCN, SEM and TEM images of (b) and (e) CN, (c) and (f) TCN and (d) and (g) AN-TCN, respectively. Reproduced with permission.140 Copyright 2022, Elsevier. | ||
 | ||
| Fig. 19 (A) (a) Representation of the formation route for mp-g-CN, SEM image of (b) CN-0, (c) CN-1, (d) CN-1.5, TEM image of (e) CN-0, (f) CN-1, (g) CN-1.5, (h) CN-2, (i) N2 adsorption–desorption isotherms, (j) and (k) pore size distribution plot of CN-0 and CN-1.5. Reproduced with permission.142 Copyright 2024, American Chemical Society. (B) (a) Schematic representation of the formation of PCNS-F, TEM and SEM images of (b) and (c) BCN, (d) and (e) PCN-F, (f) and (g) PCNS-F, (h) N2 adsorption–desorption isotherms and pore size distribution plot (inset) of all the synthesized materials (here PCN-T and PCNS-T represent the samples that were dried under temperature instead of freeze drying). Reproduced with permission.143 Copyright 2024, Elsevier. | ||
Pang and co-workers reported a facile and greener approach for the strategic synthesis of two-dimensional ultrathin porous polymer carbon nitride nanosheets (PCNS-F).143 Initially, bulk g-C3N4 (BCN) was synthesized through the thermal condensation of melamine, followed by solvothermal treatment and freeze-drying to obtain PCN-F as an intermediate (Fig. 19B-a). PCN-F was further subjected to a thermal etching process to produce PCNS-F having ultrathin and porous nanosheet surface morphology, in contrast to the bulk and stalked structure of BCN (Fig. 19B-(b–g)). Moreover, these series of synthesis steps played a crucial role in achieving the enhanced physicochemical properties and several other structural characteristics. The porosity and BET surface area were significantly improved in this method, suggesting the prominent ability of this synthesis approach to obtain a highly porous g-CN framework without using any corrosive chemicals or tedious synthesis routes (Fig. 19B-h).
 | ||
| Fig. 20 (A) (a) Diagrammatic sketch of the formation of BMNS, (b) SEM, (c) internal SEM and (d) TEM image of BMNS. Reproduced with permission.145 Copyright 2024, Elsevier. (B) Schematic illustration of the fabrication of (a) the g-C3N4-HPC composite, (b) blank g-C3N4, (c) N2 adsorption–desorption isotherms, (d) pore size distribution plot of blank g-C3N4, g-C3N4-HPC-600, g-C3N4-HPC-700, g-C3N4-HPC-800 and HPC-700, (e) and (f) SEM images of blank g-C3N4 and g-C3N4-HPC-700, (g) and (h) TEM images of blank g-C3N4 and g-C3N4-HPC-700. Reproduced with permission.146 Copyright 2022, American Chemical Society. | ||
Zhao et al. demonstrated that modifying the g-C3N4 precursor can lead to a 3D porous structure.146 Sodium alginate and melamine were employed to synthesize melamine-coated CA hydrogel beads, the real precursor for developing the 3D g-C3N4-hierarchical porous carbon composite (g-C3N4-HPC) (Fig. 20B-(a and b)). The structural features of the g-C3N4-HPC materials largely depend on the calcination temperature, which varies from 600 °C to 800 °C. Among g-C3N4-HPC-600, g-C3N4-HPC-700 and g-C3N4-HPC-800, g-C3N4-HPC-700 exhibited an exceptionally large surface area of 1435.8 m2 g−1 and a very large pore volume of 1.744 cm3 g−1 (Fig. 20B-(c and d)). Morphological investigations suggested that the neat g-C3N4 (blank g-C3N4) has a curled and agglomerated sheet structure, whereas g-C3N4-HPC-700 has a sponge-type structure decorated with numerous interconnected porous channels (Fig. 20B-(e–h)).
3.2. Functional modifications
Mesoporosity creation and various morphological modifications can shorten the diffusion length of photogenerated charge carriers, offer diffusional channels for the adsorption of reactants and desorption of products, and also provide surface-active sites for chemical reactions. However, it is necessary to modulate the band structure to maintain the indispensable overpotential necessary to target a specific and challenging chemical reaction. Functional modifications are regarded as promising strategies for improving photocatalytic efficiency by fine-tuning the band structure in mp-g-CN-based photocatalysts. The functional modifications tend to impact the physicochemical and photoelectronic properties in mp-g-CN-based photocatalysts.147 These modifications can be achieved by changing or altering the atomic contents, or functional groups or moieties attached to the framework of mp-g-CN structures. The most common strategies adopted to achieve the functional modification in mp-g-CN structures are (i) defect engineering, (ii) doping, (iii) anchoring or grafting of functional moieties through covalent linkage and (iv) hybridization with suitable nanostructure materials (Fig. 17).For example, He et al. reported a nitrogen-defective porous g-C3N4 (PDCN) by following the urea mediated supramolecular assembly approach using melamine in 1 M HCl solution.135 Fig. 21a represents the schematic illustration of the formation of PDCN where the variation of the precursor has a significant role in defect creation. They also reported a few other photocatalysts, namely, g-C3N4 (bulk g-C3N4, synthesized using melamine only), PCN (synthesized without adding HCl) and DCN (synthesized without adding urea) for the comparative study. During the hydrothermal treatment, melamine reacts with HCl. Consequently, some part of melamine is converted to cyanuric acid and NH3, and the resultant cyanuric acid forms an H-bond with the remaining melamine to form a supramolecular assembly. Moreover, the presence of urea acted as a pore-forming agent or porogen due to its decomposition to NH3 and CO2 during heat treatment. The nanostructure morphology investigation using TEM analysis suggested that g-C3N4 exhibited an aggregated structure (Fig. 21b) and DCN exhibited a rod-like structure due to self-assembly of the supramolecular intermediate formed between melamine and HCl during hydrothermal treatment (Fig. 21c). An aggregated porous fragmented structure was achieved for PCN due to urea acting as the porous template during heat treatment (Fig. 21d). However, the porous architecture formed in PCN was found to exhibit increased aggregation due to the high surface energy of the constituent fragmented particles. In contrast, PDCN exhibited a looser porous structure due to the presence of HCl and urea, which consequently created defects in the framework and resulted in minimum surface energy of the fragmented particles; hence, PDCN exhibited a less aggregated structure compared to PCN (Fig. 21e). The PXRD pattern of all the synthesized materials exhibited a less intense peak compared to that of g-C3N4, suggesting that the controlled synthesis strategy resulted in several structural changes and loss of long-range order crystal structure of the bulk phase (Fig. 21f). Furthermore, the nitrogen defect was estimated using solid-state 13C NMR analysis. All the samples displayed two types of characteristic peaks at 155.6 and 164.3 ppm corresponding to the terminal CN2(NHx) and CN3 (Fig. 21g). Also, the sideband peaks appeared at 7 and 27 ppm. It was observed that all the peaks corresponding to PDCN were broadened and weakened, which was due to the defect generation by the loss of the long-range order crystal structure of the bulk g-C3N4 phase. All the structurally modified synthesized materials exhibited a type III isotherm and an H3 hysteresis loop because of the presence of mesoporous architecture in the framework (Fig. 21h). The XPS analysis suggested that the area ratio of the ‘N’ peak corresponding to C–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C/N–(C)3 peaks for PDCN was less compared to g-C3N4, which signified the existence of ‘N’ vacancies in the nanostructure of PDCN. Moreover, the XPS and organic element analysis technique estimated a lower N/C mass ratio for PDCN compared to g-C3N4, further confirming nitrogen vacancies in PDCN. These defective and porous architectures also provide superior light absorption properties and dynamic change in the band position compared to g-C3N4. The control annealing strategy was also responsible for the formation of the ‘N’ vacancy, followed by the elimination of other nitrogen-containing moieties, such as cyanamide and amino groups, in the mp-g-CN structure.
C/N–(C)3 peaks for PDCN was less compared to g-C3N4, which signified the existence of ‘N’ vacancies in the nanostructure of PDCN. Moreover, the XPS and organic element analysis technique estimated a lower N/C mass ratio for PDCN compared to g-C3N4, further confirming nitrogen vacancies in PDCN. These defective and porous architectures also provide superior light absorption properties and dynamic change in the band position compared to g-C3N4. The control annealing strategy was also responsible for the formation of the ‘N’ vacancy, followed by the elimination of other nitrogen-containing moieties, such as cyanamide and amino groups, in the mp-g-CN structure.
 | ||
| Fig. 21 (a) Fabrication process of PDNC, TEM images of (b) g-C3N4, (c) DCN, (d) PCN, and (e) PDCN, (f) XRD, (g) solid-state 13C NMR, (h) BET isotherms. Reproduced with permission.135 Copyright 2022, American Chemical Society. | ||
Guo et al. reported an ‘N’ vacancy and cyanamide defective mp-g-CN nanostructure (NCMCN) using a multi-step annealing strategy with the co-assistance of sodium hydroxide (Fig. 22A-a).151 Bulk g-C3N4 (MCN) was synthesized by calcining melamine at 550 °C. The resultant MCN upon the controlled annealing process (at 650 °C under a N2 atmosphere) led to the loss of the terminal amino moiety in the form of NH3, creating ‘N’ vacancy modified mp-g-CN (NVMCN). Heating NVMCN with sodium hydroxide resulted in ‘N’ vacancy and cyanamide defective mp-g-CN (NCMCN). The resultant NVMCN and NCMCN exhibited distinct indications of defect creation in PXRD analysis. PXRD analysis of NVMCN and NCMCN revealed distinct signs of defect creation, as evidenced by the weaker and wider (100) and (002) planes compared to MCN, indicating a reduction in the in-plane repeating units and interplanar stacking (Fig. 22A-b). The defect creation in NVMCN and NCMCN was also revealed by FT-IR analysis, with decreasing peak intensities for –NH moieties, suggesting the existence of the ‘N’ vacancy (Fig. 22A-c). Unlike MCN and NVMCN, NCMCN exhibited the stretching vibration of cyanide, highlighting the cyanamide defective structure in NCMCN. The XPS analysis calculated the ratio of ‘C’ or ‘N’ bonded moieties to total ‘C’ or ‘N’ atoms of the samples to justify the existence of the ‘N’ vacancy in NVMCN and both ‘N’ vacancy and cyanamide defect in NVMCN respectively. Upon heat treatment, the lumpy and aggregated structure of MCN was transformed into a porous and fluffy structure in NVMCN which on further heating with sodium hydroxide resulted in highly porous NCMCN. The successive removal of NHx on multistep heat treatment enhanced porosity, as evidenced by the N2-sorption analysis (Fig. 22A-d). The ‘N’ vacancy and cyanamide defect caused a red shift in visible-light absorption and lowered the band gap (Fig. 22A-e), which caused an increase in visible-light response ability and modified the band structure. The significant reduction in PL intensity for NVMCN and NCMCN suggested that photogenerated charge recombination was markedly inhibited due to vacancy creation.
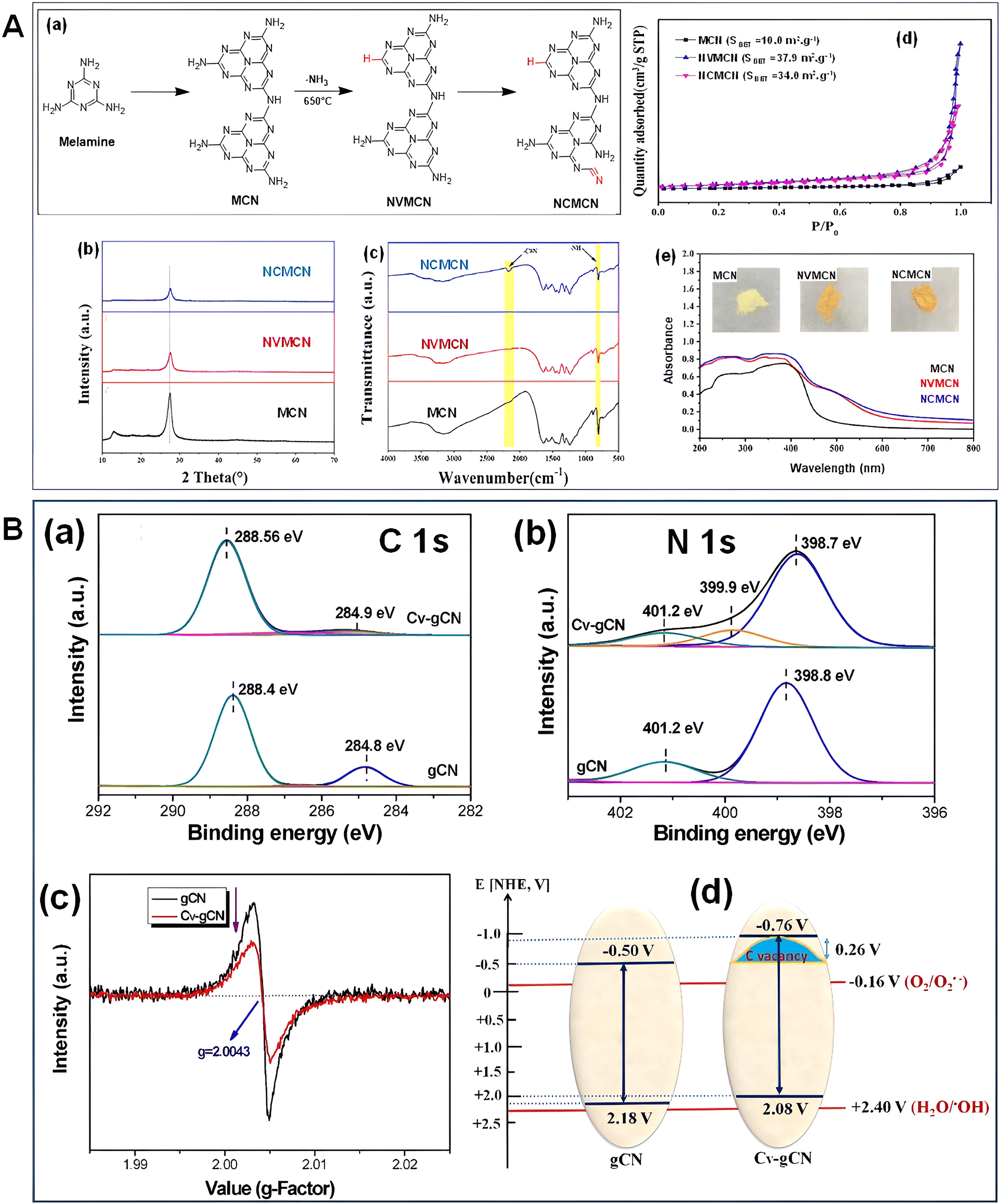 | ||
| Fig. 22 (A) (a) Illustration of the formation mechanism of MCN, NVMCN and NCMCN, (b) XRD, (c) FT-IR, (d) N2 adsorption–desorption isotherms, (e) UV-vis DRS of MCN, NVMCN, and NCMCN. Reproduced with permission.151 Copyright 2024, Elsevier (for interpretation of the references to color in this figure legend, the reader is referred to the web version of this article). (B) (a, b) XPS spectra of C1s, N1s, (c) EPR spectra, (d) band structures of gCN and Cv-gCN. Reproduced with permission.152 Copyright 2018, Elsevier. | ||
Similar to the ‘N’ vacancy, the ‘C’ vacancy also brings several advantageous properties to the mp-g-CN structure. The ‘C’ vacancy in the mp-g-CN structure can be integrated by applying the reducing atmosphere or CO2 atmosphere during the heat treatment. Li et al. reported the synthesis of carbon vacancy-rich mp-g-CN (Cv-gCN) by heating pristine g-C3N4 (gCN) with sodium bicarbonate, which provides an in situ CO2 atmosphere.152 The resultant Cv-gCN possessed a porous and loose layer structure with carbon vacancy. Therefore, Cv-gCN exhibited a very high surface area and pore volume compared to gCN. The PXRD patterns for Cv-gCN showed much lower intensity peaks with a swift shift towards higher 2θ compared to gCN, suggesting a decrement in the crystallinity due to the ‘C’ vacancy and reduction of inter-layer packing of repeat units. The XPS and EPR analysis further confirmed the existence of the ‘C’ vacancy in Cv-gCN. The lower ‘C’ to ‘N’ ratio for Cv-gCN compared to gCN suggested the existence of the ‘C’ vacancy in Cv-gCN. Moreover, the C1s peaks shifted to higher binding energy, suggesting that the remaining electrons were redistributed after the creation of carbon vacancies and hence experienced a change in the local electronic structure. The N1s species in Cv-gCN also experienced an extra peak corresponding to the amino groups because of the ‘C’ vacancy (Fig. 22B-(a and b)). The weaker EPR signal for Cv-gCN compared to gCN corroborated with the lesser number of paramagnetic carbon atoms containing unpaired electrons in the aromatic ring, further evidenced by the ‘C’ vacancy induced in Cv-gCN (Fig. 22B-c). Cv-gCN exhibited a blueshift in DRS absorption with a more negative CB position compared to gCN due to the change in the electronic structure after the creation of the ‘C’ vacancy (Fig. 22B-d).
Zhang et al. reported a templated and doping approach to fabricate tunnel-shaped phosphorus doped mp-g-CN (P-SBA-15-CN) which was synthesized by adopting SBA-15 to induce mesoporosity and phosphonitrilic chloride trimer as the phosphorus source (Fig. 23A-a).154 SBA-15 controlled the morphology, porosity, and surface area, thereby generating more active sites and enhancing the efficiency of the resulting materials. The EDX mapping obtained using HR-TEM analysis directly evidenced the phosphorus doping (Fig. 23A-(b–g)). The XPS analysis confirmed the pre-existence of the ‘N’ vacancies in the undoped mp-g-CN material, further facilitating the accommodation of phosphorus doping. The resultant phosphorus doping led to a broad and wide range of visible light absorption and also improved the separation and transportation of the photogenerated charge carriers (Fig. 23A-(h and i)).
 | ||
| Fig. 23 (A) (a) Illustrative depiction of P-SBA-15-CN synthesis, (b)–(g) TEM-EDS mapping of P-SBA-15-CN, (h) and (i) UV-Vis and band gap energy of synthesized samples. Reproduced with permission.154 Copyright 2023, Elsevier. (B) (a) Fabrication process of ultrathin 3D mp-g-CN, (b) organic elemental analysis of bulk g-C3N4 and UM3, (c) N2 adsorption–desorption analysis of all synthesized materials, (d) solid-state 13C MAS NMR of all the synthesized samples, (e) calculated structure of possible N-doping sites, (f) UV-Vis DRS, (g) band gap energies, (h) Mott–Schottky plots, (i) VB-XPS analysis, and (j) diagrammatic representation of the electronic band structure of the prepared samples; here, UMx represents the synthesized 3D mesoporous N-doped g-C3N4, where x = 0, 1, 3, 6 corresponds to the molar ratio of urea to melamine. Reproduced with permission.155 Copyright 2017, Elsevier. | ||
A 3D mesoporous nitrogen self-doped g-C3N4 material was synthesized using a precursor-recrystallization strategy reported by Tian and co-workers (Fig. 23B-a).155 Melamine underwent a pre-molecular reorganization to experience a phase transformation during the hydrothermal treatment before the calcination process, and urea served as both the porogen agent and ‘N’ precursor, resulting in the formation of ‘N’ self-doped ultrathin 3D mp-g-CN (UM3). ‘N’ doping was estimated using the organic elemental analysis by considering the N/C ratio and further confirmed by PXRD, FT-IR, and XPS analyses, which indicated that the interference of urea in the synthesis medium facilitated nitrogen doping in UM3 (Fig. 23B-b). The presence of mesopores in all the synthesized UMx samples was confirmed by the type IV BET isotherm and pore size distribution (Fig. 23B-c). 13C MAS NMR analysis and DFT calculations identified the possible doping site for ‘N’ in the heptazine unit (Fig. 23B-(d and e)). ‘N’ doping altered the local electronic structure and promoted better charge separation, which was also responsible for narrowing the band gap to utilize the maximum solar light absorption for generating the photo-excited charge carriers (Fig. 23B-(f–j)). In metal-doped mp-g-CN materials, the g-C3N4 precursors showed a high degree of polymerization due to the presence of metal at elevated temperatures; this led to higher crystallinity in the framework. Also, metal doping modifies the electronic structure and absorption edges, therefore leading to long-range visible light absorption.156,157
 | ||
| Fig. 24 Types of covalent linkages in mp-g-CN. | ||
The functional modification of mp-g-CN due to the grafting of additional functional groups is highly dependent on the synthesis condition, specifically the controlled polymerization process, which is a crucial factor. Precise copolymerization can guide intermolecular chemical interactions, facilitating the grafting of the mp-g-CN framework. As a consequence of anchoring the organic functional group, the resultant modified structure realises an extended π-conjugated field or the presence of an electron-rich heteroatom. This modification leads to an altered band position and a modulated electronic structure, which favors efficient charge separation and migration.147,158,159 A sulphonic acid grafted ordered mp-g-CN material (ompg-C3N4/SO3H) was fabricated using the covalent interaction between chloro-sulphonic acid and ompg-C3N4 synthesized using SBA-15 as the hard template.160 The successful covalent linkage of the -SO3H group was evidenced by the FT-IR and EDS analysis.
Another example of surface covalent grafting of mp-g-CN includes the fabrication of a photocatalyst composite using microtubular-shaped kapok fibre (t-KF), a class of cellulosic material, and mesoporous C-doped g-C3N4 (CCN) in the presence of succinic acid (SA) as a cross-linking agent through covalent linkage.161 FT-IR analysis revealed that the resultant CCN-SA/t-KF was constructed by the cross-linking chemical interference of the carboxylic acid group of SA with the terminal amine group in CCN and the hydroxyl group of t-KF (Fig. 25a and b). The FESEM analysis suggested the distinct integration of microtubular t-KF and sheet-like stacked flake CCN in the CCN-SA/t-KF composite. XPS analysis further demonstrated that the tertiary bridged ‘N’ atom, connected to the tri-coordinated bridge triazine rings, was substituted by ‘C’ doping in CCN and CCN-SA/t-KF (Fig. 25c and d). This facilitated the delocalization of photoinduced electrons and significantly suppressed the charge complexation. Additionally, the ‘N’ peak shifting in CCN-SA/t-KF, compared to CCN, was attributed to the changes in the chemical environment due to the composite formation (Fig. 25c). Moreover, the positive peak shifting of the (002) diffraction plane of CCN-SA/t-KF was attributed to the change in crystal lattice strain experienced due to the cross-linking of SA and integration of t-KF into the CCN nanostructure. The composite also exhibited superior light-harvesting properties by exhibiting long-range, broad absorption edges. The enhanced light absorption ability benefited the intermolecular injection of photogenerated electrons from the high electron-dense surface hydroxyl groups of t-KF to CCN, similar to the mechanism of ligand-to-metal charge transfer (LMCT).
 | ||
| Fig. 25 (a) FT-IR spectra of all the synthesized materials, (b) cross-linking interaction for the formation of the CCN-SA/t-KF hybrid, (c) and (d) XPS spectra of N1s and C1s of g-C3N4, CCN, CCN/t-KF and CCN-SA/t-KF. Reproduced with permission.161 Copyright 2023, Elsevier. | ||
Similarly, Bian and co-workers reported the post-synthetic anchoring of methoxybenzoyl (MOB) on mp-g-CN (GCN) via the amonolysis of p-anisoyl chloride with the terminal amino group of mp-g-CN (Fig. 26a).162 The resultant MOB/GCN was fabricated through the creation of an amide bond between MOB and GCN, which was evidenced by FT-IR analysis (Fig. 26b). Moreover, the XPS analysis also revealed a decrease in electron cloud density around the amide ‘N’ species of MOB/GCN, which in turn reduced the electron-donating ability of GCN, suggesting the chemical interaction between MOB and GCN. The successful composite formation led to enhanced photogenerated charge separation by suppressing their complexation and increasing their migration. This phenomenon was confirmed by the PL and photocurrent measurement (Fig. 26f-(b and c)).
 | ||
| Fig. 26 (a) Diagrammatic representation of MOB/GCN synthesis, (b) FT-IR of MOB, MOB/GCN and GCN, (c) PL, (d) transient photocurrent response of GCN and MOB/GCN. Reproduced with permission.162 Copyright 2023, Elsevier. | ||
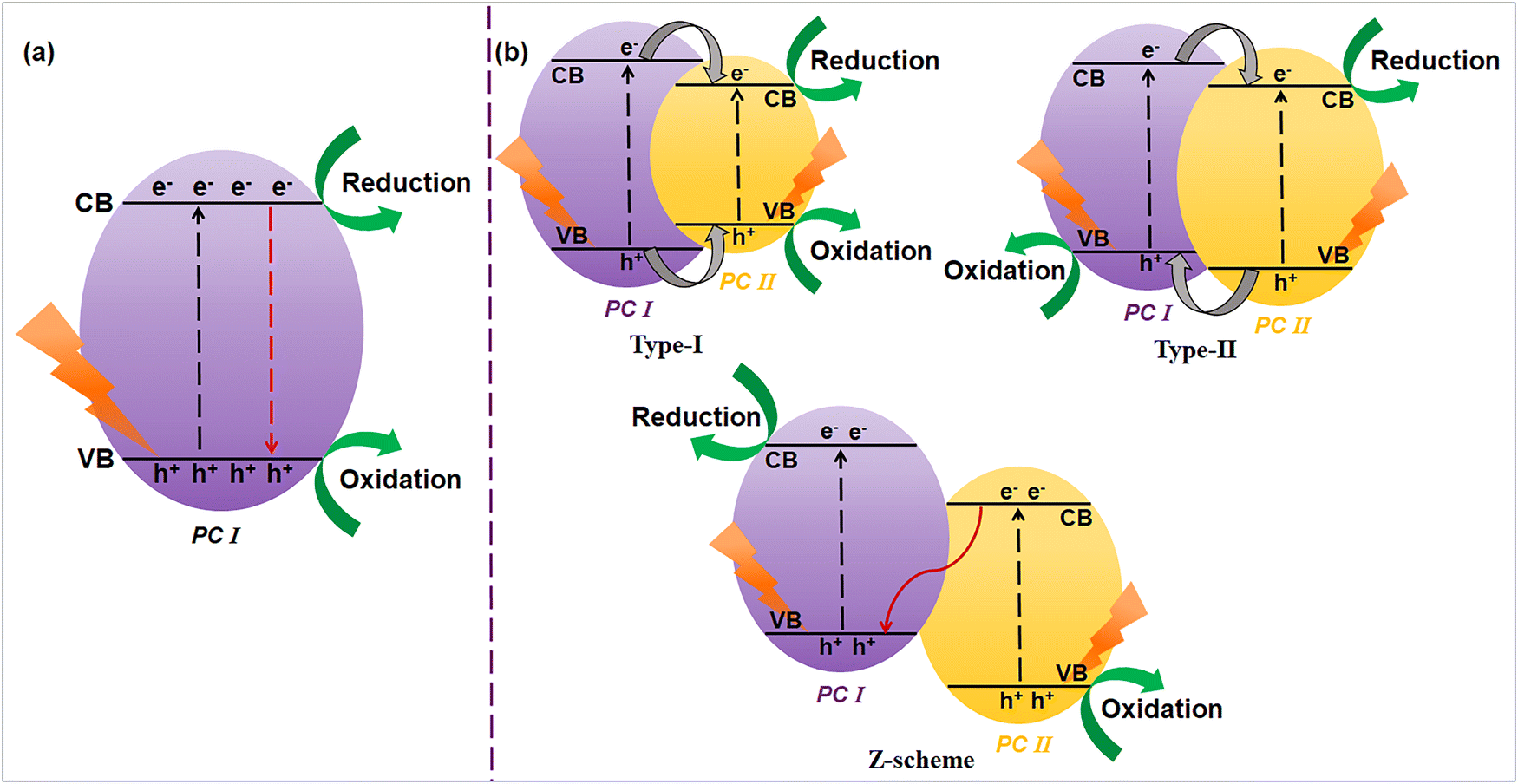 | ||
| Fig. 27 Schematic overview of (a) VB and CB levels of bare mp-g-CN (PC I), (b) Different types of mp-g-CN-based heterostructures with their respective band alignments. | ||
The hybridization strategy allows the construction of various composites of mp-g-CN-based heterojunction and Z-scheme photocatalytic materials, which experience an extrinsically modified band gap. Due to this, the actual light absorption edges induce the appropriate redox potential compared to that of bare mp-g-CN and transform the composite into a potential photoactive material. A hybrid nanocomposite of CdIn2S4/mp-g-CN photocatalytic material was fabricated by Liu et al.164 mp-g-CN was constructed using the SBA-15 hard template, and then CdIn2S4 was fabricated by the hydrothermal method. A series of mp-g-CN photocatalysts loaded with different amounts of CdIn2S4 were synthesized (CISCN10, CISCN20 and CISCN30, where 10, 20, and 30 represent the weight % of mpg-CN), and their successful incorporation was evidenced by PXRD and XPS analysis (Fig. 28a–g). XPS analysis evidenced a negative shift of C1s peaks and a positive shift of N1s peaks of the composite materials, compared to bare mpg-CN (Fig. 28c and d). The Cd3d, In3d and S2p peaks were also shifted compared to bare CdIn2S4 (Fig. 28e–g), suggesting that CdIn2S4 chemically interacted with the functional moieties of mpg-CN. Further, the intimate interfacial contact was confirmed by TEM observation (Fig. 28h and i). The light absorption edge for the composite materials showed a redshift compared to bare mpg-CN and a slight blue shift compared to bare CdIn2S4, which was due to the change in the electronic structure after the composite formation.
