
In situ QXAFS study of CO and H2 adsorption on Pt in [PtAu8(PPh3)8]-H[PMo12O40] solid†
Tomoki
Matsuyama
a,
Taishi
Suzuki
a,
Yuto
Oba
a,
Soichi
Kikkawa
 a,
Sayaka
Uchida
b,
Junya
Ohyama
c,
Kotaro
Higashi
d,
Takuma
Kaneko
d,
Kazuo
Kato
d,
Kiyofumi
Nitta
d,
Tomoya
Uruga
d,
Keisuke
Hatada
e,
Kazuki
Yoshikawa
e,
Amelie
Heilmaier
ef,
Kosuke
Suzuki
g,
Kentaro
Yonesato
g,
Kazuya
Yamaguchi
g,
Naoki
Nakatani
a,
Hideyuki
Kawasoko
ah and
Seiji
Yamazoe
*a
a,
Sayaka
Uchida
b,
Junya
Ohyama
c,
Kotaro
Higashi
d,
Takuma
Kaneko
d,
Kazuo
Kato
d,
Kiyofumi
Nitta
d,
Tomoya
Uruga
d,
Keisuke
Hatada
e,
Kazuki
Yoshikawa
e,
Amelie
Heilmaier
ef,
Kosuke
Suzuki
g,
Kentaro
Yonesato
g,
Kazuya
Yamaguchi
g,
Naoki
Nakatani
a,
Hideyuki
Kawasoko
ah and
Seiji
Yamazoe
*a
aDepartment of Chemistry, Graduate School of Science, Tokyo Metropolitan University, 1-1 Minami-osawa, Hachioji-shi, Tokyo 192-0397, Japan. E-mail: yamazoe@tmu.ac.jp
bDepartment of Basic Science, School of Arts and Sciences, The University of Tokyo, 3-8-1 Komaba, Meguro-ku, Tokyo 153-8902, Japan
cFaculty of Advanced Science and Technology, Kumamoto University, 2-39-1 Kurokami, Chuo-ku, Kumamoto-shi, Kumamoto 860-8555, Japan
dCenter for Synchrotron Radiation Research, Japan Synchrotron Radiation Research Institute (JASRI), 1-1-1, Kouto, Sayo-cho, Sayo-gun, Hyogo 679-5198, Japan
eDepartment of Physics, University of Toyama, 3190 Gofuku, Toyama 930-8555, Japan
fDepartment of Chemistry, Ludwig-Maximilians-Universität (LMU), Butenandtstr. 5-13, 81377 Munich, Germany
gDepartment of Applied Chemistry, School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan
hPrecursory Research for Embryonic Science and Technology (PRESTO), Japan Science and Technology Agency (JST), 7, Gobancho, Chiyoda-ku, Tokyo 102-0076, Japan
First published on 14th November 2024
Abstract
The adsorption behaviors of H2 and CO molecules in crown-motif [PtAu8(PPh3)8]-H[PMo12O40] (PtAu8-PMo12) solids were investigated by in situ quick-scan X-ray absorption fine structure (QXAFS) measurements with a time resolution of 0.1 s. The electronic state of Pt in PtAu8-PMo12 was drastically changed by the adsorption of H2 and CO molecules because of the formation of Pt–H2/Pt–CO interactions. H2 was adsorbed more rapidly (<0.5 s) on Pt than CO (∼2.5 s) and showed reversible adsorption/desorption behavior on Pt atoms in PtAu8-PMo12. The rapid adsorption of H2 is due to the fast diffusion of H2, which has a smaller kinetic diameter than CO, in the narrow channels between the closed voids in PtAu8-PMo12. Meanwhile, CO was irreversibly adsorbed on Pt, resulting in structural isomerization to the stable “chalice-motif” PtAu8, which was determined by XAFS analysis and density functional theory calculations. Structural isomerization was involved by pushing ligands aside to make space for CO adsorption as the void size near Pt in the crown-motif PtAu8-PMo12 was narrower than the kinetic diameter of CO.
Introduction
Metal clusters composed of <100 atoms have attracted significant attention because of their quantized electronic states and unique geometric structures that cannot be predicted from their bulk materials.1–4 The electronic state of metal clusters, which determines their functions, depends on the number of metal atoms and their geometric structures. Therefore, atomically precise synthesis of metal clusters is essential to understand the relationship between functions and structures.5,6 To date, numerous ligand-protected metal clusters composed of Au, Ag, and Cu, among others, have been synthesized in a liquid phase. For example, Au ions are reduced in the presence of different protecting ligands, like phosphine,7 thiolate,8 alkynyl,9 and N-heterocyclic carbene,10 which allows the synthesis of ligand-protected Au clusters with a highly homogeneous size distribution. Ligand-protected Au clusters have unique electronic structures depending on their size and composition, and exhibit optical properties,11,12 photoluminescence,13 magnetism,14 and redox activity.15The ligand-protected metal clusters have been applied as catalysts, such as for the selective oxidation of styrene by phosphine-protected Au11(PR3)7Cl3 and [Au11(PR3)8Cl2]+ (R = alkyl groups)16 or by thiolate-protected Au25(SR)18, Au38(SR)24 and Au144(SR)6017 as well as methane oxidation by mixed ligand-protected [Au24(PR3)10(SR)5Cl2]− and [Au25(PR3)10(SR)5Cl2]2−.18 Heterometal doping is one of the most effective methods for controlling and inducing the reactivities of ligand-protected Au clusters because metal dopants can modulate the electronic state of the clusters. The hydrogen evolution reaction has been accelerated by Pt single-atom doping to [Au25(SR)18]− because of the depletion of two valence electrons and a positive shift in reduction potential.19 Phosphine-protected MAu12(PRP)5Cl2 (M = Au, Pd, Pt, Ru, Rh, Ir) also showed a change in electronic structure depending on the central dopant.20 In particular, photoinduced intermolecular [2 + 2] cycloaddition of bisenone was shown to be promoted by Pt- and Ir-doped MAu12(PRP)5Cl2.21 Metal dopants often provide new active sites.
Among the ligand-protected metal clusters, phosphine-protected [Au9(PR3)8]3+ (Au9) with a crown-motif structure has a coordinatively unsaturated Au atom located at the center of Au9.22 Although the molecule can access the central Au in Au9, no reports have described that the molecule is activated by Au9. Meanwhile, [PtAu8(PR3)8]2+ (PtAu8) with a crown-motif structure, in which the central Au in Au9 is substituted by Pt, worked as a catalyst for the H2–D2 exchange reaction.23 In addition, PtAu8 in the solution state exhibited the nucleophilic adsorption of small molecules, such as CO.24 These reactivities have also been reported for Pt-centered Au clusters, such as [PtAu9(PR3)9]3+ and [(PR3)PtAu6(PR3)6]2+.25,26 However, the molecular adsorption on solid ligand-protected metal clusters has been limited.
