
Sulfide-mediated growth of NIR luminescent Pd/Ag atomically precise nanoclusters†
Yu-Rong
Ni
a,
Michael N.
Pillay
 a,
Tzu-Hao
Chiu
a,
Hao
Liang
b,
Samia
Kahlal
b,
Jie-Ying
Chen
c,
Yuan-Jang
Chen
c,
Jean-Yves
Saillard
*b and
C. W.
Liu
*a
a,
Tzu-Hao
Chiu
a,
Hao
Liang
b,
Samia
Kahlal
b,
Jie-Ying
Chen
c,
Yuan-Jang
Chen
c,
Jean-Yves
Saillard
*b and
C. W.
Liu
*a
aDepartment of Chemistry, National Dong Hwa University, Hualien 97401, Taiwan, Republic of China. E-mail: chenwei@gms.ndhu.edu.tw
bUniv Rennes, CNRS, ISCR-UMR 6226, F-35000 Rennes, France. E-mail: jean-yves.saillard@univ-rennes.fr
cDepartment of Chemistry, Fu Jen Catholic University, New Taipei City, 24205, Taiwan, Republic of China
First published on 4th December 2024
Abstract
An essential feature of coinage metal nanoclusters (NCs) is their photoluminescence (PL), which spans a wide range of wavelengths from visible to near-infrared regions (NIR-I/II). A key challenge for synthetic chemists is to develop materials capable of efficient spectral change with maximum efficiency. Herein, we report novel dithiolate-protected bimetallic Pd-Ag NCs of the type [PdAg16S2{S2P(OR)2}12] (R = iPr, 1Pr and iBu, 1Bu) and [Pd6Ag14S{S2P(OiBu)2}12] (2Bu). Sulfide-mediated expansions of NCs result in unique PL in the NIR-I region for 1Pr and 1Bu (λmax = 808 and 811 nm) and the NIR-II region for 2Pr (λmax = 1007) at 77 K. NIR PL enhancement largely depends on structural modification with the sulfide anions at the central position. DFT calculations indicate that the PL properties are associated with 4dπ(Pd)/3pπ(S) → 5s/5p(Ag) excitation, resulting from the existence of S–Pd(0)–S motifs in both 1 and 2. The electrochemical gaps of 1Pr, 1Bu, and 2Bu are recorded by SWV.
Introduction
Atomically precise nanoclusters (NCs) have emerged as a material of the future, offering unique advantages over their bulk counterparts. One key improvement is the ability to modulate the energy levels and tuning energy band gaps, thus influencing both optical and electrochemical performances. Photoluminescence (PL) in the near-infrared (NIR) region is highly desirable due to its minimal interaction with biological tissues, making it a safe and effective tool for various applications in the biomedical field,1,2 such as cell labeling,3 biosensing,4 and bioimaging.5–7Over the past decade, substantial progress has been made in synthesizing and characterizing coinage metal NCs with enhanced PL quantum yields (PLQYs). However, achieving PL in the NIR-II region (1000–1700 nm) remains challenging. Coinage metal NCs have shown some promise, with a few examples of gold,8–15 silver16 and copper17 reported. While noble metal-doped or alloyed Ag NCs can extend emission from the UV/vis to the NIR-I range,18–31,38–40 achieving longer wavelengths (1000–1700 nm) remains elusive. To achieve the NIR-II photoluminescence of silver NCs, we propose in this report the addition of a sulfur heteroatom into the metallic framework to induce emission at longer wavelengths. Various NC morphologies have been isolated due to the versatility of sulfide, which can occupy multiple positions within the NC, not just the central one.32,33 In several cases, the inclusion of sulfide in the NC primary sphere directs the growth of the subsequent layers and plays a crucial role in the assembly of large arrangements.34–37
Herein, we report the synthesis, composition, and optical characteristics of the selective incorporation of sulfide anions into positions of the metallic primary layer, resulting in bimetallic Pd–Ag NCs [PdAg16S2{S2P(OR)2}12] (R = iPr, 1Pr and iBu, 1Bu) and [Pd6Ag14S{S2P(OiBu)2}12] (2Bu). Single-crystal X-ray diffraction (SCXRD), X-ray photoelectron spectroscopy (XPS), ESI-MS, optical analysis, and multinuclear NMR spectroscopy determined the composition and structural properties and their electronic structures were analyzed through DFT calculations.
Results and discussion
Building upon our earlier report on sulfide-centered Pd/Ag of the type [Pd6Ag14(S){S2P(OnPr)2}12] (2Pr), which involves the cleavage of the P–S bond from the dithiophosphate and the in situ sulfide generation,19 we sought to develop a more general protocol for incorporating sulfide into NCs and evaluate its reproducibility. To achieve this, we varied the alkoxy substituent on the dithiophosphate ligand (dtp), utilizing both isopropoxy and isobutoxy derivatives.This strategy (Scheme 1) successfully yielded two novel disulfide nanoclusters, [PdAg16(S)2{S2P(OR)2}12] (1Pr, R = iPr; 1Bu, R = iBu), in addition to the sulfide-centered clusters, [Pd6Ag14(S){S2P(OiBu)2}12] (2Bu), of which the [Pd6Ag14(S){S2P(OiPr)2}12] (2Pr) relative is known.19 The products are isolated and purified by column chromatography, affording 1 and 2 in moderate yields.
 | ||
| Scheme 1 Strategic incorporation of sulfide into bimetallic NCs. | ||
The purity and composition of the isolated NCs were confirmed by mass spectrometry, Fig. 1. The mass spectrum of 1Pr exhibits a dominant peak assigned to [1Pr + Ag]+ at m/z 4563.4390 Da (calc.: m/z 4563.4375 Da), Fig. 1a. The simulated peak is given in Fig. S1,† compared with a silver and proton adduct to demonstrate the presence of a palladium atom. The dominant m/z peak for 1Bu is assigned to the silver adduct [1Bu + Ag]+ at 4899.8438 Da (calc.: m/z 4899.8140 Da), Fig. 1b and S2.† Similarly, cluster 2Bu forms a silver adduct of the type [2Bu + Ag]+ at m/z 5184.6678 Da (calc.: m/z 5184.5506 Da), Fig. 1c. The experimental and simulated isotopic distributions exhibit a strong correlation, further supporting the assigned compositions. Additionally, XPS analysis confirms the presence of Pd(0) and Ag(I) in the NCs, as evidenced by their characteristic binding energies, Fig. S3–5.† The temperature-dependent UV-vis spectra are shown in Fig. S13–15† and they were used to investigate the stability of the NCs.