 | ||
| Fig. 28 (a) XRD of all the synthesized samples, (b) XPS survey spectrum of CdIn2S4, mpg-C3N4, and CISCN20, (c) C1s and (d) N1s of mpg-C3N4 and CISCN20, (e) Cd3d, (f) In3d and (g) S2p of CdIn2S4 and CISCN20, and (h) TEM and (i) HRTEM image of CISCN20. Reproduced with permission.164 Copyright 2017, Elsevier. | ||
Ouyang et al. fabricated a Z-scheme heterojunction composed of defective mp-g-CN and a MoO3 photocatalyst.165 The schematic representation of the synthesis method showed that the calcination of dicyandiamide converted it into g-C3N4, and the combined calcination of dicyandiamide and ammonium persulfate resulted in the formation of defective mp-g-CN (D-CN). The Z-scheme heterojunction was then constructed by the calcination of D-CN and different contents of MoO3, designated as D-CNMX (X = 5, 10, 15 and 20 weight% MoO3 loading). The successful embedment of the MoO3 moiety into the mesoporous channel of D-CN and the formation of the Z-scheme heterojunction were confirmed from PXRD and FT-IR analysis (Fig. 29a and b). The XPS and EPR analysis further demonstrated that the use of ammonium persulfate induced the ‘N’ defect in D-CN. The C/N ratio from XPS analysis also suggested that certain percentage of ‘N’ atoms were missed from the D-CN framework (Fig. 29d). As a result, the extra energy states appeared below the CB position, which was highly beneficial for a longer electronic excitation process. Due to the N-defect in the D-CN framework, the adjacent sp2 carbon experienced additional unpaired electrons. As a result, D-CN exhibited a stronger EPR signal compared to g-C3N4 (Fig. 29c). The schematic model of ‘N’ defect sites in the D-CN framework is illustrated in Fig. 29e. The UV-DRS, band gap energy and associated band structure analysis suggested that the resultant D-CNMX heterojunction materials exhibited a broad light absorption spectrum, enabling the generation of more photogenerated charge pairs and their decomplexation, facilitated due to the synergistic operation of ‘N’ defect and Z-scheme heterojunction formation (Fig. 29f–i). By leveraging the several beneficial aspects of mp-g-CN, such as structural functionality and mesoporous channels, mp-g-CN can additionally act as the scaffold to entangle various nanoscopic species, such as co-catalysts and sensitizers, to harness the photoresponse properties. For instance, an organic–inorganic hybrid composite of mp-g-CN (mpg-C3N4) and polymeric cobalt phthalocyanine (CoPPc) was fabricated, which exhibited excellent photoresponse properties.166 CoPPc when chemically embedded by π–π interactions with mesoporous mpg-C3N4 (CoPPc@mpg-C3N4) by in situ polymerization boosted the charge separation, migration and generation of surface-active sites compared to bare mpg-C3N4 and the physical mixture of mesoporous-gC3N4 and cobalt phthalocyanine (CoPPc/mpg-C3N4).
 | ||
| Fig. 29 (a) XRD and (b) FT-IR spectra of all synthesized samples, (c) EPR signal and (d) C/N molar ratio of g-C3N4 and D-CN, (e) schematic depiction of possible ‘N’ defect sites in D-CN, (f) UV-vis DRS of all the samples, (g) bandgap energy, (h) VB-XPS, (i) band structure of g-C3N4, D-CN and MoO3. Reproduced with permission.165 Copyright 2023, Elsevier. | ||
The superior textural properties, optoelectronic features, and multi-functionality of the mp-g-CN material over pristine g-C3N4 stem from deliberate structural modifications, generally achieved in terms of morphological engineering (via synthesis methods) and functional tuning (via doping/defects). Further, the choice of the synthesis route (template vs. template-free) dictates the morphological structure, while post-synthetic modifications (doping, defect engineering, hybridization) tailor electronic properties. These structural modifications introduce controlled irregularities that optimize mp-g-CN as a potential photocatalyst for efficient solar energy conversion by balancing high surface area, efficient charge transport, and enhanced light absorption. Table 2 summarizes a list of mp-g-CN samples associated with various types of structural modifications, exhibiting a superior textural property.
| Photocatalyst | Synthesis method | Structural modification | BET surface area (m2 g−1) | Pore volume (cm3 g−1) | Ref. | |
|---|---|---|---|---|---|---|
| Functional modification | Morphological modification | |||||
| PHCN3 | Template-free | P-doping, N-vacancy | Micro-tube | 79.29 | 0.28 | 98 |
| CNNS | Template-free | — | Nanosheet | 212 | 1.07 | 112 |
| HACN | Template-free | O-doping | 3D interconnected hierarchical fragmented nanosheet | 54.3 | — | 136 |
| SCN | Template-free | N-vacancy | Microrod | 32.05 | — | 139 |
| CN-1.5 | Soft-template | O-doping | Nanosheet | 32.65 | 0.19 | 142 |
| PCNS-F | Template-free | C and N dual vacancy | Nanosheet | 183.3 | — | 143 |
| BMNS | Template-free | B-doping | Nanosphere | 160.78 | 0.62 | 145 |
| Cv-gCN | Soft template | C-vacancy | Nanosheet | 147 | 0.75 | 152 |
| P20-SBA-15-CN | Hard template | P-doping, N-vacancy | Nanorod | 52 | 0.14 | 154 |
| CoPPc@mpg-C3N4 | Hard template | Composite | Nanosheet | 71.6 | — | 166 |
| Cu-FeOOH/TCN | Template-free | Composite | Nanotube | 116.82 | — | 167 |
| MnFeCu/g-C3N4 NFs | Template-free | Metal doping | Nanofibre | 280 | 0.4 | 168 |
| TCN-Lc | Template-free | Edge grafting | Nanotube | 102 | — | 169 |
| TCN(NH3) | Template-free | NH3-defect | Nanotube | 135.8 | — | 170 |
| PCN1.5 | Template-free | P-doping, C-vacancy | Micro-flower | 222.12 | 1.34 | 171 |
4. DFT calculations for mp-g-CN
First-principles calculations and DFT simulation modelling provide atomic-level insights into the electronic, optical, and structural properties of mp-g-CN photocatalysts.172 DFT calculations can be used as a guiding tool for in-depth interpretation in designing efficient mp-g-C3N4-based photocatalysts. Structural relaxation and energy calculations in DFT simulations are accomplished with the Vienna ab initio simulation package (VASP) and the Perdew–Burke–Erenzerhof (PBE) exchange–correlation functional and Grimme's DFT-D3 method are used for dispersion correction.172–174 The computational studies unravel the charge carrier dynamics and electronic state by elaborating possible transfer channels. Further, the delocalization of the VB and CB level cross-layer transfer of photoexcitons can be estimated by single metal atom sites. DFT analysis can pinpoint the exact location of the doping sites, geometry, energy level, band structure, and charge dynamics to provide concrete agreement with the experimental output.172–174 GGA-PBE functional modelling is often used to predict the VB and CB level composition by calculating the density of states. For instance, in g-C3N4, the HOMO or VB level is composed of N2 orbitals, whereas the LUMO or the CB level is composed of the contribution of the C and N orbitals (Fig. 30a–c).172 The structural modification leads to achieving a final band structure which can explain the charge transfer mechanism by interpreting the synergistic effect of the Fermi level correction, work function, and built-in electric field. The adsorption energy calculation for the reactants on the pore surface is indeed a significant process for interpreting the active sites and the nature of interaction of mp-g-CN-based photocatalysts.174 In the literature, mostly monolayer models have been considered for DFT calculations, which seem uneven in real practice. Indubitably, the DFT computational analysis can critically contribute to establishing the guiding tool in the development of a new class of mp-g-CN-based materials. However, the perplexity of DFT simulation gradually interpreted with the complex compositions and large pore framework of mp-g-CN. Due to this, very limited number of DFT simulations have been reported in the literature. Kim et al. have reported a mesoporous C3N7 model based on the experimental output which is composed of tetrazine, triazole, and guanidine moieties.175 The C3N7 monolayer structure was optimized by the PBE exchange correlation functional within GGA approximation through modulating the C/N content (Fig. 30d). The positive frequencies from the phonon density of states confirm the thermodynamic stability of the model. Further partial density of states calculations found that both VB and CB were composed of the Npz orbital. The band structure analysis suggested that C3N7 is a direct semiconductor photocatalyst (Fig. 30e and f). | ||
| Fig. 30 DFT calculations using the GGA-PBE functional of (a) DOS (the dashed line represents the Fermi energy level), (b) HOMO and (c) LUMO of the g-C3N4 monolayer model. Reproduced with permission.172 Copyright 2019, Wiley-VCH. (d) Optimized model of the C3N7 monolayer, (e) PDOS and (f) band diagram of C3N7. Reproduced with permission.175 Copyright 2019, Wiley-VCH. | ||
Similarly, another report has demonstrated the 2 × 2 monolayers of g-C2N, g-C3N4, and g-C6N6 with a vacuum space of 20 Å, suggesting the mesoporous structure (Fig. 31a).176 The optimized structure was further modelled for dual B atom doping, and a negative formation energy further confirmed the thermodynamic stability of all the structures. The dual B-doped structure was optimized, and it was found that each B was connected by two bonds to an N atom and one adjacent B atom, with localized charge between the B atoms, ensuring high thermodynamic stability. Dual B-doped g-C2N and g-C6N6 exhibited the highest activity because of their high adsorption energy, optimized band structure, and mesoporous framework.
 | ||
| Fig. 31 (a) Calculated optimized models attributed to 2 × 2 monolayers of g-C2N, g-C3N4, and g-C6N6 with a vacuum space of 20 Å along the Z-axis and (b) optimized structure of dual B-doped g-C2N, g-C3N4, and g-C6N6 and related isosurface plot corresponding to the electron localization function. Reproduced with permission.176 Copyright 2023, American Chemical Society. | ||
5. Efficient solar energy conversion for sustainable fuel production over mp-g-CN-based photocatalytic materials
5.1. Principle of photocatalytic solar fuel production over semiconductor photocatalysts
A series of elementary steps direct the semiconductor-mediated artificial photosynthesis.171,172 Adequate light absorption by the semiconductor photocatalyst is the primary step. The electronic excitation from the VB to CB takes place only if the energy (hν) of incident photons is the same or higher than the bandgap energy of the semiconductor photocatalyst. This electronic excitation upon light absorption creates positively charged holes (h+) in the VB and excess electrons in the CB of the semiconductor. The subsequent step includes the shuttling of photogenerated charge carriers to the semiconductor's surface via their efficient separation and transportation. Finally, the surface redox reactions take place, where the photogenerated charge carriers on the surface of the semiconductors react with chemical species (Fig. 1). However, some of the charge pairs may exhibit volume recombination and surface recombination to retard the efficacy of the photocatalyst. The overall success relies on the extent of charge separation and their migration to the surface to execute surface redox reactions.177,178 Therefore, the photocatalytic approaches for various types of sustainable fuel production, such as H2 production, CO2 reduction, H2O2 production, N2 reduction and biomass conversion, need a fine-tuned semiconductor photocatalyst with enhanced optoelectronic properties and appropriate structural framework, which effectively generates precise and dynamic charge carriers to facilitate the desired reactions effectively. The underlying reaction steps involved in solar energy conversion entail a series of oxidation–reduction processes mediated by multiple photogenerated e− and h+ pairs. Moreover, the band edge (CB and VB) positions of the semiconductor must correspond to the potentials required to carry out the redox reactions involved in solar fuel production.37,38 Mp-g-CN-based photocatalysts display adequate characteristic features that make them one of the ideal photocatalytic systems for efficient solar fuel production.Here, we highlight the recent progress and advancement in photocatalytic solar energy conversion for sustainable fuel production over mp-g-CN-based photocatalysts. We demonstrate the fundamental principles and structural aspects to deduce the structure–activity relationship and the involved photocatalytic reaction mechanism in certain reactions such as photocatalytic H2 production, CO2 reduction, H2O2 generation, N2 reduction and lignocellulosic biomass transformation.
5.2. mp-g-CN-based photocatalytic materials for H2 production
Hydrogen fuel production using photocatalytic OWS is the simplest approach for sustainable clean energy production. Based upon the principle of natural photosynthesis, the photocatalytic OWS involves the consumption of solar energy, water and photocatalysts to produce hydrogen and oxygen without any adverse environmental impact.179,180 Thus, photocatalytic OWS is considered as the best and most highly desirable process for sustainable H2 production. However, this reaction is thermodynamically challenging as it requires Gibb's free energy of 237.2 kJ mol−1 and a minimum photon energy of 1.23 eV to initiate the splitting of a water molecule into H2 and 1/2O2.181Overall water splitting (OWS) reaction
| 2H2O + hv → 2H2 + O2, ΔG° = 237.2 kJ mol−1 | (1) |
| 4H+ + 4e− → 2H2, ΔE° = 0 V | (2) |
| 2H2O → 4H+ + 4e− + O2, ΔE° = 1.23 V | (3) |
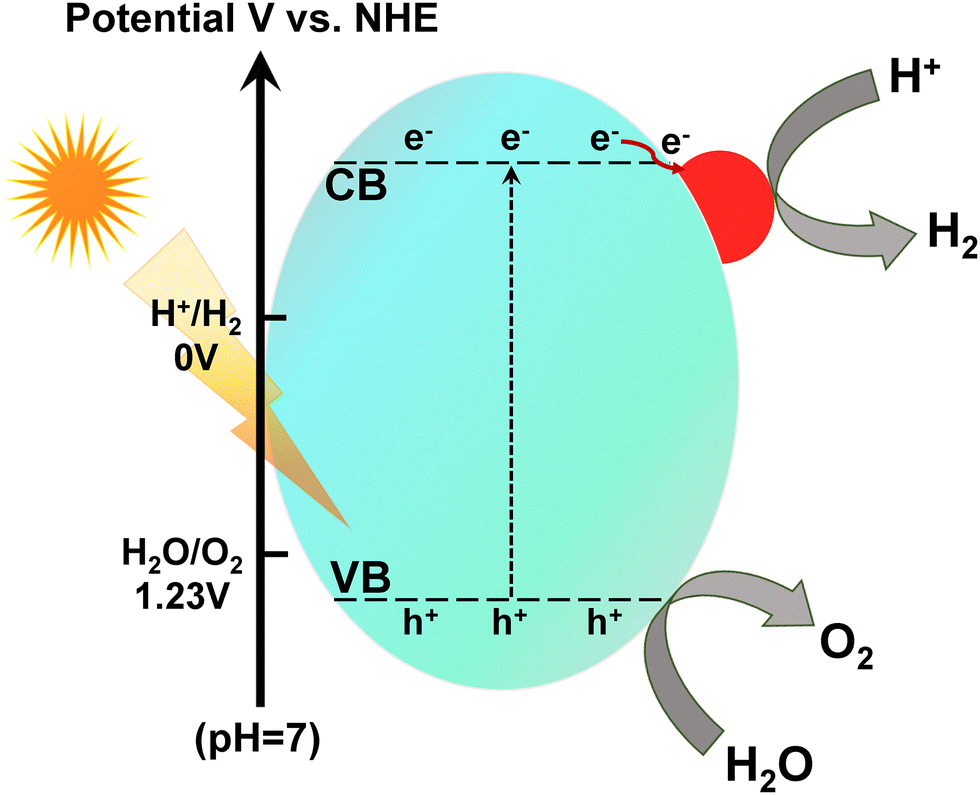 | ||
| Fig. 32 General mechanism of photocatalytic H2 evolution. | ||
The OER reaction requires comparatively much higher over-potential and a higher kinetic barrier than the HER reaction. Hence, the OWS reaction is really challenging to achieve for efficient H2 production. The e−/h+ recombination is the major cause for slowing down the kinetics in the OWS reaction. In contrast, efficient H2 production can be achieved by meticulously choosing the HER reaction and avoiding the OER reaction by quenching the photogenerated holes using various hole scavengers such as alcohols, amines, etc., as reported numerous times in the literature.179–182 The selective quenching of holes can impede the possibility of charge recombination and endorse the involvement of a greater number of photogenerated electrons in HER. Although HER provides faster kinetics for H2 production, OWS still remains the most desirable choice for sustainable H2 production. mp-g-CN-based photocatalytic materials with a CB potential more negative than the reduction potential of H+/H2 and a VB potential more positive than the oxidation potential of O2/H2O can be advantageous for the OWS reaction. In contrast, the quick recombination of the photogenerated charge carriers in bulk g-C3N4 restricts their individual availability due to their bulk recombination, which serves as a major limiting factor and hinders the photocatalytic performance for OWS reaction. However, the porous framework and large surface area in mp-g-CN materials facilitate the diffusion of photogenerated electrons and enhance the availability of reactive sites, thereby increasing the density of surface electrons and extending the charge carrier's lifetime. As a result, the diffusion of photogenerated charge carriers increases, leading to improved overall photocatalytic efficiency for OWS. For example, Chen et al. successfully fabricated an mp-g-CN photocatalyst using the SiO2 hard template.80 The confinement effect of SiO2 offered a well-ordered porous channel, leading to multiple reflections of light due to the thin pore wall enriched with numerous active sites. The mesoporous channels shortened the charge migration distance and enhanced the fast charge transfer to expedite the overall photocatalytic activity. Consistent with the prior studies, this study also observed a comparatively higher H2 prodution via HER process than OWS reaction due to the higher kinetics barrier of OWS reaction. The HER activity was tested over the resultant mp-g-CN photocatalyst in the presence of triethanolamine (TEOA) as a sacrificial agent and Pt as a co-catalyst and it was found to be 2.5 times (λ > 320 nm) and 4.7 times (λ > 420 nm) greater than that of bulk g-C3N4. However, when the same photocatalyst was used for the OWS reaction, the rate of H2 production was much lower compared to the HER reaction under both full light and visible light irradiation. Likewise, the ratio of O2/H2 formation was significantly lesser than the stoichiometry ratio (0.5) of water splitting, directly evidencing that the sluggish kinetics of the OWS reaction was mainly dominated by the slower rate of OER reaction. This suggested the inefficient H2O oxidizing ability at the VB of mp-g-CN. However, the OER activity was also enhanced by fabricating mp-g-CN with BiVO4 and Fe2+/Fe3+. In the resultant composite material, BiVO4 acted as a photoactive material to boost the OER activity, while the mesoporous channel helped to adsorb Fe2+, which plays the role of a charge carrier transfer mediator by altering the Fe2+/Fe3+ cycle. The resultant multifunctional composite material exhibited a very decent H2 production activity by OWS reaction through the successful Z-scheme mechanism. The photogenerated electron in the CB of mp-g-CN reduced H2O to H2, and the photoinduced h+ in the VB of BiVO4 simultaneously oxidized H2O to O2 and interconverted the Fe2+/Fe3+ cycle to perform the Z-scheme mechanism. In contrast, bulk g-C3N4, having stacked and agglomerated structural features, failed to adsorb Fe2+ and, thus, was unable to exhibit the Z-scheme pathway for the OWS reaction and demonstrated inefficient catalytic activity.
Another mp-g-CN photocatalyst, synthesized using template-free supramolecular assembly of melamine and cyanuric acid, was reported for the photocatalytic OWS (Fig. 33a).183 The resultant three-dimensional mp-g-CN (3D-g-C3N4 NS) photocatalyst displayed agglomerated floppy nanosheets (Fig. 33b and c). These ultrathin and crystalline nanosheets featured an interconnected open porous channel and a high specific surface area, which facilitated faster charge carrier transportation and was beneficial for the photocatalytic OWS reaction (Fig. 33d). Moreover, the band structure analysis revealed that the CB level became more negative, and the VB level aligned to become more positive compared to bulk g-C3N4 (Fig. 33e). This improved band alignment and three-dimensional interconnected mesoporous structure satisfied the thermodynamic criterion for photocatalytic OWS reaction. The photocatalytic OWS reaction was executed in the presence of Pt and IrO2 as co-catalysts, and H2 and O2 evolution rates were found to be 101.4 and 49.1 μmol g−1 h−1 under visible light, which were approximately 11.8 times greater than those of bulk g-C3N4 (Fig. 33f).
 | ||
| Fig. 33 (a) Diagrammatic sketch of the synthesis of 3D-g-C3N4 NS, (b) SEM image and (c) TEM and HRTEM image (inset) of 3D-g-C3N4 NS, (d) N2 adsorption–desorption isotherms and (e) band structures of bulk g-C3N4, g-C3N4 NS, g-C3N4 NS-1 and g-C3N4 NS-2, (f) overall water splitting rate of all prepared samples (here g-C3N4 NS refers to g-C3N4 nanosheets, 3D g-C3N4 NS-1 and 3D g-C3N4 NS-2 refer to g-C3N4 nanosheets with different amounts of melamine used in the synthesis process). Reproduced with permission.183 Copyright 2019, Elsevier. | ||
Even though the mp-g-CN framework possesses the necessary structural and optoelectronic requirements for the execution of photocatalytic OWS reaction, its successful implementation has been circumscribed and only extends to a limited number of reports in the literature due to the challenging and highly energetic reaction steps of photocatalytic OWS reaction.80,183,184 However, the photocatalytic HER reaction is more facile to occur over mp-g-CN-based photocatalysts. Therefore, several mp-g-CN-based photocatalytic systems with tunable structural features have been reported in the literature for efficient photocatalytic HER activity. For example, Obregón et al. reported an SBA-15-assisted synthesis of mp-g-CN (mpg-C3N4) for photocatalytic H2 production using 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 v/v isopropanol–water solution.185 The superior photoactivity of mpg-C3N4 was correlated with its distinct porous structure, large surface area and pore volume compared to the layered structure of bulk g-C3N4 as confirmed from SEM and TEM. SBA-15 hindered the complete condensation of the mpg-C3N4 precursor. Hence, the resultant mpg-C3N4 was associated with a decreased conjugation length and porous structure compared to the closed lamellar structure of bulk g-C3N4 (Fig. 34A-(a–d)). Thus, mpg-C3N4 displayed a hypsochromic shift in visible light absorption and experienced a strong quantum confinement effect. Based on the valence band XPS (VB XPS) analysis and band gap energies, the band structure for bulk g-C3N4 and mpg-C3N4 (Fig. 34A-(e–g)). The more negative CB value of mpg-C3N4 boosted the redox potential of the photogenerated electron, contributing to its superior H2 production capability which is 4.7 times greater than that of bulk g-C3N4. In summary, the higher H2 production rate of mpg-C3N4 can be accredited to its mesoporous structure, quantum confinement effect, and more negative CB value. Similarly, an mp-g-CN nanosheet was fabricated using a simple ultrasonic exfoliation strategy for the efficient photocatalytic HER application.186 The exfoliation of bulk g-C3N4 led to the formation of mesoporous channels with enhancement in the textural properties, thus unveiling the copious active sites for the HER process. The resultant mp-g-CN nanosheet also exhibited improved light harvesting features by multilight scattering phenomena. Also, the exfoliation process altered the band alignment by narrowing the band gap and shortening the charge carrier's transfer path in the mp-g-CN nanosheet. Therefore, the mp-g-CN nanosheet exhibited better charge separation with lower charge transfer resistance and led to 3.3 times greater H2 production than bulk g-C3N4.
:10 v/v isopropanol–water solution.185 The superior photoactivity of mpg-C3N4 was correlated with its distinct porous structure, large surface area and pore volume compared to the layered structure of bulk g-C3N4 as confirmed from SEM and TEM. SBA-15 hindered the complete condensation of the mpg-C3N4 precursor. Hence, the resultant mpg-C3N4 was associated with a decreased conjugation length and porous structure compared to the closed lamellar structure of bulk g-C3N4 (Fig. 34A-(a–d)). Thus, mpg-C3N4 displayed a hypsochromic shift in visible light absorption and experienced a strong quantum confinement effect. Based on the valence band XPS (VB XPS) analysis and band gap energies, the band structure for bulk g-C3N4 and mpg-C3N4 (Fig. 34A-(e–g)). The more negative CB value of mpg-C3N4 boosted the redox potential of the photogenerated electron, contributing to its superior H2 production capability which is 4.7 times greater than that of bulk g-C3N4. In summary, the higher H2 production rate of mpg-C3N4 can be accredited to its mesoporous structure, quantum confinement effect, and more negative CB value. Similarly, an mp-g-CN nanosheet was fabricated using a simple ultrasonic exfoliation strategy for the efficient photocatalytic HER application.186 The exfoliation of bulk g-C3N4 led to the formation of mesoporous channels with enhancement in the textural properties, thus unveiling the copious active sites for the HER process. The resultant mp-g-CN nanosheet also exhibited improved light harvesting features by multilight scattering phenomena. Also, the exfoliation process altered the band alignment by narrowing the band gap and shortening the charge carrier's transfer path in the mp-g-CN nanosheet. Therefore, the mp-g-CN nanosheet exhibited better charge separation with lower charge transfer resistance and led to 3.3 times greater H2 production than bulk g-C3N4.
 | ||
| Fig. 34 (A) (a) SEM and TEM images of (a) and (c) bulk g-C3N4, (b) and (d) mpg-C3N4, (e) and (f) VB XPS and (g) schematic band structure of bulk g-C3N4 and mpg-C3N4. Reproduced with permission.185 Copyright 2019, Elsevier. (B) (a) Illustrative depiction of the formation process of flower-like P-doped mp-g-CN, (b) band structure alignment, (c) transient photocurrent response, (d) EIS Nyquist plots, (e) photocatalytic activity and stability test of the synthesized materials. Reproduced with permission.171 Copyright 2018, The Royal Society of Chemistry. | ||
The mesoporous framework with some additional structural modification has been proven as an effective HER photocatalyst in the literature. For instance, Yang et al. reported a flower-like P-doped mp-g-CN photocatalyst (PCNx, where x = 0 g, 0.5 g, 1.0 g, 1.5 g, and 2.0 g, representing the amount of phosphoric acid added) synthesized by template-free supramolecular self-assembly of melamine and cyanuric acid with the addition of phosphoric acid as the source for P-doping (Fig. 34B-a).171 The combination of the mesoporous structure and P-doping significantly altered the band alignment, leading to a red shift in light absorption, lowering the band gap and modifying the band alignment (Fig. 34B-b). The P-doped mp-g-CN was also reported to exhibit a lower recombination rate and superior charge carrier separation and transportation compared to bulk g-C3N4 (Fig. 34B-(c and d)). All these necessary features in the resultant P-doped mp-g-CN led to an excellent HER activity, which was 24.6 times higher compared to bulk g-C3N4 with preserved activity even after the fourth consecutive cycle (Fig. 34B-e).
Recently, Guan et al. reported a hard template-assisted synthesis of a ‘N’ rich, S-doped mesoporous g-C3N5 photocatalyst for H2 production by photocatalytic HER.187 5-Amino-1,3,4-thiadiazole-2-thiol (5-ATDT) as the CN precursor, as well as the ‘S’ doping source, was used to synthesize SCN-bulk (in the absence of any template), MSCN-S (in the presence of SBA-15) and MSCN-K (in the presence of KIT-6) (Fig. 35a). The resultant MSCN-K and MSCN-S exhibited well-defined textural properties and rough, porous surfaces compared to SCN-bulk (Fig. 35b–g). The elemental composition analysis (CHNS) revealed that both samples were ‘S’ doped and had a high ‘N’ content. Further, FTIR and XPS analysis confirmed the presence of sulfite (SO) functionalization in both samples, suggesting that ‘S’ doping was also responsible for SO grafting in the mesostructure of MSCN-K and MSCN-S (Fig. 35h and i). The comparative UV and PL profile of SCN-bulk, MSCN-K and MSCN-S revealed that the light absorption efficiency was reduced after the creation of mesopores, but ‘S’ doping and SO linkage significantly improved charge separation and mitigated charge recombination (Fig. 35j–l). The combination of the high surface area, ordered porosity, ‘S’ doping and incorporation of SO moieties was reported to quench the radiative recombination of charge carriers, alter the band position and shift the CB position to more negative, ideal for ease of proton reduction and hence exhibited an excellent HER activity. The HER activity trend (Fig. 35m) was in accordance with the analysis results discussed above.
