
Holey etching strategy of siloxene nanosheets to improve the rate performance of photo-assisted Li–O2 batteries†
Wenpu
Xu
abc,
Zitai
Fu
abc,
Huanbao
Shi
abc,
Qi
Li
 *bc,
Xuexia
He
bc,
Jie
Sun
bc,
Ruibin
Jiang
bc,
Zhibin
Lei
abc and
Zong-Huai
Liu
*abc
*bc,
Xuexia
He
bc,
Jie
Sun
bc,
Ruibin
Jiang
bc,
Zhibin
Lei
abc and
Zong-Huai
Liu
*abc
aKey Laboratory of Applied Surface and Colloid Chemistry (Shaanxi Normal University), Ministry of Education, Xi'an, 710062, P. R. China. E-mail: zhliu@snnu.edu.cn
bShaanxi Key Laboratory for Advanced Energy Devices, Xi'an, 710119, P. R. China. E-mail: clliqi@snnu.edu.cn
cSchool of Materials Science and Engineering, Shaanxi Normal University, Xi'an, 710119, P. R. China
First published on 22nd November 2024
Abstract
Improving the rate performance is of great significance to achieve high-performance photo-assisted Li–O2 batteries for developing new optimized bifunctional photocatalysts. Herein, a holey etching strategy is developed to prepare porous siloxene nanosheets with a size of 10 nm and few layers (P-siloxene NSs) by a modified Ag+-assisted chemical etching method, and the optimized pore-forming conditions are: Ag+ ion concentration 0.01 mol dm−3, HF concentration 0.565 mol dm−3, and H2O2 concentration 0.327 mol dm−3. By using P-siloxene NSs with a bandgap of 2.77 eV as a novel bifunctional photo-assisted Li–O2 system, the rate performance of the assembled P-siloxene NSs photo-assisted Li–O2 batteries is clearly improved. At a current density of 0.1 mA cm−2, the system shows a low overpotential of 0.35 V, full discharge capacity of 3270 mA h g−1, and 69% round-trip efficiency at 100 cycles. In particular, at a current density of 0.8 mA cm−2, the P-siloxene NSs photo-assisted Li–O2 batteries still give a relatively good charge potential of 3.66 V and a discharge potential of 2.97 V. This work provides a new approach for improving the rate performance of photo-assisted Li–O2 systems and will open up opportunities for the high-efficiency utilization of solar energy in electric systems.
1. Introduction
As a representative of new energy storage systems, lithium-ion batteries (LIBs) have been widely used in our daily lives because of their long cycle lifetime, relatively high energy density, light weight, and environmental friendliness. However, their energy density hinders the development of LIBs as future high-energy storage devices.1 Li–O2 batteries have a ultrahigh theoretical specific capacity (3862 mA h g−1)2,3 and intrinsic utilization of abundant O2 as active materials, and have received a lot of attention because their energy density is almost 10 times higher than that of LIBs. In the discharge/charge process of the O2 cathode, the basis of the storage reaction is 2Li+ + O2 + 2e− ↔ Li2O2, and a theoretical thermodynamic standard working voltage of 2.96 V vs. Li+/Li for the formation/decomposition of Li2O2 is needed.4,5 Nevertheless, Li2O2 is insoluble and an insulating material, and these properties lead to a high charging voltage of 4–4.5 V during the oxygen evolution reaction for the charging process, and produces slow kinetics and high overpotential polarization and poor round-trip efficiency.6–9 Although numerous efforts have been made by using some traditional solid catalysts, including noble metal catalysts,10 metal oxides/sulfides,11 and soluble redox mediators,12 the unsatisfactory overpotential, poor rate performance, and potential hysteresis of these different catalysts has still not been solved.In recent years, a photo-assisted Li–O2 battery system has been constructed by introducing solar energy and directly integrating a photoelectrode as a cathode into the Li–O2 battery. This photo-assisted Li–O2 battery system can be considered as an effective strategy for achieving a significant reduction of overpotential and substantial electric energy savings.13,14 By using Fe2O3/C3N4,15 Co-TABQ,16 Au/Nv-C3N4,17 plasmonic heterojunctions,18etc., with bifunctional photocatalysts as photocathodes, some good experimental results have been achieved. In general, the theoretical discharge/charge potentials of photo-assisted Li–O2 batteries are mainly determined by the valence band (VB) and conduction band (CB) energy levels of the photoelectrodes (vs. Li+/Li), while the photoelectric properties and compatibility of the semiconductors as photocatalysts also affect the practical performance of the assembled devices.19 Moreover, the input and output electric energies of photo-assisted Li–O2 batteries are associated with the related charging and discharging plateaus, respectively, and are affected by the heavy photoexcited charge carriers and transfer barrier between the photoelectrodes employed and the Li–O2 system.20 Therefore, it is of great importance to develop new optimized bifunctional photoelectrodes for accelerating energy conversion and storage for high-efficiency photo-assisted Li–O2 batteries. In our previous work, siloxene nanosheets (siloxene NSs) with large size, few layers, and Kautsky-type structure were prepared using a modified topochemical exfoliation method, and then a bifunctional photo-assisted Li–O2 battery was assembled by using siloxene NSs as the bifunctional photocathode. The assembled bifunctional photo-assisted Li–O2 battery displayed an ultralow charging potential of 1.90 V and an ultrahigh discharge potential of 3.51 V, with good round-trip efficiency of 129% at 1 mA cm−2, an ultralong cycling life with 92% efficiency retention after 100 cycles and a fast light response, and a highly reversible capacity of 1170 mA h g−1 at 0.75 mA cm−2.21,22 Although some excellent performance results for the siloxene NSs photo-assisted Li–O2 battery have been obtained, the rate performance and full discharge capacity need to be further improved.
Two-dimensional siloxene NSs are a kind of direct bandgap semiconductor,23 and have shown good prospects for use in photocatalysts due to the quantum limiting effect produced by their nanoscale structure and the fluorescence effect produced by the Si–O–H active groups.24,25 However, the active atoms or groups of siloxene NSs are mainly distributed at the edge region, while the activity of the internal atoms or functional groups is not fully utilized because the catalytic activity mainly comes from the coordination of unsaturated atoms.26 Therefore, introducing active atoms into the interior region of siloxene NSs may be an effective way to improve the rate performance of photo-assisted Li–O2 batteries. In the present work, a holey etching strategy was developed to prepare porous siloxene nanosheets with a size of 10 nm and few layers (P-siloxene NSs) using a modified Ag+-assisted chemical etching method, and the pore-forming conditions were systematically optimized. By using P-siloxene NSs with a bandgap of 2.77 eV as a novel bifunctional photo-assisted Li–O2 photocathode, the rate performance of the assembled P-siloxene NSs photo-assisted Li–O2 batteries was notably improved. At a current density of 0.1 mA cm−2, a low overpotential of 0.35 V, full discharge capacity of 3270 mA h g−1, and 69% round-trip efficiency for 100 cycles were achieved. In particular, at a current density of 0.8 mA cm−2, the P-siloxene NSs photo-assisted Li–O2 batteries still gave a relatively good charge potential of 3.66 V and a discharge potential of 2.97 V. This work provides a new approach for improving the rate performance of the photo-assisted Li–O2 system and will open up opportunities for high-efficiency utilization of solar energy in electric systems.
2. Experimental section
2.1 Chemicals
Calcium silicide (CaSi2, 95%) was purchased from Alfa Aesar. Sodium hydroxide (NaOH), hydrochloric acid (HCl, 36%), anhydrous ethanol (CH3CH2OH), hydrofluoric acid (HF, 40%), hydrogen peroxide (H2O2, 30%), and nitric acid (HNO3, 60%) were purchased from Sinopharm Chemical Reagent Co., Ltd. Sodium dodecyl sulfate (SDS, C12H25SO4Na) was purchased from J&K Chemicals. All chemicals were used as received without further purification.2.2 Preparation of P-siloxene NSs
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 20 minutes) and subsequently freeze-dried to obtain few-layer siloxene NSs.
000 rpm for 20 minutes, followed by freeze-drying to prepare porous siloxene NSs coated with Ag nanoparticles, abbreviated as Ag/P-siloxene NSs. Then, Ag/P-siloxene NSs (5 mg) were treated with diluted HNO3 (1 mL in 20 mL anhydrous ethanol) at 50 °C for 2 hours in a dark environment, the Ag nanoparticles were removed, and porous siloxene NSs were obtained, denoted as P-siloxene NSs. By changing the concentrations of the AgNO3 solution, HF solution, and H2O2 solution, the optimal pore-formation conditions were systematically studied under otherwise identical reaction conditions using the same procedure.
000 rpm for 20 minutes) and subsequently freeze-dried to obtain few-layer siloxene NSs.
000 rpm for 20 minutes, followed by freeze-drying to prepare porous siloxene NSs coated with Ag nanoparticles, abbreviated as Ag/P-siloxene NSs. Then, Ag/P-siloxene NSs (5 mg) were treated with diluted HNO3 (1 mL in 20 mL anhydrous ethanol) at 50 °C for 2 hours in a dark environment, the Ag nanoparticles were removed, and porous siloxene NSs were obtained, denoted as P-siloxene NSs. By changing the concentrations of the AgNO3 solution, HF solution, and H2O2 solution, the optimal pore-formation conditions were systematically studied under otherwise identical reaction conditions using the same procedure.
2.3 Characterization
The X-ray diffraction (XRD) patterns of samples at different stages were determined using a MiniFlex 600 instrument (Rigaku Corporation) with Cu–Kα radiation (λ = 1.5406 Å), scanning from 5° to 80° and with an operating voltage and current of 40 kV and 15 mA, respectively. The sample morphologies were observed by scanning electron microscopy (SEM, TM-3000), field-emission scanning electron microscopy (FE-SEM, SU8020), transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM, JEM-2800). The energy dispersive X-ray spectroscopy (EDS) mappings were carried out on the transmission electron microscope (JEM-2800) with detectors. Spherical aberration corrected (AC) TEM was carried out (Titan G2 300, Thermo Fisher Scientific). The thickness and size of the siloxene NSs were measured using atomic force microscopy (AFM, Dimension ICON, Bruker) and the composition and chemical state were determined using an X-ray photoelectron spectroscopy (XPS) instrument (Kratos Analytical Ltd). The binding energy calibration was referenced to C 1s at 284.8 eV. Raman spectra were obtained on a microscopic confocal laser Raman spectrometer (Renishaw InVia). Fourier-transform infrared (FT-IR) spectroscopy was obtained by an FT-IR, Vertex70 (Bruker). N2 adsorption/desorption isotherms were obtained using a Brunauer–Emmett–Teller instrument (BET, ASAP 2460, Micromeritics). UV-visible absorption spectra were recorded on a UV-visible spectrophotometer (UV-vis, UV3600, Shimadzu). Steady-state transient fluorescence spectrometry was carried out using photoluminescence (PL) (Fluorolog-QM, HORIBA).2.4 Assembly and electrochemical measurements of Li–O2 batteries
For assembling photo-assisted Li–O2 batteries, the photoelectrodes were obtained by mixing 80 wt% of siloxene-based materials, 15 wt% of super-P, and 5 wt% of polytetrafluoroethylene (PTFE), and the mixed slurry was uniformly coated onto clean Ni foam (diameter, 19 mm). Coin cells (CR2032) with 19 holes were employed to assemble the photo-assisted Li–O2 batteries in an Ar-filled glovebox (<1 ppm of H2O and O2) with LiClO4 in DMSO (1 M) as the electrolyte, a glass fiber (Whatman) as the separator, and a high-purity Li foil (diameter, 15.8 mm) as the anode. The charging and discharging curves of the assembled devices were tested using a Solartron 1740E electrochemical workstation. The electrochemical impedance spectroscopy (EIS) curves were measured using a Solartron 1260-1460E machine, and a GELS500/350 Xe lamp (wavelength range: 200–1200 nm, Ceaulight, Beijing) with a fixed power at 500 W was used as the solar source for illumination. An optical power meter was utilized for calibrating the photodensity on the surface of the Li–O2 battery electrode to AM1.5 G, which corresponded to a photodensity of 100 mW cm−2.3. Results and discussion
Raw, layered CaSi2 possesses a hexagonal crystal structure (Fig. S1, ESI†).29 After it is treated with NaOH solution, the content of Si impurity is significantly reduced, and purified layered CaSi2 is obtained (Fig. S2, ESI†). The SEM image shows that the purified layered CaSi2 has a close-packed structural morphology with a particle size of 40 to 50 μm. EDS-mapping images indicate that Ca2+ ions and Si are uniformly distributed in the purified layered CaSi2 (Fig. S3, ESI†). The purified layered CaSi2 was stirred in concentrated HCl solution for 8 days at 0 °C in an N2 atmosphere, and a topological chemical etching reaction was carried out by removing Ca2+ ions from the interlayer on the basis of the chemical reaction CaSi2 + HCl + H2O = Si6H3(OH)3 + CaCl2 + H2, with the color of the reaction mixture completely transformed to yellowish green from black CaSi2, and the layered siloxene obtained. Comparing the XRD pattern of purified layered CaSi2, two new characteristic diffraction peaks at about 2θ = 13.3° and 27.2°, corresponding to the typical (001) and (100) lattice planes of the layered siloxane, were observed (Fig. 1a), indicating successful etching of purified layered CaSi2 into layered siloxene.21,30 In addition, a little FeSi2 was detected in the XRD pattern,31 suggesting that FeSi2 is not completely removed. Moreover, layered siloxene shows an accordion structure morphology with expanding lamellae spacing (Fig. S4a, ESI†). EDS-mapping images indicate that the Ca2+ ions are obviously reduced due removal by the topological chemical etching reaction, while Si and O elements are uniformly distributed in the layered siloxene (Fig. S4b–4e, ESI†). After, the layered siloxene was stirred in SDS/ethylene alcohol solution at 60 °C for 24 h under N2 atmosphere, followed by ultrasonic treatment for 3 h at 480 W, the layered siloxene was exfoliated into siloxene NSs and few-layer siloxene NSs were finally prepared.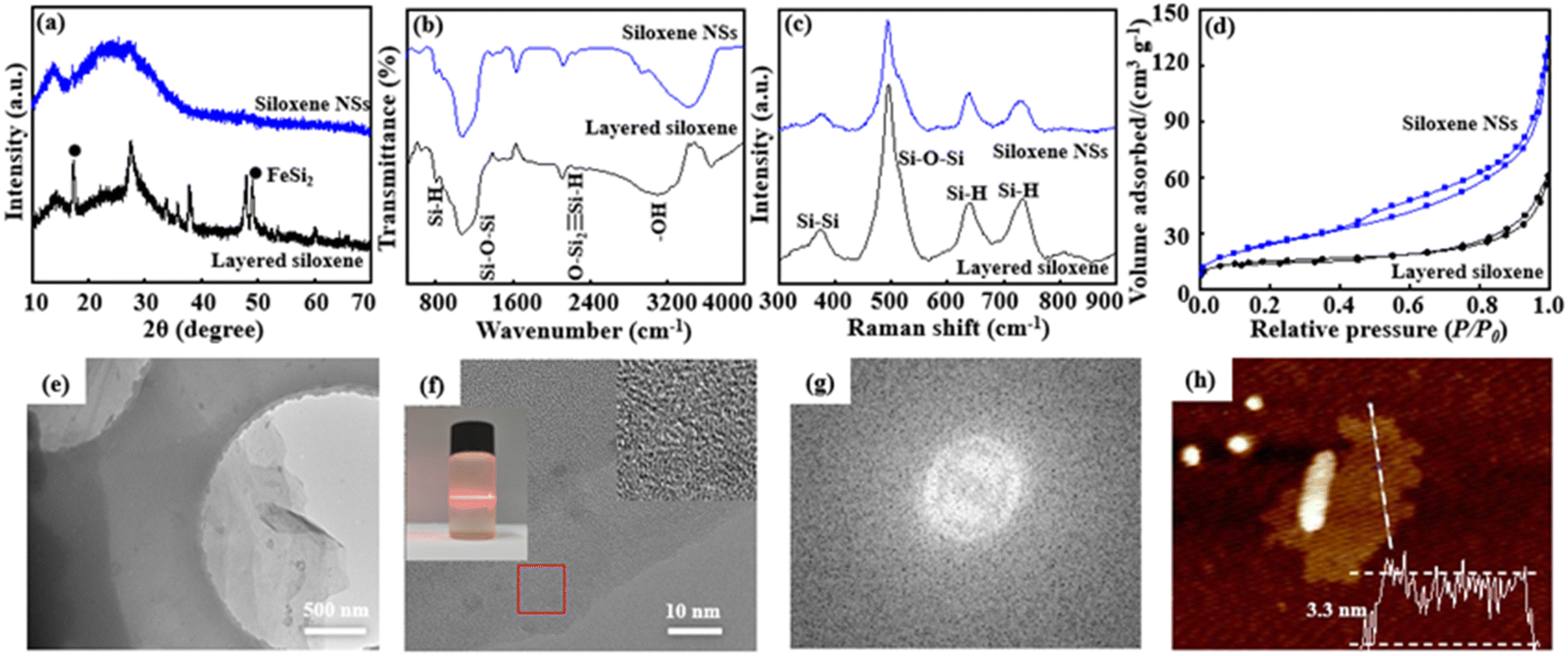 | ||
| Fig. 1 XRD patterns (a), FT-IR spectra (b), Raman spectra (c), and N2 adsorption–desorption isotherms (d) of layered siloxene and siloxene NSs. TEM image (e), HRTEM image and Tyndall effect for delaminated siloxene NSs in suspension (f). Fourier transform image (g) and AFM image and the corresponding thickness profiles (h) of siloxene NSs. | ||
Although the XRD pattern is similar to that of the layered siloxene, the broad diffraction peaks indicate that the exfoliated siloxene is composed of numerous nanometer-scale siloxene NSs. The FT-IR spectra show some evident sharp and broad peaks at 801 cm−1, 1076 cm−1, 2114 cm−1, and 3117 cm−1, attributed to the vibrations ν(Si–H), ν(Si–O–Si), ν(OSi2![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Si–H), and ν(Si–OH), respectively, and suggesting that the exfoliated siloxene NSs still possess a Kautsky-type structure (Fig. 1b).31,32 Moreover, the layered siloxene and siloxene NSs also exhibit similar Raman spectra (Fig. 1c) before and after the delaminating reaction, suggesting that the delaminating process has not caused obvious structural change. In addition, siloxene NSs have a relatively large specific surface area, indicating that the exfoliation process leads to an increase in the specific surface area (Fig. 1d).33
Si–H), and ν(Si–OH), respectively, and suggesting that the exfoliated siloxene NSs still possess a Kautsky-type structure (Fig. 1b).31,32 Moreover, the layered siloxene and siloxene NSs also exhibit similar Raman spectra (Fig. 1c) before and after the delaminating reaction, suggesting that the delaminating process has not caused obvious structural change. In addition, siloxene NSs have a relatively large specific surface area, indicating that the exfoliation process leads to an increase in the specific surface area (Fig. 1d).33
The TEM image shows that siloxene NSs have a transparent thin-film morphology, with a lateral size of about 5 to 10 μm (Fig. 1e). In addition, siloxene NSs show good dispersibility in ethyl alcohol, and an obvious Tyndall effect can be observed (insert in Fig. 1f), suggesting the typical colloid characteristics and potential for processible applications of siloxene NSs. The HR-TEM image shows that siloxene NSs have no distinct lattice fringes (Fig. 1f), and only diffraction rings without spots are observed from the Fourier transform result (Fig. 1g), further suggesting the weak crystalline characteristics, similar to amorphous structures.34,35 Furthermore, the thickness of the siloxene NSs was recorded at about 3.3 nm from the AFM image (Fig. 1h), corresponding to five layers of siloxene nanosheets of 0.63 nm thickness.23,36 The above results suggest that few-layer siloxene NSs can be obtained by a modified topochemical exfoliation technique using CaSi2 in concentrated HCl solution at 0 °C and followed by ultrasonic treatment in SDS/ethylene alcohol solution.
Inorganic nanosheets can be used to assemble optoelectronic functional materials with different structures or morphologies.37 Although the assembled optoelectronic functional materials show good performance, they are not beneficial for improving the rate capability of the optoelectronic functional materials due to poor ion transport in the vertical direction.38 Therefore, developing a holey etching strategy for inorganic nanosheets is of benefit for improving the rate efficiency for ion transport in the vertical direction. Siloxene NSs consisting of Si and O elements, in which the Si element can be etched from siloxene NSs under suitable redox media, and porous siloxene NSs, were prepared. By using a modified Ag+-assisted chemical etching method, porous siloxene NSs were prepared in a suitable solution consisting of HF, H2O2, and AgNO3. When there were no Ag+ ions in the reaction system, the siloxene NSs were reacted in HF solution or a HF + H2O2 mixed solution, and only relatively small siloxene NSs were obtained and no holey structures were formed in the siloxene NSs (Fig. 2a and b).
 | ||
| Fig. 2 TEM images of the products obtained from siloxene NSs before and after Ag+ ion adsorption under different reaction conditions: siloxene NSs dispersed in HF (a) and HF + H2O2 (b) before Ag+ ion adsorption, siloxene NSs dispersed in HF (c) and HF + H2O2 (d) after Ag+ ion adsorption. The formation process of P-siloxene NSs (e). | ||
On the other hand, when siloxene NSs are suspended in AgNO3 solution with a certain concentration in a dark environment, Ag+ ions are adsorbed onto the surface of the siloxene NSs. Then, Ag+ ions oxidize the Si element to SiO2, while Ag+ ions are reduced into Ag nanoparticles to load onto the surface of the siloxene NSs, and Ag/P-siloxene NSs are prepared. This process results in the formation of Ag-loaded porous siloxene nanosheets (Ag/P-siloxene NSs). When Ag/P-siloxene NSs are treated with HF solution for 30 minutes, Ag nanoparticles can be observed and only small holes are formed on the siloxene NSs (Fig. 2c). However, a large number of pores with a size of 10 nm are formed on the surface of the siloxene NSs and P-siloxene NSs when Ag/P-siloxene NSs are treated with a mixed solution of HF and H2O2 (Fig. 2d). These results clearly indicate that the Ag+ ions and H2O2 are key factors for formatting a large number of pores. In the presence of H2O2, Ag nanoparticles loaded onto the surface of siloxene NSs are oxidized to Ag+ ions, which subsequently oxidize Si element to SiO2. After the siloxene NSs loaded with a large amount of SiO2 are dissolved in HF solution, numerous pores with a size of 10 nm are formed and P-siloxene NSs are ultimately prepared.
Moreover, the effects of HF, H2O2, and AgNO3 concentrations on the pore formation were systematically investigated. It can be seen that the Ag+ ion concentration has an obvious influence on the pore formation at the same concentrations of HF + H2O2, with excessive (0.02 and 0.03 mol dm−3) or insufficient concentrations (0.005 mol dm−3) of Ag+ ion not being conducive to regular pore formation (Fig. S5, ESI†), and regular holey structures with a pore size of about 10 nm are obtained when the Ag+ ion concentration is 0.01 mol dm−3 in the reaction system. Meanwhile, a pore size of over 50 nm is formed on siloxene NSs in HF concentrations of 1.13 mol dm−3 when the Ag+ ion concentration is 0.01 mol dm−3 (Fig. S6, ESI†). In addition, the presence or absence of H2O2 in the reaction system is crucial for pore formation on siloxene NSs. When 0.49 mol dm−3 of H2O2 is used for the etching reaction at the same concentration of Ag+ ions (0.01 mol dm−3) and HF (0.565 mol dm−3), relatively regular pores with a size of 30 to 100 nm were observed from the TEM images (Fig. S7, ESI†). Based on the experimental results for hole formation, it can be concluded that the optimal pore-forming conditions for P-siloxene NSs are: Ag+ ion concentration of 0.01 mol dm−3, HF concentration of 0.565 mol dm−3, and H2O2 concentration of 0.327 mol dm−3. The formation process for P-siloxene NSs is illustrated in Fig. 2e.
Under optimal pore-forming conditions, P-siloxene NSs were prepared using Ag+ ions of 0.01 mol dm−3, HF of 0.565 mol dm−3, and H2O2 of 0.327 mol dm−3. Compared with the XRD pattern of siloxene NSs, P-siloxene NSs still show similar broad diffraction peaks corresponding to numerous nanometer-scale siloxene NSs, except for a small amount of impurity AgCl phase (Fig. 3a). The typical vibration peaks of ν(Si–Si), ν(Si–H), ν(Si–O–Si), and ν(OSi2Si–H), corresponding to a Kautsky-type structure, are observed in the Raman and FT-IR spectra, suggesting that the holey treatment has no effect on the structure of the siloxene NSs (Fig. 3b and c). The AFM image (Fig. 3d) and TEM image (Fig. 3e) indicate that the P-siloxene NSs have a plate-like morphology with some porosity, and the lateral dimension is about 5 μm with an average thickness of 4.2 nm. Moreover, the AC-TEM image further shows an average pore diameter of 10 nm, with the pores uniformly distributed on the siloxene NSs (Fig. 3f), and only 0.04% Ag is detected from the energy spectrum analysis of AC-TEM, suggesting that the Ag nanoparticles have nearly been removed (Fig. S8, ESI†). The XPS spectra show the characteristic binding energies for O 1s and Si 2p,34 while obvious Ag 3d spectrum peaks were not observed (Fig. S9, ESI†). The zeta potential of P-siloxene NSs suspensions with differing amounts support that the P-siloxene NSs have a negative charge,33 and the zeta potential values increase linearly with increase in the amount of P-siloxene NSs in suspension (Fig. S10, ESI†). Based on the potential advantages of few-layer and porous structures, the prepared P-siloxene NSs could be widely employed for promising applications in catalysis, electrochemical storage, and other fields.
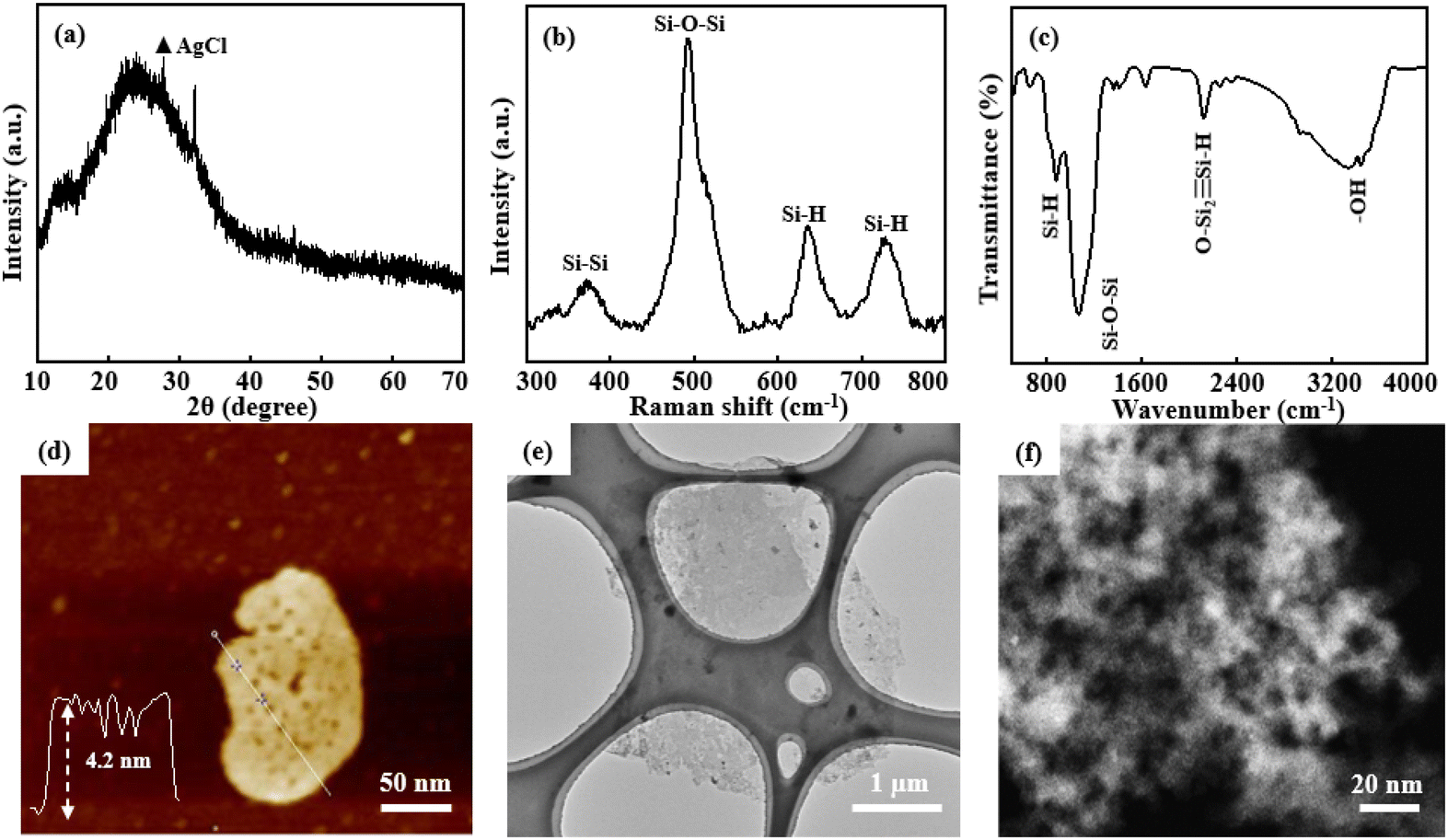 | ||
| Fig. 3 Structure and morphology characteristic of P-siloxene NSs: XRD pattern (a), Raman spectrum (b), FT-IR spectrum (c), AFM image and corresponding thickness profile (d), TEM image (e), and AC-TEM image (f). | ||