Our group focused on Au9 and heterometal-doped [MAu8(PPh3)8]2+ (MAu8, M = Pt, Pd) to investigate thermal stabilities27 and control their geometric structures using counter anions.28 Recently, crown-motif Au9 and MAu8 were synthesized by associating anionic [PMo12O40]3− (PMo12) polyoxometalate with cesium chloride crystal packing structures.27 The Au9-PMo12 and [MAu8(PPh3)8]-H[PMo12O40] (MAu8-PMo12) were thermally stable at <473 K.27 The crystal structure of PtAu8-PMo12 is shown in Fig. 1. Two types of voids of different sizes can be observed in the crystals. The large void (VL) is in the ab-plane, and PtAu8 is surrounded by 4VL. The small void (VS) is located over the Pt atom in PtAu8. H2 and CO are linear diatomic molecules of different sizes and thus can be used as probe molecules to reveal the reactivity of solid ligand-protected metal clusters in porous crystals with small voids. Interestingly, we found that small molecules, such as H2 and CO, could diffuse into the PtAu8-PMo12 solid although these voids are isolated and there is no path for the molecules to diffuse into the crystal.
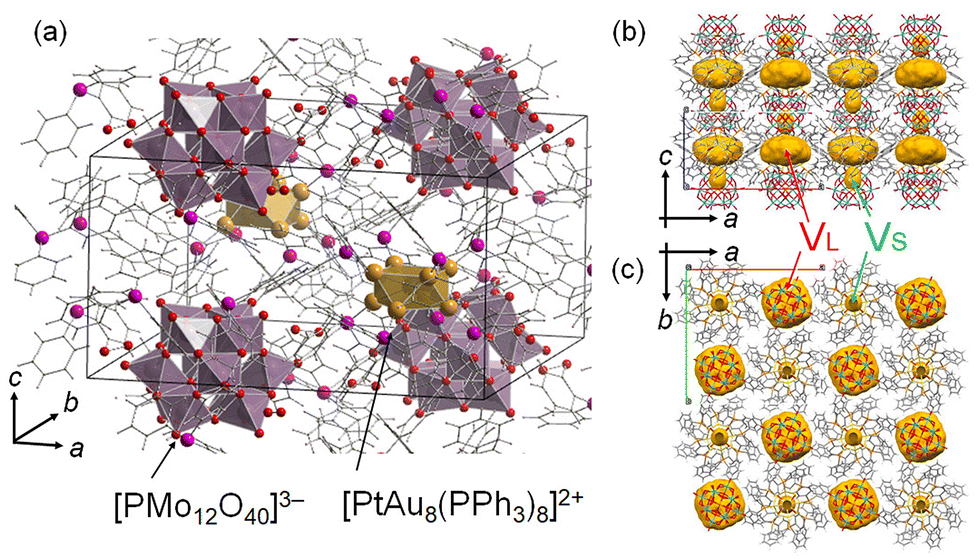 | ||
| Fig. 1 (a) Crystal structure of PtAu8-PMo12.27 Void analysis of PtAu8-PMo12 in the (b) ac-plane and (c) ab-plane. Void space (in yellow) is depicted with a probe radius of 1.2 Å using the CSD Mercury software.29 | ||
This study focuses on the H2 and CO adsorption/desorption properties of PtAu8-PMo12 solids. The interaction of the central Pt for PtAu8 with H2/CO was monitored by in situ quick-scan X-ray absorption fine structure (QXAFS) analysis because the peak intensity and energy assigned to 2p–5d electron transition were changed by the interaction of Pt with the molecule.30 We found that both H2 and CO could diffuse into PtAu8-PMo12 solid and interact with Pt of PtAu8 core, whereas Au9-PMo12, which had the same crystal structure as PtAu8-PMo12, could not interact with H2 and CO. The adsorption rate of H2 on Pt is more than four times higher than that of CO in PtAu8-PMo12, suggesting that the diffusion of molecules in PtAu8-PMo12 solids is a key process for molecular adsorptions and small H2 can pass through the narrow channels more easily than CO.
Results and discussion
The synthesis of [PtAu8(PR3)8](NO3)2 (PtAu8-NO3) was confirmed by ESI-MS and UV-Vis spectroscopy. As shown in Fig. 2a, a strong divalent molecular ion peak was observed at m/z = 1935.1. This isotopic pattern was consistent with the pattern calculated as [PtAu8(PPh3)8]2+. Fig. 2b shows the UV-Vis spectrum of the PtAu8-NO3 methanol solution. The broad band at 500–570 nm and the peaks at 428, 354, and 306 nm were in accordance with the absorption properties of the PtAu8-NO3 solution with the crown-motif structure.31PtAu8-PMo12 synthesized by anion exchange was characterized by DR-UV-Vis spectroscopy and powder XRD. A broad band at 500–570 nm and peaks at 434, 361, and 313 nm observed in the PtAu8-PMo12 solid were consistent with those of the crown-motif structure.28 The powder XRD pattern of the PtAu8-PMo12 solid had diffraction peaks at 2θ = 5.12, 7.25, and 8.09 degrees, as shown in Fig. 2c. These results indicate that the PtAu8-PMo12 solid had a CsCl-type crystal structure.28 In addition, local structures of PtAu8-PMo12 were determined by curve fitting analysis of Au L3- and Pt L3-edges EXAFS spectra at 10 K (Fig. S1 and S2†). The average coordination numbers (CNs) for Au–P (CN = 1.4 ± 0.2), Au–Pt (CN = 1.4 ± 0.1), and Au–Au bonds (CN = 2.3 ± 0.2) obtained by Au L3-edge FT-EXAFS analysis, and for Pt–Au (CN = 7.8 ± 0.3) obtained by Pt L3-edge FT-EXAFS analysis revealed that the PtAu8 in PtAu8-PMo12 had a crown-motif structure with the central atom of Pt (Tables S1 and S2†). Similar characterizations were conducted for Au9-PMo12 (Fig. S1, S3 and Table S1†). From the above results, we concluded that PtAu8-PMo12 and Au9-PMo12 solids with crown-motif structures were synthesized.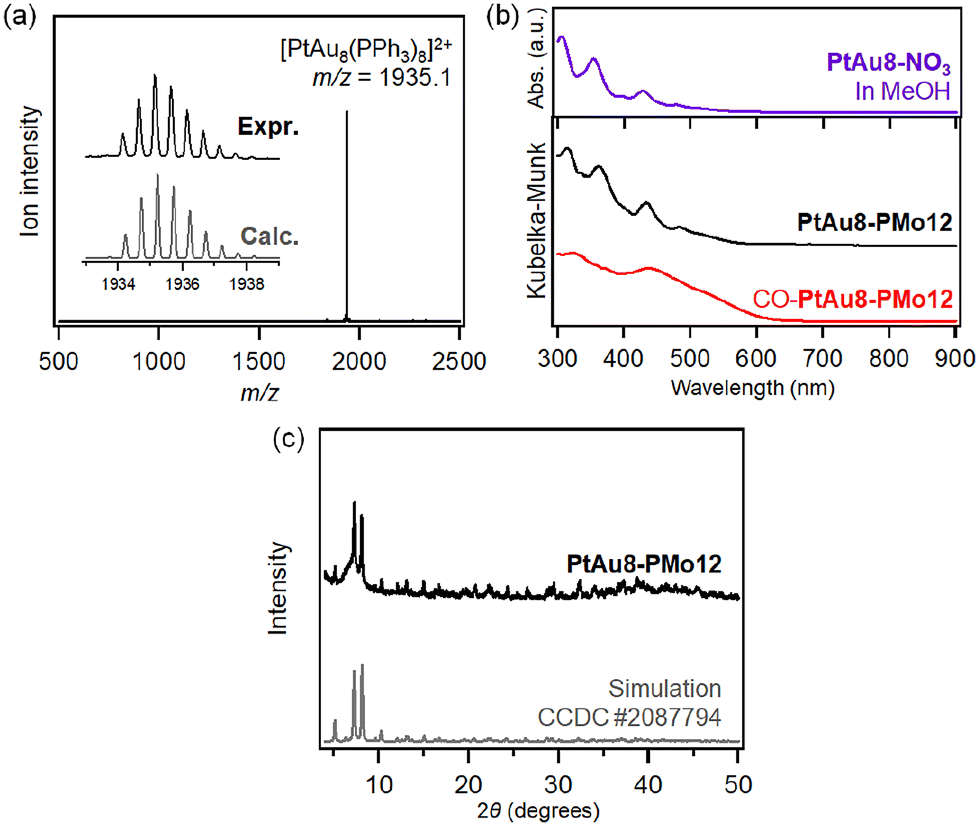 | ||
