
First-principles methods for unraveling the structure–catalytic activity relationship and mechanism of δ-MnO2-supported metal cluster catalysts in ambient temperature benzene to CO2 degradation†
Jiangmei
Yan
a,
Peng
Zhang
a,
Dan
Chen
a,
Jie
Cheng
a,
Tong
Mu
a,
Mengshan
Song
b,
Shuai
Li
b,
Hui
Zhao
b,
Guo
Chang
b,
Ruqian
Lian
 *c,
Chuangwei
Liu
*d,
Wangtu
Huo
*b and
Dongxiao
Kan
*b
*c,
Chuangwei
Liu
*d,
Wangtu
Huo
*b and
Dongxiao
Kan
*b
aKaili Catalyst & New Materials Co., Ltd, Xi'an, 710201, China
bNorthwest Institute for Non-ferrous Metal Research, Xi'an, 710016, China. E-mail: huowangtu_1988@163.com; dxkan1202@126.com
cHebei Research Center of the Basic Discipline for Computational Physics College of Physics Science and Technology, Hebei University, Baoding 071002, P. R. China. E-mail: rqlian@126.com
dDalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, 116023 China. E-mail: cwliu@dicp.ac.cn
First published on 21st March 2025
Abstract
Benzene, a volatile cyclic aromatic hydrocarbon, is frequently discharged into the environment and poses serious threats to human health. However, the effective degradation of benzene at low temperatures remains a formidable challenge in the field of environmental remediation. In this study, a series of 49 catalyst samples, spanning from single atoms to trimers and tetramers, supported on δ-MnO2 were meticulously constructed by employing the density functional theory (DFT) method. This approach facilitates an in-depth exploration of the structure–function relationship and enables the identification of prospective catalysts capable of degrading benzene into CO2 and H2O, thereby providing valuable insights for the development of efficient benzene degradation strategies at low temperatures. The obtained results unequivocally demonstrate that the separation between reaction products and reactants is substantially augmented with an increment in the number of active sites. Notably, the Ag4 tetramer exhibits a remarkable enhancement in this regard. Further in-depth analyses reveal that the superior catalytic activity of Ag4 relative to Pt4 and Pd4 tetramers can be ascribed to the closer energy alignment between the highest occupied molecular orbital (HOMO of Ag4) and the lowest unoccupied molecular orbital (LUMO of benzene), which facilitates electron transfer and reaction initiation. To assess the practical catalytic effectiveness, a detailed analysis of the reaction pathways for benzene degradation was carried out. Remarkably, the energy barrier of the rate-determining step was determined to be merely 0.71 eV at room temperature, comparable to those achieved under photocatalytic and high-temperature conditions. This finding is of great significance as it represents the first successful degradation of benzene by precisely controlling the size of metal clusters. Overall, this study not only provides valuable theoretical insights for benzene abatement but also paves the way for remediating related pollutants, heralding a new approach in the pursuit of sustainable environmental protection strategies.
1. Introduction
As one of the most prevalent volatile organic compounds (VOCs), benzene is renowned for its high toxicity, inflicting severe environmental deterioration and substantial harm to the human body upon short-term or long-term exposure.1 Nevertheless, the efficient and economical decomposition of benzene remains a formidable challenge. Over the past several decades, a plethora of methodologies for benzene elimination, such as thermal catalysis, photocatalysis, and electrocatalysis, have been widely employed.2,3 Among these, catalytic oxidation is regarded as one of the most efficacious approaches, whereby benzene can be completely converted into harmless CO2 and H2O at relatively low temperatures.4,5 However, cleaving the benzene ring under mild conditions persists as an intractable obstacle in this process.Supported noble metal nanoparticle catalysts exhibit effectiveness in benzene decomposition at low temperatures.6 Nonetheless, their extensive application is impeded by the high cost associated with them.7 Atomically dispersed catalysts (ADCs), which manipulate the configuration and composition of the central metal atom, possess advantages like high atom utilization and catalytic selectivity.8,9 ADCs manifest diverse types based on the number of central metal atoms, encompassing single-atom catalysts (SACs), dual-atom catalysts (DACs), and clusters.10–12 Among them, clusters, composed of several to tens of atoms, frequently display exceptionally high catalytic activity in comparison with nanoparticles and have achieved significant breakthroughs in VOC removal reactions.13,14
However, the diminutive size of ADCs renders them prone to agglomeration and transformation into larger nanoparticles, thereby altering the electronic structure and considerably diminishing the catalytic activity.15 Consequently, maintaining the stability and efficiency of these ADCs represents a major challenge. It has been reported that techniques of loading single atoms onto carriers, such as metal oxides, graphene, and nanoparticles, can effectively forestall the aggregation of ADCs.16,17 Zhang et al. reported that metal oxide carriers not only function as ligands for stabilizing catalytic metal centers but also directly partake in the activation of reactants and enhance the catalytic performance.18
As one kind of metal oxide, manganese dioxide (MnO2) has been regarded as an efficient and environmentally friendly catalyst for the elimination of pollutants such as CO, NO, ozone, and volatile organic compounds (VOCs).19–21 For example, ultra-thin birnessite-type manganese dioxide nanosheets (δ-MnO2) have been demonstrated to possess high activity in converting ppm-level formaldehyde into harmless carbon dioxide at room temperature.22 However, on pristine manganese oxides, the decomposition of benzene is scarcely achieved below 200 °C, with a conversion rate of merely around 40% even at a high temperature of 400 °C.23 In response to this, extensive investigations have been carried out based on layered δ-MnO2 for benzene oxidation. Chen et al. enhanced the decomposition activity of MnO2 towards benzene by modifying δ-MnO2 with Ti atoms, leading to nearly complete removal of benzene at approximately 250 °C.24 Liu et al. compared the effects of Mo6+ and Ce3+ as typical high-valence and low-valence modifiers in promoting the decomposition of benzene.25 Zhang et al. achieved remarkable performance by doping single Pt atoms into MnO2 through a hydrothermal method, which significantly boosted the catalytic activity for the degradation of toluene at low temperatures.26 Specifically, 10 ppm of toluene was completely converted into CO2 at 80 °C and fully oxidized to carbon dioxide at 220 °C. Nevertheless, although these catalysts have lowered the oxidation and degradation temperature of benzene, studies on the conversion of benzene to CO2 at room temperature remain unreported.
In the present study, first-principles methods were utilized to predict the catalytic potential of benzene degradation at room temperature on a series of δ-MnO2(001)-based atomically dispersed catalysts (ADCs). Twenty-three single-atom catalysts (SACs) and metal trimer catalysts with different work functions were investigated to explore the adsorption strengths of reactants (benzene) and products (H2O and CO2), respectively. By establishing the relationship between the catalytic activity and the amount of metal active sites, further ADCs of metal tetramers were studied to widen the gap between the adsorption energy of adsorbents and reactants. Among them, the Ag4 cluster exhibited outstanding performance with an adsorption energy gap exceeding 3 eV. Moreover, the optimal pathway for benzene degradation at room temperature was proposed, featuring a limiting step free energy barrier of only 0.71 eV, which is comparable to those reported under photocatalytic and high-temperature conditions. This work presents a novel catalytic oxidation pathway for benzene degradation and offers theoretical insights for optimizing air quality by removing VOCs.
2. Calculation method
All computational procedures were carried out employing the Vienna Ab initio Simulation Package (VASP. 6.0), which is grounded in density functional theory (DFT).27,28 To guarantee the precision of the calculations, plane-wave basis sets were implemented with a cutoff energy of 500 eV. The electron–ion core interactions were modeled by means of the Projected Augmented Wave (PAW) approach, and the General Gradient Approximation (GGA-PBE) developed by Perdew, Burke, and Ernzerhof was utilized to depict electron interactions.29,30 Brillouin zone integration was executed on a 4 × 4 × 1 grid, with a convergence criterion set at 1 × 10−5 eV. In all theoretical computations, the spin polarization method was incorporated, and the DFT-D3 method was applied for van der Waals correction.31–33 Additionally, the magnetic effects associated with δ-MnO2 were also taken into account. The spin-polarized density functional theory with Hubbard U correction (DFT+U) was used to correct the standard DFT in describing the partially filled d-states for the Mn atoms, and the effective U value was set as 5.5 eV according to the literature.34,35 Meanwhile the antiferromagnetic (AFM) configuration was adopted in the calculation as mentioned in previous studies.36,37The formation energy (Eform) of a single atom, trimer or tetramer on δ-MnO2(001) was defined according to the following formula:38
| Eform = Etotal − Esub − nEbulk/N |
The adsorption energies (Eads) were computed using the subsequent equation:39
| Eads = Etotal − Esub − Emol |
The free energy (ΔG) of each reduction step was obtained at zero bias potential using the expression:40
| ΔG = ΔE + ΔEZPE + TΔS |
3. Discussion and results
3.1 The structural stability and electronic properties of δ-MnO2(001) based SACs
The birnite type MnO2 with the (001) plane was chosen as the substrate to support ADCs and enable the degradation of benzene to CO2 and H2O at room temperature. The catalytic potential of 23 single-atom catalysts (SACs) covering a diverse range of work functions (Sc, Ti, Zr, Ni, Mn, Fe, Cu, Ag, Nb, Co, Rh, Ru, Pd, Ir, Pt, Mo, Ta, Zn, Au, V, Cr, W, and Hf) on the δ-MnO2(001) surface was initially investigated to elucidate the impact of active sites on catalytic activities. As depicted in Fig. S1,† there are two possible positions for single metal loading on the δ-MnO2(001) surface, namely the “a site” and the “b site”. Through energy comparison, it was determined that all single metals favored the “a” deposition site and coordinated with three O atoms. To assess the resistance to sintering of SACs on δ-MnO2(001), the formation energies (Eform) of the 23 types of SACs were primarily calculated. As shown in Fig. 1a and b, 15 SACs (Sc, Ti, Zr, Ni, Mn, Fe, Cu, Ag, Nb, Co, Rh, Ru, Pd, Ir, and Pt) exhibited negative Eform values, indicating high anti-sintering stabilities as illustrated in Fig. 1b. Additionally, the thermodynamic stabilities of these 15 SACs under the practical catalytic reaction temperature conditions (300 K) were simulated using the ab initio molecular dynamics (AIMD) method. It was observed that the SACs of Ag, Au, Mo, Co, Cu, Fe, Nb, Pd, and Rh maintained stable structures after 5 ps of molecular dynamics simulation (Fig. S2†). At this stage, 9 kinds of SACs possessing thermal, dynamic, and structural stability were identified, and subsequently, the structure and electronic properties of these catalysts, as well as their potential for catalyzing the degradation of benzene at room temperature, would be further explored. | ||
| Fig. 1 (a) Formation energy of single-atom catalysts (SACs) supported on δ-MnO2(001). (b) Correlation model of SACs on δ-MnO2(001). (c) Electronic localization functions of the representative SACs. (d) Bader charge between SACs and δ-MnO2(001). | ||
The chemical bonds and electronic interactions between SACs and δ-MnO2(001) could be further probed through the electronic localization function (ELF) pattern. As presented in Fig. 1c, all single metals were surrounded by a greenish electronic cloud, while the O atom was surrounded by a reddish electronic cloud. This suggested that the metal atoms donated electrons to the adjacent O atoms and formed ionic bonds. Bader analysis also provided quantitative results for the interactions between SACs and the substrate as shown in Fig. 1d, where all SACs acted as electron donors and the O atom served as the electron acceptor. The direction of electron transfer was from the single metal atom to δ-MnO2, with the number of transferred electrons exceeding 0.5 e. Notably, the interaction strength was correlated with the metal work function: the lower the metal work function, the greater the number of electrons transferred to δ-MnO2, accompanied by stronger interactions.
It is well established that an ideal catalyst should exhibit a strong adsorption strength towards reactants for effective activation and a weak adsorption strength towards products to facilitate desorption. To investigate the catalytic performance of the single-atom catalysts (SACs) in the degradation of benzene at room temperature, the adsorption properties of the reactant (benzene) and the products (CO2 and H2O) were initially examined. As illustrated in Fig. 2a, the adsorbed H2O binds to the SACs solely through its O atom, whereas multiple chemical bonds were formed between benzene, CO2, and the SACs. This maximizes the interaction, which was manifested in the charge density difference (CDD) analysis presented in Fig. 2b. The blue electron cloud surrounding benzene indicated its role as an electron donor, contributing electrons to the δ-MnO2(001)-based SACs. In contrast, CO2 and H2O capture electrons from the SACs, as evidenced by the yellow electron clouds around them. Furthermore, the Bader analysis in Fig. 2c corroborated the CDD results. The negative charge values of H2O/CO2 signify electron gain, while the positive values of benzene represent electron loss to the SACs. Notably, the aforementioned electron interactions were also correlated with the work function of the SACs. A smaller work function leads to a greater transfer of charge from the SACs to H2O and CO2. However, the interaction between benzene and the SACs follows an opposite trend. The SACs with larger work functions, such as Pd and Pt, were more prone to accepting electrons from benzene, where benzene functions as an electron donor on these SACs. The adsorption results of benzene, H2O, and CO2 on the SACs conformed to the above electron interaction principles (Fig. 2d). Specifically, a smaller work function of the SACs corresponds to a higher adsorption energy for H2O and CO2, whereas for benzene, the opposite relationship is observed. Although the difference in adsorption energy between benzene and CO2 was significant, it is challenging to separate benzene and H2O on these SACs.
 | ||
| Fig. 2 (a) Optimized structures of benzene, CO2, and H2O molecules adsorbed on δ-MnO2-supported single-atom catalysts (SACs); (b) charge density difference of benzene, CO2, and H2O adsorbates; (c) adsorption energies of benzene, CO2, and H2O on δ-MnO2-supported SACs; (d) Bader charge between the adsorbates and SACs. | ||
3.2 Active site engineering of metal clusters on δ-MnO2(001) for benzene degradation: from trimers to tetramers
To enhance the catalytic performance of ADCs, the addition of active sites was proposed to intensify the interaction between catalysts and benzene, which may widen the adsorption energy gap between reactants and products. Given the significant impact of the work function of single-atom catalysts (SACs) on the catalytic activity, 23 metal trimers (Sc3, Ti3, Zr3, Ni3, Mn3, Fe3, Cu3, Ag3, Nb3, Co3, Rh3, Ru3, Pd3, Ir3, Pt3, Mo3, Ta3, Zn3, Au3, V3, Cr3, W3, and Hf3) within different work function ranges were considered. After optimization, some of the trimers underwent distortion or oxidation, and 17 types of trimers (Ag3, Pd3, Pt3, Rh3, Ru3, Fe3, Cu3, W3, Ir3, Mo3, Au3, Cr3, V3, Nb3, Ni3, Hf3, and Ta3) exhibited structural stability (Fig. S3†). To further assess their stabilities, the formation energies were calculated, and 12 types of trimer ADCs (Ag3, Pd3, Pt3, Rh3, Ru3, Cu3, Fe3, W3, Ir3, Au3, Nb3, and Ta3) were screened out as shown in Fig. 3a. Moreover, the energy–time curves and structures obtained from molecular dynamics simulations at room temperature (300 K) for 5 ps as shown in Fig. S4† indicated that 7 trimers (Ag3, Pd3, Pt3, Rh3, Ru3, Cu3, and Fe3) exhibited potential stability. Furthermore, the AIMD simulation at 300 K was extended to 50 ps for the selected δ-MnO2-M3, which showed that all of them had high thermodynamic stability. Compared with SACs, the increase in active metal atoms in the trimers led to an increase in the number of bonds between benzene, H2O, CO2, and the catalysts, resulting in a significant increase in their adsorption energies, especially for benzene (exceeding 2 eV) as shown in Fig. 3b. Notably, with the expansion of active sites, the adsorption energy difference among H2O, CO2, and benzene on Ag3, Pd3, and Pt3 was substantially enlarged. Consequently, the products tended to desorb from the catalyst surface, thereby preventing the deactivation of the active sites.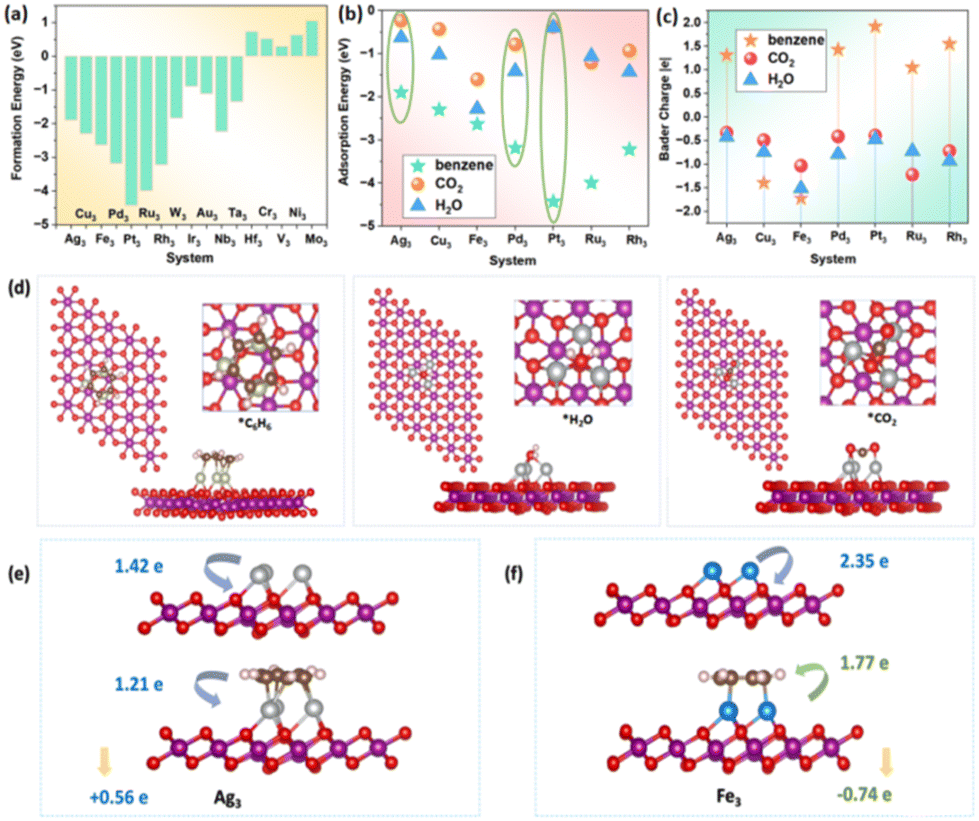 | ||
| Fig. 3 (a) Formation energy of metal trimers on δ-MnO2(001); (b) adsorption energies of benzene, CO2, and H2O on the metal trimers; (c) Bader charge of the adsorbed benzene, CO2, and H2O on the metal trimers; (d) adsorption configurations of benzene, CO2, and H2O on the metal trimers; (e) charge transfer mechanism before and after benzene adsorption on Ag3; (f) charge transfer mechanism before and after benzene adsorption on Fe3. | ||
The electronic interactions exhibited similar trends to the adsorption energies. In contrast to SACs, the charge transfer numbers between the trimers and the reactants/products gradually increased, especially for the Ag3, Pd3, and Pt3 trimers. Interestingly, both H2O and CO2 obtained electrons from SACs and trimers, but for benzene, there was a difference. From the low work function trimers (such as Cu3 and Fe3), benzene acquired electrons, while benzene lost electrons to the high work function trimers (such as Pd3 and Pt3) as shown in Fig. 3c. Furthermore, when benzene was adsorbed on trimers, the interaction between the trimer and δ-MnO2 showed two completely different patterns. For example, Pt3 gained electrons from benzene while donating 0.56 e electrons to δ-MnO2 (as shown in Fig. 3e). Conversely, Fe3 donated electrons to benzene while causing a reduction of 0.74 e electrons on δ-MnO2 (as shown in Fig. 3f).
Based on the above, the adsorption energy gap between benzene and the products was greatly increased with the increase in active sites, as benzene requires more active sites compared to CO2 and H2O. Through the trimer study, three potential single-cluster catalysts, namely Ag3, Pd3, and Pt3, were identified. To explore whether further enhancing the active sites could optimize the performance, three types of tetramer catalysts based on trimers, namely Ag4, Pd4, and Pt4, were constructed. These tetramers had two possible configurations, tetrahedron and parallelogram, as shown in Fig. S6.† Besides, the energy–time curve and structures obtained from molecular dynamics simulations for δ-MnO-M4 at room temperature over a duration of 50 ps (Fig. S7†), as well as the phonon spectrum (Fig. S8†) were calculated. The results provided robust evidence to demonstrate the stability of the system.
Through energy comparison, all of them tended to form a parallelogram on the δ-MnO2(001) surface, and their negative formation energies indicated high structural stability (Fig. 4b).
 | ||
| Fig. 4 (a) Adsorption configurations of benzene, CO2, and H2O on Ag4, Pd4, and Pt4 tetramers; (b) formation energy of Ag4, Pd4, and Pt4 tetramers on δ-MnO2; (c) adsorption energies of benzene, CO2, and H2O on Ag4, Pd4, and Pt4 tetramers; (d) lowest unoccupied molecular orbital (LUMO) and highest occupied molecular orbital (HOMO) patterns of benzene and δ-MnO2-Ag4; (e) relative positions of the LUMO of benzene and the HOMO of Ag4, Pd4, and Pt4 tetramers, respectively. | ||
The adsorption properties of benzene, H2O, and CO2 on these tetramers are presented in Fig. 4a and c. Comparatively, the tetramers had larger adsorption energies for benzene and weaker adsorption strengths for CO2 and H2O than those on the trimers. These results were consistent with expectations, where the tetramers further enhanced the adsorption energy of benzene and reduced the adsorption of CO2 and H2O. Especially on the Ag4 tetramer, the adsorption energy for benzene could reach up to 3.62 eV, while for CO2 and H2O it was less than 0.5 eV. In addition, the superiority of Ag4 over Pt4 and Pd4 was investigated through the molecular frontier orbital model. As shown in Fig. 4d and e, the lowest unoccupied molecular orbital (LUMO) of benzene was highly compatible with the highest occupied molecular orbital (HOMO) of the Ag4 cluster. Moreover, when compared with the Ag4 and Pd4 clusters, the HOMO of Ag4 was more closely aligned with the LUMO of benzene. The above analysis demonstrated that the Ag4 tetramer had outstanding advantages in the separation of benzene and its products and was a promising candidate catalyst for the degradation of benzene at room temperature.
3.3 Reaction pathway elucidation of benzene degradation on the Ag4 tetramer: a comprehensive theoretical study
To dissect the theoretical catalytic characteristics of the Ag4 tetramer during the degradation of benzene into CO2 and H2O, a comprehensive and methodical exploration into the particulars of reaction pathways was executed. The incipient oxidation of benzene could potentially transpire via three conceivable routes: activation instigated by O2 molecules, direct dehydrogenation, or combination with the oxygen atom liberated from the decomposition of O2 (Fig. 5a). Concerning the first pathway, the amalgamation between the O2 molecule and the adsorbed benzene was highly arduous due to the positive adsorption energies. Instead, the O2 molecule manifested a predilection for bonding with the Ag4 tetramer, possessing adsorption and free energy values of −0.54 eV and −0.22 eV, respectively (Fig. S9†). In the context of the dehydrogenation pathway, an additional free energy of 1.09 eV was required to propel the reaction, which was disadvantageous for the overall reaction progression. Moreover, in the O-atom binding pathway, the adsorption site of the O atom was initially scrutinized. Here, the O atom also exhibited a propensity for bonding with the Ag atom, signifying an exothermic process and a relatively diminutive reaction free energy of −0.31 eV (Fig. S10 and S11†). | ||
| Fig. 5 (a) Reaction pathway of benzene oxidation and (b) reaction pathway of benzene degradation to a pentagonal ring on δ-MnO2-supported Ag4 tetramers. | ||
Subsequently, our focus was centered on the pathways entailing benzene oxidation by O2 molecules or O atoms. Along the former pathway, the adsorbed O2 molecule abstracted an H atom from the adsorbed benzene, engendering a pair of adsorbates, C6H5 and OOH molecule, with a substantially elevated free energy value of 1.05 eV (Fig. S12†), which was inimical to a seamless reaction advancement. Up to this juncture, only the O atom binding pathway merited further scrutiny. The second step of benzene degradation via the O combination pathway was also partitioned into three alternatives, namely benzene oxygenation (addition of O1) to form C6H6 + 2O or C6H6O + O, and dehydrogenation to form C6H5 + OH. The Gibbs free energies of these three paths were −0.25 eV, 0.32 eV, and −0.11 eV, respectively, where C6H5O + OH is the easiest intermediate product of this process (Fig. S13†). The ensuing step encompassed four potential routes: further oxygenation (addition of O2) to form C6H5O + OH, C6H5 + OH + O, and C6H4 + 2OH, or dehydrogenation to form C6H4 + H2O. The corresponding reaction free energies were −0.28 eV, 0.16 eV, 0.33 eV, and 0.76 eV, respectively (Fig. S14†), among which the C6H5O + OH path was the most preferable. In the subsequent step along the C6H5O + OH path, two courses persisted: the third O (O3) binding to Pt and generating C6H5O + O + OH, or interconnecting with benzene to form C6H5O2 + OH (Fig. S15†). The free energies were 0.12 eV and −0.68 eV, respectively, intimating that the latter reaction path was more favorable. It is noteworthy that when O3 was appended to the benzene ring, three potential adsorption sites were contemplated: the ortho, meta, and para positions relative to the basal O2 (Fig. S16†). Among them, the para adsorption site was the most stable, with the lowest structural energy.
In the further degradation process of the C6H5O2 molecule, two possible paths subsisted: the fourth O (O4) combining with C6H5O2 and dehydrogenating to C6H4O2 + H2O, or O4 being captured by Ag4 and then converting C6H5O2 to C6H3O2. The reaction free energies were −0.43 eV and −0.26 eV, respectively, suggesting that the former path is more advantageous (Fig. 6a). After the first H2O molecule was desorbed, the next step, whether dehydrogenation or oxidation, remained to be ascertained. As illustrated in Fig. 6b, in the dehydrogenation path, the OH groups on benzene were cleaved, accompanied by an endothermic reaction with a free energy of 0.58 eV. In contrast, in the oxidation path, the fifth O (O5) continuously attracted the benzene ring, forming a five-membered carbon ring (C5) and a CHO molecule. This was an exothermic process, exhibiting a favorable free energy of −0.29 eV. At this stage, the hexagonal structure of the benzene ring was completely disrupted, attaining the initial accomplishment of benzene degradation at room temperature. In fact, during the step of generating C6H5O2, the conjugated π bond of the benzene ring is already perturbed, with the double C–C bond being elongated from a uniform length of 1.42 Å to 1.51 Å. Therefore, the steps of generation of C6H5O2 and C5H2O2 were both cardinal steps in the present benzene degradation work.