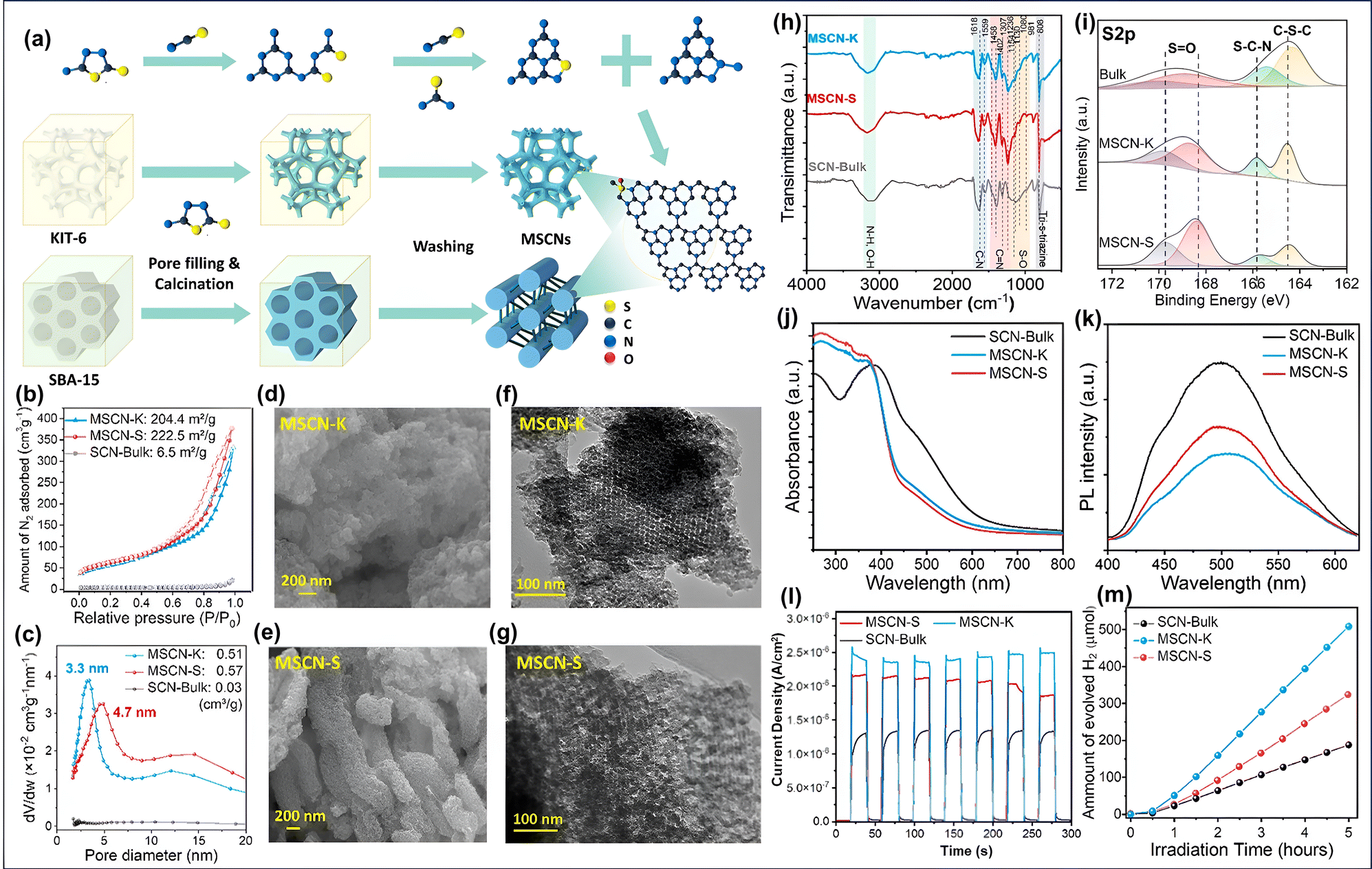 | ||
| Fig. 35 (a) Diagrammatic representation of the MSCN material, (b) BET isotherms, (c) BJH pore size distribution of MSCN samples, SEM and TEM images of (d) and (f) MSCN-K, (e) and (g) MSCN-S, (h) FT-IR spectra, (i) XPS spectra of S2p, (j) UV-vis DRS, (k) PL spectra, (l) transient photocurrent response, and (m) photocatalytic H2 evolution of SCN-bulk, MSCN-S, and MSCN-K. Reproduced with permission.187 Copyright 2024, American Chemical Society. | ||
Hitherto, several vacancy- and defect-induced mp-g-CN-based photocatalytic systems have been reported to exhibit outstanding HER activity.188–190 The vacancy or defect sites alter the band potential, create active sites for light absorption, and channelize the charge carriers for efficient HER activity. For example, an ‘N’ defect modified thin layered mp-g-CN (DTLP-CN) was synthesized using the urea and KOH thermal assisted polymerization strategy for the photoproduction of H2.116 KOH was reported to trigger the oxidative deprotonation of the –C–NH2 group in the framework, leading to the incorporation of the –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N moiety and also assisted the thermal etching process during calcination, allowing various decomposed gases to escape. As a result, numerous mesopores were formed with a decrement in layer stacking. The optimized mass ratio of urea and KOH was responsible for inducing a thin layer nanostructure, N-vacancy, –CN moiety incorporation and mesoporosity creation in DTLP-CN. The resultant uniquely designed mesoporous framework of DTLP-CN delivered better light harvesting ability due to the narrow band gap, wide absorption spectrum, better charge separation, and efficient mass transfer, which were attributed to the mesoporous and thin layer framework structure. Moreover, the ‘N’ vacancy and the –CN moiety created midgap energy levels and modified the band structure. It was also reported that more unsaturated sites with unpaired e− were formed to populate the photoexcited e− in CB levels, which enhanced the photoresponse activity. The combinational effect of all these features resulted in an excellent HER activity of 1.5557 mmol g−1 h−1 under visible light irradiation, which was 48.5 times greater than that of pristine g-C3N4. Similarly, another excellent HER photocatalyst was fabricated using ‘I’ doped and ‘N’ vacancy-modified mp-g-CN by a template-free synthesis approach.191 The synthesis methodology involved the utilization of glucose and NH4I with melamine, which released various N-rich gases during calcination and resulted in the formation of thin layered nanosheets with abundant mesopores. Furthermore, a substantial amount of ‘N’ defects and ‘I’ doping was induced in the resultant mesoporous framework. As mentioned in the previous example, the ‘N’ defect was directed to form midgap states below the CB level that functioned to momentarily trap the radiative electrons in the resultant framework. This alteration in the band structure, along with the thin layer mesoporous framework, led to a quantum confinement effect, which was responsible for better light harvesting ability as well as efficient radiative charge carrier separation. Hence, the resultant ‘I’ doped, ‘N’ vacancy-mediated mp-g-CN nanosheet was reported to exhibit a photocatalytic H2 production of 7819.2 μmol g−1 h−1 under simulated solar light irradiation, which was almost 6.5 times greater than that of bulk g-C3N4.
N moiety and also assisted the thermal etching process during calcination, allowing various decomposed gases to escape. As a result, numerous mesopores were formed with a decrement in layer stacking. The optimized mass ratio of urea and KOH was responsible for inducing a thin layer nanostructure, N-vacancy, –CN moiety incorporation and mesoporosity creation in DTLP-CN. The resultant uniquely designed mesoporous framework of DTLP-CN delivered better light harvesting ability due to the narrow band gap, wide absorption spectrum, better charge separation, and efficient mass transfer, which were attributed to the mesoporous and thin layer framework structure. Moreover, the ‘N’ vacancy and the –CN moiety created midgap energy levels and modified the band structure. It was also reported that more unsaturated sites with unpaired e− were formed to populate the photoexcited e− in CB levels, which enhanced the photoresponse activity. The combinational effect of all these features resulted in an excellent HER activity of 1.5557 mmol g−1 h−1 under visible light irradiation, which was 48.5 times greater than that of pristine g-C3N4. Similarly, another excellent HER photocatalyst was fabricated using ‘I’ doped and ‘N’ vacancy-modified mp-g-CN by a template-free synthesis approach.191 The synthesis methodology involved the utilization of glucose and NH4I with melamine, which released various N-rich gases during calcination and resulted in the formation of thin layered nanosheets with abundant mesopores. Furthermore, a substantial amount of ‘N’ defects and ‘I’ doping was induced in the resultant mesoporous framework. As mentioned in the previous example, the ‘N’ defect was directed to form midgap states below the CB level that functioned to momentarily trap the radiative electrons in the resultant framework. This alteration in the band structure, along with the thin layer mesoporous framework, led to a quantum confinement effect, which was responsible for better light harvesting ability as well as efficient radiative charge carrier separation. Hence, the resultant ‘I’ doped, ‘N’ vacancy-mediated mp-g-CN nanosheet was reported to exhibit a photocatalytic H2 production of 7819.2 μmol g−1 h−1 under simulated solar light irradiation, which was almost 6.5 times greater than that of bulk g-C3N4.
Several non-noble and noble metal-modified mp-g-CN photocatalysts have been demonstrated as excellent HER photocatalysts for efficient H2 production. For example, a Cu incorporated mp-g-CN nanotube (P-CN(Cu)) was reported to achieve an impressive H2 evolution rate of 6.53 mmol g−1 h−1 under visible light irradiation in the presence of Pt as the co-catalyst and TEOA as the sacrificial agent, which was 11.26 times greater than that of bulk g-C3N4.192 The electronic microscope investigation (Fig. 36a–d) revealed a well-defined nanotube structure of P-CN(Cu), and the X-ray absorption near-edge structure (XANES) analysis revealed Cu anchored as a single atom rather than the metallic nanoparticle form. The Cu modification enhanced the visible light absorption and modified the band alignment suitable for HER (Fig. 36e and f). Both the steady state and transient state PL analyses suggested that the modified band structure achieved due to Cu incorporation significantly impeded the charge recombination by increasing the average lifetime of the charge carrier from 4.86 ns for the sample without Cu modification (P-CN) to 14.09 ns for P-CN(Cu) (Fig. 36g and h). Moreover, the density of states and spin-density of states calculated using DFT analysis suggested that Cu single-atom doping led to the CB level shifting towards the Fermi level to enhance the charge transfer phenomena (Fig. 36i–k). Further, the Gibbs free energy for the adsorption of H* (H* was the intermediate of H2) in P-CN(Cu) was estimated to be higher than that of P-CN, which revealed that the quicker H* desorption in P-CN(Cu) facilitated the efficient H2 production near the Cu-N2 active centre (Fig. 36l). Meanwhile, the slower H* desorption because of its strong adsorption on the ‘N’ sites of P-CN resulted in sluggish H2 production.
 | ||
| Fig. 36 (a) SEM image, (b) TEM image, (c) HAADF-STEM image, (d) EDS mapping of P-CN(Cu), (e) UV-vis spectra, (f) band structure, (g) PL spectra, (h) TRPL spectra of samples, density of states of (i) P-CN, (j) P-CN(Cu), (k) spin-density of sates of P-CN(Cu), (l) Gibbs free energy profiles for H2 evolution. Reproduced with permission.192 Copyright 2023, Royal Society of Chemistry. | ||
The MNps embedded within mp-g-CN act as the co-catalyst and can efficiently improve the photocatalytic HER activity. For example, a series of MNps (Pt, Ni and Ag) as co-catalysts, when embedded within the mp-g-CN framework, demonstrated excellent H2 production.193 The entrapment of MNps inside the mesoporous channel was carried out using a multiple vacuum-induced strategy, which was proven to offer more robust composites than the normal physical agitating approach.194 The resultant MNp deposition was found to be beneficial in several ways, such as the deposited MNps being responsible for charge carrier separation by trapping the photogenerated electrons, thereby exhibiting the electron enrichment effect to boost H2 production. In this aspect, our group reported a Au Np modified mp-g-CN-SiO2 composite for the efficient H2 evolution of 844 μmol in 3 h.195 The binding efficiency of Au Nps was found to be stable due to the multi-functional effect of the g-C3N4 and mesoporous SiO2 composite. The resultant Au-modified photocatalyst was reported to exhibit enhanced light harvesting properties due to the plasmonic effect of Au Nps, and charge recombination was also significantly suppressed. Several other co-catalyst-embedded mp-g-CN photocatalysts have been reported in the literature, demonstrating their role in enhancing light harvesting, suppressing the charge recombination, and accumulating and utilizing the photogenerated electrons in photocatalytic HER for efficient H2 production.196–198
The functional modifications in mp-g-CN photocatalysts have been reported to improve the structural features to make them more viable for photocatalytic HER application. For example, Xing and co-workers reported a mesoporous ultrathin tubular g-C3N4 with edge grafted L-cysteine (TCN-Lc) constructed via supramolecular self-assembly followed by the thermal condensation method.169 The successful edge grafting of L-cysteine was estimated using PXRD, 13C NMR and XPS analysis. The (100) plane for the TCN-Lc sample almost disappeared, and the (002) plane became broad and weak, indicating structural deformation after L-cysteine was incorporated into the edge sites of the ultrathin mesoporous tubular g-C3N4, which led to disruption of the in-plane and layer order packing. The 13C NMR spectrum of TCN-Lc exhibited two additional peaks attributed to the insertion of the L-cysteine unit into the basal plane of mesoporous ultrathin tubular g-C3N4. Moreover, the two distinct peaks attributed to the heptazine unit of g-C3N4 shifted in TCN-Lc, suggesting the change in the chemical environment upon the edge effect due to the edge site insertion of L-cysteine (Fig. 37a and b). The XPS analysis revealed that the C1s peak and N1s peak of TCN-Lc shifted compared to bulk g-C3N4, indicating that the L-cysteine was covalently bonded, which resulted in high electron density around the ‘N’ sites due to the edge effect. Compared to bulk g-C3N4, TCN-Lc exhibited a red shift in visible light absorption with a broad absorption edge due to n–π* electronic transition. Consequently, the CB edge position of TCN-Lc shifted downward and constricted the band gap, thereby facilitating the transition and dissociation of photogenerated charge carriers. Moreover, the incredible photogenerated charge separation was again realized from the PL, TRPL and photocurrent measurement analysis and further validated by the DFT calculation. Gaussian 09 and DFT calculations estimated that the LUMO and HOMO arose from the contribution of C–N bonding orbitals and nitrogen pz orbitals, respectively, for BCN (Fig. 37c and d). Due to the close proximity of HOMO–LUMO lying in the same tri-s-triazine, the charge recombination became relatively much quicker in the case of BCN. Moreover, in the case of TCN-Lc, the electron cloud has been redistributed upon L-cysteine edge grafting. As a result, the HOMO was contributed by the L-cysteine unit and the adjacent tri-s-triazine ring (Fig. 37e and f). In contrast, the C–N bonding orbitals around the L-cysteine unit contributed to the LUMO. The density of states of BCN and TCN-Lc (Fig. 37g and h) suggested that an intermediate band state appeared in TCN-Lc, promoting the stronger delocalization of the charge density. Furthermore, the electron cloud was redistributed to increase the charge carrier separation efficiency in TCN-Lc (Fig. 37i). The mechanistic investigation presented in Fig. 37j suggested that the tubular porous morphology of TCN-Lc facilitated the improved light absorption and shortened the charge transfer distance. Further, the edge effect of Lc enabled in-plane directional charge transfer and accumulated a larger number of surface-active sites. Consequently, the photogenerated electrons accumulated near the Pt co-catalyst and facilitated HER, while the TEOA sacrificial agent was oxidized by holes. All these findings signified the superior photo-responsive factor of TCN-Lc which exhibited an excellent photocatalytic H2 production of 6468 μmol h−1 g−1, which was 56 times greater than that of BCN (116 μmol h−1 g−1).
 | ||
| Fig. 37 (a) and (b) Solid-state 13C NMR and XPS spectra of bulk g-C3N4 and TCN-Lc, optimized electronic structure of the HOMO and LUMO energy level of (c) and (d) BCN, (e) and (f) TCN-Lc, respectively, partial density of states of (g) BCN and (h) TCN-Lc, (i) vertical and horizontal views of the electronic charge difference profiles of TCN-Lc, (j) schematic illustration of the photocatalytic HER reaction over TCN-Lc. Reproduced with permission.169 Copyright 2022, The Royal Society of Chemistry. | ||
Furthermore, efficient HER activity can be achieved using various hybridized composite photocatalysts of mp-g-CN with different semiconductor photocatalysts. The resultant multifunctional mp-g-CN-based composite photocatalysts have been reported to exhibit essential photo-active features and improved structural properties, which are key to better photocatalytic HER activity. For instance, Wang et al. reported a porous S-doped g-C3N4/ZnIn2S4 S-scheme heterojunction composite (S-CN/ZIS), which produced 19.25 mmol g−1 h−1, which was 2.7 times greater than that of g-C3N4/ZnIn2S4 (CN/ZIS).199 S-doped g-C3N4 (S-CN) was first fabricated by a template-free method using thiourea and thioacetamide, followed by a controlled calcination process. Further, ZnIn2S4 (ZIS) was hydrothermally synthesized in the presence of S-CN to fabricate the S-CN/ZIS composite having a hollow-coral framework. The successful growth of ZIS on the surface of S-CN in S-CN/ZIS (Fig. 38a–d) formed a highly porous framework, as revealed by HR-TEM and N2 adsorption–desorption analysis. The optimum ‘S’ doping in S-CN/ZIS created lattice defects and formed C-S-C linkages by replacing the ‘N’ atom. This resulted in a red shift in light absorption compared to the neat g-C3N4 (CN), and the band gap energy was significantly altered to achieve better light harvesting. M-S measurements confirmed that S-CN/ZIS exhibited n-type semiconductor characteristics where the charge transfer process was accomplished by the S-scheme heterojunction pathway, which was confirmed from the DMPO test and in situ XPS analysis. ‘S’ doping modified the CN work function and caused a noticeable difference in the Fermi energy level of S-CN and ZIS, facilitating the formation of an effective heterojunction interface (Fig. 38e–g). Theoretical simulation modelling for the three-dimensional charge density suggested that the electron transferred from CN to ZIS in CN/ZIS and from S-CN to ZIS in S-CN/ZIS, to form an interfacial electric field (Fig. 38h–j). Notably, the interfacial electric field was stronger in the S-CN/ZIS interface due to the influence of S doping, leading to enhanced charge separation efficiency. Hence, the photogenerated e− from the CB of ZIS easily recombined with the h+ at the VB of S-CN, while the photogenerated e− at the CB of S-CN and h+ at the VB of ZIS remained separated for the surface redox reaction to execute H2 production (Fig. 38j). From deeper insight, the synergistic effect of S-doping and S-scheme heterojunction enabled the accumulation of more photoinduced e− on the S-CN surface. Further, the hydrophilic nature of the S-CN surface enhanced H2O adsorption and could be an active reservoir for the ‘H*’ species. The adsorption–desorption energy calculation suggested that the S-CN surface suitably facilitates faster adsorption and desorption of ‘H*’ species compared to CN, lowering energy barriers for hydrogen evolution. As a result, S-CN/ZIS exhibited efficient H2 production and maintained a stable photocatalytic activity over five consecutive cycles (Fig. 38k).
 | ||
| Fig. 38 TEM images of (a) S-CN, (b) CN, (c) S-CN/ZIS, (d) EDS mapping of S-CN/ZIS, work functions of (e) CN, (f) S-CN, (g) ZIS, three-dimensional charge density of (h) CN/ZIS, (i) S-CN/ZIS, (j) schematic depiction of S-CN/ZIS S-scheme heterojunction mediated H2O splitting reaction, (k) cyclic stability of S-CN/ZIS. Reproduced with permission.199 Copyright 2024, Wiley-VCH. | ||
Zeng and coworkers reported a homojunction porous CN photocatalyst by combining triazine-based and tri-s-triazine-based crystalline C3N4 through a secondary growth strategy.200 The successful formation of the interfacial junction in tri-/tri-s-tri-C3N4 was primarily confirmed from the PXRD and FT-IR analysis. The resultant material, accomplished with a type IV isotherm with a H1-type hysteresis loop, exhibited a surface area of 79.7 m2 g−1 with a distinct mesopore distribution between 2 and 18 nm. Further, nanostructure morphological analysis suggested the tri-C3N4 nanorod deposited on the lamellar tri-s-tri-C3N4 having two distinct lattice fringes corresponding to tri-C3N4 and tri-s-tri-C3N4 at the interfacial junction sites of tri-/tri-s-tri-C3N4. The resultant tri/tri-s-tri-C3N4 synthesized material exhibited an excellent photocatalytic H2 evolution performance with a rate of 144 μmol h−1, attributed to its increased crystallinity, enhanced visible light absorption, strong interfacial lattice compatibility between the two phases, and efficient separation of photogenerated charge carriers.
Similarly, Ag2MoO4/OC3N4, another example of the hybridized composite photosystem of ‘O’ doped mp-g-CN (OC3N4) and Ag2MoO4, was fabricated by a template-free calcination method followed by an in situ deposition method (Fig. 39a).201 A successful composite formation was observed during the morphological investigation and the EDX analysis (Fig. 39b–i). The fluffy and porous morphology of curly-shaped OC3N4 (Fig. 39b) accommodated the Ag2MoO4 nanoparticles on its surface as represented by the yellow circle (Fig. 39c) to form the Ag2MoO4/OC3N4 heterojunction. The band alignment analysis and work function calculation suggested that the electron flowed from OC3N4 to Ag2MoO4 until their Fermi level reached equilibrium to develop the Ag2MoO4/OC3N4 heterojunction (Fig. 39j). The electron migration experienced an interfacial electric field between the interface of OC3N4 and Ag2MoO4, which caused the band bending of their band edges (Fig. 39k). Due to band bending, the charge transfer pathway followed the S-scheme mechanism, which facilitated the recombination of e− at the CB of Ag2MoO4 and h+ at the VB of OC3N4. Hence, the h+ of Ag2MoO4 and e− of OC3N4 remained exclusively separated and were utilized for the H2 generation in the presence of TEOA (Fig. 39k). The H2 production activity of Ag2MoO4/OC3N4 reached 16619 μmol g−1, which was 1.93 and 180 times greater than that of OC3N4 and Ag2MoO4, suggesting the high photoactivity accredited to the mesoporous channel, high surface area, and unique charge transfer process.
 | ||
| Fig. 39 (a) Schematic depiction of Ag2MoO4/OC3N4 synthesis, (b) SEM, (c) TEM, (d)–(h) elemental mapping, (i) EDS profile of Ag2MoO4/OC3N4, (j) optimized work functions of OC3N4 and Ag2MoO4, (k) diagrammatic illustration of the energy level, bend bending and photocatalytic H2 production mechanism. Reproduced with permission.201 Copyright 2024, Elsevier. | ||
Besides the examples given above, substantial reports based on mp-g-CN-based photocatalyst-driven H2 production have been articulated in the literature. Table 3 demonstrates a wide range of mp-g-CN-based photocatalysts associated with diverse structural modifications, leading to the efficient production of environmentally friendly hydrogen fuel.
| Photocatalysts | Synthesis method | Types of structural modification | Types of photocatalysis | H2 production rate | Cyclic stability | Light source | Ref. |
|---|---|---|---|---|---|---|---|
| g-C3N4-15 | Hard template method | — | OWS | 81.6 μmol h−1 | 4 cycles | 300 W Xe lamp (780 nm > λ > 420 nm) | 80 |
| CN-45B-45P | Soft template method | B and P doping | HER | 9443.38 μmol h−1 g−1 | 4 cycles | 300 W Xe lamp | 89 |
| DTLP-CN | Template-free method | N-defect | HER | 1.557 mmol h−1 g−1 | 4 cycles | 500 W Xe lamp (λ > 420 nm) | 116 |
| 3D g-C3N4 NS | Template-free method | — | OWS | 101.4 μmol h−1 g−1 | More than 100 h | 300 W Xe lamp (λ ≥ 420 nm) | 183 |
| WC1−x/g-C3N4 | Template-free method | Composite | OWS | 84.1 μmol h−1 g−1 | 4 cycles | 300 W Xe lamp (λ > 420 nm) | 184 |
| CN-25 | Template-free method | N-defect and O-doping | HER | 5889.39 μmol h−1 g−1 | 4 cycles | 300 W Xe lamp (λ > 420 nm) | 189 |
| g-C3N4-GLC-NH4I | Soft-template method | I-doping and N-vacancy | HER | 7819.2 μmol h−1 g−1 | 5 cycles | 300 W Xe lamp | 185 |
| P-CN(Cu) | Template-free method | Cu-coordination and N-vacancy | HER | 6.53 mmol h−1 g−1 | 3 cycles | 300 W Xe lamp (λ > 420 nm) | 192 |
| FeTPPCl/PCN | Template-free method | Type-II composite | HER | 8301 μmol h−1 g−1 | 4 cycles | 300 W Xe lamp | 202 |
| DMC30 | Soft template method | Defect | HER | 6130.4 μmol g−1 | More than 40 h | 300 W Xe lamp (λ > 420 nm) | 203 |
| S-PDCN-3 | Template-free method | S-doping and C-vacancy | HER | 23.78 mmol g−1 h−1 | 3 cycles | 300 W Xe lamp | 204 |
| CdIn2S4/Zn–g-C3N4 | Template-free method | Composite | HER | 101.74 μmol h−1 | 4 cycles | 300 W Xe lamp | 205 |
| g-C3N4-VNs-n | Template-free method | N-defect | HER | 3.16 mmol g−1 h−1 | 5 cycles | 300 W Xe lamp (λ > 400 nm) | 206 |
| O-CNS | Template-free method | O-doping | HER | 2012.9 μmol h−1 g−1 | 3 cycles | 300 W Xe lamp (λ > 420 nm) | 207 |
| g-C3N4/CABB-10 | Template-free self-assembly method | Type-II composite | HER | 60 μmol h−1 g−1 | 300 W Xe lamp (λ ≥ 420 nm) | 208 | |
| PNCN-BNa-3 | Template-free method | B and Na co-doping | HER | 5971.51 μmol h−1 g−1 | 5 cycles | 10 W white LED lamp (λ > 420 nm) | 209 |
| CoxP/C3N4 | Hard template method | Composite | HER | 1602 μmol h−1 g−1 | — | 300 W Xe lamp | 210 |
| Cr–PHCN | Hard template method | In-plane ‘C’ ring incorporation | HER | 7581 μmol h−1 g−1 | 5 cycles | 300 W Xe lamp | 211 |
| Tm-g-C3N4 | Hard template method. | N-defect and S-doped | HER | 4441 μmol g−1 h−1 | 4 cycles | 300 W Xe lamp (λ > 400 nm) | 212 |
| PCN-CdS-5%-Ni2P | Hard template method | Composite | HER | 2905.86 μmol g−1 h−1 | 3 cycles | 300 W Xe lamp (λ ≥ 400 nm) | 213 |
| DMCN | Hard template method | Cyano group defect and N-vacancy | HER | 3.29 mmol h−1 g−1 | 5 cycles | 300 W Xe lamp (λ > 420 nm) | 214 |
5.3. mp-g-CN-based photocatalytic materials for CO2 reduction for the production of various saturated and unsaturated hydrocarbon fuels
The utilization of anthropogenic CO2 in the production of value-added fuel by harnessing solar energy is considered a potential approach to maintain carbon neutrality and mitigate carbon emissions. The successful utilization of CO2 in solar fuel production is an exciting yet challenging research topic. Several factors are associated with the low quantum efficiency of the photoconversion of CO2 to fuel. The stable linear structure of CO2 (with a high bond enthalpy of 750 kJ mol−1) having zero dipole moment hinders its adsorption/activation on the catalyst surface, which ultimately lowers its photoconversion efficiency.14–16,215 The CO2 conversion is a thermodynamically uphill process under standard conditions. Converting CO2 to various fuels, such as CH4, CH3OH, CH3CH2OH, etc., is a non-spontaneous process and hence endothermic in nature. The photoreduction of CO2 requires a high overpotential as it involves one electron reduction of the linear CO2 molecule into a bent ˙CO2− which is the rate-determining step.14,15 Therefore, the low conversion rate in photocatalytic CO2 reduction is quite acceptable in real practice. Considering a semiconductor-driven photocatalytic CO2 reduction process, the associated steps are (i) concurrent generation of photoexcited charge carriers upon light irradiation on the photocatalyst and the modulated adsorption/activation of CO2 on the photocatalyst surface, (ii) migration of light-induced charge carriers towards the photocatalyst surface, and finally (iii) the interaction of the charge carriers with the adsorbed CO2 to give the solar fuels (Fig. 40).14–16 | ||
| Fig. 40 Schematic depiction of the mechanism of photocatalytic CO2 reduction. | ||
Both CO2 and H2O, which are considered as the reactant precursors for CO2 photoreduction, adsorb more favorably on the g-C3N4 surface than the photoreduced products such as H2, CO, and CH4. This advocates that the g-C3N4 surface inevitably enables the optimum adsorption of reactant species and eases the desorption of product molecules.216 Nevertheless, the higher CB level of pristine g-C3N4 is not necessarily helpful for CO2 photoreduction. The creation of mesoporous structures can result in a comparatively more negative CB position (−1.3 to −1.5 eV), leading to multiple light scattering phenomena and better CO2 adsorption.14–16 Therefore, mp-g-CN and its allied composite structures hold modulated active sites, and appropriate band edge positions can enormously produce photogenerated charge carriers with the required potential to boost photocatalytic CO2 reduction.51,58,217,218 Besides, mp-g-CN having diffusible mesochannels and intrinsic surface basicity due to the ‘N’ linkage in the framework can efficiently adsorb and activate CO2 (due to the acidic nature of CO2), which is a prerequisite factor for efficient CO2 conversion. Hence, this can enable the generation of photoexcited electrons with different negative potentials, which facilitate the conversion of CO2 to various gaseous and liquid fuels. The product distribution and selectivity are entirely determined by the specific potential, the number of photogenerated electrons involved, and their corresponding reaction enthalpies. For instance, CO2 can be converted to CO, HCHO, CH3OH, CH4, etc., when the photogenerated electrons at the CB level possess the potential of −0.53 eV, −0.48 eV, −0.38 eV, and −0.24 eV, respectively (Fig. 40).14–16
Jia et al. reported a mp-g-CN (CPCN) fabricated via template-free supramolecular co-assembly of cyanuric acid, melamine, and glucose.219 The morphological investigations suggested that melamine produced bulk g-C3N4 (BCN) having a closed and agglomerated structure, wherein the co-assembly of melamine and cyanuric acid (PCN) resulted in a three-dimensional swollen porous network structure (Fig. 41a and b). However, the addition of glucose produced a more porous and flaky structure, as it served as a bubble template for creating mesoporosity and introducing a ‘C’ defect in the framework (Fig. 41c) The glucose-mediated-synthesized photocatalyst CPCN-2 exhibited a type IV isotherm with hysteresis, demonstrating a very high surface area of 102.5 m2 g−1 compared to PCN (42.9 m2 g−1) and BCN (8.7 m2 g−1). The substitution of the ‘N’ atom by ‘C’ doping at the edge sites of the g-C3N4 framework was responsible for the redshift in visible light absorption and lowered the bandwidth energy of CPCN-2 (Fig. 41d and e). Moreover, the successful ‘C’ doping led to extended π-delocalization, demonstrating that CPCN-2 excellently accelerated charge transfer and separation processes. The band structure analysis suggested that, the CB level CPCN-2 is more negative compared to that of BCN (Fig. 41f). Hence, CPCN-2 was found to exhibit a superior CO2 photoreduction to CO of 60.82 μmol g−1 (BCN produced only 21.89 μmol g−1) via the 2e− photoreduction process by following the below mentioned elementary reaction steps (eqn (i)–(v)).
| CO2 + H2O → H2CO3 | (i) |
| H2CO3 → HCO3− + H+ | (ii) |
| CPCN-2 + hv → CPCN-2(e− + h+) | (iii) |
| TEOA + h+ → TEOA+ | (iv) |
| CO2 + 2e− + 2H+ → CO + H2O | (v) |
 | ||
| Fig. 41 SEM of (a) BCN, (b) PCN, and (c) CPCN-2, and (d) UV-Vis, (e) band gap energy and (f) band structure analysis of the synthesized materials. Reproduced with permission.219 Copyright 2024, Elsevier. | ||
CO2 photoreduction is enhanced in mp-g-CN compared to bulk g-C3N4, as realized from the above example; however, further optimization of the electronic and optical properties, such as band engineering within the primitive mp-g-CN framework, is necessary to improve its effectiveness for CO2 photoreduction. Non-metal doping is considered a straightforward and atom-economical approach for tuning the bandgap in mp-g-CN. Non-metal doping can introduce either electron-deficient (boron doping) or electron-rich (phosphorus and sulphur doping) frameworks by interfering with empty orbitals (in the case of boron doping) or filled orbitals (in the case of phosphorus or sulphur doping), responsible for the creation of transient local energy states above the VB level and thus beneficial for broad range light absorption, easier charge separation with slow charge recombination, and improved redox properties which are believed to be the dominant factors in achieving high quantum efficiency CO2 photoreduction. For example, Zhu et al. reported a two-dimensional S-doped mp-g-CN nanosheet (S-CN) material for CO2 photoreduction to CO.220 The S-CN photocatalyst demonstrated an excellent CO production of 16.02 μmol g−1, which was 10 times greater than that of bulk-gC3N4 (bulk-CN). The PXRD pattern became narrower, less intense, and slightly shifted, suggesting that S-doping caused a substantial reduction in the layer thickness as well as in the crystallite size. S-CN exhibited a type IV isotherm and H1 hysteresis, evidencing the existence of a mesoporous framework with a specific surface area 7 times greater than that of bulk-CN. The high surface roughness value due to the mesoporous framework and thin layer thickness of S-CN was obtained from 3-dimensional structural analysis using AFM analysis, suggesting that the S-CN structure mitigated the transmission resistance for better charge separation. The mesoporosity induced in S-CN was also helpful for the efficient CO2 adsorption, as confirmed from the CO2 adsorption isotherm and DFT calculations, suggesting that the S-CN surface was more accessible for CO2 reduction reactions (Fig. 42a–c). Further, the CO-TPD analysis suggested that the S-CN surface could readily desorb CO and favorably facilitate the CO2 reduction process compared to bulk-CN (Fig. 42d). The computational analysis further estimated the electronic structure of bulk-CN and S-CN by considering the difference in the projected density of states (PDOS) and total density of states (TDOS) (Fig. 42e and f). The VB of bulk-CN was majorly comprised of the N 2p state (−2.5 eV, 0.00 eV), and the CB was comprised of the C 2p and N 2p state (0.00 eV, 5.0 eV). In contrast, the VB of S-CN was primarily comprised of the N 2p state, whereas the CB was mainly composed of the S 3p state. This led to charge rearrangement by the interference of excess electrons of S 3p and S 3s, which were excited under light irradiation to bring a more revolutionary impact in CO2 photoreduction. The charge rearrangement process was further evidenced by the charge density analysis, which revealed that the adjacent C atom of S-doping sites experienced a decrement in the charge density and received electrons from the S atom; this helped to extend the π conjugation. Hence, upon light irradiation, the S-CN surface was enriched with photogenerated electrons to facilitate CO2 photoconversion to CO. The progress of CO2 photoreduction using H2O was analyzed by in situ FT-IR analysis, suggesting that CO2 was activated by the mesoporous S-CN surface and converted to the *COOH intermediate, which further reacted with H+ and e− to produce CO (Fig. 42g).
 | ||
| Fig. 42 (a) CO2 adsorption isotherms, (b) and (c) DFT study, (d) CO-TPD spectra of the samples, (e) and (f) DOS of bulk-CN and S-CN, (g) in situ FT-IR spectra. Reproduced with permission.220 Copyright 2021, The Royal Society of Chemistry. | ||
Similarly, the effect of various non-metal doping on the photoreduction activity of CO2 to CH4 was achieved using B, O, S, and P-doped mp-g-CN.221 Various non-metal doped mp-g-CN structures were synthesized using a non-templated approach with annealing under a N2 atmosphere (Fig. 43a). The CH4 production rate was improved upon the non-metal doping, and among all the synthesized photocatalysts, S-doped mp-g-CN exhibited the highest activity (Fig. 43b). The activity trend was mainly due to the mesoporous structure, which enhanced the CO2 adsorption cum activation process and S-doping boosted the light-harvesting and charge separation phenomena. The photogenerated electrons were trapped by S-sites and were used in the 8H+/8e− reduction process for CH4 production. The elementary steps involved in CH4 production occurred via the photocatalytic reaction of CO2 with H2O (Fig. 43c).
 | ||
| Fig. 43 (a) Diagrammatic representation of the synthesis of mp-g-CN and non-metal (B, O, P, S) doped mp-g-CN, (b) photocatalytic CO2 reduction rate of the synthesized samples, (c) CH4 production mechanism via photocatalytic CO2 reduction. Reproduced with permission.221 Copyright 2022, Elsevier. | ||
Apart from non-metal doping, several strategic designs of mp-g-CN-based composite photocatalysts have been reported to improve CO2 photoreduction activity.217,218,222 Hitherto, various MNps whose work function is larger than that of mp-g-CN have acted as cocatalysts when encapsulated with mp-g-CN, forming Schottky barriers and thus efficiently causing charge separation, followed by accumulation of the photoinduced electrons on the MNp surface to expedite the CO2 photoreduction activity.222–225 For example, a 3D mp-g-CN foam and Cu Np composite exhibited excellent CO production by CO2 photoreduction.224 The efficient CO2 adsorption accomplished by the mesoporous structure and the effective charge separation by the Cu Np co-catalyst were the responsible factors for exhibiting an enhanced photoreduction activity, which was 6.34 times higher than that of pristine g-C3N4 and 2.56 times higher than that of the 3D mp-g-CN foam without Cu Np modification. Similarly, Li et al. reported the CO2 photo-reformation of CH4 to several C1 and C2+ reduced products over Ru loaded on the La-doped mp-g-CN nanosheet (Ru-LGCN).225 The Ru-LGCN photocatalyst was fabricated by thermal-oxidative exfoliation followed by an in situ photoreduction deposition method. The resultant structure was enriched with the mesoporous channel, and the Ru and La bimetallic synergistic effect contributed to an efficient CO2 reformation of CH4 to CO, CH3OH, CH3CH3, and CH3CH(OH)CH3 with an optimal yield of 401.03, 750.02, 460.56 and 460.12 μmol g−1, respectively. La-doping improved the CO2 adsorption efficacy and also channelized the charge transfer between the photocatalyst and CO2. Additionally, the Ru incorporation significantly reduced the charge recombination process and increased the photogenerated electron density for better CO2 photoreduction activity. The band edge alignment analysis using theoretical calculation revealed that the charge distribution state at the conduction band maximum (CBM) was contributed by the atomic orbital of ‘La’, whereas the valence band maximum (VBM) state was contributed by the atomic orbital of the N atom (Fig. 44A-(a–d)). Therefore, effective charge separation was possible due to La-doping and the photoexcited electrons at the CBM, which actually accumulated near La sites and were involved in the multi-step photo-reformation process. The elementary steps outlined in Fig. 44A-e show that the photogenerated h+ oxidized CH4 to the methyl radical (˙CH3) and H+. The resultant (˙CH3) active species either dimerized to form CH3CH3 or reacted with OH− and h+ to produce CH3OH. Meanwhile, CH3CH2OH was obtained from the reaction of OH− with ˙CH3CH2 (˙CH3CH2 was produced from the h+ catalyzed oxidation of CH3CH3). Similarly, radical formation and redox reaction resulted in the production of CH3CHO and CH3CH(OH)CH3 (Fig. 44A-e).
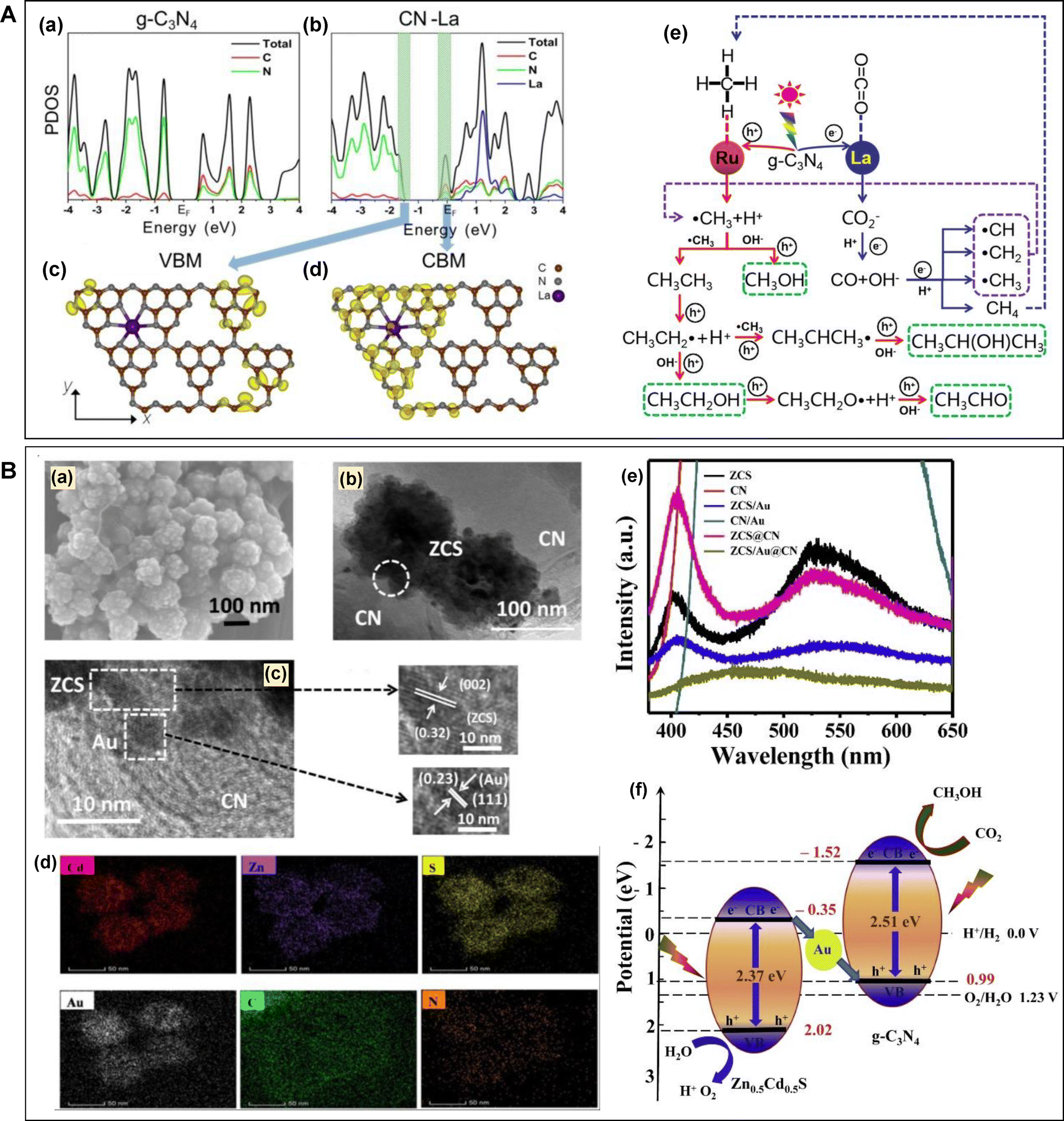 | ||
| Fig. 44 (A) (a) Representation of the electronic projected density of states (a) g-C3N4, (b) LGCN layer, top view of partial charge density with frontier states of the LGCN layer of (c) VB and (d) CB, (e) CO2 reduction pathway. Reproduced with permission.225 Copyright 2021, Wiley-VCH. (B) (a) FESEM, (b) TEM, (c) HRTEM, and (d) EDS mapping of ZCS/Au@CN, (e) PL spectra of all the prepared samples, (f) charge-transfer mechanism (Z-scheme) for photocatalytic CO2 reduction using ZCS/Au@CN. Reproduced with permission.222 Copyright 2021, Elsevier. | ||
As stated earlier, CO2 reduction is a multi-electron reduction process, where the charge separation and the band edge alignment dictate the accumulation of thermodynamically active photogenerated electrons near the CB, which is the key instructor in driving the CO2 photoreduction reaction. The mp-g-CN framework can form enormous composite photocatalysts enriched with improved light harvesting and phenomenal charge separation to achieve excellent efficiency in CO2 photoreduction. Madhusudan and co-workers reported a solid–solid Z-scheme ZnxCd1−xS/Au@g-C3N4 ternary heterojunction photocatalyst (ZCS/Au@CN) for CH3OH production via CO2 photoreduction, which was found to be 32.7 times higher than that of the neat mp-g-CN.222 ZCS/Au@CN was strategically fabricated using the photodeposition-assisted hydrothermally assembled process. The nanostructure investigation using electron microscopy analysis revealed that Au Nps were photodeposited on the external surface of ZCS, and mesoporous CN nanosheets wrapped into a thin layer around ZCS@Au to form the ZCS/Au@CN ternary composite (Fig. 44B-(a–d)). The resultant ternary photocatalyst, with its porous structure and large specific area, displayed a better CO2 adsorption and stronger visible light harvesting response. Additionally, ZCS/Au@CN facilitated efficient charge separation and fast interfacial charge migration (Fig. 44B-e). Au Nps acted as an electron mediator and enhanced photoreduction ability by exhibiting the surface plasmon resonance (SPR) effect. Several control experiments evidenced that CH3OH production was governed by a solid–solid Z-scheme mechanism (Fig. 44B-f). Under visible light irradiation, the photoexcited e− ultimately travelled from ZCS to the VB of CN through Au Nps. Moreover, due to the SPR effect, Au Nps also injected the hot electrons into the VB of CN, where the e−/h+ pair recombination occurred. Meanwhile, the photoexcited e− in the CB of CN remained separated and seamlessly participated in the 6 e−-reduction process to produce CH3OH (Fig. 44B-f).
In addition to the aforementioned examples, significant research progress has been made, showing that diverse modifications within mp-g-CN, as documented in the literature, have been adapted to achieve better efficiency in CO2 photoreduction (presented in Table 4).
| Photocatalysts | Synthesis method | Types of structural modification | CO2 reduction products | Cyclic stability | Light source | Ref. |
|---|---|---|---|---|---|---|
| CISCN-20 | Hard template method | Composite | CH3OH (42.7 μmol g−1 h−1) | 4 cycles | 300 W Xe lamp (λ > 420 nm) | 164 |
| ZCS/Au@CN | Template-free self-assembly method | Composite | CH3OH (1.31 μmol h−1 g−1) | 3 cycles | 300 W Xe lamp (λ ≥ 420 nm) | 222 |
| Ag/MCN | Hard template method | Ag loading | CO (1.66 μmol g−1 h−1) | 3 cycles | 300 W Xe lamp (850 nm ≥ λ ≥ 320 nm) | 223 |
| CPB-PCN | Template-free self-assembly method | Composite | CO (149 μmol h−1 g−1) | 3 cycles | 300 W Xe lamp (λ > 420 nm) | 226 |
| mpg-CNx|CoPPc | Hard template method | Composite | CO (11.9 μmol g−1) | 3 cycles | Xe lamp (100 mW cm−2) (λ > 400 nm) | 227 |
| 3% CdS-g-C3N4 | Hard template method | Composite | CH3OH (192.7 μmol g−1 h−1) | 5 cycles | 300 W Xe lamp | 228 |
| MCT | Template-free method | Composite | CH4 (2.117 μmol g−1 h−1) | — | 300 W Xe lamp (λ ∼ 420 nm) | 229 |
| NT/GCN-40 | Template-free method | Composite | CH3OH (13.74 μmol g−1 h−1) | 4 cycles | 300 W Xe lamp (λ > 420 nm) | 230 |
| TOCN-80 | Template-free method | Composite | CO (0.974 μmol g−1 h−1) | — | 300 W Xe lamp (λ > 420 nm) | 231 |
| PBUH/CN | Soft template method | B- and P-doping | CO (22.45 μmol g−1 h−1) | 4 cycles | 300 W high-pressure Xe lamp | 232 |
| 20CPB@TCN | Template-free method | Composite | CO (22.62 μmol g−1 h−1) | 4 cycles | 300 W Xe arc lamp (λ > 420 nm) | 233 |
| MCN-TMA-6 | Hard template method | C-doping | CO (116.6 μmol g−1) | 2 cycles | 300 W Xe lamp (λ ≥ 400) | 127 |
| 2% Cu-SCN | Template-free method | Cu modified S-doping | CH4 (2.33 μmol g−1), CO (9.6 μmol g−1) | 4 cycles | 500 W Xe arc lamp | 234 |
| AUNB/g-C3N4 | Soft template method | B-doping | CO (21.95 μmol g−1) | 5 cycles | 300 W xenon lamp (420 nm cutoff filter) | 235 |
| PCN-18A | Hard template method | Composite | CO (0.404 μmol g−1 h−1), CH4 (0.0378 μmol g−1 h−1), C2H4 (0.0481 μmol g−1 h−1) | 3 cycles | 300 W Xe lamp (λ ∼ 320–780 nm) | 236 |
| CN-AN21 | Template-free method | Ag-Ni doping | CO (77.65 μmol g−1), CH4 (17.89 μmol g−1) | 4 cycles | 300 W Xe lamp (λ > 420 nm) | 237 |
| T/U-0.65 | Template-free method | Composite | CO (21.65 μmol g−1), CH4 (73.40 μmol g−1) | 10 cycles | 300 W Xe lamp (λ ≥ 420 nm) | 238 |
| CNx-500 | Soft template method | N-defect | CH4 (9.26 μmol g−1) | 3 cycles | 500 W Xe arc lamp (λ > 400 nm) | 239 |
| OCN-Tube | Template-free method | O-doping | CH3OH (0.88 μmol g−1 h−1) | 3 cycles | 350 W Xe lamp (λ > 420 nm) | 240 |
| 12FLTC/BCN | Template-free method | Composite | CO (14.4 μmol g−1), | 3 cycles | 300 W Xe lamp (λ > 420 nm) | 241 |
5.4. mp-g-CN-based photocatalytic materials for H2O2 production
H2O2, recognized as a benign oxidant and viable liquid fuel candidate, has garnered global interest within the realm of photocatalysis owing to its synthesis from abundant natural resources, including oxygen, water, and solar energy. The photocatalytic H2O2 production by utilizing water and oxygen can be achieved via two pathways: (1) water oxidation reaction (WOR) and (2) oxygen reduction reaction (ORR) (Fig. 45). The ORR pathway holds paramount importance in photocatalytic H2O2 generation and can be achieved by two routes, i.e., (i) a direct one-step two-electron reduction process and (ii) a two-step single-electron reduction process (Fig. 45). In the case of two-electron reduction, O2 undergoes direct conversion to H2O2 by combining with two electrons and two protons. This process is also referred to as concerted 2e− ORR (eqn (4), Fig. 45). In the case of single electron reduction, O2 accepts a single electron to form a superoxide radical (O2˙−) (eqn (5), Fig. 45). Subsequently, O2˙− reacts with two protons (2H+) and an electron to form H2O2 (eqn (6), Fig. 45). Both pathways necessitate the presence of specific active sites and band structures to facilitate the production of H2O2. On the other hand, H2O2 production by WOR can be achieved via three pathways: (i) one-step two-electron pathway, (ii) two-step single electron pathway and (iii) four-electron pathway. The one-step, two-electron pathway is a thermodynamically uphill process as it is operated at a more positive oxidation potential (eqn (8), Fig. 45). The two-step single electron process involves the oxidation of H2O to a hydroxyl radical (OH˙) by photogenerated holes. Subsequently, OH˙ dimerizes to H2O2 via the 2e− WOR pathway (eqn (9) and (10), Fig. 45). The WOR pathway is a thermodynamically uphill process operated at a higher oxidative potential (eqn (8)–(10), Fig. 45) hence making it challenging to proceed. Since the WOR process is executed at a higher oxidative potential, H2O2 decomposes at this stage due to its h+ scavenger nature.28,242 Moreover, the four-electron WOR pathway is also possible (eqn (11), Fig. 45), where H2O is oxidized at a lower oxidative potential (1.23 VNHE) to generate O2 (eqn (11), Fig. 45). This generated O2 produces H2O2 by ORR (eqn (5) and (6), Fig. 45). The photocatalytic H2O2 production via ORR or WOR is further governed by the optimum adsorption of O2 (ORR) or H2O (WOR) on the photocatalyst surface and the appropriate band edge alignment of the photocatalyst.28,242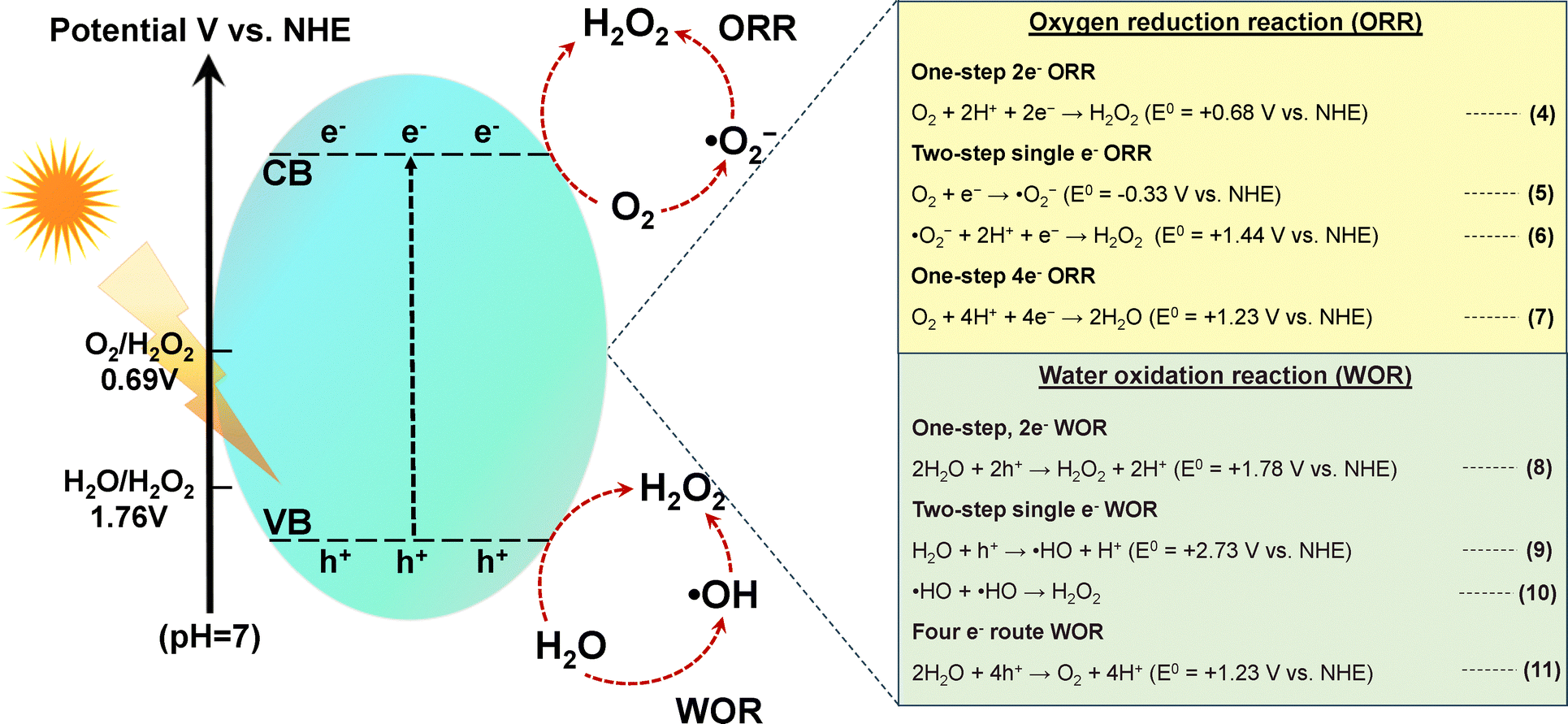 | ||
| Fig. 45 Schematic overview of the general mechanism of photocatalytic H2O2 production. | ||
mp-g-CN-based photocatalysts, with suitable active sites for O2 adsorption and activation and enhanced optical properties, have advanced the ORR process as reported in the literature.52,243 Unlike pristine g-C3N4, mp-g-CN-based photocatalysts are inherently enriched with several structural modifications like doping, vacancy and defects due to their various synthesis routes, which can further boost H2O2 production efficiency. For example, porous g-C3N4 with an inverse opal structure and C-vacancies (designated as IO CN-Cv) was constructed using monodispersed SiO2 microspheres for efficient photocatalytic H2O2 production under visible light irradiation.244 This hard template-assisted synthesis of IO CN-Cv in the ‘Ar’ atmosphere resulted in the co-existence of the inverse opal structure and C-vacancy framework. Fig. 46A-(a–c) represents the UV-Vis, bandgap energy and PL analysis of IO CN-Cv with various comparative photocatalysts synthesized in this study, such as bulk g-C3N4 (bulk CN), bulk g-C3N4 with C-vacancy (bulk CN-Cv) and g-C3N4 nanosheet (nanosheet CN). IO CN-Cv offered a larger specific surface area and a porous framework, which was reported to enhance the light absorption ability due to multiple scattering reflection phenomena (Fig. 46A-(a and b)). Both IO CN-Cv and bulk CN-Cv exhibited lower PL intensity (Fig. 46A-c), suggesting that C-vacancy improved the separation efficiency of charge carriers. Hence, the combinational effect of inverse opal structure and C-vacancy in IO CN-Cv demonstrated better visible light harvesting ability with improved charge carrier separation, resulting in superior photocurrent production and charge transfer resistance, endorsing a H2O2 production of 325.74 μM using 5% ethanol as a sacrificial agent (Fig. 46A-(d)). The ESR spin trapping experiment confirmed O2˙− involvement in H2O2 production as the active species. Fig. 46A-e demonstrates that after photoexcitation, the photoexcited h+ oxidizes ethanol to acetaldehyde and H+, while O2 is reduced to O2˙− by the photoexcited e−. The resultant O2˙− reacts with H+ to form H2O2.
 | ||
| Fig. 46 (A) (a) UV-vis DRS, (b) bandgaps, (c) PL spectra of bulk CN, bulk CN-Cv, nanosheet CN, and IO CN-Cv, (d) photocatalytic H2O2 activity with and without ethanol, (e) mechanism of photocatalytic H2O2 production over IO CN-Cv. Reproduced with permission.244 Copyright 2019, American Chemical Society. (B) SEM images of (a) CN, (b) and (c) O-CNC, TEM images of (d) CN, (e) and (f) O-CNC, (g) UV-vis DRS spectra, (h) band alignment, (i) photocatalytic H2O2 production rate, (j) mechanism of H2O2 production over O-CNC (here, O-CNC2, O-CNC4, and O-CNC6 correspond to 0.2, 0.4, and 0.6 g of oxalic acid used during the synthesis process). Reproduced with permission.101 Copyright 2021, American Chemical Society. | ||
Furthermore, mp-g-CN photocatalysts with structurally and functionally modified photosystems for excellent H2O2 production reported so far have been discussed below. Xie et al. reported a mesoporous O-doped, C-vacancy-rich g-C3N4 (O-CNC) using a template-free method by taking the MCA precursor with varying amounts of oxalic acid via a microwave synthesis approach for excellent photocatalytic H2O2 production.101 SEM and TEM analysis suggested that the oxalic acid-free photocatalyst (CN) exhibited a spherical porous structure (Fig. 46B-(a and d)); however, the addition of oxalic acid, which acted as a porogen, caused the rupture of the spherical porous balls into aggregated porous clusters (Fig. 46B-(b, c and e, f)). The addition of oxalic acid was responsible for the creation of O-doping by N-substitution and modifying the band structure. As a result, O-CNC photocatalysts showed a red shift in UV absorption with some additional absorption bands in the range of 450–500 nm due to the n–π* transition, probably due to the structural modifications after the oxalic acid introduction (Fig. 46B-g). The improved light-harvesting ability, along with enhanced charge carrier separation and transport in the O-CNC photocatalyst, was attributed to O-doping, carbon vacancies, and the mesoporous framework. As a result, the O-CNC photocatalyst exhibited an appropriate band alignment, as shown in Fig. 46B-h, to execute efficient H2O2 production by following the one electron reduction pathway, as confirmed by the p-benzoquinone (BQ) scavenging test. Therefore, O-CNC exhibited an enhanced photocatalytic H2O2 production of 2008.4 μmol h−1g−1 under simulated solar light irradiation by utilizing isopropanol (IPA) as the h+ scavenger, which was 4.4 times greater than that of pristine g-C3N4 (BCN) (Fig. 46B-i). Finally, the O-CNC with ‘O’ doping and ‘C’ vacancy garnered all the necessary features to exhibit extended light absorption to produce the photoinduced excitons. Due to the better charge separation ability of O-CNC, the photoinduced e− reduced O2 to O2˙− as the active intermediate, which was subsequently reduced to H2O2 (Fig. 46B-j).