| Fig. 2 Characterizations of PtAu8-NO3 and PtAu8-PMo12. (a) Positive-ion mode ESI-MS of the acetonitrile solution of PtAu8-NO3. (b) UV-Vis spectrum of methanolic solution of PtAu8-NO3 and DR-UV-Vis spectra of PtAu8-PMo12 and CO adsorbed PtAu8-PMo12. (c) Powder-XRD patterns of PtAu8-PMo12 and simulated PtAu8-PMo12 (CCDC 2087794). | ||
We investigated the molecular adsorption properties of crown-motif PtAu8-PMo12 using Pt L3-edge XAFS measurements. The Pt L3-edge XANES of PtAu8-PMo12 is shown in Fig. 3a. The peak appearing in the absorption edge at 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 569.6 eV was assigned to the electron transition from Pt 2p to unoccupied 5d orbitals.32 The peak energy and intensity were changed by the introduction of CO, as shown in Fig. 3a. The time course of the peak intensity at 11570 eV is shown in Fig. 3b. The peak intensity was increased by CO introduction within 2.5 s. The peak energy at the absorption edge shifted from 11569.6 eV to 11570.5 eV. These changes are due to the formation of Pt 5d–C 2p antibonding hybrid orbitals and electron donation from Pt to CO upon CO adsorption on the central Pt of the PtAu8 cluster (discussed later). No shape changes in the Pt L3-edge XANES were observed upon exposure to a vacuum after CO introduction, as shown in Fig. 3a. Meanwhile, the crown-motif Au9-PMo12, which also had a CsCl-type crystal structure (as shown in Fig. S4a†) with an Au center in the crown-motif Au9, did not adsorb CO under the same conditions because no change in Au L3-edge XANES and EXAFS was detected before and after CO introduction, as shown in Fig. S4b–d.† We also performed Pt L3-edge in situ XANES measurements for PtAu8-NO3 acetonitrile solution, of which Pt atoms reacted with H2 and CO (Fig. S5†). The XANES spectra of the solution sample showed similar changes in the white line intensity compared with that of solid PtAu8-PMo12; thus, we consider that CO and H2 adsorption on Pt occurs on the crystal surface and inside the crystal. These results suggest that the CO diffuses into solid PtAu8-PMo12 and strongly interacts with Pt of the crown-motif PtAu8.
569.6 eV was assigned to the electron transition from Pt 2p to unoccupied 5d orbitals.32 The peak energy and intensity were changed by the introduction of CO, as shown in Fig. 3a. The time course of the peak intensity at 11570 eV is shown in Fig. 3b. The peak intensity was increased by CO introduction within 2.5 s. The peak energy at the absorption edge shifted from 11569.6 eV to 11570.5 eV. These changes are due to the formation of Pt 5d–C 2p antibonding hybrid orbitals and electron donation from Pt to CO upon CO adsorption on the central Pt of the PtAu8 cluster (discussed later). No shape changes in the Pt L3-edge XANES were observed upon exposure to a vacuum after CO introduction, as shown in Fig. 3a. Meanwhile, the crown-motif Au9-PMo12, which also had a CsCl-type crystal structure (as shown in Fig. S4a†) with an Au center in the crown-motif Au9, did not adsorb CO under the same conditions because no change in Au L3-edge XANES and EXAFS was detected before and after CO introduction, as shown in Fig. S4b–d.† We also performed Pt L3-edge in situ XANES measurements for PtAu8-NO3 acetonitrile solution, of which Pt atoms reacted with H2 and CO (Fig. S5†). The XANES spectra of the solution sample showed similar changes in the white line intensity compared with that of solid PtAu8-PMo12; thus, we consider that CO and H2 adsorption on Pt occurs on the crystal surface and inside the crystal. These results suggest that the CO diffuses into solid PtAu8-PMo12 and strongly interacts with Pt of the crown-motif PtAu8.
