
High second-order nonlinear optical effect achieved by gradually decreased rotational energy barriers†
Panpan
Qiao
a,
Wentao
Yuan
a,
Qianqian
Li
*a and
Zhen
Li
 *ab
*ab
aHubei Key Lab on Organic and Polymeric Opto-Electronic Materials, Department of Chemistry, Wuhan University, Wuhan, 430072, China. E-mail: liqianqian@whu.edu.cn; lizhen@whu.edu.cn
bTaiKang Center for Life and Medical Sciences, Wuhan University, Wuhan, 430072, China
First published on 4th December 2024
Abstract
With the aim of efficiently converting the microscopic second-order nonlinear optical (NLO) effect of chromophore moieties into macroscopic NLO performance as high as possible, this work focused on the connection groups between the chromophore moieties of NLO polymers, in which alkoxy chains with different lengths and positions were systematically incorporated. The ignorable difference of the alkoxy chain from the normally utilized alkyl one directly resulted in improved macroscopic NLO performance, and d33 values increased gradually from 105 to 131/157, then to 165 pm V−1 with increasing contents of alkoxy chains, and further reached up to 178 pm V−1 with the prolonged lengths of alkoxy chains. This was mainly due to the lower rotational barriers of ether bonds than those of the commonly used alkyl chains with carbon–carbon bonds, and the isolated effect of alkoxy chains with larger sizes. This work provides a new way to achieve a high second-order NLO effect from efficient modulation of chromophore orientations by adjustment of energy barriers.
Introduction
Organic second-order nonlinear optical (NLO) materials with large macroscopic NLO efficiencies (d33), high index of refraction, and long-term alignment stability have great potential in high-speed electro-optic modulators, optical switches, frequency converters, and data storage applications.1–5 As the key component to achieve the macroscopic second-order NLO effect, organic chromophores with electron donor (D)-π bridge-electron acceptor (A) structures demonstrate a high microscopic NLO effect (β) for the strong intramolecular charge transfer (ICT) with high dipole moments. The desirable arrangement of organic chromophores is non-centrosymmetric, which can be realized by flipping the molecular orientations under the electric poling process (Fig. 1a).6–10 However, the strong dipole–dipole interactions among chromophores usually tend to the centrosymmetric arrangement, leading to largely decreased macroscopic NLO efficiencies at aggregated states.11,12 Thus, modulation of molecular arrangement is essential and crucial to the corresponding optoelectronic properties.13–16 | ||
| Fig. 1 (a) The modulation of chromophore orientation under electric polarization by the decreased energy barriers of connection moieties from alkyl to alkoxy chains with propane and dimethyl ether as the simple modes, respectively. (b) Structure diagram of target polymers and the increased trend of relative d33 values by the gradual incorporation of alkoxy chains instead of alkyl chains. | ||
Accordingly, various strategies have been explored to facilitate the modulation process: for instance, the introduction of suitable isolation groups17–22/chromophores23–28 to decrease the dipole–dipole interactions under the guidance of the site isolation principle. Indeed, this largely enhanced the macroscopic NLO effect with the combination of the high efficiency of the electric poling process, and maintainable densities of organic chromophores as efficient moieties for the NLO effect. Considering the poling process carefully, the modulation of chromophore orientation through slow rotations under electric fields is restricted not only by their strong dipole–dipole interactions, but also by the traction of the connecting groups/chains between chromophores. Generally, an increased flexibility can facilitate the rotations of chromophore moieties for decreased energy barriers, as partially proved in our previous research,29 in which we attempted to introduce ether bonds with lower rotational barriers (ΔE) into the connecting chains, after evaluating the rotational barriers of simple propane (ΔE = 13.3 kJ mol−1) and dimethyl ether (ΔE = 10.6 kJ mol−1) among various conformations (Fig. 1a).30,31 The ignorable difference of the alkoxy chain from the normally utilized alkyl one directly resulted in a largely enhanced macroscopic NLO effect. The surprising results prompted us to further investigate systematically the crucial role of alkoxy chains in the electric poling process and the final NLO effect.
Accordingly, alkoxy chains were introduced into the linkage chains of chromophores (A part) in Y-shaped monomers, or the main chains (B part) of polymers, or both A and B parts, with varied lengths (Scheme 1). The corresponding d33 values of these polymers increased gradually by 1.25-/1.28-fold, to 1.50-/1.66-fold, then to 1.57-/1.78-folds, with the analogous polymers bearing full alkyl chains (I) as the reference (Fig. 1b). This indicated that the incorporation of multiple alkoxy chains as the connection moieties of chromophores in different positions is an efficient strategy to improve the NLO performance, and the more alkoxy chains, the higher the NLO efficiencies. This was mainly due to the much easier rotation process of chromophores under electric fields, and longer chains were favorable for avoiding possible spatial restriction in the rotation process. Thus, this work systematically investigated the key role of connection moieties of chromophores in the electric poling process and NLO effect by the introduction of alkoxy chains instead of alkyl ones with different positions and lengths, and the optimal mode has been proposed to promote the development of high-efficiency organic second-order NLO materials by efficient modulation of chromophore orientations.
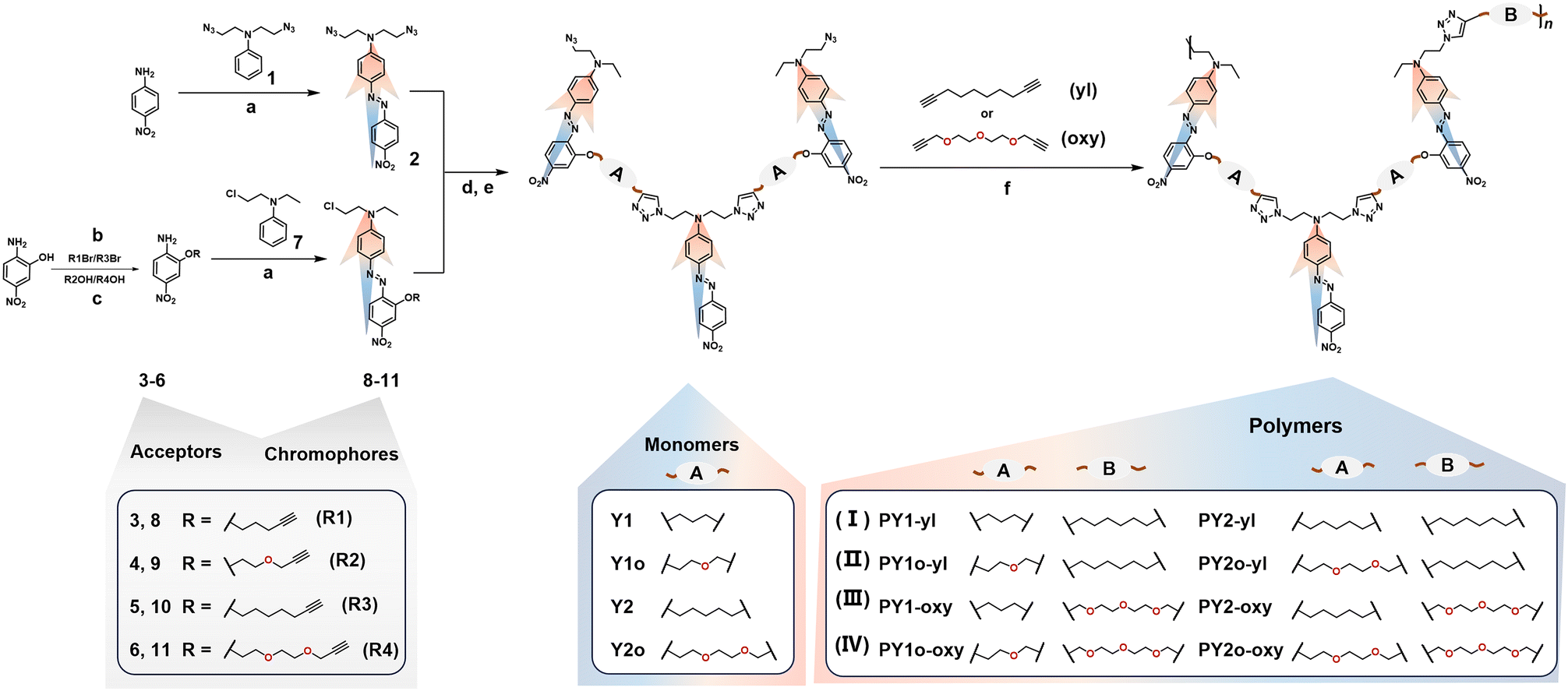 | ||
| Scheme 1 The synthetic routes of target polymers and the corresponding structure characterization. Reaction conditions: (a) HCl (38%), NaNO2/H2O, CH3CN, 0–5 °C; (b) K2CO3, DMF, 80 °C; (c) DIAD PPh3, THF, 30 °C; (d) CuSO4·H2O, VcNa, THF/H2O, 30 °C; (e) NaN3, DMF, 80 °C; (f) CuSO4·H2O, VcNa, DMF, 25 °C. | ||
Results and discussion
Design and synthesis
Eight linear–dendritic (L–D) copolymers have been designed and synthesized,32,33 in which Y-shaped monomers are the dendritic moieties, and the linear alkyl/alkoxy moieties in the main chains are the connection groups as B part (Scheme 1). The Y-shaped monomers were constructed of three nitro-azobenzene chromophores, which were also connected by linear alkyl/alkoxy moieties (A part). They were synthesized through a simple substitution reaction, azo coupling reaction34,35 and followed by “Click chemistry” reaction [Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC)] with high yields.36,37 The A parts in monomers Y1 and Y2 were alkyl chains (–C3H7 and –C5H11), and those in monomers Y1o and Y2o were the corresponding alkoxy chains (–C3H7O and –C5H11O2). Compared to the traditional linear chromophores, the “Y”-type monomers, which consisted of three nitro-azobenzene moieties as chromophores with head-to-tail linking modes, demonstrated the optimized molecular orientation with non-centrosymmetric arrangement, beneficial to the NLO effect.38–40These target polymers were polymerized through a “Click chemistry” reaction41–45 between –N3 and –![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) moieties, which has been widely used to synthesize many functional optoelectronic materials.46–51 This is mainly attributed to the advantages of the “Click chemistry” reaction, such as high selectivity and compatibility and high reactive activity with few by-products, and polymers with narrow molecular weight distribution can be obtained by simple operations of reprecipitation and Soxhlet extraction. Besides, the resultant five-member rings formed in the “Click chemistry” reaction can also act as isolation groups to reduce dipole–dipole interactions between chromophore moieties.52–54 However, due to the large steric hindrance of Y-shaped monomers, the polymerization rate was relatively slow, and larger molecular weights and higher yields can be obtained by adding catalysts in batches according to our previous work.29,55 The implosion phenomenon was observed in a short time if sufficient amounts of catalysts were added at once, along with the formation of insoluble and viscous substances on the bottom and wall of the Schlenk tube. Hence the catalysts were added to the reaction system three or four times at the same interval in this work. The detailed preparation procedures are presented in the Experimental section. These polymers were obtained in yields of 37.3%–51.5% with good solubility in common polar organic solvents, such as CH2Cl2, CHCl3, DMF, DMSO, etc. They can be divided into four types by the different connection groups among chromophores, including full alkyl chains (I), alkoxy chains in part A and alkyl chains in part B (II), alkyl chains in part A and alkoxy chains in part B (III), and full alkoxy chains (IV) (Scheme 1).
moieties, which has been widely used to synthesize many functional optoelectronic materials.46–51 This is mainly attributed to the advantages of the “Click chemistry” reaction, such as high selectivity and compatibility and high reactive activity with few by-products, and polymers with narrow molecular weight distribution can be obtained by simple operations of reprecipitation and Soxhlet extraction. Besides, the resultant five-member rings formed in the “Click chemistry” reaction can also act as isolation groups to reduce dipole–dipole interactions between chromophore moieties.52–54 However, due to the large steric hindrance of Y-shaped monomers, the polymerization rate was relatively slow, and larger molecular weights and higher yields can be obtained by adding catalysts in batches according to our previous work.29,55 The implosion phenomenon was observed in a short time if sufficient amounts of catalysts were added at once, along with the formation of insoluble and viscous substances on the bottom and wall of the Schlenk tube. Hence the catalysts were added to the reaction system three or four times at the same interval in this work. The detailed preparation procedures are presented in the Experimental section. These polymers were obtained in yields of 37.3%–51.5% with good solubility in common polar organic solvents, such as CH2Cl2, CHCl3, DMF, DMSO, etc. They can be divided into four types by the different connection groups among chromophores, including full alkyl chains (I), alkoxy chains in part A and alkyl chains in part B (II), alkyl chains in part A and alkoxy chains in part B (III), and full alkoxy chains (IV) (Scheme 1).
Characterization
The prepared Y-shaped monomers and target polymers were well characterized by Fourier transform infrared attenuated total reflection (FTIR-ATR) spectroscopy, nuclear magnetic resonance (NMR), ultraviolet-visible (UV-vis) spectroscopy, etc. Monomers Y1, Y1o, Y2 and Y2o were identified by matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry and elemental analyses (EA) (Fig. S1†). The polymers were characterized by gel permeation chromatography (GPC), and thermal analysis technologies were also employed, such as thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) (see the Experimental section and ESI† for detailed analysis). Time-dependent density functional theory (TD-DFT) calculations were performed using Gaussian 16, Revision C01.56 The optimization of molecular structure was calculated based on the M062X functional with the 6-31G(d) basis set (Fig. S2†).As a simple optical test method, FTIR-ATR spectroscopy is good for monitoring the polymerization process. The characterization peaks at 1516 cm−1 and 1335 cm−1 in FTIR-ATR spectra of monomers and polymers are attributed to the nitro groups (Fig. S3†), indicating that the nitro-based chromophores were stable during polymerization and purification procedures. It was easily observed that the peak at 2098 cm−1, which can be attributed to –N3, disappeared in the spectra of the prepared polymers, compared to those of corresponding monomers, illustrating that the polymerization process went well and the content of the terminal functional groups was much less. Successful polymerization can be further confirmed by NMR spectra. The 1H NMR spectra of polymer PY2-oxy and monomer Y2 are taken as an example to analyze the chemical shift change and peak attribution of each signal after polymerization. As shown in Fig. 2, all the signal peaks of PY2-oxy exhibit a distinct signal-broadening characteristic with maintained characteristic peaks of monomer Y2. Meanwhile, signal transitions associated with the reactive groups can be observed in the spectrum of PY2-oxy, with that of Y2 as the reference. In detail, peak (1), the signal of the hydrogen of –CH2– (3.53 ppm) linked to –N3 groups in Y2, decreased in the spectrum of PY2-oxy. Accordingly, peak (2), a hydrogen signal of –CH2– (4.59 ppm), appeared, which was linked to the triazole generated by the “Click reaction”. At the same time, peaks (3), the signals of –CH2– (3.29 ppm and 4.11 ppm) in the alkoxy chain (oxy), can be observed in the spectrum of PY2-oxy. There is no signal attributed to terminal alkyne bond in the range of 2.50–2.00 ppm, indicating that the alkoxy chains (oxy) were consumed completely in the “Click reaction” with monomer Y2. The molecular weights of target polymers were determined by GPC with a refractive index detector (DMF as an eluent and PMMA as the calibration standard). Their weight-average molecular weight (Mw) values were all higher than 104, with polydispersities (Mw/Mn, PDI) lower than 2 (Table 1). The UV-vis absorption spectra of polymers and monomers in CH2Cl2 solution are shown in Fig. 3a and 3b, and their maximum absorption wavelengths (λmax) are listed in Table 1. These absorption peaks were mainly from the ICT effect of chromophore moieties; therefore the polymers had very similar absorption features to the monomers with λmax at about 460 nm. This result showed that the alkoxy and alkyl chains as connection groups had little electronic effect on the resultant polymers and monomers. Their TGA thermograms are shown in Fig. 3c, and the 5% weight loss temperatures (Td) of polymers are listed in Table 1. All the prepared polymers had good thermal and storage stability, with Td values close to 300 °C, thanks to the superior stability of nitro-azobenzene moieties as chromophores. The glass transition temperatures (Tg) of the polymers were investigated by DSC (Fig. 3d and Table 1). The Tg of polymers decreased with the increase of the numbers (PY1-yl (97 °C) vs.PY1o-oxyl (86 °C)) and lengths (PY1-yl (97 °C) vs.PY2-yl (85 °C)) of alkoxy chains. This was directly related to the flexibility of polymer chains. Thanks to the flexibility of these polymers enhanced by the introduction and extension of the alkoxy chains with lower energy barriers, the corresponding thermal motions became easier, resulting in the decreased Tg.
 | ||
| Fig. 2 1H-NMR spectra of monomer Y2 and polymer PY2-oxy in chloroform-d, the peaks from residual solvent being marked with *. The inset is enlarged 1H-NMR spectra in the region of 3.75–3.50 ppm. | ||
 | ||
| Fig. 3 (a) UV-vis spectra of monomers Y1 and Y1o and their corresponding polymers. (b) UV-vis spectra of monomers Y2 and Y2o and their corresponding polymers. (c) TGA thermograms and (d) DSC curves of polymers measured at a heating rate of 10 °C min−1 under nitrogen. | ||
| Polymer |
M
w![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (104) a (104) |
M
w/Mna (PDI) |
T
gb (°C) |
T
dc (°C) |
λ
maxd (nm) |
l
se (nm) |
T
ef (°C) |
d
33g (pm V−1) |
d
33(∞)h (pm V−1) |
T
80%i (°C) |
Φ | N (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Determined by GPC in DMF based on calibration with PMMA. b Glass transition temperature determined by DSC analysis under nitrogen at a heating rate of 10 °C min−1. c The 5% weight loss temperature of polymers determined by TGA under nitrogen at a heating rate of 10 °C min−1. d The maximum absorption wavelength in CH2Cl2 (0.02 mg mL−1). e Film thickness. f The best poling temperature. g Second-harmonic generation (SHG) coefficient measured at a 1064 nm fundamental beam at a voltage of 7.0 kV. h The non-resonant d33 values were calculated using an approximate two-level model. i The temperature at which the SHG signals decreased to 80%. j Order parameter Φ = 1 − A1/A0, where A1 and A0 are the absorbance values of the polymer film after and before corona poling, respectively. k The loading density of the effective chromophores. | ||||||||||||
| PY1-yl | 1.62 | 1.80 | 97 | 280 | 459 | 228 | 95 | 105 | 16.2 | 100 | 0.19 | 65.9 |
| PY1o-yl | 1.97 | 1.98 | 91 | 295 | 458 | 220 | 86 | 131 | 19.4 | 93 | 0.18 | 64.4 |
| PY1-oxy | 1.93 | 1.97 | 90 | 286 | 460 | 198 | 103 | 157 | 23.4 | 98 | 0.21 | 63.7 |
| PY1o-oxy | 1.74 | 1.75 | 86 | 284 | 459 | 200 | 93 | 165 | 26.8 | 91 | 0.19 | 62.3 |
| PY2-yl | 1.86 | 1.88 | 85 | 293 | 461 | 227 | 94 | 100 | 16.0 | 93 | 0.20 | 63.3 |
| PY2o-yl | 2.27 | 1.91 | 80 | 287 | 461 | 238 | 84 | 128 | 19.0 | 90 | 0.20 | 60.6 |
| PY2-oxy | 2.24 | 1.62 | 82 | 272 | 460 | 212 | 93 | 166 | 26.9 | 84 | 0.23 | 61.2 |
| PY2o-oxy | 2.49 | 1.85 | 78 | 282 | 460 | 210 | 90 | 178 | 28.9 | 81 | 0.21 | 58.7 |
Second-order NLO effect
To evaluate the macroscopic NLO activities of the polymers, their thin films were fabricated by spin-coating. NLO coefficients were quantified as d33 values using the second-harmonic generation (SHG) technique. The method for the calculation of d33 for the poled films has been reported previously.41,57 The NLO coefficients of these polymers were measured using an SHG test system at a voltage of 7.0 kV and a 1064 nm fundamental beam.The poling curves of the SHG coefficients of polymers as a function of temperature are shown in Fig. 4a. The calculated d33 values (the macroscopic NLO activities) of the polymers PY1-yl/PY2-yl bearing full alkyl chains were 105/100 pm V−1. With the introduction of alkoxy chains into the Y dendrimer (A part) or backbone (B part), the d33 values of the polymers PY1o-yl/PY2o-yl and PY1-oxy/PY2-oxy were greatly increased. Polymers PY1o-oxy and PY2o-oxy bearing full alkoxy chains (A and B part) exhibited the highest d33 values, reaching 165 and 178 pm V−1, respectively, higher than those of most nitro-azobenzene-based polymers reported in the literatures.41,42,57–63 The results showed that the d33 values of the polymers improved with increased numbers of alkoxy chains (Table 1, and Fig. 4b), which fully demonstrated the superiority of alkoxy chains with lower rotational barriers for the modulation of chromophore arrangement.64,65 The d33(∞) values (defined as the macroscopic NLO coefficients of non-resonance enhancement) of polymers were calculated using an approximate two-level model. To a certain extent, the d33(∞) values can represent the NLO effect after deducting the fundamental frequency optical resonance, which demonstrated a similar trend to that of d33 values (Table 1). Considering the increase in d33 values carefully, the improvement of d33 value of PY2-oxy with alkoxy chains in B part (1.66-fold) was significantly higher than that of PY2o-yl with alkoxy chains in A part (1.28-fold), indicating that the flexibility of main chains represented the dominant effect. This is reasonable as the chromophores in Y-shaped monomers are connected in a head-to-tail manner, and it is supposed that the orientation of chromophores has been optimized to some extent at the beginning of the design of the Y-shaped monomers, so that the advantages of introducing flexible chains into the Y-shaped monomers are not prominent.
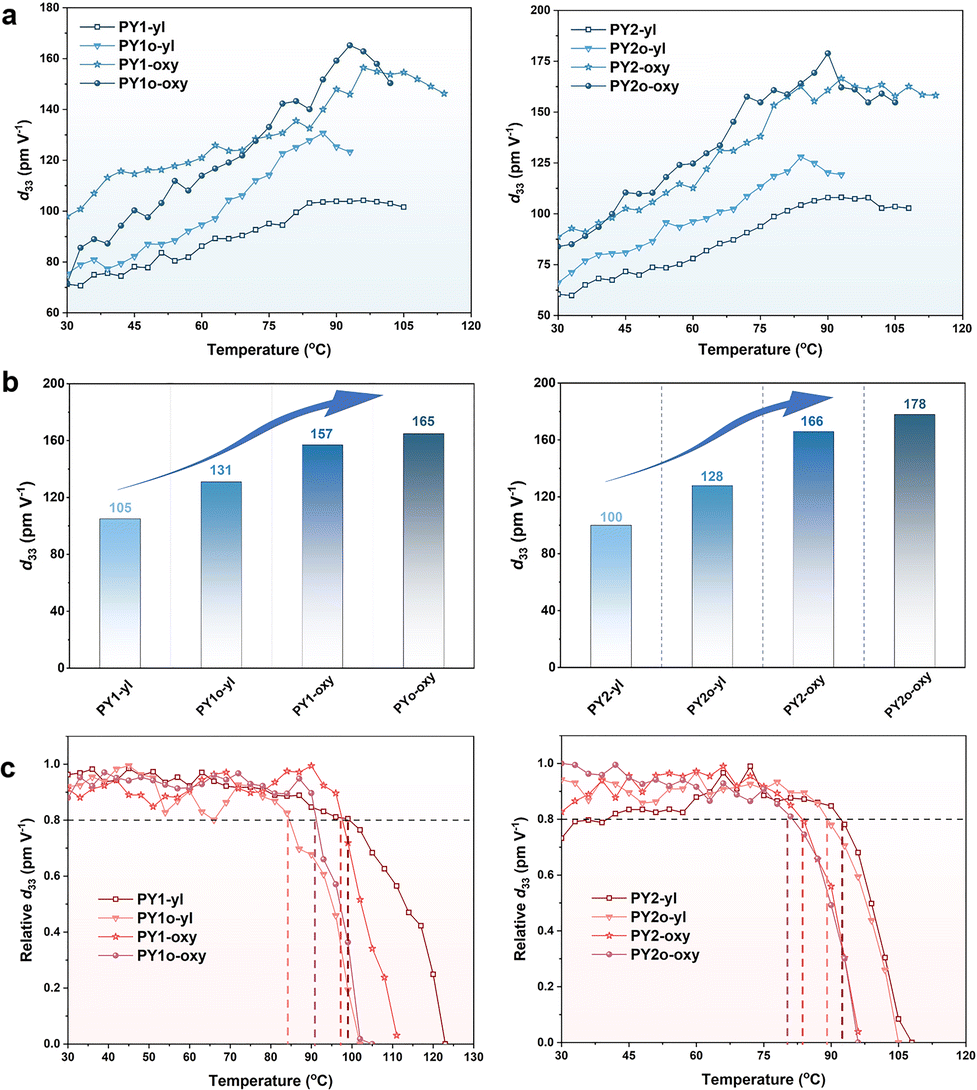 | ||
| Fig. 4 (a) Poling curves of the SHG coefficients of polymers as a function of temperature. (b) Increased d33 values due to the increased alkoxy chains. (c) Decay curves of the SHG coefficients of polymers as a function of temperature. | ||
On the other hand, the increased lengths of flexible chains may enlarge the distance of chromophores, which can weaken the electrostatic interactions among chromophores, benefiting the non-centrosymmetric arrangement during poling. For instance, as the lengths of the A part extended from –C3H7 to –C5H11, the d33 value increased from 157 pm V−1 for PY1-oxy to 166 pm V−1 for PY2-oxy. Besides, the rotational barriers of longer alkoxy chains demonstrated a larger decrease than those of shorter alkoxy chains, compared to those of similar alkyl chains. This was mainly due to the increased ratios of ether bonds with lower rotational barriers, which can be favourable to molecular rotations under electric poling to achieve the preferred molecular arrangements. Thus, the highest d33 value (178 pm V−1) was achieved by polymer PY2o-oxy bearing full alkoxy chains as the connection groups among chromophores with longer lengths, which can facilitate the rotations of chromophores under electric poling by the decreased energy barriers and diploe–dipole interactions.
In the de-poling process of these polymers (Fig. 4c), the real-time decays of their SHG signals were monitored with the temperature ranging from 30 °C to 120 °C in air at a rate of 4 °C min−1. The T80% values (defined as the temperature at which the SHG signals decreased to an initial 80%) of polymers decreased slightly with the introduction of alkoxy chains (Table 1). This is because the C–O bonds in alkoxy chains have higher activities for the possible generation of radicals, and lower rotational barriers of alkoxy chains simultaneously accelerate the relaxation process of the polymers during heating.66,67 This can be suppressed by the additional cross-linking process if needed.68–71
Conclusions
The crucial role of connection moieties, including alkyl and alkoxy chains, among chromophores in the macroscopic second-order NLO effect has been systematically investigated, and some important issues can be concluded as follows:1. Alkoxy chains as the connection moieties are preferable for the NLO effect, compared to similar alkyl moieties. This was mainly due to the decreased energy barriers of alkoxy chains, which can facilitate the rotation of chromophores during the electric poling process.
2. The effect of alkoxy chains in the main chains of polymers is more obvious, compared to those in the Y-shaped monomers. This may be attributed to the fact that the orientation of chromophores has been optimized to a certain extent in Y-shaped monomers.
3. Alkoxy chains with longer lengths can facilitate the modulation of chromophore orientation, since they can decrease the possible diploe–dipole interactions for the isolation effect with larger sizes.
In summary, this work fills the knowledge gap in the modulation of chromophore orientation through the subtle modifications of the linkage groups and provides a new idea for possibly controlling the molecular packing and aggregation at the stage of molecular design, once again demonstrating the Molecular Uniting Set Identified Characteristic (MUSIC).72,73
Experimental section
Materials
Tetrahydrofuran (THF) was dried and distilled from Na–K alloy under an atmosphere of dry nitrogen. N,N-Dimethylformamide (DMF) was deoxidized under an atmosphere of dry nitrogen. Y1, Y1o, Y2, and Y2o were prepared according to the literatures.29,55 All other reagents were used as received.Instrumentation
1H and 13C NMR spectra were recorded with a Bruker AVANCE NEO 400 MHz spectrometer using tetramethylsilane (TMS; δ = 0 ppm) as the internal standard. The FTIR-ATR spectra were recorded with a PerkinElmer-2 spectrometer in the region of 3500–500 cm−1. MALDI-TOF spectra were measured with an AB SCIEX 5800 MALDI-TOF mass spectrometer. EA was performed using an Elementar UNICUBE elemental analyzer. GPC analysis was performed using a Waters HPLC system equipped with a 2690D separation module and a 2410 refractive index detector. Polymethyl methacrylate (PMMA) was used as calibration standards and DMF was used as an eluent. UV-visible spectra were obtained using a Shimadzu UV-2700 spectrometer. TGA was performed with a Beijing Hengjiu HTG-1 thermal analyzer at a heating rate of 10 °C min−1 in nitrogen at a flow rate of 50 cm3 min−1. DSC was performed using a Mettler-Toledo DSC3 under nitrogen at a scanning rate of 10 °C min−1. The thickness of the films was measured with a profiler (Bruker DEKTAKXT).General synthesis of L–D copolymers
The monomers and the copolymer linear chains (alkyl chains or alkoxy chains) at a molar ratio of 1:1 were dissolved in DMF (the concentration of monomers was about 0.01 M) under nitrogen in a Schlenk flask at room temperature (25–30 °C). Then a certain volume of freshly prepared CuSO4 aqueous solution (0.08 M) and ascorbic acid sodium aqueous solution (NaAsc, 0.16 M) was added to the mixture using a microinjector. After each reaction time for 12 h, another batch of CuSO4 and NaAsc aqueous solution was added. The reaction should be stopped when insoluble matter was about to appear and then poured into a large amount of water. The precipitate was collected and washed several times with deionized water, anhydrous methanol, and acetone successively. The obtained solid was further purified by reprecipitation from its CH2Cl2 solution into methanol to afford a red powder.
PY1-yl. Y1 (97.8 mg, 0.08 mmol), yl (10.7 mg, 0.08 mmol), CuSO4 (100 + 50 + 50 μL), NaAsc (100 + 50 + 50 μL), in DMF (8 mL), a deep red powder (46 mg, 42.6%). Mw = 16201, Mw/Mn = 1.80 (GPC, PMMA calibration). 1H NMR (400 MHz, CDCl3, 298 K), δ (TMS, ppm): 8.18 (–ArH), 8.15–8.07 (–ArH), 7.85–7.63 (–ArH), 7.59 (–ArH), 7.57–7.51 (–ArH), 7.42 (–ArH), 7.39 (–ArH), 7.27 (–ArH), 7.20 (–ArH), 6.69–6.48 (–ArH), 4.62–4.49 (–CH2–), 4.43 (–CH2–), 4.22–4.03 (–CH2–), 3.96–3.83 (–CH2–), 3.77 (–CH2–), 3.36–3.19 (–CH2–), 3.01–2.86 (–CH2–), 2.69–2.48 (–CH2–), 2.29–2.11 (–CH2–), 1.51 (–CH2–), 1.35–1.19 (–CH2–), 1.17–1.03 (–CH2–). 13C NMR (101 MHz, CDCl3, 298 K), δ (ppm): 156.06, 155.10, 150.43, 149.69, 148.42, 148.15, 147.57, 147.25, 147.17, 146.72, 144.59, 126.24, 126.05, 124.61, 122.85, 122.55, 122.07, 117.23, 116.51, 111.31, 109.21, 68.65, 51.38, 51.18, 50.50, 47.63, 47.26, 45.54, 29.21, 28.51, 25.27, 21.89, 12.10.
PY1o-yl. Y1o (87.8 mg, 0.07 mmol), yl (9.6 mg, 0.07 mmol), CuSO4 (90 + 45 + 45 + 45 μL), NaAsc (90 + 45 + 45 + 45 μL), in DMF (7 mL), a deep red powder (50 mg, 51.5%). Mw = 19653, Mw/Mn = 1.98 (GPC, PMMA calibration). 1H NMR (400 MHz, CDCl3, 298 K), δ (TMS, ppm): 8.25–8.10 (–ArH), 7.87–7.68 (–ArH), 7.67–7.52 (–ArH), 7.28 (–ArH), 6.68–6.56 (–ArH), 6.56–6.43 (–ArH), 4.78 (–CH2–), 4.75 (–CH2–), 4.54 (–CH2–), 4.30 (–CH2–), 3.87 (–CH2–), 3.67 (–CH2–), 3.26 (–CH2–), 2.59 (–CH2–), 1.15–1.04 (–CH2–). 13C NMR (101 MHz, CDCl3, 298 K), δ (ppm): 155.99, 155.13, 150.48, 150.43, 149.50, 148.43, 148.08, 147.60, 146.75, 126.34, 126.06, 124.62, 123.89, 122.80, 122.02, 117.29, 116.85, 111.56, 111.34, 110.18, 109.74, 77.49, 68.70, 68.46, 65.01, 50.87, 50.46, 47.47, 47.14, 45.79, 45.62, 29.34, 29.18, 28.70, 28.51, 25.42, 25.29, 12.13.
PY1-oxy. Y1 (92.0 mg, 0.07 mmol), oxy (12.8 mg, 0.07 mmol), CuSO4 (90 + 50 + 85 + 30 μL), NaAsc (90 + 50 + 85 + 30μL), in DMF (7 mL), a deep red powder (50 mg, 48.1%). Mw = 19308, Mw/Mn = 1.97 (GPC, PMMA calibration). 1H NMR (400 MHz, CDCl3, 298 K), δ (TMS, ppm): 8.20–8.04 (–ArH), 7.83–7.63 (–ArH), 7.61–7.49 (–ArH), 7.37 (–ArH), 7.33 (–ArH), 4.58 (–CH2–), 4.54 (–CH2–), 4.39 (–CH2–), 4.11 (–CH2–), 3.86 (–CH2–), 3.74 (–CH2–), 3.51 (–CH2–), 3.27 (–CH2–), 2.91 (–CH2–), 2.19 (–CH2–), 1.18–0.94 (–CH2–). 13C NMR (101 MHz, CDCl3, 298 K), δ (ppm): 156.08, 155.09, 150.37, 149.77, 148.22, 147.66, 147.21, 146.69, 145.32, 144.62, 126.19, 126.02, 124.61, 123.98, 122.86, 122.51, 117.25, 116.52, 111.87, 111.65, 111.40, 109.25, 70.49, 70.45, 69.50, 69.46, 68.63, 64.43, 53.50, 51.45, 51.14, 50.33, 47.64, 47.29, 45.68, 45.57, 28.63, 28.50, 21.97, 21.86, 12.11.
PY1o-oxy. Y1o (125.5 mg, 0.10 mmol), oxy (18.2 mg, 0.10 mmol), CuSO4 (130 + 65 + 130 + 65 μL), NaAsc (130 + 65 + 130 + 65 μL), in DMF (10 mL), a deep red powder (56 mg, 38.9%). Mw = 17414, Mw/Mn = 1.75 (GPC, PMMA calibration). 1H NMR (400 MHz, CDCl3, 298 K), δ (TMS, ppm): 8.31–8.24 (–ArH), 8.24–8.11 (–ArH), 7.87–7.70 (–ArH), 7.69–7.58 (–ArH), 7.54 (–ArH), 6.67–6.58 (–ArH), 6.53–6.45 (–ArH), 4.79 (–CH2–), 4.73 (–CH2–), 4.61 (–CH2–), 4.54 (–CH2–), 4.36–4.25 (–CH2–), 3.85 (–CH2–), 3.73–3.48 (–CH2–), 3.36–3.18 (–CH2–), 1.16–1.03 (–CH2–). 13C NMR (101 MHz, CDCl3, 298 K), δ (ppm): 156.09, 155.16, 150.40, 148.25, 147.80, 146.75, 145.36, 144.62, 144.54, 126.33, 126.08, 124.69, 123.93, 123.79, 122.85, 117.35, 116.91, 111.62, 111.44, 109.69, 70.52, 70.47, 69.96, 69.54, 68.51, 65.12, 64.47, 50.84, 50.35, 47.53, 47.12, 45.66, 12.15.
PY2-yl. Y2 (89.5 mg, 0.07 mmol), yl (9.7 mg, 0.07 mmol), CuSO4 (90 + 45 + 45 + 45 μL), NaAsc (90 + 45 + 45 + 45 μL), in DMF (7 mL), a deep red powder (38 mg, 38.3%). Mw = 18624, Mw/Mn = 1.88 (GPC, PMMA calibration). 1H NMR (400 MHz, CDCl3, 298 K), δ (TMS, ppm): 8.24–8.11 (–ArH), 7.89–7.69 (–ArH), 7.68–7.55 (–ArH), 7.29–7.23 (–ArH), 6.71–6.64 (–ArH), 6.63–6.54 (–ArH), 4.57 (–CH2–), 4.37 (–CH2–), 4.14 (–CH2–), 3.90 (–CH2–), 3.71 (–CH2–), 3.63–3.49 (–CH2–), 3.27 (–CH2–), 2.75–2.68 (–CH2–), 2.66–2.57 (–CH2–), 1.89 (–CH2–), 1.71 (–CH2–), 1.55 (–CH2–), 1.36–1.19 (–CH2–), 1.16–1.05 (–CH2–). 13C NMR (101 MHz, CDCl3, 298 K), δ (ppm): 156.09, 155.39, 155.34, 150.34, 148.43, 148.35, 148.28, 146.73, 144.65, 126.26, 126.11, 124.62, 122.83, 122.02, 117.18, 116.38, 111.70, 111.51, 111.41, 109.20, 69.82, 51.22, 50.53, 48.96, 47.53, 47.15, 45.73, 29.14, 28.67, 25.48, 12.13.
PY2o-yl. Y2o (134.3 mg, 0.10 mmol), yl (13.8 mg, 0.10 mmol), CuSO4 (130 + 65 + 65 + 65 μL), NaAsc (130 + 65 + 65 + 65 μL), in DMF (10 mL), a deep red powder (60 mg, 40.5%). Mw = 22683, Mw/Mn = 1.91 (GPC, PMMA calibration). 1H NMR (400 MHz, CDCl3, 298 K), δ (TMS, ppm): 8.34–8.11 (–ArH), 7.93–7.68 (–ArH), 7.66–7.47 (–ArH), 7.29 (–ArH), 6.62 (–ArH), 4.63 (–CH2–), 4.55 (–CH2–), 4.34 (–CH2–), 3.90 (–CH2–), 3.73 (–CH2–), 3.63 (–CH2–), 3.25 (–CH2–), 2.62 (–CH2–), 1.29 (–CH2–), 1.10 (–CH2–). 13C NMR (101 MHz, CDCl3, 298 K), δ (ppm): 156.16, 155.19, 150.42, 148.42, 148.22, 148.11, 146.94, 145.47, 144.66, 144.54, 126.31, 126.12, 124.69, 123.90, 122.93, 117.40, 116.87, 111.77, 111.33, 110.22, 77.40, 77.08, 76.76, 70.99, 70.15, 69.69, 69.53, 64.51, 51.20, 50.48, 47.50, 47.04, 45.73, 29.24, 28.60, 25.36, 12.10.
PY2-oxy. Y2 (153.5 mg, 0.12 mmol), oxy (22.2 mg, 0.12 mmol), CuSO4 (225 + 150 + 75 μL), NaAsc (225 + 150 + 75 μL), in DMF (12 mL), a deep red powder (60 mg, 37.5%). Mw = 22392, Mw/Mn = 1.62 (GPC, PMMA calibration). 1H NMR (400 MHz, CDCl3, 298 K), δ (TMS, ppm): 1 8.27–8.23 (–ArH), 8.21–8.11 (–ArH), 7.87–7.70 (–ArH), 7.67–7.59 (–ArH), 7.23 (–ArH), 6.74–6.64 (–ArH), 6.62–6.53 (–ArH), 4.67–4.50 (–CH2–), 4.41–4.25 (–CH2–), 4.22–4.08 (–CH2–), 3.96–3.82 (–CH2–), 3.76–3.50 (–CH2–), 3.37–3.24 (–CH2–), 2.77–2.62 (–CH2–), 1.88 (–CH2–), 1.71 (–CH2–), 1.54 (–CH2–), 1.19–0.98 (–CH2–). 13C NMR (101 MHz, CDCl3, 298 K), δ (ppm): 156.18, 155.40, 155.34, 150.28, 149.49, 148.36, 147.76, 146.72, 145.31, 144.70, 126.21, 124.68, 123.89, 122.86, 122.04, 117.20, 116.42, 111.74, 111.48, 109.25, 70.49, 69.81, 69.55, 64.49, 51.18, 50.41, 47.59, 47.13, 45.73, 29.13, 28.76, 8.66, 25.77, 25.48, 12.14.
PY2o-oxy. Y2o (161.2 mg, 0.12 mmol), oxy (22.2 mg, 0.12 mmol), CuSO4 (225 + 150 + 75 + 75 μL), NaAsc (225 + 150 + 75 + 75 μL), in DMF (12 mL), a deep red powder (80 mg, 43.7%). Mw = 24876, Mw/Mn = 1.85 (GPC, PMMA calibration). 1H NMR (400 MHz, CDCl3, 298 K), δ (TMS, ppm): 8.39–8.14 (–ArH), 7.94–7.70 (–ArH), 7.68–7.49 (–ArH), 6.71–6.55 (–ArH), 4.62 (–CH2–), 4.57 (–CH2–), 4.34 (–CH2–), 4.26 (–CH2–), 3.89 (–CH2–), 3.72 (–CH2–), 3.63 (–CH2–), 3.58 (–CH2–), 3.27 (–CH2–), 1.09 (–CH2–). 13C NMR (101 MHz, CDCl3, 298 K), δ (ppm): 156.18, 155.13, 150.38, 149.22, 148.24, 147.82, 146.83, 145.40, 145.25, 144.55, 126.30, 126.12, 124.69, 123.96, 122.92, 117.40, 116.86, 111.76, 109.70, 70.97, 70.49, 70.10, 69.66, 69.55, 64.46, 51.23, 50.32, 47.57, 47.03, 45.73, 12.12.
Preparation of polymer thin films
The polymers were dissolved in dichloromethane (concentration 30 mg mL−1), and the solutions were fully oscillated and left to rest overnight so that the polymers were completely swelled and dissolved. Then the solutions were filtered through a 0.22 μm syringe filters and spin-coated onto the non-conductive side of indium–tin–oxide (ITO)-coated glass substrates, which were cleaned by ultrasonic treatment in different solvents of water, acetone, deionized water, DMF, and THF before use. Residual solvent was removed by heating the films in a vacuum oven at 40 °C. The thicknesses of the prepared films were usually about 200 nm, which can not only ensure unbroken films under high voltages but also achieve a high poling efficiency.NLO measurement of poled films
The second-order NLO efficiencies of the compounds were measured by an in situ SHG experiment using a closed temperature-controlled oven with optical windows and three-needle electrodes. The films were kept at 45° to the incident beam and poled inside the oven, and the SHG intensity was monitored simultaneously. The poling temperatures of the films were different (Table 1). They shared the same other poling conditions: voltage, 7.0 kV at the needlepoint; gap distance, 0.8 cm. The SHG measurements were carried out using an Nd:YAG laser operating at a 10 Hz repetition rate and an 8 ns pulse width at 1064 nm. A Y-cut quartz crystal served as the reference.After poling, the de-poling curves of second-order NLO materials were obtained as follows: poled films were heated gradually from room temperature until no obvious signals were observed in the absence of an applied electric field. The heating rate was 4 °C min−1.
Author contributions
Q.L. and Z.L.: conceptualization, funding acquisition, project administration, supervision, writing – review & editing; P.Q.: formal analysis, investigation, validation, visualization, writing – original draft; W.Y.: formal analysis.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22235006 and 22122504) and Foundation of Hubei Scientific Committee (2022BAA015 and 2022EHB010). We are grateful to National Supercomputing Centre in Shenzhen and Beijing Parallel Technology for theoretical calculation support.References
- S. R. Marder, B. Kippelen, A. K. Y. Jen and N. Peyghambarian, Nature, 1997, 388, 845–851 Search PubMed.
- L. R. Dalton, P. A. Sullivan and D. H. Bale, Chem. Rev., 2010, 110, 25–55 Search PubMed.
- D. M. Burland, R. D. Miller and C. A. Walsh, Chem. Rev., 1994, 94, 31–75 Search PubMed.
- Y. Shi, C. Zhang, H. Zhang, J. H. Bechtel, L. R. Dalton, B. H. Robinson and W. H. Steier, Science, 2000, 288, 119–122 Search PubMed.
- Y. Bai, N. Song, J. P. Gao, X. Sun, X. Wang, G. Yu and Z. Y. Wang, J. Am. Chem. Soc., 2005, 127, 2060–2061 Search PubMed.
- A. W. Harper, S. Sun, L. R. Dalton, S. M. Garner, A. Chen, S. Kalluri, W. H. Steier and B. H. Robinson, J. Opt. Soc. Am. B, 1998, 15, 329–337 Search PubMed.
- B. H. Robinson, L. R. Dalton, A. W. Harper, A. Ren, F. Wang, C. Zhang, G. Todorova, M. Lee, R. Aniszfeld, S. Garner, A. Chen, W. H. Steier, S. Houbrecht, A. Persoons, I. Ledoux, J. Zyss and A. K. Y. Jen, Chem. Phys., 1999, 245, 35–50 Search PubMed.
- Q. Li and Z. Li, Acc. Chem. Res., 2020, 53, 962–973 Search PubMed.
- A. Huang, Q. Li and Z. Li, Chin. J. Chem., 2022, 40, 2356–2370 Search PubMed.
- S. Li, Q. Li and Z. Li, Prog. Chem., 2022, 34, 1554–1575 Search PubMed.
- D. Yu, A. Gharavi and L. Yu, J. Am. Chem. Soc., 1995, 117, 11680–11686 Search PubMed.
- W. H. Steier, A. Chen, S. S. Lee, S. Garner, H. Zhang, V. Chuyanov, L. R. Dalton, F. Wang, A. S. Ren, C. Zhang, G. Todorova, A. Harper, H. R. Fetterman, D. Chen, A. Udupa, D. Bhattacharya and B. Tsap, Chem. Phys., 1999, 245, 487–506 Search PubMed.
- Y. Yue, B. Zheng, M. Liu, Y. Chen, L. Huo, J. Wang and L. Jiang, Sci. China Mater., 2022, 65, 3402–3410 Search PubMed.
- Y. Fan, Q. Li and Z. Li, Sci. China: Chem., 2023, 66, 2930–2940 Search PubMed.
- Y. Gao, J. Lu, Q. Liao, S. Li, Q. Li and Z. Li, Natl. Sci. Rev., 2023, 10, nwad239 Search PubMed.
- J. Ni, Q. Zang, H. Zhang, J. Z. Sun and B. Z. Tang, Sci. China Mater., 2023, 66, 1959–1967 Search PubMed.
- Z. Li, Z. Li, C. Di, Z. Zhu, Q. Li, Q. Zeng, K. Zhang, Y. Liu, C. Ye and J. Qin, Macromolecules, 2006, 39, 6951–6961 Search PubMed.
- T. D. Kim, J. W. Kang, J. Luo, S. H. Jang, J. W. Ka, N. Tucker, J. B. Benedict, L. R. Dalton, T. Gray, R. M. Overney, D. H. Park, W. N. Herman and A. K. Y. Jen, J. Am. Chem. Soc., 2007, 129, 488–489 Search PubMed.
- Z. Shi, J. Luo, S. Huang, X. H. Zhou, T.-D. Kim, Y. J. Cheng, B. M. Polishak, T. R. Younkin, B. A. Block and A. K. Y. Jen, Chem. Mater., 2008, 20, 6372–6377 Search PubMed.
- Z. Li, Q. Li and J. Qin, Polym. Chem., 2011, 2, 2723–2740 Search PubMed.
- W. Wu, C. Ye, J. Qin and Z. Li, Polym. Chem., 2013, 4, 2361–2370 Search PubMed.
- R. Tang, H. Chen, S. Zhou, B. Liu, D. Gao, H. Zeng and Z. Li, Polym. Chem., 2015, 6, 6680–6688 Search PubMed.
- W. Wu, C. Ye, G. Yu, Y. Liu, J. Qin and Z. Li, Chem. – Eur. J., 2012, 18, 4426–4434 Search PubMed.
- W. Wu, Y. Fu, C. Wang, Z. Xu, C. Ye, J. Qin and Z. Li, Chin. J. Polym. Sci., 2013, 31, 1415–1423 Search PubMed.
- W. Wu, S. Xin, Z. Xu, C. Ye, J. Qin and Z. Li, Polym. Chem., 2013, 4, 3196–3203 Search PubMed.
- W. Wu, C. Ye, J. Qin and Z. Li, Chem. – Asian J., 2013, 8, 1836–1846 Search PubMed.
- P. Qiao, Q. Li and Z. Li, Chin. Sci. Bull., 2023, 68, 4015–4034 Search PubMed.
- A. Rahman, W. Zhang, S. Bo, A. Ali, R. Wahab, K. Shehzad and F. Liu, Dyes Pigm., 2024, 231, 112379 Search PubMed.
- K. Wang, X. Deng, Q. Li and Z. Li, Polym. Chem., 2023, 14, 2205–2211 Search PubMed.
- R. A. Buono, R. J. Zauhar and C. A. Venanzi, J. Mol. Struct., 1996, 370, 97–133 Search PubMed.
- E. Sabbaghian, S. Mehdipour Ataei, S. Jalilian, M. Esfahanizadeh, A. M. Salehi, F. Khodabakhshi and E. Jalalian, Polym. Adv. Technol., 2015, 26, 1–9 Search PubMed.
- F. Wurm and H. Frey, Prog. Polym. Sci., 2011, 36, 1–52 Search PubMed.
- H. Sun, F. M. Haque, Y. Zhang, A. Commisso, M. A. Mohamed, M. Tsianou, H. Cui, S. M. Grayson and C. Cheng, Angew. Chem., Int. Ed., 2019, 58, 10572–10576 Search PubMed.
- Z. Zhu, Z. Li, Y. Tan, Z. Li, Q. Li, Q. Zeng, C. Ye and J. Qin, Polymer, 2006, 47, 7881–7888 Search PubMed.
- J. Xie, L. Hu, W. Shi, X. Deng, Z. Cao and Q. Shen, Polym. Int., 2008, 57, 965–974 Search PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 Search PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 Search PubMed.
- H. Sun, S. Zhang and V. Percec, Chem. Soc. Rev., 2015, 44, 3900–3923 Search PubMed.
- L. Xu, J. Zhang, L. Yin, X. Long, W. Zhang and Q. Zhang, J. Mater. Chem. C, 2020, 8, 6342–6349 Search PubMed.
- L. Xu, W. Lin, B. Huang, J. Zhang, X. Long, W. Zhang and Q. Zhang, J. Mater. Chem. C, 2021, 9, 1520–1536 Search PubMed.
- Q. Zeng, Z. a. Li, Z. Li, C. Ye, J. Qin and B. Z. Tang, Macromolecules, 2007, 40, 5634–5637 Search PubMed.
- Z. Li, W. Wu, C. Ye, J. Qin and Z. Li, Phys. Chem. B, 2009, 113, 14943–14949 Search PubMed.
- G. Liu, Q. Liao, H. Deng, W. Zhao, P. Chen, R. Tang, Q. Li and Z. Li, J. Mater. Chem. C, 2019, 7, 7344–7351 Search PubMed.
- R. Tang, S.-M. Zhou, Z. Cheng, H. Chen, L. Deng, Q. Peng and Z. Li, CCS Chem., 2020, 2, 1040–1048 Search PubMed.
- X. Deng, K. Wang, Q. Li and Z. Li, Mater. Chem. Front., 2022, 6, 3349–3358 Search PubMed.
- Z. Li, G. Yu, P. Hu, C. Ye, Y. Liu, J. Qin and Z. Li, Macromolecules, 2009, 42, 1589–1596 Search PubMed.
- J. Liu, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2009, 109, 5799–5867 Search PubMed.
- A. Qin, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2010, 39, 2522–2544 Search PubMed.
- C. Cabanetos, E. Blart, Y. Pellegrin, V. Montembault, L. Fontaine, F. Adamietz, V. Rodriguez and F. Odobel, Eur. Polym. J., 2012, 48, 116–126 Search PubMed.
- W. Wu and Z. Li, Polym. Chem., 2014, 5, 5100–5108 Search PubMed.
- D. Huang, Y. Liu, A. Qin and B. Z. Tang, Polym. Chem., 2018, 9, 2853–2867 Search PubMed.
- W. Wu, G. Xu, C. Li, G. Yu, Y. Liu, C. Ye, J. Qin and Z. Li, Chem. – Eur. J., 2013, 19, 6874–6888 Search PubMed.
- Z. Li, W. Wu, Q. Li, G. Yu, L. Xiao, Y. Liu, C. Ye, J. Qin and Z. Li, Angew. Chem., Int. Ed., 2010, 49, 2763–2767 Search PubMed.
- Z. Li, G. Yu, W. Wu, Y. Liu, C. Ye, J. Qin and Z. Li, Macromolecules, 2009, 42, 3864–3868 Search PubMed.
- R. Tang, H. Chen, S. Zhou, W. Xiang, X. Tang, B. Liu, Y. Dong, H. Zeng and Z. Li, Polym. Chem., 2015, 6, 5580–5589 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- Z. Li, Q. Zeng, Z. Li, S. Dong, Z. Zhu, Q. Li, C. Ye, C. a. Di, Y. Liu and J. Qin, Macromolecules, 2006, 39, 8544–8546 Search PubMed.
- Z. Li, P. Hu, G. Yu, W. Zhang, Z. Jiang, Y. Liu, C. Ye, J. Qin and Z. Li, Chem. Chem. Phys., 2009, 11, 1220–1226 Search PubMed.
- Z. Li, G. Qiu, C. Ye, J. Qin and Z. Li, Dyes Pigm., 2012, 94, 16–22 Search PubMed.
- Z. Li, W. Wu, G. Yu, Y. Liu, C. Ye, J. Qin and Z. Li, ACS Appl. Mater. Interfaces, 2009, 1, 856–863 Search PubMed.
- W. Wu, Q. Huang, G. Qiu, C. Ye, J. Qin and Z. Li, J. Mater. Chem. C, 2012, 22, 18486–18495 Search PubMed.
- W. Wu, Q. Huang, C. Zhong, C. Ye, J. Qin and Z. Li, Polymer, 2013, 54, 5655–5664 Search PubMed.
- W. Wu, C. Ye, J. Qin and Z. Li, Chem. – Asian J., 2013, 8, 1836–1846 Search PubMed.
- G. Ungar, J. Stejny, A. Keller, I. Bidd and M. C. Whiting, Science, 1985, 229, 386–389 Search PubMed.
- A. Keller, Rep. Prog. Phys., 1968, 31, 623–704 Search PubMed.
- C. Xu, B. Wu, M. W. Becker, L. R. Dalton, P. M. Ranon, Y. Shi and W. H. Steier, Chem. Mater., 1993, 5, 1439–1444 Search PubMed.
- P. A. Sullivan, A. J. P. Akelaitis, S. K. Lee, G. McGrew, S. K. Lee, D. H. Choi and L. R. Dalton, Chem. Mater., 2006, 18, 344–351 Search PubMed.
- Z. Cheng, R. Tang, R. Wang, Y. Xie, P. Chen, G. Liu and Z. Li, Polym. Chem., 2018, 9, 3522–3527 Search PubMed.
- R. Wang, Z. Cheng, X. Deng, W. Zhao, Q. Li and Z. Li, J. Mater. Chem. C, 2020, 8, 6380–6387 Search PubMed.
- Y. Tian, Y. He, P. Liu, H. Zhang, Q. Zheng, J. Liu, L. Xiao, X. Wang, Y. Ao and M. Li, J. Mater. Sci., 2021, 56, 5910–5923 Search PubMed.
- X. You, P. Wang, Y. Tan, Y. Li, J. Wang, Z. Li, Y. Ao and M. Li, Mater. Today Chem., 2024, 37, 101971 Search PubMed.
- Q. Liao, Q. Li and Z. Li, Adv. Mater., 2023, 35, 2306617 Search PubMed.
- J. Yang, X. Zhen, B. Wang, X. Gao, Z. Ren, J. Wang, Y. Xie, J. Li, Q. Peng, K. Pu and Z. Li, Nat. Commun., 2018, 9, 840 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The detailed synthesis and MALDI-TOF spectra of Y-shaped monomers; FTIR-ATR spectra, 1H NMR spectra, and 13C NMR spectra of monomers and polymers. See DOI: https://doi.org/10.1039/d4py01238k |
| This journal is © The Royal Society of Chemistry 2025 |