
Tannic acid salt-modified CoFe-layered double hydroxide boosts stable seawater oxidation at an industrial-level current density†
Zhengwei
Cai‡
a,
Yaxin
Guo‡
a,
Chaoxin
Yang
 a,
Zixiao
Li
a,
Shengjun
Sun
a,
Meng
Yue
a,
Xiaoyan
Wang
a,
Min
Zhang
a,
Hefeng
Wang
a,
Yongchao
Yao
b,
Dongdong
Zheng
a,
Asmaa
Farouk
c,
Fatma A.
Ibrahim
c,
Yanqin
Lv
*a,
Xuping
Sun
*ab and
Bo
Tang
*ad
a,
Zixiao
Li
a,
Shengjun
Sun
a,
Meng
Yue
a,
Xiaoyan
Wang
a,
Min
Zhang
a,
Hefeng
Wang
a,
Yongchao
Yao
b,
Dongdong
Zheng
a,
Asmaa
Farouk
c,
Fatma A.
Ibrahim
c,
Yanqin
Lv
*a,
Xuping
Sun
*ab and
Bo
Tang
*ad
aCollege of Chemistry, Chemical Engineering and Materials Science, Shandong Normal University, Jinan 250014, Shandong, China. E-mail: yanqinLv@sdnu.edu.cn; xpsun@uestc.edu.cn; tangb@sdnu.edu.cn
bCenter for High Altitude Medicine, West China Hospital, Sichuan University, Chengdu 610041, Sichuan, China
cDepartment of Chemistry, College of Science, King Khalid University, 61413 Abha, Saudi Arabia
dLaoshan Laboratory, Qingdao 266237, Shandong, China
First published on 18th November 2024
Abstract
Seawater electrolysis for green hydrogen production is a promising approach toward achieving carbon neutrality. However, the abundance of Cl− in seawater can severely corrode catalytic sites, significantly reducing the lifespan of seawater electrolysis systems. Herein, we present metal ion-chelated tannic acid nanoparticles anchored on the CoFe layered double hydroxide nanosheet array on nickel foam (CoFe LDH@CoFe-TA/NF), synthesized via an interfacial coordination assembly method, serving as an efficient and stable electrocatalyst for alkaline seawater oxidation (ASO). The formed CoFe-TA nanoparticles promote the transformation of Co3+ into the more robust acid Co4+, thereby favoring the adsorption of the hard base OH− rather than the soft base Cl−. In addition, the CoFe-TA ligand network effectively inhibits metal ion leaching and stabilizes active sites. As a result, the CoFe LDH@CoFe-TA/NF electrode requires a low overpotential of only 379 mV to obtain a current density of 1000 mA cm−2 in 1 M KOH + seawater. Furthermore, the electrode also shows a stable operation for 450 h at an industrial-level current density, underscoring its potential for sustainable energy applications.
10th anniversary statementOur team has been working on the rational design, structural modulation, and catalytic and sensing applications of functional nanomaterials. Inorganic Chemistry Frontiers provides us with an excellent platform to demonstrate our recent research advances in inorganic chemistry and related fields. It is our great pleasure to have published 37 papers since its launch and also my honor to be invited to be one of the guest editors for the themed collection on nitrogen-cycle electrocatalysis. ICF is developing rapidly, and I anticipate ICF to publish more excellent papers and continue to increase its influence in the future! |
Hydrogen (H2) is widely recognized as a super-versatile energy carrier for a carbon-neutral and sustainable future, which is due to its non-polluting emission and high energy density.1–3 Water electrolysis is a sustainable and cost-effective method for producing high-purity H2, utilizing waste heat or renewable yet intermittent sources like wind tidal and wind energy.4,5 However, the large-scale implementation of water-splitting technologies leads to significant freshwater consumption, which puts immense pressure on precious water resources.6,7 Seawater is widely regarded as a promising feedstock for water electrolysis, given that it accounts for 96.5% of the Earth's water resources and also distributes more even than freshwater.8–12 Nevertheless, this process is primarily limited by the oxygen evolution reaction (OER) at the anode. Specifically, the chlorine evolution reaction (CER) competes with the OER due to the faster reaction rate of the CER.13,14 In particular, both the chloride ions (Cl−) and their derivatives (Cl2/HClO/ClO−) would continuously and aggressively corrode the electrode during the seawater oxidation process, thus reducing the lifespan of seawater electrolysis systems.15,16 Therefore, the research and design of chlorine-resistant catalysts are of importance for seawater oxidation.
Transition metal-based electrocatalysts have garnered significant interest due to their abundant availability, low cost, and high catalytic performance.17,18 Among these catalysts, CoFe layered double hydroxide (CoFe LDH) has gained significant attention as an excellent OER catalyst due to its unique structure, surface hydroxyl groups, and tunable properties.19,20 Unfortunately, the products of the CER (Cl2/HClO/ClO−) can corrode the catalyst, leading to poor anode durability and thereby limiting its industrial application.21,22 To address this challenge, several effective approaches have been proposed, such as the anion protection strategy and inert material coating strategy.23–28 Although significant progress has been made in seawater electrolysis, the reported strategies still face certain limitations. For instance, the limited coverage and electrostatic repulsion of anions lead to deficiencies in long-term resistance to Cl− corrosion at industrial-level current densities (j).29,30 Previous research has shown that the in situ formation of a metal oxide/oxy(hydroxide) layer on the anode surface during the OER process can enhance the catalyst's resistance to chlorine corrosion.31,32 Tannic acid (TA) is an eco-friendly natural polyphenol with a strong ability to coordinate with metal ions, enabling coordination compounds of TA and metal ions to easily form coatings on various substrates.33 Additionally, studies have shown that the TA–Fe ligand network can effectively prevent the dissolution of Fe ions and rapidly promote the formation of active oxy(hydroxide) phases, thereby enhancing the catalytic activity and stability during alkaline freshwater oxidation.34 Therefore, the preparation of the TA salt-modified CoFe LDH is expected to promote the in situ formation of a metal oxide/oxy(hydroxide) layer, enhancing the chlorine resistance of the catalyst at ampere-level j during the alkaline seawater oxidation (ASO) process.
In this study, a surface modification strategy has been developed to enhance the corrosion resistance of the catalyst by constructing CoFe-TA nanoparticles on the CoFe LDH surface supported by nickel foam (CoFe LDH@CoFe-TA/NF). CoFe-TA nanoparticles, synthesized via an interfacial self-assembly method, are dispersed on the CoFe LDH surface, promoting rapid in situ formation of a CoFe oxy(hydroxide) layer, resulting in excellent OER activity and chlorine resistance. Moreover, the CoFe-TA ligand network effectively prevents the dissolution of Co and Fe ions, further enhancing the catalyst's stability during the ASO process. The CoFe LDH@CoFe-TA/NF requires an overpotential (η) of 379 mV to reach 1000 mA cm−2 for ASO. It also demonstrates sustained stability for up to 450 h at 1000 mA cm−2. After long-term stability testing, ultraviolet-visible (UV-vis) spectroscopy analysis shows that almost no active chlorine is detected in the electrolyte. Notably, the CoFe LDH@CoFe-TA/NF exhibits high stability at 1000 mA cm−2 in an alkaline electrolyte with a high Cl− concentration (1 M KOH + 2 M NaCl). These results demonstrated a larger practical application perspective for such materials for seawater electrolysis.
CoFe LDH@CoFe-TA/NF was synthesized using a straightforward hydrothermal process followed by a soaking treatment (Fig. 1a). In comparison with the typical diffraction peaks in the X-ray diffraction (XRD) pattern of CoFe LDH (JCPDS No. 50-0235), relatively weak diffraction peaks can be observed after the interfacial coordination (Fig. 1b). This confirms that the interfacial coordination did not change the structure of CoFe LDH/NF.35,36 Additionally, two prominent peaks at 44.50°, 51.85°, and 76.37° can be attributed to the NF. As can be seen in the SEM images, CoFe LDH@CoFe-TA/NF shows a rough nanosheet structure (Fig. 1c and d), while CoFe LDH/NF shows a smooth nanosheet structure (Fig. S1†). Fig. 1e shows the corresponding energy dispersive X-ray (EDX) elemental mapping images of CoFe LDH@CoFe-TA/NF, which reveal the homogenous distributions of the Co, Fe, C, and O elements. The transmission electron microscopy (TEM) image of CoFe LDH@CoFe-TA shows that the nanoparticles are uniformly distributed on the nanosheet (Fig. 1f). Furthermore, the high-resolution TEM (HRTEM) image (Fig. 1g) of CoFe LDH@CoFe-TA shows a lattice spacing of 0.24 nm, which is indexed to the (104) plane of CoFe LDH. In addition, a distinct amorphous region is visible in the yellow line area, providing evidence for the formation of the amorphous phase of CoFe-TA nanoparticles.
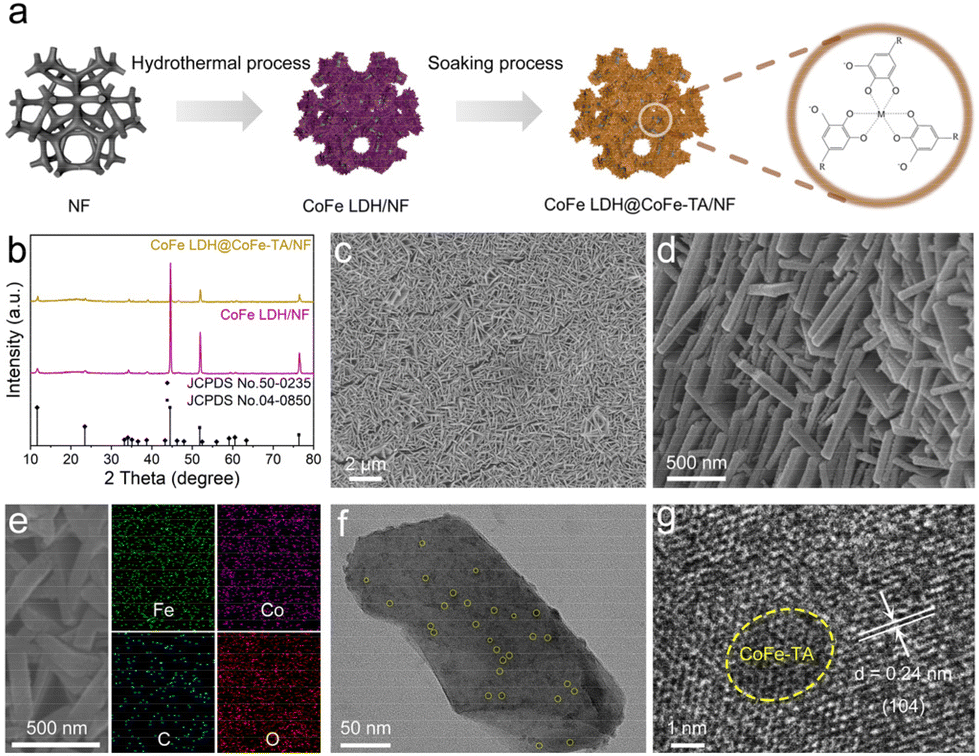 | ||
| Fig. 1 (a) The schematic diagram for the fabrication of CoFe LDH@CoFe-TA/NF. (b) XRD patterns of CoFe LDH@CoFe-TA/NF and CoFe LDH/NF. (c) Low- and (d) high-magnification SEM images of CoFe LDH@CoFe-TA/NF. (e) SEM and the corresponding EDX elemental mapping images of CoFe LDH@CoFe-TA/NF. (f) TEM and (g) HRTEM images of CoFe LDH@CoFe-TA. | ||
Furthermore, X-ray photoelectron spectroscopy (XPS) is used to characterize the surface electronic structure and the interactions between TA and CoFe LDH. The XPS spectra of both samples show peaks of Co, Fe, O, and C (Fig. 2a). Additionally, the high-resolution Fe 2p XPS spectrum reveals two distinct peaks corresponding to 2p3/2 and 2p1/2, located at 712.72 and 725.92 eV, respectively (Fig. 2b).37 Compared with CoFe LDH, the Fe 2p peak for CoFe LDH@CoFe-TA shifts to higher binding energy, which can be attributed to the transfer of electrons from Fe to TA.34 In the Co 2p region, the peaks at 798.94 and 784.68 eV correspond to Co2+, while the peaks at 797.10 and 781.39 eV correspond to Co3+. The slight positive shifts observed for CoFe LDH@CoFe-TA indicate the interaction between the Co and Fe species (Fig. 2c).38,39 For C 1s spectra of CoFe LDH@CoFe-TA and CoFe LDH (Fig. 2d), the C content of CoFe LDH@CoFe-TA is obviously more than that of CoFe LDH. Moreover, the contents of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (288.85 eV) and C–O (286.50 eV) are significantly increased.21 Furthermore, in the O 1s region, the peaks at 530.12, 530.99, 531.61, and 532.51 eV are ascribed to M–O, CO, –OH, and C–O, respectively (Fig. 2e).35,40 The presence of the CO peak indicates the successful preparation of CoFe-TA nanoparticles. This result agrees well with the results from Fourier transform infrared spectroscopy (FT-IR). FTIR spectra (Fig. 2f) show that the peaks of CoFe LDH@CoFe-TA at 1032, 1190, and 1350 cm−1 correspond to the C–O stretching vibration of the phenolic hydroxyl group, while the peak at 1698 cm−1 corresponds to the CO stretching vibration of the ester group in the TA molecule.41
O (288.85 eV) and C–O (286.50 eV) are significantly increased.21 Furthermore, in the O 1s region, the peaks at 530.12, 530.99, 531.61, and 532.51 eV are ascribed to M–O, CO, –OH, and C–O, respectively (Fig. 2e).35,40 The presence of the CO peak indicates the successful preparation of CoFe-TA nanoparticles. This result agrees well with the results from Fourier transform infrared spectroscopy (FT-IR). FTIR spectra (Fig. 2f) show that the peaks of CoFe LDH@CoFe-TA at 1032, 1190, and 1350 cm−1 correspond to the C–O stretching vibration of the phenolic hydroxyl group, while the peak at 1698 cm−1 corresponds to the CO stretching vibration of the ester group in the TA molecule.41
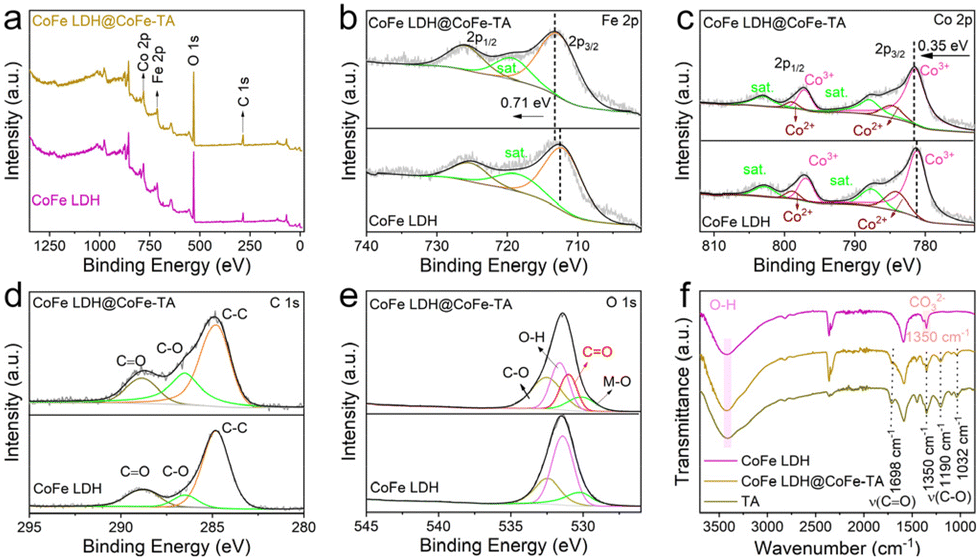 | ||
| Fig. 2 (a) Survey spectra of XPS for CoFe LDH@CoFe-TA and CoFe LDH. High-resolution XPS spectra of CoFe LDH@CoFe-TA and CoFe LDH in the (b) Fe 2p, (c) Co 2p, (d) C 1s, and (e) O 1s regions. (f) FT-IR spectra of CoFe LDH@CoFe-TA, CoFe LDH, and TA. | ||
Furthermore, the OER performance of different catalysts was measured in 1 M KOH. The linear scanning voltammetry (LSV) curves of different catalysts show the superior catalytic performance of CoFe LDH@CoFe-TA/NF, compared with the RuO2/NF (a benchmark OER electrocatalyst) (Fig. 3a). CoFe LDH@CoFe-TA/NF only needs a η of 310 and 347 mV to achieve a j of 500 and 1000 mA cm−2, respectively, which are lower than those for the RuO2/NF. As can be seen in Fig. 3b, the Tafel slopes of different materials follow the order: 32.71 < 33.68 < 86.22 < 171.58 mV dec−1, indicating that CoFe LDH@CoFe-TA/NF has advantages in reaction kinetics. Moreover, electrochemical impedance spectroscopy is used for the characterization of the charge transfer properties of the electrocatalysts. As shown in Fig. 3c, CoFe LDH@CoFe-TA/NF shows a lower charge-transfer resistance (Rct) value (4.76 Ω) than CoFe LDH/NF (6.30 Ω), which indicates that CoFe LDH@CoFe-TA/NF has faster OER kinetics than CoFe LDH/NF. In addition, in situ Nyquist and Bode plots are recorded from 1.42 V to 1.47 V versus the reversible hydrogen electrode (RHE) to confirm the conductivity and interfacial charge transfer of the catalysts.42,43 Over the recorded voltage range, CoFe LDH@CoFe-TA/NF shows smaller Rct values than CoFe LDH/NF (Fig. 3d), implying an easy proton transport and indicating good reaction kinetics. Also, CoFe LDH@CoFe-TA/NF shows a lower phase angle than CoFe LDH/NF in the high-frequency region (Fig. 3e), which confirms its better internal electron conduction. Furthermore, the phase angle of CoFe LDH@CoFe-TA/NF decreases sharply with an increase of the voltage in the low-frequency region (Fig. 3f), indicating that the metal oxide/oxy(hydroxide) layer can promote the charge transport in the interfacial reactions. In addition, the stability of CoFe LDH@CoFe-TA/NF is confirmed from the multistep chronopotentiometric curve (Fig. S2†), which shows that the CoFe LDH@CoFe-TA/NF remains at a nearly constant potential value during 600 s and for various j values, indicating an excellent mass transfer and mechanical stability of CoFe LDH@CoFe-TA/NF. In addition, a small attenuation is observed in the LSV curve after 3000 cyclic voltammetry (CV) cycles, which also highlights the stability of the CoFe LDH@CoFe-TA/NF (Fig. S3†).
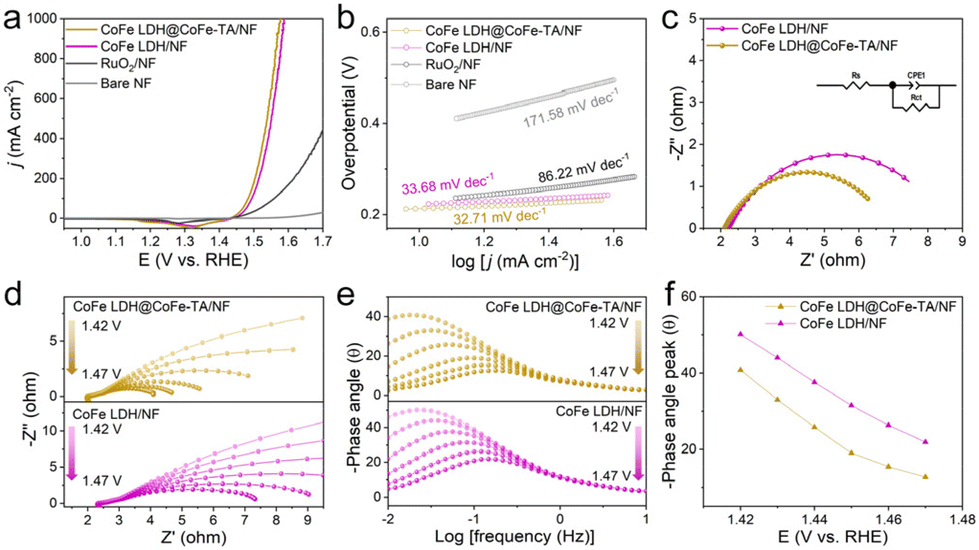 | ||
| Fig. 3 (a) LSV curves and (b) corresponding Tafel plots of CoFe LDH@CoFe-TA/NF, CoFe LDH/NF, RuO2/NF, and bare NF in 1 M KOH. (c) Nyquist plots of CoFe LDH@CoFe-TA/NF and CoFe LDH/NF. (d) Nyquist plots and (e) Bode plots of CoFe LDH@CoFe-TA/NF and CoFe LDH/NF at various potentials vs. RHE. (f) Phase peak angles of different prepared samples at 1.42–1.47 V (vs. RHE). | ||
Given the excellent OER catalytic performance of CoFe LDH@CoFe-TA/NF in alkaline freshwater, we further investigated its OER activity in various electrolytes, including simulated alkaline seawater (1 M KOH + 0.5 M NaCl) and alkaline seawater (1 M KOH + seawater) (Fig. 4a). The results show that CoFe LDH@CoFe-TA/NF displays excellent OER activity in all three electrolyte solutions. Notably, the η of the catalyst is presented in Fig. 4b, illustrating that CoFe LDH@CoFe-TA/NF in 1 M KOH + seawater requires a η of 267, 363, and 379 mV to achieve a j of 100, 500, and 1000 mA cm−2, respectively. As seen in Fig. S4,† CoFe LDH@CoFe-TA/NF still shows better activity than CoFe LDH/NF in 1 M KOH + seawater. In addition, as demonstrated in Fig. 4c, CoFe LDH@CoFe-TA/NF is superior in comparison with many other reported catalysts in terms of catalytic activity. Moreover, the turnover frequency (TOF) values of the catalysts can be calculated to evaluate the intrinsic catalytic activity and instantaneous efficiency during the ASO process. CoFe LDH@CoFe-TA/NF shows higher TOF values than CoFe LDH/NF, which confirms the superiority of CoFe LDH@CoFe-TA/NF (Fig. S5†).44,45 Moreover, the electrochemical activation energy (Ea) is a well-known empirical parameter in chemical kinetics, which demonstrates the intrinsic activity of the catalyst. Ea is obtained from the LSV curves at different temperatures (Fig. S6†). Ea values of 40.15 and 69.39 kJ mol−1 were obtained for CoFe LDH@CoFe-TA/NF and CoFe LDH/NF, respectively, revealing that CoFe LDH@CoFe-TA/NF has higher intrinsic activity in alkaline seawater.46,47
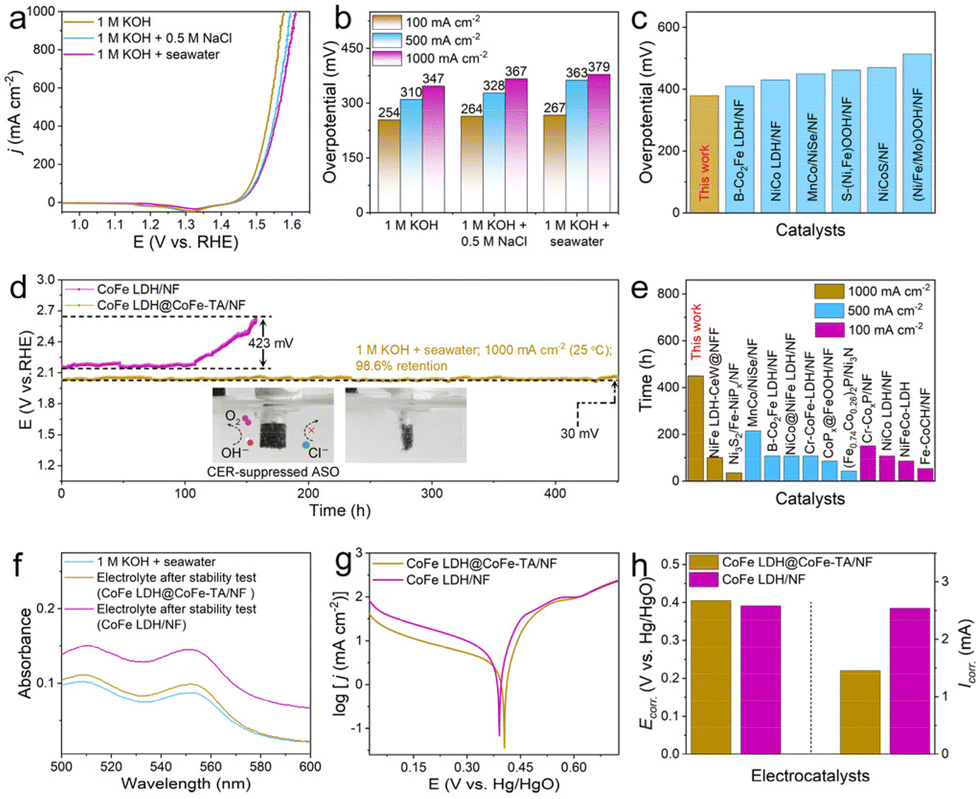 | ||
| Fig. 4 (a) LSV curves and (b) corresponding overpotential comparison of CoFe LDH@CoFe-TA/NF in different electrolytes. (c) Comparison of the overpotentials between CoFe LDH@CoFe-TA/NF and other reported electrocatalysts at 1000 mA cm−2 in 1 M KOH + seawater. (d) Chronopotentiometry curves of CoFe LDH@CoFe-TA/NF and CoFe LDH/NF without iR compensation in 1 M KOH + seawater (insets: digital images of CoFe LDH@CoFe-TA/NF during the ASO process). (e) Comparison of the stability of CoFe LDH@CoFe-TA/NF in 1 M KOH + seawater with other reported electrocatalysts. (f) UV-vis absorption spectra of the electrolytes after the stability test at 1000 mA cm−2. (g) Corrosion polarization curves and (h) corresponding Ecorr. and Icorr. of CoFe LDH@CoFe-TA/NF and CoFe LDH/NF in 1 M KOH + seawater. | ||
Most importantly, CoFe LDH@CoFe-TA/NF not only exhibits excellent OER activity but also demonstrates remarkable stability. It operates steadily in alkaline seawater for 450 h at a j of 1000 mA cm−2, maintaining an intact morphology, whereas CoFe LDH/NF shows severe degradation and a corroded morphology after only 120 h (Fig. 4d and S7†). Moreover, Fig. S8† demonstrates that the crystal phase of CoFe LDH@CoFe-TA/NF remains almost unchanged, while Fig. S9† shows that its morphology is well preserved, indicating that CoFe-TA nanoparticles effectively inhibit metal element release and repel Cl−. In addition, CoFe LDH@CoFe-TA/NF is also found to be superior to other reported catalysts (Fig. 4e and Table S1†). It can be ascribed to the reconfiguration of the amorphous CoFe-TA nanoparticles and the generation of CoFe oxy(hydroxide) layers on the anode surface. Subsequently, to assess resistance to chlorine corrosion, the concentration of active chlorine in the electrolyte after electrolysis was measured using the N,N-diethyl-p-phenylenediamine method. As shown in Fig. 4f and S10,† the concentration of active chlorine in the electrolyte of CoFe LDH@CoFe-TA/NF is lower than that in the electrolyte of CoFe LDH/NF. We also detected the presence of hypochlorite in the solution after stability testing using the iodometric titration method and colorimetric paper. As shown in Fig. S11,† the electrolyte after the stability test (CoFe LDH/NF) exhibits a noticeable yellow color after adding KI, indicating the formation of ClO−. Titration with sodium thiosulfate revealed a ClO− concentration of 3.4 mmol L−1. Notably, the amount of ClO− in the electrolyte after the stability test (CoFe LDH@CoFe-TA/NF) was extremely low (Fig. S12†). Furthermore, the color of the test paper (Fig. S13†) did not change, suggesting that hypochlorite was not produced during the ASO process. In addition, the corrosion polarization curves further support these findings. Fig. 4g and h show that the corrosion potential (Ecorr.) of CoFe LDH@CoFe-TA/NF is higher, and the corrosion current (Icorr.) is lower than those of CoFe LDH/NF in alkaline seawater, indicating stronger chloride corrosion resistance for CoFe LDH@CoFe-TA/NF. In addition, we tested the Co and Fe content in the electrolyte by inductively coupled plasma optical emission spectroscopy. Notably, the results (Table S2†) show that Co and Fe leaching from CoFe LDH@CoFe-TA/NF is considerably lower than that from CoFe LDH/NF, indicating that the CoFe-TA nanoparticles effectively mitigate Co and Fe leaching, thereby enhancing the stability of the electrode. In practical applications, however, the process of seawater electrolysis is accompanied by the accumulation of chloride salts due to the continuous addition of seawater. Therefore, the durability of CoFe LDH@CoFe-TA/NF is investigated with 1 M KOH + 2 M NaCl as the electrolyte. As depicted in Fig. S14,† it lasted for up to 50 h, which reveals its corrosion resistance. After the drainage test at a current density of up to 1000 mA cm−2 (Fig. S15a†), the amount of gas collected at the anode closely matched the theoretical oxygen production values (Fig. S15b†) during the ASO process, with a faradaic efficiency (FE) of approximately 99%.
To gain further insights into the mechanism for Cl−-blocking, we conducted in situ Raman spectroscopy studies to monitor the generation and evolution of surface species. Raman spectra of CoFe LDH/NF and CoFe LDH@CoFe-TA/NF in alkaline seawater (Fig. S16†) were collected at oxidation potentials ranging from 1.1 V to 1.8 V. In Fig. S16,† two distinct Raman peaks at 443.8 and 520.3 cm−1,48 associated with the Co–O bond, gradually shift to 462.5 and 558.1 cm−1, respectively, as the voltage increases, indicating the progressive formation of CoOOH.49 Compared to CoFe LDH/NF, CoFe LDH@CoFe-TA/NF displays CoOOH characteristic peaks with stronger diffraction at lower voltages, suggesting that CoFe-TA particles can more rapidly promote active site generation and facilitate the complete conversion of CoOOH. In contrast, the signal of CoOOH in CoFe LDH/NF was notably weaker at high voltage, suggesting that CoOOH was present in minimal quantities. The generated CoOOH not only exhibits selective performance for the OER, but it also effectively mitigates chloride corrosion during the oxidation of seawater. To further analyze the mechanism, we performed CV curve tests. The CoFe LDH@CoFe-TA/NF CV curves (Fig. S17†) showed the presence of anodic peaks corresponding to the change in the valence state of the Co species, which were the Co2+/Co3+ redox pairs (<1.4 V) and the Co3+/Co4+ redox pairs (>1.4 V), respectively.50 The CV curves of both CoFe LDH@CoFe-TA/NF and CoFe LDH/NF show two redox pairs, suggesting the formation and further conversion of CoOOH to Co4+ species. The lower Co3+/4+ conversion potential of CoFe LDH@CoFe-TA/NF in comparison with CoFe LDH/NF suggests that Co4+ sites are more easily formed on the surface of CoFe LDH@CoFe-TA/NF. The CoFe-TA promotes the transformation of Co3+ into the more robust acid Co4+, thereby favoring the adsorption of the hard base OH− over the soft base Cl−.51 This selectivity not only enhances the efficiency of the OER, but also effectively suppresses the undesired CER.
To more accurately assess the prospects for practical application of the catalyst, test data from self-assembled electrolyzers are provided. An alkaline seawater anion exchange membrane electrolyzer was constructed, employing Pt/C/NF as the cathode and CoFe LDH@CoFe-TA/NF as the anode. A self-assembled Pt/C/NF∥RuO2/NF electrolyzer was used for comparison. The Pt/C/NF∥CoFe LDH@CoFe-TA/NF electrolyzer achieves a remarkable efficiency, requiring only 3.0 V to reach a j of 1000 mA cm−2. In contrast, the Pt/C/NF∥RuO2/NF electrolyzer only attains 422 mA cm−2 at the same voltage (see Fig. S18a and b†). Furthermore, the Pt/C/NF∥CoFe LDH@CoFe-TA/NF configuration demonstrates stable electrolysis performance, sustaining a j of 300 mA cm−2 for over 100 hours (Fig. S18c†). This suggests that the CoFe LDH@CoFe-TA/NF electrocatalyst can facilitate the electrolysis process with greater efficiency and stability while requiring a lower energy input, highlighting its potential for industrial applications.
In summary, anchoring CoFe-TA nanoparticles on CoFe LDH/NF surfaces is an effective strategy to improve the corrosion resistance of the CoFe LDH, ensuring a stable ASO performance. Remarkably, CoFe LDH@CoFe-TA/NF requires a low η of 379 mV to achieve a j of 1000 mA cm−2 and exhibits a stable operation for 450 h in alkaline seawater. This performance can be attributed to the CoFe-TA, which promotes the formation of metal oxide/oxy(hydroxide) layers, thereby improving OER activity. Furthermore, the CoFe-TA promotes the transformation of Co3+ into the more robust acid Co4+, thereby favoring the adsorption of the hard base OH− over the soft base Cl−. Moreover, the CoFe-TA ligand network can effectively prevent the dissolution of Co and Fe ions, which further improves the stability of the catalyst during the ASO process. Overall, this work not only introduces an efficient and stable electrocatalyst for ASO, but also paves the way for utilizing cost-effective, biomass-derived substances as innovative modifiers to improve the durability of anode materials under industrial-level j.
Data availability
All data can be obtained from the corresponding authors upon request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors extend their appreciation to the Deanship of Scientific Research at King Khalid University for funding support through a large group research project under Grant No. RGP2/119/45.References
- A. Goldthau, The G20 must govern the shift to low-carbon energy, Nature, 2017, 546, 203–205 CrossRef CAS PubMed.
- S. J. Davis, N. S. Lewis, M. Shaner, S. Aggarwal, D. Arent, I. L. Azevedo, S. M. Benson, T. Bradley, J. Brouwer, Y. M. Chiang, C. T. M. Clack, A. Cohen, S. Doig, J. Edmonds, P. Fennell, C. B. Field, B. Hannegan, B. M. Hodge, M. I. Hoffert, E. Ingersoll, P. Jaramillo, K. S. Lackner, K. J. Mach, M. Mastrandrea, J. Ogden, P. F. Peterson, D. L. Sanchez, D. Sperling, J. Stagner, J. E. Trancik, C. J. Yang and K. Caldeira, Net-zero emissions energy systems, Science, 2018, 360, eaas9793 CrossRef PubMed.
- J. A. Turner, Sustainable hydrogen production, Science, 2004, 305, 972–974 CrossRef CAS PubMed.
- W. Xu, Y. Wang, J. Dang, X. Zhang, W. Li and J. Zhang, Synergistic promotion of the oxygen evolution reaction by Co and Fe dual-doping of NiS2, Inorg. Chem. Front., 2024, 11, 5866–5875 RSC.
- J. Liang, J. Li, H. Dong, Z. Li, X. He, Y. Wang, Y. Yao, Y. Ren, S. Sun, Y. Luo, D. Zheng, J. Li, Q. Liu, F. Luo, T. Wu, G. Chen, X. Sun and B. Tang, Aqueous alternating electrolysis prolongs electrode lifespans under harsh operation conditions, Nat. Commun., 2024, 15, 6208 CrossRef CAS PubMed.
- S. L. Postel, Entering an era of water scarcity: The challenges ahead, Ecol. Appl., 2000, 10, 941–948 CrossRef.
- R. Sheibani, R. Shamsi and M. Sheibani, Social consequences of Iran's water crisis, Science, 2023, 382, 164 CrossRef PubMed.
- J. Liang, Z. Li, X. He, Y. Luo, D. Zheng, Y. Wang, T. Li, B. Ying, S. Sun, Z. Cai, Q. Liu, B. Tang and X. Sun, Electrocatalytic seawater splitting: Nice designs, advanced strategies, challenges and perspectives, Mater. Today, 2023, 69, 193–235 CrossRef CAS.
- J. Guo, Y. Zheng, Z. Hu, C. Zheng, J. Mao, K. Du, M. Jaroniec, S. Z. Qiao and T. Ling, Direct seawater electrolysis by adjusting the local reaction environment of a catalyst, Nat. Energy, 2023, 8, 264–272 CAS.
- Y. Kuang, M. J. Kenney, Y. Meng, W.-H. Hung, Y. Liu, J. E. Huang, R. Prasanna, P. Li, Y. Li, L. Wang, M.-C. Lin, M. D. McGehee, X. Sun and H. Dai, Solar-driven, highly sustained splitting of seawater into hydrogen and oxygen fuels, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 6624–6629 CrossRef CAS PubMed.
- J. Liang, Z. Cai, Z. Li, Y. Yao, Y. Luo, S. Sun, D. Zheng, Q. Liu, X. Sun and B. Tang, Efficient bubble/precipitate traffic enables stable seawater reduction electrocatalysis at industrial-level current densities, Nat. Commun., 2024, 15, 2950 CrossRef CAS PubMed.
- H. You, D. Wu, D. Si, M. Cao, F. Sun, H. Zhang, H. Wang, T.-F. Liu and R. Cao, Monolayer NiIr-layered double hydroxide as a long-lived efficient oxygen evolution catalyst for seawater splitting, J. Am. Chem. Soc., 2022, 144, 9254–9263 CrossRef CAS PubMed.
- S. Dresp, F. Dionigi, M. Klingenhof and P. Strasser, Direct electrolytic splitting of seawater: Opportunities and challenges, ACS Energy Lett., 2019, 4, 933–942 CrossRef CAS.
- O. F. Aldosari, I. Hussain and Z. Malaibari, Emerging trends of electrocatalytic technologies for renewable hydrogen energy from seawater: Recent advances, challenges, and techno-feasible assessment, J. Energy Chem., 2023, 80, 658–688 CrossRef.
- G. Liu, Oxygen evolution reaction electrocatalysts for seawater splitting: A review, J. Electroanal. Chem., 2022, 923, 116805 CrossRef CAS.
- F. Zhang, L. Yu, L. Wu, D. Luo and Z. Ren, Rational design of oxygen evolution reaction catalysts for seawater electrolysis, Trends Chem., 2021, 3, 485–498 CrossRef CAS.
- X. Wang, M. Geng, S. Sun, Q. Xiang, S. Dong, K. Dong, Y. Yao, Y. Wang, Y. Yang, Y. Luo, D. Zheng, Q. Liu, J. Hu, Q. Wu, X. Sun and B. Tang, Recent advances of bifunctional electrocatalysts and electrolyzers for overall seawater splitting, J. Mater. Chem. A, 2024, 12, 634–656 RSC.
- Z. Cai, J. Liang, Z. Li, T. Yan, C. Yang, S. Sun, M. Yue, X. Liu, T. Xie, Y. Wang, T. Li, Y. Luo, D. Zheng, Q. Liu, J. Zhao, X. Sun and B. Tang, Stabilizing NiFe sites by high-dispersity of nanosized and anionic Cr species toward durable seawater oxidation, Nat. Commun., 2024, 15, 6624 CrossRef CAS PubMed.
- R. Fan, C. Liu, Z. Li, H. Huang, J. Feng, Z. Li and Z. Zou, Ultrastable electrocatalytic seawater splitting at ampere-level current density, Nat. Sustain., 2024, 7, 158–167 CrossRef.
- Y. Yao, C. Yang, S. Sun, H. Zhang, M. Geng, X. He, K. Dong, Y. Luo, D. Zheng, W. Zhuang, S. Alfaifi, A. Farouk, M. S. Hamdy, B. Tang, S. Zhu, X. Sun and W. Hu, Boosting alkaline seawater oxidation of CoFe-layered double hydroxide nanosheet array by Cr doping, Small, 2024, 20, 2307294 CrossRef CAS PubMed.
- D. Wang, L. Liu, Y. Liu, W. Luo, Y. Xie, T. Xiao, Y. Wang, Z. Song, H. Zhang and X. Wang, Hierarchical superaerophobic nanoarray electrode (FeOOH/CoFe LDH) as an efficient oxygen evolution reaction catalyst for alkaline seawater electrolysis, ACS Sustainable Chem. Eng., 2023, 11, 16479–16490 CrossRef CAS.
- F. Cheng, X. Feng, X. Chen, W. Lin, J. Rong and W. Yang, Synergistic action of Co-Fe layered double hydroxide electrocatalyst and multiple ions of sea salt for efficient seawater oxidation at near-neutral pH, Electrochim. Acta, 2017, 251, 336–343 CrossRef CAS.
- X. Wang, Z. Li, S. Sun, H. Sun, C. Yang, Z. Cai, H. Zhang, M. Yue, M. Zhang, H. Wang, Y. Yao, Q. Liu, L. Li, W. Chu, J. Hu, X. Sun and B. Tang, Oxalate anions-intercalated NiFe layered double hydroxide as a highly active and stable electrocatalyst for alkaline seawater oxidation, J. Colloid Interface Sci., 2024, 662, 596–603 CrossRef CAS PubMed.
- P. Guo, D. Liu and R. Wu, Recent progress in design strategy of anode for seawater electrolysis, Small Struct., 2023, 4, 2300192 CrossRef CAS.
- H. J. Song, H. Yoon, B. Ju, D. Y. Lee and D. W. Kim, Electrocatalytic selective oxygen evolution of carbon-coated Na2Co1−xFexP2O7 nanoparticles for alkaline seawater electrolysis, ACS Catal., 2020, 10, 702–709 CrossRef CAS.
- Z. Wang, C. Wang, L. Ye, X. Liu, L. Xin, Y. Yang, L. Wang, W. Hou, Y. Wen and T. Zhan, MnOx film-coated NiFe-LDH nanosheets on Ni foam as selective oxygen evolution electrocatalysts for alkaline seawater oxidation, Inorg. Chem., 2022, 61, 15256–15265 CrossRef CAS PubMed.
- C. Huang, Z. Wang, S. Cheng, Y. Liu, B. Deng, S. Xu, L. Yu and Y. Yu, Challenges and strategies of chlorine inhibition in anode systems for seawater electrolysis, Sci. China: Chem., 2024, 67, 3198–3208 CrossRef CAS.
- Z. Li, Y. Yao, S. Sun, J. Liang, S. Hong, H. Zhang, C. Yang, X. Zhang, Z. Cai, J. Li, Y. Ren, Y. Luo, D. Zheng, X. He, Q. Liu, Y. Wang, F. Gong, X. Sun and B. Tang, Carbon oxyanion self-transformation on NiFe oxalates enables long-term ampere-level current density seawater oxidation, Angew. Chem., Int. Ed., 2024, 63, e202316522 CrossRef CAS PubMed.
- Y. Xin, Q. Hua, C. Li, H. Zhu, L. Gao, X. Ren, P. Yang and A. Liu, Enhancing electrochemical performance and corrosion resistance of nickel-based catalysts in seawater electrolysis: Focusing on OER and HER, J. Mater. Chem. A, 2024, 12, 23147–23178 RSC.
- T. U. Haq and Y. Haik, Strategies of anode design for seawater electrolysis: Recent development and future perspective, Small Sci., 2022, 2, 2200030 CrossRef CAS.
- L. Yu, Q. Zhu, S. Song, B. McElhenny, D. Wang, C. Wu, Z. Qin, J. Bao, Y. Yu, S. Chen and Z. Ren, Non-noble metal-nitride based electrocatalysts for high-performance alkaline seawater electrolysis, Nat. Commun., 2019, 10, 5106 CrossRef PubMed.
- R. Fan, X. Zhang, N. Yu, F. Wang, H. Zhao, X. Liu, Q. Lv, D. Liu, Y. Chai and B. Dong, Rapid “self-healing” behavior induced by chloride anions to renew the Fe-Ni(oxy)hydroxide surface for long-term alkaline seawater electrolysis, Inorg. Chem. Front., 2022, 9, 4216–4224 RSC.
- Y. Wang, S. Chen, S. Zhao, Q. Chen and J. Zhang, Interfacial coordination assembly of tannic acid with metal ions on three-dimensional nickel hydroxide nanowalls for efficient water splitting, J. Mater. Chem. A, 2020, 8, 15845–15852 RSC.
- Y. Zhou, R. Fan, Y. Cao, H. Wang, B. Dong, H. Zhao, F. Wang, J. Yu and Y. Chai, Oriented and robust anchoring of Fe via anodic interfacial coordination assembly on ultrathin Co hydroxides for efficient water oxidation, Nanoscale, 2021, 13, 13463–13472 RSC.
- X. Song, Y. Zhao, F. Zhang, J. Ni, Z. Zhang, Z. Tan, X. Wang and Y. Li, Coupling plant polyphenol coordination assembly with Co(OH)2 to enhance electrocatalytic performance towards oxygen evolution reaction, Nanomaterials, 2022, 12, 3972 CrossRef CAS PubMed.
- X. Sun, R. Wang, Q. Wang and K. Ostrikov, Interface-engineered urchin-like CoFe-layered double hydroxide for high-efficiency electrocatalytic oxygen evolution, Inorg. Chem. Front., 2024, 11, 1458–1471 RSC.
- W. Liu, K. Jiang, Y. Hu, Q. Li, Y. Deng, J. Bao and Y. Lei, Zr-doped CoFe-layered double hydroxides for highly efficient seawater electrolysis, J. Colloid Interface Sci., 2021, 604, 767–775 CrossRef CAS PubMed.
- W. Zhang, D. Zheng, H. Wang and S. Yao, Water splitting of nickel foam-supported CuCo2S4@CoFe-LDH efficient bifunctional catalysts, J. Solid State Chem., 2024, 333, 124600 CrossRef CAS.
- W. Zhu, X. Wang, Y. Zhu, L. Fang, C. Yao, X. Song, H. Chen, X. Wang and G. Zhu, Amorphous Fe/Co-based tannic acid salts as robust oxygen evolution pre-catalysts, New J. Chem., 2023, 47, 19087–19095 RSC.
- Y. Hu, W. Liu, K. Jiang, L. Xu, M. Guan, J. Bao, H. Ji and H. Li, Constructing a CeO2−x@CoFe-layered double hydroxide heterostructure as an improved electrocatalyst for highly efficient water oxidation, Inorg. Chem. Front., 2020, 7, 4461–4468 RSC.
- G. Zhou, Y. Ma, C. Gu, J. Yang, H. Pang, J. Li, L. Xu and Y. Tang, Fe incorporation-induced electronic modification of Co-tannic acid complex nanoflowers for high-performance water oxidation, Inorg. Chem. Front., 2022, 9, 1091–1099 RSC.
- X. Chen, Q. Wang, Y. Cheng, H. Xing, J. Li, X. Zhu, L. Ma, Y. Li and D. Liu, S-doping triggers redox reactivities of both iron and lattice oxygen in FeOOH for low-cost and high-performance water oxidation, Adv. Funct. Mater., 2022, 32, 2112674 CrossRef CAS.
- H. Jia, N. Yao, Y. Jin, L. Wu, J. Zhu and W. Luo, Stabilizing atomic Ru species in conjugated sp2 carbon-linked covalent organic framework for acidic water oxidation, Nat. Commun., 2024, 15, 5419 CrossRef CAS PubMed.
- R. Chen, S. Hung, D. Zhou, J. Gao, C. Yang, H. Tao, H. B. Yang, L. Zhang, L. Zhang, Q. Xiong, H. M. Chen and B. Liu, Layered structure causes bulk NiFe layered double hydroxide unstable in alkaline oxygen evolution reaction, Adv. Mater., 2019, 31, 1903909 CrossRef CAS PubMed.
- F. Lyu, Q. Wang, S. M. Choi and Y. Yin, Noble-metal-free electrocatalysts for oxygen evolution, Small, 2019, 15, 1804201 CrossRef PubMed.
- Y. Duan, Z. Yu, S. Hu, X. Zheng, C. Zhang, H. Ding, B. Hu, Q. Fu, Z. Yu, X. Zheng, J. Zhu, M. Gao and S. Yu, Scaled-up synthesis of amorphous NiFeMo oxides and their rapid surface reconstruction for superior oxygen evolution catalysis, Angew. Chem., Int. Ed., 2019, 58, 15772–15777 CrossRef CAS PubMed.
- M. Wang, C. Dong, Y. Huang, Y. Li and S. Shen, Electronic structure evolution in tricomponent metal phosphides with reduced activation energy for efficient electrocatalytic oxygen evolution, Small, 2018, 14, 1801756 CrossRef PubMed.
- D.-H. Kim and Y.-K. Lee, Understanding highly active and durable Fe-decorated Co(OH)2 catalysts in alkaline oxygen evolution reaction by in situ XANES studies, Chem. Eng. J., 2024, 490, 151701 CrossRef CAS.
- Y. Deng, Y. Cao, Y. Xia, X. Xi, Y. Wang, W. Jiang, D. Yang, A. Dong and T. Li, Self-templated synthesis of CoFeP@C cage-in-cage superlattices for enhanced electrocatalytic water splitting, Adv. Energy Mater., 2022, 12, 2202394 CrossRef CAS.
- A. Moysiadou, S. Lee, C.-S. Hsu, H. M. Chen and X. Hu, Mechanism of oxygen evolution catalyzed by cobalt oxyhydroxide: Cobalt superoxide species as a key intermediate and dioxygen release as a rate-determining step, J. Am. Chem. Soc., 2020, 142, 11901–11914 CrossRef CAS PubMed.
- X. Wang, H. Hu, J. Song, J. Ma, H. Du, J. J. Wang, M. Wang, W. Chen, Y. Zhou, J. Wang, M. Yang and L. Zhang, Surface anticorrosion engineering by polyphosphate oxyanions for durable seawater oxidation, Adv. Energy Mater., 2024 DOI:10.1002/aenm.202402883.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section and supplementary figures. See DOI: https://doi.org/10.1039/d4qi02404d |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2025 |