In order to widen the promising applications of the prepared P-siloxene NSs, their semiconductor properties were evaluated compared with layered siloxene and siloxene NSs. Ultraviolet–visible spectra show that the absorption range is blue-shifted, due to the quantum size effect when layered siloxene is exfoliated into siloxene NSs and followed with porous treatment (Fig. 4a), and the maximum absorption edge wavelengths of the three samples were 549 nm, 509 nm, and 487 nm, respectively. The bandgaps of the three samples were measured as 2.44 eV, 2.59 eV, and 2.77 eV by UV-vis spectroscopy, respectively, while the EVB was obtained by ultraviolet photoelectron spectroscopy (UPS) characterization. Thus the ECB can be acquired by subtracting the bandgap value from the EVB value. Tauc plots show that P-siloxene NSs are a direct bandgap semiconductor, and the bandgap increases to 2.77 eV from 2.59 eV due to the introduction of defects (Fig. 4b).33,39 Moreover, steady-state PL spectra further support the blue shift of the absorption range (Fig. 4c). In addition, UPS measurements also suggest that P-siloxene NSs have a relatively high VB and low CB due to the quantum size effect (Fig. S11, ESI†). Although the porosity causes a certain blue shift in the absorption range of the prepared P-siloxene NSs, an obvious decrease of the average fluorescence lifetime is observed from the average fluorescence spectra (Fig. 4d). Based on the calculated equations,23,40 the average fluorescence lifetime gradually reduces to 0.96 ns from 4.04 ns for layered siloxane, and to 1.16 ns for siloxene NSs after the holey strategy treatment, suggesting that the prepared P-siloxene NSs may be beneficial for electronic transmission and could be applied to Li–O2 batteries as photocatalysts.
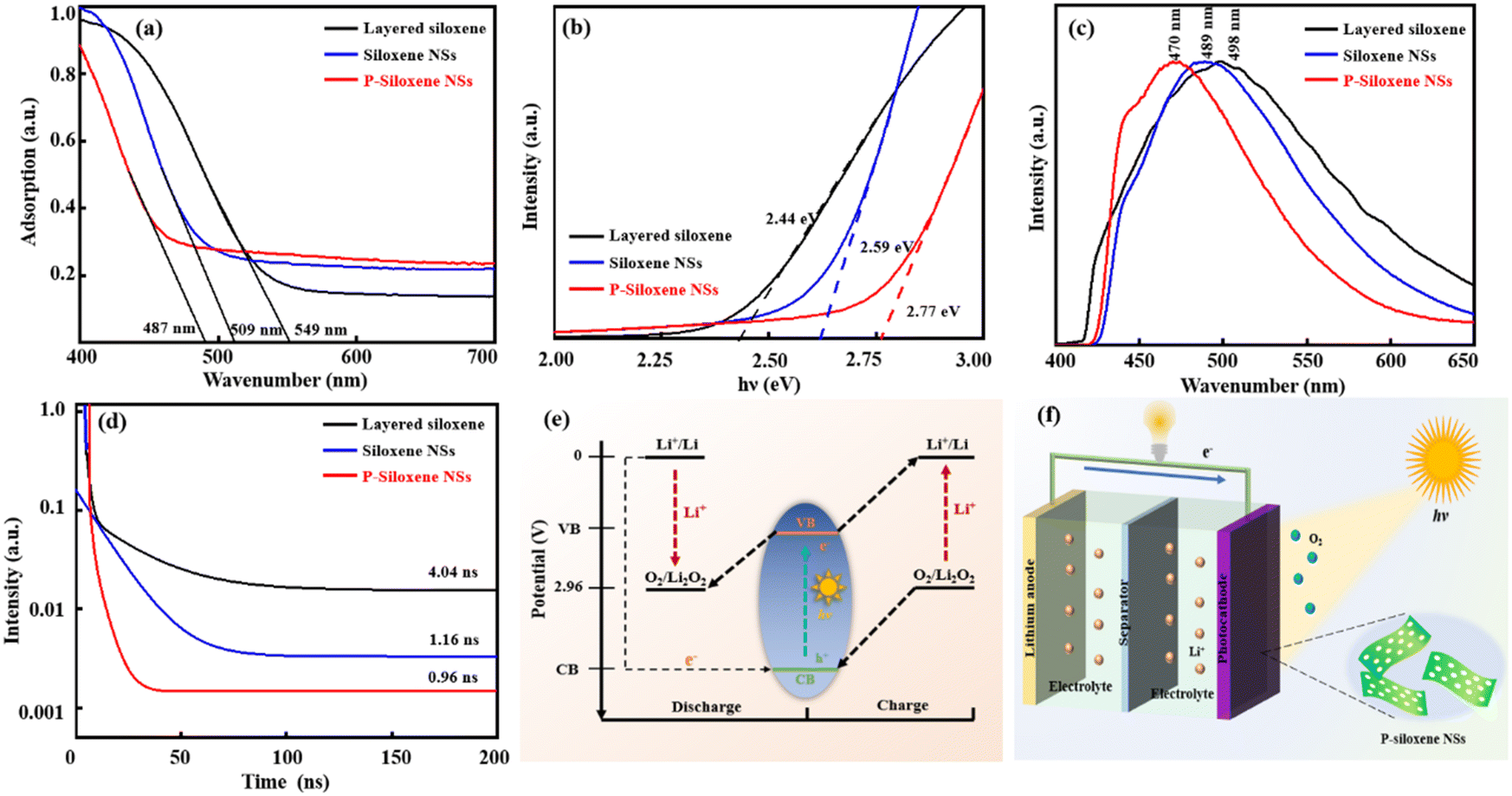 | ||
| Fig. 4 Semiconductor property characteristics of layered siloxene, siloxene NSs, and P-siloxene NSs: UV-vis spectra (a), Tauc plots (b), steady-state fluorescence spectra (c), fluorescence lifetime and fitting curves (d), band structure (e), and the schematic diagram of photo-assisted Li–O2 batteries consisting of Li anode, electrolyte and P-siloxene NSs photocathode (f). | ||
The potential (vs. Li+/Li) of O2/Li2O2 is outlined in Fig. 4e. This demonstrates that the prepared P-siloxene NSs can be used as photoelectrodes to participate in the oxygen evolution reaction (OER) and oxygen reduction reaction (ORR) of Li–O2 batteries because the VB potential is higher than the oxidation potential of the O2/Li2O2 couple, while the CB potential is below the reduction potential for O2 to Li2O2.41,42 Therefore, P-siloxene NSs can be excited by photo energy to generate holes and electrons, and can be used as bifunctional photoelectrodes to participate in the OER and ORR in Li–O2 batteries. A typical photo-assisted Li–O2 battery was constructed by utilizing the metal Li as the anode, a non-aqueous electrolyte and Ni foam coated with the prepared P-siloxene NSs as the porous O2 cathode and photoelectrode, and the working principle of the assembled photo-assisted Li–O2 battery is displayed in Fig. 4f. During the discharge process under illumination, the excited electrons on the P-siloxene NSs photocathode can induce a reduction from O2 to Li2O2 (2Li+ + O2 + 2e−(CB) → Li2O2), and the photoexcited holes detained in the VB are reduced by the electrons from the external circuit. On the other hand, the photo-assisted charging represents a reversible process compared to the photo-assisted discharging. An oxidation reaction (2h+(VB) + Li2O2 → 2Li+ + O2) occurs on the P-siloxene NSs photocathode, while the photoexcited electrons transfer to the Li anode via the external circuit. Therefore, the photo-assisted discharge and charge potentials (3.05 V and 3.4 V) of the assembled Li–O2 batteries will be determined and influenced by the CB and VB potentials (vs. Li+/Li) of the P-siloxene NSs photoelectrode, on the basis of the above unique bifunctional storage mechanism upon illumination.
Photo-assisted charge/discharge could be one of the best strategies to reduce the overpotential in Li–O2 batteries. A 2032-type coin Li–O2 battery was assembled using Ni foam-coated P-siloxene NSs as the porous O2 cathode and photoelectrode, and Li metal as anode, and the photoelectric conversion and storage performance were systematically evaluated using a 500-W Xe lamp as the photo source for illumination testing. Compared with the EIS of the assembled devices using Ni foam coated with layered siloxene and siloxene NSs as porous O2 cathodes, the charge transfer resistance (Rct) of the assembled device, using Ni foam-coated P-siloxene NSs as the porous O2 cathode, was the lowest, and the linear slope was the largest in the low frequency region, indicating that the assembled device shows good ion dynamics with illumination and the ion transfer barrier is effectively reduced by the holey etching process (Fig. 5a).43 In addition, the charge transfer resistance (Rct) of the P-siloxene NSs electrode in a three-electrode system also gave similar results, suggesting that the holey etching strategy is favorable for ion transfer in the electrode (Fig. S12, ESI†).
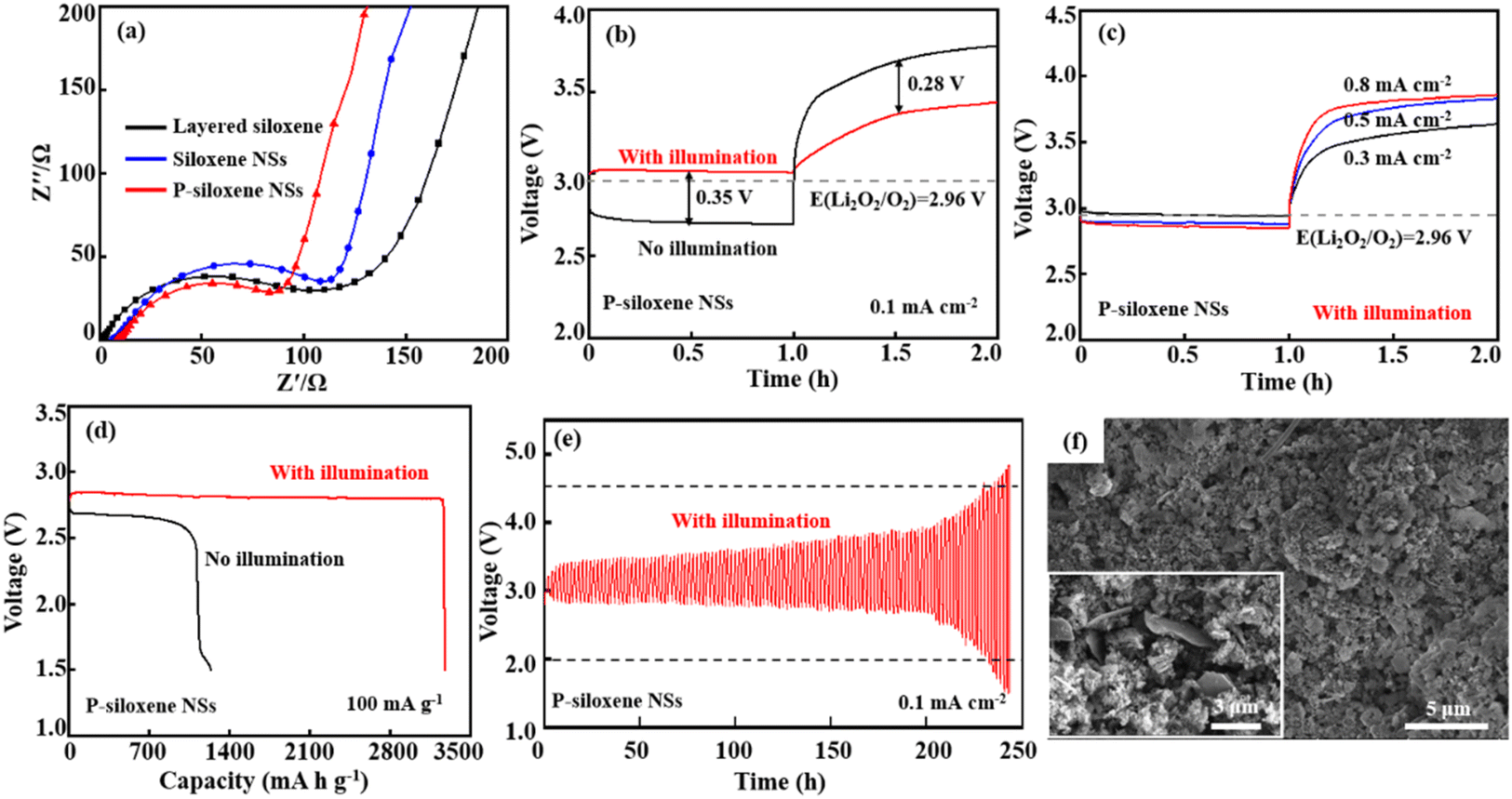 | ||
| Fig. 5 Performance characteristic of P-siloxene NSs-based photo-assisted Li–O2 batteries with and without illumination: EIS spectra (a). Discharge and charge curves at 0.1 mA cm−2 (b), the rate performance at different current densities (c), full discharge capacity (d), cycle stability (e), and SEM images before (inserted) and after discharge for 120 cycles (f) of P-siloxene NSs@Ni foam photocathode. | ||
The comparison of the discharge and charge curves for P-siloxene NSs photo-assisted Li–O2 battery is presented in Fig. 5b with and without illumination. It can be seen that the photo-assisted discharge potential reaches 3.05 V at 0.1 mA cm−2 under illumination, which is higher than the discharging potential without illumination (2.70 V). Compared to that of the 2032-type coin Li–O2 battery assembled using Ni foam-coated layered siloxene and siloxene NSs as the porous O2 cathode and photoelectrode, a significant improvement in the discharging potential is obtained for the P-siloxene NSs photoelectrode (Fig. S13, ESI†). Meanwhile, the charging voltage of the P-siloxene NSs photo-assisted Li–O2 battery is 3.40 V, with a 0.35 V lower overpotential between the discharging potential and charge potential and a high round-trip efficiency of 90% was achieved. Moreover, the electrochemical performance of P-siloxene NSs obtained at 0.005 mol L−1 and 0.02 mol L−1 for Ag+ ions and 0.452 mol L−1 and 0.753 mol L−1 for HF concentrations are shown in Fig. S14 (ESI†). These experimental results indicate that the size effect has a significant impact on the photocatalytic performance of the obtained products,42,44 suggesting that siloxene-based photocatalysts can effectively inhibit the aggregation of the discharge product Li2O2 by illumination, accelerate the generation of photogenerated electrons in the catalytic reaction, and improve the reaction kinetics process.