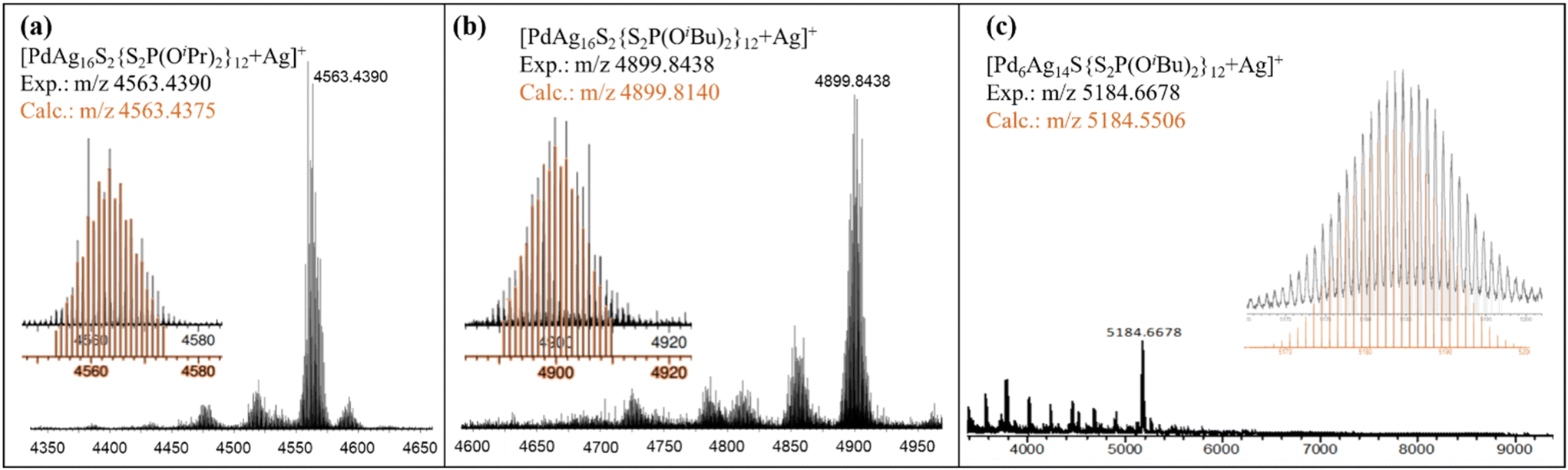 | ||
| Fig. 1 ESI-TOF-MS of spectra for 1Pr (a), 1Bu (b) and 2Bu (c). Experimental (black) and simulated (orange). | ||
Structural analysis
The atomic arrangements of 1Pr and 1Bu were determined by SCXRD analysis and their refinement details are given in Table S1.† Selected interatomic distances are given in Table 1. 1Pr and 1Bu crystallize in two different space groups, with 1Pr being solvated by a dichloromethane molecule. Otherwise, they are isostructural (apart from small differences in the ligand coordination modes, which are discussed later). Thus, the following discussion is based on 1Bu (see Fig. 2). Its highly unique core structure features a Pd-centered Ag10 pentagonal antiprism kernel, with two sulfides capping each of the pentagonal faces (Fig. 2a). It can thus be seen also as a Pd-centered Ag10S2 icosahedron with approximate D5d symmetry and can be compared with typical species having Pd@Ag12 icosahedral cores.21 Substituting two vertices with sulfur atoms results in a large contraction compared to the regular Ag12 icosahedron, with Pd⋯S distances (ca. 2.30 Å) much shorter than the Pd⋯Ag in Pd@Ag12 species (ca. 2.90 Å).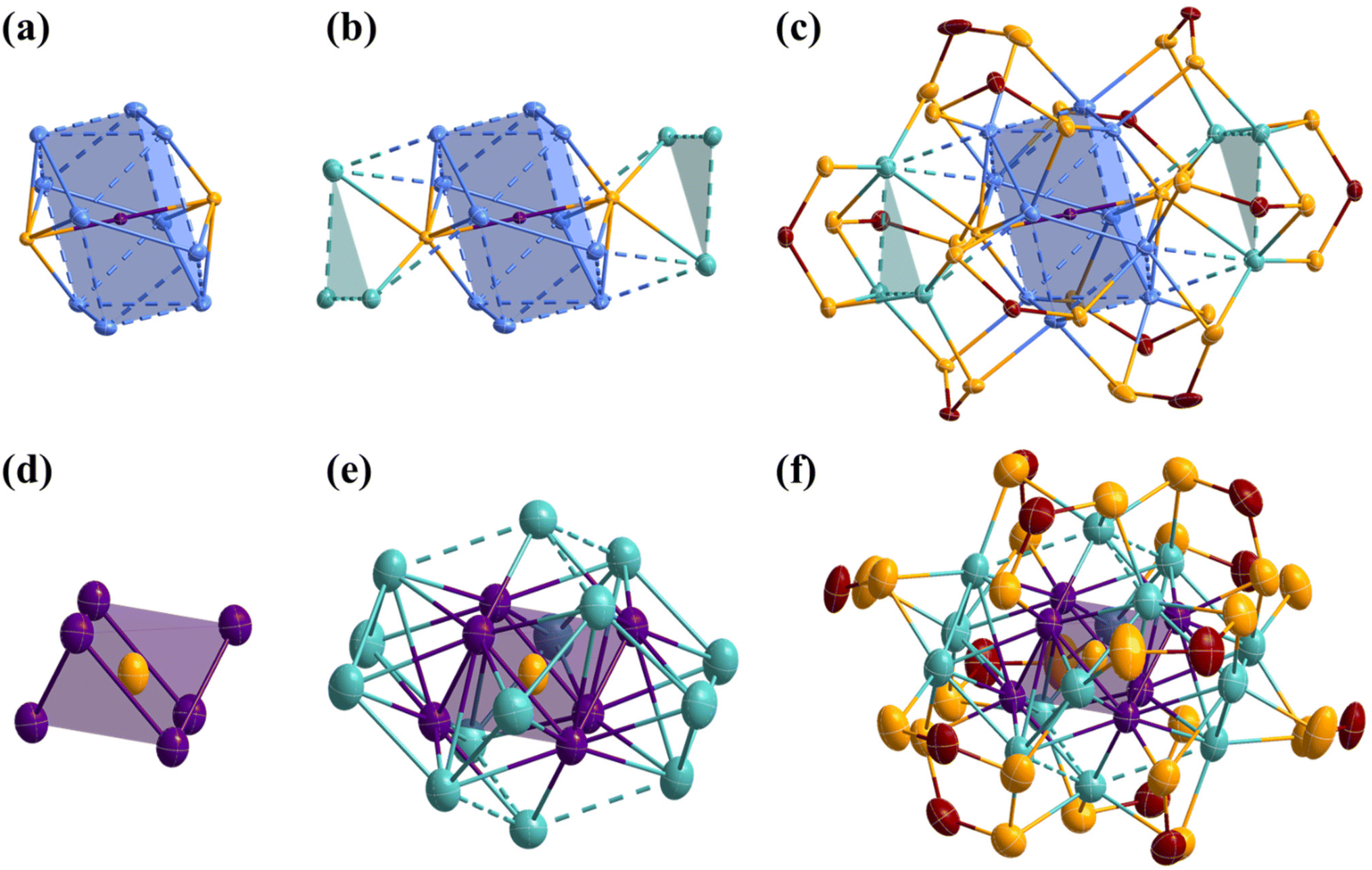 | ||
| Fig. 2 The SCXRD structure of 1Bu and 2Bu. (a) The Pd@Ag10S2 kernel of 1Bu. (b) The two Ag3 motifs capping the cluster core of 1Bu. (c) Total structure of 1Bu with ellipsoids drawn at 35%, H and C atoms omitted for clarity. (d) The S@Pd6 core of 2Bu. (e) The S@Pd6@Ag14 framework of 2Bu. (f) Total structure of 2Bu with ellipsoids drawn at 35%, H and C atoms are omitted for clarity (color code: Pd, purple; Agker, blue; Agcap, green; S, yellow; P, maroon). | ||
| 1Pr, X-ray | 1Bu, X-ray | 1, DFT | 2Bu, X-ray | 2, DFT | |
|---|---|---|---|---|---|
| Pd–S | 2.296(8) | 2.297(3) | 2.336 [0.351] | 2.2600(11) | 2.301 [0.260] |
| Agker–S | 2.7582(9) | 2.718(3) | 2.752 [0.132] | — | — |
| Agcap–S | 2.9168(9) | 2.8265(3) | 3.040 [0.041] | — | — |
| Pd–Ag | 2.8863(3) | 2.8950(9) | 2.916 [0.066] | 2.9071(16) | 2.997 [0.060] |
| Pd–Pd | — | — | — | 2.9433(14) | 2.985 [0.074] |
| Agker–Agker | 3.0431(4) | 3.0478(14) | 3.124 [0.032] | — | — |
| Agker–Agcap | 3.1199(4) | 3.1454(14) | 3.215 [0.022] | — | — |
| Agcap–Agcap | 3.1758(5) | 3.2196(15) | 3.482 [0.010] | 3.039(18) | 3.131 [0.040] |