In another study, Zhou and co-workers synthesized an ultrathin g-C3N4 nanosheet featuring a hierarchical mesoporous framework with P-doping (P-mMCNNS-25) by a one-step calcination method, which offered numerous active reaction sites and diffusion channels, thereby facilitating H2O2 generation in the liquid–solid–gas three-phase reaction interface.245 The PL emission band for P-mMCNNS-25 showed a blue shift (Fig. 47a) indicative of a reduced band gap, corresponding to the quantum confinement effect, where the CB position shifted to −0.32 eV and the VB position shifted to 2.39 eV. UV-DRS analysis and band gap energy analysis revealed a significant enhancement in the light-harvesting ability in P-mMCNNS-25 compared to bulk CN (Fig. 47b and c). Based on the optoelectronic results and VB XPS analysis, a band structure was proposed, as shown in Fig. 47d. First-principles calculations for the DFT study estimated the band structure by considering the band position, and in the projected band structure for bulk CN (Fig. 47e) the red color (bright) represents the contribution of C atoms, while the blue color (dark) corresponds to the contribution of N atoms. The partial charge density regions corresponding to the VB and CB were mostly developed by the C atoms and N atoms, respectively (Fig. 47f and g). Similarly, the modified band structure and the corresponding partial charge density regions of P-mMCNNS-25 resulted from the reduced band gap and P doping (Fig. 47h–j). The synergetic effect of several structural advancements, enhanced optoelectronic features, hierarchical mesoporous structure and phosphorus doping was responsible for the narrow band gap of P-mMCNNS-25. Due to this, it was difficult to achieve the reduction potential of O2/O2˙−; hence unlike pristine g-C3N4, P-mMCNNS-25 preferred the one-step 2e− reduction pathway over the two-step single-electron reduction pathway for H2O2 production. The DMPO spin-trapping ESR technique and radical-trapping experiments further validated the involvement of an efficient and faster one-step 2e− reduction pathway mechanism for P-mMCNNS-25 exhibiting an excellent H2O2 yield of 1083 μmol g−1 h−1.
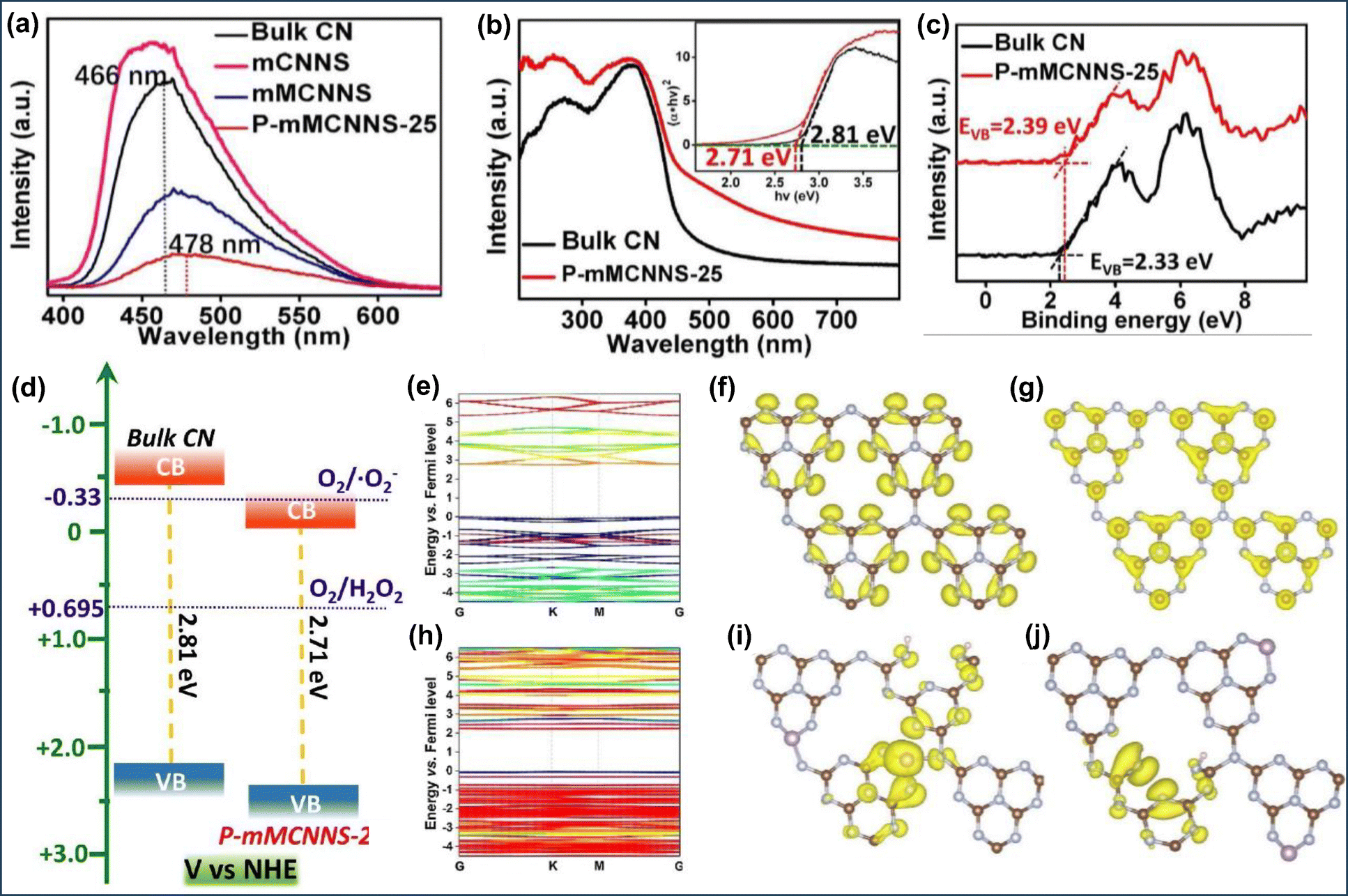 | ||
| Fig. 47 (a) PL spectra of all the prepared samples (here, mCNNS represents mesoporous g-C3N4 nanosheets and mMCNNS represents hierarchical porous g-C3N4 nanosheets), (b) UV-vis DRS and bandgap energy (inset), (c) VB XPS, (d) schematic of band structure alignment of bulk CN and P-mMCNNS-25, (e) band structure of bulk CN by DFT calculations, partial charge density of (f) and (g) VB and CB of bulk CN, (h) band structure of P-mMCNNS-25 by DFT calculations, partial charge density of (i) and (j) VB and CB of P-mMCNNS-25. Reproduced with permission.245 Copyright 2020, Elsevier. | ||
The interfacial contact within the mp-g-CN-based heterojunction provides adequate active site density to accelerate photocatalytic H2O2 production. For example, a Ti3C2 MXene/mp-g-CN interfacial Schottky junction (TC/pCN-X, where X = 1, 2, and 5, representing the mass ratio of Ti3C2 to porous g-C3N4) was fabricated via facile electrostatic self-assembly of Ti3C2 and porous g-C3N4, as reported by Yang and co-workers.246 The effective Schottky junction formed due to the strong coupling between the negatively charged surface of the Ti3C2 nanosheet and the positively charged surface of the porous g-C3N4 nanosheet as determined from the morphological investigation using electron microscopy analysis (Fig. 48a–c). FESEM EDS profiling suggested the absence of any additional impurities and the existence of the respective elements in the TC/pCN composite (Fig. 48d). The XPS analysis showed a significant peak shifting in the binding energies of the constituent elements, confirming the strong interaction of Ti3C2 nanosheets and g-C3N4 porous nanosheets in TC/pCN-2. Due to the strong interaction and Schottky junction formation, the visible light absorption capability was significantly improved. The slower charge recombination, longer lifetime of charge carriers, highest transient photocurrent response and better interfacial charge transfer phenomena were recorded for TC/pCN-2, suggesting that the optimum content of Ti3C2 successfully constructed a robust Schottky heterojunction (Fig. 48e–h). Further, the efficient spatial charge separation in TC/pCN-2 facilitated the two-step single electron reduction of O2 to O2˙−, as confirmed by the superoxide detection experiments (DMPO and NBT) and rotating disk electrode measurement, thereby augmenting the H2O2 production rate to 2.20 μmol L−1 min−1 under visible light irradiation (λ > 420 nm). A more detailed mechanistic investigation was proposed based on the work function estimation. The interfacial contact of pCN and Ti3C2 facilitated the flow of e− from pCN to Ti3C2 due to the lower work function of pCN, achieving Fermi level equilibrium. As a result, an upward band bending was experienced, resulting in the formation of a Schottky barrier. Consequently, the photoexcited e− in Ti3C2 couldn’t bounce back to pCN and remained separated, thereby boosting the photoreduction process as shown in Fig. 48qi.
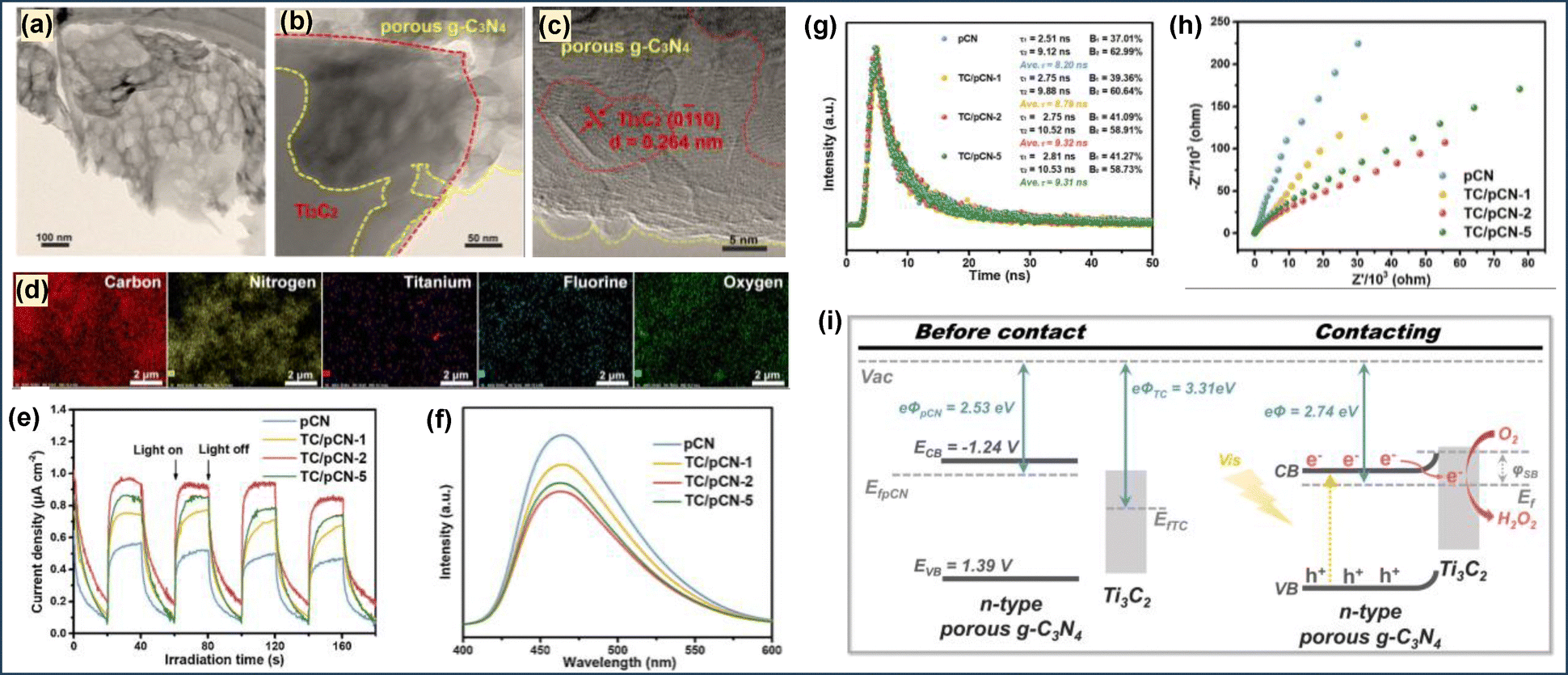 | ||
| Fig. 48 TEM images of (a) pCN and (b) TC/pCN, (c) HRTEM and (d) FESEM EDS mapping of TC/pCN, (e) transient photocurrent density, (f) PL, (g) TRPL, (h) EIS spectra of the synthesized samples, (i) photocatalytic mechanism of the TC/pCN Schottky heterojunction. Reproduced with permission.246 Copyright 2019, Elsevier. | ||
The addition of a co-catalyst to the structurally modified mp-g-CN photocatalyst enhances charge separation, accelerates the oxygen reduction reaction, and promotes the formation of the O2˙− active intermediate, thereby significantly boosting H2O2 production.247,248 Du et al. synthesized a Ni-loaded hollow one-dimensional oxygen-doped mesoporous nanotube (Ni/OtCN) using a template-free supramolecular self-assembly approach, comprising four steps, as shown in Fig. 49a.247 Ni/OtCN exhibited a porous and tubular structure having homogeneous dispersion of Ni Nps, as visualized by SEM, TEM, and HR-TEM analysis (Fig. 49b–d), and was characterized with a high surface area of 124 m2 g−1. The oxygen doping by substituting the ‘N’ atom and subsequent Ni loading lowered the band gap, improved the light-harvesting phenomena through wider light absorption, and was responsible for higher photocurrent production and efficient charge carrier separation. Further, the high surface area porous framework enriched with abundant active sites provides multiple diffusion channels for efficient O2 adsorption. Therefore, Ni/OtCN, with its beneficial structural characteristics, experienced a reduced kinetic threshold for H2O2 production by promoting the 2e− reduction process in the ORR pathway. The rotating ring-disk electrode (RRDE) analysis estimated that the ORR reaction was accomplished with the 2e−-transfer process, which was further validated by DFT calculations. The free energy calculations (ΔG) suggested that bCN exhibited the strongest adsorption of intermediates and hence showed the lowest kinetics. The ‘O’ doping and Ni deposition reduced the ΔG value to execute a 2e− reduction of the *OOH intermediate to form H2O2 (Fig. 49e). The H2O2 production activity trend further validated the influential role of oxygen doing, mesoporous channels, and Ni loading in Ni/OtCN, accomplishing an excellent evolution rate of 2464 μmol g−1 h−1. During the photocatalysis process, the photoexcited electrons in the CB of Ni/OtCN continuously migrated to the Fermi level, forming O2*− as an active intermediate to facilitate H2O2 formation exclusively. Several mp-g-CN-based photocatalytic materials with notable structural modifications for efficient H2O2 production are presented in Table 5.
 | ||
| Fig. 49 (a) Diagrammatic representation of the preparation of Ni/OtCN, (b) SEM, (c) TEM, (d) HRTEM image of Ni/OtCN, (e) free energy diagram for ORR. Reproduced with permission.247 Copyright 2022, Elsevier. | ||
| Photocatalysts | Synthesis method | Types of structural modification | H2O2 production rate | Cyclic stability | Light source | Ref. |
|---|---|---|---|---|---|---|
| IO CN-Cv | Hard template method | C vacancy | 325.74 μM | 5 cycles | 300 W Xe lamp (λ ≥ 420 nm) | 244 |
| P-mMCNNS-25 | Bubble template method | P-doping | 1083 μmol g−1 h−1 | 5 cycles | Simulated sunlight (filter: AM1.5) | 245 |
| 0.05 K-CN | Template-free method | KBr doping | 26.0 mmol g−1 h−1 | 3 cycles | 10 W Blue light lamp (λ ∼ 420–430 nm) | 249 |
| 2D-B-CN | Template-free method | B-doping | 4240.3 μM g−1 h−1 | 10 cycles | 20 W neon lamp (λ ∼ 400–800 nm) | 250 |
| CNPC | Template-free method | Homojunction composite | 642.54μM |
4 cycles | 300 W Xe lamp (λ > 420 nm) | 251 |
| KDCN-0.2 | Template-free method | K and CN modification | 278.9 μmol L−1 h−1 | 4 cycles | 300 W Xe lamp (λ ∼ 320–780 nm) | 252 |
| GCN | Hard template method | Surface defect | 92 μmol | — | 2 kW Xe lamp (λ > 420 nm) | 253 |
| ZnO/g-C3N4 | Template-free method | Composite | 1544 μmol L−1 h−1 | 4 cycles | 300 W Xe lamp (λ > 350 nm) | 254 |
| B-CNT, P-CNT, and S-CNT | Template-free method | B, S, P-doping | 42.31 μM min−1, 24.95 μM min−1, 24.22 μM min−1 | — | 300 W Xe lamp | 255 |
| LCHF@EDS-1 | Template-free method | Composite | 25.40 mmol g−1 h−1 | 10 cycles | 300 W Xe arc lamp (λ > 420 nm) | 256 |
| MCN | Template-free method | Doping | 1147.03 μM | 4 cycles | 50 W halogen lamp | 257 |
| Bi4O5Br2/g-C3N4 | Template-free method | Composite | 124 μM | 5 cycles | 300 W Xe lamp (λ > 420 nm) | 258 |
5.5. mp-g-CN-based photocatalytic materials for N2 photoreduction
The NRR can be achieved via two fundamental mechanisms, i.e., dissociative and associative mechanisms.259–261 In the dissociative mechanism, the NN bond decomposes on the photocatalyst surface to generate an *N adsorbed intermediate, which subsequently undergoes hydrogenation to NH3. Conversely, the associative mechanism occurs via two distinct pathways: the associative alternative pathway and the associative distal pathway. In the associative alternative pathway, both nitrogen atoms of the adsorbed NN are alternately protonated, leading to the release of NH3 molecules simultaneously. In contrast, the associative distal pathway involves the initial hydrogenation of the nitrogen atom farthest from the catalyst surface, as illustrated in Fig. 50a. As reported in numerous studies, the photocatalytic NRR predominantly follows the associative mechanism.259–261 The photocatalytic NRR to ammonia using various photocatalysts is considered an optimal process and currently holds a keen research interest. In this process, H+ and e− act as driving forces to produce NH3 through a competitive reaction between water splitting and N2 reduction (eqn (12)–(14)).23,24 Hence, it is obvious to compromise with the low quantum efficiency in NH3 production. Therefore, selecting a photocatalyst that can efficiently facilitate NRR over water splitting is crucial.| 2H2O + 4h+ → -O2 + 4H+ | (12) |
| N2 + 6H+ + 6e− → 2NH3 | (13) |
| 1/2N2 + 3/2H2O → NH3 + 3/4O2 | (14) |
 | ||
| Fig. 50 (a) Proposed mechanism of NRR on the surface of a heterogeneous catalyst, (b) schematic depiction of photocatalytic NRR. | ||
The key step in photocatalytic NRR is the adsorption cum activation of NN and achieving an appropriate band structure of the photocatalyst that facilitates the NRR over the OWS reaction. The N2 adsorption and activation process requires a high energy input due to the inert nature of N2, high dissociative enthalpy (941 kJ mol−1) of the strong nonpolar NN bond, high ionization energy (15.58 eV), and poor proton affinity (−1.90 eV). Upon light irradiation, the first photocatalytic step in NRR involves the oxidation of water to H+ and O2 at the VB of the semiconductor photocatalyst.23,262 Here, water acts as a hole scavenger as well as a proton donor for NH3 production. The in situ generated H+ from the hole-mediated oxidation of H2O at the VB can either react with the adsorbed cum activated N2 to produce NH3 and several other nitrogen compounds through a multi-electron reduction process or simply reduce to H2 at the CB (Fig. 50b).23,24,262 Hence, the photocatalytic NRR is a kinetically challenging process and requires an enormous amount of energy. The implementation of appropriate photocatalysts, such as functionally rich and porous modified g-C3N4 featuring optimized band structures, can lower the kinetic barrier, promote the chemisorption of molecular N2 and consequently improve the efficiency of photocatalytic NRR. Recently, several mp-g-CN-based photocatalysts have been employed for photocatalytic NH3 production.262–264
The doping of the ‘B’ like electron-deficient heteroatom in the mp-g-CN framework can fine-tune the electronic structure and thereby facilitate the NN bond adsorption and activation due to its Lewis base characteristics. Moreover, B doping also empowers the CB of the resultant photocatalyst to facilitate the multi-reduction process essential for photocatalytic NRR. For example, a ‘B’ doped mp-g-CN photocatalyst (2 wt% B/g-C3N4) was reported for photocatalytic N2 fixation with an NH3 production of 81.56 mol L−1 h−1, which was 2.44 times higher than that of g-C3N4.265 From the SEM images (Fig. 51a and b), 2 wt% B/g-C3N4 comprised the porous nanosheets assembled with a non-uniform stacked multilayer porous framework. 2 wt% B/g-C3N4 exhibited a redshift in light absorption with a lowered bandgap energy. Further, the ‘B’ doping caused the availability of a more significant number of unpaired electrons in the framework, in both the dark and light conditions. This structural modification was reported to expand the light transmission path length, improve the light-harvesting phenomena and reduce the charge transfer process to slow down the charge recombination. Thus, 2 wt% B/g-C3N4 exhibited better photocurrent density with minimum charge transfer resistance, beneficial for higher NH3 production compared to other photocatalysts. The mechanistic aspects of the photocatalytic NRR of 2 wt% B/g-C3N4 initiated with the H2O oxidation near its VB to produce O2 and H+ (Fig. 51c). Additionally, the unsaturated nature of the valence electrons of ‘B’ atoms weakens the NN, simultaneously enhancing the charge carrier separation. This process increased the population of photoexcited electrons at the CB of 2 wt% B/g-C3N4, consequently enabling the coupling of H+ with the activated and adsorbed NN to significantly produce NH3.
 | ||
| Fig. 51 SEM images of (a) g-C3N4 and (b) 2 wt% B/g-C3N4, (c) photocatalytic NRR activity of various synthesized photocatalysts. Reproduced with permission.265 Copyright 2023, Elsevier. | ||
Similarly, another example of B-doped mp-g-CN (BCN) photocatalyst was reported to enhance the adsorption and activation of the N2 molecule and smoothly drive the photocatalytic NRR.266 BCN was fabricated using a metal templated strategy followed by acid etching to attain a hollow tubular structure (Fig. 52a). The porous BCN framework allowed efficient diffusion of N2, making it accessible at the ‘B’ sites. The presence of B–N–C and B–N bonds created an e−-deficient BCN framework, which efficiently polarized the NN bond via the σ-donation and π-back donation process (Fig. 52b and c). The B-doping finely tuned the band structure of BCN, leading to significant improvements in light absorption and charge separation (Fig. 52d and e). During the aqueous-mediated photocatalytic N2 reduction, the consumption of photoexcited charge carriers led to the production of O2 and NH3, with a record yield of 1.2 mmol h−1 g−1 NH3 production. The 15N2 isotope labelling experiment was further conducted to confirm that NH3 was purely achieved from the N2 feed gas and not from the framework decomposition. The in situ DRIFT analysis revealed that the N2 adsorption (N–N adsorption peak at 1109 cm−1) on BCN-4 became more prominent in the presence of moisture (OH stretching band at 3678 cm−1) and light (Fig. 52f and g). The band at 1453 cm−1, corresponding to H–N–H bending vibration, appeared upon light irradiation, suggesting the seamless desorption of NH3 from the surface of BCN-4. In contrast, BCN-0 didn’t possess such type of band, highlighting the decisive role of B-doping in N2 photofixation. Additionally, the DFT calculations estimated that the B-doping sites reasonably enhanced the adsorption and activation of NN, forming the *N2 active intermediate, which was then easily protonated, boosting the NH3 production via the photocatalytic NRR. The transformation of *N2 to *NNH active species was the kinetic rate-limiting step in BCN-4. The B-doping in BCN-4 lowered the Gibbs free energy (ΔG) for N2 adsorption to −1.11eV, whereas BCN-0 corroborated with the ΔG value of +0.66 eV (Fig. 52h and i). This was attributed to the inconsistent chemisorption of N2 on BCN-0, which led to poor N2 fixation performance. Overall, the BCN framework exhibited poor water adsorption, better N2 adsorption, and more efficient NH3 desorption, making it more favorable for driving the photocatalytic NRR over photocatalytic OWS.
 | ||
| Fig. 52 (a) Schematic depiction of BCN fabrication, schematic depiction of N2 adsorption and activation at (b) transition metal and (c) BCN, (d) UV-vis DRS, (e) PL of BCN-0 and BCN-4, in situ DRIFT spectra of (f) BCN-0 and (g) BCN-4, DFT analysis of the NRR pathway on (h) BCN-0 and (i) BCN-4. Reproduced with permission.266 Copyright 2023, Elsevier. | ||
The substitution of the ‘C’ atom in the mp-g-CN framework with the ‘P’ atom can create abundant P-N active sites and facilitate NRR activity. For example, optimum doping of ‘P’ in the porous g-C3N4 nanosheet (PCN0.6) was accomplished by one-step thermal polymerization of urea and phosphonitrilic chloride, which was reported to improve N2 photo fixation significantly.267 A series of PCNx were prepared (where x represents the amount of phosphonitrilic chloride, x = 0.1, 0.3, 0.6, 0.9, and 1.2 g). Thermal decomposition of phosphonitrilic chloride dictated the P-doping and induced the porous architecture in PCNx. The successful substitution of the ‘C’ atom with ‘P’ created P-N electron transfer bridging sites, resulting in redshift and broadened light absorption with decreased band gap compared to g-C3N4 (denoted as CN) (Fig. 53A-a). The effect of ‘P’ doping in the band structure was further investigated using density of states analysis (DOS), which suggested that the VBM of CN and PCN0.6 was constructed by N atoms, while the CBM of CN was composed of a combination of C and N atoms. Similarly, the CBM of PCN0.6 was constructed by the combination of C, N, and P atoms (Fig. 53A-(b and c)). As a result, PCN0.6 exhibited reduced bandgap energies with a more negative CB potential, thereby associated with a favorable electronic band structure, resulting in enhanced photocurrent production and charge separation, favorable for the photocatalytic NRR (Fig. 53A-(d–f)). The spatial charge density was estimated by DFT calculations, which suggested that the electron distribution in PCN0.6 was more localized due to ‘P’ doping. As a result, the redox site was more dominantly separated in PCN0.6 compared to CN. Consequently, the P-doping sites acted as photoinduced electron reservoirs and efficiently facilitated charge separation. The charge density difference of the N2 adsorbed CN and PCN0.6 surface was further calculated by DFT analysis, which suggested that, ‘C’ atoms in CN served as the active sites for N2 adsorption, while in P-CN.6, the P atoms more preferentially adsorbed N2 with a very negative adsorption energy compared to the ‘C’ atom (Fig. 53A-(g and h)). The N2 TPD experiment also suggested that the N2 chemisorption significantly improved after P-doping due to electron distribution occurring between the P-doping site and adsorbed N2 (Fig. 53A-i). This electron-sharing process debilitated the binding energy of N2, and the Bader charge analysis estimated a greater number of electrons transferred from PCN0.6 to N2 compared to that for CN. Therefore, PCN0.6 possessed all the characteristic features for efficient photocatalytic NRR performance, achieving an NH3 production rate of 3110 μg L−1 h−1 g−1, which is 15 times greater than that of CN.
 | ||
| Fig. 53 (A) (a) UV-vis spectra, total DOS and partial DOS of (b) CN and (c) PCN0.6, (d) band structure, (e) transient photocurrent, (f) PL of CN and PCN0.6, calculation of the charge density difference of N2 adsorbed on the (002) facet of (g) CN and (h) PCN0.6, (i) N2-TPD of CN and PCN0.6. Reproduced with permission.267 Copyright 2024, Elsevier. (B) (a) UV-vis-DRS, (b) PL spectra, (c) transient photocurrent response, (d) EIS spectra of CN, VN-CN, Ni3B/VN-CN, and (e) proposed mechanism over Ni3B/VN-CN for photocatalytic NRR. Reproduced with permission.268 Copyright 2023, American Chemical Society (for interpretation of the references to color in this figure legend, the reader is referred to the web version of this article). | ||
The synergistic effect of both co-catalyst loading and defect engineering can have a promotional impact on the photocatalytic NRR. Zhang et al. reported a bubble template approach to fabricate an ‘N’-vacancy induced mp-g-CN (VN-CN) coupled with the Ni3B co-catalyst.268 The introduction of the ‘N’-vacancy and porous framework in the resultant photocatalyst Ni3B/VN-CN led to a redshift in light absorption and reduced bandgap compared to g-C3N4 (CN) (Fig. 53B-a). Further, the intimate contact of Ni3B and VN-CN in Ni3B/VN-CN formed an interfacial Schottky junction, which enhanced the light absorption ability, reduced charge recombination, and facilitated charge carrier separation and transportation (Fig. 53B-b). As a result, Ni3B/VN-CN exhibited higher photocurrent production with the smallest charge migration resistance (Fig. 53B-(c and d)). The charge transfer mechanism was investigated using the DFT analysis, which suggested that the higher work function (Φ) of Ni3B facilitated the flow of electrons from VN-CN to Ni3B until the Fermi level equilibrium. As a result, an internal electric field was developed at the interface of Ni3B and VN-CN and formed a Schottky junction. Therefore, during the photocatalytic NRR, photogenerated e− from the CB of VN-CN migrated to the Fermi level of Ni3B, where the N2 preferentially adsorbed and ultimately reduced to NH3 with a production rate of 7.68 mM g−1 h−1, which was 6.7 times greater than that of CN (Fig. 53B-e).
Various MNp-doped mp-g-CN-based photocatalysts have been demonstrated to significantly expedite the photocatalytic NRR significantly. Gao et al. reported the successful fabrication of copper-doped porous g-C3N4 (Cu@pg-C3N4) through a method involving dicyandiamide and nitric acid treatment driven supramolecular self-assembly, with the ethylenediamine tetraacetic acid copper disodium complex (Cu-EDTA) serving as both Cu precursor and CN framework modifier.269 Compared to bulk g-C3N4, the surface area of Cu@pg-C3N4 was significantly improved from 5.69 m2 g−1 to 75.76 m2 g−1, whereas the Cu-free porous g-C3N4 (pg-C3N4) displayed a surface area of 58.72 m2 g−1 (Fig. 54a). This suggested that Cu incorporation increased the adsorptive sites, which was further clarified by N2 TPD analysis (Fig. 54b). Cu sites served as the active sites for the physisorption of molecular N2, thereby accessing the abundant N2 molecules near these active sites (Cu) for the photocatalytic NRR. The porous framework intrinsically accommodated Cu-doping and offered a strong interaction of Cu sites and the C–N framework, as confirmed by the peak shifting in PXRD observations. Again, due to Cu incorporation and the porous nature of the material, the light absorption ability significantly improved and showed a redshift in the UV-Vis absorption with a decreased band gap (Fig. 54c and d). The suitable band structure, slower charge recombination process, higher photocurrent production response, and better charge transfer efficiency suggested that Cu incorporation played a crucial role in elevating the overall efficiency of Cu@pg-C3N4 compared to the other competent photocatalysts (Fig. 54e and f). The synergistic effect of the mesoporous functional structure and Cu incorporation drove the aqueous-mediated NRR reaction, as summarized below (eqn (vi)–(x)):
| pg-C3N4 + hv → pg-C3N4(h+ + e−) | (vi) |
| pg-C3N4(e−) + Cu → Cu(e−) | (vii) |
| 2H2O + 4h+ → 4H+ + O2 | (viii) |
| N2 + 6e− + 6H+ → 2NH3 | (ix) |
| NH3 + H2O → NH4+ + OH− | (x) |
 | ||
| Fig. 54 (a) N2 adsorption–desorption isotherms, (b) N2-TPD profiles, (c) UV-Vis DRS analysis, (d) band gap energies, (e) steady-state PL profile, (f) transient photocurrent analysis of the synthesized photocatalysts, and (g) N2 photofixation mechanism over Cu@pg-C3N4. Reproduced with permission.269 Copyright 2023, Wiley-VCH. | ||
Upon light irradiation, the spatially distributed Cu in Cu@pg-C3N4 trapped the photogenerated e−, while the photogenerated h+ oxidized H2O to generate H+. The photofixation of N2 was driven by the electron-rich Cu sites, efficiently producing NH4+ with a production rate of 150.47 μmol L−1, which was 20 times higher than that of bulk g-C3N4 (Fig. 54g).
Unlike photocatalytic H2 production, CO2 photoreduction and photocatalytic H2O2 production, there are comparatively limited reports on the photocatalytic NRR over mp-g-CN-based photocatalysts. Some recently reported mp-g-CN-based systems for efficient photocatalytic NRR activity are summarized in Table 6.
| Photocatalysts | Synthesis method | Types of structural modification | NH3 production rate | Cyclic stability | Light source | Ref. |
|---|---|---|---|---|---|---|
| B/g-C3N4 | Template-free method | B-doping | 164 μmol g−1 h−1 | 3 cycles | Visible light (λ > 420 nm) | 265 |
| PCN0.6 | Template-free method | P-doping | 3110 μg L−1 h−1 g−1 | 4 cycles | 300 W Xenon lamp (λ > 420 nm) | 267 |
| Ni3B/VN-CN | Temple-free self-assembly method | Composite | 7.68 mM g−1 h−1 | 5 cycles | 300 W Xe lamp | 269 |
| BCN | Template-free method | B-doping | 313.9 μmol h−1 g−1 | 5 cycles | 250 W Xe lamp (λ > 400 nm) | 270 |
| PF-g-C3N4 | Template-free method | N-vacancy | 315.54 μmol L−1 h−1 g−1 | 3 cycles | 300 W Xe lamp | 271 |
| HGCN | Template-free method | C-vacancy | 25.54 mg L−1 h−1 g−1 | — | 300 W Xe lamp (λ >420 nm) | 272 |
| GF-500 | Template-free method | Composite | 47.9 mg L−1 h−1 | 3 cycles | 300 W high-pressure Xe lamp | 273 |
| PDI/10H-CNv | Template-free method | Composite | 519.2 μmol h−1 g−1 | 8 cycles | 300 W Xe lamp | 274 |
| PCN-V | Template-free method | P-doping, N-vacancy | 9.3 μmol | — | 2 kW Xe lamp (λ > 420 nm) | 275 |
5.6. mp-g-CN-based photocatalytic materials for the production of liquid fuels using valorisation of lignocellulosic biomass
Lignocellulosic biomass mainly refers to organic residuals of wood, grass, and agricultural crop byproducts. Similar to other renewable precursors, lignocellulosic biomass is a promising feedstock for the production of various liquid-gaseous biofuels along with platform chemical commodities.31–36 Fig. 55 represents the complex structure of various building blocks of lignocellulosic biomass, mainly comprised of cellulose, hemicellulose, and lignin. All these building blocks mostly have C–C and C–O linkages, which need to be deconstructed in an efficient manner to produce various biofuels and bioproducts.31–36,276 Both cellulose and hemicellulose are linked via 1,4-β-glycosidic linkages and serve as precursors for several platform compounds, including sugars, furan derivatives such as furfural (FUR), furfuryl alcohol (FOL), 5-hydroxymethyl furfural (HMF), 2,5-furandicarboxylic acid (FDCA), 2,5-diformyl furan (DFF), 2-methyl furan (MF), levulinic acid/esters (LA/LEs), gammavalerolactone (GVL), etc.31–34 Similarly, lignin is made of phenylpropanoid building blocks that can produce various lignin oils, such as coniferyl alcohol, p-coumaryl alcohol, sinapyl alcohol, vanillin, syringaldehyde, etc., and lignin model compounds, and is characterized by aromatic building blocks having β-O-4, α-O-4, and 4-O-5 type of C–C and C–O linkages.31–34,276 The depolymerization followed by deoxygenation of these complex structures of the pre-treated lignocellulose biomass should necessarily be carried out to obtain value-added commodities, which is a challenging and energy-intensive process. In the literature, various biological and chemocatalytic approaches have been reported for utilizing lignocellulosic biomass and its model compounds to produce various fuel additives and chemical commodities.276,277 | ||
| Fig. 55 Schematic overview of the lignocellulosic biomass structure and its three main components. | ||
The high oxygen content in biofuels prevents their blending with transportation fuels. Therefore, various reductive catalytic approaches have been employed to deoxygenate the bio-oils and biofuels, making them potential for practical applications.32,278 A schematic presentation in Fig. 56 describes the systematic catalytic transformation of lignocellulosic biomass moieties for the production of bioproducts, diesel-like hydrocarbons and fuels. These catalytic processes typically require harsh reaction conditions and noble metal catalysts.35 Alternatively, photo-reformation of lignocellulosic biomass is a sustainable process that has been proven to bring tremendous revolutionary aspects in biorefineries and is only limited to a few developed countries.35,36,279 The photocatalytic valorization process has been successfully conducted on various biomass moieties for the production of liquid biofuels and bio-diesel through C–C coupling, decarboxylation, hydrogenolysis, hydrodeoxygenation of biomass-derived compounds or model compounds and transesterification of crude bio-oil.35,36,280 Photocatalytic lignocellulosic biomass transformation follows similar fundamental photocatalytic reaction steps as discussed in Fig. 1 and Section 5. During the photo-reformation of lignocellulosic biomass, reactive oxygen species, such as hydroxyl radicals, superoxide radicals, and singlet oxygen, are often generated. These active intermediates play a crucial role in valorizing lignocellulosic biomass moieties into various desired products.36 Furthermore, the lignocellulosic biomass can be mineralized to produce syngas (CO + H2) via the photo-reformation process. However, over-oxidation can lead to the production of CO2, which is undesirable. This issue can be mitigated by implementing strategic measures in photocatalyst design and modulating their active sites effectively.281
 | ||
| Fig. 56 Diagrammatic illustration of the stepwise chemical transformation pathway for the conversion of lignocellulosic biomass moieties to various value-added bioproducts and biofuels. | ||
Herein, the attention focuses on the solar energy-assisted transformation of lignocellulosic biomass feedstock into various sustainable fuel precursors and fuel additives using mp-g-CN-based photocatalytic systems. For example, the selective photooxidation of HMF to DFF was achieved using a modified and porous g-C3N4 photocatalyst.282 The pristine g-C3N4 (designated as g-C3N4-M) was synthesized by the calcination of melamine, followed by structural modification through water treatment. The resultant modified structure (g-C3N4) exhibited enhanced porosity and surface area, enriched with active sites beneficial for a very high degree of photoactivity. TEM analysis found that the closed sheet-like morphology of g-C3N4-M turned into a more open porous framework after the modification. As a result, g-C3N4 exhibited a higher surface area and pore volume than g-C3N4-M (Fig. 57a and b). The light absorption edge also exhibited a red shift after modification, suggesting improved light harvesting dominated by the improved textural properties and structural modification. Thus, g-C3N4 enriched with high surface area, porosity, enhanced light harvesting ability and improved charge recombination exclusively achieved a selectivity of 85.6% for DFF with a HMF conversion of 31.2% in the presence of O2 oxidant (Fig. 57c). Based on the scavenging tests and controlled experiments, the photoexcited e− reduced O2 to O2˙−, while the basicity of g-C3N4 deprotonated HMF and formed the corresponding alkoxide anion, which further reacted with photogenerated h+ to form a radical intermediate (Fig. 57d). This radical intermediate was further consumed to form DFF and FDCA (overoxidation product).
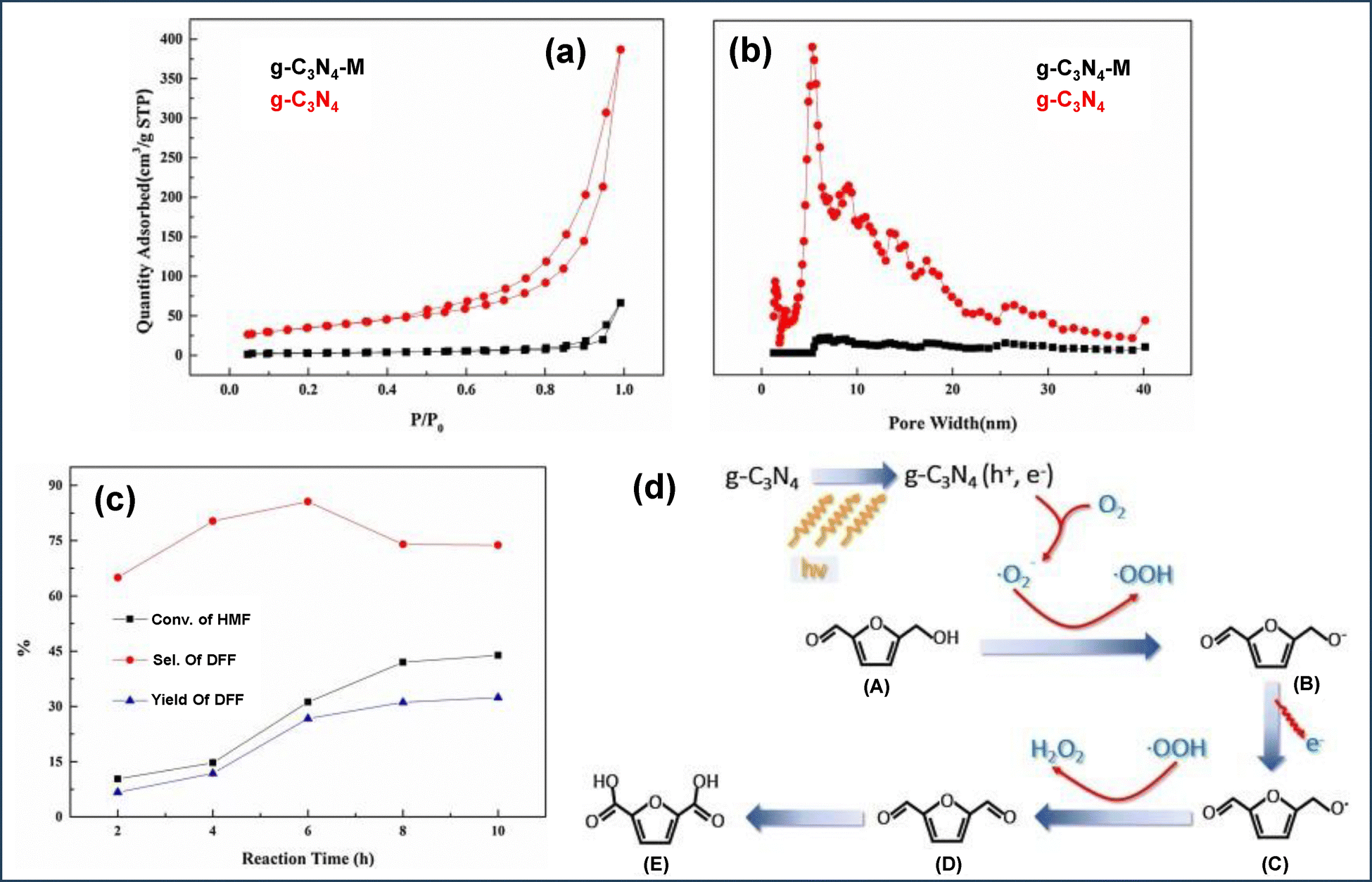 | ||
| Fig. 57 (a) BET isotherms, (b) pore size distribution plot of the samples, (c) time-dependent photocatalytic activity of g-C3N4-M, (d) mechanism for the photooxidation of HMF to DFF using g-C3N4-M. Reproduced with permission.282 Copyright 2017, Elsevier. | ||
Another example of HMF to DFF conversion was achieved over a mesoporous Z-scheme heterojunction of g-C3N4 and BiOCl.283 The SEM and EDS mapping suggested a successful close contact interface of g-C3N4 and BiOCl in the g-C3N4/BiOCl Z-scheme heterojunction (Fig. 58a–i). Moreover, XPS analysis found that the binding energies corresponding to C, N and O in the g-C3N4/BiOCl composite significantly shifted to higher values while the binding energies of Bi and Cl shifted to lower values which can be attributed to the electron transfer from g-C3N4 to BiOCl due to the higher Fermi energy level of g-C3N4 than BiOCl. Furthermore, the band alignment analysis suggested the creation of a compact and intact Z-type heterojunction interface with staggered energy band topologies in the g-C3N4/BiOCl composite, beneficial for enhanced light absorption and significant charge separation. The resultant Z-scheme g-C3N4/BiOCl composite also displayed higher photocurrent production and charge transfer efficiency than that of neat BiOCl and g-C3N4. Therefore, the g-C3N4/BiOCl composite efficiently facilitated the photocatalytic oxidation of HMF to DFF compared to g-C3N4 and BiOCl (Fig. 58j). The highest photocatalytic activity was achieved in acetonitrile solvent due to higher O2 solubility and low polarity (Fig. 58k). The band structure analysis revealed the formation of the Z-scheme heterojunction at the interface of BiOCl and g-C3N4. Therefore, during the photooxidation process, the photogenerated e− of BiOCl recombined with the photogenerated h+ of g-C3N4 while the photoexcited e− of g-C3N4 and the photoexcited h+ of BiOCl remained separate for redox reaction as shown in Fig. 58l. Further, the higher VB edge potential of BiOCl facilitated the HMF oxidation while O2 was reduced to O2˙− at the CB of g-C3N4.
 | ||
| Fig. 58 SEM image of (a) BiOCl, (b) g-C3N4, (c) g-C3N4/BiOCl, (d)–(i) EDS mapping of g-C3N4/BiOCl, (j) photocatalytic activity of various synthesized catalysts, (k) effect of solvents in the photocatalytic oxidation reaction over g-C3N4/BiOCl, (l) Z-scheme charge carrier separation mechanism for photocatalytic oxidation reaction. Reproduced with permission.283 Copyright 2024, Elsevier. | ||
The full utilization of lignocellulosic biomass moieties for the production of various value-added fuel precursors and chemicals requires a highly precise band engineering in the photocatalyst and advanced process engineering to execute the photo-valorization process. Ku et al. reported a ‘S’-doped and –CN group grafted mp-g-CN (MCSCN-75) photocatalyst through a template-free thermal decomposition of urea and trithiocyanuric acid for the photovalorization of waste Camellia oleifera shell (WCOS) lignin to monophenolic compounds via the cleavage of Cα–Cβ.284 A step-wise fabrication process of MCSCN-75 is presented in Fig. 59a. N2 sorption analysis suggested that MCSCN-75 was enriched with well-defined mesopores and high surface area compared to bulk g-C3N4 (Fig. 59b and c). FT-IR analysis indicated an asymmetric stretching vibration of –CN at 2170 cm−1 in MCSCN-75, confirming the successful –CN grafting in MCSCN-75 (Fig. 59d). MCSCN-75 exhibited a redshift in absorption, reduced bandgap, and enhanced light absorption ability due to extended conjugation from –CN grafting and ‘S’-doping. Due to the appropriate and improved photoresponse features, MCSCN-75 showed higher photogenerated charge carrier production, separation, and migration. Therefore, MCSCN-75 efficiently carried out the photovalorization of WCOS waste (Fig. 59e), producing various important value-added compounds like vanillin, syringaldehyde, etc. (Fig. 59f) compared to g-C3N4 (Fig. 59g).
 | ||
| Fig. 59 (a) Schematic depiction of the fabrication of MCSCN, (b) N2 adsorption–desorption isotherms, (c) pore-size distribution plot, (d) FT-IR, (e) the proposed structure of waste Camellia oleifera shell (WCOS) lignin, (f) GC–MS chromatogram of the reaction mixture, (g) photoactivity of bulk g-C3N4 and MCSCN-75. Reproduced with permission.284 Copyright 2022, Elsevier. | ||
Another photocatalytic lignocellulosic transformation involved the photooxidation of guaiacol, a lignin model compound, using the mp-g-CN photocatalyst in the presence of molecular O2 as a green oxidant.285 This process achieved an 82% conversion, yielding p-benzoquinone (59%), catechol (27%) and pyrogallol (6%) as the primary products. The VB-XPS analysis and the respective band structure of mp-g-CN revealed its ability to efficiently generate various reactive oxygen species (ROS), which facilitated the photooxidation process (Fig. 60a and b). The ˙OH DMPO spin trapping ESR analysis in operando mode confirmed the formation of the DMPO-˙OH complex and evidenced the photooxidation of guaiacol driven by the ˙OH radical. This photooxidation process was initiated by the attack of the ˙OH radical at the methoxy group of guaiacol to produce p-benzoquinone as the major product; however, the possibility of the attack of the ˙OH radical at the hydroxide centre was ruled out, as phenol as the photooxidation product was not observed in the reaction mixture (Fig. 60c).
 | ||
| Fig. 60 (a) VB-XPS analysis, (b) band structure and mechanism of light-driven production of ROS, (c) reaction pathway for the photooxidation of guaiacol. Reproduced with permission.285 Copyright 2021, Elsevier. | ||
Solar-assisted hydrogen production via photo-reformation of lignocellulosic biomass is an interesting research topic which serves as an alternative to OWS and HER.281 The H2 production via photo-reformation of lignocellulosic biomass in the oxidation-half reaction is energetically more favourable, as the oxidation potential required for this process is lower than that needed for the oxidation of water. The photo-reformation of lignocellulosic biomass also offers simultaneous production of sustainable H2 and value-added bioproducts due to the utilization of photogenerated e− and h+, respectively. The photogenerated h+ oxidizes the lignocellulosic biomass moieties, while the photogenerated e− reacts with the aqueous H+ to produce H2 (Fig. 61).286 For example, Xie et al. reported a carbon ring incorporated two-dimensional porous g-C3N4 (g-C3N4–C0.4) photocatalyst for photo-reformation of α-cellulose under near-infrared (NIR) irradiation.287 Fig. 62a represents a schematic depiction of the fabrication of g-C3N4–C0.4, achieved by the dehydration and subsequent facile polymerization of melamine and glucose. The successful incorporation of the C–C–C conjugated carbon ring in g-C3N4–C0.4 was confirmed by 13C NMR, XPS, N2 adsorption analysis, etc. A distinct single peak in the 13C NMR of g-C3N4–C0.4 at 57 ppm was attributed to the presence of the C–C–C aromatic ring in the g-C3N4–C0.4 nanostructure (Fig. 62b). The C-ring incorporation intercepted the C–N framework growth to prevent the formation of bulk-g-C3N4, thereby creating abundant adsorptive sites to increase the specific surface area of g-C3N4–C0.4 compared to g-C3N4 (Fig. 62c). The TEM images of g-C3N4 were associated with closely stacked nanosheets, whereas g-C3N4–C0.4 possessed small-sized porous nanosheets (Fig. 62d–g). Due to this, the photogenerated charge carrier transfer distance shortened and exposed more surface-active sites in g-C3N4–C0.4. The optical absorption analysis suggested that g-C3N4–C0.4 exhibited redshift and enhanced NIR light absorption ability compared to g-C3N4 (Fig. 62h). The sluggish charge carrier recombination property, greater lifetime of charge carriers and slower charge transfer resistance attributed to the C-ring incorporation were beneficial for achieving superior photoresponse ability in g-C3N4–C0.4 compared to g-C3N4 (Fig. 62i–k). The appropriate band structure of g-C3N4–C0.4 was characterized using MS measurement, VB-XPS analysis, and bandgap measurement, which suggested the formation of an intermediate band (IB) in g-C3N4–C0.4 due to the presence of a conjugated C–C–C ring. This ring, associated with plenty of unpaired e−, as confirmed by the EPR analysis (Fig. 62l), enabled fast photoinduced e− transfer flow and constructed the polarization of charge density distribution to separate the charge carriers efficiently (Fig. 62l–o). Hence, compared to g-C3N4, g-C3N4–C0.4 favourably displayed a higher photocurrent production under the NIR region to accomplish the photo-reformation process. Moreover, the DMPO scavenging test confirmed that the OH˙ radical and O2˙− reactive intermediates were actively engaged in the photo-reformation of α-cellulose to exclusively produce H2 with a production rate of 2.1 μmol h−1 along with several bioproducts such as mannose, galacturonic acid, glucuronic acid, galactose, lactic acid, formic acid, and HMF.
 | ||
| Fig. 61 Schematic illustration of the photo-reformation of lignocellulosic biomass moieties for the simultaneous production of H2 and value-added bioproducts. | ||
 | ||
| Fig. 62 (a) Schematic representation of g-C3N4–C0.4 synthesis, (b) solid-state 13C NMR, (c) BET isotherm and pore-size distribution (inset) of g-C3N4 and g-C3N4–C0.4, TEM images of (d) and (e) g-C3N4, (f) and (g) g-C3N4–C0.4, (h) UV-Vis-NIR DRS, (i) PL, (j) TRPL, (k) EIS spectra, (l) EPR spectra of the synthesized materials, (m) band alignment of g-C3N4 and carbon, (n) and (o) charge transfer mechanism and estimated charge transfer pathway of g –C3N4–C0.4. Reproduced with permission.287 Copyright 2023, American Chemical Society (for interpretation of the references to color in this figure legend, the reader is referred to the web version of this article). | ||
Biomass model compounds can also serve as alternatives to traditional photogenerated h+ sacrificial agents such as triethanolamine, methanol, etc., usually utilized in the photocatalytic HER.281,286 This approach offers a sustainable and meaningful strategy for producing value-added products (H2 and bio-products) by fully utilizing the photoinduced charge carriers. For example, a 2D/2D LaVO4/g-C3N4 (LaVO4/CN) photocatalyst was fabricated for the simultaneous oxidation of furfuryl alcohol and H2 production under visible light irradiation.288 This 2D/2D heterojunction was synthesized via a self-assembly approach, and the regular interfacial contact of LaVO4 and g-C3N4 was visualized in the LaVO4/CN heterojunction. Various composites with different LaVO4 loadings (x% LaVO4/CN, where x = 5, 10, 15, 20, and 25) were prepared and characterized. TEM, HR-TEM and PXRD analyses demonstrated a strong and intimate interaction of LaVO4 and g-C3N4 in the LaVO4/CN heterojunction (Fig. 63a–e). LaVO4/CN composites displayed superior light harvesting ability compared to individual LaVO4 and g-C3N4 (Fig. 61g) and significantly diminished the charge carrier recombination rate (Fig. 63f and g). These improved optoelectronic properties in LaVO4/CN composites triggered better photocurrent production and efficient charge carrier separation with lower charge transfer resistance. The charge transfer dynamics was analysed by TRPL analysis, which suggested that the shorter fluorescence lifetime of LaVO4/CN corresponded to nonradiative electronic charge transfer between LaVO4 and CN, favoring the efficient migration and maximum utilization of the charge carriers (Fig. 63h). The Z-scheme LaVO4/CN heterojunction featured a staggered energy band structure between LaVO4 and g-C3N4 components, where the Fermi energy level of LaVO4 was positioned far below that of g-C3N4 (Fig. 63i). Charge separation was facilitated by the formation of an internal electric field at the interface, where photogenerated e− in the CB of LaVO4 recombined with photogenerated h+ in the VB of g-CN. Consequently, h+ in the VB of LaVO4 and e− in the CB of g-C3N4 exclusively separated and remained available to execute the redox reaction. This mechanism enabled the oxidation of furfuryl alcohol to furfural at the VB of LaVO4 and the reduction of water to produce H2 at the CB of g-C3N4, with the assistance of a Pt co-catalyst (Fig. 63i).
 | ||
| Fig. 63 TEM images of (a) CN, (b) LaVO4, (c) LaVO4/CN, (d) HRTEM of LaVO4/CN, (e) XRD, (f) PL, (g) UV-vis, (h) TRPL, (i) Z-scheme photocatalytic mechanism of LaVO4/CN for H2 and furfural production. Reproduced with permission.288 Copyright 2022, Elsevier. | ||
The simultaneous production of various valuable platform chemicals/fuel additives along with the H2 production via photo-reformation of lignocellulosic biomass encounters several bottlenecks such as product selectivity, separation, diffusion, and catalyst deactivation, which consequently requires more attention for its practicality.
Based on the above-given examples in the solar energy conversion for sustainable fuel production using mp-g-CN-based photocatalysts, the ideal design principles of mp-g-CN photocatalysts are primarily associated with the synchronous effect of improved textural properties, optimized electronic structure and facile redox reactivities. In a deeper context, extended light absorption, enhanced charge separation, and creating exposed surface-active sites without affecting the structural integrity are the key points for designing the most efficient mp-g-CN-based photocatalysts for solar fuel production. The hierarchical mesoporous framework with a narrow bandgap in mp-g-CN, exhibiting multiple scattering of light, is of particular interest for enhancing light-harvesting ability. Moreover, the mesoporous architecture and high surface area impart accessible active sites and promote better mass transport. Proton adsorption and H2 desorption processes are exclusively dominated in the mesoporous framework of mp-g-CN-based systems, hence facilitating the efficient photocatalytic H2 production. Various synthesis approaches can lead to mp-g-CN with a variety of nanostructures, such as 2D and 3D interconnected porous networks, which shortens the charge diffusion path, preventing fast charge recombination. However, the template-free synthesis approach is currently blooming as an emerging technique for cost-effective large-scale mp-g-CN production. Further, on advocating the most appealing features in terms of advanced modification strategies, the electron orbital regulation due to the heteroatom doping and defect engineering modifies the hybridization state in the mp-g-CN framework, resulting in fine-tuning of the band alignment and electronic properties, narrowing the band gap and creating mid-gap states. Further, the spin state regulation by N-vacancy or metal doping maintains the spin dynamics, boosting the charge separation and kinetics of photoactivity in solar energy conversion. The alteration of the chemical and physical state by surface functionalization, such as –NH2, –OH, –CN, –COOH, etc., can modify the microenvironment of active sites in mp-g-CN, resulting in better surface reactivity by the promotional effect of adsorption of reactants and intermediates, directional charge transfer behavior to reduce recombination, optimum hydrophilicity to prevent photocorrosion and favoring co-catalyst integration by acting as the anchoring sites for efficient solar fuel production. The functional groups, such as –SO3H and –COOH, grafted on mp-g-CN based photocatalysts can significantly boost the acid-catalyzed biofuel production. The appropriate heterojunction construction can significantly improve the spatial charge separation, boost the charge utilization and escalate the redox potential to achieve efficient photoredox activity in solar fuel production. Due to the optimum porosity and surface area, the strategic composite of the mp-g-CN photocatalyst with redox mediators or OER catalysts such as BiVO4 or MnOx can significantly enhance the H2O splitting reaction by promoting the OER for achieving higher H2 production. Overall, it is absolutely required to judiciously install the multifunctionality in the mp-g-CN framework, depending upon the science and challenges associated with a specific type of application. Therefore, appropriate design principles and advanced structural modifications of mp-g-CN collectively underpin its potential in solar energy conversion for efficient solar fuel production.
6. Summary, challenges and future prospects
This review underscores mp-g-CN as a transformative photocatalytic platform for solar energy conversion, addressing the intrinsic limitations of pristine g-C3N4. Several research findings demonstrate that the porous framework in mp-g-CN based photocatalysts enables many characteristic features to enhance the photoresponsive factors by many fold by enhancing the light-harvesting phenomena via multiple scattering effects, shortening the migration path of charge carriers, optimizing the band alignment, and creating an electron transport system, through the quantum confinement effect. All these integrated features can seamlessly trigger the surface redox reaction to achieve a reasonable quantum efficiency over mp-g-CN-based photocatalysts. By realizing the importance of this hot topic, we anticipate that the demonstration of current research activities in the present critical review would essentially expedite future advancements in this fast-developing field. Overall, we provide an inclusive estimation of the rising of mp-g-CN-based photocatalytic materials and the ongoing scenario of their synthesis approaches with structural and functional modifications, aiming to achieve an effective solution for semiconductor-based photocatalysis in terms of solar energy production. We believe the summary of the collection of various effective strategies for synergistically regulating the material characteristic features of mp-g-CN can open a new horizon to fabricate more committed g-C3N4-based photocatalysts for highly efficient solar-energy conversion.We have discussed several fundamental and advanced synthesis methodologies for the development of mp-g-CN photocatalysts for solar energy conversion. Then we have summarized that the hard template-assisted synthesis method provides superior structural control, yielding an ordered and precisely defined porous mp-g-CN relative to soft template approaches. Furthermore, supramolecular self-assembly has emerged as the predominant strategy, surpassing conventional template-assisted methods by offering sustainable and hazardous chemical-free methods for synthesizing mp-g-CN with tailored dimensions and morphology. We have then highlighted the impact of various structural modification strategies to optimize the electronic properties by controlling dimension and morphology, doping and defect engineering, surface functionalization, and forming composite heterojunctions. Such adjustments can efficiently improve the light harvesting ability and charge carrier separation of mp-g-CN-based photocatalysts. Thereafter, we have systematically discussed the various aspects of sustainable solar energy production from natural gases and lignocellulosic biomass as renewable precursors. The systematic investigation of the current research progress of mp-g-CN photocatalysts deals with the method of fabrication, important characterization to determine the structural features, optoelectronic properties, active site identification, reaction pathways, and involved mechanism investigations for solar energy production applications. We have observed that compared to several existing modified g-C3N4 photocatalysts, either structurally or functionally modified mp-g-CN photocatalysts have shown excellent responses towards visible light absorption and efficient charge separation ability. To advance mp-g-CN-based photocatalytic materials for artificial photosynthesis, future research should focus on advancements in their fabrication and execution as discussed below:
• As the research fraternity is continuously focused on the development of environmental sustainability by carbon capture and clean energy production using photocatalysis, g-C3N4-based materials have been considered as the best choice to achieve the goal towards sustainable energy production using artificial photosynthesis. Therefore, continuous efforts should be made to explore more research in the development and advancement of mp-g-CN-based photocatalytic systems.
