
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnique NiCo bimetal boosting 98% CH4 selectivity and high catalysis stability for photothermal CO2 hydrogenation†
Yubin Zheng‡
a,
Zhe Lu‡ b,
Zhisheng Shi*c,
Lu Wangb,
Shuyuan Yangd,
Runzhi Caoa,
Gao Wa*e,
Xin Zhou*df,
Yong Yangg,
Chong Shenga,
Yong Zhou*ab and
Zhigang Zouab
b,
Zhisheng Shi*c,
Lu Wangb,
Shuyuan Yangd,
Runzhi Caoa,
Gao Wa*e,
Xin Zhou*df,
Yong Yangg,
Chong Shenga,
Yong Zhou*ab and
Zhigang Zouab
aKey Laboratory of Modern Acoustics (MOE), Institute of Acoustics, School of Physics, Jiangsu Key Laboratory of Nanotechnology, Eco-materials and Renewable Energy Research Center (ERERC), National Laboratory of Solid-State Microstructures, Collaborative Innovation Center of Advanced Microstructures, Nanjing University, Nanjing, Jiangsu 210093, P. R. China. E-mail: zhouyong1999@nju.edu.cn
bSchool of Science and Engineering, The Chinese University of Hong Kong, Shenzhen, Shenzhen 518172, P. R. China
cSchool of Chemical and Environmental Engineering, Anhui Polytechnic University Wuhu, Anhui, 241000, P. R. China. E-mail: shizhisheng@ahpu.edu.cn
dCollege of Environment and Chemical Engineering, Dalian University, Dalian 116622, P. R. China
eSchool of Physical Science and Technology, Tiangong University, Tianjin 300387, P.R. China. E-mail: gaowa@tiangong.edu.cn
fInterdisciplinary Research Center for Biology and Chemistry, Liaoning Normal University, Dalian 116029, P. R. China. E-mail: xzhou@lnnu.edu.cn
gSchool of Chemistry and Chemical Engineering, Nanjing University of Science and Technology, Nanjing 210094, China. E-mail: yychem@njust.edu.cn
First published on 31st January 2025
Abstract
A highly dispersed NiCo alloy catalyst derived from NiCo bimetallic–organic framework nanosheets was synthesized for efficient photothermal catalysis for CO2 hydrogenation to methane. The NiCo bimetal catalyst achieves a CH4 production rate of 55.60 mmol g−1 h−1 with only a 18.82% decline in performance after 86 hours under atmospheric pressure at selected 290 °C and continuous flow reaction. In situ diffuse reflectance infrared Fourier transform spectroscopy (in situ DRIFTS) reveals the synergistic and complementary roles of light and heat in photothermal catalysis. The thermochemical process drives the reverse water–gas shift reaction to form a *CO intermediate, a key intermediate for the formation of CH4, and light irradiation-generating strong near field from the surface plasma resonance of NiCo bimetals promotes the subsequent *CO hydrogenation step to form *CHO, the rate-determining step for the hydrogenation of CO2 into CH4. Meanwhile, density functional theory (DFT) calculations suggest that the NiCo bimetallic catalyst can also lower the energy barrier of *CO hydrogenation, facilitating the formation of *CHO.
1. Introduction
Harnessing solar energy to drive the conversion of CO2 into high-value-added hydrocarbons is considered an effective method for achieving carbon neutrality, thereby helping in mitigating the greenhouse effect.1–3 However, this approach typically utilizes only a small fraction of the solar spectrum captured by semiconductors, specifically the ultraviolet and visible light portions, which together account for 48% of the total solar energy.4–6 As a result, it often yields low production rates at the μmol gcat−1 h−1 scale.7 In contrast, photothermal catalysis, which integrates photochemical and thermochemical processes by utilizing an external heat source, has the potential to overcome the limitations of low efficiency in purely light-driven processes and the harsh reaction conditions associated with thermal catalysis.8–10 Photothermally driven CO2 conversion thus enables achieving in general production rates in the mmol gcat−1 h−1 range, three orders of magnitude higher than those of conventional photochemical reactions.10–12 Elucidation of the synergistic roles of light and heat in the catalytic process and uncovering of the catalytic transformation mechanisms are important for advancing the achievement of the dual carbon strategy goals.13,14 The CO2 hydrogenation reaction usually undergoes the reverse water–gas shift (RWGS) reaction (CO2 + H2 → CO + H2O) and the Sabatier reaction (CO2 + 4H2 → CH4 + 2H2O), respectively.15,16 Depending on the specific application scenario, CO2 hydrogenation is typically desired to selectively convert into either CO or CH4. However, the concurrent occurrence of both reactions leads to reduced selectivity, making this goal still challenging.17,18In the past decade, metal alloy catalysts have shown potential industrial applications in CO2 hydrogenation reactions.19,20 Specifically, the synergistic effects introduced by different metal elements in alloy catalysts have effectively enhanced catalytic yields and optimized selectivity in CO2 hydrogenation.21 Among these, selection of the appropriate metal composition and distribution is crucial for achieving high selectivity.22,23 Notably, non-precious nickel-based catalysts have garnered significant attention due to their excellent cost-to-performance ratio and high selectivity for methane.24–26 However, the limitations of nickel-based catalysts in terms of low-temperature catalytic activity and dispersibility restrict their potential applications.27,28 The construction of alloy catalysts provides a promising approach to enhancing the activity of nickel-based catalysts.29 Specifically, the strong C–O dissociation ability of cobalt metal at lower temperatures, combined with its atomic number being similar to that of nickel, makes it a feasible choice for a bimetallic alloy when combined with Ni.30–32 Additionally, to improve the dispersibility of NiCo alloy catalysts, carbon-based materials (such as graphene, carbon nitride, g-C3N4, etc.) with high specific surface areas can provide abundant loading sites for NiCo alloy particles, thus reducing particle agglomeration during the reaction.33–35 Although alloy catalysts have shown unique advantages in CO2 hydrogenation, the complexity of the catalytic conversion mechanisms means that the relationship between the atomic structure and performance still requires continuous exploration.36
In this study, highly dispersed NiCo alloy particles in the carbon matrix were successfully prepared through annealing two-dimensional NiCo metal–organic framework nanosheets (NiCo-MOFNs) under an inert atmosphere for efficient photothermal catalysis of CO2 hydro-methanation. The resulting NiCo/C catalyst achieved a methane production rate and selectivity of 55.60 mmol g−1 h−1 and 98.01%, respectively, and a CO2 conversion rate of 42.34% under flowing photothermal conditions. A decline of only 18.82% in the catalysis performance after 86 hours demonstrates excellent catalytic stability. In situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) reveals the synergistic and complementary roles of light and heat in photothermal catalysis. The thermochemical process drives the reverse water–gas shift (RWGS) reaction to form *CO, a key intermediate for the formation of CH4, and light illumination-generating strong near field from the surface plasma resonance (SPR) of NiCo bimetals promotes the subsequent *CO hydrogenation step to *CHO, the rate-determining step for the hydrogenation of CO2 into CH4. Meanwhile, density functional theory (DFT) calculations suggest that the NiCo bimetallic catalyst can also lower the energy barrier for the *CO hydrogenation, facilitating the formation of *CHO.
2. Experimental section
2.1. Materials
All the chemicals were employed without further treatment. NiCl2·6H2O (99.9%), CoCl2·H2O (99.99%), N,N-dimethylformamide (DMF) and triethylamine (TEA) were bought from Aladdin Chemical Co., Ltd. Benzenedicarboxylic acid (BDC) was bought from Sigma-Aldrich Co., Ltd. Absolute ethyl alcohol was bought from Sinopharm Chemical Reagent Co., Ltd.2.2. Preparation of samples
2.3. Characterization
The powder X-ray diffraction (XRD) patterns of the samples were collected on a Bruker-D8 Advance Powder X-ray diffractometer (40 kV, 40 mA) using Ni-filtered Cu Kα radiation in the 2θ range from 20 to 70° at a scan rate of 0.1° per second. The morphologies of the samples were observed using a scanning electron microscope (SEM, Zeiss Gemini 500) and a transmission electron microscope (TEM, FE TecnaiG2 F20S-TWIN) coupled with an energy-dispersive X-ray spectrometer. The metal content of the samples was quantified by inductively coupled plasma optical emission spectrometry (ICP-OES, Analytik PQ9100). The X-ray photoelectron spectroscopy (XPS) measurements were performed on a PHI5000 VersaProbe.2.4. Photo-/thermal-/photothermal catalytic performance test
The CO2 hydrogenation reaction was carried out in a fixed-bed flow reactor under atmospheric pressure. A total of 200 mg of catalyst was homogeneously dispersed in 500 mg of quartz sand. The mixed catalyst was pressed and subsequently ground (20–40 mesh). The above catalysts were uniformly dispersed on a quartz microfiber filter. Before the reaction, the catalyst was ex situ reduced in flowing H2/N2 (H2/N2 = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9) at 50 ml min−1 and 350 °C for 2 h. After the reduced catalyst cooled down to room temperature, the reactor was flushed with Ar for 1 h. Then, The CO2 hydrogenation reaction was carried out in the reaction gas (CO2/H2/N2 = 1:3:1) of 50 ml min−1. A 300 W Xe lamp (300–2500 nm) was used as the light source (PLS-XSE300+). In the photocatalytic test, a Xe lamp was used as a light source at room temperature. In the thermal catalytic test, the Xe lamp was removed and the reactor was heated to 290 °C. In the photothermal catalysis test, a Xe lamp was used in conjunction with an external heat source. The effluent gas was analysed online by gas chromatography (GC, Fuli GC9790II).
:9) at 50 ml min−1 and 350 °C for 2 h. After the reduced catalyst cooled down to room temperature, the reactor was flushed with Ar for 1 h. Then, The CO2 hydrogenation reaction was carried out in the reaction gas (CO2/H2/N2 = 1:3:1) of 50 ml min−1. A 300 W Xe lamp (300–2500 nm) was used as the light source (PLS-XSE300+). In the photocatalytic test, a Xe lamp was used as a light source at room temperature. In the thermal catalytic test, the Xe lamp was removed and the reactor was heated to 290 °C. In the photothermal catalysis test, a Xe lamp was used in conjunction with an external heat source. The effluent gas was analysed online by gas chromatography (GC, Fuli GC9790II).
2.5. In situ DRIFTS analysis
In situ diffuse reflectance infrared Fourier transform spectroscopy was performed on a Thermo Nicolet iSS0 FTIR spectrometer with a high-precision mercury–cadmium–telluride (MCT) detector cooled with liquid nitrogen. Before collecting the spectra, the samples were warmed to 350 °C with He (15 sccm), followed by switching to 11 vol% H2/He and pre-reduction for 1 h. The samples were then cooled down to 290 °C with He (for solely photocatalytic experiments, cooled down to room temperature) and stabilised for 10 min, at which point the background spectra were collected. Finally, the reaction gas (CO2/H2 = 1:3) was introduced into the reactor (4 sccm), with or without a light source selected according to the reaction conditions, and spectra were collected.
2.6. Numerical simulation
The electric field was simulated by the finite-difference time-domain (FDTD, Lumerical FDTD solutions) method with perfectly matched layer boundary conditions. The dielectric constants of Ni, Co and NiCo ware taken from the literature.382.7. DFT simulation
All the DFT computational calculations were carried out with Vienna ab initio simulation package (VASP),39,40 and the projector-augmented wave (PAW) method is used to describe interactions of electrons and ions. The exchange–correlation potential is represented by the generalized gradient approximation (GGA) as parameterized by Perdew–Burke–Ernzerhof (PBE).41 The valence electrons are described with a plane wave basis set, and a plane-wave cutoff energy of 400 eV is used in the calculations.The per unit cell of Ni, Co and Ni–Co is a cubic crystal with the space group of Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, fully optimized with the Monkhorst–Pack type k-point mesh of 9 × 9 × 9. The lattice parameters of Ni bulk are a = b = c = 3.515 Å, α = β = γ = 90°, those of Co bulk are a = b = c = 3.512 Å, α = β = 90°, and those of Co–Ni alloy bulk are a = b = c = 3.534 Å, α = β = γ = 90°.
m, fully optimized with the Monkhorst–Pack type k-point mesh of 9 × 9 × 9. The lattice parameters of Ni bulk are a = b = c = 3.515 Å, α = β = γ = 90°, those of Co bulk are a = b = c = 3.512 Å, α = β = 90°, and those of Co–Ni alloy bulk are a = b = c = 3.534 Å, α = β = γ = 90°.
All the models are constructed with 2 × 2 × 2 supercells of the (111) facet containing five layers to simulate the surface of Co, Ni and Ni–Co alloy, and the two layers of atoms at the bottom were fixed. A vacuum layer of 15 Å was added to avoid the interaction between separate periodic slabs for the surface calculations. The k-point sampling was set to 3 × 3 × 1 to perform the surface calculations. When the residual forces were less than 0.01 eV A−1, the convergence of optimizing the atomic positions and cell parameters was reached, and the electronic energy was converged to 10−5 eV. Grimme’ s scheme (DFT + D3)42 was adopted to treat the van der Waals (vdW) interactions between the adopted species and surfaces.
The free energy change (ΔG) of each elementary step in the CO2RR can be obtained by using the formula: ΔG = ΔE + ΔZPE − TΔS + eU, where ΔE is the reaction energy of the reactant and product species adsorbed on the catalyst directly obtained from DFT computations; ΔZPE and TΔS represent the differences in zero-point energy and entropy, respectively, between the adsorbed species and the gas phase molecules at 298.15 K, which can be calculated from the vibrational frequencies. U is the applied potential. According to the obtained free energy change of each elementary step, the limiting potential (UL) was further computed as follows: 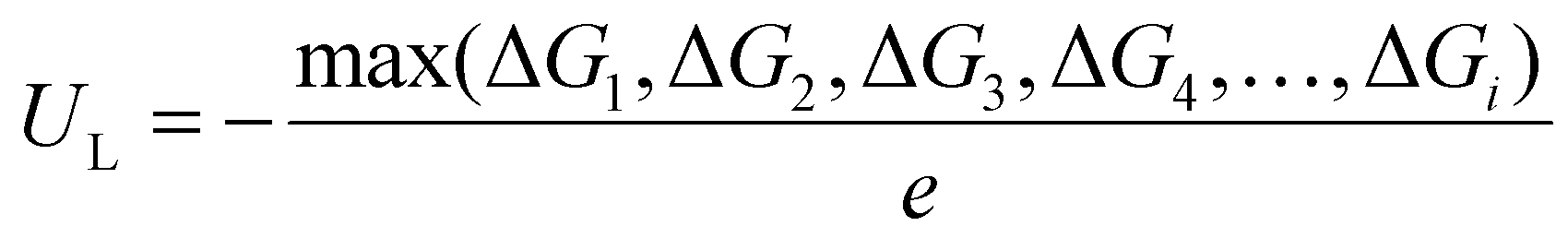 .43
.43
3. Results and discussion
3.1. Characterization of samples
The synthetic process of NiCo/C has been described in the Experimental section in detail. Briefly, the sheet-like NiCo-MOFN precursor was prepared by the coordination of Ni2+ and Co2+ and benzenedicarboxylic acid (BDC) in binary DMF-ethanol solution at room temperature, which is characterized using X-ray diffraction (XRD) (Fig. S2†), scanning electron microscopy (SEM) (Fig. S3†), and transmission electron microscopy (TEM) analyses (Fig. S4†). Subsequently, the resulting NiCo-MOFNs were pyrolyzed under an N2 atmosphere to form the NiCo/C catalyst. For comparison, Ni or Co was also added separately during the precursor preparation to obtain Ni-MOFNs and Co-MOFNs, which were then subjected to the same annealing process to generate Co/C and Ni/C, respectively. Thermogravimetric analysis (TGA) was used to collect data on the pyrolysis process of metal–MOFNs from 50 °C to 800 °C to understand the formation mechanism of this metal alloy catalyst (Fig. S5†). It was found that the metal–MOFN undergoes significant pyrolysis in N2 after 600 °C, converting the organic components into carbon matrices, and the metal elements are transferred into metal nanoparticles.The XRD patterns of various Ni and Co-based catalysts show the presence of a cubic phase of metal nanoparticles with no obvious diffraction peaks corresponding to metallic oxides (Fig. S6†). The diffraction peaks at 44.39° and 51.72° can be indexed to the cubic phase (111) and (200) crystal planes of the NiCo bimetal, respectively. The diffraction peaks of Co/C, NiCo/C and Ni/C are gradually shifted to large angles, implying the formation of NiCo alloy nanoparticles. No diffraction peaks associated with the carbon matrix were observed, implying a high degree of disorder in the carbon matrix. The graphitization degree of the carbon matrix in the NiCo/C catalyst was assessed by Raman spectroscopy (Fig. S7†). The intensity ratio of the disordered graphite (D band) at 1350 cm−1 and the E2g mode of graphite (G band) at 1588 cm−1 (ID/IG) is 1.03, confirming the high degree of disorder of the carbon matrix in the synthesized catalyst.26
The transmission electron microscopy (TEM) image of NiCo/C shows that NiCo bimetal particles were uniformly dispersed in the carbon matrix with an average particle size of ∼9–10 nm (Fig. 1a and Fig. S8†). The high-resolution TEM image displays the lattice fringes with an interlayer distance of 0.21 nm, being assigned to the (111) crystal plane of NiCo alloy particles (Fig. 1b and c). The high-angle annular dark-field scanning TEM (HAADF-STEM) image (Fig. 1d) and the corresponding energy-dispersive X-ray spectroscopy (EDS) mapping images (Fig. 1e–g) confirm the co-existence and uniform distribution of C, Ni, and Co elements. Inductively coupled plasma optical emission spectrometry (ICP-OES) verifies the atomic ratios of Ni to Co of 1.056:1 in the NiCo alloy, close to that in the MOFN precursors. The metal loadings of Ni/C, Co/C and NiCo/C were 85.02 wt%, 78.70 wt% and 82.01 wt%, respectively.
 | ||
| Fig. 1 (a) TEM images of NiCo/C. (b) and (c) HRTEM images of NiCo/C. (d) HAADF-STEM images of NiCo/C. EDS mapping images of (e) C, (f) Ni and (g) Co for NiCo/C. | ||
X-ray photoelectron spectroscopy (XPS) spectra of NiCo/C confirm the presence of C, O, Ni, and Co (Fig. S9†). In the C 1s spectra, three main peaks of the synthesized catalysts at 284.3, 285.1 and 288.6 eV corresponded to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C sp2, C–C sp3 and CO, respectively (Fig. S10†).44 Two peaks at 852.30 eV and 854.78 eV for the Ni 2p spectra can be ascribed to metallic Ni0 and Nix+, respectively (Fig. 2a), and the peaks at 777.82 eV and 780.25 eV in the Co 2p spectra to metallic Co0 and Cox+, respectively (Fig. 2b).45,46 The presence of Nix+ and Cox+ may originate from partial surface oxidation of the metallic Ni and Co in air. The binding energy of Ni0 in NiCo/C decreases relative to Ni/C, while that of Co0 increases, compared to Co/C, which implies electron donation from Co to Ni in NiCo/C.
C sp2, C–C sp3 and CO, respectively (Fig. S10†).44 Two peaks at 852.30 eV and 854.78 eV for the Ni 2p spectra can be ascribed to metallic Ni0 and Nix+, respectively (Fig. 2a), and the peaks at 777.82 eV and 780.25 eV in the Co 2p spectra to metallic Co0 and Cox+, respectively (Fig. 2b).45,46 The presence of Nix+ and Cox+ may originate from partial surface oxidation of the metallic Ni and Co in air. The binding energy of Ni0 in NiCo/C decreases relative to Ni/C, while that of Co0 increases, compared to Co/C, which implies electron donation from Co to Ni in NiCo/C.
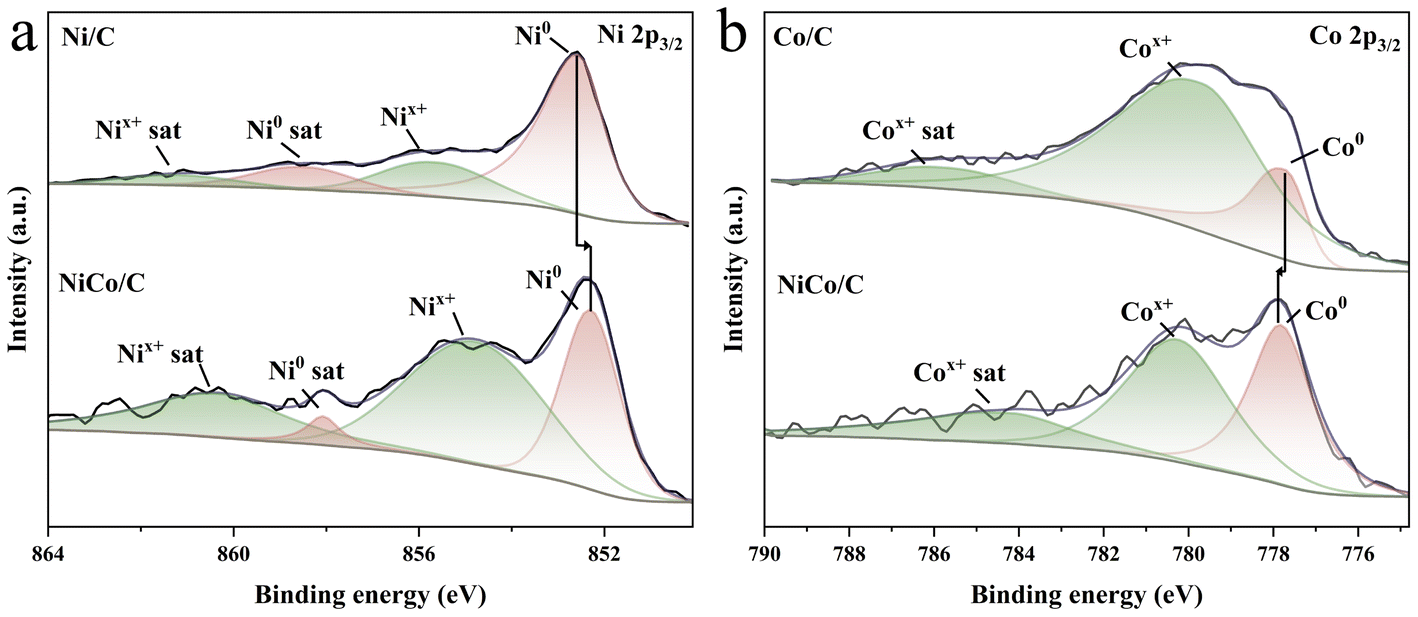 | ||
| Fig. 2 (a) Ni 2p3/2 core-level XPS spectra of Ni/C and NiCo/C. (b) Co 2p3/2 core-level XPS spectra of Co/C and NiCo/C. | ||
3.2. Photothermal catalytic CO2 hydrogenation performance
The catalytic performance of photothermal CO2 hydrogenation was evaluated in a flow reactor at selected 290 °C, powered by an external heat source. CH4 was detected as the main product for all Ni/C, Co/C, and NiCo/C with the production rates of 4.17, 8.85, and 55.60 mmol g−1 h−1, respectively (Fig. 3a). The superior catalysis performance of NiCo/C relative to Ni/C and Co/C demonstrates the necessity of the NiCo bimetallic synergism for the CO2 hydrogenation reaction. To the best of our knowledge, the CH4 yield on the present NiCo/C in the flow reaction is at the top among those of most reported metal-based catalysts (see Table S1†). In addition, CO was also produced on Co/C and NiCo/C with the rates of 2.38 and 0.96 mmol g−1 h−1, respectively (Fig. 3b), and absent on Ni/C. The CH4 selectivity of NiCo/C was detected to be 98.1%, close to 99.27% for Co/C, and much higher than 77.02% for Ni/C (Fig. 3b). The CO2 conversion rate of NiCo/C reaches as high as 42.34%, much higher than 3.14% for Ni/C and 8.58% for Co/C. After more than 86 hours of flow catalysis, the CO2 conversion rate still remained around 34.87%, with only a 18.82% decline, demonstrating high stability of the NiCo/C catalyst (Fig. 3c). The isotopic 13CO2 experiment confirms that the carbon source for the CO2 hydro-methanation reaction originates from the input CO2 gas (Fig. S11†). It should be mentioned that light illumination alone is just slightly capable of driving the present CO2 hydro-methanation reaction, achieving negligible amounts of products and a CO2 conversion rate of 0.02% despite the surface temperatures reaching 177 °C after 5 min-light illumination from photo-induced heat (Fig. S12†). Compared to the monometallic Ni and Co samples, the catalyst surface temperatures of the bimetallic NiCo alloys were elevated by 52 and 47 °C, respectively. Meanwhile, with purely thermal catalysis at 290 °C under dark conditions, the CO2 conversion rate and methane yield of NiCo/C were detected to be 0.18% and 0.21 mmol g−1 h−1 (Fig. 3d), respectively, indicating that thermal catalytic CO2 hydrogenation on NiCo/C can occur, but still with relatively low efficiency. It definitely verifies the synergistic effect of light and heat in triggering the photothermal reaction.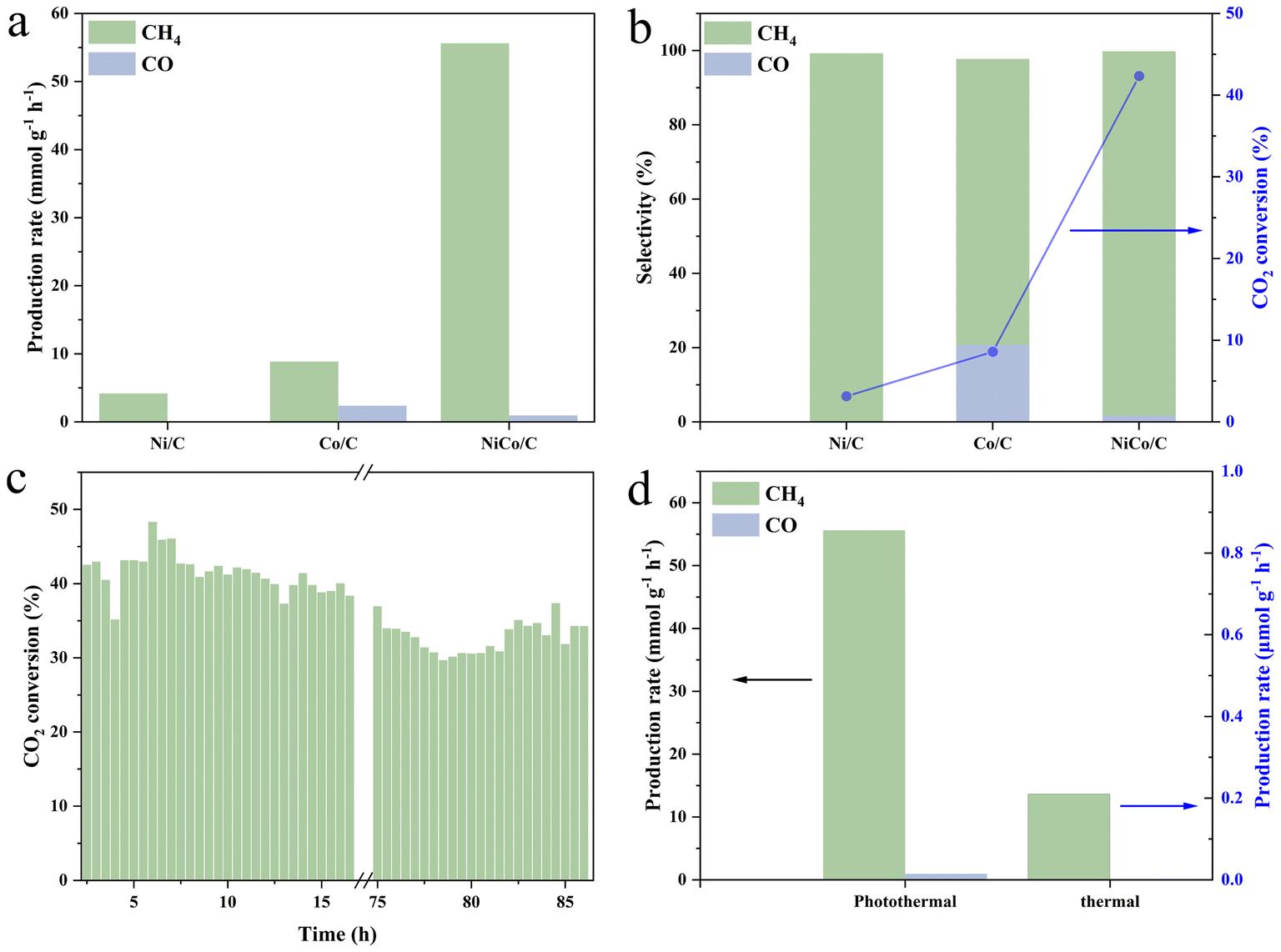 | ||
| Fig. 3 (a) Production rate of CH4 and CO of Ni/C, Co/C and NiCo/C catalysts at 290 °C under light irradiation. (b) Product selectivities and CO2 conversion rates of Ni/C, Co/C and NiCo/C catalysts at 290 °C under light irradiation. (c) Production rates of CH4 and CO of the NiCo/C catalyst under different conditions. (d) CO2 conversion of the NiCo/C catalyst during long-time reaction at 290 °C under light irradiation. | ||
3.3. Mechanism of carbon dioxide hydro-methanation reaction
Fig. 4a shows the comparative in situ DRIFTS spectra of NiCo/C after 60 minutes of reaction under solely light-driven, thermal catalytic, and photothermal conditions, and the corresponding time-dependent spectra are shown in Fig. S13.† The absorption peaks at 1266/1518 cm−1 and 1699 cm−1 were attributed to the formation of monodentate carbonate (m-*CO32−) and ˙CO2− species, respectively, indicating no significant difference in CO2-activated precursor species on NiCo alloys under different conditions.47,48 Additionally, the adsorption peak at 1649 cm−1 was assigned to *COOH, suggesting that the carboxyl pathway is the main reaction route.49 The *CO peaks at 2074, 2113, and 2177 cm−1 were observed under both thermally and photothermally catalysed conditions, indicating that the CO2 hydrogenation-to-methane reaction on NiCo alloys under these conditions involves the RWGS process.50 The peaks at 2965 cm−1 and 3014 cm−1, associated with the C–H stretching vibrations of CHx and CH4, respectively, were observed under both photothermal and thermal catalytic conditions, but not under solely light-driven conditions.51,52 Additionally, the peak related to *CHO at 1059/1086 cm−1 was detected under solely light-driven and photothermal conditions, and absent under purely thermal catalysis process.29,53 This suggests that light illumination may typically promote the formation of *CHO intermediates. This promotion possibly originates from a light-generating strong near field from the surface plasma resonance (SPR) on NiCo bimetal nanoparticles, which may activate the intermediate species.10,53 Furthermore, in situ DRIFTS experiments were also performed on monometallic Ni and Co catalysts (Fig. S14†) for the same photothermal CO2 hydrogenation reaction. While the characteristic peaks were found almost consistent with those observed in the NiCo alloy, the peaks of *CHO on both Ni and Co were significantly weaker than those on the NiCo alloy (Fig. 4b). This indicates that the synergistic effect of the bimetallic Ni and Co facilitates the hydrogenation of *CO to *CHO. The finite-difference time-domain (FDTD) method was employed to simulate SPR near-field enhancement around Ni, Co, and NiCo. The near-field electric field strength of the bimetallic NiCo catalysts is indeed significantly higher than that of the monometallic Ni and Co catalysts (Fig. S15–17†).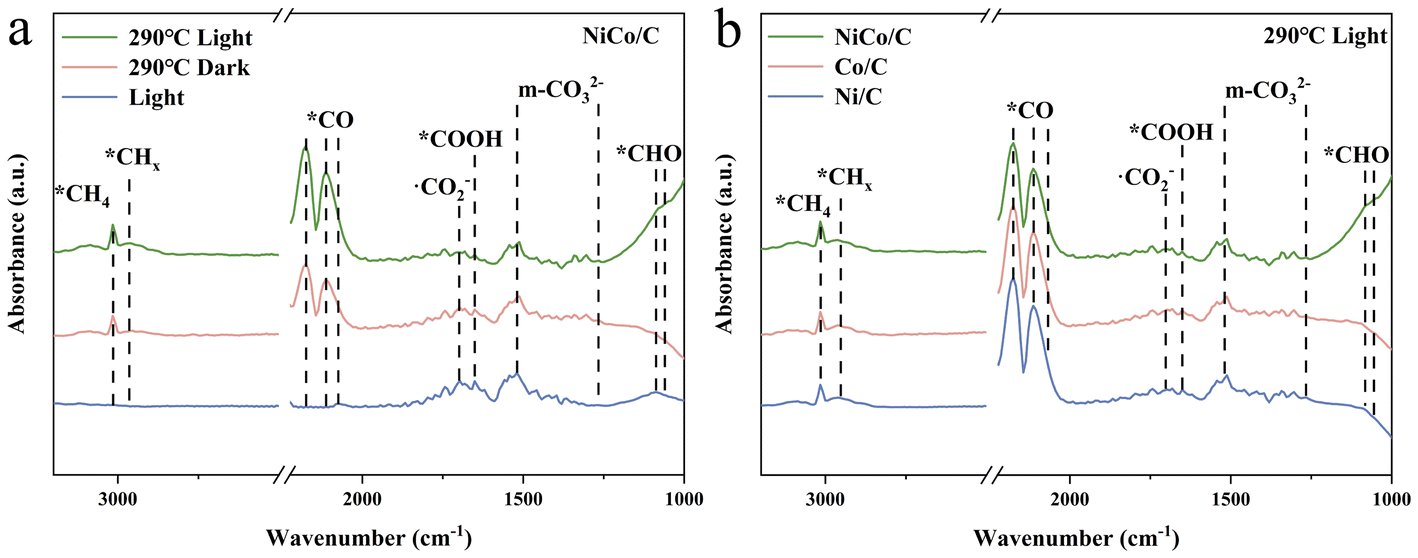 | ||
| Fig. 4 (a) In situ DRIFTS spectra of the NiCo/C catalyst under solely light-driven, thermal catalytic (at 290 °C in dark), and photo-thermal conditions for 60 min. (b) In situ DRIFTS spectra of Ni/C, Co/C and NiCo/C catalysts at 290 °C under light irradiation for 60 min. | ||
DFT calculations were conducted to investigate the adsorption and activation processes of CO2 on Ni (111), Co (111), and Ni–Co (111). The free energy changes for each reaction step are shown in Fig. 5a, and the optimized configurations of the key intermediates of Ni–Co (111) are depicted in Fig. 5b. The optimized configurations of the involved intermediates of Ni (111), Co (111), and Ni–Co (111) are shown in Fig. S18–20.† The calculated CO2 hydrogenation-to-methane mechanisms reveal that one oxygen atom of CO2 is hydrogenated to form *COOH species with free energy changes of −0.23 eV and −0.10 eV on Ni (111) and Co (111), respectively (Table S2†), while a slight free energy increase of 0.01 eV is observed on Ni–Co (111). Subsequently, the generated *COOH intermediate reacts with a second (H+ + e−) pair to form *CO and H2O. However, as the rate-determining step, the further hydrogenation of the *CO intermediate to form *CHO species requires a relatively high energy input: 1.06 eV for Ni (111), 0.98 eV for Co (111), and 0.82 eV for Ni–Co (111). Due to the significantly lower energy input (0.82 eV) required by the NiCo alloy catalyst for the hydrogenation of *CO to *CHO, the formation of *CHO intermediates is promoted, consistent with the stronger characteristic *CHO FTIR signal.
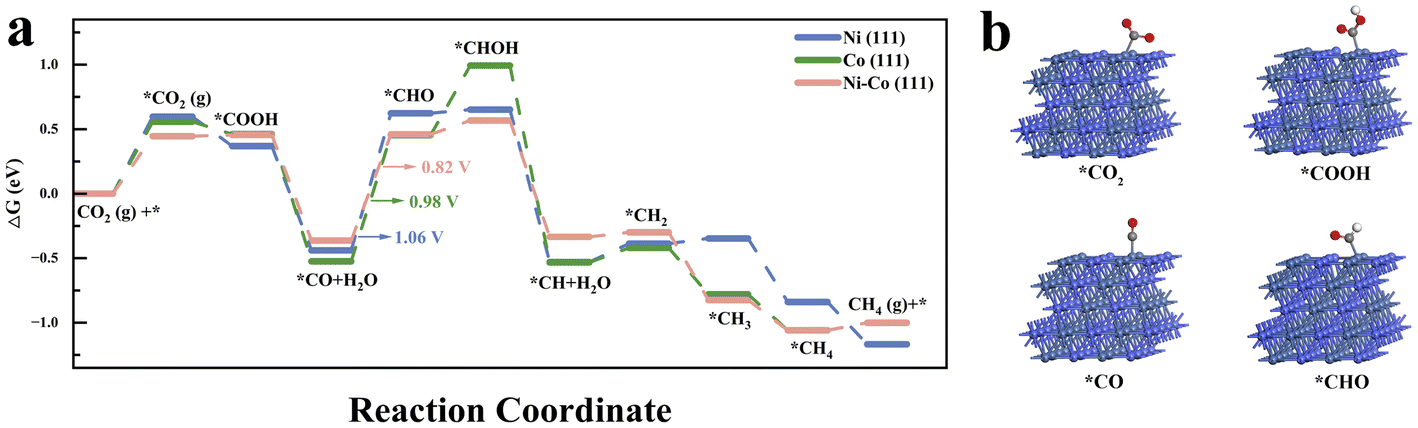 | ||
| Fig. 5 (a) CO2 hydro-methanation pathway on the Ni (111), Co (111) and Ni–Co (111) interface. (b) The optimized configurations of the key intermediates of Ni–Co (111). The blue, purple, grey, red and white spheres represent Ni, Co, C, O and H atoms, respectively. | ||
4. Conclusions
The unique NiCo/C catalyst was synthesized for photothermally catalysing CO2 hydrogenation to methane under a flow reactor with high efficiency and stability. The photochemical and thermochemical processes are aligned in the formation of CO2-activated precursors and the reactive carboxylate pathway. The individual contributions of the photochemical and thermochemical processes are reflected in the fact that the thermochemical process introduces the RWGS process, and light illumination promotes the formation of the reaction intermediate *CHO. This study provides valuable insights into the photothermal synergy and bimetallic cooperation in photothermal catalytic reactions.Author contributions
Yubin Zheng: writing – original draft, investigation, methodology, data curation and formal analysis. Zhe Lu: data curation, formal analysis and Validation. Zhisheng Shi: data curation and formal analysis. Shuyuan Yang: methodology, investigation and calculations. Runzhi Cao: methodology, investigation and calculations. Gao Wa: writing – review and editing and Investigation. Xin Zhou: methodology, investigation and calculations. Yong Yang: methodology and investigation. Chong Sheng: methodology, investigation and calculations. Yong Zhou: writing – review and editing, supervision, resources, funding acquisition, and formal analysis. Zhigang Zou: supervision and resources.Data availability
All data included in this study are available upon request by contact with the corresponding author.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work is financially sponsored by the National Natural Science Foundation of China (No. 22472002, 22372017, and 22202152), NSF of Jiangsu Province (No. BK20220006), the Program for Guangdong Introducing Innovative and Entrepreneurial Team (2019ZL08L101), the University Development Fund (UDF01001159), Cross-Subject Project of Chemistry Discipline of Yangzhou University (yzuxk202014), and the Project of Key Laboratory of Organic Synthesis of Jiangsu Province, College of Chemistry, Chemical Engineering and Materials Science, Soochow University (No. KJS2328).References
- K. Marvel, B. I. Cook, C. J. W. Bonfils, P. J. Durack, J. E. Smerdon and A. P. Williams, Nature, 2019, 569, 59–65 CrossRef CAS PubMed.
- E. Gong, S. Ali, C. B. Hiragond, H. S. Kim, N. S. Powar, D. Kim, H. Kim and S.-I. In, Energy Environ. Sci., 2022, 15, 880–937 RSC.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS.
- K. Sun, Y. Qian and H.-L. Jiang, Angew. Chem., Int. Ed., 2023, 62, e202217565 CrossRef CAS.
- B. Zhu, J. Sun, Y. Zhao, L. Zhang and J. Yu, Adv. Mater., 2024, 36, 2310600 CrossRef CAS.
- Q. Zhu, Q. Xu, M. Du, X. Zeng, G. Zhong, B. Qiu and J. Zhang, Adv. Mater., 2022, 34, 2202929 CrossRef CAS.
- W. Tu, Y. Zhou and Z. Zou, Adv. Mater., 2014, 26, 4607–4626 CrossRef CAS PubMed.
- F. Zheng, T. Lin, K. Wang, Y. Wang and G. Li, Nano Res., 2023, 16, 12919–12935 CrossRef CAS.
- X. Li, Y. Chen, Y. Tao, L. Shen, Z. Xu, Z. Bian and H. Li, Chem Catal., 2022, 2, 1315–1345 CrossRef CAS.
- Y. Tang, T. Zhao, H. Han, Z. Yang, J. Liu, X. Wen and F. Wang, Adv. Sci., 2023, 10, 2300122 CrossRef CAS PubMed.
- L. Wang, Y. Dong, T. Yan, Z. Hu, F. M. Ali, D. M. Meira, P. N. Duchesne, J. Y. Y. Loh, C. Qiu, E. E. Storey, Y. Xu, W. Sun, M. Ghoussoub, N. P. Kherani, A. S. Helmy and G. A. Ozin, Nat. Commun., 2020, 11, 2432 CrossRef CAS.
- M. Cai, Z. Wu, Z. Li, L. Wang, W. Sun, A. A. Tountas, C. Li, S. Wang, K. Feng, A.-B. Xu, S. Tang, A. Tavasoli, M. Peng, W. Liu, A. S. Helmy, L. He, G. A. Ozin and X. Zhang, Nat. Energy, 2021, 6, 807–814 CrossRef CAS.
- S. Fang and Y. H. Hu, Chem. Soc. Rev., 2022, 51, 3609–3647 RSC.
- L. Zhou, D. F. Swearer, C. Zhang, H. Robatjazi, H. Zhao, L. Henderson, L. Dong, P. Christopher, E. A. Carter, P. Nordlander and N. J. Halas, Science, 2018, 362, 69–72 CrossRef CAS PubMed.
- Y. Choi, G. D. Sim, U. Jung, Y. Park, M. H. Youn, D. H. Chun, G. B. Rhim, K. Y. Kim and K. Y. Koo, Chem. Eng. J., 2024, 492, 152283 CrossRef CAS.
- X. Li, C. Wang and J. Tang, Nat. Rev. Mater., 2022, 7, 617–632 CrossRef CAS.
- Y. Xie, J. Chen, X. Wu, J. Wen, R. Zhao, Z. Li, G. Tian, Q. Zhang, P. Ning and J. Hao, ACS Catal., 2022, 12, 10587–10602 CrossRef CAS.
- H. Jiang, L. Wang, H. Kaneko, R. Gu, G. Su, L. Li, J. Zhang, H. Song, F. Zhu, A. Yamaguchi, J. Xu, F. Liu, M. Miyauchi, W. Ding and M. Zhong, Nat. Catal., 2023, 6, 519–530 CrossRef CAS.
- S. Rawalekar and T. Mokari, Adv. Energy Mater., 2013, 3, 12–27 CrossRef CAS.
- D. Mateo, J. L. Cerrillo, S. Durini and J. Gascon, Chem. Soc. Rev., 2022, 51, 1547–1547 RSC.
- J. Zhao, R. Shi, G. I. N. Waterhouse and T. Zhang, Nano Energy, 2022, 102, 107650 CrossRef CAS.
- H. Xie, J. Wang, K. Ithisuphalap, G. Wu and Q. Li, J. Energy Chem., 2017, 26, 1039–1049 CrossRef.
- M. Humayun, C. Wang and W. Luo, Small Methods, 2022, 6, 2101395 CrossRef CAS.
- M. Sayed, J. Yu, G. Liu and M. Jaroniec, Chem. Rev., 2022, 122, 10484–10537 CrossRef CAS.
- L. Shen, J. Xu, M. Zhu and Y.-F. Han, ACS Catal., 2020, 10, 14581–14591 CrossRef CAS.
- I. S. Khan, D. Mateo, G. Shterk, T. Shoinkhorova, D. Poloneeva, L. Garzón-Tovar and J. Gascon, Angew. Chem., Int. Ed., 2021, 60, 26476–26482 CrossRef CAS.
- J. Zeng, K. Lu, J. Zhang, Y. Sun, Z. Chang, J. Li, B. Dai, F. Yu, J. Li and J. Liu, Int. J. Hydrogen Energy, 2022, 47, 2234–2244 CrossRef CAS.
- R. Ye, L. Ma, X. Hong, T. R. Reina, W. Luo, L. Kang, G. Feng, R. Zhang, M. Fan, R. Zhang and J. Liu, Angew. Chem., Int. Ed., 2024, 63, e202317669 CrossRef CAS PubMed.
- H. Huang, J. Zhao, H. Guo, B. Weng, H. Zhang, R. A. Saha, M. Zhang, F. Lai, Y. Zhou, R.-Z. Juan, P.-C. Chen, S. Wang, J. A. Steele, F. Zhong, T. Liu, J. Hofkens, Y.-M. Zheng, J. Long and M. B. J. Roeffaers, Adv. Mater., 2024, 36, 2313209 CrossRef CAS.
- Y. Tang, H. Wang, C. Guo, Z. Yang, T. Zhao, J. Liu, Y. Jiang, W. Wang, Q. Zhang, D. Wu, Y. Zhao, X.-D. Wen and F. Wang, ACS Nano, 2024, 18, 11449–11461 CrossRef CAS.
- Z. Li, J. Chen, Y. Xie, J. Wen, H. Weng, M. Wang, J. Zhang, J. Cao, G. Tian, Q. Zhang and P. Ning, J. Energy Chem., 2024, 91, 213–225 CrossRef CAS.
- I. C. T. Have, J. J. G. Kromwijk, M. Monai, D. Ferri, E. B. Sterk, F. Meirer and B. M. Weckhuysen, Nat. Commun., 2022, 13, 324 CrossRef CAS PubMed.
- S. N. Talapaneni, G. Singh, I. Y. Kim, K. AlBahily, A. A. H. Al-Muhtaseb, A. S. Karakoti, E. Tavakkoli and A. Vinu, Adv. Mater., 2020, 32, 1904635 CrossRef CAS.
- S. Wang, X. Zhang, Y. Tang, S. Hao, S. Zheng, J. Qiao, Z. Wang, L. Wu, J. Liu and F. Wang, Carbon, 2024, 216, 118528 CrossRef CAS.
- P. Li, L. Liu, W. An, H. Wang, H. Guo, Y. Liang and W. Cui, Appl. Catal., B, 2020, 266, 118618 CrossRef CAS.
- Y. Dong, W. Zhang, Z. Hu, Y. H. Ng, Z. Wei, Y. Liu, J. Deng, H. Dai and L. Jing, Appl. Catal., B, 2024, 357, 124347 CrossRef CAS.
- B. Han, X. Ou, Z. Deng, Y. Song, C. Tian, H. Deng, Y.-J. Xu and Z. Lin, Angew. Chem., Int. Ed., 2018, 57, 16811–16815 CrossRef CAS PubMed.
- P. B. Johnson and R. W. Christy, Phys. Rev. B, 1974, 9, 5056–5070 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B:Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B:Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS.
- T. Zhao, T. Yan, Y. Sun, Z. Wang, Q. Cai, J. Zhao and Z. Chen, J. Mater. Chem. A, 2023, 11, 19444–19454 RSC.
- L. Zhang, G. Chen, M. N. Hedhili, H. Zhang and P. Wang, Nanoscale, 2012, 4, 7038–7045 RSC.
- S. Wu, Y. Li, Q. Zhang, Q. Hu, J. Wu, C. Zhou and X. Zhao, Adv. Energy Mater., 2020, 10, 2002602 CrossRef CAS.
- B. Qi, W. Chang, Q. Xu, L. Jiang, S. An, J.-F. Chu and Y.-F. Song, ACS Appl. Mater. Interfaces, 2023, 15, 12078–12087 CrossRef CAS PubMed.
- C. Ding, L. Yang, X. Lu, H. Chi, Y. Yang, J. Yuan, X. Wang, X. Wu, Y. Zhang, Y. Zhou and Z. Zou, Adv. Sci., 2024, 11, 2406329 CrossRef CAS.
- L. Cheng, X. Yue, L. Wang, D. Zhang, P. Zhang, J. Fan and Q. Xiang, Adv. Mater., 2021, 33, 2105135 CrossRef CAS.
- Y. Wang, J. Yang, Y. Sun, D. Ye, B. Shan, S. C. E. Tsang and X. Tu, Chem, 2024, 10, 2590–2606 CAS.
- X. Y. Zhang, Z. X. Lou, J. Chen, Y. Liu, X. Wu, J. Y. Zhao, H. Y. Yuan, M. Zhu, S. Dai, H. F. Wang, C. Sun, P. F. Liu and H. G. Yang, Nat. Commun., 2023, 14, 7681 CrossRef CAS PubMed.
- W.-G. Pan, C.-F. Li, Z.-R. Zhang, T. Wu and R.-T. Guo, Appl. Catal., B, 2024, 343, 123492 CrossRef CAS.
- Z. Xie, H. Luo, S. Xu, L. Li and W. Shi, Adv. Funct. Mater., 2024, 34, 2313886 CrossRef CAS.
- J. Zhang, K. Xie, Y. Jiang, M. Li, X. Tan, Y. Yang, X. Zhao, L. Wang, Y. Wang, X. Wang, Y. Zhu, H. Chen, M. Wu, H. Sun and S. Wang, ACS Catal., 2023, 13, 10855–10865 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr05471g |
| ‡ These authors contributed equally to this work and should be considered co-first authors. |
| This journal is © The Royal Society of Chemistry 2025 |