Additionally, the P-siloxene NSs photo-assisted Li–O2 battery system shows good reversibility and excellent rate performance (Fig. 5c). Compared with the increase of current densities, the overpotential gradually increases to 0.95 V at 0.8 mA cm−2 from 0.35 V at 0.1 mA cm−2, suggesting that the P-siloxene NSs photo-assisted Li–O2 battery system has good rate performance. Moreover, the P-siloxene NSs photo-assisted Li–O2 battery system effectively reduces polarization in the ORR process due to the rapid transport of the photogenerated electrons, making it exhibit a more stable plateau during the discharge process, with no significant voltage drop.43 Under a current density of 100 mA g−1, the P-siloxene NSs photo-assisted Li–O2 battery system exhibited a complete discharge capacity of 3270 mA h g−1, which is significantly higher than the 1130 mA h g−1 under conditions of no illumination (Fig. 5d). After complete discharge, the Li2O2 intermediate product exhibits a typical disc-like stacking morphology (Fig. S15, ESI†), consistent with previous studies.45,46 Moreover, this battery system maintains stability over 120 cycles, and the round-trip efficiency can reach 69% for 100 cycles (Fig. 5e), which is also significantly higher than for the layered siloxene and siloxene NSs photo-assisted Li–O2 battery systems (Fig. S16, ESI†). A comparison of photoelectric properties between P-siloxene NSs and other photocatalysts is shown in Table S1 (ESI†). After the discharge cycling, flower-like particles corresponding to the typical morphology of Li2O2 particles are generated on the surface of P-siloxene NSs,47,48 which is obviously different from the morphology of the P-siloxene NSs photo-assisted O2 cathode before discharge cycling (insert in Fig. 5f), clearly suggesting that the P-siloxene NSs photocathode enables the fast photo-response with illumination. These results indicate that a large number of active sites are formed on the P-siloxene NSs by the etching process, and they can generate photogenerated electrons for participation in the photocatalytic reaction under illumination, and thus present a fast photo-response and good round-trip efficiency upon illumination.
4. Conclusions
Using a modified Ag+-assisted chemical etching method, P-siloxene NSs with a lateral size of 5 to 10 μm, a pore size of 10 nm, and few layers were prepared by suspending siloxene NSs in AgNO3 solution of 0.01 mol dm−3 at 50 °C for 30 min in a dark environment, followed by addition of a mixed solution of HF and H2O2. The P-siloxene NSs exhibit a bandgap of 2.77 eV, better ion dynamics, and an appropriate potential level, and can be used as a bifunctional photo-assisted catalyst in Li–O2 batteries, to improve the rate performance. Under illumination, P-siloxene NSs can provide numerous active sites for the O2 cathode, and the photoexcited electrons and holes can stimulate the formation and decomposition of Li2O2 during the discharge and charge processes. The assembled siloxene NSs photo-assisted Li–O2 battery shows a low overpotential of 0.35 V, full discharge capacity of 3270 mA h g−1, and 69% round-trip efficiency for 100 cycles at a current density of 0.1 mA cm−2, and it still exhibits a relatively good charge/discharge potential at an increased current density of 0.8 mA cm−2. This work presents a new strategy that will open up opportunities for highly efficient conversion of solar energy into electric systems.Author contributions
Wenpu Xu: conceptualization, data curation, formal analysis, investigation, methodology, software, visualization and writing original draft. Zitai Fu: formal analysis, software, validation and visualization. Huanbao Shi: formal analysis, software, validation and visualization. Qi Li: formal analysis, software and visualization. Xuexia He: formal analysis, software and visualization. Jie Sun: software, validation and visualization. Ruibin Jiang: software and visualization. Zhibin Lei: conceptualization, software and Validation. Zong-Huai Liu: conceptualization, formal analysis, funding acquisition, methodology, project administration, supervision & writing original draft.Data availability
The authors are unable or have chosen not to specify which data have been used.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (51772182, 51902193), the 111 Project, Shaanxi “Three Qin Scholars” Innovation Team, and the Fundamental Research Funds for the Central Universities (GK202103057).References
- X. Shen, H. Liu, X.-B. Cheng, C. Yan and J.-Q. Huang, Energy Storage Mater., 2018, 12, 161–175 CrossRef.
- G. Girishkumar, B. McCloskey, A. C. Luntz, S. Swanson and W. Wilcke, J. Phys. Chem. Lett., 2010, 1, 2193–2203 CrossRef CAS.
- R. Padbury and X. Zhang, J. Power Sources, 2011, 196, 4436–4444 CrossRef CAS.
- D. Geng, N. Ding, T. S. A. Hor, S. W. Chien, Z. Liu, D. Wuu, X. Sun and Y. Zong, Adv. Energy Mater., 2016, 6, 1502164 CrossRef.
- W.-J. Kwak, Rosy, D. Sharon, C. Xia, H. Kim, L. R. Johnson, P. G. Bruce, L. F. Nazar, Y.-K. Sun and A. A. Frimer, Chem. Rev., 2020, 120, 6626–6683 CrossRef CAS PubMed.
- M. Tułodziecki, G. M. Leverick, C. V. Amanchukwu, Y. Katayama, D. G. Kwabi, F. Bardé, P. T. Hammond and Y. Shao-Horn, Energy Environ. Sci., 2017, 10, 1828–1842 RSC.
- F. Wu, Y. Xing, X. Zeng, Y. Yuan, X. Zhang, R. Shahbazian-Yassar, J. Wen, D. J. Miller, L. Li and R. Chen, Adv. Funct. Mater., 2016, 26, 7626–7633 Search PubMed.
- A. Dutta, R. A. Wong, W. Park, K. Yamanaka, T. Ohta, Y. Jung and H. R. Byon, Nat. Commun., 2018, 9, 680 CrossRef.
- S.-M. Xu, X. Liang, X.-Y. Wu, S.-L. Zhao, J. Chen, K.-X. Wang and J.-S. Chen, Nat. Commun., 2019, 10, 5810 CAS.
- R. Gao, Z. Li, X. Zhang, J. Zhang, Z. Hu and X. Liu, ACS Catal., 2016, 6, 400–406 CAS.
- J. G. Kim, Y. Kim, Y. Noh, S. Lee, Y. Kim and W. B. Kim, ACS Appl. Mater. Interfaces, 2018, 10, 5429–5439 CAS.
- J. B. Park, S. H. Lee, H. G. Jung, D. Aurbach and Y. K. Sun, Adv. Mater., 2018, 30, 1704162 Search PubMed.
- X.-X. Wang, D.-H. Guan, C.-L. Miao, D.-C. Kong, L.-J. Zheng and J.-J. Xu, J. Am. Chem. Soc., 2023, 145, 5718–5729 Search PubMed.
- P. Tan, X. Xiao, Y. Dai, C. Cheng and M. Ni, Renewable Sustainable Energy Rev., 2020, 127, 109877 CAS.
- Z. Zhu, Q. Lv, Y. Ni, S. Gao, J. Geng, J. Liang and F. Li, Angew. Chem., 2022, 134, e202116699 CrossRef.
- Q. Lv, Z. Zhu, S. Zhao, L. Wang, Q. Zhao, F. Li, L. A. Archer and J. Chen, J. Am. Chem. Soc., 2021, 143, 1941–1947 CrossRef CAS.
- Z. Zhu, Y. Ni, Q. Lv, J. Geng, W. Xie, F. Li and J. Chen, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, 2024619118 CrossRef.
- Y. Feng, H. Xue, T. Wang, H. Gong, B. Gao, W. Xia, C. Jiang, J. Li, X. Huang and J. He, ACS Sustainable Chem. Eng., 2019, 7, 5931–5939 CrossRef CAS.
- M. Li, X. Wang, F. Li, L. Zheng, J. Xu and J. Yu, Adv. Mater., 2020, 32, 1907098 CrossRef CAS.
- Y. Wei, Z. Zhou and R. Long, J. Phys. Chem. Lett., 2017, 8, 4522–4529 CrossRef CAS.
- H. Nakano, M. Ishii and H. Nakamura, Chem. Commun., 2005, 2945–2947 RSC.
- C. Jia, F. Zhang, L. She, Q. Li, X. He, J. Sun, Z. Lei and Z. H. Liu, Angew. Chem., Int. Ed., 2021, 60, 11257–11261 CrossRef CAS PubMed.
- S. Li, H. Wang, D. Li, X. Zhang, Y. Wang, J. Xie, J. Wang, Y. Tian, W. Ni and Y. Xie, J. Mater. Chem. A, 2016, 4, 15841–15844 RSC.
- X. Yan, W. Sun, L. Fan, P. N. Duchesne, W. Wang, C. Kubel, D. Wang, S. G. H. Kumar, Y. F. Li, A. Tavasoli, T. E. Wood, D. L. H. Hung, L. Wan, L. Wang, R. Song, J. Guo, I. Gourevich, F. M. Ali, J. Lu, R. Li, B. D. Hatton and G. A. Ozin, Nat. Commun., 2019, 10, 2608 CrossRef PubMed.
- R. Fu, K. Zhang, R. P. Zaccaria, H. Huang, Y. Xia and Z. Liu, Nano Energy, 2017, 39, 546–553 CrossRef CAS.
- S. Hu and M. Zhu, ChemCatChem, 2019, 11, 6147 CrossRef CAS.
- Y. Qin and A. R. Kamali, J. Alloys Compd., 2021, 888, 161506 CrossRef CAS.
- Q. Guo, Y. Han, N. Chen and L. Qu, ACS Energy Lett., 2021, 6, 1786–1794 CrossRef CAS.
- P. Pazhamalai, K. Krishnamoorthy, S. Sahoo, V. K. Mariappan and S.-J. Kim, ACS Appl. Mater. Interfaces, 2018, 11, 624–633 CrossRef.
- J. Ji, S. Mazinani, E. Ahmed, Y. J. Chew and D. Mattia, J. Membr. Sci., 2021, 635, 119447 Search PubMed.
- J. Luo, D. J. Arnot, S. T. King, A. Kingan, A. Nicoll, X. Tong, D. C. Bock, E. S. Takeuchi, A. C. Marschilok, S. Yan, L. Wang and K. J. Takeuchi, ACS Appl. Mater. Interfaces, 2023, 15, 24306–24318 CrossRef CAS.
- S. B. Ko, Y. Sun, G. Park, H. J. Choi, J. G. Kim, J. B. Kim, H. J. Jung, G. S. Lee, S. Hong, S. P. Sasikala and S. O. Kim, ACS Appl. Mater. Interfaces, 2023, 15, 32707–32716 Search PubMed.
- K. Krishnamoorthy, P. Pazhamalai and S.-J. Kim, Energy Environ. Sci., 2018, 11, 1595–1602 RSC.
- L. Zhou, Y. Wang, X. Xu, W. Lei, J. Huang, L. Chen, L. Zhu and Z. Ye, J. Colloid Interface Sci., 2020, 579, 205–211 Search PubMed.
- H. Li, X. Duan, X. Wu, X. Zhuang, H. Zhou, Q. Zhang, X. Zhu, W. Hu, P. Ren and P. Guo, J. Am. Chem. Soc., 2014, 136, 3756–3759 Search PubMed.
- Y. Li, Z. Y. Fu and B. L. Su, Adv. Funct. Mater., 2012, 22, 4634–4667 Search PubMed.
- H. J. Kang, J. W. Park, H. J. Hwang, H. Kim, K. S. Jang, X. Ji, H. J. Kim, W. B. Im and Y. S. Jun, Carbon Energy, 2021, 3, 976–990 Search PubMed.
- B. J. Ryan, M. P. Hanrahan, Y. Wang, U. Ramesh, C. K. Nyamekye, R. D. Nelson, Z. Liu, C. Huang, B. Whitehead and J. Wang, Chem. Mater., 2019, 32, 795–804 Search PubMed.
- C. Jia, F. Zhang, N. Zhang, Q. Li, X. He, J. Sun, R. Jiang, Z. Lei and Z.-H. Liu, ACS Nano, 2023, 17, 1713–1722 Search PubMed.
- Y. Feng, H. Xue, T. Wang, H. Gong, B. Gao, W. Xia, C. Jiang, J. Li, X. Huang and J. He, ACS Sustainable Chem. Eng., 2019, 7, 5931–5939 CrossRef CAS.
- R. Liu, S. Yao and Y. Shen, J. Mol. Liq., 2020, 320, 114481 CrossRef CAS.
- M. Li, X. Wang, F. Li, L. Zheng, J. Xu and J. Yu, Adv. Mater., 2020, 32, 1907098 CrossRef CAS.
- H. Yu, D. Liu, Z. Fu, S. Wang, X. Zuo, X. Feng and Y. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202401272 CrossRef CAS PubMed.
- Y. Qiao, Y. Liu, K. Jiang, X. Li, Y. He, Q. Li, S. Wu and H. Zhou, Small Methods, 2018, 2, 1700284 CrossRef.
- Q. Cui, Y. Zhang, S. Ma and Z. Peng, Sci. Bull., 2015, 60, 1227–1234 CrossRef CAS.
- C. Xia, M. Waletzko, L. Chen, K. Peppler, P. J. Klar and J. Janek, ACS Appl. Mater. Interfaces, 2014, 6, 12083–12092 CrossRef CAS.
- A. D. S. McWilliams, C. Martínez-Jiménez, A. M. Ya’akobi, C. J. Ginestra, Y. Talmon, M. Pasquali and A. A. Martí, ACS Appl. Nano Mater., 2021, 4, 142–151 CrossRef.
- T. Fang, H. Liu, X. Luo, M. Sun, W. Peng, Y. Li, F. Zhang and X. Fan, Small, 2024, 2309600 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr03850a |
| This journal is © The Royal Society of Chemistry 2025 |