
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tailoring reactive handles on the surface of nanoparticles for covalent conjugation of biomolecules†
Francesca
Mazzotta
a,
Sharafudheen Pottanam
Chali
a,
Ingo
Lieberwirth
 a,
Calum T. J.
Ferguson
ab and
Katharina
Landfester
*a
a,
Calum T. J.
Ferguson
ab and
Katharina
Landfester
*a
aMax Planck Institute for Polymer Research, Department of Physical Chemistry of Polymers, Ackermannweg 10, 55128 Mainz, Germany
bSchool of Chemistry, University of Birmingham, Edgbaston, BirminghamB15 2TT, UK. E-mail: ferguson@mpip-mainz.mpg.de
First published on 27th November 2024
Abstract
Surface modification of nanoparticles involves numerous types of active molecules such as DNA, antibodies, enzymes, or carbohydrates. These modifications usually require reactive handles like amines, carboxylic acids, azides, etc. on the nanoparticles. In this work, utilizing poly-benzyl methacrylate based nanoparticles as a model nanoparticle system, the number of functional groups was tuned with functional comonomers, amino ethyl methacrylate for the amino groups or methyl methacrylate for the carboxylic groups. Herein a systematic study is presented where the functional groups in the nanoparticles are differentiated between total, visible and accessible functional groups. The concentration of each type of functional group is compared using various methods. Polymers synthesized using free radical polymerization were analyzed using 1H-NMR spectroscopy to obtain the total number of functional groups. Via a miniemulsion–solvent evaporation technique, these polymers were used to synthesize the nanoparticles. Zeta potential, pH value and particle charge detection measurements were used to determine the number of visible functional groups. The number of accessible functional groups was quantified by conjugating small dyes and fluorescence measurements were directly executed on the system under investigation, hence eliminating errors associated with indirect measurements and detecting very low concentrations (e.g. 80 nM). Lastly, human serum albumin was conjugated to investigate the effect of a bulky molecule on the accessibility of these reactive handles.
Introduction
Surface modification of nanoparticles (NPs) is of great interest in order to create novel and functional nanomaterials. Their application extends from the biomedical field1,2 to materials science.3 NPs have shown their potential in nanomedicine owing to their ability to encapsulate therapeutic molecules protecting them from binding to the plasma protein, suppressing the clearance rate, increasing biodistribution, enhancing tissue uptake and so on.4 For the targeted delivery of cargoes, the NP surface is often functionalized with molecules such as antibodies,5–8 enzymes,9–12 carbohydrates,13,14 peptides,15,16 or other small molecules.17 For this purpose, precise control of the active molecules on the NP surface is required. These ligands are primarily introduced by using covalent conjugation exploiting reactive handles such as amines,2 carboxylic acids,18 azides,19 maleimides,20 or alkynes,5 which can be directly incorporated on the NP surface during the synthesis process. Various conjugation strategies can be applied to couple molecules of interest. A cheap and commonly implemented reaction is amide bond formation by using EDC/NHS (N-ethyl-N′-(3-dimethylaminopropyl)carbodiimide/N-hydroxysuccinimide) chemistry to either conjugate amines to carboxylic functionalized surfaces or vice versa, making this a versatile tool for the coupling of many molecules, for example, proteins.2,21 Therefore, the evaluation of FGs on the NP surface is of utmost importance for their successful application.There are various strategies to study and characterize both surface modified and unmodified NPs. The number of FGs on the unmodified NPs can be distinguished between the total (e.g. in the whole particle), the visible (e.g. those that affect the physiochemical properties of the NPs) and the accessible (e.g. the reactive handles that can be used for subsequent conjugation) amounts. The total number of FGs can be determined in a straightforward way via NMR spectroscopy.22,23 A reliable strategy to determine the visible groups is to use electrochemical tools, such as potentiometric titrations, which can use the smallest probes available (protons or hydroxide ions) to quantify all the surface charges associated with the FGs. However, these methods require tedious work and long equilibration times.22 Optical, photometric and fluorimetric techniques, such as the toluidine blue assay, can also be used.22–24 While these methods report on the visible groups, analyzing reactive handles is based on indirect measurements. As such, they require multiple steps which increase the error in the final results. Quantification can be achieved by conjugating a dye and measuring the unbound fraction. Alternatively, Moser et al.25 reported cleavable probes for amine and carboxylic acid quantification.
For protein modified NPs, the quantification of surface conjugation can be done by using standard protein assays such as the BCA,26 Bradford and Pierce5 assays. However, their result can often be subject to errors associated with interfering substances.27 Additionally, depending on the type of protein, its characteristics can be used for quantification purposes: for enzymes, their enzymatic activity,28 while for antibodies, their interaction with antigens.29 Instead, for carbohydrate-conjugated NPs, the primary quantification strategy is based on thermogravimetric analysis.14,30 Lastly, the development of super resolution microscopy has generated quantification methods such as quantitative PAINT (points accumulation for imaging in nanoscale topography), which are capable of analyzing single NPs, thus incorporating particle distribution information.6,12,28,30
While the particles can be characterized utilizing the above-mentioned methods, to the best of our knowledge, there are no reports regarding two very important aspects that affect the final product: (1) the bulkiness of the active molecules and (2) the number of reactive handles on the NP surface. Herein, we report a systematic study on the characterization of NPs from the raw material to the final product by tuning and investigating the number of total, visible and accessible FGs (amine and carboxylic groups) on polymeric NPs. For this purpose, statistical polymers were synthesized with precise functional monomer composition and NMR spectroscopy was used to quantify the total number of FGs. The polymers were then used to synthesize NPs via the miniemulsion–solvent evaporation technique. Next, the visible FGs were investigated using a quick potentiometric tool: particle charge detection (PCD). The accessible FGs were determined by applying the commonly used EDC/NHS reaction to couple a small fluorescent molecule. The conjugated dye is directly quantified on the system under investigation by drying the polymeric particles and subsequently dissolving them in an organic solvent. In this way, common errors associated with indirect measurements (e.g. supernatant studies) were avoided. Finally, we conjugated a model biomacromolecule, human serum albumin, to analyze the effect of a bulky protein as an active molecule.
Results and discussion
Polymeric NPs were synthesized to obtain reactive surfaces containing amino (NH2) or carboxylic (COOH) functional groups (FGs). The synthesis of the starting material was tuned to vary the FG content. For this purpose, via free radical polymerization, statistical linear copolymers composed of benzyl methacrylate (BzMA) as the primary monomer (99 or 90 wt%) and amino ethyl methacrylate (AEMA) or methyl methacrylate (MAA) as functional comonomers (1 or 10 wt%) were synthesized.To improve monomer conversion, the more commonly used styrene monomer was substituted with BzMA due to styrene's higher activation energy.31 P(BzMA-co-AEMA) (Fig. 1) was synthesized by solubilizing AEMA in MeOH and subsequently adding an acetone solution containing BzMA and the initiator. The mixture of MeOH and acetone as solvents was necessary to efficiently combine the hydrophilic solid form of AEMA and the hydrophobic BzMA. P(BzMA-co-MAA) (Fig. 1) was more easily obtained by the polymerization of BzMA and MAA in DMSO along with the initiator. P(BzMA-co-AEMA) with 1 wt% and 10 wt% functional monomers was used to synthesize, respectively, the NH2-NPs-1% and NH2-NPs-10% systems. Similarly, the COOH-NPs-1% and COOH-NPs-10% systems were obtained using the P(BzMA-co-MAA) polymer. Given the synthesis protocol implemented in this work, the final polymer composition, investigated via1H-NMR spectroscopy, can be related to the total amount of FGs in the NPs. Specifically, the molar ratio between the BzMA monomer and the functional monomer (AEMA or MAA) in the final and purified polymer was used to determine the molar concentration (details in the ESI†).
 | ||
| Fig. 1 Polymer synthesis scheme for P(BzMA-co-AEMA) and P(BzMA-co-MAA) and NP formation. The polymer's final composition was calculated based on mol% while the nomenclature is associated with the initial wt% of the monomers added. | ||
To obtain the NPs, the polymers were dissolved in ethyl acetate and emulsified with an aqueous solution containing the non-ionic surfactant Lutensol AT50, which is necessary to stabilize the NPs at increased ionic strength. After the miniemulsion and solvent evaporation process (Fig. S7†), the NPs were purified and characterized via DLS and TEM. All NPs showed a narrow size distribution (Fig. 2A and E) and a spherical morphology (Fig. 2B, C, F and G) with a diameter (Dh) of approximately 200 nm. However, for the NH2-NPs-10% system, additional polymer (450 mg vs. 150 mg) and reduced surfactant (0.1 vs. 0.4 wt%) (Table 1) were required to increase the particle size to 200 nm. Such necessity could be associated with the intrinsic nature of the polymer: the amino group in P(BzMA-co-AEMA) is charged making the polymer present strong hydrophilicity that allows the charged FGs to reach the NP surface.32 In turn, the NPs present electrostatic stabilization in addition to the steric one; consequentially this could reduce the particle size. An initial detection of the visible FGs was done with ξ-potential and pH measurements of a 0.1 wt% particle sample (Table 1) which confirm the presence of the FGs and their different concentrations. When comparing NPs-1% and NPs-10%, for NH2-NPs there is an increase in ξ-potential from 32.5 mV to 38.8 mV, and a decrease in the pH value, from 4.7 to 4.3. Conversely, for COOH-NPs, there is a decrease in both the ξ-potential and pH value which vary from −15.4 mV and pH 6.1 to −26.1 mV and pH 5.7.
 | ||
| Fig. 2 Chromatogram of (A) NH2-NPs and (E) COOH-NPs. Representative TEM images of NH2-NPs-1% (B) and NH2-NPs-10% (C) and COOH-NPs-1% (F) and COOH-NPs-10% (G). Quantification of FGs via NMR spectroscopy for the total information, PCD (particle charge detection) for the visible amount and fluorescence methods for accessibility studies for NH2-NPs (D) and COOH-NPs (H). The black bars represent the error on each measurement. The detailed values are reported in Table S3.† | ||
| NPs | FGs % | Polymer (mg) | Lutensol AT50 (wt%) | D h (nm) | PDI | ξ (mV) | pH |
|---|---|---|---|---|---|---|---|
| a Hydrodynamic radius measured via DLS. | |||||||
| NH2 | 10 | 450 | 0.1 | 277 | 0.099 | 38.8 | 4.3 |
| 1 | 150 | 0.4 | 199 | 0.111 | 32.5 | 4.7 | |
| COOH | 10 | 150 | 0.1 | 207 | 0.034 | −26.1 | 5.7 |
| 1 | 150 | 0.1 | 206 | 0.031 | −15.4 | 6.1 | |
The quantification of visible groups at the particle's surface was performed using particle charge detection (PCD). To ensure the polymers were in the required charged state, the solution's pH value was adjusted to pH 2.5 for NH2-NPs and pH 8 for COOH-NPs. The systems were then titrated with the oppositely charged polyelectrolyte: sodium polyethylene sulfonate (PES-Na) for NH2-NPs and poly(diallyldimethylammonium chloride) (PDADMAC) for COOH-NPs. The results (Fig. 2D and H) indicate a difference in the numbers calculated via NMR spectroscopy; for all systems, the visible groups are less than the total ones, which is intrinsically due to the diversity of the two methods: while PCD analyzes only the NP surface, NMR studies the whole system thus being unable to differentiate between the FGs trapped in the NP core and those present on the surface. Moreover, the values of NH2-NPs, with an 182 and 24 μM amino concentration, are closer to the total amount compared to COOH-NPs, due to the protonated state of P(BzMA-co-AEMA). The strong hydrophilicity of the charged amino groups in the polymer introduces a driving force for the FGs to reach the NP surface during the synthesis, thus allowing this system to approach the maximum values. Contrarily, the carboxylic groups in the P(BzMA-co-MAA) polymer are not or not entirely in their charged form, which reduces the group's hydrophilicity trapping the FGs within the particle's core, thus detecting lower concentrations: 53 and 24 μM. The latter leads to an additional observation: COOH-NPs-10% and COOH-NPs-1% lack the 10-fold decrease which, contrarily, is observed for NH2-NPs. It should be taken into consideration that the PCD technique assumes accessibility of all FGs33 and the possibility of simplex formation between the polyelectrolyte and all the FGs on the NP surface.34 However, some might be trapped in sterically less available conformations, hampering proper PCD interpretation. Such trapping is representative of NPs derived from statistical copolymers that form a “shell-like” surface layer.32 This shell, by lacking a single FG-layer conformation, traps some of the FGs, hampering proper PCD interpretation. The results, therefore, could be associated with the structural difference of the functional monomers: AEMA has a short linker while MAA does not. Such chemical structure can cause the carboxylic groups to be closer to the NP surface generating constraints and limited access, thus hampering detection.
Next, the focus was shifted to analyzing how many of these groups are accessible for chemical reactions with small ligands. Here we used model molecules (∼400 g mol−1), fluorescent probes, and either amino modified bodipy (BDP FL amine) or free acid CY3, for the covalent conjugation on the reactive handles via EDC/NHS chemistry. Common strategies to quantify the labeled particles include either flow cytometry or analysis of the supernatant solution obtained during the purifying steps. The first methodology cannot be implemented as the NP size is below the required value, and thus cannot be detected by the instrument. Contrarily, the supernatant assay could be applied; however, the method introduces errors associated with indirect measurement approaches such as repetitive pipetting which could cause imprecision in the collected volume necessary for the final analysis. Thus, in this work, the conjugated dye was quantified directly on the system under investigation. To do so, the purified fluorescently labeled particles were dried and dissolved in an organic solvent to record the fluorescence spectrum. The latter was then compared to a calibration curve of the dye in the same organic solvent and the amount of dye conjugated to the polymer (polymer–dye system) was calculated from this curve. The solubilization of the particle–dye can allow the polymer–dye, of which the NPs are composed, (i) to have increased mobility compared to the particle–dye, (ii) to reduce the environmental and solvent effect (iii) and to remove NP scattering, all of which hamper the final fluorescent signal. To use this method, the fluorescence spectra obtained need to be compared to that of the free dye to ensure a lack of spectral shape difference (Fig. S10 and S13†), which could indicate that the dye's properties have changed and thus the fluorescence is no longer linked to the original calibration curve. In this regard, it was important to use the BDP FL amine dye compared to the cheaper and more commonly used fluorescein due to large spectral differences between the fluorescein and the polymer–fluorescein system (Fig. S11†).35–37
The results, reported in Fig. 3, are obtained by conjugating a 0.1 wt% solution of NPs with varying concentrations of the dye to produce a titration curve. In all the NP systems, a plateau was reached within the margin of error obtained from the experiments. For NH2-NPs the plateau is more evident due to a strong increase in the bound dye with an increase in the concentration of the dye. Contrarily, for COOH-NPs the curve is noisier and requires increased dye concentrations (a minimum of 150 μM for COOH-NPs vs. a maximum of 110 μM for NH2-NPs) to saturate. Such noise could be associated with the small (less than 0.1 μM) bound concentrations detected in these systems, which would be difficult to measure with a supernatant assay due to the limited difference in the concentration between the added dye (e.g. 200 μM) and the unbound one (e.g. 199.8 μM), a difference that could be masked by human errors (e.g. small pipetting mistakes).
 | ||
| Fig. 3 Fluorescence titration curves obtained via conjugation of CY3 on (A) NH2-NPs-1% and (B) NH2-NPs-10% and BDP on (C) COOH-NPs-1% and (D) COOH-NPs-10%. The plots report triplicate measurements and, in black, the average result used for the quantification. | ||
The final plateaus are employed to quantify the total number of accessible groups, which, as expected, demonstrate an increase when comparing NPs-1% with NPs-10%. The NPs yielded concentrations of 0.007 μM and 0.022 μM for COOH-NPs, whereas for NH2-NPs concentrations of 0.7 μM and 17 μM were observed. The higher concentration for NH2-NPs could be associated with the presence of more visible FGs, as measured with PCD, but could also be indicative of the ability of the carboxylic group on the CY3 dye, which is a small and mobile molecule, to easily interact with the coupling reagents (EDC/NHS) and in turn be activated for amide bond formation. Contrarily, on COOH-NPs the carboxylic groups are constrained to the NP surface which can cause mobility hampering and steric hindrance both of which limit the carboxylic activation thus reducing the final conjugation product.
In the specific case of NH2-NPs, the fluorescamine assay is often implemented to quantify the amino groups on different types of NPs such as silica NPs1,2 liposomes38 or protein nanocapsules.4 This reaction can be of high interest due to its fast execution time; thus, it was tested with NH2-NPs. The method required some adaptation as in this system the fluorescence of the fluorescamine-NPs was much stronger than the corresponding calibration curve (Fig. S15A†). Therefore, for PBzMA based NPs, this assay is associated with errors due to the use of DMSO as the solvent for fluorescamine, which can alter the surface morphology and thus the number of visible groups. Additionally, to quantify the number of functional groups that can be used for further surface modification (accessible groups), there is a need for a versatile reaction, such as the EDC/NHS reaction. Hence, the fluorescamine assay can be used for a quick (30 min) overview of NH2-NPs; however, it does not give information about either the visible or the accessible FGs.
For all described methods, experimental and mathematical errors can arise (details in the ESI†). Specifically, the experimental errors are those associated with the intrinsic nature of the method applied. Such errors affect the experimentally determined value. On the other hand, mathematical errors are those that arise from applying a conversion from the experimental value to a concentration value. For instance, the experimental error will be associated with the fluorescence detection limit in the plate reader and will affect the fluorescence intensity. The mathematical error will be converting the fluorescence intensity into dye concentration based on a calibration curve. Therefore, quantification by performing triplicate measurement is of utmost importance to investigate error effects. In addition to experimental and mathematical errors, quantification differences could also arise from reaction conditions. Such an effect plays a predominant role in accessible FG quantification where variation in temperature, pH buffer, or reaction time can strongly affect the reaction's yield (Fig. S9†).
Lastly, human serum albumin (HSA) was conjugated to analyze the effect of bulky molecules (66 kg mol−1) on the NP coverage. Protein quantification is often performed using the Pierce, Bradford or BCA assay. However, the quantification can be strongly influenced and hampered by interfering substances in solution such as PBS and MES buffers, and thus is incompatible with the detection of NPs (Fig. S12†). Therefore, in this work the previously described dye quantification protocol was adapted for protein detection. HSA was fluorescently labeled with rhodamine B to be treated as a bulky dye molecule and conjugated to COOH-NPs. Unfortunately, the polymer–HSA–rhodamine spectra in DMSO showed an 11 nm red-shift (Fig. S17†) compared to the reference (HSA–rhodamine in DMSO); thus the protein quantification based on the calibration curve was not possible. Nevertheless, the fluorescence intensity can be utilized to qualitatively compare NPs-1% and NPs-10%. For NPs-1%, the fluorescence was approximately 2 times lower than that in NPs-10% (Fig. 4). The value is consistent with the difference in the conjugated dye and in the PCD results. The interesting aspect is that an increase in the probe size (from 425 g mol−1 to 66![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) strongly modifies the steric hindrance contribution to the conjugation; thus, the large probe (e.g. protein) should have less space for interaction compared to the small probe (e.g. dye). Given two NPs with the same morphology and size, as for the systems herein presented, the surface area should be fully covered thus conjugating approximately 4000 proteins (calculations in the ESI†) and obtaining a similar fluorescence intensity. However, based on the dye quantification results, 720 and 220 FGs per particle were measured on NPs-10% and NPs-1%, respectively (calculations in the ESI†). These smaller numbers prevent the NPs from being completely covered by HSA and can explain the detected difference in fluorescence intensity. The results emphasize the importance of quantifying the available reactive handles, whereas the bulkiness of the probe is of secondary importance. Therefore, thoroughly investigating the morphology and characteristics of the unmodified NPs is crucial before conjugating the larger active molecules to them.
000 g mol−1) strongly modifies the steric hindrance contribution to the conjugation; thus, the large probe (e.g. protein) should have less space for interaction compared to the small probe (e.g. dye). Given two NPs with the same morphology and size, as for the systems herein presented, the surface area should be fully covered thus conjugating approximately 4000 proteins (calculations in the ESI†) and obtaining a similar fluorescence intensity. However, based on the dye quantification results, 720 and 220 FGs per particle were measured on NPs-10% and NPs-1%, respectively (calculations in the ESI†). These smaller numbers prevent the NPs from being completely covered by HSA and can explain the detected difference in fluorescence intensity. The results emphasize the importance of quantifying the available reactive handles, whereas the bulkiness of the probe is of secondary importance. Therefore, thoroughly investigating the morphology and characteristics of the unmodified NPs is crucial before conjugating the larger active molecules to them.
 | ||
| Fig. 4 (A) Schematic representation of HSA (PDB ID 1AO6)39 conjugation on COOH-NPs. (B) Fluorescence intensity at the plateau for both NPs-10% and NPs-1%. The black line represents the error derived from triplicate measurements. | ||
Conclusions
A detailed investigation of the total, visible and accessible FGs on the surface of NPs was described in this work. Statistical copolymers with 1% or 10% AEMA or MAA functional monomers were used in the miniemulsion–solvent evaporation method to obtain NPs with NH2 or COOH FGs. While the total number of FGs was analyzed via NMR spectroscopy, the visible FGs were quantified via particle charge detection. To determine the amount of accessible FGs, a small fluorescent dye was conjugated on the NPs and the fluorescence was measured after drying and subsequently dissolving the NPs in an organic solvent. The evaluation was directly performed on the systems to overcome errors associated with indirect quantification studies which could overlook small dye concentrations (e.g. 80 nM). The conjugation was based on EDC/NHS chemistry that can be translated to other reactions (e.g. click chemistry) as well. Additionally, this method can be applied to any aqueous NP (e.g. polymer NP, liposomes, etc.) with different types of FGs, thus allowing proper characterization of both the unmodified and modified NPs. The results showed a subsequent decrease in the amount of FGs from the total to the visible and finally the accessible ones. To investigate the effect of molecule size on the accessibility of reactive handles, HSA was conjugated to the NPs. We observed that with increased bulkiness the quantity of conjugated molecules remained dependent on the number of available FGs. The quantification results presented in this study might be influenced by additional experimental conditions, such as the pH value of the solution or saline conditions. Consequently, there are numerous additional experimental conditions that could be investigated. Nevertheless, the result presented focus on the importance of tuning the number and location of reactive handles.Experimental
Materials
Benzyl methacrylate (BzMA, Sigma-Aldrich, 96%), methyl methacrylate (MAA, Merck, 99%), 2-amino-ethyl methacrylate hydrochloride (AEMA, Sigma-Aldrich, 90%), azobisisobutyronitrile (AIBN), Lutensol AT50 (poly(ethylene glycol)-hexadecyl ether), dimethyl sulfoxide (DMSO), tetrahydrofuran (THF), ethanol (EtOH), methanol (MeOH), acetone, ethyl acetate, dimethylformamide (DMF), N-ethyl-N′-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC–HCl), N-hydroxy sulfosuccinimide (sulfo-NHS), poly(diallyldimethylammonium chloride) (PDADMAC), sodium polyethylene sulfonate (PES-Na), and human serum albumin (HSA, Sigma-Aldrich). Fluorescamine, 6-amino-fluorescein (ANF, Sigma-Aldrich), amino derivative of boron dipyrromethene FL dye (BDP, Lumiprobe), free Cyanine3 carboxylic acid (CY3, Lumiprobe), and rhodamine B isocyanate (Sigma-Aldrich). BzMA and MAA were purified using an aluminum oxide column prior to use.Polymer synthesis
Polymers were synthesized via free radical polymerization. The carboxylic functionalized polymers were prepared by adding MAA (1 or 10 wt%) and BzMA (99 or 90 wt%) in a 15 mL glass vial with DMSO (4 mL) and AIBN (1 wt% of the monomers). The amino functionalized polymers were obtained by mixing AEMA (1 or 10 wt%) in MeOH (1 mL) with BzMA (99 or 90 wt%) in acetone (4 mL) in a 15 mL glass vial with AIBN (1 wt% of the monomers). All samples were degassed with nitrogen flow for 10 min under vigorous stirring and the reactions were activated by heating to 60 °C in an oil bath for 24 h. Subsequently the product was analyzed via1H-NMR spectroscopy and purified via precipitation in MeOH (P(BzMa-co-MAA)) or EtOH (P(BzMa-co-AEMA)), and dried at 40 °C in a vacuum oven; these steps were repeated three times. The final polymer was analyzed via1H-NMR spectroscopy (Fig. S1–S5†) and via GPC (Fig. S2†) using DMF as the eluent and polystyrene as the reference. The polymer yields are around ∼35% (P(BzMa-co-AEMA)) and ∼85% (P(BzMA-co-MAA)).Nanoparticle synthesis and characterization
Carboxylic (COOH-NPs) and amino functionalized (NH2-NPs) NPs were obtained based on a previously reported protocol.40 Briefly, the polymer was dissolved in 3 mL of EA and mixed with 6 mL of 0.1 wt% or 0.4 wt% Lutensol AT50. The solution was stirred with a T25 Ultra-Turrax for 1 min at 5 krpm and ultrasonicated with a Branson Ultrasonics Sonifier for 3 min (pulsed 20 s on, 10 s off) with a ½′ tip at 70% amplitude. The miniemulsion was left to evaporate for 16 h at 40 °C at 1 krpm on a heat plate (Fig. S7†); the obtained NPs were centrifuged for 20 min at 2 krpm and the supernatant was collected. The solid content of the NPs was determined gravimetrically by drying 100 μL of the particle solution. NP size (Dh) was measured by means of dynamic light scattering (DLS) at a fixed angle of 175° using a Malvern Zeta Nanosizer (Malvern Instruments GmbH, Herrenberg, Germany). The same instrument was used to measure the ξ-potential by diluting 5 μL of NPs with 2 mL of a 0.1 M KCl solution. TEM (transmission electron microscopy) samples were prepared by drop casting 5 μL of diluted NP solution onto a carbon coated copper grid, back-blotting with Whatman paper excess solution and lastly depositing a 2 nm layer of carbon. Grids were imaged using an FEI Tecnai (120 kV) transmission electron microscope with a 4k × 4k Gatan CCD camera at a low electron dose.Particle charge detection
10 mL of a 0.1 wt% solution of the NP samples was prepared in MilliQ water and the pH value was adjusted to 2.5 or 8 with HCl or NaCl respectively for NH2-NPs and COOH-NPs. The solution was then introduced into a PCD 02 (Mütek GmbH, Herrsching, Germany) and titrated using a 702 SM Titrino automated titrator (Metrohm AG, Herisau, Switzerland) with 1 mM solution of either PES-Na or PDADMAC respectively for NH2-NPs and COOH-NPs; each sample was analyzed in triplicate. As a result, the volume of the titrant necessary to reach the point of zero charge of the sample was detected (VTitrant) and used to calculate a molar concentration of FGs (CFGs) in solution as follows (eqn (1)):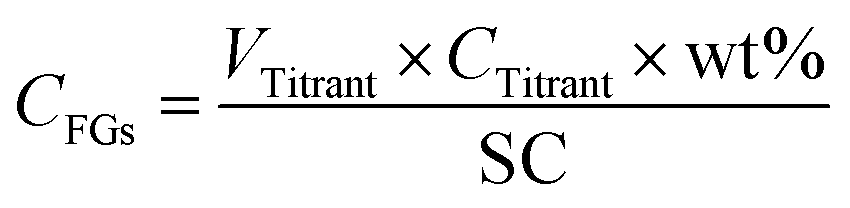 | (1) |
EDC–NHS chemistry on COOH-NPs
COOH-NPs were conjugated with BDP or HSA via a two-step reaction. 0.1 wt% solution of COOH-NPs was activated using EDC–HCl (14.4 mM) and sulfo-NHS (14.4 mM) in 200 μL of 10 mM PBS pH 5.4 for 10 min. Subsequently, the NPs were centrifuged for 10 min at 4 °C with a 15.5 krcf; the supernatant was discarded and the pellets were quickly redissolved in 10 mM PBS at pH 8 with the desired concentration of amino groups. Finally, the samples were left to react for 2.5 h at room temperature after which the product was isolated by repeated centrifugation (10 min at 4 °C with a 15.5 krcf 4 times) and resuspension in 200 μL of a 0.1 wt% Lutensol AT50 solution. In the last purification step, the pellets were left to dry at 40 °C and dissolved in 200 μL DMSO. The fluorescence spectrum was recorded with an Infinite M1000 plate reader (Tecan, Männedorf, Switzerland) (λexcitation = 430 nm, λemission = 490–550 nm). The schematic illustration and details can be found in the ESI (Fig. S8†).EDC–NHS chemistry on NH2-NPs
NH2-NPs were covalently conjugated via EDC/NHS chemistry similarly to the above-described protocol. EDC–HCl (1.4 mM) and sulfo-NHS (1.4 mM) were dissolved in a final volume of 20 μL of a 10 mM PBS pH 5.4 solution and mixed with various concentrations of CY3 or HSA. The sample was mixed on a moving plate for 10 min. Subsequently, 180 μL of a 0.11 wt% solution of NH2-NPs in 10 mM PBS at pH 8 were added and the reaction was carried out at room temperature for 2.5 h. The product was isolated by repeated centrifugation (10 min at 4 °C with a 15.5 krcf 4 times) and resuspension in 200 μL of a 0.1 wt% Lutensol AT50 solution. In the last purification step, the pellets were left to dry and dissolved in a 1:1 DMSO:THF solution to record the fluorescence spectrum (λexcitation = 550 nm, λemission = 570–660 nm). The schematic illustration and details can be found in the ESI (Fig. S12†).
Fluorescamine titration of NH2-NPs
NH2-NPs were characterized via adaptation of the fluorescamine assay.42 To 100 μL of a 0.01 wt% solution of NH2-NPs in 10 mM PBS buffer at pH 8, 0.5 μL of a fluorescamine stock solution (5 or 0.5 mM in DMSO) were added sequentially, stirred for 1 min and the fluorescence spectrum (λexcitation = 400 nm, λemission = 425–525 nm) was recorded at each step until there was no evident fluorescence intensity change. The schematic illustration and details can be found in Fig. S14.†Author contributions
FM performed the experiments. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the Max Planck Society for financial support. The authors acknowledge funding from the European Union's Horizon 2020 Research and Innovation Program under the Marie Skłodowska-Curie program (ITN SUPERCOL, Grant Agreement 860914). For the preparation of graphical representations https://www.Biorender.com was used.References
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Nat. Rev. Drug Discovery, 2021, 20, 101–124 CrossRef CAS.
- Ł. Richter, C. A. Stevens, P. J. Silva, L. R. Julià, C. Malinverni, L. Wei, M. Łoś and F. Stellacci, ACS Nano, 2022, 16, 18990–19001 CrossRef.
- Y. Wang, Y. Wang, X. Zheng, É. Ducrot, J. S. Yodh, M. Weck and D. J. Pine, Nat. Commun., 2015, 6, 7253 CrossRef CAS PubMed.
- S. Pottanam Chali and B. J. Ravoo, Angew. Chem., Int. Ed., 2020, 59, 2962–2972 CAS.
- M. Brückner, M. Fichter, R. da Costa Marques, K. Landfester and V. Mailänder, Pharmaceutics, 2022, 14, 1614 Search PubMed.
- E. Archontakis, L. Woythe, B. van Hoof and L. Albertazzi, Nanoscale Adv., 2022, 4, 4402–4409 CAS.
- J. Simon, M. Fichter, G. Kuhn, M. Brückner, C. Kappel, J. Schunke, T. Klaus, S. Grabbe, K. Landfester and V. Mailänder, Nano Today, 2022, 43, 101375 CAS.
- B. Iyisan, J. Simon, Y. Avlasevich, S. Baluschev, V. Mailaender and K. Landfester, ACS Appl. Bio Mater., 2022, 5, 622–629 CAS.
- O. Ilic, I. Kaminer, B. Zhen, O. D. Miller, H. Buljan and M. Soljačić, 2017 Conf. Lasers Electro-Optics, CLEO 2017 - Proc., 2017, 2017-January, 1–2.
- M. Wan, T. Li, H. Chen, C. Mao and J. Shen, Angew. Chem., Int. Ed., 2021, 60, 13158–13176 CAS.
- Y. Tu, F. Peng, A. A. M. André, Y. Men, M. Srinivas and D. A. Wilson, ACS Nano, 2017, 11, 1957–1963 CAS.
- T. Patiño, N. Feiner-Gracia, X. Arqué, A. Miguel-López, A. Jannasch, T. Stumpp, E. Schäffer, L. Albertazzi and S. Sánchez, J. Am. Chem. Soc., 2018, 140, 7896–7903 CrossRef PubMed.
- A. T. Thodikayil, S. Sharma and S. Saha, ACS Appl. Bio Mater., 2021, 4, 2907–2940 CrossRef CAS.
- L. B. Capeletti, J. F. A. de Oliveira, L. M. D. Loiola, F. E. Galdino, D. E. da Silva Santos, T. A. Soares, R. de Oliveira Freitas and M. B. Cardoso, Adv. Funct. Mater., 2019, 29, 1–11 Search PubMed.
- S. Asati, V. Pandey and V. Soni, Int. J. Pept. Res. Ther., 2019, 25, 49–65 CrossRef CAS.
- N. Graf, D. R. Bielenberg, N. Kolishetti, C. Muus, J. Banyard, O. C. Farokhzad and S. J. Lippard, ACS Nano, 2012, 6, 4530–4539 CrossRef CAS PubMed.
- K. Ulbrich, K. Holá, V. Šubr, A. Bakandritsos, J. Tuček and R. Zbořil, Chem. Rev., 2016, 116, 5338–5431 CAS.
- M. Maddahfar, S. Wen, S. M. Hosseinpour Mashkani, L. Zhang, O. Shimoni, M. Stenzel, J. Zhou, B. Fazekas De St Groth and D. Jin, Bioconjugate Chem., 2021, 32, 1146–1155 CAS.
- J. Wan, Y. Li, K. Jin, J. Guo, J. Xu and C. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 23717–23725 CAS.
- T. Andrian, S. Pujals and L. Albertazzi, Nanoscale Adv., 2021, 3, 6876–6881 CAS.
- A. J. Sivaram, A. Wardiana, C. B. Howard, S. M. Mahler and K. J. Thurecht, Adv. Healthcare Mater., 2018, 7, 1–25 Search PubMed.
- D. Geißler, N. Nirmalananthan-Budau, L. Scholtz, I. Tavernaro and U. Resch-Genger, Microchim. Acta, 2021, 188, 321 Search PubMed.
- Y. Sun, F. Kunc, V. Balhara, B. Coleman, O. Kodra, M. Raza, M. Chen, A. Brinkmann, G. P. Lopinski and L. J. Johnston, Nanoscale Adv., 2019, 1, 1598–1607 RSC.
- A. Hennig, H. Borcherding, C. Jaeger, S. Hatami, C. Würth, A. Hoffmann, K. Hoffmann, T. Thiele, U. Schedler and U. Resch-Genger, J. Am. Chem. Soc., 2012, 134, 8268–8276 CrossRef CAS PubMed.
- M. Moser, N. Nirmalananthan, T. Behnke, D. Geißler and U. Resch-Genger, Anal. Chem., 2018, 90, 5887–5895 CrossRef CAS PubMed.
- M. T. Hussain, N. Forbes and Y. Perrie, Pharmaceutics, 2019, 11, 9–11 CrossRef.
- J. E. Noble and M. J. A. Bailey, Methods in enzymology, 2009, pp. 73–95 Search PubMed.
- L. Woythe, N. B. Tito and L. Albertazzi, Adv. Drug Delivery Rev., 2021, 169, 1–21 CrossRef CAS PubMed.
- E. S. Day, L. R. Bickford, J. H. Slater, N. S. Riggall, R. A. Drezek and J. L. West, Int. J. Nanomed., 2010, 5, 445 CAS.
- J. Stein, F. Stehr, P. Schueler, P. Blumhardt, F. Schueder, J. Mücksch, R. Jungmann and P. Schwille, Nano Lett., 2019, 19, 8182–8190 CAS.
- S. Beuermann and M. Buback, Prog. Polym. Sci., 2002, 27, 191–254 CAS.
- T. J. Neal, A. J. Parnell, S. M. King, D. L. Beattie, M. W. Murray, N. S. J. Williams, S. N. Emmett, S. P. Armes, S. G. Spain and O. O. Mykhaylyk, Macromolecules, 2021, 54, 1425–1440 CrossRef CAS PubMed.
- S. Schwarz, H. M. Buchhammer, K. Lunkwitz and H. J. Jacobasch, Colloids Surf., A, 1998, 140, 377–384 CrossRef CAS.
- R. H. Müller and G. E. Hildebrand, Paperb. APV.
- F. Grzegorzewski, A. Benhaim, Y. Itzhaik Alkotzer, E. Zelinger, N. Yaakov and G. Mechrez, Polymers, 2019, 11, 1480 CrossRef CAS PubMed.
- E. Y. Liu, S. Jung and H. Yi, Langmuir, 2016, 32, 11043–11054 CrossRef CAS.
- F. Novio, J. Lorenzo, F. Nador, K. Wnuk and D. Ruiz-Molina, Chem. – Eur. J., 2014, 20, 15443–15450 CrossRef CAS PubMed.
- Y. Wang, Y. Wang, X. Zheng, É. Ducrot, J. S. Yodh, M. Weck and D. J. Pine, Nat. Commun., 2015, 6, 7253, DOI:10.1038/ncomms8253.
- S. Sugio, A. Kashima, S. Mochizuki, M. Noda and K. Kobayashi, Protein Eng., 1999, 12, 439–446 CrossRef CAS PubMed.
- M. Urban, B. Freisinger, O. Ghazy, R. Staff, K. Landfester, D. Crespy and A. Musyanovych, Macromolecules, 2014, 47, 7194–7199 CAS.
- A. Musyanovych, R. Rossmanith, C. Tontsch and K. Landfester, Langmuir, 2007, 23, 5367–5376 CrossRef CAS PubMed.
- A. Musyanovych and H. J. P. Adler, Langmuir, 2005, 21, 2209–2217 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py01119h |
| This journal is © The Royal Society of Chemistry 2025 |