Furthermore, the differential bonding between Pd–S and Ag–S is crucial for stabilizing this unique kernel configuration, which is unlikely to form in the absence of the central Pd atom. Another structural distinctive feature of 1 is the arrangement of the secondary silver layer around the Pd@Ag10S2 core. The six peripheral silver atoms are arranged into two triangular Ag3 capping motifs, with only two atoms per triangle in bonding contacts with the Ag10S2 icosahedron (Fig. 2b). These triangular arrays are unprecedented in silver-rich NCs, where capping atoms usually occupy symmetrically favored positions dictated by the ligand shell. Consequently, the Agcap⋯Agcap distances in these peripheral motifs are notably longer than the Agker⋯Agker distances within the kernel. Two ligand coordination modes, six η4 (μ2, μ2), and six η3 (μ2, μ1), are observed in 1Pr. In contrast, 1Bu exhibits three coordination modes, namely two η5 (μ3, μ2), four η4 (μ2, μ2), and six η3 (μ2, μ1). The increase in the denticity in 1Bu occurs for two ligands bridging the triangular Ag3 motif to the kernel. This is accommodated by the adaptability of the sulfide, which is μ7 in 1Bu compared to μ8 in 1Pr. Notably, two of the six η3 (μ2, μ1) in both 1Pr and 1Bu are located at unique positions on the triangular Ag3 motifs of the nanocluster, where they do not interact with the central core (Fig. 2c).
The SCXRD structure of 2Bu is shown in Fig. 2 and selected metrical data are given in Table 1. 2Bu is found isostructural with the previously reported 2Pr.19 It shows a central sulfur atom directing the NC shape through its encapsulation inside a Pd6Ag2 rhombohedron (distorted cube), the latter lying within an Ag6 octahedron. This S@(Pd6Ag2)@Ag6 framework is surrounded by six capping Ag atoms and twelve dtp ligands. The latter exhibit two coordination modes, namely six η3 (μ2, μ1) and six η4 (μ2, μ2) (Fig. 2f). The whole structure is of approximate S6 symmetry. Each Pd atom in 2Bu is coordinated to the central sulfide ion and an S atom from a dtp ligand in a nearly linear fashion (S–Pd–S = 177°). In addition to two sulfur atoms, six Ag atoms and two Pd atoms complete the coordination sphere around each Pd atom. Notably, one of the Pd–Ag contacts is particularly long (∼3.2 Å). The completely different metallic frameworks in 1 and 2 underscore the structural diversity achievable through ligand manipulation and the impact of varying metal ratios within these systems.
The 1H NMR data confirm the presence of the isopropyl or isobutyl group in compounds 1 and 2, as shown in Fig. S6c, d and S7b.† The 1Pr31P NMR spectrum shows three distinct phosphorus environments identified by resonances at 105.31, 104.35, and 100.72 ppm. In contrast, 1Bu displays four resonances at 108.92, 106.27, 106.00, and 102.36 ppm (Fig. S6a and b†), indicating an additional coordination mode in 1Bu, in accordance with its solid-state structure. In the case of 2Bu, two 31P NMR resonances are observed at 102.63 and 100.58 ppm, in agreement with the S6 symmetry observed in the solid state being preserved in solution (Fig. S7a).†
Optical properties
NCs, due to their small size, typically fall below the size regime where surface plasmon resonance dominates their optical properties. However, subtle changes can significantly impact the discrete energy levels of these NCs. In general, as the size of the NC increases, the absorption spectra typically shift to longer wavelengths, attributed to the quantum confinement effect, wherein the energy levels become less discrete as the NC grows. However, larger NCs often exhibit lower photoluminescence quantum yields due to an increased opportunity for nonradiative decay processes. The optical spectra are shown in Fig. 3, and detailed values are compared in Table 2. Compounds 1Pr and 1Bu have similar absorption profiles with peaks observed at 365 and 366 nm (Fig. 3a). They emit in the NIR-I region, with peaks centered at 808 and 811 nm at 77 K, and emission lifetimes of 92 and 72 μs, respectively (Fig. 3b and S8a and b).† Compound 2Bu features a relatively broader absorption profile covering a range of 375–630 nm (Fig. 3a). Importantly, 2Bu exhibits unique NIR-II emission at ambient and low temperatures, with peaks centered at λem = 1034 nm (298 K) and λem = 1004 nm (77 K) (Fig. 3b). A change in the alkyl substituent in 2Pr, results in peaks centered at λem = 1046 nm (298 K) and λem = 1007 nm (77 K). Variations in the alkyl substituents of the ligand do not significantly influence the emission profile or lifetime. However, an increase in the size of the alkyl groups leads to an increase in the PLQY. This enhancement is possibly due to the increased electron-donating ability of the larger alkyl substituents, facilitating the emission process. The PLQYs for this series of NIR emissive compounds at ambient and low temperatures are listed in Table 2. The emission lifetimes of 2Pr and 2Bu increase at low temperatures, consistent with a spin-forbidden transition (Fig. S9).† A comparative analysis of emission maxima for doped Ag-rich nanoclusters reveals that NC 2 exhibits a significantly redshifted emission wavelength compared to previously reported alloys25–30 and monometallic nanoclusters31 (Fig. 3c).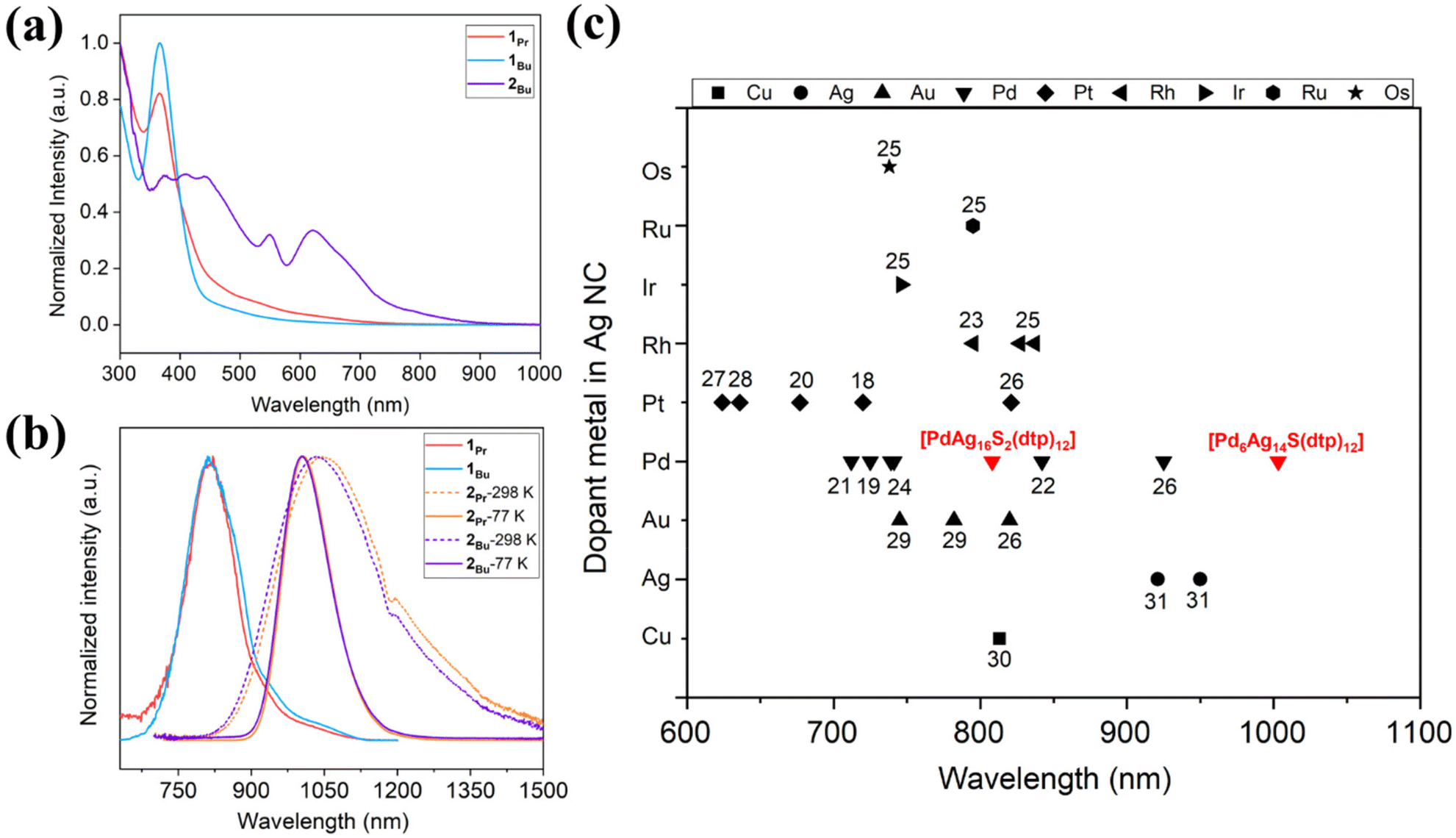 | ||
| Fig. 3 (a) The absorption spectra for 1Pr, 1Bu, and 2Bu recorded in 2-MeTHF. (b) Emission spectra for 1Pr (λem = 808 nm), 1Bu (λem = 811 nm), 2Pr (λem = 1007 nm) and 2Bu (λem = 1004 nm) in 2-MeTHF at 77 K. (c) Comparison of λem for doped silver rich NC with entries labeled by the corresponding references. | ||
| NC | λ max (nm) | λ em (nm) | τ (μs) |
Φ
em![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (%) a (%) |
k
ob (μs−1) |
k
rc (μs−1) |
k
nrd (μs−1) |
|
|---|---|---|---|---|---|---|---|---|
| a Φ em = emission quantum yield (%). b k o = mean excited-state decay rate constant = 1/lifetime (τ). c k r = radiative constant = Φem × 0.01 × ko. d k nr = nonradiative constant = ko − kr. | ||||||||
| 298 K | 1Pr | 365 | — | — | — | — | — | — |
| 1Bu | 366 | — | — | — | — | — | — | |
| 2Pr | 368, 408, 443, 548, 623 | 1046 | 0.0437 | 0.025 | 22.88 | 0.0057 | 22.87 | |
| 2Bu | 374, 413, 446, 545, 625 | 1034 | 0.127 | 0.294 | 7.87 | 0.023 | 7.85 | |
| 77 K | 1Pr | 808 | 92.3 | — | — | — | — | |
| 1Bu | 811 | 71.9 | — | — | — | — | ||
| 2Pr | 1007 | 21.9 | 9.04 | 0.046 | 0.0041 | 0.042 | ||
| 2Bu | 1004 | 20.6 | 14 | 0.049 | 0.0068 | 0.042 | ||
The cyclic voltammetry (CV) and the square wave voltammetry (SWV) of compounds 1 and 2 were recorded in organic solvent (THF and CH2Cl2) with 0.1 M [nBuN][PF6] as the supporting electrolyte. The results are summarized in Table 3 and Fig. 4 and S10–12.† At low temperature (233 K), the CV of 1Pr exhibits an irreversible reduction process (Epc) at −1.23 V (vs. Fc+/Fc) and three irreversible oxidation processes (Epa) at 0.17, 0.55 and 0.766 V, respectively (Fig. S10†). The SWV measurements confirm these redox processes, with Epc = −1.17, −1.51, and −1.86 V and Epa = 0.25, 0.55, 0.94, and 1.38 V at 298 K (Fig. 4a). 1Bu displays a similar pattern in both CV and SWV at 233 and 298 K in THF (Fig. S11†). The electrochemical HOMO–LUMO gap, which is calculated to be 1.40 eV, is different between O1 and R1. The results of 2Bu are shown at 298 and 233 K in Fig. 4b and S12.† Its calculated electrochemical HOMO–LUMO gap is 1.13 eV.
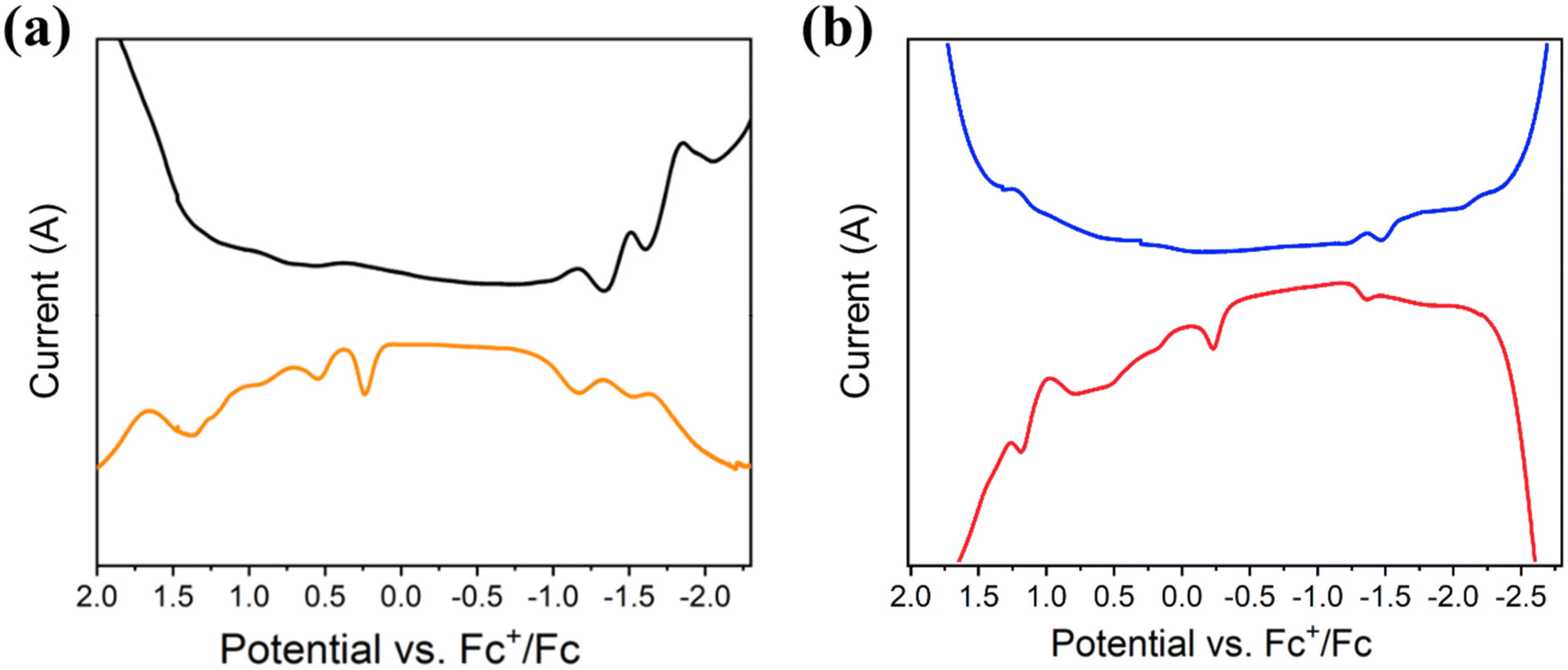 | ||
| Fig. 4 Square wave voltammetry of 1Pr (a) and 2Bu (b) recorded in THF and CH2Cl2, at 298 K containing 0.1 M [nBuN][PF6] under a N2 atmosphere. | ||
| NC |
E
pa
|
E
pc
|
E
Eg
|
E
Og
|
E
PLg
|
|---|---|---|---|---|---|
| a Values determined from SWV spectra. b E Eg = Epa − Epc. c E Og = 1240/λmax, λmax is the lowest electronic transition in absorption wavelength. d E PLg = 1240/λem, λem is the centered PL wavelength. | |||||
| 1Pr (298 K) | +0.25, +0.55, +0.94, +1.38 | −1.17, −1.51, −1.86 | 1.42 | 3.40 | — |
| 1Pr (233 K) | +0.28, +0.61, +0.97 | −1.35, −1.78 | 1.63 | — | 1.53 |
| 1Bu (298 K) | +0.18, +0.41, +1.12 | −1.22, −1.91 | 1.40 | 3.39 | — |
| 1Bu (233 K) | +0.25, +0.95 | −1.41, −1.89 | 1.66 | — | 1.52 |
| 2Bu (298 K) | −0.23, +0.21, +0.78, +1.19 | −1.36, −1.63, −2.20 | 1.13 | 1.98 | 1.20 |
| 2Bu (233 K) | +0.01, +0.65, +1.09, +1.40 | −1.52, −1.67, −2.04 | 1.49 | — | 1.24 |
Bonding analysis and DFT investigation
The DFT-optimized geometry carried out on a simplified model for 1, [PdAg16S2(S2PH2)12], is in good agreement with its SCXRD structure (see Table 1 and corresponding xyz file in the ESI†). Analysis of its electronic structure supports the first-approximation view of a linear [PdS2]4− complex stabilized by a [Ag16(S2PH2)12]4+ cage, of which 12 Ag+ are in contact with the encapsulated [PdS2]4−. The NAO atomic charges (Table S2†) are consistent with Pd(0) and Ag(I) metal centers.19 Not considering the metal–metal contacts, 14 among the 16 Ag atoms are bonded to three dtp sulfur atoms, making them locally 16-electron centers. 4 of them not in contact with Pd (labelled Agcap) achieve a stable, nearly planar, coordination mode, whereas 10 of them in contact with Pd are somewhat pyramidalized to allow some 4d(Pd) → 5s/5p(Ag) electron transfer. Only 2 among the 16 Ag atoms are tetracoordinated, with a trigonal-planar coordination mode to three dtp sulfur atoms, to which a weaker bonding contact with one sulfide should be added. This bonding description is supported by the average Wiberg bond indices (WBI) reported in Table S2,† notably the large Pd–Ssulfide value (0.351), the Agker⋯Agker and Agker⋯Agcap values (0.032 and 0.022, respectively) characteristic of d10⋯d10 metallophilic interactions, and the somewhat larger Pd–Ag values (0.066) indicative of some weak covalent character in addition to metallophilicity. The orbital diagram of 1 is shown in Fig. S16.† It features two highest occupied orbitals derived from the two HOMOs of the hypothetical [PdS2]4−, with 4dπ(Pd) contribution mixed in an antibonding way with 3pπ(S). The lowest vacant orbitals are of 4s/4p(Ag) dominant character originating from the 16-electron Ag centers.The TD-DFT-simulated UV-vis spectrum of 1 is shown in Fig. S18.† It shows a low-energy band at 502 nm, which is a combination of the HOMO → LUMO+1, HOMO−1 → L and HOMO−1 → LUMO+1 transitions, thus of 4dπ(Pd)/3pπ(S) → 5s/5p(Ag) nature. This low-energy peak is hardly distinguishable in the experimental spectra of 1Pr and 1Bu (Fig. 3a) and we believe that its computed oscillator strength is significantly overestimated in our TD-DFT calculations. The high-energy peak involves lower 4d(Ag) levels and the LUMO and LUMO+1, thus is of 4d(Ag) → 4s/4p(Ag) nature.
Although its composition and 3D architecture look fairly different from that of 1, the bonding in 2, whose simplified [Pd6Ag14(S)(S2PH2)12] model we previously investigated,19 presents important similarities with that in 1. Whereas 1 possesses a central linear S–Pd(0)–S motif, 2 shows six of them, sharing the central sulfide and making a flattened octahedron. Discarding the hypercoordinated nature of the sulfide, the six S–Pd(0)–S motifs are stable 14-electron centers, while the silver atoms are all tricoordinated 16-electron centers, of which those in contact with Pd are involved in (moderate) 4d(Pd) → 5s/5p(Ag) electron transfer. As in 1, the whole architecture is maintained by the bidentate dtp ligands. The orbital diagram of 2 (Fig. S17†) shows highest occupied levels which can be roughly described as combinations of the two π-type HOMOs of the six S–Pd(0)–S motifs of 4dπ(Pd) and 3pπ(S) character, as in (1). Because of the existence of Pd⋯Pd contacts (Table 1), the 2 × 6 occupied combinations develop a “band” according to their (rather weak) amount of Pd⋯Pd bonding/antibonding character, of which the highest are (moderately) Pd⋯Pd antibonding, thus tending to limit the width of the HOMO–LUMO separation.
The TD-DFT-simulated UV-vis spectra of 1 and 2 (Fig. S18†) are in a good agreement with their experimental counterpart (Fig. 3a). They show two major bands which are of similar nature. Their major high-energy band is of 4d(Ag/Pd) → ligand character. The oscillator strength of lowest energy bands (of 4dπ(Pd)/3pπ(S) → 5s/5p(Ag) nature) are overestimated with respect to experiment. In fact, in the case of 1, this computed low-energy band (502 nm, Fig. S18†) can be associated with transitions buried in the tails observed around 400–500 nm in the 1Pr and 1Bu experimental spectra (Fig. 3a). In the case of 2, this band, also of 4dπ(Pd)/3pπ(S) → 5s/5p(Ag) nature, results from a transition from the degenerate HOMO−1/HOMO−2 eu level to the degenerate lowest unoccupied eg level (LUMO/LUMO+1). Although of similar nature, the low-energy band in 2 is red-shifted as compared to that computed for 1. We suggest that this is the same transition which is responsible for the NIR-II phosphorescence of 2. The large Stokes shift experimentally observed can be rationalized from the fact that the eu → eg transition in 2 corresponds to the depopulation of a Pd⋯Pd antibonding level and the occupation of an Ag⋯Ag bonding level, with the consequence of a non-negligible stabilization of the corresponding excited state, a situation which does not exist in 1.
Conclusion
This study has successfully demonstrated the strategic synthesis of sulfide-doped nanoclusters (NCs) by modulating the reaction pH with triethylamine, a method that effectively generates sulfide ions from dithiophosphate ligands. Notably, the isolation of 1Pr and 1Bu, which exhibit the first silver-rich icosahedron containing two sulfur heteroatoms at its vertices, resulted in the appearance of photoluminescence in the NIR-I region. Furthermore, the sulfide-centered NC 2Bu exhibits remarkable NIR-II photoluminescence at both ambient and low temperatures, highlighting the potential of this synthetic strategy for producing rare NIR emissive nanoclusters. Given that the NIR-II emission originates from a PdS2 → Ag transition, the targeted incorporation of additional Pd and S atoms into silver NCs is a promising strategy for further enhancing their optical properties. This study sheds light on the intricate relationship between size, structure, and optical properties in NCs, emphasizing the importance of precise control over these parameters. This finding and our controlled synthesis and ligand modification capabilities open exciting avenues for designing new materials with tailored optical responses. These advancements hold promise for applications in bioimaging, sensing, optoelectronics, and beyond.Author contributions
Y.-R. N., T.-H. C., and M. N. P.: investigation, data curation, formal analysis, methodology, and writing. J. Y. C., Y.-J. C., H. L., and S. K.: data curation and formal analysis. J.-Y. S.: data curation, formal analysis, and writing. C. W. L.: supervision, writing, conceptualization, project administration, and resources.Data availability
Crystallographic data for [1Pr, 1Bu, 2Bu] have been deposited at the Cambridge Crystallographic Data Centre under [CCDC 2379720–2379722].† Computational details (including coordinate file), X-ray structure analyses, NMR spectra, electrochemical data, ESI-MS data, and luminescence decay curves are available in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science and Technology Council of Taiwan (113-2123-M-259-001) and the GENCI French National Computer Resource Center (A0030807367).References
- Y. Qin, X. Chen, Y. Gui, H. Wang, B. Z. Tang and D. Wang, J. Am. Chem. Soc., 2022, 144, 12825–12833 CrossRef CAS PubMed.
- A. Baghdasaryan, F. Wang, F. Ren, Z. Ma, J. Li, X. Zhou, L. Grigoryan, C. Xu and H. Dai, Nat. Commun., 2022, 13, 5613 CrossRef CAS PubMed.
- M. A. H. Muhammed, P. K. Verma, S. K. Pal, R. C. A. Kumar, S. Paul, R. V. Omkumar and P. Thalappil, Chem. – Eur. J., 2009, 15, 10110–10120 CrossRef CAS PubMed.
- Y. Xiao, Z. Wu, Q. Yao and J. Xie, Aggregate, 2021, 2, 114–132 CrossRef CAS.
- Z. Luo, K. Zheng and J. Xie, Chem. Commun., 2014, 50, 5143–5155 RSC.
- G. Yang, X. Pan, W. Feng, Q. Yao, F. Jiang, F. Du, X. Zhou, J. Xie and X. Yuan, ACS Nano, 2023, 17, 15605–15614 CrossRef CAS.
- X. Song, W. Zhu, X. Ge, R. Li, S. Li, X. Chen, J. Song, J. Xie, X. Chen and H. Yang, Angew. Chem., Int. Ed., 2021, 60, 1306–1312 CAS.
- Z. Liu, L. Luo and R. Jin, Adv. Mater., 2024, 36, 2309073 CAS.
- Z. Liu, L. Luo, J. Kong, E. Kahng, M. Zhou and R. Jin, Nanoscale, 2024, 16, 7419–7426 RSC.
- H. Qian, M. Zhu, E. Lanni, Y. Zhu, M. E. Bier and R. Jin, J. Phys. Chem. C, 2009, 113, 17599–17603 CrossRef CAS.
- A. Das, T. Li, K. Nobusada, Q. Zeng, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2012, 134, 20286–20289 CrossRef CAS.
- R. Jin, C. Liu, S. Zhao, A. Das, H. Xing, C. Gayathri, Y. Xing, N. L. Rosi, R. R. Gil and R. Jin, ACS Nano, 2015, 9, 8530–8536 Search PubMed.
- X. Wan, W. W. Xu, S. Yuan, Y. Gao, X. Zeng and Q. Wang, Angew. Chem., 2015, 127, 9819–9822 Search PubMed.
- L. Luo, Z. Liu, X. Du and R. Jin, J. Am. Chem. Soc., 2022, 144, 19243–19247 Search PubMed.
- X. Fu, X. Lin, X. Ren, R. Wu, C. Liu and J. Huang, Nanoscale, 2020, 12, 11825–11829 Search PubMed.
- M. S. Bootharaju, S. Lee, G. Deng, S. Malola, W. Baek, H. Häkkinen, N. Zheng and T. Hyeon, Angew. Chem., Int. Ed., 2021, 60, 9038–9044 Search PubMed.
- T. Jia, Z. J. Guan, C. Zhang, X. Z. Zhu, Y. X. Chen, Q. Zhang, Y. Yang and D. Sun, J. Am. Chem. Soc., 2023, 145, 10355–10363 CrossRef CAS.
- M. S. Bootharaju, S. M. Kozlov, Z. Cao, M. Harb, M. R. Parida, M. N. Hedhili, O. F. Mohammed, O. M. Bakr, L. Cavallo and J. M. Basset, Nanoscale, 2017, 9, 9529–9536 Search PubMed.
- S. K. Barik, T.-H. Chiu, Y. C. Liu, M. H. Chiang, F. Gam, I. Chantrenne, S. Kahlal, J. Y. Saillard and C. W. Liu, Nanoscale, 2019, 11, 14581–14586 RSC.
- T.-H. Chiu, J.-H. Liao, F. Gam, I. Chantrenne, S. Kahlal, J. Y. Saillard and C. W. Liu, J. Am. Chem. Soc., 2019, 141, 12957–12961 CrossRef CAS PubMed.
- S. K. Barik, C. Y. Chen, T.-H. Chiu, Y.-R. Ni, F. Gam, I. Chantrenne, S. Kahlal, J. Y. Saillard and C. W. Liu, Commun. Chem., 2022, 5, 151 CrossRef.
- Y.-R. Ni, M. N. Pillay, T.-H. Chiu, Y. Y. Wu, S. Kahlal, J. Y. Saillard and C. W. Liu, Chem. – Eur. J., 2023, 9, e202300730 CrossRef PubMed.
- T.-H. Chiu, J.-H. Liao, Y. Y. Wu, J. Y. Chen, Y. J. Chen, X. Wang, S. Kahlal, J. Y. Saillard and C. W. Liu, J. Am. Chem. Soc., 2023, 145, 16739–16747 CrossRef CAS PubMed.
- Y.-R. Ni, M. N. Pillay, T.-H. Chiu, J. Rajaram, Y. Y. Wu, S. Kahlal, J. Y. Saillard and C. W. Liu, Inorg. Chem., 2024, 63, 2766–2775 CAS.
- H. Yi, S. Song, S. M. Han, J. Lee, W. Kim, E. Sim and D. Lee, Angew. Chem., Int. Ed., 2023, 62, e202302591 CAS.
- X. Liu, J. Yuan, C. Yao, J. Chen, L. Li, X. Bao, J. Yang and Z. Wu, J. Phys. Chem. C, 2017, 121, 13848–13853 CAS.
- T.-H. Chiu, J.-H. Liao, F. Gam, I. Chantrenne, S. Kahlal, J. Y. Saillard and C. W. Liu, Nanoscale, 2021, 13, 12143–12148 CAS.
- T.-H. Chiu, J.-H. Liao, F. Gam, Y. Y. Wu, X. Wang, S. Kahlal, J. Y. Saillard and C. W. Liu, J. Am. Chem. Soc., 2022, 144, 10599–10607 CrossRef CAS.
- Y.-R. Lin, P. V. V. N. Kishore, J.-H. Liao, S. Kahlal, Y.-C. Liu, M. H. Chiang, J. Y. Saillard and C. W. Liu, Nanoscale, 2018, 10, 6855–6860 RSC.
- W. J. Yen, J.-H. Liao, T.-H. Chiu, Y. S. Wen and C. W. Liu, Inorg. Chem., 2022, 61, 6695–6700 CrossRef CAS PubMed.
- R. S. Dhayal, Y. R. Lin, J.-H. Liao, Y. J. Chen, Y. C. Liu, M. H. Chiang, S. Kahlal, J. Y. Saillard and C. W. Liu, Chem. – Eur. J., 2016, 29, 9943–9947 CrossRef PubMed.
- H. W. Chang, R. Y. Shiu, C. S. Fang, J.-H. Liao, P. V. V. N. Kishore, S. Kahlal, J. Y. Saillard and C. W. Liu, J. Cluster Sci., 2017, 28, 679–694 CrossRef CAS.
- N. Yoshinari, Z. L. Goo, K. Nomura and T. Konno, Inorg. Chem., 2023, 62, 9291–9294 CAS.
- Y. M. Su, Z. Z. Cao, L. Feng, Q. W. Xue, C. H. Tung, Z. Y. Gao and D. Sun, Small, 2022, 18, 2104524 CAS.
- W. H. Wu, H. M. Zeng, Z. N. Yu, C. Wang, Z. G. Jiang and C. H. Zhan, Chem. Commun., 2021, 57, 13337–13340 CAS.
- Z. A. Nan, Y. Xiao, X. Y. Liu, T. Wang, X. L. Cheng, Y. Yang, Z. Lei and Q. M. Wang, Chem. Commun., 2019, 55, 6771–6774 RSC.
- S. Jin, S. Wang, Y. Song, M. Zhou, J. Zhong, J. Zhang, A. Xia, Y. Pei, M. Chen, P. Li and M. Zhu, J. Am. Chem. Soc., 2014, 136, 15559–15565 CrossRef CAS PubMed.
- W. D. Tian, W. D. Si, S. Havenridge, C. Zhang, Z. Wang, C. M. Aikens, C. H. Tung and D. Sun, Sci. Bull., 2024, 69, 40–48 CrossRef CAS.
- W. D. Si, C. Zhang, M. Zhou, Z. Wang, L. Feng, C. H. Tung and D. Sun, Sci. Adv., 2024, 10, eadm6928 CrossRef CAS.
- Z. Wang, Y. Wang, C. Zhang, Y. J. Zhu, K. P. Song, C. M. Aikens, C. H. Tung and D. Sun, Natl. Sci. Rev., 2024, 11, nwae192 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2379720–2379722. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4nr04136d |
| This journal is © The Royal Society of Chemistry 2025 |