 | ||
| Fig. 3 (a) Pt L3-edge XANES spectra of PtAu8-PMo12, CO-introduced PtAu8-PMo12, and after evacuation. (b) Time course of peak intensity at 11570 eV in Pt L3-edge XANES of PtAu8-PMo12 during CO introduction. (c) Optimized structure of CO-PtAu8 by DFT calculation. (d) Pt L3-edge XANES spectra of PtAu8-PMo12, H2-introduced PtAu8-PMo12, and after evacuation. (e) Time course of peak intensity at 11570 eV in Pt L3-edge XANES of PtAu8-PMo12 during H2 introduction. (f) Optimized structure of H2-PtAu8 by DFT calculation. | ||
Because it is known that CO adsorbed on PtAu8 in solution to form [Pt(CO)Au8(PPh3)8]2+,24 the structure of PtAu8-PMo12 after CO introduction (CO-PtAu8-PMo12) was investigated by ESI-MS, UV-Vis, XAFS, and DFT calculations. The ESI-MS spectrum of CO-PtAu8-PMo12 is shown in Fig. S6.† The [(CO)PtAu8(PPh3)8]2+ was detected, suggesting that CO adsorbed on PtAu8 in a solid form. The DR-UV-Vis spectra of PtAu8-PMo12 before and after CO introduction indicated that the optical properties were changed by CO introduction (Fig. 2b), and a peak at 430 nm and shoulder peak at 520 nm appeared. The optical properties of CO-PtAu8-PMo12 resembled those of [Pt(CO)Au8(PPh3)8]2+ in an ethanol solution, as shown in Fig. S7.† The stable structure of CO-adsorbed PtAu8 (CO-PtAu8) calculated by DFT is shown in Fig. 3c. The stable structure, with a “chalice-motif” structure, was in accordance with [Pt(CO)Au8(PPh3)8]2+ in solution, as reported previously.24 The absorption peaks of CO-PtAu8-PMo12 at 430 and 530 nm were reproduced by TDDFT calculations using the calculated CO-PtAu8 structure, as shown in Fig. S7.† Au L3- and Pt L3-edge EXAFS and FT-EXAFS revealed that the crown-motif structure of PtAu8-PMo12 was changed by CO introduction, as shown in Fig. S8.† Curve fitting analysis revealed that CNs and the bonding distances of Au–P (1.0 ± 0.2, 2.23 ± 0.03 Å), Au–Pt (1.3 ± 0.1, 2.69 ± 0.02 Å), and Au–Au (3.6 ± 0.7, 2.93 ± 0.11 Å) in Au L3-edge and CNs and the bonding distances of Pt–C (1.3 ± 0.3, 1.81 ± 0.10 Å), Pt–Au (8.0 ± 0.4, 2.64 ± 0.03 Å) in Pt L3-edge of CO-PtAu8-PMo12 agreed with those obtained from the calculated CO-PtAu8, as shown in Tables S1, S2 and S3.† In addition, the absorption edge peak appearing in the Pt L3-edge XANES shifted to a high-energy region by CO adsorption (Fig. 3a). The simulated Pt L3-edge XANES of the crown-motif PtAu8 and the chalice-motif CO-PtAu8 (using the structure in Fig. 3c) revealed that the peaks in the absorption edge of CO-PtAu8 appeared at a higher-energy region than those of the crown-motif PtAu8 (Fig. S9†). These results indicate that CO diffused into the PtAu8-PMo12 solid and that one CO was adsorbed on the central Pt of PtAu8.
H2 adsorption behavior was also investigated by Pt L3-edge XANES of PtAu8-PMo12 solids. The Pt L3-edge XANES spectra of PtAu8-PMo12 before and after H2 introduction are shown in Fig. 3d. The peak at 11569.6 eV was slightly shifted to 11570.2 eV and increased by H2 introduction because of the formation of the hybrid orbital of Pt 5d–H 1s.30 The time course of peak intensity at 11570 eV is shown in Fig. 3e. Interestingly, the peak intensity was immediately changed within 500 ms by H2 introduction, which was faster than that of CO adsorption. The Pt L3-edge XANES of PtAu8-PMo12 returned to its original state after H2 evacuation, as shown in Fig. 3d. After H2 evacuation, H2 was reintroduced into PtAu8-PMo12. The H2 again interacted with Pt within 500 ms, and the Pt L3-edge XANES of the second H2-adsorbed PtAu8-PMo12 was in good agreement with that of the first one. The changes in Pt L3-edge XANES shape corresponding to the H2 adsorption and desorption were repeated four times, as shown in Fig. S10.† Meanwhile, the crown-motif Au9-PMo12, as shown in Fig. S4,† did not interact with H2 under the same conditions because no change in Au L3-edge XANES was detected before and after H2 introduction, as shown in Fig. S11.† Therefore, we found that H2 adsorption/desorption proceeded reversibly on Pt in PtAu8-PMo12.
The structure of H2-adsorbed PtAu8 in PtAu8-PMo12 was investigated by Au L3-edge EXAFS, as shown in Fig. S12.† Changes in the EXAFS oscillations of PtAu8-PMo12 before and after H2 adsorption were not observed. The DFT calculation of H2 adsorption on Pt at end-on while maintaining the crown-motif PtAu8 in Fig. 3f also demonstrated that there was no conformational change due to H2 adsorption.
Next, the diffusion of CO and H2 molecules into PtAu8-PMo12 solids is discussed. PtAu8-PMo12 possesses two types of closed voids, VL (8.9 Å × 8.9 Å × 5.3 Å) and VS (3.1 Å × 3.1 Å × 4.0 Å), as shown in Fig. 1 and S13.† The large void, VL, is in the ab-plane, and PtAu8 is surrounded by four VL. The small void, VS, is located over the Pt atom in PtAu8. Considering that the kinetic diameters of H2 and CO, which are empirically used for gas adsorption, are 2.89 Å and 3.76 Å, respectively,33VL has a sufficient size for both H2 and CO, whereas VS can only accept H2. Interestingly, the neighbouring VL–VL in the ab-plane and VL–VS in the ac- or bc-plane are connected by a narrow channel (diameter of 1.6–2.2 Å), as shown in Fig. 4. Uchida's group reported that an ionic crystal composed of polyoxometalate and metal complex could absorb Cs+ (ion radius: 3.1 Å) by the diffusion between the closed voids due to the flexibility of the crystal lattice.34 Identically, H2 and CO can reach Pt of PtAu8 by diffusing between the closed voids through the narrow channels, as shown in Fig. 4, owing to the flexibility of the ligands of PtAu8. After the molecules diffuse into the VL in the ab-plane through narrow path 1 in Fig. 4, some of the molecules move to VS through path 2 in Fig. 4. In the case of H2 adsorption/desorption, the VS over PtAu8, which is the void through which Pt is accessed, is larger than H2. Therefore, H2 adsorbs/desorbs on/from Pt without structural changes. Pignolet's group reported that the H2–D2 exchange reaction proceeded on the Pt of PtAu8.23 However, they did not determine the adsorption site of H2 experimentally or mention the H2 diffusion into a solid. We demonstrated that H2 diffused into the PtAu8-PMo12 solid and was adsorbed on Pt in PtAu8 without structural change.
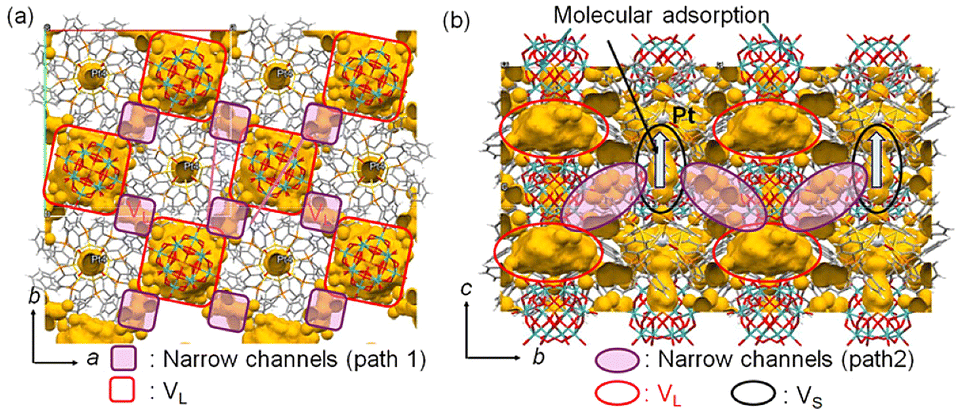 | ||
| Fig. 4 Void spaces and narrow channels in PtAu8-PMo12: views of (a) ab- and (b) bc-planes. A probe radius of 0.8 Å was used to visualize the narrow channels. | ||
We also demonstrated that CO, which is larger than H2, diffused into the PtAu8-PMo12 solid and adsorbed on Pt, despite the VS being smaller than a CO molecule. Because the adsorbed CO did not desorb under vacuum conditions (Fig. 3a), CO was strongly adsorbed on Pt. Therefore, it is expected that the crown-motif structure isomerized to the chalice-motif structure to make space to adsorb CO on Pt, as shown in Fig. 3c. Steggerda's group reported that CO reacted with PtAu8 in solution to obtain the chalice-motif [Pt(CO)Au8(PPh3)8]2+.24 In addition, the structural isomerization from crown-motif to chalice-motif is achieved by small changes in cluster size and (3AuPPh3)–Pt–(3AuPPh3) angle, as shown in Scheme S1.† Therefore, the chalice-motif [Pt(CO)Au8(PPh3)8]2+ can be formed by CO adsorption on crown-motif PtAu8-PMo12 with structural isomerization even in the solid phase.
The in situ QXAFS measurements revealed that H2 adsorption on Pt of PtAu8-PMo12 occurred faster than CO adsorption, as shown in Fig. 3b and e. The adsorption rate depends on the adsorption energy and/or diffusion path. The adsorption/desorption studies clearly showed that the interaction between H2 and Pt was weaker than that between CO and Pt because H2 was reversibly adsorbed/desorbed, whereas CO was not desorbed under the same conditions. The H2 adsorption energy (difference in the Gibbs free energies) on Pt of the crown-motif PtAu8 was calculated to be a positive value (0.33 eV) due to the effect of entropy because the difference in the adsorption enthalpies was −0.035 eV, which was similar to H2 adsorption energies on metal clusters.35 The Pt–H bond length of 2.902 Å in H2-PtAu8 is longer than the Pt–H bond (1.646 Å) in hydride-adsorbed H-PtAu836 calculated by DFT, as shown in Fig. S14.† Therefore, the interaction between H2 and PtAu8 is weak, and adsorbed H2 is desorbed under vacuum conditions. The reason that the CO did not desorb from PtAu8-PMo12 under vacuum conditions is that the CO adsorption energy was calculated to be −0.73 eV, which means that CO-PtAu8 is more stable than crown-motif PtAu8 with CO and is the driving force for structural isomerization. The calculated Pt-CO length of 1.91 Å in CO-PtAu8 is in the range of typical coordination bonds. Therefore, CO adsorption occurs as chemisorption, while H2 adsorption occurs as physisorption. These results clearly explained the trends between H2 and CO adsorption/desorption although the H2 adsorption rate was not explained by the adsorption energy.
The molecular diffusion in a solid depends on the diffusion path, void size, and molecular size. From the above discussion, there is no difference in the diffusion path through which the two molecules diffuse. However, molecules must pass through the narrow channels (paths 1 and 2) with a diameter of 1.6–2.2 Å to reach the Pt sites in PtAu8-PMo12. Because the kinetic diameter of H2 (2.89 Å) is smaller than that of CO (3.76 Å), H2 diffuses in the narrow channel more easily than CO. In addition, CO needs to push the ligands aside to make space for adsorption on Pt because the VS is narrower than the kinetic diameter of CO. We concluded that the molecular adsorption rate in PtAu8-PMo12 depends on the diffusion between closed voids and that the smaller molecule, H2, shows more rapid adsorption on Pt in PtAu8-PMo12 than CO, which must make space for adsorption by pushing ligands aside.
This work indicates that the gas diffusion rates of H2 and CO of PtAu8-PMo12 differ due to the size of the molecules and voids. In addition, we demonstrated that the optical properties of the clusters could be changed by CO adsorption with structural isomerization. The void size was controlled by the combination of cationic ligand-protected metal clusters and anionic metal oxide clusters with different sizes and compositions. Thus, the void-engineering of composite clusters and/or control of optical properties by molecular adsorption leads to the development of gas separation and/or gas sensors.
Conclusions
In this study, we investigated the molecular adsorption behaviors of crown-motif PtAu8-PMo12 solid by in situ QXAFS measurements with a time resolution of 0.1 s. Pt L3-edge XANES spectra showed that CO was irreversibly adsorbed on the Pt site with structural isomerization to the chalice-motif structure in solid form. Meanwhile, H2 was reversibly adsorbed/desorbed on Pt without a structural change. The adsorption rate of H2 (<0.5 s) on Pt in PtAu8-PMo12 was faster than that of CO (∼2.5 s), whereas the interaction of Pt–H2 was weaker than that of Pt–CO. The void analysis revealed that PtAu8-PMo12 with CsCl-type crystal packing has two types of closed voids with a large void, VL, being in the ab-plane and PtAu8 being surrounded by four VL, and a small void, VS, being located over the Pt atom in PtAu8. H2/CO could diffuse between the closed voids through the narrow channels (1.6–2.2 Å) of PtAu8-PMo12 and is adsorbed on Pt from the VS space. Because the kinetic diameter of H2 (2.89 Å) is smaller than that of CO (3.76 Å), H2 diffuses in the narrow channel more easily than CO. In addition, CO needs to push the ligands aside to create space for adsorption on Pt. From these results, we concluded that the molecular adsorption rate is governed by the diffusion process of molecules in PtAu8-PMo12. Therefore, the smaller molecule, H2, shows more rapid adsorption on Pt in PtAu8-PMo12 compared with CO.Experimental
Chemicals
All reagents used in this study were commercially available. Hydrogen tetrachloroaurate(III) tetrahydrate (HAuCl4·4H2O, 99.0%) was purchased from Kanto Chemical Co., Inc. Tetrabutyl ammonium bromide {TBABr: [N(C4H9)4]Br, >98.0%}, triethylamine [TEA: N(C2H5)3, >99.0%], and tetrakis(triphenylphosphine)platinum(0) [Pt(PPh3)4, >97.0%] were purchased from Tokyo Chemical Industry Co., Ltd. Sodium borohydride (NaBH4, 95.0%), triphenylphosphine (PPh3, 97.0%), phosphomolybdic acid hydrate {H3[PMo12O40]·nH2O, >95.0%}, and acetic anhydride [(CH3CO)2O, >97.0%] were purchased from Wako Pure Chemical Industry.Synthesis of [Au9(PPh3)8](NO3)3 (Au9-NO3)
Undoped Au9-NO3 was prepared in accordance with the literature.37 First, 0.6 mmol of Au(PPh3)NO3, synthesized by anion exchange from Au(PPh3)Cl, was dispersed in 24 mL of ethanol. 0.2 mmol of NaBH4 dissolved in 14 mL of ethanol solution was added to this suspension dropwise. After stirring at room temperature for 2 h, the brown solution was filtered through a membrane filter (pore size = 0.20 μm). The filtrate was evaporated to dryness and redissolved in dichloromethane (5 mL). The solution was filtered and dried, and the precipitate was washed with tetrahydrofuran and hexane. Finally, it was vacuum dried to produce a green powder.Synthesis of [PtAu8(PPh3)8](NO3)2 (PtAu8-NO3)
PtAu8-NO3 was synthesized in accordance with the literature.31 Here, 0.2 mmol of Pt(PPh3)3 obtained by heat treatment of Pt(PPh3)427 was placed in a glass container with 1.0 mmol of Au(PPh3)NO3. Next, 20 mL of THF was added, and a small amount of H2 gas was bubbled while stirring at room temperature. Note that an excessive flow rate results in the formation of [PtAu6(PPh3)7](NO3)2 as a byproduct. The red-orange precipitate was gathered by centrifugation (2500 rpm) and washed with a mixture of a small amount of dichloromethane and a large amount of diethyl ether three times. After vacuum drying, 169 mg of red-orange powder was dissolved in 10 mL of dichloromethane with 52 mg of Au(PPh3)NO3. Then, 0.2 mmol of TEA was added to the solution with stirring. After 24 h, this solution was evaporated and washed with diethyl ether. Finally, a brown solid was obtained after vacuum drying.
Synthesis of TBA3[PMo12O40] (TBA-PMo12)
TBA-PMo12 was synthesized by cation exchange reaction.37 Here, 0.3 mmol of H3[PMo12O40]·nH2O was dissolved in 50 mL of pure water. Next, 1.2 mmol of solid form TBABr was added to this solution. After stirring for 0.5 h at room temperature, the yellow precipitate was collected by centrifugation (3500 rpm) and washed with pure water. The precipitate was then reprecipitated using 5 mL of acetone and 45 mL of hexane. Finally, it was vacuum dried to produce yellow powder.Synthesis of Au9-PMo12 and PtAu8-PMo12
Au9-PMo12 and PtAu8-PMo12 were synthesized in the same way as previously reported.28 Here, 0.016 mmol of TBA-PMo12 dissolved in 20 mL of acetonitrile was added at room temperature to 0.010 mmol of Au9-NO3/PtAu8-NO3 solution (40 mL of acetonitrile). After 30 min of stirring, the precipitate was collected by centrifugation (2500 rpm) and washed with acetonitrile three times. Finally, it was vacuum-dried to give Au9-PMo12 and PtAu8-PMo12 solids.Characterizations
The synthesized ligand-protected metal clusters, polyoxometalates, and their composites were analyzed by ultraviolet-visible (UV-Vis) spectroscopy, electrospray ionization (ESI) mass spectrometry, and powder X-ray diffraction (XRD). The UV-Vis spectra of PtAu8-NO3 and Au9-NO3 in methanol and ethanol solutions of CO-adsorbed PtAu8, which were synthesized by reacting CO with PtAu8-NO3 in ethanol, were measured in transmittance mode (V-770; Jasco). The UV-Vis spectra of the solid states of PtAu8-PMo12, Au9-PMo12, and CO-adsorbed PtAu8-PMo12 were recorded using an integrating sphere unit in diffuse reflectance (DR) mode. The ESI mass spectra of PtAu8-NO3 and Au9-NO3 in acetonitrile were measured in positive-ion mode using a time-of-flight mass spectrometer (microTOF-II; Bruker). The powder XRD patterns of the composite metal clusters were determined using a powder X-ray diffractometer with Cu Kα radiation (MiniFlex600; Rigaku).The XAFS spectra of PtAu8-PMo12 and Au9-PMo12 were measured at the BL01B1 beamline of the SPring-8 facility operated by the Japan Synchrotron Radiation Research Institute. Si (111) double-crystal monochromators were employed to obtain the incident X-ray beam for Pt L3- and Au L3-edge XAFS measurements. All solid cluster samples were mixed with boron nitride as a diluent using an agate mortar. These mixtures were pressed into pellets. For Pt L3- and Au L3-edge XAFS measurements at 10 K, the pellets were mounted in a copper holder attached to a cryostat. To investigate the molecular adsorption behavior, quick-scan XAFS measurements (QXAFS) under transmission mode were performed at the BL36XU beamline of SPring-8 with a time resolution of 100 ms.38 A Si (220) channel-cut crystal monochromator was used to obtain the incident X-rays. Pt L3- and Au L3-edge in situ XAFS measurements were carried out using the pelletized samples mounted on an in situ cell connected to a gas-introducing apparatus (Fig. S15†). Approximately 15 mg solid cluster samples were mixed with 50 mg of boron nitride and pressed into φ7 pellet. To maintain gas diffusion in the solid cluster samples, the pellets were crushed into granules and re-pelletized. For gas adsorption tests, 100% H2 and CO were used, and sufficient amounts of gas were introduced to match the quantity of clusters present. After the sample was evacuated, 100% H2 or 100% CO was introduced immediately using a gas valve. The XAFS data were analyzed using the xTunes software39 for both X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS). The k3-weighted χ spectra (EXAFS spectra) in the k ranges of 3.0–9.0 Å−1 for the Pt L3-edge and 3.0–18.0 Å−1 for the Au L3-edge were Fourier transformed into r space (FT-EXAFS). The curve fitting analysis was conducted using FT-EXAFS spectra in the r range of 1.8–3.2 Å for the Pt L3-edge and 1.65–2.95 Å for the Au L3-edge.
Computational method
Density functional theory (DFT) calculations were conducted using the Gaussian 16 program.40 The geometries and electronic energies of PtAu8, and CO-, H2-, and H (hydride)-adsorbed PtAu8 (abbreviated as CO-PtAu8, H2-PtAu8, and H-PtAu8, respectively) were calculated using the B3LYP functional. LanL2DZ basis set was used for Au and Pt atoms and 6-31G* basis set was used for C, H, O, and P atoms. Frequency analyses were performed for PtAu8, CO-PtAu8 and H2-PtAu8 to confirm that no imaginary frequencies were obtained at the optimized geometries. The optical properties of CO-PtAu8 were also calculated by applying the time-dependent (TD) DFT method, solving 200 states, with the same functional and basis sets used in the ground state calculations. H2/CO adsorption energies were obtained in the difference of Gibbs free energies between H2/CO + PtAu8 and H2/CO-PtAu8 where the electronic energies were replaced by calculations using B3LYP-D3 functional with the same basis sets to incorporate dispersion interactions.Pt L3-edge XANES calculations were performed with the Full Potential Multiple Scattering (FPMS) code41 that employed the scattering potential without spherical potential approximation (Muffin-tin approximation). This code is based on Multiple-Scattering theory, which uses a multi-center expansion in spherical harmonics and a numerical solution of the local Schrödinger equation on each atomic site. In the overall calculation, lmax is set at 8. H atoms bonded to the C atoms of the protecting ligands were not considered in the calculations because their contributions to the calculated XANES spectrum are negligible due to their long distance to the absorbing Pt atom (more than 4 Å) and their small scattering power. However, closer H atoms, as in the case of adsorption, were considered and demonstrated an effect on the spectra. Energy-dependent complex Hedin-Lundqvist potential42,43 was used as the optical potential.
Author contributions
S. Y. guided the whole experiment and conceived the idea. T. M. and S. K. drafted the manuscript. T. M. and T. S. conducted the experiments, Y. O. developed analytical software for XAFS. U. S. carried out void analysis. S. K., J. O., and K. K. designed in situ cell for QXAFS. K. Hi., T. K. and T. U. designed the optical setup including measurement system for in situ QXAFS apparatus in SPring-8. K. N. gave some advice for structural analysis using low-temperature XAFS. K. Ha., Ka. Yo. and A. H. carried out XANES simulation. N. N. conducted energy calculation of clusters and gave some advice for DFT calculation results. K. S., Ke. Yo., K. Ya., and H. K. supported in situ XAFS measurements and discussed molecular diffusion. All the authors contributed to the final polishing of the manuscript.Data availability
The data supporting this manuscript is available in the ESI† and are available on request. Crystallographic data for PtAu8-PMo12 has been deposited at the CCDC 2087794.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was financially supported by NEDO (JPNP14004), JSPS KAKENHI (No. 22K14543, 24K01259, 24K17562, 24H02210, 24H02211, and 24H02217), Tokyo Metropolitan Government Advanced Research (R3-1), Tokyo Human Resources Fund for City Diplomacy, and Tokyo Metropolitan University Research Fund for Young Scientists. The synchrotron radiation experiment was performed in SPring-8 under the approval of the Japan Synchrotron Radiation Research Institute (JASRI) as 202407, 2023A1326, 2022B1259, 2021B1380.References
- E. L. Albright, T. I. Levchenko, V. K. Kulkarni, A. I. Sullivan, J. F. DeJesus, S. Malola, S. Takano, M. Nambo, K. Stamplecoskie, H. Häkkinen, T. Tsukuda and C. M. Crudden, J. Am. Chem. Soc., 2024, 146, 5759–5780 CrossRef CAS PubMed.
- S. Li, N.-N. Li, X.-Y. Dong, S.-Q. Zang and T. C. W. Mak, Chem. Rev., 2024, 124, 7262–7378 CrossRef CAS PubMed.
- Y. Horita, M. Ishimi and Y. Negishi, Sci. Technol. Adv. Mater., 2023, 24, 2203832 CrossRef PubMed.
- R. Jin, G. Li, S. Sharma, Y. Li and X. Du, Chem. Rev., 2021, 121, 567–648 CrossRef CAS PubMed.
- K. J. Taylor, C. L. Pettiette-Hall, O. Cheshnovsky and R. E. Smalley, J. Chem. Phys., 1992, 96, 3319–3329 CrossRef CAS.
- B. E. Salisbury, W. T. Wallace and R. L. Whetten, Chem. Phys., 2000, 262, 131–141 CrossRef CAS.
- R. H. Adnan, J. M. L. Madridejos, A. S. Alotabi, G. F. Metha and G. G. Andersson, Adv. Sci., 2022, 9, 2105692 CrossRef CAS PubMed.
- S. Hossain, D. Hirayama, A. Ikeda, M. Ishimi, S. Funaki, A. Samanta, T. Kawawaki and Y. Negishi, Aggregate, 2023, 4, e255 CrossRef CAS.
- M.-M. Zhang, X.-Y. Dong, Y.-J. Wang, S.-Q. Zang and T. C. W. Mak, Coord. Chem. Rev., 2022, 453, 214315 CrossRef CAS.
- C. A. Smith, M. R. Narouz, P. A. Lummis, I. Singh, A. Nazemi, C.-H. Li and C. M. Crudden, Chem. Rev., 2019, 119, 4986–5056 CrossRef CAS PubMed.
- J. Olesiak-Banska, M. Waszkielewicz, P. Obstarczyk and M. Samoc, Chem. Soc. Rev., 2019, 48, 4087–4117 RSC.
- Y. Shichibu and K. Konishi, ChemNanoMat, 2022, 8, e202200194 CrossRef CAS.
- Z. Liu, L. Luo and R. Jin, Adv. Mater., 2024, 36, 2309073 CrossRef CAS PubMed.
- J. Foxley and K. L. Knappenberger Jr, Annu. Rev. Phys. Chem., 2023, 74, 53–72 CrossRef CAS PubMed.
- K. Kwak and D. Lee, Acc. Chem. Res., 2019, 52, 12–22 CrossRef CAS PubMed.
- L. Wang, J. Peng, Z. Tang, X. Kang, M. Fu and S. Chen, Appl. Catal., 2018, 557, 1–6 CrossRef CAS.
- Y. Zhu, H. Qian, M. Zhu and R. Jin, Adv. Mater., 2010, 22, 1915–1920 CrossRef CAS PubMed.
- X. Cai, G. Saranya, K. Shen, M. Chen, R. Si, W. Ding and Y. Zhu, Angew. Chem., Int. Ed., 2019, 58, 9964–9968 CrossRef CAS PubMed.
- K. Kwak, W. Choi, Q. Tang, M. Kim, Y. Lee, D.-E. Jiang and D. Lee, Nat. Commun., 2017, 8, 14723 CrossRef PubMed.
- S. Takano, H. Hirai, T. Nakashima, T. Iwasa, T. Taketsugu and T. Tsukuda, J. Am. Chem. Soc., 2021, 143, 10560–10564 CrossRef CAS PubMed.
- H. Hirai, S. Takano, T. Nakashima, T. Iwasa, T. Taketsugu and T. Tsukuda, Angew. Chem., Int. Ed., 2022, 61, e202207290 CrossRef CAS PubMed.
- M. Schulz-Dobrick and M. Jansen, Eur. J. Inorg. Chem., 2006, 4498–4502 CrossRef CAS.
- M. A. Aubart and L. H. Pignolet, J. Am. Chem. Soc., 1992, 114, 7901–7903 CrossRef CAS.
- R. P. F. Kanters, P. P. J. Schlebos, J. J. Bour, W. P. Bosman, H. J. Behm and J. J. Steggerda, Inorg. Chem., 1988, 27, 4034–4037 CrossRef CAS.
- D. A. Krogstad, W. V. Konze and L. H. Pignolet, Inorg. Chem., 1996, 35, 6763–6771 CrossRef CAS PubMed.
- L. N. Ito, J. D. Sweet, A. M. Mueting, L. H. Pignolet, M. F. J. Schoondergang and J. J. Steggerda, Inorg. Chem., 1989, 28, 3696–3701 CrossRef CAS.
- T. Matsuyama, S. Kikkawa, Y. Fujiki, M. Tsukada, H. Takaya, N. Yasuda, K. Nitta, N. Nakatani, Y. Negishi and S. Yamazoe, J. Chem. Phys., 2021, 155, 044307–044314 CrossRef CAS PubMed.
- Y. Fujiki, T. Matsuyama, S. Kikkawa, J. Hirayama, H. Takaya, N. Nakatani, N. Yasuda, K. Nitta, Y. Negishi and S. Yamazoe, Commun. Chem., 2023, 6, 129 CrossRef CAS PubMed.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS PubMed.
- S. N. Reifsnyder, M. M. Otten, D. E. Sayers and H. H. Lamb, J. Phys. Chem. B, 1997, 101, 4972–4977 CrossRef CAS.
- J. J. Bour, R. P. F. Kanters, P. P. J. Schlebos, W. P. Bosman, H. Behm, P. T. Beurskens and J. J. Steggerda, Recl. Trav. Chim. Pays-Bas, 1987, 106, 157–158 CrossRef CAS.
- P. Glatzel, J. Singh, K. O. Kvashnina and J. A. van Bokhoven, J. Am. Chem. Soc., 2010, 132, 2555–2557 CrossRef CAS PubMed.
- N. Mehio, S. Dai and D.-E. Jiang, J. Phys. Chem. A, 2014, 118, 1150–1154 CrossRef CAS PubMed.
- S. Seino, R. Kawahara, Y. Ogasawara, N. Mizuno and S. Uchida, Angew. Chem., Int. Ed., 2016, 55, 3987–3991 CrossRef CAS PubMed.
- S. M. Perera, S. R. Hettiarachchi and J. W. Hewage, ACS Omega, 2022, 7, 2316–2330 CrossRef CAS PubMed.
- J. J. Bour, P. P. J. Schlebos, R. P. F. Kanters, M. F. J. Schoondergang, H. Addens, A. Overweg and J. J. Steggerda, Inorg. Chim. Acta, 1991, 181, 195–200 CrossRef CAS.
- T. Matsuyama, J. Hirayama, Y. Fujiki, S. Kikkawa, W. Kurashige, H. Asakura, N. Kawamura, Y. Negishi, N. Nakatani, K. Hatada, F. Ota and S. Yamazoe, J. Phys. Chem. C, 2021, 125, 3143–3149 CrossRef CAS.
- T. Uruga, M. Tada, O. Sekizawa, Y. Takagi, T. Yokoyama and Y. Iwasawa, Chem. Rec., 2019, 19, 1444–1456 CrossRef CAS PubMed.
- H. Asakura, S. Yamazoe, T. Misumi, A. Fujita, T. Tsukuda and T. Tanaka, Radiat. Phys. Chem., 2020, 175, 108270–108273 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision B.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- K. Hatada, K. Hayakawa, M. Benfatto and C. R. Natoli, J. Phys.: Condens. Matter, 2010, 22, 185501 CrossRef PubMed.
- L. Hedin and S. Lundqvist, Solid State Phys., 1970, 23, 1–181 Search PubMed.
- L. Hedin and B. I. Lundqvist, J. Phys. C: Solid State Phys., 1971, 4, 2064 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S15, Scheme S1 and Tables S1–S3. See DOI: https://doi.org/10.1039/d4nr03785e |
| This journal is © The Royal Society of Chemistry 2025 |