 | ||
| Fig. 6 (a) Reaction paths and Gibbs free energy of the fifth step in benzene degradation: combination of the fourth oxygen (O4) with C6H5O2 to generate C6H4O2 + H2O or capture of O4 by Ag4, leading to the conversion of C6H5O2 to C6H3O2; (b) reaction paths and Gibbs free energy of the sixth step in benzene degradation: dehydrogenation or oxidation (addition of O5) process; (c) overall reaction pathways and Gibbs free energy of benzene degradation to CO2 and H2O on the Ag4 tetramer catalyst. | ||
Based on the above exploration, the reaction pathway for the cleavage of the benzene ring on the Ag4 tetramer at room temperature was initially elucidated. Subsequently, a detailed study on the complete degradation process of benzene to CO2 and H2O was conducted. As depicted in Fig. 6c, the process involved benzene adsorption (forming *C6H6), oxidation and dehydrogenation (forming *C6H5O), disruption of the conjugated π bond (forming *C6H5O2), decomposition of the hexagonal structure (forming *C5H2O2 + CHO), further oxidation and dehydrogenation to *C5HO3 + CO on the Ag4 tetramer, followed by the generation of the first CO2. Subsequently, the C5 moiety was disrupted (*C5H2O2), with the second CO2 being desorbed from the catalyst and a carbon chain structure (*C4HO2) being formed. As the continuous release of CO2 proceeded, the carbon chain was transformed into *C3HO2 and then *C2HO2. During this process, the excess hydrogen was converted into water, which was released from the catalyst, and the adsorbed C2HO2 molecules underwent conversion to C2O3 and CO as intermediates, which were ultimately converted into CO2. It was observed that six endothermic reactions occurred throughout the entire process of benzene degradation at room temperature. Among them, the generation of *C4HO3 from *C4HO2 was the rate-determining step, with a reaction free energy of 0.71 eV. This indicated that the catalytic activity of benzene degradation on δ-MnO2-Ag4 at room temperature was comparable to those under photocatalytic and high-temperature conditions from a theoretical perspective.41,42 This study holds significant importance as it provides novel insights into the mechanism and kinetics of benzene degradation at room temperature, potentially facilitating the development of more efficient and environmentally friendly catalytic systems for pollutant elimination.
To thoroughly explore the fundamental reasons for the high catalytic activity of catalysts, we conducted in-depth electronic analyses on a series of δ-MnO2-M3 (Ag3, Pd3, Pt3, Cu3, Fe3, Rh3, and Ru3) and δ-MnO2-M4 (Ag4, Pd4, and Pt4) catalyst systems. Firstly, the projected densities of states of δ-MnO2-Ag4, δ-MnO2-Pd4, and δ-MnO2-Pt4 before and after benzene adsorption were calculated and are presented in Fig. S17.† As shown, there was a significant hybridization between the C 2p orbitals of benzene molecules and the d-electron orbitals of metal atoms, resulting in a strong resonance phenomenon. It indicated that throughout the entire catalytic reaction process, the d-electron orbitals of metals played a decisive role in the activation of benzene and are a key factor influencing catalytic activity. To further realize the determining reasons of the high catalytic activity, the work functions, d-band centers of δ-MnO2-M3 and δ-MnO2-M4, and the electronegativities of the relevant metals were summarized. The results in Fig. S18 and 19† showed that the work function and the d-band center values of δ-MnO2-Ag4 as well as the electronegativity of Ag were all in the middle position. Thus, δ-MnO2-Ag4 may possess a moderate adsorption energy for reactants (benzene) and products (H2O and CO2). This moderate adsorption energy ensures that the adsorption of products on the catalyst surface is neither too strong, thus avoiding the problem of difficult desorption, nor too weak, guaranteeing the effective activation of benzene molecules. It is precisely this optimal adsorption characteristic that endows δ-MnO2-Ag4 with relatively ideal catalytic activity, enabling it to exhibit excellent performance in the catalytic reaction. Through the above-mentioned supplementary calculations and analyses, we explored the reasons for the high catalytic activity of catalysts not only from a phenomenological level but also from the essential levels of electronic structure and chemical properties.
4. Conclusions
In this study, a comprehensive investigation was conducted on the catalytic properties of 49 types of ADCs (spanning from a single atom to a tetramer) supported on δ-MnO2(001) with the objective of identifying promising candidates to facilitate benzene degradation at room temperature. The specific research conclusions are as follows:1. Among the 23 SAC samples, 9 SACs were selected based on their high structural and thermodynamic stabilities. However, these SACs demonstrated relatively suboptimal separation capabilities between reactants and products, as the adsorption energies of benzene, CO2, and H2O were in close proximity.
2. To widen the adsorption energy gap between reactants and products, 23 metal trimers, which possess more active sites and enhanced metal utilization, were constructed. The results indicated that with the increment of active sites, the potential catalytic performance was improved, especially for the Ag3, Pd3, and Pt3 trimers. Moreover, when the active sites were further augmented to form tetramers, the separation effect became even more pronounced, which could be ascribed to the stronger bonding interactions between benzene and the metal clusters.
3. The Ag4 cluster emerged as the most promising candidate, exhibiting an adsorption energy gap exceeding 3 eV. This property not only enables the complete activation of reactants but also facilitates the smooth desorption of products. The superiority of the Ag4 cluster over the Pt4 and Pd4 clusters can be attributed to the closer alignment of the highest occupied molecular orbital (HOMO) of Ag4 with the lowest unoccupied molecular orbital (LUMO) of benzene.
4. The catalytic performance and reaction pathways of benzene degradation on the Ag4 cluster were thoroughly explored at room temperature. An optimal pathway for the degradation of benzene to CO2 and H2O was proposed. The key intermediates in this process were identified as C6H5O2 and C5H2O2, with a rate-limiting step free energy barrier of merely 0.71 eV. These results are comparable to the catalytic activities reported under photocatalytic and high-temperature conditions.
In summary, this work has uncovered a novel catalytic oxidation pathway for benzene degradation and has successfully accomplished the conversion of benzene to CO2 at room temperature, thereby providing valuable theoretical insights and laying a solid foundation for future research in this field.
Data availability
The data supporting this article have been included as part of the ESI.† And no primary research results, software or code have been included in this work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by the National & Local Joint Engineering Research Center for New Noble Metal Catalysts R&D Technology; the Shaanxi Key Laboratory of Catalytic Materials and Technology and the Xian Key Laboratory of Catalytic Materials and Technology; and the National Key R&D and the Ministry of Program of China (2022YFA1503504). The authors gratefully acknowledge the support of the NSFC (Grant No. 22303068, 62204207), the Natural Science Basic Research Program of Shaanxi Province (2023-JC-QN-0123, 2024JC-YBQN-0079), the Natural Science Basic Research Program of Hebei Province (B2024201089), the Qinchuangyuan High-level Innovation and Entrepreneurship Talent Projects (QCYRCXM-2023-004), the Xi'an Youth Talent Lifting Program (959202413034) and the Northwest Institute for Nonferrous Metal Research Institute-Controlled Fund (YK2114).References
- C. J. Jiang, D. D. Li, P. Y. Zhang, J. G. Li, J. Wang and J. G. Yu, Formaldehyde and volatile organic compound (VOC) emissions from particleboard: identification of odorous compounds and effects of heat treatment, Build. Environ., 2017, 117, 118–126 Search PubMed.
- L. Kirkeskov, T. Witterseh, L. W. Funch, E. Kristiansen, L. Molhave, M. K. Hansen and B. B. Knudsen, Health evaluation of volatile organic compound (VOC) emission from exotic wood products, Indoor Air, 2009, 19, 45–57 CAS.
- C. Vogt, S. Kleinsteuber and H. H. Richnow, Anaerobic benzene degradation by bacteria, Microb. Biotechnol., 2011, 4, 710–724 Search PubMed.
- M. E. Caldwell and J. M. Suflita, Detection of phenol and benzoate as intermediates of anaerobic benzene biodegradation under different terminal electron-accepting conditions, Environ. Sci. Technol., 2000, 34, 1216–1220 CrossRef CAS.
- R. Chakraborty and J. D. Coates, Hydroxylation and carboxylation-Two crucial steps of anaerobic benzene degradation by Dechloromonas strain RCB, Appl. Environ. Microbiol., 2005, 71, 5427–5432 CrossRef CAS PubMed.
- H. Zhang, S. Sui, X. Zheng, R. Cao and P. Zhang, One-pot synthesis of atomically dispersed Pt on MnO2 for efficient catalytic decomposition of toluene at low temperatures, Appl. Catal., B, 2019, 257, 117878 CrossRef CAS.
- H. Huang, Y. Xu, Q. Feng and D. Y. C. Leung, Low temperature catalytic oxidation of volatile organic compounds: a review, Catal. Sci. Technol., 2015, 5, 2649–2669 RSC.
- J. E. Post, Manganese oxide minerals: crystal structures and economic and environmental significance, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 3447–3454 CrossRef CAS PubMed.
- S. Grangeon, A. Manceau, J. Guilhermet, A. C. Gaillot, M. Lanson and B. Lanson, Zn sorption modifies dynamically the layer and interlayer structure of vernadite, Geochim. Cosmochim. Acta, 2012, 85, 302–313 CrossRef CAS.
- M. Zeng, Y. Li, F. Liu, Y. Yang, M. Mao and X. Zhao, Cu doped OL-1 nanoflower: A UV vis-infrared light-driven catalyst for gas-phase environmental purification with very high efficiency, Appl. Catal., B, 2017, 200, 521–529 CrossRef CAS.
- L. Zhu, J. Wang, S. Rong, H. Wang and P. Zhang, Cerium modified birnessite-type MnO2 for gaseous formaldehyde oxidation at low temperature, Appl. Catal., B, 2017, 211, 212–221 CrossRef CAS.
- G. G. Yadav, J. W. Gallaway, D. E. Turney, M. Nyce, J. Huang, X. Wei and S. Banerjee, Regenerable Cu-intercalated MnO2 layered cathode for highly cyclable energy dense batteries, Nat. Commun., 2017, 8, 14424–14432 CrossRef PubMed.
- Y. Liu, W. Yang, P. Zhang and J. Zhang, Nitric acid-treated birnessite-type MnO2:An efficient and hydrophobic material for humid ozone decomposition, Appl. Surf. Sci., 2018, 442, 640–649 CrossRef CAS.
- F. Liu, S. Rong, P. Zhang and L. Gao, One-step synthesis of nanocarbon-decorated MnO2 with superior activity for indoor formaldehyde removal at room temperature, Appl. Catal., B, 2018, 235, 158–167 CrossRef CAS.
- J. Hou, Y. Li, M. Mao, L. Ren and X. Zhao, Tremendous effect of the morphology of birnessite-type manganese oxide nanostructures on catalytic activity, ACS Appl. Mater. Interfaces, 2014, 6, 14981–14987 CrossRef CAS PubMed.
- H. Yan, X. Zhao and N. Guo, et al., Atomic engineering of high-density isolated Co atoms on graphene with proximal-atom controlled reaction selectivity, Nat. Commun., 2018, 9, 3197 CrossRef PubMed.
- T. Zhang, J. Zhang, P. Derreumaux and Y. Mu, J. Phys. Chem. B, 2013, 15, 3993–4002 CrossRef PubMed.
- Y. Li, Y. Zhang, K. Qian and W. Huang, Metal -Support Interactions in Metal/Oxide Catalysts and Oxide -Metal Interactions in Oxide/Metal Inverse Catalysts, ACS Catal., 2022, 12, 1268–1287 CrossRef CAS.
- M. Wang, P. Zhang, J. Li and C. Jiang, The effects of Mn loading on the structure and ozone decomposition activity of MnOx supported on activated carbon, Chin. J. Catal., 2014, 35, 335–341 CrossRef CAS.
- J. Ma, C. Wang and H. He, Transition metal doped cryptomelane-type manganese oxide catalysts for ozone decomposition, Appl. Catal., B, 2017, 201, 503–510 CrossRef CAS.
- J. Li and Y. Yu, Enhanced catalytic performance of pillared δ-MnO2 with enlarged layer spaces for lithium -and sodium -oxygen batteries: a theoretical investigation, Nanoscale, 2021, 13, 20637–20648 RSC.
- Y. Liu, W. Zong, H. Zhou, D. Wang, R. Cao, J. Zhan, L. Liu and B. W.-L. Jang, Tuning the interlayer cations of birnessite-type MnO2 to enhance its oxidation ability for gaseous benzene with water resistance, Catal. Sci. Technol., 2018, 8, 5344–5358 RSC.
- Y. Liu, H. Zhou, R. Cao, X. Liu, P. Zhang, J. Zhan and L. Liu, Facile and green synthetic strategy of birnessite-type MnO2 with high efficiency for airborne benzene removal at low temperatures, Appl. Catal., B, 2019, 245, 569–582 CrossRef CAS.
- D. Li, W. Li, Y. Deng, X. Wu, N. Han and Y. Chen, Effective Ti Doping of δ-MnO2 via Anion Route for Highly Active Catalytic Combustion of Benzene, J. Phys. Chem. C, 2016, 19, 10275–10282 CrossRef.
- Y. Liu, H. Zhou, R. Cao, T. Sun, W. Zong, J. Zhan and L. Liu, Different behaviors of birnessite-type MnO2 modified by Ce and Mo for removing carcinogenic airborne benzene, Mater. Chem. Phys., 2019, 221, 457–466 CrossRef CAS.
- H. Zhang, S. Sui, X. Zheng, R. Cao and P. Zhang, One-pot synthesis of atomically dispersed Pt on MnO2 for efficient catalytic decomposition of toluene at low temperatures, Appl. Catal., B, 2019, 257, 117878 CrossRef CAS.
- R. Fei, W. Kang and L. Yang, Ferroelectricity and Phase Transitions in Monolayer Group-IV Monochalcogenides, Phys. Rev. Lett., 2016, 117, 097601 CrossRef PubMed.
- H. Zhang, K. Haule and D. Vanderbilt, Metal-Insulator Transition and Topological, Phys. Rev. Lett., 2017, 118, 026404 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- C. Juan, M. Jorge, L. José, S. B. Trickey and V. Alberto, A PW91-like exchange with a simple analytical form, Chem. Phys. Lett., 2016, 651, 268–273 CrossRef.
- P. E. Blöchl and O. K. Andersen, Improved tetrahedron method for Brillouin-zone integrations, Phys. Rev. B:Condens. Matter Mater. Phys., 1994, 49, 16223 CrossRef PubMed.
- H. J. Monkhorst and J. D. Pack, Special points for Brillouin-zone integrations, Phys. Rev. B:Condens. Matter Mater. Phys., 1976, 13, 5188 CrossRef.
- J. Hostaš and J. Řezáč, Accurate DFT-D3 Calculations in a Small Basis Set, J. Chem. Theory Comput., 2017, 13, 3575–3585 CrossRef PubMed.
- G. K. H. Madsen and P. Novák, Charge order in magnetite: An LDA+U study, Europhys. Lett., 2005, 69, 777–783 CrossRef CAS.
- D. A. Tompsett, D. S. Middlemiss and M. S. Islam, Importance of anisotropic Coulomb interactions and exchange to the band gap and antiferromagnetism of β-MnO2 from DFT+U, Phys. Rev. B:Condens. Matter Mater. Phys., 2012, 86, 205126 CrossRef.
- Y. Noda, K. Ohno and S. Nakamura, Momentum-dependent band spin splitting in semiconducting MnO2: a density functional calculation, Phys. Chem. Chem. Phys., 2016, 18, 13294–13303 RSC.
- M. Shenggui, Y. Xue, J. Xia, C. Wanglai, J. Wenju and W. Hualin, First principles calculation of mechanical, dynamical and thermodynamic properties of MnO2 with four crystal phases, J. Alloys Compd., 2021, 852, 157007 CrossRef.
- M. H. Naik and M. Jain, CoFFEE: Corrections For Formation Energy and Eigenvalues for charged defect simulations, Comput. Phys. Commun., 2018, 226, 114–126 CrossRef CAS.
- F. Calle-Vallejo, D. Loffreda and M. Koper, et al., Introducing structural sensitivity into adsorption–energy scaling relations by means of coordination numbers, Nat. Chem., 2015, 7, 403–410 CAS.
- C. J. Bartel, S. L. Millican and A. M. Deml, et al., Physical descriptor for the Gibbs energy of inorganic crystalline solids and temperature-dependent materials chemistry, Nat. Commun., 2018, 9, 4168 Search PubMed.
- M. Dingren, Y. Liu, S. Zhongyi and C. Yang, Photo-catalytic degradation mechanism of benzene over ZnWO4: Revealing the synergistic effects of Na-doping and oxygen vacancies, Chem. Eng. J., 2021, 405, 126538 Search PubMed.
- M. Wei, S. Wu, Q. Mao, Y. Wang, G. Guo and D. Zhang, The oxidation mechanism investigation of benzene catalyzed by palladium nanoparticle: A ReaxFF molecular dynamics, Fuel, 2020, 275, 117989 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr05227g |
| This journal is © The Royal Society of Chemistry 2025 |