• Even though porosity engineering and structural modifications enhance the light-harvesting properties, developing mp-g-CN-based materials with a broader range of light absorption ability remains critical to maximize solar light utilization. Moreover, researchers are actively working on advancing the porous g-C3N4 photocatalysts by fine-tuning from the molecular level to achieve an optimized mp-g-CN framework, aiming to improve physicochemical and optoelectronic properties that can suppress charge recombination and maintain photostability over repeated cycles. However, it remains a formidable challenge to achieve; therefore, the active researchers must carry forward these studies further in advancing the mp-g-CN-based photocatalysts.
• It still remains unclear as to the type of specific modification the mp-g-CN photocatalysts need to target a particular type of photocatalytic application. Since it is an ever-growing research field, a plethora of articles have been published over the past few decades. Hence, it demands a precise database and simulation methods ensuring structure controllability, photoactivity, and product selectivity, which will help the future researchers to think precisely for advancing and strengthening the research input.
• mp-g-CN photocatalysts synthesized by template-assisted and non-templated approaches exhibit different dimensions and morphology. Hence, it is necessary to develop simulation methods and modelling to gain a deeper understanding of further modification and execution. This approach can provide a direction for the rational design of highly efficient mp-g-CN-based photocatalytic materials.
• Currently, researchers are more focused on developing mp-g-CN using template-free synthesis strategies and avoiding the multiple-step associated template-assisted strategies. However, it can open a new opportunity to advance the current template-free synthesis route by rationalizing the g-CN precursors in order to develop mp-gCN materials with reproducible textural properties. Large-scale production of mp-g-CN-based catalysts and laboratory-to-industry technology transformation for their practical implementation towards solar energy production involve several key factors such as cost-effective synthesis strategies in pilot plants, process designing, appropriate photoreactor modelling, minimizing the environmental detriments, and maintaining atom economy based upon the existing infrastructure.
• It can be noted that the use of sacrificial reagents for various artificial photosynthesis applications, like OWS and other solar energy production, creates economic barriers for their practical implementation. Hence, it is imperative to develop multifunctional mp-g-CN-based photocatalysts that can enable seamless solar energy production without using sacrificial agents and additives.
• mp-g-CN-based photocatalysts can efficiently mimic natural photosynthesis. The Lewis base nature and porosity of mp-g-CN enable enhanced CO2 adsorption. Additionally, the structural properties of mp-g-CN allow for the incorporation of multifunctionality by embedding or grafting active moieties into its mesochannels, which can improve the H2O splitting reaction. Furthermore, the photoexcited active intermediates generated from the H2O splitting reaction can interact with the adsorbed and activated CO2, facilitating the formation of carbon-based fuels.
• The lignocellulosic biomass transformation requires precise and modulated active sites for the activation of lignocellulosic biomass moieties. The active sites, typically the acidic and basic sites, can be easily transcribed into mp-g-CN-based photocatalysts to activate the lignocellulosic biomass moieties. Additionally, the porous channel is beneficial for mass diffusion, and the CN framework provides Lewis basicity, which can promote esterification reactions, biomass condensation reactions, isomerization, and cyclization reactions. Since this is a new research area, only a limited studies have been carried out so far for lignocellulosic biomass transformation using mp-g-CN-based photocatalysts. However, the strategic development and modifications of mp-g-CN can lead to the development of highly promising photocatalysts for various challenging biomass transformations.
• Although porous g-C3N4 can more efficiently develop composite photocatalysts compared to other modified g-C3N4 analogues, continuous effort should focus on the novel fabrication techniques to ensure the photostability and heterogeneity of the mp-g-CN-based composites. For instance, metal sulphide-based composites exhibit a strong photocorrosion effect due to the oxidation of S2− by photoexcited holes, which can be resolved by making an appropriate heterojunction structure (S-scheme or Z-scheme) that can channelize the charge carriers and mitigate the photocorrosion. Similarly, doping, vacancy, and defect engineering in the mp-g-CN framework can improve the charge mobility and prevent the oxidative damage of the composite photocatalyst.
• Integrating structurally and functionally modified mp-g-CN-based photocatalytic materials with solar cell technologies in tandem or integrated systems can be an interesting aspect of renewable energy production using a sustainable approach.
• Being a nitrogen-rich compound and porous framework with a high surface area, mp-g-CN can be explored in electrocatalysis to further increase its horizon in sustainable energy production. The hierarchical porosity and high surface area enable faster diffusion and accessible active site distribution. The pyridinic ‘N’ atom in the framework, having a strong electron-withdrawing ability, acts as the active site in the electrocatalytic process, such as ORR, HER, H2O splitting, and fuel cell application. The robustness of mp-g-CN-based materials contributes to their long-term catalytic stability since the electrocatalytic processes are carried out in acidic or alkaline media. Additionally, mp-g-CN can be ideal for the intercalation of Li+, as well as serve as a good separator and anode electrode for lithium-ion batteries. However, the poor electrical conductivity and sluggish charge transfer conductivity are the main limitations which push back its substantial usage in various electrocatalytic processes. Also, limited systematic investigations were unable to provide a clear depiction of the potential challenges and prospects of mp-g-CN-based materials in electrocatalysis.
• Finally, the future prospects for mp-g-CN photocatalysts are promising due to the current research scenario. This topic has the potential to carve out a significant space among the research fraternity, encompassing multidisciplinary research; hence, it encourages more collaborative research among different disciplines, from science to engineering.
Data availability
This review article only utilizes data assessment of the published articles by various researchers in this domain, which can be found from the sources referenced in the manuscript. No new data were generated during the preparation of this review. The required copyright permission has also been taken.Conflicts of interest
There are no conflicts to declare.Acknowledgements
PD is thankful to the Siksha ‘O’ Anusandhan (Deemed to be University) management for providing the necessary facilities and their constant support and motivation.References
- B. H. Kreps, Am. J. Econ. Sociol., 2020, 79, 695–717 CrossRef.
- T. Ahmad and D. Zhang, Energy Rep., 2020, 6, 1973–1991 CrossRef.
- D. Gielen, R. Gorini, N. Wagner, R. Leme, L. Gutierrez, G. Prakash, E. Asmelash, L. Janeiro, G. Gallina, G. Vale and L. Sani, Global energy transformation: a roadmap to 2050, 2019.
- J. L. Holechek, H. M. E. Geli, M. N. Sawalhah and R. Valdez, Sustainability, 2022, 14, 4792 CrossRef.
- S. Parida and D. P. Dutta, in Biorenewable Nanocomposite Materials, Vol. 1: Electrocatalysts and Energy Storage, ed. D. Pathania and L. Singh, ACS Symposium Series, American Chemical Society, Washington, DC, 2022, vol. 1410, pp. 307–366 Search PubMed.
- K. Kaygusuz, Renewable Sustainable Energy Rev., 2012, 16, 1116–1126 CrossRef.
- N. S. Lewis, Science, 2016, 351, aad1920 CrossRef PubMed.
- K. Kohse-Hoeinghaus, Chem. Rev., 2023, 123, 5139–5219 CrossRef CAS PubMed.
- M. J. B. Kabeyi and O. A. Olanrewaju, Front. Energy Res., 2022, 9, 743114 CrossRef.
- P. J. Megía, A. J. Vizcaíno, J. A. Calles and A. Carrero, Energy Fuels, 2021, 35, 16403–16415 CrossRef.
- H. Blanco and A. Faaij, Renewable Sustainable Energy Rev., 2018, 81, 1049–1086 CrossRef.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
- H. Wang, Q. Guo, H. Zhang and C. Zuo, Carbon Resour. Convers., 2024, 7, 100263 CrossRef CAS.
- A. Behera, A. K. Kar and R. Srivastava, Mater. Horiz., 2022, 9, 607–639 RSC.
- S. A. Fors and C. A. Malapit, ACS Catal., 2023, 13, 4231–4249 CrossRef CAS.
- R. J. Detz, J. N. H. Reek and B. C. C. van der Zwaan, Energy Environ. Sci., 2018, 11, 1653–1669 RSC.
- A. Hayat, M. Sohail, J. Ali Shah Syed, A. G. Al-Sehemi, M. H. Mohammed, A. A. Al-Ghamdi, T. Taha, H. Salem AlSalem, A. M. Alenad and M. A. Amin, Chem. Rec., 2022, 22, e202100310 CrossRef CAS PubMed.
- P. Kuang, M. Sayed, J. Fan, B. Cheng and J. Yu, Adv. Energy Mater., 2020, 10, 1903802 CrossRef CAS.
- S. Adhikari, S. Fernando, S. R. Gwaltney, S. Filipto, R. Markbricka, P. Steele and A. Haryanto, Int. J. Hydrogen Energy, 2007, 32, 2875–2880 CrossRef CAS.
- P. Zhou, I. A. Navid, Y. Ma, Y. Xiao, P. Wang, Z. Ye, B. Zhou, K. Sun and Z. Mi, Nature, 2023, 613, 66–70 CrossRef CAS PubMed.
- M. S. Reza, N. B. H. Ahmad, S. Afroze, J. Taweekun, M. Sharifpur and A. K. Azad, Chem. Eng. Technol., 2022, 46, 420–434 CrossRef.
- P. K. Panda, B. Sahoo and S. Ramakrishna, Energy Technol., 2023, 11, 2201434 CrossRef.
- G. Zhang, Y. Li, C. He, X. Ren, P. Zhang and H. Mi, Adv. Energy Mater., 2021, 11, 2003294 CrossRef CAS.
- S. Lin, X. Zhang, L. Chen, Q. Zhang, L. Ma and J. Liu, Green Chem., 2022, 24, 9003–9026 RSC.
- J. G. Chen, R. M. Crooks, L. C. Seefeldt, K. L. Bren, R. M. Bullock, M. Y. Darensbourg, P. L. Holland, B. Hoffman, M. J. Janik, A. K. Jones, M. G. Kanatzidis, P. King, K. M. Lancaster, S. V. Lymar, P. Pfromm, W. F. Schneider and R. R. Schrock, Science, 2018, 360, eaar6611 CrossRef PubMed.
- J. Humphreys, R. Lan and S. Tao, Adv. Energy Sustainability Res., 2021, 2, 2000043 CrossRef CAS.
- C. M. Goodwin, P. Lömker, D. Degerman, B. Davies, M. Shipilin, F. Garcia-Martinez, S. Koroidov, J. Katja Mathiesen, R. Rameshan, G. L. S. Rodrigues, C. Schlueter, P. Amann and A. Nilsson, Nature, 2024, 625, 282–286 CrossRef CAS PubMed.
- L. Wang, J. Zhang, Y. Zhang, H. Yu, Y. Qu and J. Yu, Small, 2022, 18, 2104561 CrossRef CAS PubMed.
- H. Hou, X. Zeng and X. Zhang, Angew. Chem., Int. Ed., 2020, 59, 17356–17376 CrossRef CAS PubMed.
- E. Anetjärvi, E. Vakkilainen and K. Melin, Energy, 2023, 276, 127202 CrossRef.
- S. Wang, A. Cheng, F. Liu, J. Zhang, T. Xia, X. Zeng, W. Fan and Y. Zhang, Ind. Chem. Mater., 2023, 1, 188–206 RSC.
- S. Shylesh, A. A. Gokhale, C. R. Ho and A. T. Bell, Acc. Chem. Res., 2017, 50, 2589–2597 CrossRef CAS PubMed.
- C. Z. Li, X. C. Zhao, A. Q. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed.
- G. W. Huber, P. O'Connor and A. Corma, Appl. Catal., A, 2007, 329, 120–129 CrossRef CAS.
- N. Keller, J. Ivanez, J. Highfield and A. M. Ruppert, Appl. Catal., B, 2021, 296, 120320 CrossRef CAS.
- H. Zhao, J. Liu, N. Zhong, S. Larter, Y. Li, M. G. Kibria, B.-L. Su, Z. Chen and J. Hu, Adv. Energy Mater., 2023, 13, 2300257 CrossRef CAS.
- S. K. Lee, A. Mills and C. O'Rourke, Chem. Soc. Rev., 2017, 46, 4877–4894 RSC.
- F. Wang, Q. Li and D. Xu, Adv. Energy Mater., 2017, 7, 1700529 CrossRef.
- J. Fu, J. Yu, C. Jiang and B. Cheng, Adv. Energy Mater., 2018, 8, 1701503 CrossRef.
- W. J. Ong, L. L. Tan, Y. H. Ng, S. T. Yong and S. P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS.
- S. Patnaik, S. Martha, S. Acharya and K. Parida, Inorg. Chem. Front., 2016, 3, 336–347 RSC.
- B. P. Mishra and K. Parida, J. Mater. Chem. A, 2021, 9, 10039–10080 RSC.
- S. Patnaik, A. Behera and K. Parida, Catal. Sci. Technol., 2021, 11, 6018–6040 RSC.
- S. Patnaik, D. P. Sahoo and K. Parida, Renewable Sustainable Energy Rev., 2018, 82, 1297–1312 CrossRef CAS.
- P. Mishra, A. Behera, D. Kandi and K. M. Parida, Nanoscale Adv., 2019, 1, 1864–1879 RSC.
- S. K. Sahoo, L. Acharya, L. Biswal, P. Priyadarshini and K. Parida, Inorg. Chem. Front., 2024, 11, 491–4973 RSC.
- J. Wen, J. Xie, X. Chen and X. Li, Appl. Surf. Sci., 2017, 391, 72–123 CrossRef CAS.
- L. Wang, K. Wang, T. He, Y. Zhao, H. Song and H. Wang, ACS Sustainable Chem. Eng., 2020, 8, 16048–16085 CrossRef CAS.
- Y. Li, X. Li, H. Zhanga and Q. Xiang, Nanoscale Horiz., 2020, 5, 765–786 RSC.
- S. Samanta and R. Srivastava, Mater. Adv., 2020, 1, 1506–1545 RSC.
- R. Ghalta and R. Srivastava, Catal. Sci. Technol., 2023, 13, 1541–1557 RSC.
- H. Dhiman, R. Ghalta, R. Bal and R. Srivastava, ACS Appl. Nano Mater., 2024, 21, 24819–24832 CrossRef.
- R. Ghalta and R. Srivastava, Sustainable Energy Fuels, 2023, 7, 1707–1723 RSC.
- R. S. de Almeida Ribeiro, L. E. Monteiro Ferreira, V. Rossa, C. G. Lima, M. W. Paixaõ, R. S. Varma and T. de Melo Lima, ChemSusChem, 2020, 13, 3992–4004 CrossRef CAS PubMed.
- B. D. Zdravkov, J. J. Čermák, M. Šefara and J. Janků, Cent. Eur. J. Chem., 2007, 5, 385–395 CAS.
- B. C. Pan, Y. Xiong, A. M. Li, J. L. Chen, Q. X. Zhang and X. Y. Jin, React. Funct. Polym., 2002, 53, 63–72 CrossRef CAS.
- Y. Ma, H. Li, J. Liu and D. Zhao, Nat. Rev. Chem., 2024, 8, 915–931 CrossRef PubMed.
- A. K. Kar and R. Srivastava, ACS Sustainable Chem. Eng., 2019, 7, 13136–13147 CrossRef CAS.
- S. L. Suib, Chem. Rec., 2017, 17, 1169–1183 CrossRef CAS PubMed.
- R. J. White, R. Luque, V. L. Budarin, J. H. Clark and D. J. Macquarrie, Chem. Soc. Rev., 2009, 38, 481–494 RSC.
- J. Wang and S. Wang, Coord. Chem. Rev., 2022, 453, 214338 CrossRef CAS.
- N. S. N. Hasnan, M. A. Mohamed and Z. A. Mohd Hir, Adv. Mater. Technol., 2022, 7, 2100993 CrossRef CAS.
- M. Ismael and Y. Wu, Sustainable Energy Fuels, 2019, 3, 2907–2925 RSC.
- S. Patnaik, D. P. Sahoo and K. Parida, Carbon, 2021, 172, 682–711 CrossRef CAS.
- M. A. Khan, S. Mutahir, I. Shaheen, Y. Qunhui, M. Bououdina and M. Humayun, Coord. Chem. Rev., 2025, 522, 216227 Search PubMed.
- Y. Liu, M. Tayyab, W. Pei, L. Zhou, J. Lei, L. Wang, Y. Liu and J. Zhang, Small, 2023, 19, 2208117 CrossRef CAS PubMed.
- K. S. Lakhi, D. H. Park, K. Al-Bahily, W. Cha, B. Viswanathan, J.-H. Choy and A. Vinu, Chem. Soc. Rev., 2017, 46, 72–101 RSC.
- S. Sun and S. Liang, Nanoscale, 2017, 9, 10544–10578 RSC.
- J. Dong, Y. Zhang, M. I. Hussain, W. Zhou, Y. Chen and L. N. Wang, Nanomaterials, 2022, 12, 1–35 Search PubMed.
- D. A. Giannakoudakis, N. A. Travlou, J. Secor and T. J. Bandosz, Small, 2017, 13, 1601758 CrossRef PubMed.
- H. Wang, Y. Fu, X. Liu, R. Yang, Y. Hu, D. Liu, J. Wan and Z. Zeng, Sens. Actuators, B, 2023, 377, 132796 CrossRef CAS.
- L. Chen, Z. Yang and B. Chen, Energy Fuels, 2021, 35, 10746–10755 CrossRef CAS.
- Z. Yang, Y. Zhang and Z. Schnepp, J. Mater. Chem. A, 2015, 3, 14081–14092 RSC.
- X. Huo, H. Yi, Y. Fu, Z. An, L. Qin, X. Liu, B. Li, S. Liu, L. Li, M. Zhang, F. Xu, G. Zeng and C. Lai, Environ. Sci.: Nano, 2021, 8, 1835–1862 RSC.
- Y. Li and J. Shi, Adv. Mater., 2014, 26, 3176–3205 CrossRef CAS PubMed.
- A. Vinu, K. Ariga, T. Mori, T. Nakanishi, S. Hishita, D. Golberg and Y. Bando, Adv. Mater., 2005, 17, 1648 CrossRef CAS.
- M. Groenewolt and M. Antonietti, Adv. Mater., 2005, 17, 1789–1792 CrossRef CAS.
- F. Goettmann, A. Fischer, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2006, 45, 4467–4471 CrossRef CAS PubMed.
- J. Liu, J. Yan, H. Ji, Y. Xu, L. Huang, Y. Li, Y. Song, Q. Zhang, H. Xu and H. Li, Mater. Sci. Semicond. Process., 2016, 46, 59–68 CrossRef CAS.
- W. Chen, M. Liu, X. Li and L. Mao, Appl. Surf. Sci., 2020, 512, 145782 CrossRef CAS.
- W. Wang, Z. Shu, J. Zhou, T. Li, P. Duan, Z. Zhao, Y. Tan, C. Xie and S. Cui, Appl. Clay Sci., 2018, 158, 143–149 CrossRef CAS.
- Z. Yang, Z. Xing, Q. Feng, H. Jiang, J. Zhang, Y. Xiao, Z. Li, P. Chen and W. Zhou, J. Colloid Interface Sci., 2020, 568, 255–263 CrossRef CAS PubMed.
- X. Ren, X. Zhang, R. Guo, X. Li, Y. Peng, X. Zhao and X. Pu, Sep. Purif. Technol., 2021, 270, 118797 CrossRef CAS.
- H. K. Bisoyi and S. Kumar, Chem. Soc. Rev., 2011, 40, 306–319 RSC.
- H. Yan, Chem. Commun., 2012, 48, 3430–3432 RSC.
- A. K. Kar, A. Chauhan and R. Srivastava, Sustainable Energy Fuels, 2023, 7, 1855–1869 RSC.
- X. L. Wang, Q. Liu, Q. Yang, Z. G. Zhang and X. M. Fang, Carbon, 2018, 136, 103–112 CrossRef CAS.
- S. Zhao, Y. W. Zhang, Y. M. Zhou, Y. Y. Wang, K. B. Qiu, C. Zhang, J. S. Fang and X. L. Sheng, Carbon, 2018, 126, 247–256 CrossRef CAS.
- Q. Wang, Y. Hu, D. Zeng, L. Tian, J. Song, E. Cui, H. Yuan and R. Guan, J. Solid State Chem., 2023, 319, 123805 CrossRef CAS.
- K. Zhao, I. Khan, K. Qi, Y. Liu and A. Khataee, Mater. Chem. Phys., 2020, 253, 123322 CrossRef CAS.
- S. Zhao, Y. Zhang, Y. Wang, Y. Zhou, K. Qiu, C. Zhang, J. Fang and X. Sheng, J. Power Sources, 2017, 370, 106–113 CrossRef CAS.
- M. Zhang, J. Xu, R. Zong and Y. Zhu, Appl. Catal., B, 2014, 147, 229–235 CrossRef CAS.
- A. N. Kadam, H. Kim and S. W. Lee, Ceram. Int., 2020, 46, 28481–28489 CrossRef CAS.
- F. He, G. Chen, Y. S. Zhou, Y. G. Yu, Y. Zheng and S. E. Hao, Chem. Commun., 2015, 51, 16244–16246 RSC.
- X. Fu, X. Hu, Z. Yan, K. Lei, F. Li, F. Cheng and J. Chen, Chem. Commun., 2016, 52, 1725–1728 RSC.
- B. L. Phoon, C. W. Lai, G. T. Pan, T. C. K. Yang and J. C. Juan, Nanomaterials, 2021, 11, 2041 CrossRef CAS PubMed.
- P. Sharma, P. P. Sarngan, A. Lakshmanan and D. Sarkar, J. Mater. Sci.: Mater. Electron., 2022, 33, 9116–9125 CrossRef CAS.
- Z. Ya, X. Jiang, P. Wang, J. Cai, Q. Wang, H. Xie, S. Xiang, T. Wang and D. Cai, Small, 2023, 19, 2208254 CrossRef CAS PubMed.
- S. Zhang, C. An, R. Zhang, D. Kong, D. Xu and S. Zhang, Langmuir, 2024, 40, 6453–6462 CrossRef CAS PubMed.
- L. Chen, S. Yang, W. Qi, Q. Zhang, J. Zhu and P. Zhao, ACS Appl. Mater. Interfaces, 2021, 13, 49993–50004 CrossRef CAS PubMed.
- H. Xie, Y. M. Zheng, X. L. Guo, Y. Y. Liu, Z. Zhang, J. J. Zhao, W. J. Zhang, Y. X. Wang and Y. Huang, ACS Sustainable Chem. Eng., 2021, 9, 6788–6798 CrossRef CAS.
- X. J. Lu, C. Z. Yuan, S. Chen, J. H. Li, I. Ullah, M. Qi and A. W. Xu, Langmuir, 2024, 40, 11067–11077 CrossRef PubMed.
- H. Luo, T. Shan, J. Zhou, L. Huang, L. Chen, R. Sa, Y. Yamauchi, J. You, Y. Asakura, Z. Yuan and H. Xiao, Appl. Catal., B, 2023, 337, 122933 CrossRef CAS.
- X. Sun, Y. Shi, J. Lu, W. Shi and F. Guo, Appl. Surf. Sci., 2022, 606, 154841 CrossRef CAS.
- B. Shen, Z. H. Hong, Y. L. Chen, B. Z. Lin and B. F. Gao, Mater. Lett., 2014, 118, 208–211 CrossRef CAS.
- X. Chen, L. Cheng, Y. Yang, X. Chen, F. Chen and W. Lu, ACS Sustainable Chem. Eng., 2023, 11, 1518–1526 CrossRef CAS.
- L. Chen, M. A. Maigbay, M. Li and X. Qiu, Adv. Powder Mater., 2023, 1, 100150 Search PubMed.
- J. Pei, H. Li, S. Zhuang, D. Zhang and D. Yu, Catalysts, 2023, 13, 1402 CrossRef CAS.
- A. V. Abega, C. Marchal, M.-A. Dziurla, N. C. B. Dantio and D. Robert, Mater. Chem. Phys., 2023, 304, 127803 CrossRef CAS.
- A. Hayat, M. Sohail, A. El Jery, K. M. Al-Zaydi, K. F. Alshammari, J. Khan and M. Z. Ansari, Chem. Rec., 2023, 23, e202200171 CrossRef CAS PubMed.
- S. Kumar, T. Surendar, B. Kumar, A. Baruah and V. Shanker, RSC Adv., 2014, 4, 8132–8137 RSC.
- S. Stefa, M. Griniezaki, M. Dimitropoulos, G. Paterakis, C. Galiotis, G. Kiriakidis, E. Klontzas, M. Konsolakis and V. Binas, ACS Appl. Nano Mater., 2023, 6, 1732–1743 CrossRef CAS.
- X. Dong and F. Cheng, J. Mater. Chem. A, 2015, 3, 23642–23652 RSC.
- S. Meng, X. Jiang and Z. Nan, Surf. Interfaces, 2023, 42, 103390 CrossRef CAS.
- Y. Liu, S. Shen, Z. Li, D. Ma, G. Xu and B. Fang, Mater. Charact., 2021, 174, 111031 CrossRef CAS.
- Y. Huang, J. Liu, C. Zhao, X. Jia, M. Ma, Y. Qian, C. Yang, K. Liu, F. Tan, Z. Wang, X. Li, S. Qu and Z. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 52603–52614 CrossRef CAS PubMed.
- H. A. El-Sabban, S. Y. Attia, M. A. Diab and S. G. Mohamed, J. Energy Storage, 2023, 60, 106593 CrossRef.
- A. K. Adepu, R. Anumula and V. Narayanan, Microporous Mesoporous Mater., 2017, 247, 86–94 CrossRef CAS.
- W. Liu, M. Sun, Z. Ding, Q. Zeng, Y. Zheng, W. Sun and X. Meng, Sep. Purif. Technol., 2022, 282, 120097 CrossRef CAS.
- Q. Li, J. Yang, D. Feng, Z. Wu, Q. Wu, S. S. Park, C. S. Ha and D. Zhao, Nano Res., 2010, 3, 632 CrossRef CAS.
- S. N. Talapaneni, G. P. Mane, A. Mano, C. Anand, D. S. Dhawale, T. Mori and A. Vinu, ChemSusChem, 2012, 5, 700 CrossRef CAS PubMed.
- S. N. Talapaneni, S. Anandan, G. P. Mane, C. Anand, D. S. Dhawale, S. Varghese, A. Mano, T. Mori and A. Vinu, J. Mater. Chem., 2012, 22, 9831 RSC.
- G. P. Mane, D. S. Dhawale, C. Anand, K. Ariga, Q. Ji, M. A. Wahab, T. Mori and A. Vinu, J. Mater. Chem. A, 2013, 1, 2913 RSC.
- V. K. Tomer, R. Malik and K. Kailasam, ACS Omega, 2017, 2, 3658–3668 CrossRef CAS PubMed.
- B. Antil, R. Ranjan, C. S. Gopinath and S. Deka, J. Mater. Chem. A, 2020, 8, 13328–13339 RSC.
- P. Wang, S. Wei, Y. Han, S. Lin, L. Zhang, Q. Li, Y. Xu, L. Lian, Y. Zhou, M. Song and W. Zhuang, Langmuir, 2024, 40, 14045–14056 CrossRef CAS PubMed.
- A. M. Sadanandan, M. Fawaz, N. P. Dharmarajan, M. Huš, G. Singh, C. I. Sathish, B. Likozar, Z. Li, A. M. Ruban, C. H. Jeon and J. H. Yang, Appl. Catal., B, 2025, 362, 124701 CrossRef CAS.
- Y. Wang, J. Zhang, X. Wang, M. Antonietti and H. Li, Angew. Chem., Int. Ed., 2010, 49, 3356–3359 CrossRef CAS.
- W. Shen, L. Ren, H. Zhou, S. Zhang and W. Fan, J. Mater. Chem., 2011, 21, 3890–3894 RSC.
- Q. Yan, C. Zhao, L. Zhang, Y. Hou, S. Wang, P. Dong, F. Lin and Y. Wang, ACS Sustainable Chem. Eng., 2019, 7, 3866–3874 CrossRef CAS.
- A. Ahmed, P. John, M. H. Nawaz, A. Hayat and M. Nasir, ACS Appl. Nano Mater., 2019, 2, 5156–5168 CrossRef CAS.
- W. Iqbal, B. Yang, X. Zhao, M. Waqas, M. Rauf, C. Guo, J. Zhang and Y. Mao, Appl. Catal., A, 2019, 573, 13–21 CrossRef CAS.
- J. Shen, H. Yang, Q. Shen, Y. Feng, Q. Cai and H. Yang, Eur. J. Inorg. Chem., 2015, 2611–2618 CrossRef CAS.
- J. Xu, Z. Wang and Y. Zhu, ACS Appl. Mater. Interfaces, 2017, 9, 27727–27735 CrossRef CAS PubMed.
- F. He, S. Cheng, H. Song, C. Zhao, J. Zhang, S. Wang and H. Sun, Langmuir, 2022, 38, 828–837 CrossRef CAS PubMed.
- L. Lei, H. Fan, Y. J. Li, L. J. Miao and W. Wang, ACS Appl. Mater. Interfaces, 2024, 16, 14809–14821 CrossRef CAS PubMed.
- S. Patnaik, B. P. Mishra and K. Parida, Catal. Sci. Technol., 2021, 11, 7505–7524 RSC.
- W. Wang, Q. Niu, G. Zeng, C. Zhang, D. Huang, B. Shao, C. Zhou, Y. Yang, Y. Liu, H. Guo, W. Xiong, L. Lei, S. Liu, H. Yi, S. Chen and X. Tang, Appl. Catal., B, 2020, 273, 119051 CrossRef CAS.
- R. C. Pawar, S. Kang, J. H. Park, J. Kim, S. Ahn and C. S. Lee, Sci. Rep., 2016, 6, 31147 CrossRef CAS PubMed.
- T. Wu, Z. Liu, B. Shao, Q. Liang, Q. He, Y. Pan, X. Zhang, Y. Liu, J. Sun and S. Gong, Chem. Eng. J., 2022, 447, 137332 CrossRef CAS.
- Q. Su, Y. Li, R. Hu, F. Song, S. Liu, C. Guo, S. Zhu, W. Liu and J. Pan, Adv. Sustainable Syst., 2020, 4, 2000130 CrossRef CAS.
- D. Lan, H. Zhu, J. Zhang, C. Xie, F. Wang, Y. Zheng, Z. Guo, M. Xu and T. Wu, Ind. Eng. Chem. Res., 2024, 63, 3491–3503 CrossRef CAS.
- L. Pang, C. Jia, A. Gao, G. Zhang, S. Zhao, J. Cui and Y. Yan, Colloids Surf., A, 2024, 697, 134339 CrossRef CAS.
- A. Anus and S. Park, Chem. Eng. J., 2024, 486, 150213 CrossRef CAS.
- Y. Han, M. Chao, C. Luo and L. Yan, J. Colloid Interface Sci., 2024, 657, 309–319 CrossRef CAS.
- Z. Zhao, X. Wang, S. Wang, Z. Xiao, S. Zhai, J. Ma, X. Dong, H. Sun and Q. An, Langmuir, 2022, 38, 11054–11067 CrossRef CAS.
- N. Wang, L. Cheng, Y. L. Liao and Q. J. Xiang, Small, 2023, 19, 2300109 CrossRef CAS.
- N. S. N. Hasnan, M. A. Mohamed and Z. A. Mohd Hir, Adv. Mater. Technol., 2022, 7, 2100993 CrossRef CAS.
- X. Yu, S. F. Ng, L. K. Putri, L. L. Tan, A. R. Mohamed and W. J. Ong, Small, 2021, 17, e2006851 CrossRef PubMed.
- X. Xu, Y. Xu, Y. Liang, H. Long, D. Chen, H. Hu and J. Z. Ou, Mater. Chem. Front., 2022, 6, 3143–3173 RSC.
- Y. Guo, G. Liu, W. Yin, Y. Zhang and L. Shi, Fuel, 2024, 362, 130743 CrossRef CAS.
- Y. Li, W. Ho, K. Lv, B. Zhu and S. C. Lee, Appl. Surf. Sci., 2018, 430, 380–389 CrossRef CAS.
- F. He, G. Chen, Y. Yu, Y. Zhou, Y. Zheng and S. Hao, Chem. Commun., 2015, 51, 425–427 RSC.
- H. Zhang, X. Hu, Y. Tang, H. Zhang and K. Li, Microporous Mesoporous Mater., 2023, 360, 112733 CrossRef CAS.
- N. Tian, Y. Zhang, X. Li, K. Xiao, X. Du, F. Dong, G. I. Waterhouse, T. Zhang and H. Huang, Nano Energy, 2017, 38, 72–81 CrossRef CAS.
- G. Li, R. Wang, B. Wang and J. Zhang, Appl. Surf. Sci., 2020, 517, 146212 CrossRef CAS.
- G. Li, B. Wang, J. Zhang, R. Wang and H. Liu, Chem. Eng. J., 2020, 391, 123500 CrossRef CAS.
- M. Majdoub, Z. Anfar and A. Amedlous, ACS Nano, 2020, 14, 12390–12469 CrossRef CAS PubMed.
- T. Fidan, M. Torabfam, Q. Saleem, C. Wang, H. Kurt, M. Yüce, J. Tang and M. K. Bayazit, Adv. Energy Sustainability Res., 2021, 2, 2000073 CrossRef CAS.
- M. A. D. Fard, H. Ghafuri and A. Rashidizadeh, Microporous Mesoporous Mater., 2019, 274, 83–93 CrossRef.
- N. S. N. Hasnan, M. A. Mohamed, N. A. Nordin, W. N. R. W. Ishak and M. B. Kassim, Carbohydr. Polym., 2023, 317, 121096 CrossRef CAS PubMed.
- C. Bian, B. Zhou, F. Mo, X. Liu, P. Sun and X. Dong, Sep. Purif. Technol., 2023, 325, 124556 CrossRef CAS.
- H. Mao, Q. Zhang, F. Cheng, Z. Feng, Y. Hua, S. Zuo, A. Cui and C. Yao, Ind. Eng. Chem. Res., 2022, 61, 8895–8907 CrossRef CAS.
- H. Liu, Z. Zhang, J. Meng and J. Zhang, Mol. Catal., 2017, 430, 9–19 CrossRef CAS.
- Y. Ouyang, H. Jiang, H. Zhong, G. Li, X. Ji, Q. Jin, W. Xue, X. Hu and M. Zhu, Chem. Eng. J., 2023, 464, 142542 CrossRef CAS.
- H. Li, W. Xu, J. Qian and T. T. Li, Chem. Commun., 2021, 57, 6987–6990 RSC.
- J. Tang, R. P. Xu, G. Z. Sui, D. X. Guo, Z. L. Zhao, S. S. Fu, X. Yang, Y. Li and J. L. Li, Small, 2023, 19, 2208232 CrossRef CAS.
- B. Salah, A. Abdelgawad, Q. Lu, A. K. Ipadeola, R. Luque and K. Eid, Green Chem., 2023, 25, 6032–6040 RSC.
- W. Xing, F. Ma, Z. Li, A. Wang, M. Liu, J. Han, G. Wu and W. Tu, J. Mater. Chem. A, 2022, 10, 18333–18342 RSC.
- Z. Mo, X. Zhu, Z. Jiang, Y. Song, D. Liu, H. Li, X. Yang, Y. She, Y. Lei, S. Yuan, H. Li, L. Song, Q. Yan and H. Xu, Appl. Catal., B, 2019, 256, 117854 CrossRef CAS.
- H. Yang, Y. Zhou, Y. Wang, S. Hu, B. Wang, Q. Liao, H. Li, J. Bao, G. Ge and S. Jia, J. Mater. Chem. A, 2018, 6, 16485–16494 RSC.
- C. Zhu, B. Cheng, L. Y. Zhang and J. G. Yu, Carbon Energy, 2019, 1, 32–56 CrossRef.
- S. Adekoya, S. Qian, X. Gu, W. Wen, D. Li, J. Ma and S. Zhang, Nano-Micro Lett., 2021, 13, 13–57 CrossRef PubMed.
- B. C. Zhu, L. Y. Zhang, D. F. Xu, B. Cheng and J. G. Yu, J. CO2 Util., 2017, 21, 327–335 Search PubMed.
- Y. Kim, S. Kim, S. Premkumar, J. H. Yang, S. Umapathy and A. Vinu, Small, 2020, 16, 1903572 CrossRef PubMed.
- M. A. Dar, Inorg. Chem., 2023, 62, 13672–13679 CrossRef CAS PubMed.
- M. Z. Rahman and C. B. Mullins, Acc. Chem. Res., 2019, 52, 248–257 CrossRef CAS PubMed.
- N. Serpone and A. V. Emeline, J. Phys. Chem. Lett., 2012, 3, 673–677 CrossRef CAS PubMed.
- Y. Miseki and K. Sayama, Adv. Energy Mater., 2019, 9, 1801294 CrossRef.
- A. Mishra, A. Mehta, S. Basu, N. Shetti, K. Reddy and T. Aminabhavi, Carbon, 2019, 149, 693–721 CrossRef CAS.
- V. Hasija, A. Kumar, A. Sudhaik, P. Raizada, P. Singh, Q. Van Le, T. Le and V. Nguyen, Environ. Chem. Lett., 2021, 19, 2941–2966 CrossRef CAS.
- A. Augustin, P. Ganguly, S. Shenoy, C. Chuaicham, S. C. Pillai, K. Sasaki, A. F. Lee and K. Sekar, Adv. Sustainable Syst., 2024, 8, 2400321 CrossRef CAS.
- X. Chen, R. Shi, Q. Chen, Z. Zhang, W. Jiang, Y. Zhu and T. Zhang, Nano Energy, 2019, 59, 644–650 Search PubMed.
- Y. Xiong, Y. Chen, N. Yang, C. Jin and Q. Sun, Sol. RRL, 2019, 3, 1800341 CrossRef.
- S. Obregón, A. Vázquez, M. A. Ruíz-Gómez and V. Rodríguez-González, Appl. Surf. Sci., 2019, 488, 205–212 CrossRef.
- Y. Liu, S. Shen, Z. Li, D. Ma, G. Xu and B. Fang, Mater. Charact., 2021, 174, 111031 CrossRef CAS.
- X. Guan, X. Zhang, Z. Li, S. Deshpande, M. Fawaz, N. P. Dharmarajan, C.-H. Lin, Z. Lei, L. Hu, J.-K. Huang, P. Kumar, Z. Sun, S. Chakraborty and A. Vinu, Chem. Mater., 2024, 36, 4511–4520 CrossRef CAS.
- M. Rezaei, A. Nezamzadeh-Ejhieh and A. R. Massah, Energy Fuels, 2024, 38, 8406–8436 Search PubMed.
- X. Liu, X. Pan, Y. Qiu, J. Li, X. Ma and D. Li, Langmuir, 2023, 39, 11294–11303 CrossRef CAS PubMed.
- A. Kumar, P. Raizada, A. Hosseini-Bandegharaei, V. K. Thakur, V. Nguyen and P. Singh, J. Mater. Chem. A, 2021, 9, 111–153 Search PubMed.
- W. Iqbal, B. Yang, X. Zhao, M. Rauf, I. M. A. Mohamed, J. Zhang and Y. Mao, Catal. Sci. Technol., 2020, 10, 549–559 RSC.
- Z. Su, J. Zhang, Z. Tan, J. Hu, F. Zhang, R. Duan, L. Yao, B. Han, Y. Zhao and Y. Yang, Green Chem., 2023, 25, 2577–2582 RSC.
- J. Huang, J. Liu, J. Yan, C. Wang, T. Fei, H. Ji, Y. Song, C. Ding, C. Liu, H. Xu and H. Li, Colloids Surf., A, 2020, 585, 124067 CrossRef CAS.
- N. Wang, Q. Sun and J. Yu, Adv. Mater., 2019, 31, 1803966 CrossRef PubMed.
- S. Patnaik, D. P. Sahoo and K. Parida, Mater. Today, 2021, 35, 247–251 CAS.
- C. Li, H. Wu, Y. Du, S. Xi, H. Dong, S. Wang and Y. Wang, ACS Sustainable Chem. Eng., 2020, 8, 12934–12943 CrossRef CAS.
- P. Zhang, F. Wang, Y. Qin and N. Wang, ACS Appl. Nano Mater., 2020, 3, 7847–7857 CrossRef CAS.
- P. Wen, K. Zhao, H. Li, J. Li, J. Li, Q. Ma, S. M. Geyer, L. Jiang and Y. Qiu, J. Mater. Chem. A, 2020, 8, 2995–3004 RSC.
- T. Wang, X. Pan, M. He, L. Kang and W. Ma, Adv. Sci., 2024, 11, 2403771 CrossRef CAS PubMed.
- Z. Zeng, H. Yu, X. Quan, S. Chen and S. Zhang, Appl. Catal., B, 2018, 227, 153–160 CrossRef CAS.
- M. Jing, A. Zhang, Q. Zhang, B. Weng, F. Ni, F. Meng, Y. Mei and S. Pang, Int. J. Hydrogen Energy, 2024, 79, 118–129 CrossRef CAS.
- M. Humayun, H. Ullah, C. Hu, M. Tian, W. Pi, Y. Zhang, W. Luo and C. Wang, ACS Appl. Mater. Interfaces, 2023, 15, 14481–14494 CAS.
- C. Yang, Y. Chen, T. Chen, J. Low, S. Rajendran, Z. Zeng, X. Zhang and J. Qin, Fuel, 2023, 337, 126894 CrossRef CAS.
- Y. Tian, D. Zeng, T. Shen, R. Guan and W. Shi, Fuel, 2024, 357, 129927 CrossRef CAS.
- S. Tian, H. Ren, W. Sun, Y. Song, H. Ge, A. Yang, W. Zheng and Y. Zhao, New J. Chem., 2024, 48, 3695–3706 RSC.
- J. Hu, H. Liu, C. Sun, L. Wu and F. Jiao, Ind. Eng. Chem. Res., 2024, 63, 2665–2675 CrossRef CAS.
- T. Jia, J. Li, Z. Deng, D. Yu and J. H. Lee, Materials, 2024, 17, 3345 CrossRef CAS PubMed.
- K. Song, J. Gou, L. Yang and C. Zeng, Catal. Lett., 2023, 153, 534–543 CrossRef CAS.
- X. H. Chi, F. J. Liu, Y. Gao, J. Song, R. F. Guan and H. B. Yuan, Appl. Surf. Sci., 2022, 576, 151837 CrossRef CAS.
- J. Zhang, L. Zhang, C. Mao, R. Gu, W. Wang, Y. Wang, Z. Zhou, B. Yan, L. Bi, Q. Fu and Y. Zhu, Eur. J. Inorg. Chem., 2023, e202200609 CrossRef CAS.
- X. Wu, H. Fan, W. Wang, L. Lei, X. Chang and L. Ma, J. Mater. Chem. A, 2022, 10, 17817–17826 RSC.
- Y. Liu, Y. Zhang and L. Shi, Colloids Surf., A, 2022, 641, 128577 CrossRef CAS.
- Q. Gong, S. Cao, Y. Zhou, R. Wang and W. Jiao, Int. J. Hydrogen Energy, 2021, 46, 21442–21453 CrossRef CAS.
- G. Liu, L. Shi, L. Yao and L. Cui, Int. J. Energy Res., 2021, 45, 10478–10488 CrossRef CAS.
- X. Chang, T. Wang and J. Gong, Energy Environ. Sci., 2016, 9, 2177–2196 RSC.
- B. C. Zhu, L. Y. Zhang, D. F. Xu, B. Cheng and J. G. Yu, J. CO2 Util., 2017, 21, 327–335 CrossRef CAS.
- S. N. Talapaneni, G. Singh, I. Y. Kim, K. AlBahily, A. H. Al-Muhtaseb, A. S. Karakoti, E. Tavakkoli and A. Vinu, Adv. Mater., 2020, 32, 1904635 CrossRef CAS PubMed.
- Q. Q. Lu, K. Eid, W. P. Li, A. M. Abdullah, G. B. Xu and R. S. Varma, Green Chem., 2021, 23, 5394–5428 RSC.
- Y. Jia, X. Tong, H. Zhou, J. Zhang, Y. Chen, L. Zhang, Y. Yang and X. Ji, J. Alloys Compd., 2024, 987, 174217 CrossRef CAS.
- Z. Zhu, Z. Liu, X. Tang, K. Reeti, P. Huo, J. W. C. Wong and J. Zhao, Catal. Sci. Technol., 2021, 11, 1725–1736 RSC.
- M. Arumugam, M. Tahir and P. Praserthdam, Chemosphere, 2022, 286, 131765 CrossRef CAS PubMed.
- P. Madhusudan, R. Shi, S. Xiang, M. Jin, B. N. Chandrashekar, J. Wang, W. Wang, O. Peng, A. Amini and C. Cheng, Appl. Catal., B, 2021, 282, 119600 CrossRef CAS.
- Q. Heng, B. Wang, X. Fan, W. Chen, X. Li, L. Mao and W. Shangguan, J. Alloys Compd., 2022, 920, 165945 CrossRef CAS.
- Z. Sun, W. Fang, L. Zhao and H. Wang, Appl. Surf. Sci., 2020, 504, 144347 CrossRef CAS.
- N. Li, X. Li, R. Pan, M. Cheng, J. Guan, J. Zhou, M. Liu, J. Tang and D. Jing, ChemPhotoChem, 2021, 5, 748–757 Search PubMed.
- M. Ou, W. G. Tu, S. M. Yin, W. N. Xing, S. Y. Wu, H. J. Wang, S. P. Wan, Q. Zhong and R. Xu, Angew. Chem., Int. Ed., 2018, 57, 13570–13574 Search PubMed.
- S. Roy and E. Reisner, Angew. Chem., Int. Ed., 2019, 58, 12180–12184 Search PubMed.
- I. A. Mkhalid, R. M. Mohamed, A. A. Ismail and M. Alhaddad, Ceram. Int., 2021, 47, 17210–17219 Search PubMed.
- X. Li, Y. Bai, X. Shi, J. Huang, K. Zhang, R. Wang and L. Ye, Appl. Surf. Sci., 2021, 546, 149111 Search PubMed.
- H. Guo, S. Wan, Y. Wang, W. Ma, Q. Zhong and J. Ding, Chem. Eng. J., 2021, 412, 128646 CrossRef CAS.
- Q. Wang, L. Zhang, Y. Guo, M. Shen, M. Wang, B. Li and J. Shi, Chem. Eng. J., 2020, 396, 125347 CrossRef CAS.
- M. K. Hussien, A. Sabbah, M. Qorbani, R. Putikam, S. Kholimatussadiah, D. M. Tzou, M. H. Elsayed, Y. J. Lu, Y. Y. Wang, X. H. Lee, T. Y. Lin, N. Q. Thang, H. L. Wu, S. C. Haw, K. C. Wu, M. C. Lin, K. H. Chen and L. C. Chen, Small, 2024, 20, e2400724 CrossRef PubMed.
- D. Li, K. Wang, Y. Chen, R. Li, J. Tang, Y. Zhao, W. Guo, Q. Chen and Y. Li, ChemSusChem, 2024, e202401638 Search PubMed.
- N. Ojha, A. Bajpai and S. Kumar, Catal. Sci. Technol., 2019, 9, 4598–4613 RSC.
- M. K. Hussien, A. Sabbah, M. Qorbani, M. H. Elsayed, P. Raghunath, T.-Y. Lin, S. Quadir, H.-Y. Wang, H.-L. Wu, D.-L. M. Tzou, M.-C. Lin, P.-W. Chung, H.-H. Chou, L.-C. Chen and K.-H. Chen, Chem. Eng. J., 2022, 430, 132853 CrossRef.
- Z. Fang, Q. Wang, X. Zhao, Y. Li, W. Zhang and D. Zhang, J. Environ. Chem. Eng., 2023, 11, 109478 CrossRef CAS.
- F. A. Qaraah, S. A. Mahyoub, H. Shen, X. Yin, A. Salah, S. A. Onaizi, Q. A. Drmosh and F. Xin, Carbon, 2025, 232, 119735 CrossRef CAS.
- H. Liu, Y. Yang, C. Guo and Y. Zhou, Catal. Sci. Technol., 2024, 14, 5938–5948 RSC.
- J. Y. Tang, X. Y. Kong, B. J. Ng, Y. H. Chew, A. R. Mohamed and S. P. Chai, Catal. Sci. Technol., 2019, 9, 2335–2343 RSC.
- J. Fu, B. Zhu, C. Jiang, B. Cheng, W. You and J. Yu, Small, 2017, 13, 1603938 CrossRef PubMed.
- H. Wang, Q. Tang and Z. Wu, ACS Sustainable Chem. Eng., 2021, 9, 8425–8434 CrossRef CAS.
- Z. Yong and T. Ma, Angew. Chem., Int. Ed., 2023, 135, e202308980 Search PubMed.
- H. T. Vuong, D. P. Bui, D. V. Nguyen, L. Pho Phuong, P. P. Duc Minh, T. Do Dat and N. H. Hieu, ChemPhotoChem, 2023, 7, e202200299 CrossRef CAS.
- J. Lei, B. Chen, W. Lv, L. Zhou, L. Wang, Y. Liu and J. Zhang, ACS Sustainable Chem. Eng., 2019, 7, 16467–16473 Search PubMed.
- L. Zhou, J. Feng, B. Qiu, Y. Zhou, J. Lei, M. Xing, L. Wang, Y. Zhou, Y. Liu and J. Zhang, Appl. Catal., B, 2020, 267, 118396 CrossRef CAS.
- Y. Yang, Z. Zeng, G. Zeng, D. Huang, R. Xiao, C. Zhang, C. Zhou, W. Xiong, W. Wang, M. Cheng, W. Xue, H. Guo, X. Tang and D. He, Appl. Catal., B, 2019, 258, 117956 CrossRef CAS.
- R. Du, K. Xiao, B. Li, X. Han, C. Zhang, X. Wang, Y. Zuo, P. Guardia, J. Li, J. Chen, J. Arbiol and A. Cabot, Chem. Eng. J., 2022, 441, 135999 Search PubMed.
- T. Song, X. Zhang, K. Matras-Postolek and P. Yang, J. Environ. Chem. Eng., 2022, 10, 108747 CrossRef CAS.
- Z. Liu, X. Wang, P. Zhang, H. Zhang, S. Wang, G. Jiao, W. Wu and M. Wu, Chem. – Eur. J., 2025, e202404003 CrossRef CAS PubMed.
- Y. Ding, S. Maitra, C. Wang, R. Zheng, M. Zhang, T. Barakat, S. Roy, J. Liu, Y. Li, T. Hasan and B.-L. Su, J. Energy Chem., 2022, 70, 236–247 CrossRef CAS.
- R. Tang, D. Gong, Y. Zhou, Y. Deng, C. Feng, S. Xiong, Y. Huang, G. Peng, L. Li and Z. Zhou, Appl. Catal., B, 2022, 303, 120929 Search PubMed.
- Y. Pan, X. Liu, W. Zhang, B. Shao, Z. Liu, Q. Liang, T. Wu, Q. He, J. Huang, Z. Peng, Y. Liu and C. Zhao, Chem. Eng. J., 2022, 427, 132032 Search PubMed.
- Y. Shiraishi, Y. Kofuji, H. Sakamoto, S. Tanaka, S. Ichikawa and T. Hirai, ACS Catal., 2015, 5, 3058–3066 Search PubMed.
- B. Liu, C. Bie, Y. Zhang, L. Wang, Y. Li and J. Yu, Langmuir, 2021, 37, 14114–14124 CrossRef CAS PubMed.
- Y. Liu, Y. Zheng, W. Zhang, Z. Peng, H. Xie, Y. Wang and Y. Huang, J. Mater. Sci. Technol., 2021, 95, 127–135 CrossRef CAS.
- R. Pan, W. Lv, X. Ge, X. Huang, Q. Hu, K. Song, Q. Liu, H. Xie, B. Wu and J. Yuan, Adv. Funct. Mater., 2025, 35, 2414193 Search PubMed.
- H.-T. Vuong, D.-V. Nguyen, P. P. Ly, P. D. M. Phan, T. D. Nguyen, D. D. Tran, P. T. Mai and N. H. Hieu, ACS Appl. Nano Mater., 2022, 6, 664–676 CrossRef.
- X. Zhao, Y. You, S. Huang, Y. Wu, Y. Ma, G. Zhang and Z. Zhang, Appl. Catal., B, 2020, 278, 119251 CrossRef CAS.
- K. Ithisuphalap, H. Zhang, L. Guo, Q. Yang, H. Yang and G. Wu, Small Methods, 2019, 3, 1800352 CrossRef.
- T. Žibert, B. Likozar and M. Huš, ChemSusChem, 2024, 17, e202301730 CrossRef PubMed.
- D. Ješić, B. Pomeroy, K. M. Kamal, Z. Kovačič, M. Huš and B. Likozar, Adv. Energy Sustainability Res., 2024, 5, 2400083 CrossRef.
- Z. Wang, X. Hu, Z. Liu, G. Zou, G. Wang and K. Zhang, ACS Catal., 2019, 9, 10260–10278 CrossRef CAS.
- L. Li, P. Zhang, N. Li, T. Reyila, Y. Yu, X. Su, C. Peng and L. Han, J. Environ. Chem. Eng., 2024, 12, 112142 CrossRef CAS.
- L. Zhang, S. Q. Hou, T. Y. Wang, S. X. Liu, X. C. Gao, C. Y. Wang and G. X. Wang, Small, 2022, 18, 2202252 CrossRef CAS PubMed.
- L. Bao, B. Yuan and Y. J. Yuan, Chem. Phys. Lett., 2023, 828, 140715 CrossRef CAS.
- S. Ajmal, A. Rasheed, N. Q. Tran, X. Shao, Y. Hwang, V. Q. Bui, Y. D. Kim, J. Kim and H. Lee, Appl. Catal., B, 2023, 321, 122070 CrossRef CAS.
- H. Dang, Q. Li, M. Li, Y. Han, M. Shao and Q. Liu, Appl. Surf. Sci., 2024, 653, 159307 CrossRef CAS.
- Q. Zhang, Q. Li, H. Li, X. Shi, Y. Zhou, Q. Ye, R. Yang, D. Li and D. Jiang, Inorg. Chem., 2023, 62, 12138–12147 CrossRef CAS PubMed.
- P. Gao, X. Rong, S. Zhang, T. Sun, J. Liu, X. Qiu, X. Zhou, Z. Wu and J. Wei, Environ. Prog. Sustainable Energy, 2024, 43, e14246 CrossRef CAS.
- W. Wang, H. Zhou, Y. Liu, S. Zhang, Y. Zhang, G. Wang, H. Zhang and H. Zhao, Small, 2020, 16, 1906880 CrossRef CAS PubMed.
- Y. Li, M. Ti, D. Zhao, Y. Zhang, L. Wu and Y. He, J. Alloys Compd., 2021, 870, 159298 CrossRef CAS.
- J. Ge, L. Zhang, J. Xu, Y. Liu, D. Jiang and P. Du, Chin. Chem. Lett., 2020, 31, 792–796 CrossRef CAS.
- S. Liu, S. Wang, Y. Jiang, Z. Zhao, G. Jiang and Z. Sun, Chem. Eng. J., 2019, 373, 572–579 CrossRef CAS.
- D. Cui, X. Yang, Y. Liu, M. Li, C. Wang and F. Li, Sep. Purif. Technol., 2025, 356, 130035 CrossRef CAS.
- Y. Shiraishi, S. Shiota, Y. Kofuji, M. Hashimoto, K. Chishiro, H. Hirakawa, S. Tanaka, S. Ichikawa and T. Hirai, ACS Appl. Energy Mater., 2018, 1, 4169–4177 CrossRef CAS.
- S. Wang, A. Cheng, F. Liu, J. Zhang, T. Xia, X. Zeng, W. Fan and Y. Zhang, Ind. Chem. Mater., 2023, 1, 188–206 RSC.
- Z. Chen, Y. G. Wang, H. N. Cheng and H. B. Zhou, J. Chem. Technol. Biotechnol., 2023, 98, 331–345 CrossRef CAS.
- L. Qu, X. Jiang, Z. Zhang, X. Zhang, G. Song, H. Wang, Y. Yuan and Y. Chang, Green Chem., 2021, 23, 9348–9376 Search PubMed.
- X. Q. Tan, W. Mo, A. R. Mohamed and W. J. Ong, J. Cleaner Prod., 2024, 436, 140270 CrossRef CAS.
- X. Wu, N. Luo, S. Xie, H. Zhang, Q. Zhang, F. Wang and Y. Wang, Chem. Soc. Rev., 2020, 49, 6198–6223 RSC.
- S. P. Shelake, D. N. Sutar, B. M. Abraham, T. Banerjee, A. V. S. Sainath and U. Pal, Adv. Funct. Mater., 2024, 2403795 CrossRef CAS.
- Q. Wu, Y. He, H. Zhang, Z. Feng, Y. Wu and T. Wu, Mol. Catal., 2017, 436, 10–18 CrossRef CAS.
- X. D. Zhou and Y. H. Tian, J. Photochem. Photobiol., A, 2024, 454, 115728 CrossRef CAS.
- C. H. Ku, K. X. Li, H. Q. Guo, Q. Wu and L. S. Yan, Appl. Surf. Sci., 2022, 592, 153266 CrossRef CAS.
- S. D. Rojas, N. E.-Villalobos, R. Salazar, N. Escalona, D. Contreras, V. Melin, M. A. L.-Bercero, M. S.-Arenillas, E. Vergara, L. C.-Jensen and J. R.-Becerra, J. Photochem. Photobiol., 2021, 421, 113513 Search PubMed.
- D. Zhou, D. Li, S. Yuan and Z. Chen, Energy Fuels, 2022, 36, 10721–10731 CrossRef CAS.
- Z. K. Xie, Y. J. Jia, Y. Y. Huang, D. B. Xu, X. J. Wu, M. Chen and W. D. Shi, ACS Catal., 2023, 13, 13768–13776 CrossRef CAS.
- X. Li, J. Hu, T. Yang, X. Yang, J. Qu and C. M. Li, Nano Energy, 2022, 92, 106714 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |