
A dual-initiating organic frustrated Lewis pair catalyst for living polymerizations of (bio)acrylates to facilitate the synthesis of metal-free multiblock copolymers†
Zhen-Hua
Zhang‡
ab,
Yuyang
Chen‡
b,
Yuesheng
Li
 a and
Miao
Hong
*bc
a and
Miao
Hong
*bc
aTianjin Key Lab of Composite & Functional Materials, School of Materials Science and Engineering, Tianjin University, Tianjin 300072, China
bState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, China. E-mail: miaohong@sioc.ac.cn
cSchool of Chemistry and Materials Sciences, Hangzhou Institute for Advanced Study, University of Chinese Academy of Sciences, 1 Sub-lane Xiangshan, Hangzhou 310024, China
First published on 17th December 2024
Abstract
The development of powerful catalysis or methodologies for the synthesis of fundamentally and technologically important multiblock copolymers is a perpetual task. Dual-initiating Lewis pair (LP) catalysts for living/controlled polymerizations of methacrylates have been demonstrated to be an effective approach towards all-acrylic multiblock copolymers. However, the utilization of metal-based Lewis acids (LAs) and complicated Lewis bases (LBs) that typically require laborious syntheses are essential. This article reports the first organic dual-initiating frustrated Lewis pair (FLP) catalysts consisting of tris(2,4-difluorophenyl)borane [B(2,4-F2C6H3)3] LA and commercially available 1,2-bis[(di-tert-butylphosphino)methyl]benzene (DPtBu2) LB, which can mediate efficient and living polymerizations of various (bio)acrylates. Accordingly, all-acrylic multiblock copolymers (up to hepta blocks) with well-defined structures can be readily prepared via sequential monomer addition, thus successfully establishing DPtBu2/B(2,4-F2C6H3)3 FLP as a green/sustainable and user-friendly polymerization catalyst for the convenient synthesis of metal-free all-acrylic multiblock copolymers.
Introduction
Lewis pair polymerization (LPP) is a newly emerging polymerization methodology which is catalyzed by a frustrated Lewis pair (FLP) or a classical Lewis adduct (CLA) that can dissociate into an FLP in the presence of a suitable solvent or monomer.1–5 Inspired by FLP chemistry for cooperative activation of small molecules,6,7 Chen and coworkers reported the first LPP in 2010 through uncovering highly efficient polymerization of methyl methacrylate (MMA) and γ-methyl-α-methylene-γ-butyrolactone (MMBL) by a Lewis pair (LP) catalyst consisting of Al(C6F5)3 Lewis acid (LA) and PtBu3 or N-heterocyclic carbene Lewis base (LB).8Subsequently, the contributions from the groups of Chen,8–15 Zhang,16–27 Lu,28–30 Xu,31–35 Hong,36–41 Zhu,42,43 Rieger,44,45 Takasu,46,47 and Matsuoka48–51etc. have demonstrated LPP to be a very powerful methodology which not only enables the efficient and/or living polymerizations of various polar vinyl monomers [e.g. methacrylates,8,16,17,28 acrylates,39,47,48,50–52 acrylamides,20,49 vinylpyridine,10,34 and dialkyl vinylphosphonate44,45], but also achieves chemoselective polymerizations of polar divinyl monomers,21,29–31 regioselective polymerizations of (E,E)-alkyl sorbates,19 stereoselective polymerizations of methacrylates,35,36 and topology-controlled polymerizations (e.g. cyclic polymers).13,14,23,46,47 Moreover, in response to the ever-growing demand for sustainable polymers, the scope of polar vinyl monomers has also been extended to biomass-derived vinyl monomers, such as lignin-based methacrylates,22,25 methyl crotonate,15 (E,E)-alkyl sorbates,19,46 methylene butyrolactones,8,9,40,41 and cellulose-derived β-angelica lactone.37,38 The successes achieved in the field of LPPs are closely related to the unique ability of both unquenched LB and LA sites of LP catalysts to cooperatively activate monomers as well as to stabilize the propagating active species to suppress chain transfer and termination side reactions.1–5
On the other hand, the development of powerful catalysis or methodologies for the synthesis of multiblock copolymers is a perpetual fundamental task because these copolymers can lead to the formation of highly ordered materials with unique functions and advanced properties (e.g. thermoplastic elastomers (TPEs), nanotechnology etc.).53–55 Recently, the development of dual-initiating LP catalysts for controlled/living polymerizations has been demonstrated to be an efficient approach towards all-acrylic multiblock copolymers. In 2021, Zhang and co-workers developed a new FLP catalyst comprising a sterically encumbered methylaluminum bis(2,6-di-tert-butyl-4-methylphenoxide) [MeAl(BHT)2] LA and bifunctional organophosphorus superbase (μHex[P(NIiPr)Ph]2, Scheme 1, top), which established the first dual-initiating and living LPP for the successful preparation of triblock TPEs containing soft midblock poly(2-ethyoxyethyl methacrylate) and hard end block PMMBL.24 Subsequently, through the integration of the unique compounded sequence control (CSC) strategy of LPP and a dual-initiating FLP catalyst consisting of iBu2Al(BHT) LA and pyridinylidenaminophosphine (μOct[P(mMPy)Ph2]2, Scheme 1, top), one-step and one-pot block copolymerizations from lignin-based syringyl methacrylate and n-butyl acrylate (nBA) monomer mixtures to triblock TPEs25 as well as from MMA, MMBL, nBA, and t-butyl acrylate (tBA) monomer mixtures to sequence-controlled multiblock copolymers (up to hepta blocks)26 have been achieved by Zhang et al. In 2023, up to undecablock all-acrylic copolymers can be synthesized with a three-step monomer mixture feeding by Zhu, Zhang, and coworkers via the development of a new dual-initiating CLA catalyst based on MeAl(BHT)2 LA and bis-phosphonium ylide LB (Scheme 1, top).43 Very recently, Zhang's group designed novel tetraphenylethylene-linked N-heterocyclic olefins (NHOs) as dual- and tetra-functional LBs (Scheme 1, top). By combining with iBuAl(BHT)2 LA, the resultant LPs can yield sequence-controlled linear and four-arm star triblock TPEs via one-step and one-pot block copolymerizations.27
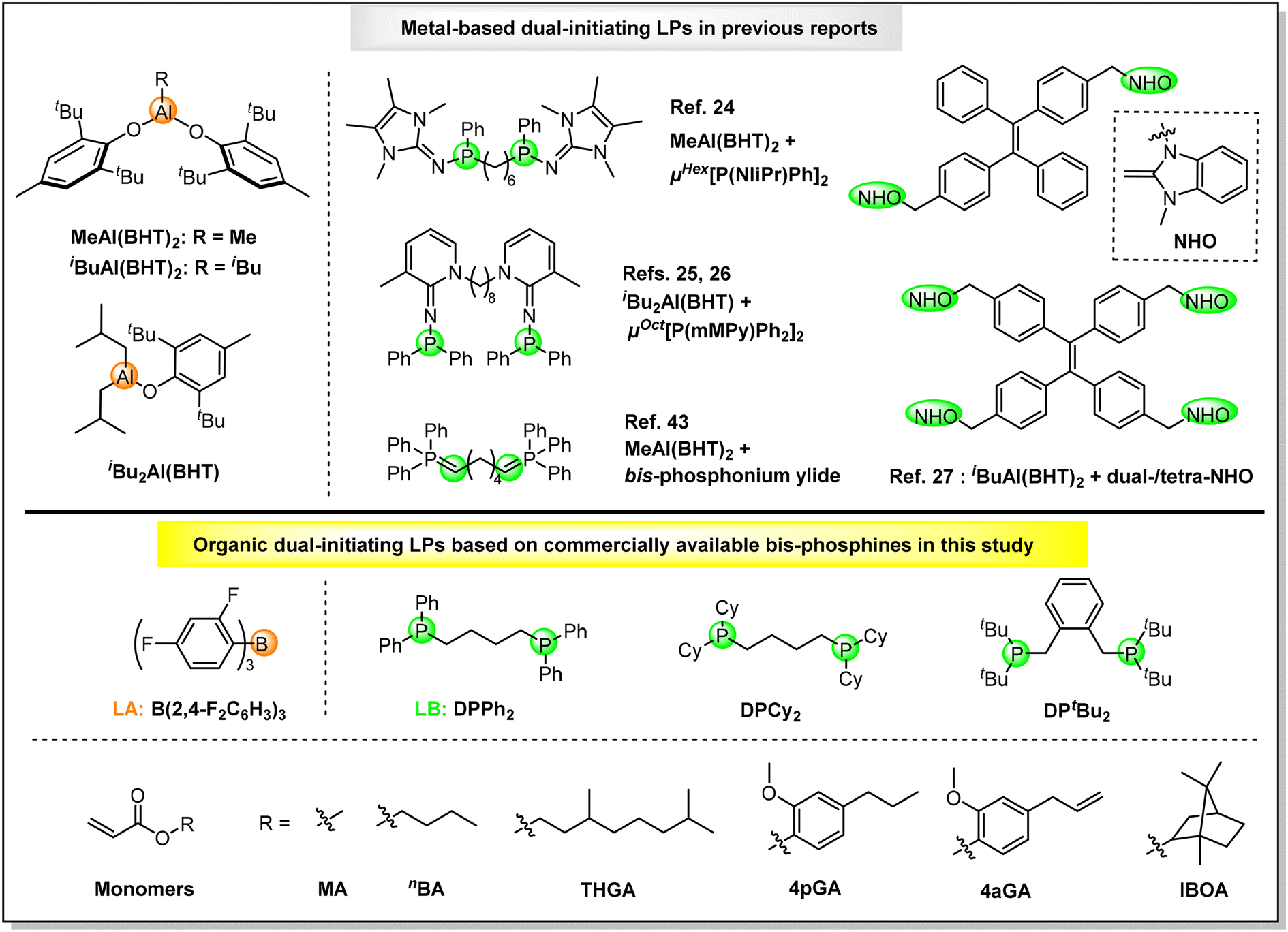 | ||
| Scheme 1 Structures of metal-based dual-initiating LPs in previous reports (top) and organic dual-initiating LPs and monomers investigated in this study (bottom). | ||
As revealed by above overview, although several dual-initiating LP catalysts have been developed for the synthesis of all-acrylic multiblock copolymers, air/moisture sensitive metal-based LAs and complicated LBs that typically require laborious syntheses are essential.56–58 Compared with metal-based catalysts, organic catalysts are, in general, relatively non-toxic, environmentally benign, air/moisture stable and readily available, which have been grown into a preferred method when metal-free products are of primary concern (e.g. biomedical and microelectronic applications).52–54 To make dual-initiating LPs truly green/sustainable and user-friendly polymerization catalysts for multiblock copolymers, it is highly desirable to develop metal-free LAs and commercially available dual-initiating LBs. Recently, we have developed a thermally-stable and air/moisture-tolerant organic LP catalyst consisting of tris(2,4-difluorophenyl)borane LA [B(2,4-F2C6H3)3, Scheme 1, bottom] and triisobutylphosphine (PiBu3) LB, which enabled the first efficient and living LPPs of acrylates at ambient and high temperatures (25–80 °C).39 Herein, commercially available bisphosphines, including 1,4-bis(diphenylphosphino)butane (DPPh2), 1,4-bis(dicyclohexylphosphino)butane (DPCy2), and 1,2-bis[(di-tert-butylphosphino)methyl]benzene (DPtBu2) (Scheme 1, bottom), have been utilized as LBs in this study. In combination with B(2,4-F2C6H3)3 LA, organic dual-initiating LPs have been constructed for the first time. Investigations into the polymerization behaviors reveal that B(2,4-F2C6H3)3/DPtBu2 is a new robust dual-initiating organic FLP which can mediate efficient and living LPPs of (bio)acrylates, completely chemoselective LPP of divinyl acrylic monomer, and also enable the convenient synthesis of metal-free all-acrylic multiblock copolymers.
Experimental
Materials
All syntheses and manipulations of air- and moisture-sensitive materials were carried out in flamed Schlenk-type glassware on a dual-manifold Schlenk line or in an argon-filled glovebox. HPLC-grade organic solvents were first sparged extensively with nitrogen during the filling of 20 L solvent reservoirs and then dried by passage through activated alumina [for dichloromethane (DCM) and tetrahydrofuran (THF)] followed by passage through Q-5 supported copper catalyst stainless steel columns [for toluene (TOL) and hexane]. Methyl acrylate (MA) and n-butyl acrylate (nBA) were purchased from TCI Chemical Co., which were dried over CaH2 overnight followed by vacuum distillation. Further purification of acrylate monomers involved titration with tri(n-octyl)aluminum (Sigma-Aldrich Co.) to a yellow end point, followed by distillation under reduced pressure. Isobornyl acrylate (IBOA) was purchased from TCI Chemical Co., while tetrahydrogeraniol acrylate (THGA), 4-propylguaiacol acrylate (4pGA), and 4-allylguaiacol acrylate (4aGA) were synthesized according to a literature procedure.59 The obtained four biomass-derived acrylate monomers were dissolved in toluene, dried over CaH2 overnight, and then filtered in an argon-filled glovebox. The concentrations of these monomers were determined by quantitative 1H NMR spectra. All purified monomers were stored in brown bottles inside a glovebox freezer at −30 °C for further use. 1,4-Bis(diphenylphosphino)butane (DPPh2), 1,2-bis[(di-tert-butylphosphino)methyl]benzene (DPtBu2), and 1,4-bis(dicyclohexylphosphino)butane (DPCy2) were purchased from Sigma-Aldrich, which were dissolved in toluene and dried over CaH2 overnight and then filtered in an argon-filled glovebox. The solvent was removed under vacuum, and the residue was dried for 12 h under vacuum. The dried bis-phosphines were stored in brown bottles inside a glovebox freezer at −30 °C for further use. B(2,4-F2C6H3)3 was synthesized according to a literature procedure.39Stoichiometric reaction of B(2,4-F2C6H3)3 with bis-phosphines
In a typical procedure, a Teflon-valve-sealed J. Young-type NMR tube was charged with B(2,4-F2C6H3)3 (35.0 mg, 0.1 mmol) in the glovebox. A solution of bis-phosphine (0.05 mmol) in 0.50 mL of benzene-d6 or toluene-d8 was added to the above NMR tube via a pipette at RT, and the resulting mixture was then taken out of the glovebox for NMR analysis.General polymerization procedures
Polymerizations were performed in 20 mL glass reactors inside the glovebox at room temperature (RT). A predetermined amount of LA was first dissolved in a predetermined amount of monomer and solvent inside a glovebox. The mixture was stirred for 10 minutes, and then the polymerization was started by rapid addition of a predetermined amount of Lewis base LB in the solvent via a gastight syringe to the above mixture under vigorous stirring. After the measured time interval, a 0.2 mL aliquot was taken from the reaction mixture via a syringe and quickly quenched into a 2 mL septum-sealed vial containing 0.4 mL of “wet” CDCl3 mixed with 250 ppm of benzoic acid; the quenched aliquots were later analyzed by 1H NMR spectroscopy to obtain the percent monomer conversion data. After a desired period of time, the polymerization was immediately quenched by the addition of 5 mL of methanol acidified with HCl (5%). The quenched mixture was precipitated into a 100 mL mixed solvent of water and methanol (Vwater/Vmethanol = 1/5), filtered and dried in a vacuum oven at 80 °C to a constant weight.Polymer characterization
NMR spectra were recorded on a Varian 400 MHz spectrometer. Chemical shifts for 1H NMR and 13C NMR spectra were referenced to internal solvent resonances and were reported as parts per million relative to SiMe4. Polymer number average molecular weight (Mn) and molecular weight distributions (Đ = Mw/Mn) were measured by gel permeation chromatography (GPC) analyses carried out at 40 °C and a flow rate of 1.0 mL min−1, with DMF containing a small amount of LiBr (20 mmol L−1) or THF as the eluent on a Waters E2695 GPC instrument equipped with a Wyatt OPTILAB® T-Rex refractive-index detector, a Wyatt DAWN HELEOS II multi (18)-angle light scattering detector, one Agilent Plgel 5 μm guard column, and two Agilent Plgel 5 μm Mixed-C columns (Polymer Laboratories; linear range of MW = 200–2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000). Glass-transition temperatures (Tgs) were measured by differential scanning calorimetry (DSC) on a DSC Q2500, TA Instruments. All Tg values were obtained from the second scan after the thermal history was removed from the first scan. The first heating rate was 10 °C min−1, while the cooling rate was 10 °C min−1 and the second heating rate was 5 °C min−1. The onset decomposition temperatures (Td, defined at 5% weight loss) of the polymers were measured by thermal gravimetric analysis (TGA) on a TGA550, TA Instruments. Polymer samples were heated from ambient temperatures to 700 °C at a rate of 20 °C min−1.
000000). Glass-transition temperatures (Tgs) were measured by differential scanning calorimetry (DSC) on a DSC Q2500, TA Instruments. All Tg values were obtained from the second scan after the thermal history was removed from the first scan. The first heating rate was 10 °C min−1, while the cooling rate was 10 °C min−1 and the second heating rate was 5 °C min−1. The onset decomposition temperatures (Td, defined at 5% weight loss) of the polymers were measured by thermal gravimetric analysis (TGA) on a TGA550, TA Instruments. Polymer samples were heated from ambient temperatures to 700 °C at a rate of 20 °C min−1.
The isolated low molecular weight samples were analyzed by matrix assisted laser desorption/ionization time-of-flight mass spectroscopy (MALDI–TOF MS); the experiment was performed on a Bruker Reflex II mass spectrometer operated in positive ion, reflector mode using a Nd:YAG laser at 355 nm and 25 kV accelerating voltage. A thin layer of a 1% NaI solution was first deposited on the target plate, followed by 0.6 μl of both the sample and matrix (dithranol, 20 mg mL−1 in methanol, 10% AAC). External calibration was done using a peptide calibration mixture (4 to 6 peptides) on a spot adjacent to the sample.
Results and discussion
Investigations into the interaction between bis-phosphine and B(2,4-F2C6H3)3
Considering that the interaction between LA and LB has a significant influence on polymerization activity and initiation efficiency (I*), the interactions between B(2,4-F2C6H3)3 and three bis-phosphines were first investigated in our initial study. As shown in Fig. 1A–D and S1, S2 and S4,† the stoichiometric NMR reactions clearly reveal that DPPh2 and DPCy2 form CLAs with B(2,4-F2C6H3)3, as evidenced by the notable shifts of chemical resonances after the reactions. For example, the chemical resonances of free bis-phosphines (DPPh2: −16.42 ppm, DPCy2: −6.10 ppm) in 31P NMR spectra shift to the downfield [DPPh2 → B(2,4-F2C6H3)3: 8.07 ppm, DPCy2 → B(2,4-F2C6H3)3: 3.12 ppm] after the reactions while the shift of the chemical resonances of the B(2,4-F2C6H3)3 moiety in CLAs can also be observed in 19F NMR spectra [B(2,4-F2C6H3)3: −94.45, −102.66 ppm; DPPh2 → B(2,4-F2C6H3)3: −89.92, −112.50 ppm; DPCy2 → B(2,4-F2C6H3)3: −88.93, −112.73 ppm]. In sharp contrast, in the stoichiometric NMR reaction of B(2,4-F2C6H3)3 and DPtBu2 (Figs. 1E, F and S3, S4†), there were no detectable changes of chemical resonances in 1H, 11B, 31P, and 19F NMR spectra before and after mixing B(2,4-F2C6H3)3 and DPtBu2, indicating that DPtBu2/B(2,4-F2C6H3)3 is a non-interacting FLP.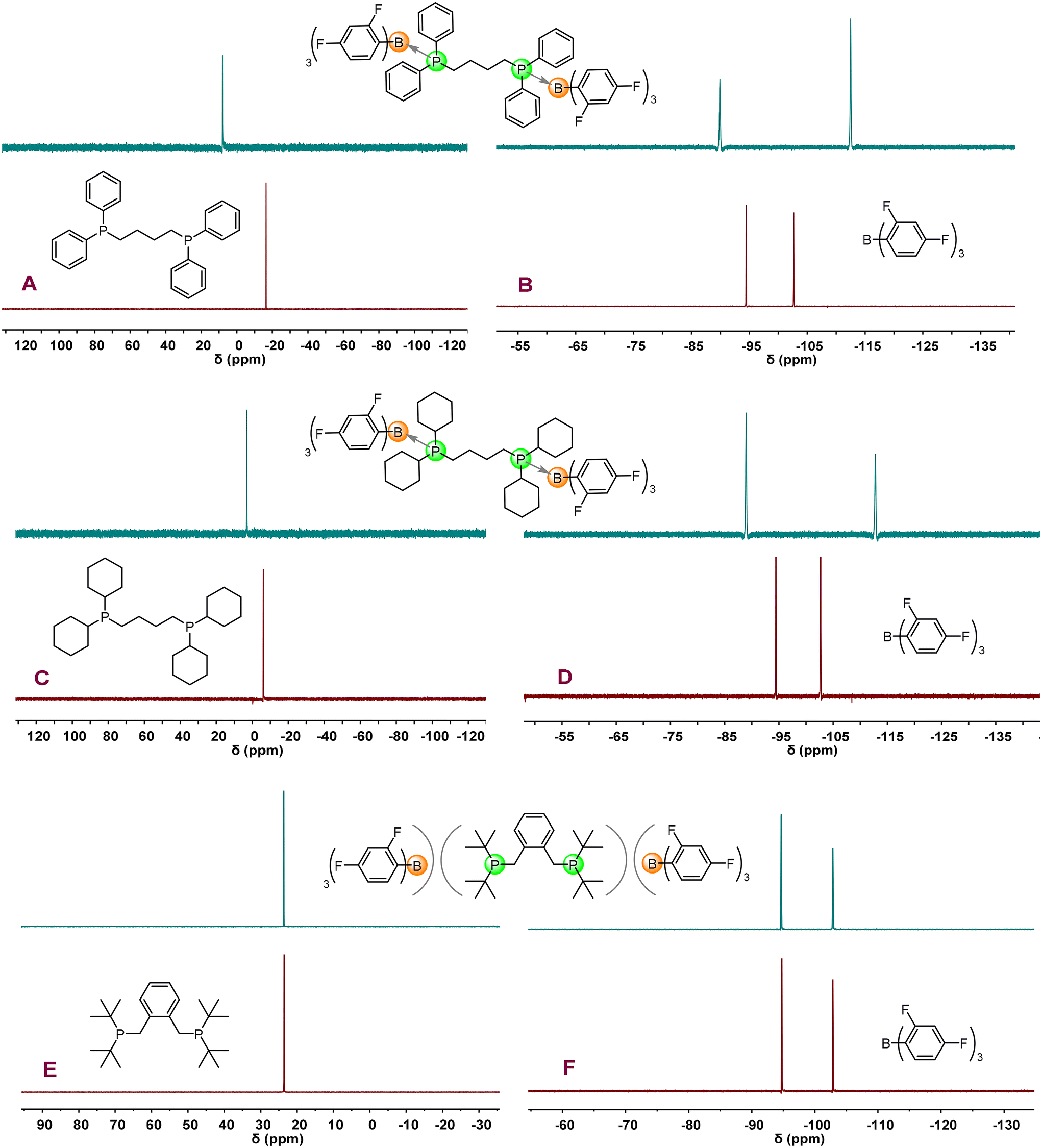 | ||
| Fig. 1 31P NMR spectra of DPPh2 → B(2,4-F2C6H3)3 CLA, DPCy2 → B(2,4-F2C6H3)3 CLA, DPtBu2/B(2,4-F2C6H3)3 FLP, and their corresponding bis-phosphines (A, C and E), as well as the 19F NMR spectra of the above LPs and B(2,4-F2C6H3)3 (B, D and F) (A–D: toluene-d8; E and F: benzene-d6). | ||
Polymerization characteristics of methyl acrylate catalyzed by dual-initiating organic LPs
The above three dual-initiating organic LPs were first employed as the catalysts for the polymerizations of methyl acrylate (MA). With a [MA]0:[LA]0:[LB]0 ratio of 200:4:1, both DPPh2 → B(2,4-F2C6H3)3 and DPCy2 → B(2,4-F2C6H3)3 CLAs showed a high degree of control over the polymerizations at room temperature (RT), which afforded PMAs with extremely narrow dispersities (Đ = 1.01) and gave rise to high I* values of 85.0–86.4% (Table 1, runs 1 and 2). It is interesting that these dual-initiating CLAs can achieve high I*s in this study, since LPPs by CLA catalysts in previous reports8–10,28 generally led to low to moderate I*s because of the irreversible formation of CLAs that is difficult to release free LBs and LAs for initiating polymerization. Such results reveal that both DPPh2 → B(2,4-F2C6H3)3 and DPCy2 → B(2,4-F2C6H3)3 CLAs can readily dissociate into FLPs in the presence of a monomer to provide sufficient orthogonal reactivity for fast and efficient initiation. The relatively weak Lewis acidity of B(2,4-F2C6H3)3,39 in comparison with commonly used LAs [e.g. B(C6F5)3, Al(C6F5)3, and MeAl(BHT)2], should be responsible for weak LA–LB interactions and thereby facile dissociation. However, in contrast to the corresponding mono-initiating PPh3 → B(2,4-F2C6H3)3 and PCy3 → B(2,4-F2C6H3)3 CLAs (90–240 min, Conv. >99%, Table 1, runs 3 and 4), these dual-initiating CLAs exhibited much lower polymerization activity, which required 1080–1440 min to consume all MA monomers. A slow propagation process is speculated to be the main reason, which might be caused by the increase of steric hindrance generated from the other propagation site that hampers the activation and conjugate addition. When switching from DPPh2 and DPCy2 with a flexible –(CH2)4– linkage between two phosphine moieties to rigid aryl-linked DPtBu2, the activity in DPtBu2/B(2,4-F2C6H3)3 FLP-mediated polymerization was significantly enhanced where quantitative monomer conversion was accomplished within 120 min (Table 1, run 5). More importantly, a high degree of control over the polymerization was well maintained, which afforded a PMA with a very low Đ of 1.02 at a high I* value of 90.2%.
| Run | LB | M. | [M.]0:[LA]0:[LB]0 |
[M.]0 (mol L−1) | Time (min) | Conv.b (%) | M n (kg mol−1) | Đ | I*d (%) |
|---|---|---|---|---|---|---|---|---|---|
| a The polymerizations were carried out by premixing LA and the monomer (M.) first followed by adding LB unless otherwise noted: LB = 0.01 mmol, Temp. = 25 °C, toluene as the solvent. b Monomer conversion measured by 1H NMR spectroscopy. c Number-average molecular weight (Mn) and molecular weight distribution (Đ = Mw/Mn) determined by GPC at 40 °C in DMF coupled with a multi(18)-angle light scattering detector and a refractive index detector. d Initiation efficiency (I*)% = Mn(calcd)/Mn(exptl) × 100, where Mn(calcd) = [MW(M.)] × ([M.]0/[LB]0) × conversion% + MW(LB) + MW(end groups). e Initiation efficiency (I*)% = Mn(calcd)/Mn(exptl) × 100, where Mn(calcd) = [MW(M.)] × ([M.]0/[LB]0) × conversion% + MW(end groups). f THF as the eluent in GPC measurement. | |||||||||
| 1 | DPPh2 | MA | 200:4:1 |
1.0 | 1440 | >99 | 20.4 | 1.01 | 86.4 |
| 2 | DPCy2 | MA | 200:4:1 |
1.0 | 1080 | >99 | 20.8 | 1.01 | 85.0 |
| 3e | PPh3 | MA | 100:2:1 |
1.0 | 240 | >99 | 40.7 | 1.06 | 21.7 |
| 4e | PCy3 | MA | 100:2:1 |
1.0 | 90 | >99 | 9.30 | 1.02 | 95.5 |
| 5 | DPtBu2 | MA | 200:4:1 |
1.0 | 120 | >99 | 19.5 | 1.02 | 90.2 |
| 6 | DPtBu2 | MA | 400:4:1 |
1.5 | 210 | >99 | 35.1 | 1.03 | 99.7 |
| 7 | DPtBu2 | MA | 800:4:1 |
2.0 | 300 | >99 | 71.4 | 1.02 | 97.0 |
| 8 | DPtBu2 | MA | 1200:4:1 |
2.5 | 420 | >99 | 101.7 | 1.01 | 101.0 |
| 9 | DPtBu2 | n BA | 200:4:1 |
1.0 | 30 | >99 | 30.2 | 1.09 | 86.1 |
| 10 | DPtBu2 | n BA | 1200:4:1 |
2.5 | 120 | >99 | 180.3 | 1.01 | 85.4 |
| 11 | DPtBu2 | 4pGA | 200:4:1 |
1.0 | 40 | >99 | 46.9 | 1.07 | 96.1 |
| 12 | DPtBu2 | 4pGA | 800:4:1 |
1.5 | 150 | >99 | 217.7 | 1.17 | 81.0 |
| 13 | DPtBu2 | 4aGA | 200:4:1 |
1.0 | 40 | >99 | 44.8 | 1.08 | 98.2 |
| 14 | DPtBu2 | 4aGA | 800:4:1 |
1.5 | 150 | >99 | 216.2 | 1.15 | 80.1 |
| 15 | DPtBu2 | THGA | 200:4:1 |
1.0 | 50 | >99 | 43.4f | 1.08 | 98.5 |
| 16 | DPtBu2 | THGA | 800:4:1 |
1.5 | 240 | >99 | 175.1f | 1.21 | 97.1 |
| 17 | DPtBu2 | IBOA | 200:4:1 |
1.0 | 120 | >99 | 52.9f | 1.12 | 80.0 |
| 18 | DPtBu2 | IBOA | 800:4:1 |
1.5 | 300 | >99 | 170.0f | 1.13 | 98.1 |
To verify the living nature of DPtBu2/B(2,4-F2C6H3)3 FLP-mediated polymerization, a series of experiments were performed as follows. First, as shown in Fig. 2A, when gradually increasing the ratio of [MA]0:[B(2,4-F2C6H3)3]0:[DPtBu2]0 from 200:4:1 to 1200:4:1, a linear increase of the Mns of PMAs can be observed with predictable Mn values (I* = 90.2–101.0%) and very low dispersities (Đ = 1.01–1.03) (Table 1, runs 5–8). Noteworthily, even though a large excess of 1200 equivalents of MA were employed, quantitative monomer conversion can also be accomplished within a desirable polymerization time (420 min, Table 1, run 8), producing a PMA with a high Mn of 101.7 kg mol−1 and an extremely low Đ of 1.01. Second, keeping [MA]0:[B(2,4-F2C6H3)3]0:[DPtBu2]0 at a fixed ratio of 400:4:1, the Mn of PMA increases linearly with an increase of monomer conversion, while extremely low Đ values (1.03–1.08) are maintained (Fig. 2B). GPC curves of these samples gradually shift to the higher molecular-weight (MW) region with the increase of the monomer-to-initiator ratio and monomer conversion (Fig. 2C and D) and all show very narrow and unimodal distributions. Third, after the first batch of MA (200 equiv.) was completely converted into PMA, the resulting unquenched system was held at RT for 1 h and then was able to repolymerize immediately once another batch of MA (200 equiv.) was added. The resultant sample showed a narrow and unimodal GPC curve without a detectable low-MW tail (Fig. 2E), and its Mn (34.4 kg mol−1, Đ = 1.02, I* = 101.1%) was very close to that of the PMA (Mn = 35.1 kg mol−1, Đ = 1.03, I* = 99.7%) produced by the polymerization at a [MA]0/[B(2,4-F2C6H3)3]0/[DPtBu2]0 ratio of 400:4:1, clearly demonstrating the successful chain extension. Based on the above three key pieces of evidence, the living characteristic of the DPtBu2/B(2,4-F2C6H3)3 FLP-mediated polymerization of MA has been unequivocally established.
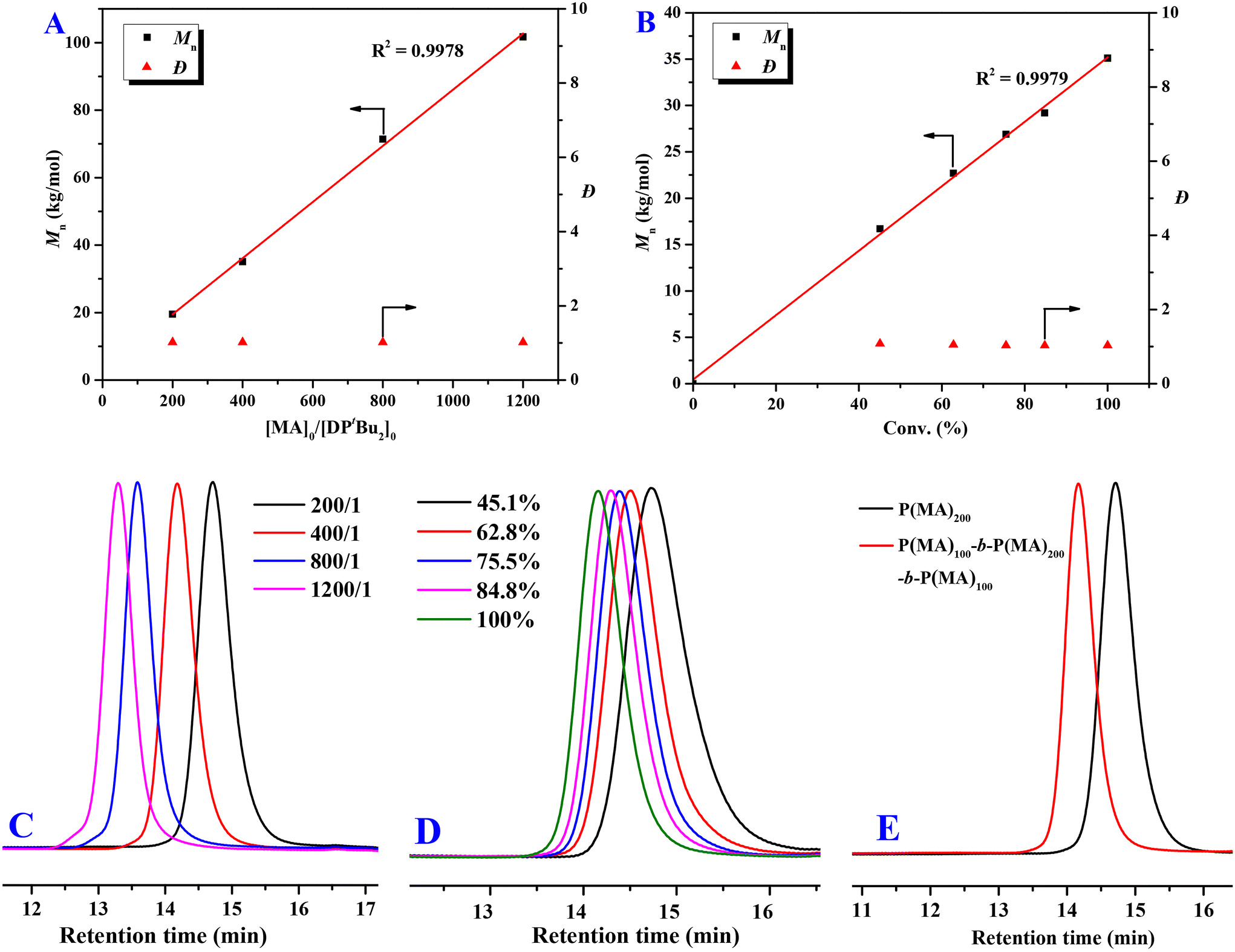 | ||
| Fig. 2 (A) Plots of Mn and Đ for the PMA vs. the [MA]0:[DPtBu2]0 ratio (Table 1, runs 5–8); (B) plots of Mn and Đ for the PMA vs. monomer conversion (%) (Conv. % = 45.1, 62.8, 75.5, 84.8, 100%; Mn = 16.7, 22.7, 26.8, 29.2, 35.1 kg mol−1; Đ = 1.08, 1.05, 1.03, 1.04, 1.03); (C) GPC curves of PMAs produced at different [MA]0:[DPtBu2]0 ratios (Table 1, runs 5–8); (D) GPC curves of PMAs produced at different monomer conversions ([MA]0:[DPtBu2]0 = 400:1); (E) GPC curves of PMA samples obtained by the chain extension experiment: Mn = 19.5 kg mol−1, Đ = 1.02 (first batch) and Mn = 34.4 kg mol−1, Đ = 1.02 (second batch after 1 h). | ||
Matrix-assisted laser desorption/ionization time-of-flight mass spectroscopy (MALDI-TOF MS) analysis of low-MW PMA was also performed. As shown in Fig. 3, only one set of molecular ion peaks was observed, corresponding to one type of polymer chains starting with the –(tBu)PCH2ArCH2P(tBu)– moiety formed by dual initiation from both phosphine sites of DPtBu2 LB and the subsequent elimination of two tert-butyl groups from –(tBu)2P+CH2ArCH2P+(tBu)2– during the MALDI-TOF MS measurement. After the quenching process, two H-terminated chain ends were generated. This result further confirms that DPtBu2/B(2,4-F2C6H3)3 FLP-mediated polymerization proceeds in a living manner without any backbiting termination and chain transfer events and, more importantly, clearly demonstrates an exclusive dual initiation process that facilitates the synthesis of multi-block copolymers.
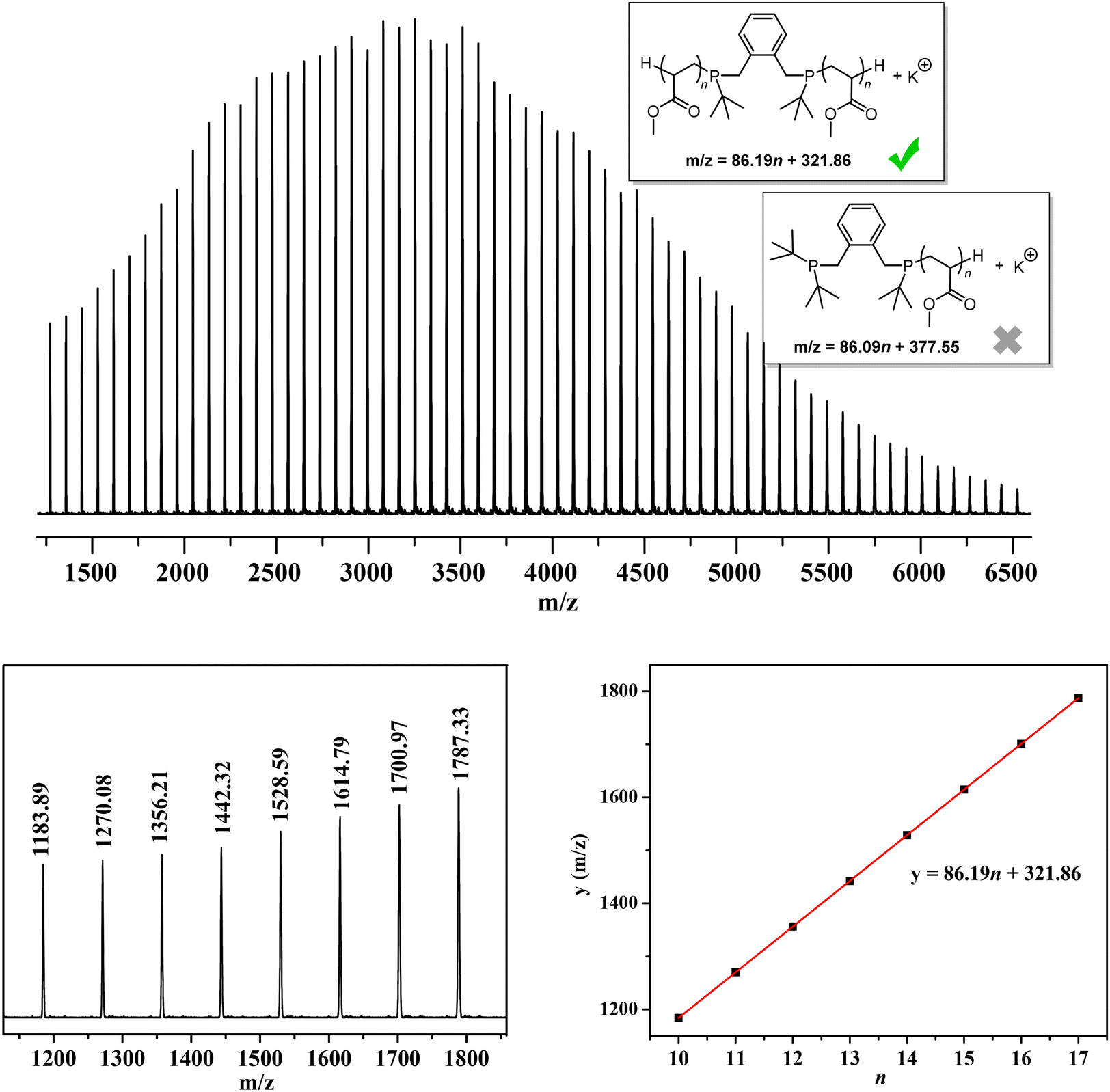 | ||
| Fig. 3 MALDI-TOF MS spectrum of low-MW PMAs and plots of m/z values (y) vs. the number of MS repeat units (n) for molecular ion peaks. | ||
DPtBu2/B(2,4-F2C6H3)3 FLP-mediated living polymerizations of (bio)acrylates to all-acrylic multiblock copolymers
Having established the DPtBu2/B(2,4-F2C6H3)3 FLP as the best dual-initiating catalyst for efficient and living polymerization of MA, the scope of monomers was further explored. Again, the polymerization of n-butyl acrylate (nBA) by the DPtBu2/B(2,4-F2C6H3)3 FLP still proceeded in a living manner, affording PnBAs with very low Đs of 1.01–1.09 at high I* values of 85.4–86.1% (Table 1, runs 9 and 10). It is worth noting that this FLP is especially effective for nBA polymerization, where 1200 equivalents of nBA were rapidly polymerized within 120 min (Table 1, run 10), which was 3 times faster than MA polymerization under the same conditions (Table 1, run 8).Besides, bio-based acrylates were also utilized as the monomers, including tetrahydrogeraniol acrylate (THGA), 4-propylguaiacol acrylate (4pGA), 4-allylguaiacol acrylate (4aGA) and isobornyl acrylate (IBOA) (Scheme 1). Among them, IBOA is commercially available, while THGA, 4pGA, and 4aGA can be synthesized via a one-step reaction of acryloyl chloride and biomass-derived alcohols with high yields (71.2–82.1%) (Fig. S5–S10†). Presumably due to the bulkier side groups of these monomers, their polymerizations by the DPtBu2/B(2,4-F2C6H3)3 FLP were relatively slower than nBA (Table 1, runs 11–18). At a low monomer loading (200:4:1), living polymerizations of these monomers can also be achieved. High to quantitative I* values (80.0–98.5%) and narrow unimodal distributions (Đ = 1.07–1.21) were well-maintained. However, in the cases of high-monomer-loading LPPs (800:4:1), a significant increase of the viscosity of the polymerization mixture was observed at the late stage of polymerization. Therefore, decreasing the monomer concentration from 2.0 to 1.5 M was necessary to relieve unfavorable monomer diffusion at the late stage of polymerization. Accordingly, upon the employment of dilute conditions, controlled polymerizations at a high monomer loading can also be achieved, producing high-Mn bio-based polyacrylates (170.0–217.7 kg mol−1) with relatively narrow dispersities (Đs: 1.13–1.21). The structures of these bio-based polyacrylates were fully characterized by NMR spectra (Fig. S11–S18†). Of particular note is the completely chemoselective polymerization of 4aGA rendered by the DPtBu2/B(2,4-F2C6H3)3 FLP, as evidenced by the complete disappearance of acrylic double bonds at 5.94–6.02, 6.31–6.39, and 6.59–6.63 ppm after the polymerization while the allyl groups at 5.92 and 5.08–5.14 ppm remain unreacted (Fig. 4). As a result, soluble P4aGA without any cross-linking was obtained with pendant double bonds on the side groups, which thus offers great potential in the synthesis of functional or advanced acrylic polymers.
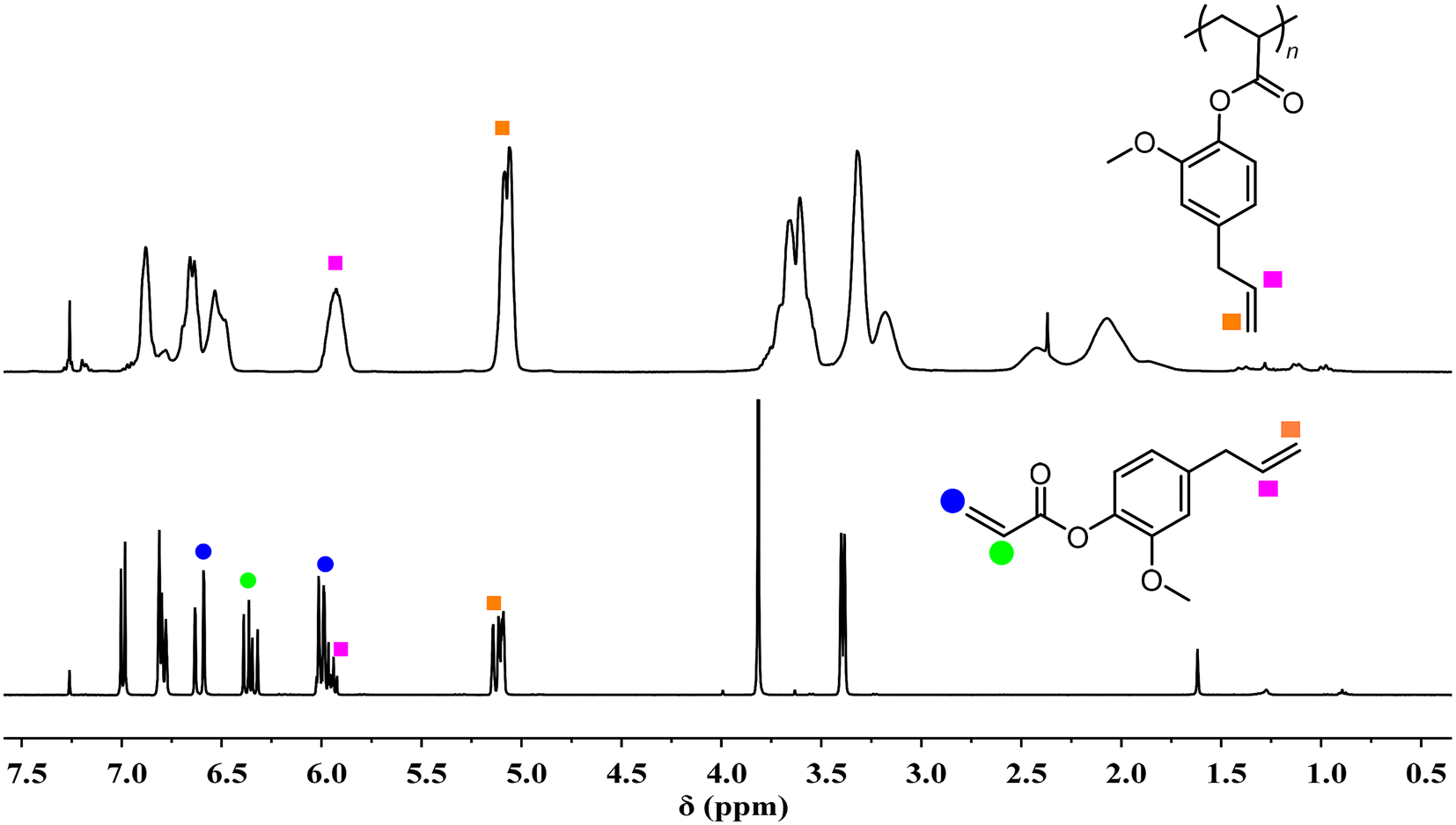 | ||
| Fig. 4 1H NMR spectra (CDCl3, RT): (top) P4aGA produced by the DPtBu2/B(2,4-F2C6H3)3 FLP (Table 1, run 13); (bottom) 4aGA monomer. | ||
The unique living and dual-initiating nature of organic DPtBu2/B(2,4-F2C6H3)3 FLP-mediated polymerizations of acrylates thereby enabled the convenient synthesis of metal-free multiblock copolymers via sequential monomer addition. Accordingly, besides PnBA-b-PMA-b-PnBA and bio-based PIBOA-b-PTHGA-b-PIBOA triblock copolymers, P4pGA-b-PnBA-b-PMA-b-PnBA-b-P4pGA pentablock and P4aGA-b-P4pGA-b-PnBA-b-PMA-b-PnBA-b-P4pGA-b-P4aGA heptablock copolymers can also be readily prepared using three and four steps of comonomer additions, respectively. As shown in Fig. 5, even though the unquenched polymerization mixture was held for 30 min before the addition of the next batch of comonomers, GPC curves of the resultant samples maintained very narrow and unimodal distributions without any detectable low-MW tails and gradually shifted to higher MW regions with the increase of block numbers, confirming the livingness/stability of the propagating species that ensures the successful block copolymerizations. It is worth pointing out that there was essentially no effect of comonomer addition order on the polymerization outcome. For example, identical MWs and distributions can be achieved in the block copolymerization of nBA and MA with different monomer addition orders (PnBA-b-PMA-b-PnBA: Mn = 40.5 kg mol−1, Đ = 1.03, Fig. 5A; PMA-b-PnBA-b-PMA: Mn = 38.4 kg mol−1, Đ = 1.09, Fig. S19†).
 | ||
| Fig. 5 (A) GPC curves of the PMA homopolymer (Mn = 19.5 kg mol−1, Đ = 1.02), the PnBA-b-PMA-b-PnBA triblock (Mn = 40.5 kg mol−1, Đ = 1.03), the P4pGA-b-PnBA-b-PMA-b-PnBA-b-P4pGA pentablock (Mn = 102.3 kg mol−1, Đ = 1.05), and P4aGA-b-P4pGA-b-PnBA-b-PMA-b-PnBA-b-P4pGA-b-P4aGA (Mn = 158.5 kg mol−1, Đ = 1.07) heptablock copolymers; (B) GPC curves of the PTHGA homopolymer (Mn = 43.4 kg mol−1, Đ = 1.08) and the PIBOA-b-PTHGA-b-PIBOA triblock copolymer (Mn = 78.4 kg mol−1, Đ = 1.18). | ||
The thermal properties of these bio-based acrylic homopolymers and all-acrylic multiblock copolymers were examined by differential scanning calorimetry (DSC) and thermal gravimetric analysis (TGA). The second heating scans of DSC curves of bio-based acrylic homopolymers (Fig. 6A) show glass transition temperatures (Tgs) that vary over a large range: PTHGA with flexible side groups exhibits a low Tg of −60.0 °C, while P4pGA (58.6 °C), P4aGA (58.1 °C), and PIBOA (99.1 °C) with rigid side groups possess high Tgs. In the case of multiblock copolymers (Fig. 6B), multi-Tgs corresponding to different blocks can be observed, which demonstrates the microphase separation between these blocks and thus indicates the potential applications of these multiblock copolymers in TPEs and self-assembling nanomaterials. For example, the P4pGA-b-PnBA-b-PMA-b-PnBA-b-P4pGA pentablock shows Tg values of −42.0, 20.0, and 56.6 °C, corresponding to PnBA, PMA, and P4pGA blocks, respectively.
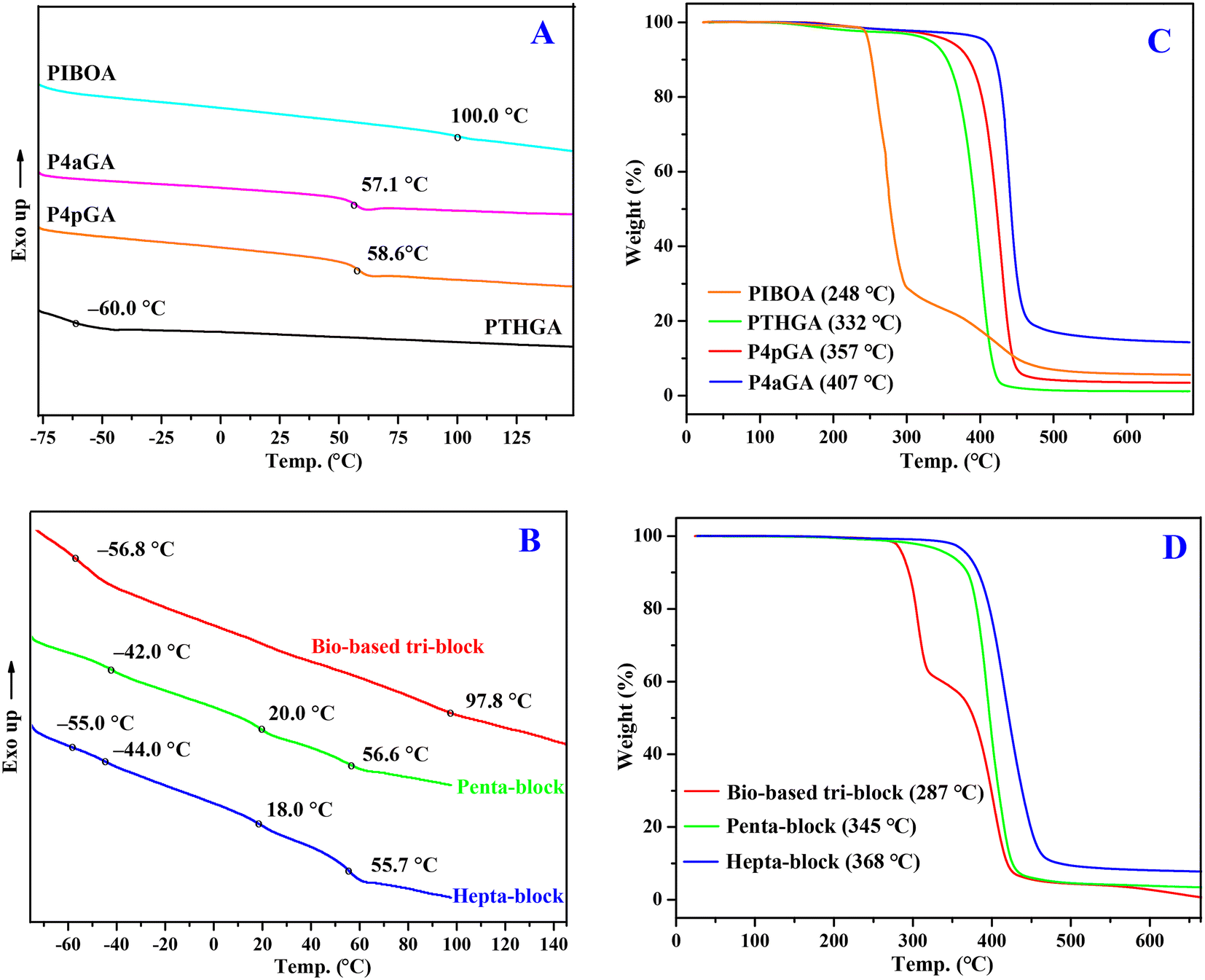 | ||
| Fig. 6 Overlays of the second heating scan DSC curves (A) and TGA curves (C) of PTHGA, P4pGA, P4aGA, and PIBOA homopolymers as well as overlays of the second heating scan DSC curves (B) and TGA curves (D) of the PIBOA-b-THGA-b-PIBOA triblock, the P4pGA-b-PnBA-b-PMA-b-PnBA-b-P4pGA pentablock, and P4aGA-b-P4pGA-b-PnBA-b-PMA-b-PnBA-b-P4pGA-b-P4aGA heptablock copolymers. | ||
As shown in Fig. 6C, TGA analysis reveals that bio-based PTHGA, P4pGA, and P4aGA homopolymers exhibit high thermal stability by showing a sharp, one-step degradation profile with a high onset degradation temperature (Td, defined by the temperatures of 5% weight loss) ranging from 332 to 407 °C. It should be pointed out that noticeable carbonaceous material remained after the TGA measurement of P4aGA (ca. 15% mass), which should be attributed to the formation of a cross-linked network structure during the heating caused by the allyl group. In contrast, the thermal stability of PIBOA is relatively low with a Td of 248 °C, presumably due to the good leaving ability of the bulky side group. Moreover, a two-step degradation profile is observed for PIBOA, including the leaving of side groups and subsequently thermal degradation of backbones. In the case of multiblock copolymers (Fig. 6D), their Td values are somewhere between high-Td and low-Td blocks. Taking the bio-based PIBOA-b-PTHGA-b-PIBOA triblock copolymer as an example, its Td value (287 °C) is higher than the corresponding PIBOA homopolymer (248 °C) but lower than the PTHGA homopolymer (332 °C). Except for the bio-based triblock copolymer that exhibits a two-step degradation profile, both penta- and heptablock copolymers show a one-step degradation profile, indicating a similar thermal degradation behavior between blocks.
Conclusions
In summary, organic dual-initiating LP catalysts consisting of B(2,4-F2C6H3)3 LA and three commercially available bisphosphine LBs have been reported for the first time in this study. All these three LP catalysts exhibit high control over the polymerizations of MA but show different polymerization activities: DPPh2 → B(2,4-F2C6H3)3 and DPCy2 → B(2,4-F2C6H3)3 CLAs based on bisphosphines with flexible –(CH2)4– linkages have relatively low polymerization activity, featured with fast initiation but slow propagation, while the rigid aryl-linked DPtBu2/B(2,4-F2C6H3)3 FLP can mediate efficient polymerizations with significantly enhanced activity. The living characteristic of DPtBu2/B(2,4-F2C6H3)3-mediated LPPs of MA has been unequivocally established by the linear increase of polymer Mnvs. monomer conversion and the monomer-to-initiator ratio with predictable polymer Mns (up to 101.7 kg mol−1), extremely low Đs (1.01–1.03), and high to near quantitative I*s (90.2–101.0%) as well as successful chain extension. Besides commonly used MA and nBA, the (chemoselective) polymerizations of bio-based acrylates (THGA, 4pGA, 4aGA and IBOA) by the DPtBu2/B(2,4-F2C6H3)3 FLP also proceed in a living manner. Accordingly, multiblock copolymers (up to hepta blocks) with well-defined structures can be readily prepared via sequential monomer addition. The notable features of the DPtBu2/B(2,4-F2C6H3)3 FLP, including the utilization of organic LA and commercially available LB as well as its living and dual-initiating nature, thus successfully establish it as a green/sustainable and user-friendly polymerization catalyst for the convenient synthesis of metal-free all-acrylic multiblock copolymers. The existence of microphase separation as evidenced by multi-Tgs indicates the potential applications of these multiblock copolymers in TPEs and self-assembling nanomaterials, which are currently in progress.Data availability
The data supporting this article can be found in the tables and figures of the manuscript and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (2021YFA1501700), the National Natural Science Foundation of China (Grant No. 22471286), the Science and Technology Commission of Shanghai Municipality (Grant No. 22ZR1481900 and 23XD1424600), the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDB0610000), and the CAS Project for Young Scientists in Basic Research (Grant No. YSBR-094).References
- M. Hong, J. Chen and E. Y.-X. Chen, Chem. Rev., 2018, 118, 10551–10616 CrossRef CAS.
- M. Hong, Frustrated Lewis Pairs, Molecular Catalysis 2, Springer Cham, Switzerland, 2021, pp. 283–317 Search PubMed.
- M. L. McGraw and E. Y.-X. Chen, Macromolecules, 2020, 53, 6102–6122 CrossRef CAS.
- W. Zhao, J. He and Y. Zhang, Sci. Bull., 2019, 64, 1830–1840 CrossRef CAS.
- W. Zhao, J. He and Y. Zhang, Sci. China: Chem., 2023, 66, 2256–2266 CrossRef CAS.
- G. C. Welch, R. R. S. Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 CrossRef CAS.
- D. W. Stephan, Science, 2016, 354, aaf7229 CrossRef PubMed.
- Y. Zhang, G. M. Miyake and E. Y.-X. Chen, Angew. Chem., Int. Ed., 2010, 49, 10158–10162 CrossRef CAS PubMed.
- T. Xu and E. Y.-X. Chen, J. Am. Chem. Soc., 2014, 136, 1774–1777 CrossRef CAS.
- J. He, Y. Zhang, L. Falivene, L. Caporaso, L. Cavallo and E. Y.-X. Chen, Macromolecules, 2014, 47, 7765–7774 CrossRef CAS.
- M. L. McGraw, R. W. Clarke and E. Y.-X. Chen, J. Am. Chem. Soc., 2020, 142, 5969–5973 CrossRef.
- L. T. Reilly, M. L. McGraw, F. D. Eckstrom, R. W. Clarke, K. A. Franklin, E. R. Chokkapu, L. Cavallo, L. Falivene and E. Y.-X. Chen, J. Am. Chem. Soc., 2022, 144, 23572–22358 CrossRef CAS.
- M. L. McGraw, R. W. Clarke and E. Y.-X. Chen, J. Am. Chem. Soc., 2021, 143, 3318–3322 CrossRef CAS PubMed.
- R. W. Clarke, M. R. Caputo, L. P. Fonseca, M. L. McGraw, L. T. Reilly, K. A. Franklin, A. J. Müller and E. Y.-X. Chen, J. Am. Chem. Soc., 2024, 146, 4930–4941 CrossRef CAS PubMed.
- M. McGraw and E. Y.-X. Chen, ACS Catal., 2018, 8, 9877–9887 CrossRef CAS.
- Q. Wang, W. Zhao, S. Zhang, J. He, Y. Zhang and E. Y.-X. Chen, ACS Catal., 2018, 8, 3571–3578 CrossRef CAS.
- Y. Bai, J. He and Y. Zhang, Angew. Chem., Int. Ed., 2018, 57, 17230–17234 CrossRef CAS PubMed.
- Y. Bai, H. Wang, J. He and Y. Zhang, Angew. Chem., Int. Ed., 2020, 59, 11613–11619 CrossRef CAS PubMed.
- W. Zhao, F. Li, C. Li, J. He, Y. Zhang and C. Chen, Angew. Chem., Int. Ed., 2021, 60, 24306–24311 CrossRef CAS PubMed.
- H. Wang, Q. Wang, J. He and Y. Zhang, Polym. Chem., 2019, 10, 3597–3603 RSC.
- W. Zhao, Q. Wang, J. He and Y. Zhang, Polym. Chem., 2019, 10, 4328–4335 RSC.
- Y. Wan, W. Zhao, H. Zhao, M. Zhou, J. He and Y. Zhang, Macromolecules, 2023, 56, 7763–7770 CrossRef CAS.
- Y. Song, J. He, Y. Zhang, R. A. Gilsdorf and E. Y.-X. Chen, Nat. Chem., 2023, 15, 366–376 CrossRef CAS.
- Y. Bai, H. Wang, J. He, Y. Zhang and E. Y.-X. Chen, Nat. Commun., 2021, 12, 4874 CrossRef CAS.
- Y. Wan, J. He, Y. Zhang and E. Y.-X. Chen, Angew. Chem., Int. Ed., 2022, 61, e202114946 CrossRef CAS.
- Y. Wan, J. He and Y. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202218248 CrossRef CAS PubMed.
- C. Li, W. Zhao, J. He and Y. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202401265 CrossRef CAS PubMed.
- Y.-B. Jia, Y.-B. Wang, W.-M. Ren, T. Xu, J. Wang and X.-B. Lu, Macromolecules, 2014, 47, 1966–1972 CrossRef CAS.
- Y.-B. Jia, W.-M. Ren, S.-J. Liu, T. Xu, Y.-B. Wang and X.-B. Lu, ACS Macro Lett., 2014, 3, 896–899 CrossRef CAS.
- P. Zhang, H. Zhou and X.-B. Lu, Macromolecules, 2019, 52, 4520–4525 CrossRef CAS.
- P. Xu, L. Wu, L. Dong and X. Xu, Molecules, 2018, 23, 360 CrossRef PubMed.
- P. Xu and X. Xu, ACS Catal., 2018, 8, 198–202 CrossRef CAS.
- P. Xu, Y. Yao and X. Xu, Chem. – Eur. J., 2017, 23, 1263–1267 CrossRef CAS.
- Y. Su, Y. Zhao, H. Zhang, Y. Luo and X. Xu, Macromolecules, 2021, 54, 7724–7731 CrossRef CAS.
- Y. Zhou, S. Jiang and X. Xu, Chin. J. Chem., 2021, 39, 149–156 CrossRef CAS.
- X. Wang and M. Hong, Macromolecules, 2020, 53, 4659–4669 CrossRef CAS.
- X.-J. Wang and M. Hong, Angew. Chem., Int. Ed., 2020, 59, 2664–2668 CrossRef CAS PubMed.
- X. Wang, Y. Zhang and M. Hong, Polym. Chem., 2023, 14, 3286–3293 RSC.
- Z.-H. Zhang, X. Wang, X.-J. Wang, Y. Li and M. Hong, Macromolecules, 2021, 54, 8495–8502 CrossRef CAS.
- Z.-H. Zhang, X. Wang, B. Weng, Y. Zhang, G. Zhang and M. Hong, ACS Polym. Au, 2022, 2, 266–274 CrossRef CAS.
- Z. Zhang, X. Wu and M. Hong, Acta Polym. Sin., 2022, 53, 1150–1160 CAS.
- Z.-K. Chen, W.-C. Zhao, Y.-L. Zhao, C.-L. Liu, L.-Y. Jiang, Y.-T. Zhang and H.-P. Zhu, Chin. J. Polym. Sci., 2024, 42, 159–167 CrossRef CAS.
- C. Liu, W. Zhao, L. Li, Z. Chen, Y. Zhao, Y. Zhang and H. Zhu, Macromolecules, 2024, 57, 110–121 CrossRef.
- M. Weger, R. K. Grötsch, M. G. Knaus, M. M. Giuman, D. C. Mayer, P. J. Altmann, E. Mossou, B. Dittrich, A. Pöthig and B. Rieger, Angew. Chem., Int. Ed., 2019, 58, 9797–9801 CrossRef CAS PubMed.
- M. G. M. Knaus, M. M. Giuman, A. Pöthig and B. Rieger, J. Am. Chem. Soc., 2016, 138, 7776–7781 CrossRef CAS.
- Y. Hosoi, A. Takasu, S. Matsuoka and M. Hayashi, J. Am. Chem. Soc., 2017, 139, 15005–15012 CrossRef CAS.
- H. Yato, K. Oto, A. Takasu and M. Higuchi, RSC Adv., 2023, 13, 13616–13623 RSC.
- R. Akita, M. Horibe, K. Yamamoto and S. Matsuoka, Macromolecules, 2024, 57, 4926–4936 CrossRef CAS.
- R. Akita and S. Matsuoka, ACS Macro Lett., 2024, 13, 1272–1278 CrossRef CAS PubMed.
- Y. Naganawa, K. Mori, S. Matsuoka and M. Suzuki, Polym. J., 2024, 56, 145–151 CrossRef CAS.
- Z. Wang, X. Zhang, H. Liang, M. Xian and X. Wang, Polym. Chem., 2020, 11, 5526–5533 RSC.
- J. Chen and E. Y.-X. Chen, Isr. J. Chem., 2015, 55, 216–225 CrossRef CAS.
- F. S. Bates, M. A. Hillmyer, T. P. Lodge, C. M. Bates, K. T. Delaney and G. H. Fredrickson, Science, 2012, 336, 434–440 CrossRef CAS.
- N. G. Engelis, A. Anastasaki, G. Nurumbetov, N. P. Truong, V. Nikolaou, A. Shegiwal, M. R. Whittaker, T. P. Davis and D. M. Haddleton, Nat. Chem., 2017, 9, 171–178 CrossRef CAS PubMed.
- A. Anastasaki, B. Oschmann, J. Willenbacher, A. Melker, M. H. C. van Son, N. P. Truong, M. W. Schulze, E. H. Discekici, A. J. McGrath, T. P. Davis, C. M. Bates and C. J. Hawker, Angew. Chem., Int. Ed., 2017, 56, 14483–14487 CrossRef CAS PubMed.
- N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813–5840 CrossRef CAS.
- S. Naumann and A. P. Dove, Polym. Chem., 2015, 6, 3185–3200 RSC.
- W. N. Ottou, H. Sardon, D. Mecerreyes, J. Vignolle and D. Taton, Prog. Polym. Sci., 2016, 56, 64–115 CrossRef CAS.
- S. Noppalit, A. Simula, N. Ballard, X. Callies, J. M. Asua and L. Billon, Biomacromolecules, 2019, 20, 2241–2251 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py01067a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |